
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural guidelines for stabilization of α-helical coiled coils via PEG stapling†
Qiang
Xiao
a,
Zachary B.
Jones
a,
Samantha C.
Hatfield
a,
Dallin S.
Ashton
a,
Nicholas A.
Dalley
a,
Cody D.
Dyer
a,
Judah L.
Evangelista
b and
Joshua L.
Price
 *a
*a
aDepartment of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, USA. E-mail: jlprice@chem.byu.edu
bDepartment of Biochemistry, University of Utah School of Medicine, Salt Lake City, Utah 84112, USA
First published on 26th July 2022
Abstract
Macrocyclization or stapling is one of the most well-known and generally applicable strategies for enhancing peptide/protein conformational stability and target binding affinity. However, there are limited structure- or sequence-based guidelines for the incorporation of optimal interhelical staples within coiled coils: the location and length of an interhelical staple is either arbitrarily chosen or requires significant optimization. Here we explore the impact of interhelical PEG stapling on the conformational stability and proteolytic resistance of a model disulfide-bound heterodimeric coiled coil. We demonstrate that (1) interhelical PEG staples are more stabilizing when placed farther from an existing disulfide crosslink; (2) e/g′ staples are more stabilizing than f/b′ or b/c′ staples; (3) PEG staples between different positions have different optimal staple lengths; (4) PEG stapling tolerates variation in the structure of the PEG linker and in the mode of conjugation; and (5) the guidelines developed here enable the rational design of a stabilized PEG-stapled HER-2 affibody with enhanced conformational stability and proteolytic resistance.
1. Introduction
Macrocyclization or stapling is one of the most well-known and generally applicable strategies for enhancing peptide/protein conformational stability and target binding affinity.1–4 Staples preorganize the peptide/protein into a conformation that resembles the folded or bound state by crosslinking two groups that are close to each other in the folded or bound conformation but not in the unfolded or unbound conformation. This covalent constraint prepays part of the energetic cost of folding/binding via a combination of entropic and enthalpic effects.Advances in chemoselective biorthogonal reactions,5–8 chemical protein synthesis,9–11 and expression of proteins with unnatural amino acids12–14 have enabled peptide/protein stapling via multiple site-specific strategies. Among the most important of these are thiol alkyl-15–19 or arylation,20 olefin metathesis,21–26 and azide/alkyne cycloaddition.27–33 We recently showed that stapling via olefin metathesis vs. the copper-catalyzed azide–alkyne cycloaddition (CuAAC) provide similar increases in the conformational stability of WW, a β-sheet miniprotein derived from the WW domain of the human protein Pin1.34,35 We observed similar levels of stabilization for staples comprised of discrete polyethylene glycol oligomers (i.e., PEG staples) vs. conventional hydrocarbon staples. The most important determinant of PEG-staple-based stabilization in WW is that the two crosslinked groups be far apart in primary sequence but close together in the folded tertiary structure. Presumably this arrangement provides optimal restriction of the conformational freedom of the unfolded ensemble without substantially perturbing the folded state; the resulting destabilization of the unfolded state relative to the folded state provides a more favorable free energy of folding.
Early stapling efforts focused on stabilizing α-helical secondary structure in short peptides.15,21,22,36–43 Others have expanded this approach more recently to α-helical coiled-coil tertiary/quaternary structure. Coiled-coil primary sequence consists of a seven-residue repeating unit in which non-polar residues occupy the a- and d-positions within an abcdefg heptad; polar and/or charged residue occupy the other positions. Peptides whose sequences follow these patterns are globally amphipathic in an α-helical conformation, with non-polar a- and d-residues aligned along the same face of the helix. Burial of these a- and d-residues via “knobs-into-holes” packing at the interhelical interface provides the major driving force for coiled-coil self-association. The e- and g-positions flank the interhelical interface and often engage in complementary electrostatic interactions (i.e., salt bridges). The identify of these a-, d-, e-, and g-residues can control oligomerization state (dimer, trimer, tetramer, etc.); homo- vs. heteroassociation; and helical orientation (parallel vs. antiparallel).44–47
Arora and coworkers recently substituted a bis-triazole staple for an interhelical e/e′ salt bridge within a designed antiparallel coiled-coil heterodimer comprised of nine-residue subunits.48 They similarly substituted a bis-thioether or bis-triazole staple for an interhelical e/g′ salt bridge within related parallel coiled-coil heterodimers comprised of 10- or 14-residue subunits.49 These staples enabled a surprising amount of helicity in such short peptides and the resulting stabilized coiled coils were subsequently useful as scaffolds for rational design of protein–protein interaction inhibitors. Liu, Jiang, and coworkers substituted each of three identical interhelical e/g′ Glu–Lys salt bridges with an interhelical Glu–Lys isopeptide staple within a trimeric coiled coil derived from the N-terminal domain of HIV-1 gp41.50 The resulting stapled variant was resistant to proteolysis, aggregation, and thermal denaturation. However, in each of these cases, the precise energetic contribution of the staple to coiled-coil conformational stability was not explored in detail.
Karlström and coworkers used interhelical thioether staples between a chloroacetamide-modified Lys and a nearby Cys to stabilize three proteins that adopt similar monomeric helix-bundle tertiary structures comprised of three α-helices: the albumin binding domain (ABD) of streptococcal protein G,51 a HER2 affibody (HER2a)52 and an EGFR affibody (EGFRa).53 Within each protein, they identified a location where a Cys-Lys staple substantially increases melting temperature (by 5–10 °C). However, Cys-Lys staples at other locations were strongly destabilizing, for reasons that remain unclear. Grossmann and coworkers54 used a novel tris-electrophile to cross-link non-native Cys residues within the helix-bundle KIX domain. The resulting bicyclic KIX variant bound its partner MLL with similar affinity as its non-stapled counterpart, but had a much higher melting temperature, indicating substantial increase to conformational stability.
Despite these advances, there are limited structure- or sequence-based guidelines for the incorporation of optimal interhelical staples within coiled coils; the location and length of an interhelical staple is either arbitrarily chosen or requires significant optimization.48 Here we explore the impact of interhelical PEG staples of different lengths and at various solvent-exposed locations on conformational stability and resistance to proteolysis within a model disulfide-bound heterodimeric coiled coil. We demonstrate staple-based stabilization depends strongly on the location of the PEG staple, and that stapling tolerates substantial variations in the structure of the PEG linker with mono- and bis-triazole linkages providing comparable levels of stabilization. Finally, we use the guidelines developed here to generate a stabilized PEG-stapled variant of a HER-2 affibody.
2. Results and discussion
We recently explored the impact of interhelical PEG stapling on the conformational stability of a previously characterized coiled-coil tertiary structure in which acidic peptide A and basic peptide B are connected via a disulfide bond to form monomeric two-helix parallel coiled coil dA/B (Fig. 1).55 We prepared non-stapled disulfide-bound variant d27e/29g′-z4x by (1) replacing e-position Glu27 in subunit A with z4, an Asn derivative in which the side-chain amide nitrogen has been modified with an azide-terminated PEG oligomer comprised of four ethylene oxide units; and (2) replacing g-position Lys29′ in subunit B with propargylglycine x. (Fig. 2A and B). We then prepared PEG-stapled variant sd27e/29g′-z4x from d27e/29g′-z4xvia CuAAC, which connects the azide of z4 to the alkyne of xvia a triazole linkage (Fig. 2C and D).35 Our approach differs from that of Arora and coworkers48,49 in the structure of the z4x staple and its placement between e- and g′-positions that are not involved in a salt bridge with each other: in the structure of parent compound dA/B, Glu27 is involved in a salt bridge with Lys22′, whereas Lys29′ is involved in a salt bridge with Glu34.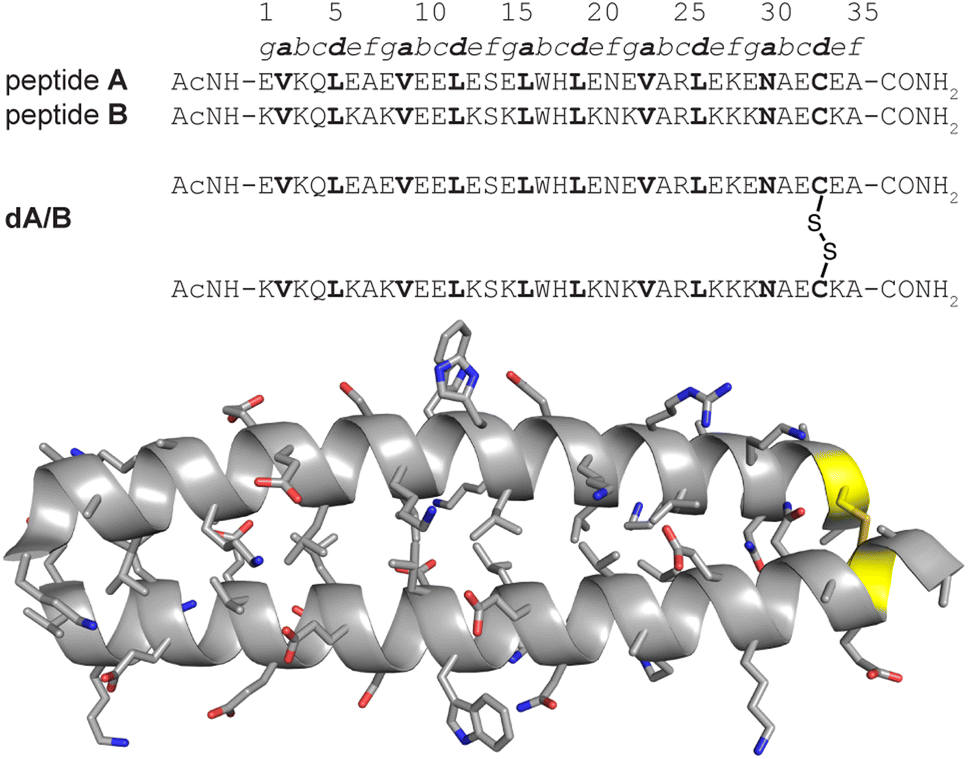 | ||
| Fig. 1 Sequences of acidic monomer A; basic monomer B; and disulfide-bound heterodimer dA/B. Also shown is the ribbon diagram of dA/B (PDB ID: 1KD9) with side chains shown as sticks and Cys33–Cys33′ disulfide highlighted in yellow. | ||
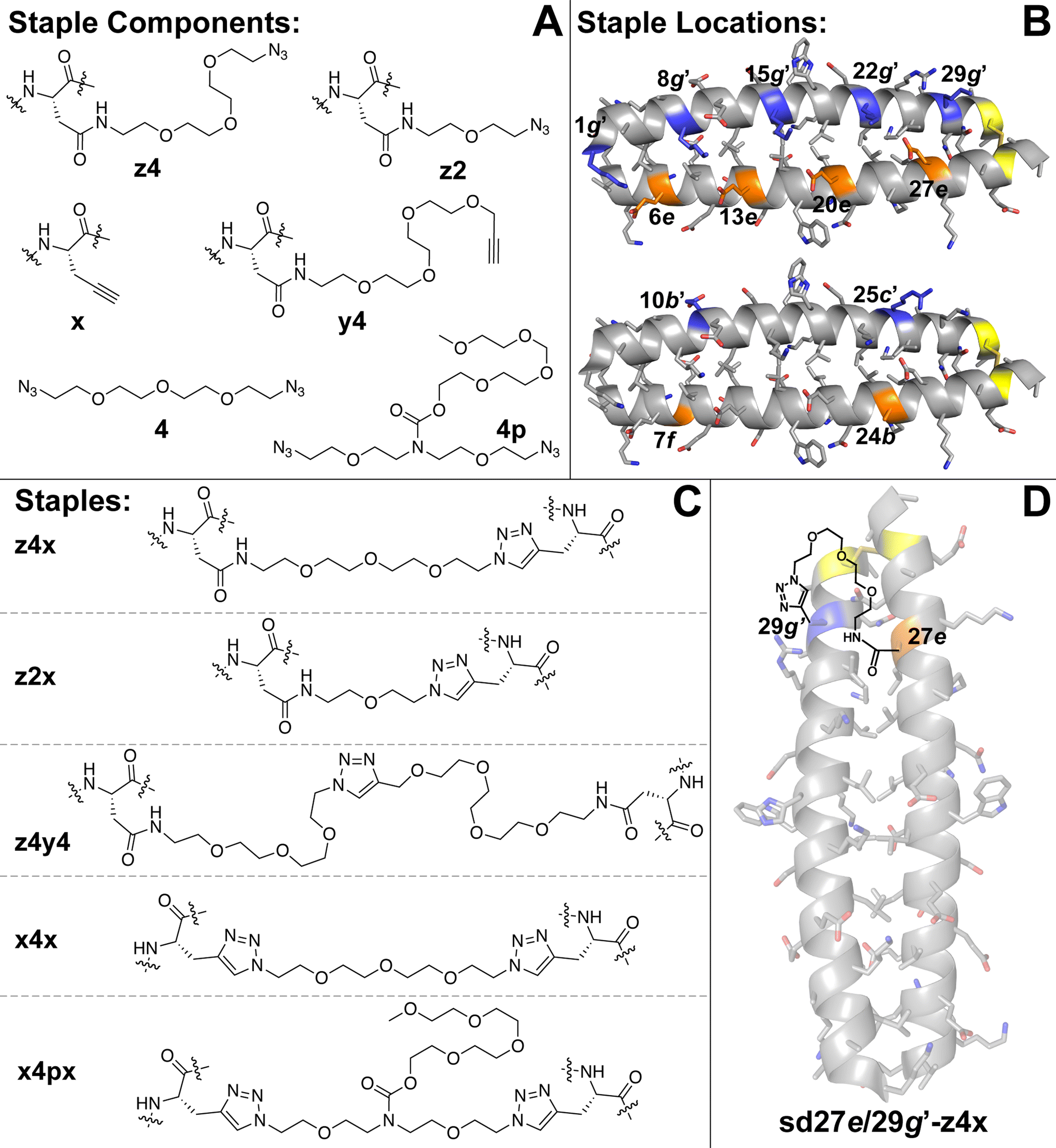 | ||
| Fig. 2 (A) Structures of staple components, including three Asn derivatives in which the side-chain amide nitrogen has been modified with azide-terminated four- (z4) or two-unit PEGs or with an alkyne-terminated four-unit PEG (y4). Also shown is propargylglycine (x), four-unit bis-azido PEG (4) and four-unit bis-azido PEG with branching PEG carbamate (4p). (B) Locations within subunits A and B where we incorporated staple components are highlighted in orange and blue, respectively, on the ribbon diagram of coiled coil dA/B (PDB: 1KD9) and are labelled according to their numbered heptad position within the sequence. (C) Structures of staples z4x, z2x, z4y4, x4x, and x4px, formed from via CuAAC from the indicated components. (D) Structure of stapled disulfide-bound variant sd27e/29g′-z4x. | ||
We used the z4x staple because modelling suggested that it would readily span the distance between positions 27e and 29g′ (9.2 Å, based on the distance between side-chain centers of mass at corresponding positions in the crystal structure of dA/B; PDB ID 1KD9). Briefly, we generated a model for the z4x staple in GaussView 6.0 based on the structure shown in Fig. 2C, but with a single N′-acetyl amino acid N-methyl amide on either end of the staple. We then optimized this model structure in Gaussian 16 using density functional theory (APFD) calculations with the 6-31G+d,p basis set (see electronic ESI† for details). We used the distance between the β-carbons on either end of the staple as an estimate of the distance that could be comfortably spanned by the z4x staple. The calculated length of the z4x staple is 18.5 Å (Table 1), which, we hypothesized, would be more than sufficient to span the 9.2 Å between positions 27e and 29g.
| Protein | T m (°C) | Impact of stapling | Distance between staple positions (Å) | Calculated staple length (Å) | ||
|---|---|---|---|---|---|---|
| ΔΔG (kcal mol−1) | ΔΔH (kcal mol−1) | −TΔΔS (kcal mol−1) | ||||
| a Distance between staple positions for each variant were calculated by measuring the distance between the centers of mass of the corresponding side chains in the crystal structure of the parent disulfide-bound coiled-coil heterodimer dA/B (PDB ID: 1KD9). Calculated staple length measured from β-carbon to β-carbon within model staple structures (see ESI) optimized in Gaussian 16 using density functional theory APFD and the 6-31G+d,p basis set. ΔΔG, ΔΔH, and −TΔΔS values for each variant are given ± std. error in kcal mol−1 at the melting temperature of its corresponding non-stapled counterpart at 15 μM protein concentration in 20 mM sodium phosphate buffer (pH 7)+4.0 M GdnHCl, except for affibody a, non-stapled a8/42-xx, and stapled sa8/42-x4x, which were characterized without denaturant. | ||||||
| d27e/29g′-z4x | 41.1 ± 0.2 | |||||
| sd27e/29g′-z4x | 48.2 ± 0.1 | −0.65 ± 0.02 | 1.3 ± 0.6 | −1.9 ± 0.6 | 9.2 | 18.5 |
| d20e/22g′-z4x | 41.8 ± 0.2 | |||||
| sd20e/22g′-z4x | 54.3 ± 0.1 | −1.09 ± 0.02 | 1.6 ± 0.6 | −2.7 ± 0.6 | 9.2 | 18.5 |
| d13e/15g′-z4x | 42.4 ± 0.1 | |||||
| sd13e/15g′-z4x | 57.7 ± 0.1 | −1.33 ± 0.02 | 2.1 ± 0.5 | −3.4 ± 0.5 | 9.7 | 18.5 |
| d6e/8g′-z4x | 39.5 ± 0.2 | |||||
| sd6e/8g′-z4x | 69.1 ± 0.1 | −2.53 ± 0.04 | −1.9 ± 0.6 | −0.7 ± 0.6 | 10.6 | 18.5 |
| d27e/22g′-z4x | 43.2 ± 0.1 | |||||
| sd27e/22g′-z4x | 63.6 ± 0.1 | 12.01 ± 0.02 | −2.7 ± 0.5 | 0.6 ± 0.5 | 6.3 | 18.5 |
| d6e/1g′-z4x | 45.0 ± 0.1 | |||||
| sd6e/1g′-z4x | 73.7 ± 0.2 | −2.30 ± 0.04 | 1.7 ± 0.6 | −4.0 ± 0.6 | 6.0 | 18.5 |
| d24b/25c′-z4x | 43.4 ± 0.1 | |||||
| sd24b/25c′-z4x | 33.0 ± 0.2 | 0.65 ± 0.02 | 8.1 ± 0.5 | −7.4 ± 0.5 | 14.3 | 18.5 |
| d7f/10b′-z4x | 42.6 ± 0.2 | |||||
| sd7f/10b′-z4x | 51.4 ± 0.3 | −0.61 ± 0.03 | 8.8 ± 0.9 | −9.4 ± 0.9 | 15.6 | 18.5 |
| d24b/25c′-z4y4 | 46.5 ± 0.1 | |||||
| sd24b/25c′-z4y4 | 44.5 ± 0.2 | 0.17 ± 0.02 | 0.8 ± 0.6 | −0.6 ± 0.6 | 14.3 | 28.5 |
| d7f/10b′-z4y4 | 43.7 ± 0.2 | |||||
| sd7f/10b′-z4y4 | 54.2 ± 0.2 | −0.68 ± 0.01 | 6.6 ± 0.6 | −7.2 ± 0.6 | 15.6 | 28.5 |
| d27e/29g′-z2x | 38.8 ± 0.2 | |||||
| sd27e/29g′-z2x | 33.7 ± 0.1 | 0.31 ± 0.01 | 6.1 ± 0.4 | −5.8 ± 0.4 | 9.2 | 8.1 |
| d27e/22g′-z2x | 43.4 ± 0.1 | |||||
| sd27e/22g′-z2x | 64.9 ± 0.3 | −2.04 ± 0.03 | −0.5 ± 0.6 | −1.5 ± 0.6 | 6.3 | 8.1 |
| d27e/29g′-xx | 39.8 ± 0.2 | |||||
| sd27e/29g′-x4x | 52.9 ± 0.2 | −1.08 ± 0.03 | −2.0 ± 0.5 | 0.9 ± 0.5 | 9.2 | 19.3 |
| sd27e/29g′-x4px | 53.4 ± 0.1 | −1.21 ± 0.02 | −4.1 ± 0.5 | 2.9 ± 0.5 | ||
| affibody a | 66.4 ± 0.2 | |||||
| a8/42-xx | 60.9 ± 0.1 | 0.64 ± 0.02 | −2.1 ± 1.1 | 2.8 ± 1.1 | 7.4 | 19.3 |
| sa8/42-x4x | 76.1 ± 0.1 | −1.09 ± 0.03 | 3.5 ± 1.3 | −4.6 ± 1.3 | ||
Variable temperature circular dichroism (CD) experiments in 20 mM sodium phosphate buffer (pH 7) with 4 M guanidinium chloride revealed that stapled sd27e/29g′-z4x is −0.65 ± 0.02 kcal mol−1 more stable than its non-stapled counterpart due to a favourable entropic effect (−TΔΔS = −1.9 ± 0.06 kcal mol−1) offset by an unfavourable enthalpic effect (ΔΔH = 1.3 ± 0.6 kcal mol−1). We used denaturant because we were otherwise unable to observe complete or nearly complete thermal unfolding transitions for these and other variants. These observations are consistent with the expectation that stapling limits the conformational freedom of the unfolded ensemble, thereby decreasing the entropic cost of folding.
2.1 Location of staple relative to existing disulfide bridge
The z4x PEG staple in variant sd27e/29g′-z4x is close to the Cys33–Cys33′ disulfide in both primary sequence and folded tertiary structure (Fig. 2D). We wondered whether the z4x PEG staple might provide superior stabilization between analogous non-salt-bridged e- and g′-positions farther away from the disulfide (e.g., 20e/22g′; 13e/15g′; or 6e/8g′). To test this hypothesis, we prepared non-stapled disulfide-bound variants d20e/22g′-z4x, d13e/15g′-z4x, and d6e/8g′-z4x (in which z4 occupies e-positions 20, 13, and 6, respectively, whereas x occupies g-positions 22′, 15′, and 8′, respectively) along with their PEG-stapled counterparts sd20e/22g′-z4x, sd13e/15g′-z4x, and sd6e/8g′-z4x. Staple components, locations, and structures are shown in Fig. 2A–C; see the ESI† for a more detailed representation of the sequence and structure of each variant. In each case, modelling suggested that the z4x staple would be more than sufficient for spanning the distance between staple positions (Table 1). As before, we assessed the conformational stability of the PEG-stapled variants relative to their non-stapled counterparts using variable temperature CD experiments in 20 mM sodium phosphate buffer (pH 7) with 4 M guanidinium chloride (Table 1).The stabilizing impact of the z4x PEG staple increases linearly with increasing distance from the Cys33–Cys33 disulfide: sd20e/22g′-z4x, sd13e/15g′-z4x, and sd6e/8g′-z4x are −1.09 ± 0.02, −1.33 ± 0.02, and −2.53 ± 0.04 kcal mol−1 more stable, respectively, than their non-stapled counterparts. These observations are congruent with our previous studies35 in the context of the WW and SH3 domains: a PEG staple yields the greatest energetic benefit when placed between positions close in tertiary structure, but distant from each other in primary sequence or (in this case) from the nearest disulfide crosslink. Consistent with our previous observations for the z4x staple at 27e/29g′, ΔΔG values associated with the z4x staples at 20e/22g′ and at 13e/15g′ come from favourable entropic terms, which become more favourable with increasing distance from the Cys33–Cys33′ disulfide bond (Table 1). Interestingly, the ΔΔG value for the z4x staple at 6e/8g′ has the smallest favourable entropic term of the series (−TΔΔS = −0.7 ± 0.6 kcal mol−1), along with a substantial favourable enthalpic term (ΔΔH = −1.9 ± 0.6 kcal mol−1). Interpreting these observations can be difficult due to entropy/enthalpy compensation; however, it is possible that the long-range covalent constraint provided by the z4x staple at 6e/8g′ strengthens existing enthalpically favourable interactions within the coiled coil (e.g., intrahelical i-to-i + 4 hydrogen bonding; interhelical salt bridges).
2.2 Staples between salt-bridged vs. non-salt-bridged e and g positions
We next wondered whether z4x PEG staples (18.5 Å long) might provide similar levels of stabilization at salt-bridged positions 27e/22g′ or 6e/1g′ (6.3 and 6.0 Å apart, respectively) as we observed above for non-salt-bridged positions 27e/29g′ or 6e/8g′. We explored this possibility by preparing non-stapled variants d27e/22g′-z4x and d6e/1g′-z4x (in which z4 occupies e-positions 27 and 6, whereas x occupies g-positions 22′ and 1g′ respectively), and their stapled counterparts sd27e/22g′-z4x and sd6e/1g′-z4x (Fig. 2B–D). Stapled variant sd27e/22g′-z4x is −1.93 ± 0.02 kcal mol−1 more stable than its non-stapled counterpart (Table 1), an effect driven unexpectedly by a favourable enthalpic term (ΔΔH = −3.4 ± 0.5 kcal mol−1) offset by an unfavourable entropic term (−TΔΔS = 1.5 ± 0.5 kcal mol−1). In contrast, sd6e/1g′-z4x is −2.30 ± 0.04 kcal mol−1 more stable than its non-stapled counterpart due to a favourable entropic term (−TΔΔS = −4.0 ± 0.6 kcal mol−1), offset by an unfavourable enthalpic term (ΔΔH = 1.7 ± 0.6 kcal mol−1). The ΔΔG value for the z4x staple at 27e/22g′ is much more favourable than we observed previously at 27e/29g′ (−1.93 ± 0.03 kcal mol−1vs. −0.65 kcal mol−1). In contrast, the ΔΔG values for the z4x staples at 6e/1g′ vs. 6e/8g′ are similar (−2.30 ± 0.05 kcal mol−1vs. −2.53 ± 0.04 kcal mol−1). Placing the z4x staple at salt-bridged vs. non-salt-bridged e- and g′-positions appears to matter more at locations closer to the Cys33–Cys33′ disulfide. Alternatively, it is possible that we have reached an upper limit for staple-based stabilization of a disulfide-bound coiled-coil heterodimer.2.3 Impact of f/b′ vs. b/c′ PEG Staples
Next, we wondered whether z4x staples between other solvent exposed positions (i.e., b, c, and f, Fig. 2B) might provide similar levels of stabilization as we observed for the e/g′ staples described above. Residues at b- and c′-positions or at f- and b′-positions are generally farther apart in space than e- and g′-positions and are oriented away from instead of toward the interhelical interface. We wondered whether these differences might attenuate the impact of stapling. Accordingly, we prepared variants d7f/10b′-z4x and d24b/25c′-z4x (in which z4 occupies position 7f and position 24b, whereas x occupies position 10b′ and position 25c′, respectively) along with their stapled counterparts sd7f/10b′-z4x and sd24b/25c′-z4x (Fig. 2B and C). In both cases, we expected the distance between staple positions (15.6 Å for 7f/10b′; 14.3 Å for 24b/25c′) to be near the upper limit of what can be comfortably spanned by the z4x staple.Stapled variant sd7f/10b′-z4x is −0.65 ± 0.03 kcal mol−1 more stable than its non-stapled counterpart (Table 1). This is a much smaller level of stabilization than we observed for the z4x staples at 6e/8g′ or 6e/1g′, which are similarly distant from the disulfide bridge, possibly indicating that z4x staples between f- and b′-positions are less stabilizing than between e- and g′-positions. Stapled variant sd24b/25c′-z4x is 0.74 ± 0.02 kcal mol−1 less stable than its non-stapled counterpart (Table 1), a substantial destabilization that contrasts with the stabilizing impact of the z4x staples at 27e/29g′ or 27e/22g′, which are similarly distant from the Cys33–Cys33′ disulfide.
2.4 Impact of changes in staple length/structure
We wondered whether poor performance of the z4x staples at 7f/10b′ and at 24b/25c′ might reflect the increased distances between f- and b′-positions (15.6 Å) or between b- and c′-positions (14.3 Å) relative to e- and g′-positions (6.3–9.2 Å). To test this hypothesis, we prepared variants d7f/10b′-z4y4 and d24b/25c′-z4y4 (in which z4 occupies positions 7f or 24b, whereas y4 occupies positions 10b′ and 25c′, respectively) and their stapled counterparts sd7f/10b′-z4y4 and sd24b/25c′-z4y4 (Fig. 2B and C). The z4y4 staple has eight ethylene oxide units, whereas the z4x staple has only four; indeed, modelling suggests that the z4y4 staple can comfortably span a much longer distance (28.5 Å) than the z4x staple (see ESI†). The impact of the eight-unit z4y4 staple on the stability of variant sd7f/10b′-z4y4 relative to its non-stapled counterpart (ΔΔG = −0.68 ± 0.01 kcal mol−1) is indistinguishable from that of the four-unit z4x staple at the same positions. This observation suggests that if a staple is already sufficiently long to span the distance between positions, additional increases in length will not improve staple-based stabilization. In contrast, variant sd24b/25c′-z4y4 is 0.17 ± 0.02 kcal mol−1 less stable than its non-stapled counterpart, a smaller increment of destabilization than we observed above for the four-unit z4x staple at 24b/25c′. This observation suggests that stapling between some positions is intrinsically destabilizing in a way that increased staple length cannot compensate for. In any case, the z4y4 staples at 7f/10b and at 24b/25c′ provided inferior stabilization relative to the z4x staples at the e/g′-positions described above.We wondered whether we might enhance the favourable impact of stapling between at 27e/29g′ or at 27e/22g′ by truncating the z4x staple from four ethylene oxide units to two. Accordingly, we prepared variants d27e/29g′-z2x and d27e/29g′-z2x (in which two-unit azide-terminated Asn derivative z2 occupies position 27e, whereas x occupies positions 29g′ vs. 22g′, respectively), and their stapled counterparts sd27e/29g′-z2x and sd27e/29g′-z2x (Fig. 2B and C). The impact of the z2x staple at 27e/29g′ (ΔΔG = 0.31 ± 0.01 kcal mol−1) is much less favourable than that of the z4x staple (ΔΔG = −0.65 ± 0.02 kcal mol−1). This effect is driven by an unfavourable enthalpic term (ΔΔH = 6.1 ± 0.4 kcal mol−1), potentially indicating that the two-unit staple disrupts favourable interactions or introduces unfavourable contacts within the coiled coil. Presumably, this reflects the longer distance between 27e/29g′ (9.2 Å) relative to the length of the shorter z2x staple (calculated length = 8.1 Å; Table 1). In contrast, the impact of the z2x staple at 27e/22g′ (ΔΔG = −1.95 ± 0.04 kcal mol−1) is indistinguishable from that of the z4x staple (ΔΔG = −1.93 ± 0.03 kcal mol−1), an effect driven similarly by enthalpy (ΔΔHf = −1.5 ± 0.6 kcal mol−1), with a nominally favourable entropic contribution (−TΔΔSf = −0.4 ± 0.6 kcal mol−1). This observation is consistent with the shorter distance between 27e/22g′ (6.3 Å) relative to the length of the z2x staple (8.1 Å). More generally, it is possible that salt-bridged e/g′-positions are more tolerant of shorter staples than are non-salt-bridged e/g′-positions, due to the closer proximity of the salt-bridged e/g′-positions.
In preparing the variants above, we incorporated the z4, z2, y4, and x staple components at the indicated positions by solid phase peptide synthesis, which becomes progressively less efficient for larger proteins. In contrast, staple component x (i.e., propargylglycine) can be incorporated into expressed proteins as a methionine surrogate.56 We envisioned that stapling of two x residues with a bis-azido PEG might be easier to implement in larger proteins than the z4x staple. However, we wondered whether such a staple would have a similar impact on coiled-coil conformational stability as we observed for the z4x staples. To explore this possibility, we prepared variant d27e/29g′-xx (in which x occupies both positions 27e and 29g′). We then reacted d27e/29g′-xx with four-unit bis-azido PEG 4 (Fig. 2A) via CuAAC to give stapled variant sd27e/29g′-x4x (Fig. 2C; calculated staple length = 19.3 Å). Variant sd27e/29g′-x4x is −1.08 ± 0.03 kcal mol−1 more stable than its non-stapled counterpart, a modestly larger increment of stabilization than we observed above for the four-unit z4x staple between the same positions. Interestingly, the stabilizing impact of the x4x staple comes from a favourable enthalpic term, offset by a nominally unfavourable entropic term, suggesting that the precise origins of staple-based stabilization might be different for the four-unit z4xvs.x4x staples.
The modular nature of the x4x staple allows us to consider attaching additional groups to the staple, thereby combining the benefits of PEG stapling with additional functionalities (e.g., longer PEG chains; fluorophores, etc.). We explored this possibility by preparing branched PEG bis-azide 4p, in which the central oxygen of the four-unit PEG bis-azide has been replaced with nitrogen, which was subsequently conjugated to an additional linear four-unit PEG via a carbamate linkage (Fig. 2A). Stapling of d27e/29g′-xx with PEG bis-azide 4pvia CuAAC resulted in variant sd27e/29g′-x4px. Variant sd27e/29g′-x4px is −1.21 ± 0.02 kcal mol−1 more stable than non-stapled d27e/29g′-xx. The observation that staples derived from the branched vs. linear PEG bis-azides provide similar benefits to conformational stability suggests that one can incorporate additional functional groups within the PEG staple without disrupting staple-based stabilization.
2.5 Impact of PEG stapling on proteolytic resistance
We previously showed that PEG staple-based increases in WW conformational stability are associated with increased levels of protection from proteolysis.35 We wondered whether this would be true for the PEG-stapled coiled-coil variants described above. We explored this possibility by exposing 15 μM solutions of stapled variants sd27e/29g′-z4x, sd6e/8g′-z4x, sd27e/22g′-z4x, sd24b/25c′-z4x, sd27e/29g′-z2x, sd27e/22g′-z2x, sd27e/29g′-x4x, and their non-stapled counterparts to proteinase K (17 μg mL−1) and monitoring the amount of full-length protein remaining in solution at regular intervals by analytical HPLC. We fit the resulting data for each variant to a monoexponential decay function to obtain apparent proteolysis rate constants k. The results of this analysis are shown in Fig. 3A. All the PEG-stapled variants with improved thermodynamic stability showed enhanced proteolytic resistance, whereas PEG-stapled variants with compromised conformational stability (e.g., sd24b/25c′-z4x) were more vulnerable to proteolysis. For each variant, we calculated a proteolytic resistance factor r, which is the ratio between the apparent rate constant k for a PEG-stapled variant to that of its non-stapled counterpart. Staples with smaller r values provide better protection from proteolysis. We then plotted the natural logarithm of r against the corresponding ΔΔG values for each stapled variant relative to its non-stapled counterpart (Fig. 3B). The natural logarithm of r varies linearly with ΔΔG (R2 = 0.60), indicating that staples that better enhance conformational stability generally provide better protection from proteolysis. Interestingly, the x4xvs. z2x staples between positions 27e and 29g′ provide similar levels of proteolytic resistance even though they have substantially different impacts on conformational stability (x4x is stabilizing, whereas z2x is not), suggesting that staple location is a more important determinant of proteolytic resistance than is staple length (Fig. 3C).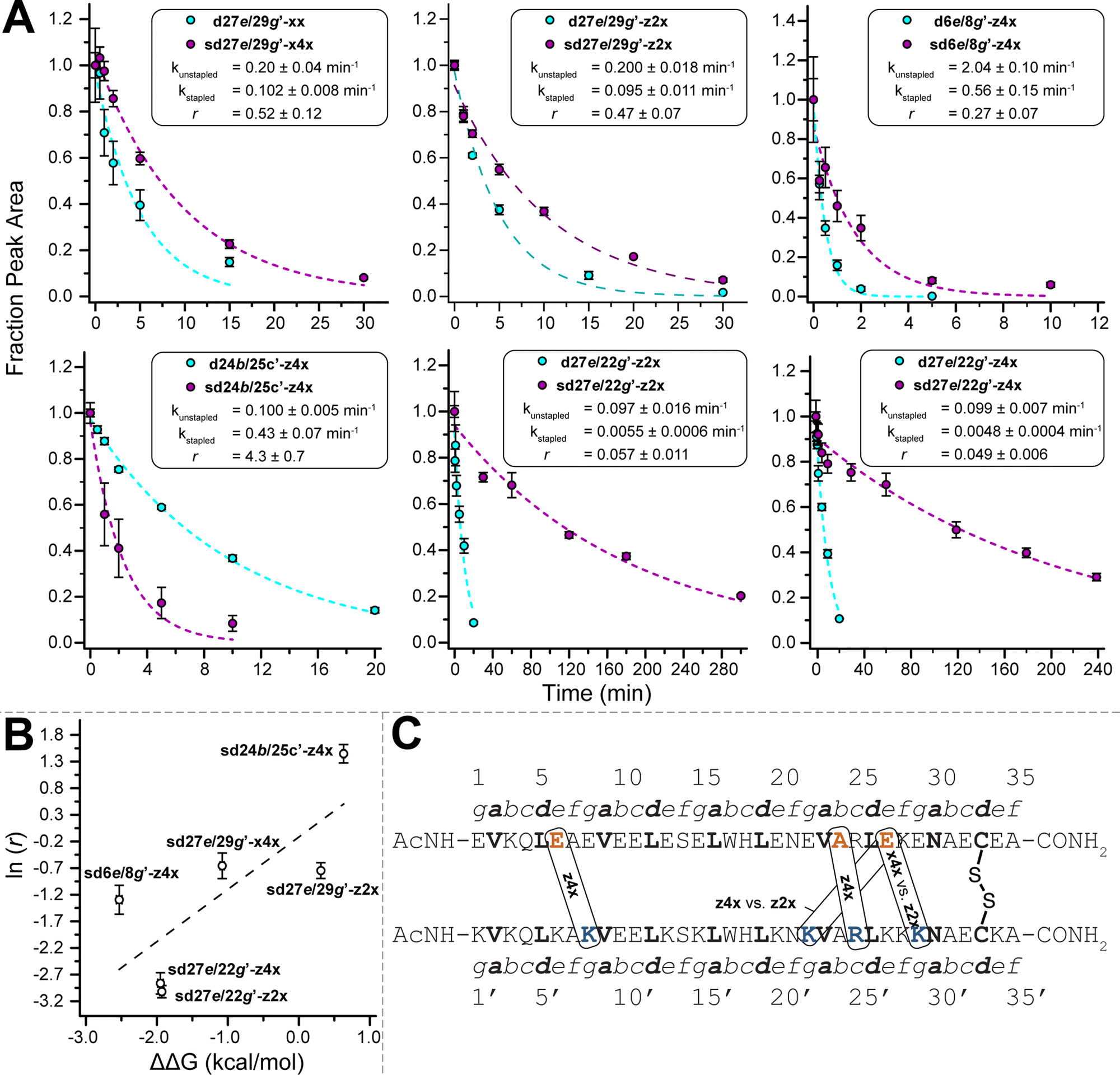 | ||
Fig. 3 (A) Proteolysis of selected unstapled and stapled variants by proteinase K (17 μg mL−1) at 15 μM protein concentration in 20 mM sodium phosphate buffer (pH 7) as monitored by HPLC. Data points represent the average of three replicate experiments for unstapled variants (cyan) and for stapled variants (magenta). Coloured dotted lines represent fits of the data for each variant to a mono-exponential decay function, which was used to determine apparent proteolysis rate constants k and rate constant ratios r. (B) The impact of PEG-stapling on the conformational stability of these variants (ΔΔG) plotted against the natural logarithm of r. Black dotted line represents fit of the ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (r) vs. ΔΔG data to a linear equation. Slope = 0.98 ± 0.40; R2 = 0.60. (C) Location and identity of the staples investigated here within the disulfide-bound coiled coil. (r) vs. ΔΔG data to a linear equation. Slope = 0.98 ± 0.40; R2 = 0.60. (C) Location and identity of the staples investigated here within the disulfide-bound coiled coil. | ||
2.6 Application of PEG-stapling in a HER2 affibody
Finally, we applied the PEG bis-azide stapling strategy to HER2 affibody a,57 which adopts a monomeric helix-bundle conformation comprised of three α-helices. This tertiary structure is closely reminiscent of a trimeric coiled coil, though its sequence does not strictly follow the canonical pattern of the heptad repeat nor does it appear to engage in knobs-into-holes packing. However, positions 8 and 42 (Fig. 4) roughly correspond to the g- and e-positions we stapled in the dA/B coiled coil above; they are solvent exposed; and are similarly close to each other in tertiary structure (∼7.4 Å between side-chain centers of mass) but are far apart in primary sequence. We prepared non-stapled affibody variant a8/42-xx (in which x occupies both positions 8 and 42). We then reacted a8/42-xx with four-unit PEG bis-azide 4 to give stapled variant sa8/42-x4x. We also prepared the unmodified parent HER2 affibody a, in which Glu and Ala occupy positions 8 and 42, respectively. PEG-stapled affibody sa8/42-x4x is −1.09 ± 0.02 kcal mol−1 more stable than the native affibody a and is −1.60 ± 0.03 kcal mol−1 more stable than non-stapled a8/42-xx (chemical denaturant was unnecessary for variable temperature CD experiments on the affibody and its derivatives). Proteolysis assays analogous to those described above indicate that stapled sa8/42-x4x is nine-times more resistant to proteolysis than parent affibody a and seventeen-times more resistant to proteolysis than non-stapled a8/42-xx (Fig. 4). Fluorescence polarization direct binding assays reveal that the unstapled affibody variant binds the extracellular domain of the HER2 protein with a dissociation constant (Kd) of 0.38 ± 0.05 nM, whereas the Kd for the stapled variant (0.69 ± 0.10 nM) is only slightly higher than that of its unstapled counterpart (see ESI†), suggesting that the enhanced proteolytic resistance conferred by PEG-stapling is not accompanied by dramatic decreases in binding affinity.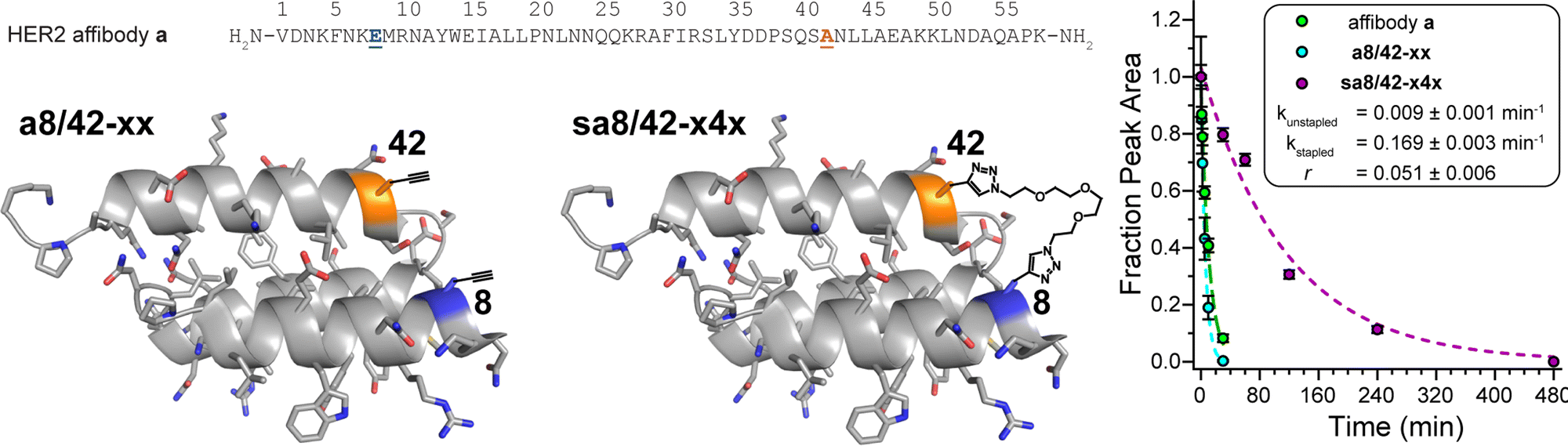 | ||
| Fig. 4 Sequence of HER2 affibody a. Also shown are structures of variant a8/42-xx and its stapled counterpart sa8/42-x4x drawn on the ribbon diagrams of parent affibody a (PDB: 3MZW); locations where we incorporated staple component x or staple x4x are highlighted in orange and blue and are labelled according to their numbered heptad positions within the sequence. Also shown are proteolysis data for affibody a (green), unstapled variant a8/42-xx (cyan), and stapled sa8/42-x4x (magenta) in proteinase K (17 mg mL−1) at 15 μM protein concentration in 20 mM sodium phosphate buffer (pH 7) as monitored by HPLC. Data points represent the average of three replicate experiments. Colored dotted lines represent fits of the data for each variant to a mono-exponential decay function, which we used to calculate apparent proteolysis rate constants k and rate constant ratio r. | ||
3. Conclusions
Here we have explored the impact of PEG stapling on the conformational and proteolytic stability of a disulfide-bonded α-helical coiled-coil heterodimer. Our observations provide important insights into the structural determinants of staple-based stabilization within coiled coils. Interhelical PEG staples (1) are more stabilizing when placed farther from an existing disulfide crosslink; (2) are more stabilizing between salt-bridged e- and g′-positions than between non-salt-bridged e- and g′-positions; (3) are more stabilizing between e- and g-positions generally than between f- and b′- or b- and c′-positions; and (4) appear to be most stabilizing when the calculated staple length exceeds the distance between staple positions by a reasonable margin. We also found that the PEG-staple is tolerant of additional functional groups within the staple: in fact, an appended branching PEG increased the stabilizing impact of the staple by a modest amount. Finally, we demonstrated that stapling does not significantly compromise binding affinity in a HER2 affibody, whilst conferring substantial proteolytic resistance. Our observations now enable the rational design of PEG-stapled α-helical peptide/protein tertiary structures with predictably enhanced conformational stability and proteolytic resistance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NIH Grant number 2R15GM116055-02. The authors acknowledge Profs. Michael S. Kay and Debra M. Eckert at the University of Utah for assistance with fluorescence polarization experiments.References
- D. J. Cram, Angew. Chem., Int. Ed. Engl., 1986, 25, 1039–1057 CrossRef.
- J. M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89–112 CrossRef.
- C. J. Pedersen, Angew. Chem., Int. Ed. Engl., 1988, 27, 1021–1027 CrossRef.
- V. J. Hruby, Life Sci., 1982, 31, 189–199 CrossRef CAS.
- H. Y. Chow, Y. Zhang, E. Matheson and X. Li, Chem. Rev., 2019, 119, 9971–10001 CrossRef CAS PubMed.
- T. K. Tiefenbrunn and P. E. Dawson, Biopolymers, 2010, 94, 95–106 CrossRef CAS PubMed.
- C. P. R. Hackenberger and D. Schwarzer, Angew. Chem., Int. Ed., 2008, 47, 10030–10074 CrossRef CAS PubMed.
- J. N. deGruyter, L. R. Malins and P. S. Baran, Biochemistry, 2017, 56, 3863–3873 CrossRef CAS PubMed.
- D. Schumacher and C. P. R. Hackenberger, Curr. Opin. Chem. Biol., 2014, 22, 62–69 CrossRef CAS PubMed.
- J. M. Chalker, Chem. Biol. Drug Des., 2013, 81, 122–135 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clarklewis and S. B. H. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- B. C. Bundy and J. R. Swartz, Bioconjugate Chem., 2010, 21, 255–263 CrossRef CAS PubMed.
- J. T. Ngo and D. A. Tirrell, Acc. Chem. Res., 2011, 44, 677–685 CrossRef CAS PubMed.
- L. Wang, A. Brock, B. Herberich and P. G. Schultz, Science, 2001, 292, 498–500 CrossRef CAS PubMed.
- F. M. Brunel and P. E. Dawson, Chem. Commun., 2005, 2552–2554, 10.1039/B419015G.
- F. Zhang, O. Sadovski, S. J. Xin and G. A. Woolley, J. Am. Chem. Soc., 2007, 129, 14154–14155 CrossRef CAS PubMed.
- N. Bionda, A. L. Cryan and R. Fasan, ACS Chem. Biol., 2014, 9, 2008–2013 CrossRef CAS PubMed.
- L. Peraro, T. R. Siegert and J. A. Kritzer, in Peptide, Protein and Enzyme Design, ed. V. L. Pecoraro, 2016, vol. 580, pp. 303–332 Search PubMed.
- E. J. Moore, D. Zorine, W. A. Hansen, S. D. Khare and R. Fasan, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12472–12477 CrossRef CAS PubMed.
- A. J. Rojas, C. Zhang, E. V. Vinogradova, N. H. Buchwald, J. Reilly, B. L. Pentelute and S. L. Buchwald, Chem. Sci., 2017, 8, 4257–4263 RSC.
- H. E. Blackwell and R. H. Grubbs, Angew. Chem., Int. Ed., 1998, 37, 3281–3284 CrossRef CAS PubMed.
- C. E. Schafmeister, J. Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891–5892 CrossRef CAS.
- L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305, 1466–1470 CrossRef CAS PubMed.
- R. N. Chapman, G. Dimartino and P. S. Arora, J. Am. Chem. Soc., 2004, 126, 12252–12253 CrossRef CAS PubMed.
- A. Patgiri, A. L. Jochim and P. S. Arora, Acc. Chem. Res., 2008, 41, 1289–1300 CrossRef CAS PubMed.
- G. L. Verdine and G. J. Hilinski, Methods Enzymol., 2012, 503, 3–33 CAS.
- M. Roice, I. Johannsen and M. Meldal, QSAR Comb. Sci., 2004, 23, 662–673 CrossRef CAS.
- S. Cantel, A. Le Chevalier Isaad, M. Scrima, J. J. Levy, R. D. DiMarchi, P. Rovero, J. A. Halperin, A. M. D’Ursi, A. M. Papini and M. Chorev, J. Org. Chem., 2008, 73, 5663–5674 CrossRef CAS PubMed.
- Y. H. Lau, Y. T. Wu, P. de Andrade, W. R. J. D. Galloway and D. R. Spring, Nat. Protoc., 2015, 10, 585–594 CrossRef CAS PubMed.
- C. M. Haney, H. M. Werner, J. J. McKay and W. S. Horne, Org. Biomol. Chem., 2016, 14, 5768–5773 RSC.
- P. T. Tran, C. O. Larsen, T. Rondbjerg, M. De Foresta, M. B. A. Kunze, A. Marek, J. H. Loper, L. E. Boyhus, A. Knuhtsen, K. Lindorff-Larsen and D. S. Pedersen, Chem. – Eur. J., 2017, 23, 3490–3495 CrossRef CAS PubMed.
- M. Scrima, A. Le Chevalier-Isaad, P. Rovero, A. M. Papini, M. Chorev and A. M. D'Ursi, Eur. J. Org. Chem., 2010, 446–457 CrossRef CAS.
- S. A. Kawamoto, A. Coleska, X. Ran, H. Yi, C.-Y. Yang and S. Wang, J. Med. Chem., 2012, 55, 1137–1146 CrossRef CAS PubMed.
- Q. Xiao, N. A. Becar, N. P. Brown, M. S. Smith, K. L. Stern, S. R. E. Draper, K. P. Thompson and J. L. Price, Org. Biomol. Chem., 2018, 16, 8933–8939 RSC.
- Q. Xiao, D. S. Ashton, Z. B. Jones, K. P. Thompson and J. L. Price, RSC Chem. Biol., 2020, 1, 273–280 RSC.
- G. Osapay and J. W. Taylor, J. Am. Chem. Soc., 1992, 114, 6966–6973 CrossRef CAS.
- M. Chorev, E. Roubini, R. L. Mckee, S. W. Gibbons, M. E. Goldman, M. P. Caulfield and M. Rosenblatt, Biochemistry, 1991, 30, 5968–5974 CrossRef CAS PubMed.
- D. Y. Jackson, D. S. King, J. Chmielewski, S. Singh and P. G. Schultz, J. Am. Chem. Soc., 1991, 113, 9391–9392 CrossRef CAS.
- A. M. Leduc, J. O. Trent, J. L. Wittliff, K. S. Bramlett, S. L. Briggs, N. Y. Chirgadze, Y. Wang, T. P. Burris and A. F. Spatola, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11273–11278 CrossRef CAS PubMed.
- F. M. Brunel, M. B. Zwick, R. M. F. Cardoso, J. D. Nelson, I. A. Wilson, D. R. Burton and P. E. Dawson, J. Virol., 2006, 80, 1680–1687 CrossRef CAS PubMed.
- J. R. Kumita, O. S. Smart and G. A. Woolley, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3803–3808 CrossRef CAS PubMed.
- Y. Wang and D. H.-C. Chou, Angew. Chem., Int. Ed., 2015, 54, 10931–10934 CrossRef CAS PubMed.
- H. Jo, N. Meinhardt, Y. Wu, S. Kulkarni, X. Hu, K. E. Low, P. L. Davies, W. F. DeGrado and D. C. Greenbaum, J. Am. Chem. Soc., 2012, 134, 17704–17713 CrossRef CAS PubMed.
- F. H. C. Crick, Acta Crystallogr., 1953, 6, 689–697 CrossRef CAS.
- P. Burkhard, J. Stetefeld and S. V. Strelkov, Trends Cell Biol., 2001, 11, 82–88 CrossRef CAS PubMed.
- D. N. Woolfson, Adv. Protein Chem., 2005, 70, 79–112 CrossRef CAS PubMed.
- A. N. Lupas and M. Gruber, Adv. Protein Chem., 2005, 70, 37–78 CrossRef CAS.
- M. G. Wuo, A. B. Mahon and P. S. Arora, J. Am. Chem. Soc., 2015, 137, 11618–11621 CrossRef CAS PubMed.
- M. G. Wuo, S. Hong, A. Singh and P. S. Arora, J. Am. Chem. Soc., 2018, 140, 16284–16290 CrossRef CAS PubMed.
- C. Wang, X. Li, F. Yu, L. Lu, X. Jiang, X. Xu, H. Wang, W. Lai, T. Zhang, Z. Zhang, L. Ye, S. Jiang and K. Liu, Sci. Rep., 2016, 6, 32161 CrossRef CAS PubMed.
- J. Lindgren and A. Eriksson Karlström, ChemBioChem, 2014, 15, 2132–2138 CrossRef CAS PubMed.
- T. Ekblad, V. Tolmachev, A. Orlova, C. Lendel, L. Abrahmsén and A. E. Karlström, Pept. Sci., 2009, 92, 116–123 CrossRef CAS PubMed.
- A. Nilsson, J. Lindgren and A. Eriksson Karlström, ChemBioChem, 2017, 18, 2056–2062 CrossRef CAS PubMed.
- M. Pelay-Gimeno, T. Bange, S. Hennig and T. N. Grossmann, Angew. Chem., Int. Ed., 2018, 57, 11164–11170 CrossRef CAS PubMed.
- A. E. Keating, V. N. Malashkevich, B. Tidor and P. S. Kim, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14825–14830 CrossRef CAS PubMed.
- F. Truong, T. H. Yoo, T. J. Lampo and D. A. Tirrell, J. Am. Chem. Soc., 2012, 134, 8551–8556 CrossRef CAS PubMed.
- C. Eigenbrot, M. Ultsch, A. Dubnovitsky, L. Abrahmsén and T. Härd, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 15039–15044 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods; compound characterization data, including mass spectra, HPLC chromatograms, and NMR spectra where applicable; CD spectra; global fits of variable temperature CD data; proteolysis assay data. See DOI: https://doi.org/10.1039/d1cb00237f |
| This journal is © The Royal Society of Chemistry 2022 |