
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence6″-Modifed α-GalCer-peptide conjugate vaccine candidates protect against liver-stage malaria†
Michael A.
Meijlink‡
a,
Yu Cheng
Chua‡
 b,
Susanna T. S.
Chan
a,
Regan J.
Anderson
a,
Matthew W.
Rosenberg
a,
Anton
Cozijnsen
c,
Vanessa
Mollard
c,
Geoffrey I.
McFadden
c,
Sarah L.
Draper
a,
Lauren E.
Holz
b,
Ian F.
Hermans
de,
William R.
Heath§
b,
Gavin F.
Painter§
*ae and
Benjamin J.
Compton§
*a
b,
Susanna T. S.
Chan
a,
Regan J.
Anderson
a,
Matthew W.
Rosenberg
a,
Anton
Cozijnsen
c,
Vanessa
Mollard
c,
Geoffrey I.
McFadden
c,
Sarah L.
Draper
a,
Lauren E.
Holz
b,
Ian F.
Hermans
de,
William R.
Heath§
b,
Gavin F.
Painter§
*ae and
Benjamin J.
Compton§
*a
aFerrier Research Institute, Victoria University of Wellington, Lower Hutt, New Zealand. E-mail: benji.compton@vuw.ac.nz; gavin.painter@vuw.ac.nz
bDepartment of Microbiology and Immunology, Doherty Institute for Infection and Immunity, University of Melbourne, Melbourne, VIC, Australia
cSchool of BioSciences, University of Melbourntie, Parkville, VIC, Australia
dMalaghan Institute of Medical Research, Wellington, New Zealand
eAvalia Immunotherapies Limited, Lower Hutt, New Zealand
First published on 2nd March 2022
Abstract
Self-adjuvanting vaccines consisting of peptide epitopes conjugated to immune adjuvants are a powerful way of generating antigen-specific immune responses. We previously showed that a Plasmodium-derived peptide conjugated to a rearranged form of α-galactosylceramide (α-GalCer) could stimulate liver-resident memory T (TRM) cells that were effective killers of liver-stage Plasmodium berghei ANKA (Pba)-infected cells. To investigate if similar or even superior TRM responses can be induced by modifying the α-GalCer adjuvant, we created new conjugate vaccine cadidates by attaching an immunogenic Plasmodium-derived peptide antigen to 6″-substituted α-GalCer analogues. Vaccine synthesis involved developing an efficient route to α-galactosylphytosphingosine (α-GalPhs), from which the prototypical iNKT cell agonist, α-GalCer, and its 6″-deoxy-6″-thio and -amino analogues were derived. Attaching a cathepsin B-cleavable linker to the 6″-modified α-GalCer created pro-adjuvants bearing a pendant ketone group available for peptide conjugation. Optimized reaction conditions were developed that allow for the efficient conjugation of peptide antigens to the pro-adjuvants via oxime ligation to create new glycolipid-peptide (GLP) conjugate vaccines. A single dose of the vaccine candidates induced acute NKT and Plasmodium-specific CD8+ T cell responses that generated potent hepatic TRM responses in mice. Our findings demonstrate that attaching antigenic peptides to 6″-modifed α-GalCer generates powerful self-adjuvanting conjugate vaccine candidates that could potentially control hepatotropic infections such as liver-stage malaria.
Introduction
Immunomodulatory glycolipids play important roles in immunosurveillance and pathogen clearance. Through their agonist activity on cellular receptors, these compounds stimulate the release of signalling molecules that have a multitude of downstream effects including immune cell recruitment, cellular differentiation and proliferation, maturation and activation. As such, harnessing the immunostimulatory properties of glycolipids is widely recognised as holding tremendous potential for developing vaccines to both treat and prevent communicable and non-communicable diseases.A particularly potent and well-studied example of an immune-activating glycolipid is α-galactosylceramide (α-GalCer, KRN7000), a synthetic analogue of a natural product isolated from the marine sponge Agelas mauritianus.1 Since its first report in 1995,2 α-GalCer has been used in over 30 clinical trials3 and has been the focus of an extensive list of publications that describe an important chemical aspect or biological function of the parent compound. Whereas most immune-activating (glyco)lipid classes function by directly binding specific pattern recognition receptors within or on antigen presenting cells (i.e. toll- and Nod-like receptors, C-type lectins), α-GalCer derives its adjuvant activity by functioning as an agonist for type I (or “invariant”) natural killer T (iNKT) cells. Unlike conventional T cells, which exhibit considerable diversity in their antigen receptors and require several days of proliferation to generate sufficient effector function, iNKT cells express largely invariant receptors, are found in large numbers, and respond to antigen within hours of stimulation. Given this capacity to mobilise a strong and rapid response, iNKT cells straddle the boundary between innate and adaptive immunity. The antigen receptors expressed by iNKT cells recognise glycolipids (such as α-GalCer) displayed by the cell surface presenting molecule CD1d. Upon activation, iNKT cells rapidly release potent cytokines and direct molecular signals that activate antigen presenting cells leading to their improved capacity to stimulate T cells and drive antibody production. Because CD1d is non-polymorphic and iNKT cells are found in all humans, α-GalCer is an appealing adjuvant for the development of new vaccines.
Investigations in animal models have shown that α-GalCer boosts immune responses when co-administered with antigen, either as peptide,4 protein5,6 or through mRNA7–9 translation. Studies where α-GalCer is covalently bound to antigen consistently demonstrate superior T cell and antibody responses compared to admixture comparators.10,11 This can be attributed to the close temporal release of both components inside the same antigen presenting cell, thereby focusing iNKT-derived stimulatory signals on the cells engaged in driving adaptive responses.
However, the possible points for attaching peptide antigens to α-GalCer appear limited as the galactosyl and ceramide hydroxyls function as important H-bond donors when bound within the CD1d-T cell receptor (TCR) complex,12–14 perhaps with the exception of the 6″-position, which appears solvent exposed in X-ray crystal structures. An elegant way to overcome this limitation was through migration of the ceramide acyl chain of α-GalCer to the neighboring 4-OH allowing the phytosphingosine nitrogen atom to be further modified with an immolative linker capable of undergoing chemoselective peptide conjugation.10 Upon intracellular processing by antigen presenting cells, such as dendritic cells, the linker underwent enzymatic cleavage followed by facile O → N acyl migration co-releasing the active constituents (i.e. α-GalCer and peptide antigen).10 Conjugates of this nature (Fig. 1) have been shown to induce potent antigen-specific T cell responses with therapeutic potential for treating cancer15,16 and infectious diseases17 including malaria.18
 | ||
| Fig. 1 Self-adjuvanting peptide-conjugate vaccine designs based on α-GalCer illustrating the ‘migration’ approach (previous work) and the 6″-modifed approach (this work). | ||
In this work, we explore an alternative vaccine design that takes advantage of the galactose-6-OH (Fig. 1), which is considered non-essential for binding of the CD1d:α-GalCer complex to the TCR.14 Previously reported 6″-modified α-GalCer analogues include the amino,19 azido,20 thio,21 hydro (deoxy),22 and uronic acid23 derivatives, which have been further modified to produce a range of ether,19 ester,24 amido,25–29 carbamoyl,26 carbamido,27 triazole,20,30–32 disulfide21 and thioether21 analogues, many of which display potent immunostimulatory activities that are biased for either a Th119,20,22,23,26,27,30,32 or Th219,20,25 response compared to α-GalCer. Notable among these 6″-modified α-GalCer analogues are ABX196 and PBS-57, both of which are 6″-NHAc derivatives with disparate N-acyl ceramide chains. ABX196 has been advanced into clinical trials for hepatocellular carcinoma as a combination therapy with the checkpoint inhibitor nivolumab33 while PBS-57 is used for the creation of CD1d monomer or tetramer reagents for the detection of NKT cells.34 In light of these successes, we viewed the simple 6″-deoxy-6″-thio and -amino α-GalCer analogues as attractive adjuvants for glycolipid-peptide (GLP)-based vaccine candidates due to their structural similarity to α-GalCer and the fact that these heteroatoms offer a chemically orthogonal site for peptide antigen attachment. Herein, we report the synthesis of these 6″-modified derivatives, the appendage of an enzymatically labile self-immolative linker to the N- and S-atoms, and their subsequent conjugation to a peptide antigen. Head-to-head in vivo testing of the two vaccines against the original migrated design enabled us to probe the immunological implications of attaching peptide antigen via the phytosphingosine portion of the molecule (i.e. the ‘migration’ approach) versus appending it at the sugar head group (i.e. the ‘6″-modifed’ approach), in particular, the ability of the vaccine candidates to generate antigen-specific memory T cell responses in the liver and their ability to protect against Plasmodium sporozoite challenge.
Results and discussion
Vaccine synthesis
Of the many reported syntheses of α-GalCer (reviewed in Banchet-Cadeddu et al.35), Gervay-Hague's galactosyl iodide mediated glycosylation36 offers an elegant approach that is both high yielding and stereoselective. The high α-selectivity obtained from this reaction is due to the in situ formation of a β-galactosyl iodide and its subsequent displacement by a phytosphingosine acceptor. Because the donor can be easily prepared from D-galactose in one step which does not require chromatographic purification, this synthetic approach appears amenable to scale-up, an important consideration for GMP manufacture.The synthesis of α-GalCer and its 6″-modified analogues commenced from commercially available N-Boc-protected phytosphingosine 1 which was converted into the acetonide-containing acceptor (4) in three steps via temporary protection of the primary hydroxy group as a silyl ether (Scheme 1). This route provides the glycosyl acceptor in 55% overall yield (>95% pure) and, importantly, requires only one simple chromatography purification step. Glycosylation with excess per-TMS-protected galactose 5 in the presence of iodotrimethylsilane (TMSI), tetrabutylammonium iodide (TBAI) and N,N-diisopropylethylamine (DIPEA) converted 4 into the desired α-linked product after 48 h at ambient temperature. Monitoring the glycosylation reaction by HPLC-MS, we observed that freshly prepared TMSI was superior to a commercially acquired solution of TMSI in dichloromethane whereby the former reagent effected the glycosylation in 87% compared to 76% for the latter. Treating the crude product with tetrabutylammonium fluoride (TBAF) in tetrahydrofuran (THF) gave 6 in 74% isolated yield (based on a 2.2 mmol reaction of 4). The feasibility of scaling up this reaction was demonstrated by performing the reaction on 11 mmol scale to afford multi-gram quantities of 6 in 64% yield. Removal of the acid-labile protecting groups with 40% trifluoracetic acid in dichloromethane gave α-galactosylphytosphingosine (α-GalPhs, 7). N-Acylation of 7 with cerotic acid, pre-activated as a mixed carbonic anhydride, followed by per-O-acetylation afforded a protected form of α-GalCer (8) that is easily purified from low level contaminants on scale by silica gel chromatography. Zemplén deacetylation followed by precipitation from hot ethanol afforded gram quantities α-GalCer (9) in good overall yield.
 | ||
| Scheme 1 Synthetic route to α-GalCer (9) and its 6″-deoxy-6″-azido analogue (13). | ||
Access to the O-6″ position was achieved by selective sulfonation of the primary hydroxyl group in 6 using the sterically hindered 2,4,6-triisopropylbenzenesulfonyl chloride (TPSCl).21 Subsequent per-O-acetylation was performed to improve the separation of this compound from small amounts of contaminating bis-sulfonated materials. Azide displacement at C-6″ followed by removal of the protecting groups afforded 12 which was N-acylated to give 6″-azido-6″-deoxy-α-GalCer (13). Palladium-mediated reduction of 13 to 6″-amino-6″-deoxy-α-GalCer (14) gave a product that was poorly soluble in most common organic solvents and mixtures thereof with the apparent exception being hexafluoroisopropanol (HFIP). To avoid the difficulties surrounding its handling, 13 was reduced using Pd/C and H2, and the resulting amine was carried forward in the synthesis without purification. Here, filtration of the catalyst and swapping of the solvents from dichloromethane/methanol to pyridine provided a suspension of crude 14 which, upon reaction with the pre-activated dipeptide-ketone linker 17,37 afforded the pro-adjuvant 18 in readiness for peptide conjugation (Scheme 2).
 | ||
| Scheme 2 Synthetic route to the pro-adjuvant 18 from 6″-azido-6″-deoxy-α-GalCer and immolative dipeptide linker. | ||
Various linker strategies have been developed for α-GalCer-based self-adjuvanting conjugate vaccine candidates that allow release of the glycolipid adjuvant and its antigen cargo inside the same immune cell. Examples include that of Seeberger and De Libero et al.11 who attached a synthetic amine-equipped α-GalCer derivative non-selectively to the capsular polysaccharide of Streptococcus pneumoniae through the action of cyanogen bromide. The resulting isourea, N-substituted imidocarbonate and N-substituted carbamate linkages were believed to degrade upon exposure to an acidic environment. Another approach has been to attach peptide antigen to 6″-deoxy-6″-thio-α-GalCer via maleimido and disulfide linkages that exploit glutathione-mediated release mechanisms.21 For this work, our vaccine design employs the versatile p-aminobenzyl-citrulline-valine (PAB-Cit-Val) linker38 which provides a substrate for proteolytic cathepsin enzymes (cysteine proteases which are found at high levels inside immune cells). Recognition and cleavage of the citrulline carboxylamide bond in 18 (or its peptide conjugate) would result in a facile 1,6-elimination cascade releasing a molecule of CO2, azaquinone methide and the adjuvant, 6″-amino-6″-deoxy-α-GalCer (14). Prior to the synthesising the full GLP-vaccine candidates, susceptibility of the PAB-Cit-Val linker to human liver-derived cathepsin B was investigated in a simplified system whereby the pro-adjuvant 18 was exposed to the enzyme for 24 h at 37 °C in a mixture of HFIP/DMSO in ammonium acetate buffer. Following the reaction by HPLC-MS revealed that after 1 h, 4 h and 24 h, approximately 44%, 17% and 4% (respectively) of 18 remained whilst the cleaved products 8-oxononanoyl-valine-citrulline (Cit-Val-Non) and 6″-amino-6″-deoxy-α-GalCer (14) were detected (ESI,† Fig. S1).
Having demonstrated the enzyme-mediated release of 6″-amino-6″-deoxy-α-GalCer from its pro-adjuvant form, we were interested to see if this same strategy could be applied to the potent iNKT cell agonist, 6″-deoxy-6″-thio-α-GalCer.21 To the best of our knowledge, this would be the first use of a self-immolative p-aminobenzyl linker to release a thiol-containing drug. Towards this, we embarked on the synthesis of 6″-deoxy-6″-thio-α-GalCer and its conversion to the corresponding thiocarbonate-containing pro-adjuvant.
Starting from the advanced intermediate 10, thioacetate displacement of the triisopropylbenzenesulfonyl group was effected by heating in DMF with potassium thioacetate (KSAc) affording 19 in high yield (Scheme 3). Deacetylation was achieved by treating 19 with catalytic NaOMe in methanol to afford the crude product as an 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 mixture of thiol and disulfide. Subsequent treatment with 50% TFA/dichloromethane in the presence of water and anisole removed the acetonide and N-Boc protecting groups without altering the thiol/disulfide ratio. Reduction of this mixture to the sulfhydryl product (20) was achieved by treating the mixture with TCEP in methanol/water and the resulting product was isolated using RP-C18 chromatography. Protection of the thiol as the pyridyl disulfide permitted N-acylation with cerotic acid to give 22. TCEP-mediated reductive cleavage of the pyridyl disulfide group gave 23 which was subsequently reacted with the PAB-Cit-Val-ketone linker (17) under basic conditions to afford the thiocarbonate-containing pro-adjuvant 24.
:15 mixture of thiol and disulfide. Subsequent treatment with 50% TFA/dichloromethane in the presence of water and anisole removed the acetonide and N-Boc protecting groups without altering the thiol/disulfide ratio. Reduction of this mixture to the sulfhydryl product (20) was achieved by treating the mixture with TCEP in methanol/water and the resulting product was isolated using RP-C18 chromatography. Protection of the thiol as the pyridyl disulfide permitted N-acylation with cerotic acid to give 22. TCEP-mediated reductive cleavage of the pyridyl disulfide group gave 23 which was subsequently reacted with the PAB-Cit-Val-ketone linker (17) under basic conditions to afford the thiocarbonate-containing pro-adjuvant 24.
 | ||
| Scheme 3 Synthetic route to 6″-deoxy-6″-thio-α-GalCer (23) and its pro-adjuvant form (24). | ||
Incubating 24 with cathepsin B and following the reaction by HPLC-MS revealed the pro-adjuvant cleaved to its respective constituents (Cit-Val-Non and 23) as intended with approximately 34% of 24 remaining after 24 h. (ESI,† Fig. S2). When cathepsin B was omitted, pro-adjuvant 24 remained intact demonstrating that thiocarbonate cleavage was enzyme mediated and not the result of direct hydrolysis. Although 24 appears more resistant to cathepsin B activity than 18in vitro, work by Dubowchik and Firestone38 suggests that other intracellular enzymes can also cause linker immolation in vivo thus we considered that the utility of this design was best examined in a functional assay.
With the 6″-modified pro-adjuvants 18 and 24 in hand, the next step was to conjugate these substrates with peptide antigen to prepare the GLP-conjugate vaccine candidates. The antigen selected for this work was the short peptide epitope NVFDFNNL. Identified from the Plasmodium berghei ANKA ribosomal protein (PbRPL6), the NVFDFNNL sequence (PbRPL6120–127) has previously been shown to be an excellent antigen for vaccination against liver-stage malaria.39 To investigate if this epitope could also induce antigen-specific CD8+ liver TRM cells when attached to our 6″-modified α-GalCer pro-adjuvants, the epitope was modified by adopting a similar design to what has been previously shown to work for peptide-conjugate vaccines.18 Here, the CD8 epitope was adorned with naturally flanking amino acid residues – ST- (N-terminus) and -S (C-terminus) from the protein antigen and the N-terminal – FFRK – proteasomal cleavage sequence, separated by a tri-alanine (-AAA-) spacer.
To create the GLP vaccine candidates, our strategy was to synthesise the modified peptide antigen with an N-terminal aminooxy acetyl residue (25) and attach it to the pro-adjuvants (18 and 24) via chemoselective oxime ligation. The oxime linkage was chosen for its relative hydrolytic stability40 and its ‘amide-like’ small chemical footprint. Initial attempts to form the oxime bond employed conditions (4:2:3 THF/MeOH/300 mM aniline buffer at pH 4.1) which were successful for the synthesis of other GLP-vaccine candidates.18 Unfortunately, 18 was sparingly soluble in these reaction conditions, even at temperatures approaching 50 °C, resulting in poor conversion into the desired vaccine product. Because HFIP efficiently solvates 18 and has inherent acidity, we next evaluated this as a suitable solvent for oxime ligation. Pleasingly, HFIP efficiently dissolved both the pro-adjuvant (18) and modified peptide (25) while the addition of aniline (100 mM) catalysed the reaction such that it was complete within 48 h at ambient temperature. Swapping the aniline for the purportedly superior catalyst m-phenylenediamine41 (100 mM in HFIP) failed to improve the reaction kinetics. Because HFIP readily dissolves arylamine catalysts of oxime ligation, a high concentration of these can be easily achieved using this system. Increasing the concentration of aniline from 100 to 500 mM in HFIP whilst maintaining the pH at ∼5 by the addition of TFA, reliably afforded the desired oxime products with no obvious side reactions (as determined by HPLC-MS) within 18 h at 35 °C. Employing these optimized conditions, the pro-adjuvants 18 and 24 were reacted with peptide 25 (1.2–1.5 equiv.) to give conjugates 26 and 27 respectively (Scheme 4). The excess peptide and aniline catalyst were removed by preparative HPLC affording the GLP-vaccine products (>98% purity by HPLC) for biological testing.
 | ||
| Scheme 4 Formation of self-adjuvanting antimalarial vaccine candidates 26 and 27. | ||
Biological studies
The 6″-modified α-GalCer GLP vaccine candidates (26 and 27) were assessed for their ability to induce memory T cell responses, particularly those capable of preventing malaria. These responses can involve several T cell subsets, including two that recirculate around the body termed central memory T (TCM) and effector memory T (TEM) cells and a subset that resides permanently within tissues, termed resident memory T (TRM) cells. While TRM cells can be found in a number of tissues, those localised in the liver are crucial for controlling liver-stage infection by Plasmodium parasites that cause malaria.42 C57BL/6 (B6) mice were immunised intravenously with 26 or 27 or with our previously reported immunogenic vaccine candidate, 28 (Fig. 2), which is based on the migrated α-GalCer design and known to efficiently generate TRM cell responses in the liver.18 After 30 days, to allow formation of memory responses, antigen-specific T cells in the liver and spleen were enumerated by flow cytometry and assessed for key surface markers to determine their memory T cell subset composition and their antigen specificity. A H-2Kb tetramer (tet) containing the NVFDFNNL (NVF) peptide was used to identify T cells specific for the vaccine antigen (Fig. 3). This analysis revealed that both 6″-modified vaccine candidates efficiently generated NVF-specific (tet+) CD8+ TRM cells in the liver (Fig. 3A, B), with vaccine candidate 26 inducing about twice as many as the control 28. Significantly more NVF-specific memory T cells were also generated in the spleens of mice vaccinated with 26 compared to 28 (Fig. 3C), largely as a consequence of an increase in splenic TRM cell numbers. Overall, these results showed that both 26 and 27 were immunogenic and capable of inducing liver TRM cells, with the 6″-amino-6″-deoxy-α-GalCer conjugate (26), capable of generating the highest number of these cells.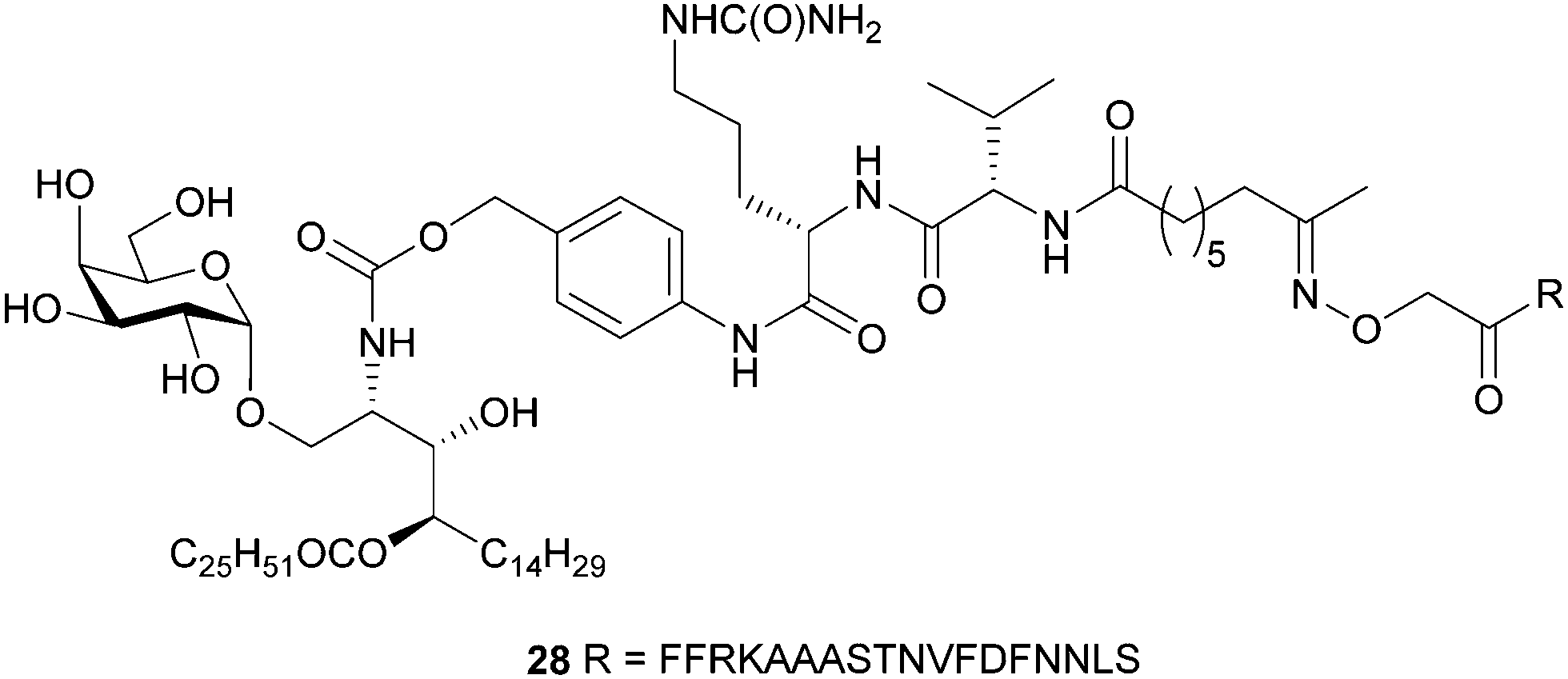 | ||
| Fig. 2 Antimalarial GLP-vaccine (28) employing the ‘migrated’ α-GalCer design. | ||
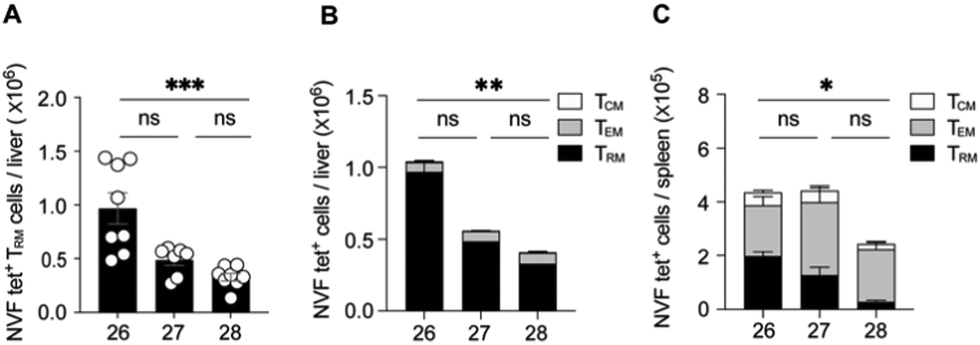 | ||
| Fig. 3 Conjugate vaccines comprising 6″-modified α-GalCer generate large numbers of liver TRM cells. B6 mice were vaccinated intravenously with vaccines 26, 27 or 28 and examined for NVF-specific memory T cell responses >30 days later by flow cytometry after staining single cell suspensions with NVF-tetramers (NVF-tet) and various surface markers. (A) The number of NVF-tet+ CD8+ TRM (CD8+CD44+CD69+CD62L−) cells in the liver in individual mice (circles). Mean (black bar). (B and C) The number of NVF-tet+ TRM (black), TEM (grey; CD8+CD44+CD69−CD62L−) and TCM (white, CD8+CD44+CD69−CD62L+) memory CD8+ T cells in the liver (B) and spleen (C). Data are pooled from two independent experiments using 3–4 mice per group for each experiment. Error bars represent mean ± SEM. Numbers of NVF-tet+ CD8+ TRM cells in (A) were log-transformed and compared using a Kruskal–Wallis test and Dunn's multiple comparisons test. Total numbers of NVF-tet+ CD8+ memory cells in (B and C) were log-transformed and compared using a Kruskal-Wallis test and Dunn's multiple comparisons test. ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001. | ||
To determine whether the greater ability of 26 to generate liver TRM cells was due to an enhanced ability to activate NKT cells, the NKT cell responses of mice vaccinated with 26, 27 or 28 were assessed shortly after vaccination (day 7) (Fig. 4). This revealed that, relative to 28, both 26 and 27 induced significantly more NKT cells in the liver and spleen (Fig. 4A, B and ESI† Fig. S3A, SB), most likely through proliferation. Phenotypic analysis showed that all three vaccine candidates stimulated both splenic and liver NKT cells to downregulate surface CD69 and NK1.1, indicating NKT cell activation, with greater downregulation of CD69 after immunization with 26 and 27 relative to 28 (Fig. 4C–F and ESI,† Fig. S3C, D). Examination of CD8+ T cell responses at this early time point (day 7) showed similar numbers of NVF-specific effector T cells in the spleens and livers of mice vaccinated with all three compounds (Fig. 4G, H and ESI,† Fig. S3E, F), indicating no obvious difference in priming at this early stage. Together, these data suggest that while vaccine candidate 26 induced about 2-fold more liver TRM cells than 28, this difference could not be unequivocally attributed to better NKT cell activation as vaccine candidate 27 also showed improved NKT cell activation but did not generate significantly more liver TRM cells. Clearly, all three vaccine candidates efficiently activate NKT cells and are capable of excellent generation of liver TRM cells.
 | ||
| Fig. 4 Vaccine candidates 26 and 27 induce stronger NKT cell activation than their migrated α-GalCer counterpart. B6 mice were immunised intravenously with 26, 27 or 28 and then examined for acute NKT and NVF-specific T cell responses at day 7. (A and B) Enumeration of NKT cells in the spleen (A) and liver (B). (C and D) Expression of CD69 on NKT cells in the spleen (C) and (D) liver. (E and F) Expression of NK1.1 on NKT cells in the spleen (E) and liver (F). (G and H) The number of NVF-tet+ CD44+ CD8+ T cells in the spleen (G) and liver (H). Data are pooled from two independent experiments using 3 mice per group for each experiment. Error bars represent mean ± SEM. Groups in (A, B, G and H) were log-transformed and compared by one-way ANOVA with Tukey's multiple comparison post-test. Groups in (C-F) were compared by one-way ANOVA with Tukey's multiple comparison post-test. ns, not significant, *P < 0.05, **P < 0.01, ****P < 0.0001. Nv, naïve. | ||
Although 26 induced superior numbers of memory T cells than the previous best vaccine candidate, 28, it was unclear whether these cells were functional. To test function, we assessed whether liver TRM cells generated by 26, like those of 28, could mediate protection against malaria. B6 mice were immunised with 26 or 28 and then left for 30 days before assessing T cell numbers in the liver in some mice from each group and then challenging the remaining mice with 200 PbA sporozoites (Fig. 5). This confirmed that vaccine candidate 26 generated more liver TRM cells than 28 (Fig. 5A) and showed that all mice were protected from a 200 PbA sporozoite challenge (Fig. 5B and ESI,† Fig. S4A), indicating that TRM cells induced by 26 were indeed functional. To further assess function, surviving mice were rechallenged with a higher dose of 3000 PbA sporozoites, which showed that all mice immunized with 26 were protected from this higher dose (Fig. 5B and ESI,† Fig. S4B). Overall, these data indicated that vaccine 26 is highly effective at generating liver TRM cells and inducing protective immunity against malaria.
 | ||
| Fig. 5 Compound 26 generates large numbers of liver TRM cells that protect mice from malaria. B6 mice were immunised intravenously with vaccines 26 or 28 and, after 30 days, were assessed by T cell enumeration or parasite challenge. (A) Livers of vaccinated mice were enumerated for NVF-specific memory T cell responses by flow cytometry after staining single cell suspensions with NVF-tet and various surface markers. Error bars represent mean ± SEM. Total numbers of NVF-tet+ CD8+ memory cells were log-transformed and compared by one-way ANOVA with Tukey's multiple comparison post-test. *P < 0.05, ****P < 0.0001. Note that the bar for naïve (Nv) group shown in (A) is not visible due to low cell counts (<1600 cells). (B) Vaccinated mice were challenged intravenously with 200 PbA sporozoites and parasitemia measured up to day 12 post-infection. Mice showing no parasitemia were recorded as protected and were rechallenged intravenously with 3000 PbA sporozoites 21 days after the first challenge and monitored for parasitemia for 12 days. The percentage of mice that were protected from both 200 and 3000 sporozoites is shown in grey; the percentage only protected from 200 sporozoites shown in white; the percentage unprotected shown in black. Data are pooled from two independent experiments using 2–5 mice per group for each experiment. Numbers above bars indicate the number of mice that were protected against the initial 200 sporozoite challenge over total number of mice challenged. | ||
Conclusions
This work provides a succinct synthetic route to the potent iNKT cell agonist α-GalCer and its 6″-deoxy-6″-thio and 6″-amino analogues. Attaching a traceless linker to the 6″-S or -N atoms of these glycolipids produced pro-adjuvants of 6″-substituted α-GalCer that were readily cleaved by human cathepsin B in vitro with the self-immolating thiocarbonate version being the first example of its kind. Oxime ligation of peptide antigen to the pro-adjuvants was expedited by developing optimized HFIP-based reaction conditions. The resulting glycolipid-peptide conjugates were shown to co-stimulate NKT cells and peptide-specific CD8+ T cells generating high levels of TRM cells in the liver with our lead 6″-amino-6″-deoxy-α-GalCer conjugate vaccine candidate inducing protective immunity against malaria in a Plasmodium sporozoite challenge model.Data availability
Full experimental details and compound characterisation are available in the ESI.†Author contributions
M. A. M., R. J. A., M. W. R., S. L. D., and B. J. C. synthesized the compounds. Y. C. C., S. T. S. C., and L. E. H. performed the experiments and analysed the data. A. C., V. M., and G. I. M. provided reagents. B. J. C., G. F. P., I. F. H., and W. R. H. conceived the experiments. B. J. C., Y. C. C., and W. R. H. wrote the manuscript and all authors contributed to its reviewing and editing.Conflicts of interest
The authors declare the following competing financial interest(s): G. F. P. and I. F. H. are the chief technical officer and chief scientific officer (respectively) of Avalia Immunotherapies Limited, and W. R. H. is a member of its Scientific Advisory Board. Avalia holds exclusive, worldwide license to patents related to aspects of the chemical design reported here. R. J. A., I. F. H., G. F. P., and B. J. C. are inventors on a granted patent (U.S. patent no. 10046046, granted 14 August 2018). B.J.C., R.J.A., I.F.H., G. F. P., L. E. H., and W. R. H. are named inventors on a PCT application (PCTNZ2020050048) submitted by Victoria University of Wellington, Malcorp Biodiscoveries Limited, and Victoria Link Limited that covers the production of tissue-resident memory T cells with glycolipid-peptide vaccines.Acknowledgements
This work was supported by funding from the Health Research Council of New Zealand (Contract HRC 20/569), New Zealand Ministry of Business Innovation and Employment (Contract RTVU1603), and the National Health and Medical Research Council of Australia (Program grant 1113292 and Fellowship 1154457). We thank members of Godfrey laboratory (University of Melbourne, Australia) for CD1d tetramers.Notes and references
- T. Natori, M. Morita, K. Akimoto and Y. Koezuka, Agelasphins, Novel Antitumor and Immunostimulatory Cerebrosides from the Marine Sponge Agelas-Mauritianus, Tetrahedron, 1994, 50(9), 2771–2784 CrossRef CAS.
- M. Morita, K. Motoki, K. Akimoto, T. Natori, T. Sakai, E. Sawa, K. Yamaji, Y. Koezuka, E. Kobayashi and H. Fukushima, Structure-Activity Relationship of Alpha-Galactosylceramides against B16-Bearing Mice, J. Med. Chem., 1995, 38(12), 2176–2187 CrossRef CAS PubMed.
- Y. T. Zhang, R. Springfield, S. Y. Chen, X. Li, X. T. Feng, R. Moshirian, R. R. Yang and W. M. Yuan, alpha-GalCer and iNKT Cell-Based Cancer Immunotherapy: Realizing the Therapeutic Potentials, Front. Immunol., 2019, 10, 1126 CrossRef CAS PubMed.
- A. N. Courtney, P. N. Nehete, B. P. Nehete, P. Thapa, D. Zhou and K. J. Sastry, Alpha-galactosylceramide is an effective mucosal adjuvant for repeated intranasal or oral delivery of HIV peptide antigens., Vaccine, 2009, 27(25–26), 3335–3341 CrossRef CAS PubMed.
- S. Fujii, K. Shimizu, C. Smith, L. Bonifaz and R. M. Steinman, Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein, J. Exp. Med., 2003, 198(2), 267–279 CrossRef CAS PubMed.
- N. Singh, S. Hong, D. C. Scherer, I. Serizawa, N. Burdin, M. Kronenberg, Y. Koezuka and L. Van Kaer, Cutting edge: activation of NK T cells by CD1d and alpha-galactosylceramide directs conventional T cells to the acquisition of a Th2 phenotype, J. Immunol., 1999, 163(5), 2373–2377 CAS.
- S. Fujii, A. Goto and K. Shimizu, Antigen mRNA-transfected, allogeneic fibroblasts loaded with NKT-cell ligand confer antitumor immunity, Blood, 2009, 113(18), 4262–4272 CrossRef CAS PubMed.
- R. Verbeke, I. Lentacker, K. Breckpot, J. Janssens, S. Van Calenbergh, S. C. De Smedt and H. Dewitte, Broadening the Message: A Nanovaccine Co-loaded with Messenger RNA and alpha-GalCer Induces Antitumor Immunity through Conventional and Natural Killer T Cells, ACS Nano, 2019, 13(2), 1655–1669 CAS.
- M. L. Guevara, Z. Jilesen, D. Stojdl and S. Persano, Codelivery of mRNA with alpha-Galactosylceramide Using a New Lipopolyplex Formulation Induces a Strong Antitumor Response upon Intravenous Administration, ACS Omega, 2019, 4(8), 13015–13026 CrossRef CAS PubMed.
- R. J. Anderson, C. W. Tang, N. J. Daniels, B. J. Compton, C. M. Hayman, K. A. Johnston, D. A. Knight, O. Gasser, H. C. Poyntz, P. M. Ferguson, D. S. Larsen, F. Ronchese, G. F. Painter and I. F. Hermans, A self-adjuvanting vaccine induces cytotoxic T lymphocytes that suppress allergy, Nat. Chem. Biol., 2014, 10(11), 943–949 CrossRef CAS PubMed.
- M. Cavallari, P. Stallforth, A. Kalinichenko, D. C. K. Rathwell, T. M. A. Gronewold, A. Adibekian, L. Mori, R. Landmann, P. H. Seeberger and G. De Libero, A semisynthetic carbohydrate-lipid vaccine that protects against S-pneumoniae in mice, Nat. Chem. Biol., 2014, 10(11), 950–956 CrossRef CAS PubMed.
- M. Koch, V. S. Stronge, D. Shepherd, S. D. Gadola, B. Mathew, G. Ritter, A. R. Fersht, G. S. Besra, R. R. Schmidt, E. Y. Jones and V. Cerundolo, The crystal structure of human CD1d with and without alpha-galactosylceramide, Nat. Immunol., 2005, 6(8), 819–826 CrossRef CAS PubMed.
- D. M. Zajonc, C. Cantu, J. Mattner, D. P. Zhou, P. B. Savage, A. Bendelac, I. A. Wilson and L. Teyton, Structure and function of a potent agonist for the semi-invariant natural killer T cell receptor, Nat. Immunol., 2005, 6(8), 810–818 CrossRef CAS PubMed.
- N. A. Borg, K. S. Wun, L. Kjer-Nielsen, M. C. J. Wilce, D. G. Pellicci, R. Koh, G. S. Besra, M. Bharadwaj, D. I. Godfrey, J. McCluskey and J. Rossjohn, CD1d-lipid-antigen recognition by the semi-invariant NKT T-cell receptor, Nature, 2007, 448(7149), 44–49 CrossRef CAS PubMed.
- C. Grasso, C. S. Field, C. W. Tang, P. M. Ferguson, B. J. Compton, R. J. Anderson, G. F. Painter, R. Weinkove, I. F. Hermans and M. V. Berridge, Vaccines adjuvanted with an NKT cell agonist induce effective T-cell responses in models of CNS lymphoma, Immunotherapy, 2020, 12(6), 395–406 CrossRef CAS PubMed.
- R. J. Anderson, B. J. Compton, C. W. Tang, A. Authier-Hall, C. M. Hayman, G. W. Swinerd, R. Kowalczyk, P. Harris, M. A. Brimble, D. S. Larsen, O. Gasser, R. Weinkove, I. F. Hermans and G. F. Painter, NKT cell-dependent glycolipid-peptide vaccines with potent anti-tumour activity, Chem. Sci., 2015, 6(9), 5120–5127 RSC.
- R. J. Anderson, J. Li, L. Kedzierski, B. J. Compton, C. M. Hayman, T. L. Osmond, C. W. Tang, K. J. Farrand, H. F. Koay, C. F. D. S. E. Almeida, L. R. Holz, G. M. Williams, M. A. Brimble, Z. F. Wang, M. Koutsakos, K. Kedzierska, D. I. Godfrey, I. F. Hermans, S. J. Turner and G. F. Painter, Augmenting Influenza-Specific T Cell Memory Generation with a Natural Killer T Cell-Dependent Glycolipid-Peptide Vaccine, ACS Chem. Biol., 2017, 12(11), 2898–2905 CrossRef CAS PubMed.
- L. E. Holz, Y. C. Chua, M. N. de Menezes, R. J. Anderson, S. L. Draper, B. J. Compton, S. T. S. Chan, J. Mathew, J. Li, L. Kedzierski, Z. F. Wang, L. Beattie, M. H. Enders, S. Ghilas, R. May, T. M. Steiner, J. Lange, D. Fernandez-Ruiz, A. M. Valencia-Hernandez, T. L. Osmond, K. J. Farrand, R. Seneviratna, C. F. Almeida, K. M. Tullett, P. Bertolino, D. G. Bowen, A. Cozijnsen, V. Mollard, G. I. McFadden, I. Caminschi, M. H. Lahoud, K. Kedzierska, S. J. Turner, D. I. Godfrey, I. F. Hermans, G. F. Painter and W. R. Heath, Glycolipid-peptide vaccination induces liver-resident memory CD8(+) T cells that protect against rodent malaria, Sci. Immunol., 2020, 5(48), eaaz8035 CrossRef CAS PubMed.
- J. T. Hung, R. C. Sawant, J. C. Chen, Y. F. Yen, W. S. Chen, A. L. Yu and S. Y. Luo, Design and synthesis of galactose-6-OH-modified alpha-galactosyl ceramide analogues with Th2-biased immune responses, RSC Adv., 2014, 4(88), 47341–47356 RSC.
- P. J. Jervis, L. M. Graham, E. L. Foster, L. R. Cox, S. A. Porcelli and G. S. Besra, New CD1d agonists: Synthesis and biological activity of 6″-triazole-substituted alpha-galactosyl cramides, Bioorg. Med. Chem. Lett., 2012, 22(13), 4348–4352 CrossRef CAS PubMed.
- B. J. Compton, C. W. Tang, K. A. Johnston, T. L. Osmond, C. M. Hayman, D. S. Larsen, I. F. Hermans and G. F. Painter, Synthesis and Activity of 6″-Deoxy-6″-thio-alpha-GalCer and Peptide Conjugates, Org. Lett., 2015, 17(24), 5954–5957 CrossRef CAS PubMed.
- T. Tashiro, R. Nakagawa, T. Shigeura, H. Watarai, M. Taniguchi and K. Mori, RCAI-61 and related 6′-modified analogs of KRN7000: Their synthesis and bioactivity for mouse lymphocytes to produce interferon-gamma in vivo, Bioorg. Med. Chem., 2013, 21(11), 3066–3079 CrossRef CAS PubMed.
- Y. J. Chang, J. R. Huang, Y. C. Tsai, J. T. Hung, D. Wu, M. Fujio, C. H. Wong and A. L. Yu, Potent immune-modulating and anticancer effects of NKT cell stimulatory glycolipids, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(25), 10299–10304 CrossRef CAS PubMed.
- D. Chennamadhavuni, N. A. Saavedra-Avila, L. J. Carreno, M. J. Guberman-Pfeffer, P. Arora, Y. Q. Tang, R. Pryce, H. F. Koay, D. I. Godfrey, S. Keshipeddy, S. K. Richardson, S. Sundararaj, J. H. Lo, X. S. Wen, J. A. Gascon, W. M. Yuan, J. Rossjohn, J. Le Nours, S. A. Porcelli and A. R. Howell, Dual Modifications of alpha-Galactosylceramide Synergize to Promote Activation of Human Invariant Natural Killer T Cells and Stimulate Anti-tumor Immunity, Cell Chem. Biol., 2018, 25(7), 571–584 CrossRef CAS PubMed.
- T. Ebensen, C. Link, P. Riese, K. Schulze, M. Morr and C. A. Guzman, A pegylated derivative of alpha-galactosylceramide exhibits improved biological properties, J. Immunol., 2007, 179(4), 2065–2073 CrossRef CAS PubMed.
- J. Guillaume, N. Pauwels, S. Aspeslagh, D. M. Zajonc, D. Elewaut and S. Van Calenbergh, Synthesis of C-5″ and C-6″-modified alpha-GalCer analogues as iNKT-cell agonists, Bioorg. Med. Chem., 2015, 23(13), 3175–3182 CrossRef CAS PubMed.
- M. Trappeniers, K. Van Beneden, T. Decruy, U. Hillaert, B. Linclau, D. Elewaut and S. Van Calenbergh, 6′-Derivatised alpha-GalCer Analogues Capable of Inducing Strong CD1d-Mediated Th1-Biased NKT Cell Responses in Mice, J. Am. Chem. Soc., 2008, 130(49), 16468–16469 CrossRef CAS PubMed.
- M. H. Hsieh, J. T. Hung, Y. W. Liw, Y. J. Lu, C. H. Wong, A. L. Yu and P. H. Liang, Synthesis and Evaluation of Acyl-Chain- and Galactose-6″'-Modified Analogues of alpha-GalCer for NKT Cell Activation, ChemBioChem, 2012, 13(11), 1689–1697 CrossRef CAS PubMed.
- X. T. Zhou, C. Forestier, R. D. Goff, C. H. Li, L. Teyton, A. Bendelac and P. B. Savage, Synthesis and NKT cell stimulating properties of fluorophore- and biotin-appended 6″-amino-6″-deoxy-galactosylceramides, Org. Lett., 2002, 4(8), 1267–1270 CrossRef CAS PubMed.
- W. W. Ma, J. J. Bi, C. F. Zhao, Z. G. Zhang, T. X. Liu and G. S. Zhang, Synthesis and biological activities of amino acids functionalized alpha-GalCer analogues, Bioorg. Med. Chem., 2020, 28(1), 115141 CrossRef CAS PubMed.
- P. J. Jervis, M. Moulis, J. P. Jukes, H. Ghadbane, L. R. Cox, V. Cerundolo and G. S. Besra, Towards multivalent CD1d ligands: synthesis and biological activity of homodimeric alpha-galactosyl ceramide analogues., Carbohydr. Res., 2012, 356, 152–162 CrossRef CAS PubMed.
- N. Pauwels, S. Aspeslagh, D. Elewaut and S. Van Calenbergh, Synthesis of 6″-triazole-substituted alpha-GalCer analogues as potent iNKT cell stimulating ligands, Bioorg. Med. Chem., 2012, 20(24), 7149–7154 CrossRef CAS PubMed.
- A Phase 1-2 Study of ABX196 in Combination With Nivolumab in Patients With Hepatocellular Carcinoma. https://clinicaltrials.gov/ct2/show/NCT03897543.
- Y. Liu, R. D. Goff, D. P. Zhou, J. Mattner, B. A. Sullivan, A. Khurana, C. Cantu, E. V. Ravkov, C. C. Lbegbu, J. D. Altman, L. Teyton, A. BendelaC and P. B. Savage, A modified alpha-galactosyl ceramide for staining and stimulating natural killer T cells, J. Immunol. Methods, 2006, 312(1–2), 34–39 CrossRef CAS PubMed.
- A. Banchet-Cadeddu, E. Henon, M. Dauchez, J. H. Renault, F. Monneaux and A. Haudrechy, The stimulating adventure of KRN 7000, Org. Biomol. Chem., 2011, 9(9), 3080–3104 RSC.
- M. Schombs, F. E. Park, W. J. Du, S. S. Kulkarni and J. Gervay-Hague, One-Pot Syntheses of Immunostimulatory Glyeolipids, J. Org. Chem., 2010, 75(15), 4891–4898 CrossRef CAS PubMed.
- R. J. Anderson; G. F. Painter; I. F. Hermans; O. S. Gasser and R. Weinkove, MAIT Cell Agonists, WO2019058289, 2018 Search PubMed.
- G. M. Dubowchik, R. A. Firestone, L. Padilla, D. Willner, S. J. Hofstead, K. Mosure, J. O. Knipe, S. J. Lasch and P. A. Trail, Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: model studies of enzymatic drug release and antigen-specific in vitro anticancer activity, Bioconjugate Chem., 2002, 13(4), 855–869 CrossRef CAS PubMed.
- A. M. Valencia-Hernandez, W. Y. Ng, N. Ghazanfari, S. Ghilas, M. N. de Menezes, L. E. Holz, C. Huang, K. English, M. Naung, P. S. Tan, K. M. Tullett, T. M. Steiner, M. H. Enders, L. Beattie, Y. C. Chua, C. M. Jones, A. Cozijnsen, V. Mollard, Y. P. Cai, D. G. Bowen, A. W. Purcell, N. L. La Gruta, J. A. Villadangos, T. de Koning-Ward, A. E. Barry, W. Barchet, I. A. Cockburn, G. I. McFadden, S. Gras, M. H. Lahoud, P. Bertolino, R. B. Schittenhelm, I. Caminschi, W. R. Heath and D. Fernandez-Ruiz, A Natural Peptide Antigen within the Plasmodium Ribosomal Protein RPL6 Confers Liver T-RM Cell-Mediated Immunity against Malaria in Mice, Cell Host Microbe, 2020, 27(6), 950–962 CrossRef CAS PubMed.
- J. Kalia and R. T. Raines, Hydrolytic stability of hydrazones and oximes., Angew. Chem., Int. Ed., 2008, 47(39), 7523–7526 CrossRef CAS PubMed.
- M. Rashidian, M. M. Mahmoodi, R. Shah, J. K. Dozier, C. R. Wagner and M. D. Distefano, A Highly Efficient Catalyst for Oxime Ligation and Hydrazone-Oxime Exchange Suitable for Bioconjugation, Bioconjugate Chem., 2013, 24(3), 333–342 CrossRef CAS PubMed.
- D. Fernandez-Ruiz, W. Y. Ng, L. E. Holz, J. Z. Ma, A. Zaid, Y. C. Wong, L. S. Lau, V. Mollard, A. Cozijnsen, N. Collins, J. Li, G. M. Davey, Y. Kato, S. Devi, R. Skandari, M. Pauley, J. H. Manton, D. I. Godfrey, A. Braun, S. S. Tay, P. S. Tan, D. G. Bowen, F. Koch-Nolte, B. Rissiek, F. R. Carbone, B. S. Crabb, M. Lahoud, I. A. Cockburn, S. N. Mueller, P. Bertolino, G. I. McFadden, I. Caminschi and W. R. Heath, Liver-Resident Memory CD8(+) T Cells Form a Front-Line Defense against Malaria Liver-Stage Infection, Immunity, 2016, 45(4), 889–902 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00251a |
| ‡ These authors contributed equally to this work as co-first authors. |
| § These authors contributed equally to this work as co-senior authors. |
| This journal is © The Royal Society of Chemistry 2022 |