
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of novel druggable pockets on polyomavirus VP1 through crystallographic fragment-based screening to develop capsid assembly inhibitors†
Evgenii M.
Osipov‡
 a,
Ali H.
Munawar‡
abc,
Steven
Beelen
a,
Daren
Fearon
de,
Alice
Douangamath
de,
Conor
Wild
fg,
Stephen D.
Weeks
ac,
Arthur
Van Aerschot
h,
Frank
von Delft
defij and
Sergei V.
Strelkov
*a
a,
Ali H.
Munawar‡
abc,
Steven
Beelen
a,
Daren
Fearon
de,
Alice
Douangamath
de,
Conor
Wild
fg,
Stephen D.
Weeks
ac,
Arthur
Van Aerschot
h,
Frank
von Delft
defij and
Sergei V.
Strelkov
*a
aBiocrystallography, KU Leuven, Herestraat 49, Leuven, Belgium. E-mail: sergei.strelkov@kuleuven.be
bOrthogon Therapeutics LLC, 45 Dan Road Suite 126, Canton, MA 02021, USA
cPledge Tx B.V., Gaston Geenslaan 1, Leuven, Belgium
dDiamond Light Source Ltd., Harwell Science and Innovation Campus, Didcot, UK
eResearch Complex at Harwell, Harwell Science and Innovation Campus, Didcot OX11 0FA, UK
fCentre for Medicines Discovery, University of Oxford, South Parks Road, Headington OX3 7DQ, UK
gDepartment of Statistics, University of Oxford, 29 St Giles', Oxford OX1 3LB, UK
hMedicinal Chemistry, Rega Institute for Medical Research, KU Leuven, Herestraat 49, Leuven, Belgium
iStructural Genomics Consortium, University of Oxford, Old Road Campus, Roosevelt Drive, Headington OX3 7DQ, UK
jDepartment of Biochemistry, University of Johannesburg, Auckland Park 2006, South Africa
First published on 29th April 2022
Abstract
Polyomaviruses are a family of ubiquitous double-stranded DNA viruses many of which are human pathogens. These include BK polyomavirus which causes severe urinary tract infection in immunocompromised patients and Merkel cell polyomavirus associated with aggressive cancers. The small genome of polyomaviruses lacks conventional drug targets, and no specific drugs are available at present. Here we focus on the main structural protein VP1 of BK polyomavirus which is responsible for icosahedral capsid formation. To provide a foundation towards rational drug design, we crystallized truncated VP1 pentamers and subjected them to a high-throughput screening for binding drug-like fragments through a direct X-ray analysis. To enable a highly performant screening, rigorous optimization of the crystallographic pipeline and processing with the latest generation PanDDA2 software were necessary. As a result, a total of 144 binding hits were established. Importantly, the hits are well clustered in six surface pockets. Three pockets are located on the outside of the pentamer and map on the regions where the ‘invading’ C-terminal arm of another pentamer is attached upon capsid assembly. Another set of three pockets is situated within the wide pore along the five-fold axis of the VP1 pentamer. These pockets are situated at the interaction interface with the minor capsid protein VP2 which is indispensable for normal functioning of the virus. Here we systematically analyse the three outside pockets which are highly conserved across various polyomaviruses, while point mutations in these pockets are detrimental for viral replication. We show that one of the pockets can accommodate antipsychotic drug trifluoperazine. For each pocket, we derive pharmacophore features which enable the design of small molecules preventing the interaction between VP1 pentamers and therefore inhibiting capsid assembly. Our data lay a foundation towards a rational development of first-in-class drugs targeting polyomavirus capsid.
Introduction
Polyomaviridae (PyV) is a family of double-stranded DNA viruses related to the archetypical simian virus SV40. This family includes about a dozen human pathogens which cause a latent infection in 30% to 90% of the world's population but pose a distinct threat to immunocompromised individuals.1 Such patients can develop a range of PyV-associated diseases which affect the urinary tract, brain and skin, including cancers. In particular, more than 80% of the world population is serologically positive to BK polyomavirus.2 Primary BKPyV infection is typically mild but results in the virus permanently settling in epithelial cells of urinary tract. In immunosuppressed patients such as undergoing renal or bone marrow transplantation, BKPyV can cause nephropathy or haemorrhagic cystitis, respectively.3 Statistically, acute disease develops in about 5% of donor kidney recipients which typically results in allograft loss.2 The closely related JC polyomavirus is a CNS tropic pathogen which can re-activate upon immunosuppression, causing a devastating demyelinating disease called progressive multifocal leukoencephalopathy.4 Other polyomaviruses such as Merkel cell polyomavirus have been linked to aggressive forms of skin cancer.5 BKPyV infection has also been associated with increased cancer risk, which is due to the ability of its large tumour antigen to bind and inhibit tumour suppressor proteins like p53 and retinoblastoma protein.6As systematically reviewed very recently,1 no effective drugs are available to date against any of the polyomaviruses, despite multiple compounds being evaluated in preclinical and clinical studies. While cidofovir (a general nucleoside analogue active against a broad range of DNA viruses) has shown activity against BKPyV infection, this drug causes severe nephrotoxicity and induces renal failure in approximately 30% of patients.7 At present, the only way to prevent the progression of the disease is to discontinue immunosuppression, which has limited therapeutic applicability due to the risk of allograft rejection.8
Rational search for drugs against polyomaviruses is complicated by a limited choice of druggable targets. The polyomaviridae genome is small (∼5100 bp) and encodes three structural proteins VP1, VP2 and VP3, agnoprotein and large and small tumour antigens. VP1 is the main building block of the viral capsid. Besides its key structural role, VP1 mediates the interactions with the host cell early in the viral life cycle and facilitates the viral genome packaging into mature virions.9–11 Individual VP1s readily form pentamers through a tight association of their central core domains, which contain a β-structured jelly roll motif supplemented with multiple surface loops and shorter α-helices. The globular core domain is flanked by flexible N- and C-terminal arms which enable further association of the pentamers (Fig. 1A and B). In particular, the ‘invading’ C-terminal arms of one pentamer form specific interactions with neighbouring pentamers in the assembled capsid, as was initially revealed by the crystal structure of the SV40 capsid12 and confirmed through cryo-EM studies of BKPyV.9,13 Moreover, the importance of the C-terminal arm attachment for polyomavirus assembly was highlighted through mutagenesis studies.14–17 The capsid assembly depends on pH, ionic strength and the presence of Ca2+ ions.18 In the optimal conditions, 72 pentamers assemble into icosahedral capsid with T = 7 d symmetry.12 Besides bona fide capsids, VP1 pentamers can also assemble into aberrant T = 1 particles, including a 12-mer icosahedron and a 24-mer octahedron, which are unable to package viral DNA.18,19 The minor capsid protein VP2 and its truncated version VP3 bind in the central pore of VP1 pentamer. While not essential for capsid assembly, VP2 and VP3 are involved in the translocation of viral DNA into nucleus after host cell entry.20,21 Studies on ΔVP2/ΔVP3 mutants of murine polyomavirus indicated that the absence of VP2 delayed synthesis of viral DNA.22
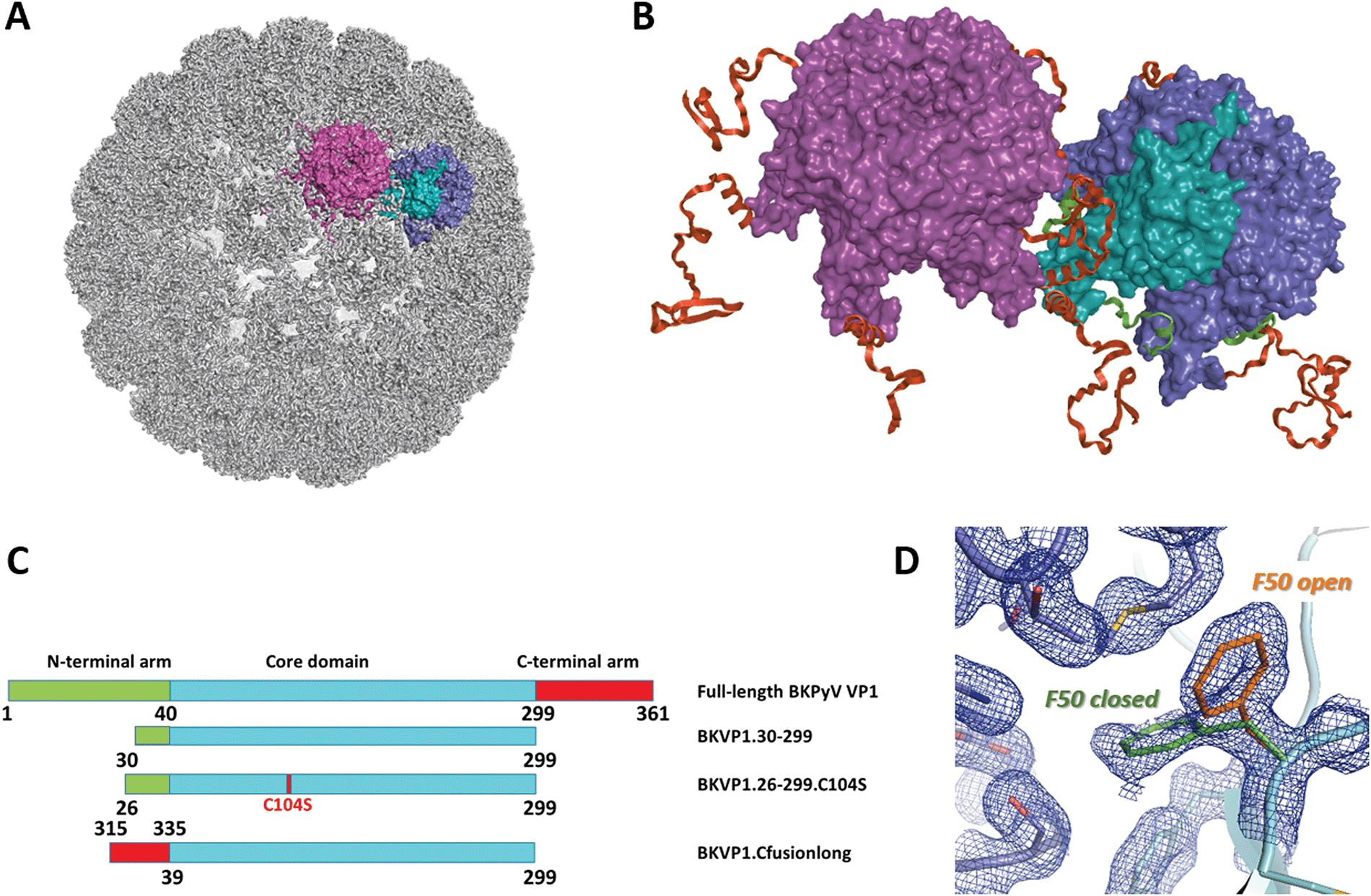 | ||
| Fig. 1 Polyomavirus capsid assembly. (A) Cryo-EM structure of the BK virus-like particle.9 The surfaces of two adjacent VP1 pentamers are shown in purple and blue/cyan. A single monomer is highlighted in cyan. (B) Close-up view revealing the interactions between two pentamers. The N-terminal arms (residues 1–39) and C-terminal arms (residues 300–361) of VP1 monomers are coloured green and red, respectively. (C) Schematic diagrams of the full-length BKPyV VP1 and the main constructs used here for crystallographic studies. (D) Two conformations of the F50 residue observed in the crystal structure of BKVP1.30-299 construct at 1.44 Å resolution prior to drug-like fragment soaking. Two VP1 monomers are coloured cyan and blue. Open and closed conformations of F50 are coloured orange and green, respectively. 2Fo-Fc electron density map contoured at 0.7σ is shown as a blue mesh. | ||
In the absence of more traditional drug targets such as proteases or polymerases, developing drugs that interfere with polyomavirus capsid assembly is a promising option. Specifically, disrupting the normal protein-protein interfaces (PPIs) either between the VP1 subunits or VP1/VP2 contacts can be considered. In the past, some efforts in this direction were undertaken. In particular, the exposed part of VP1 carries a sialic acid binding pocket which plays a key role in host cell recognition. Disruption of this interaction was suggested as a possible drug design strategy.23–25 Recently, a VP2-mimicking peptide was shown to bind to VP1 pentamers and produce an antiviral effect at nanomolar concentration, but the exact mechanism of its action remained not fully understood.26
PPIs are generally known to be challenging as drug targets, but efficient targeting is possible if sufficiently deep druggable pockets are present on one of the partners.27 More recently, considerable progress in targeting PPIs has been achieved through fragment-based screening (FBS).27,28 This approach utilizes small drug-like compound libraries with a molecular weight typically not exceeding 300 Da. Due to their relatively simple structure, such fragments can sample chemical space more efficiently compared to full-sized compounds. Compared to high-throughput screening with larger compounds, the fragment libraries are much smaller in size (∼1000 compounds) but provide higher hit rates. At the same time, small fragments typically have low affinities (Kd > 1 mM) and are thus difficult to detect using standard assays.29,30 X-ray crystallography-based fragment screening (FBS-X) was shown to offer the highest sensitivity and lowest false-positives compared to more traditional screening using other biophysical methods.31
Recently, multiple methodological improvements have increased the throughput of FBS-X, including the development of bright synchrotron sources, efficient libraries,32–35 and automated crystal soaking, X-ray data collection and processing.36,37 As a result, X-ray crystallography can now be used as a primary fragment screening technique. The efficiency of FBS-X was very recently demonstrated with multiple SARS-CoV-2 targets, including helicase (non-structural protein (Nsp) 13), main protease (Nsp5) and the macrodomain of Nsp3.38–40
Here we present an FBS-X centred study where we show for the first time that VP1 of BKPyV carries multiple druggable pockets that can be used for the development of capsid assembly inhibitors. Specifically, we could establish three novel pockets which are located on the outside surface of the pentamer and are critically involved in the interactions between the core pentamer and the ‘invading’ C-terminal arms. Blocking each of these pockets through rationally designed inhibitors should interfere with essential PPIs and thus compromise the capsid assembly. We argue that high conservation of these pockets between BKPyV and related human pathogens including JCPyV opens up a perspective of developing potent drugs against a range of polyomaviruses.
Results and discussion
Preliminary FBS-X screening prompted optimization of crystallographic system
Initially we prepared a truncated variant of BKPyV capsid protein VP1 containing residues 30 to 299 (Fig. 1C), which essentially corresponds to the globular core domain (residues 41 to 299). A close construct was previously shown to form pentamers in solution and was amenable to X-ray crystallography.41 Upon crystallization screening and refinement, well-diffracting crystals could be obtained (Table S1, ESI†). These crystals were used to establish the structure of unliganded VP1 core domain at 1.44 Å resolution (Table S2, ESI†), which is an improvement over the 1.7 Å structure established previously (PDB entry 4mj0).41 The structure contains one pentamer per asymmetric unit (ASU).Next, we performed a large-scale FBS-X screening using the XChem facility at Diamond synchrotron.42 The obtained crystals could withstand soaking of drug-like fragments at up to 60 mM in the presence of 30% DMSO for one hour without visual deterioration. We screened a total of 1132 fragments which included the complete 776 original Diamond-SGC-Poised library (DSPL)43 and portions of the Maybridge and Edelris fragment libraries. Eventually, six binders could be identified through PanDDA analysis36 and later confirmed by difference electron density maps of Polder type.44
The relatively low success rate of this preliminary screening was due to several factors. First, after soaking only a fraction of the crystals (350 of 1132) diffracted sufficiently well (beyond 2.5 Å resolution), while the experiment was only performed once. Second, it was realized that, upon preparation of the large batch of crystals towards FBS-X, the BKVP1.30-299 construct formed crystals in two space groups P21212 or P212121, with one or two pentamers per ASU respectively, from the same crystallization conditions. The switch between the two crystal forms was difficult to control, even after further attempts to refine the conditions. Both limited resolution and in part the space group instability limited the performance of the computational pipeline used for fragment placement. In addition, in both crystal forms, the N-terminal part (residues 30–39) of selected protein chains was flanking one of the fragment-binding hotspots (pocket F50, see below). This situation (Fig. S1A, ESI†) led to artifactual binding of some fragments in the pocket, which had to be discarded.
Search for the best crystallization construct
Limited success of the initial FBS-X studies prompted us to optimize the BKVP1 construct used. In an attempt to remove unwanted interactions of residues 30–39, we created shorter constructs corresponding to residues 40–299 and 43–299, i.e. starting right with the first β-strand of the VP1 core, but both constructs were found to be insoluble upon overexpression. Eventually, an optimal solution could be found by using the construct 26–299 i.e. just four residues longer than the original 30–299 construct. Crystallization of the optimized construct involved microseeding with a suspension of crushed crystals, which enabled a reliable growth of new macroscopic crystals within several days. The crystals consistently diffracted to at least 1.7 Å resolution, had a space group P21 with two pentamers per ASU and were suitable for soaking drug-like fragments in presence of 30% DMSO. In the resulting structure, the N-termini of all ten chains had the same conformation and located at a distance from the β-strand formed by residues 44 to 52 (Fig. S1A, ESI†).A further improvement of the BKV1.26-299 construct was achieved through introducing a point mutation C104S. This mutation corresponds to removal of a cysteine residue located in the loop CD (residues 98–106) which forms the ‘feet’ of the pentamer located deep into the assembled capsid.12 Indeed, even though a reducing agent was added to crystallization drops upon setup, its capacity reduced as a matter of a few days, leading to a likely oxidation of the surface-exposed C104. We have found that the C104S mutation improved the reliability of crystallization and helped preserving the best diffraction quality of the crystals for several months, which is an asset while performing a large-scale FBS-X.45 The optimized BKV1.26-299.C104S construct crystallized isomorphously with the wild-type construct and was used to refine the structure of the core BKVP1 pentamer at 1.47 Å resolution (Table S2, ESI†).
Main FBS-X screening
Next, we proceeded with the main screening at the XChem facility using the optimized BKVP1.26-299.C104S construct crystals and a new generation of the DSPL library named DSiP43 with 768 compounds. The crystals withstood the soaking procedure remarkably well, as 634 soaked crystals (83%) still diffracted beyond 2.5 Å resolution, and a vast majority of those could be used for further crystallographic analysis (Fig. S2, ESI†). However, a search for bound fragments using the established PanDDA algorithm44 failed to capture a number of hits which were apparent in the standard Fo-Fc maps. We hypothesized that the reason for this poor performance was heterogeneity of the crystals. To circumvent this problem, we resorted to clustering analysis of electron density as implemented in the new developmental version of the algorithm, named PanDDA2. This analysis revealed the existence of three clusters of datasets (Fig. S3, ESI†). The differences between the clusters are linked to both the differences in the packing of VP1 pentamers in the crystals and local changes such as distinct conformations of the CD loop. After re-processing with PanDDA2, 124 datasets (19.5% of all well-diffracting crystals) were found to contain a bound ligand (Table 1). One fragment binder had already been detected in the preliminary XChem screening, but its pose could be further refined using a new dataset with improved resolution (Fig. S4, ESI†).| Protein construct | Facility | Library | No. of comps. screened | No. of datasets with resolution better than 2.5 Å | No. of bindersa (hit rate, %) | |
|---|---|---|---|---|---|---|
| a Hit rate defined as the percentage of binders in the total number of fragment-soaked crystals diffracting to better than 2.5 Å resolution. | ||||||
| Preliminary screening | BKVP1.30-299 | XChem | Complete DSPL43 + select Maybridge and Edelris fragments | 1132 | 350 | 6 (1.7) |
| Main screening | BKVP1.26-299.C104S | XChem | DSiP library based on DSPL | 768 | 634 | 124 (19.5) |
| Manual | Subset of brominated FragLites33 | 22 | 21 | 15 (71) | ||
In addition, a subselection of 22 compounds from the FragLites library (31 compounds)33 was used in a manual screening. We restricted ourselves to brominated fragments since higher hit rates had been reported for these compounds in the original publication33 compared to their iodinated counterparts. In addition, one iodinated fragment, 4-iodopyrazole, was included due to its prominent binding promiscuity.32 One particular advantage of these compounds was that they could be reliably located using the anomalous signal from the halogen atom. As the result, a very high hit rate of 71% could be achieved (Table 1). Many brominated fragments bound to multiple sites on the pentamer.
Overview of fragment-binding sites
A total of 144 drug-like fragment hits were identified. Typically, the compounds could be placed in equivalent pockets in all or most of the five or ten protein chains present in the ASU of the crystals (Table S2, ESI†). Comparison across different chains provided an additional means to verify the correct ligand pose. Overall, 27 fragments (19% of all binders) located at crystal lattice interfaces between different pentamers and were excluded from further consideration as crystallographic artefacts.In total, 76 fragments (53%) were observed to bind on the outer side of the VP1 pentamer rather than in its central pore (Fig. 2A). Notably, the majority of these fragments (69 out of 76) cluster in three distinct pockets. All pockets were populated by fragments from both DSPL/DSiP and FragLite libraries. We labelled these three pockets by the residues at which they are centered, namely F50, R92 and M109. The site F50 spans two subunits of the pentamer, while the other two sites fully map on a single monomer. The main features of the three binding pockets are provided in Table 2. In particular, the druggability scores of the three pockets as provided by the Sitemap algorithm 46 are similar to scores of PPIs previously shown to be amenable to small-molecule inhibition.47,48
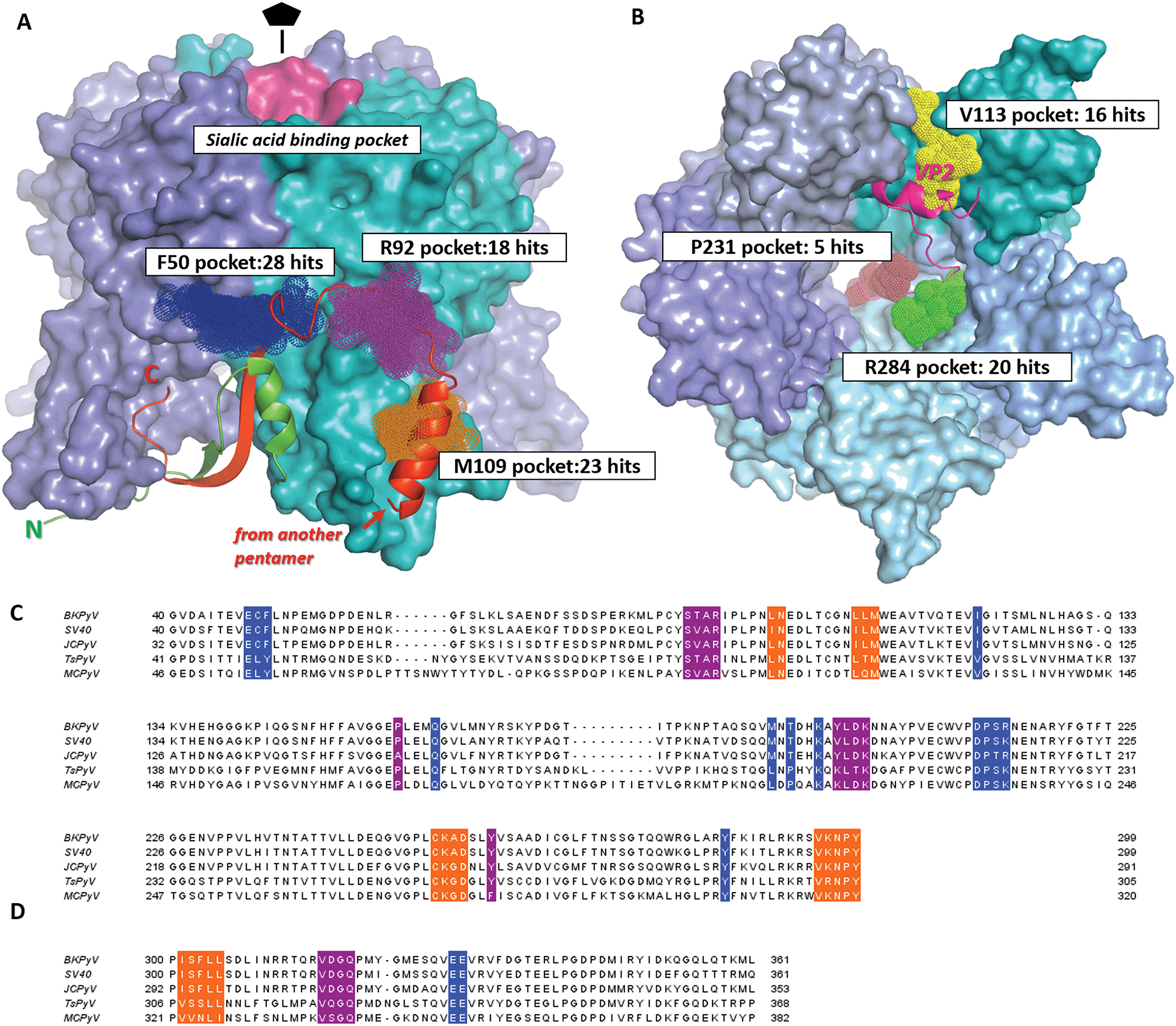 | ||
| Fig. 2 Drug-like fragment binding sites on the surface of BKVP1 pentamer identified through FBS-X. (A) Superposition of all obtained fragment binders on the outer side of the pentamer in sites F50, R92 and M109 are shown as dark blue, purple and orange dotted spheres, respectively. The core domains of the pentamer are shown as a molecular surface, with monomers in distinct colours. The N-terminal arm (residues 10–40) of the monomer shown in cyan is shown as a green ribbon. The ‘invading’ C-terminal arm from an adjacent pentamer in the assembled capsid8 is shown as a red ribbon. The sialic acid binding pocket is coloured pink. (B) Superposition of all obtained fragment binders within the pentamer pore in sites V113, R284 and P231 shown as yellow, green and brown dotted spheres, respectively. A view along the five-fold axis is shown. The bound C-terminal segment of VP2 as established through co-crystallization studies50 is depicted as a fuchsia ribbon. (C) Amino acid sequence alignment for the core domains of VP1 from main pathogenic human polyomaviruses including BK (BKPyV) (Uniprot P03088), JC (JCPyV) (Uniprot P03089), Trichodysplasia spinulosa (TsPyV) (Uniprot E2ESL7) and Merkel cell polyomavirus (MCPyV) (Uniprot C0JPK). A closely related simian virus 40 (SV40) (Uniprot P03087) is also included. Residues forming the fragment-binding pockets F50, R92 and M109 are highlighted in the same colour as in panel A. (D) Alignment of the C-terminal arms of VP1. The regions interacting with the three pockets are shown in the corresponding colour. | ||
| Name | Residues | Seq. identitya | No. of residues | Pocket features | Druggabi-lity scoreb | No. of fragment hitsc | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| H-Bond donors | H-Bond acceptors | Charged groups | Aromatic residues | Relative hydropho-bicityb | ||||||
| a Sequence identity for the given site. The value was produced by taking the sequence identity for the site residues between BKPyV and each of four related polyomaviruses (SV40, JCPyV, TsPyV and MCPyV, see Fig. 2), and taking the mean of the four values. b Properties calculated in SiteMap.46 Briefly, the relative hydrophobicity is based on calibration with data from ref. 55. Average hydrophobicity of the calibration dataset was 1.6. Druggability score aims to evaluate the prospect of a given site for future drug development. Scores above 1 are typically obtained for deep hydrophobic pockets (e.g. in cytochrome P450). Scores below 0.63 indicate shallow and hydrophilic, poorly druggable sites. c Some successful fragments (Table 1) were seen to bind in more than one site simultaneously. | ||||||||||
| F50 | Chain A: 48, 49, 50, 120, 285 | 0.73 | 13 | Nζ K194 | Oε E48, Oγ S213 | K194, R214 | F50, Y285 | 1.95 | 0.76 | 28 |
| Chain B: 162, 189, 191, 194, 211, 12, 213, 214 | ||||||||||
| R92 | 90, 91, 92, 158, 196, 197, 198, 199, 260 | 0.77 | 9 | Nε R92, N N200, Oη Y260 | O T90, O L197, O D198 | R92, D198, K199 | Y260, Y196 | 0.53 | 0.74 | 18 |
| M109 | 98, 99, 107, 108, 109, 254, 255, 256, 257, 295, 296, 297, 298 | 0.87 | 13 | Nδ Asn99, N A256 | Oδ D257, water molecule | K255, D257 | 1.87 | 0.84 | 23 | |
Our most significant finding is that all three binding sites F50, R92 and M109 match the interaction interface with the ‘invading’ C-terminal arm of another pentamer (Fig. 2A). As explained above, such interaction plays a key role in polyomavirus capsid assembly.9,49 We have further analysed the amino-acid sequence conservation of the three binding pockets across a range of VP1s from pathogenic polyomaviruses (Table 2 and Fig. 2C). All pockets have a significantly higher sequence conservation (73–87% identity between BKPyV and related viruses; see footnote of Table 2 for definition) compared to the complete VP1 sequence (65% identity). This correlates with the functional role of these pockets towards the C-terminal arm binding which is a common assembly mechanism for all polyomaviruses. Many key residues involved in ligand binding, such as K194 in the F50 site, are strictly conserved across all species. Such highly conserved, functional pockets are especially attractive towards the development of small-molecule capsid assembly inhibitors, since such drugs are likely to inactivate a range of pathogenic polyomaviruses and should be less prone to resistance development.
In addition, numerous further binders (41 fragments or 28% of the total number of hits) were located within the central pore of the VP1 pentamer (Fig. 2B). Just like the fragments binding at the outer surface of the pentamer, these hits localise to three distinct pockets centred at residues V113, P231 and R284 respectively. Remarkably, these pockets correspond to the interaction interface with the minor capsid protein VP2, as observed in the cryo-EM structure of BKV virions9 and in the X-ray structures of both murine polyomavirus VP1 complexed with truncated VP250 and BKPyV VP1 complexed with a thirteen-residue long VP2 derived peptide.26 Hence the three pockets in the pentamer pore are also likely to be relevant for capsid assembly and function, and should be explored towards rational drug design. A detailed analysis of these pockets and fragments binding therein will be presented elsewhere.
Finally, we detected three further fragments that bind in a small surface pocket known to be involved in sialic acid binding and hence host receptor recognition24,41,51 (Fig. 2A). This pocket has been previously suggested to be suitable for drug design.25 However, this pocket is prone to mutations and highly variable across different polyomaviruses.24,52 The higher conservation of the assembly-relevant pockets F50, R92 and M109 that we describe in detail below makes them potentially better targets.
F50 site is an inducible pocket involved in capsid assembly
Binding site F50 is a hydrophobic pocket located at the interface of two VP1 monomers within the pentamer (Fig. 2A and 3A). It is highly conserved across several pathogenic human polyomaviruses (Fig. 2C). In particular, only a single amino-acid difference in this pocket is seen for JCPyV where the residue S213 is replaced by a threonine. Interestingly, due to the ability of the side chain of F50 to accommodate two different conformations this pocket can be either ‘open’ or ‘closed’. For instance, in the pentamer seen in our high-resolution unliganded structure of the BKVP1.30-299 construct, the F50 pocket is closed in four chains while in the fifth chain the F50 residue is split between two alternative conformations (Fig. 1D). Also, in the previously established VP1 pentamer structures of BKPyV (PDB code 4mj0)41 and the closely related SV40 (PDB code 3bwq)23 this pocket was seen in the closed state. In the crystals of VP1 pentamers of other polyomaviruses, the pocket is open in all or at least some chains in the ASU (PDB codes 4 × 17 and 4wdy).53,54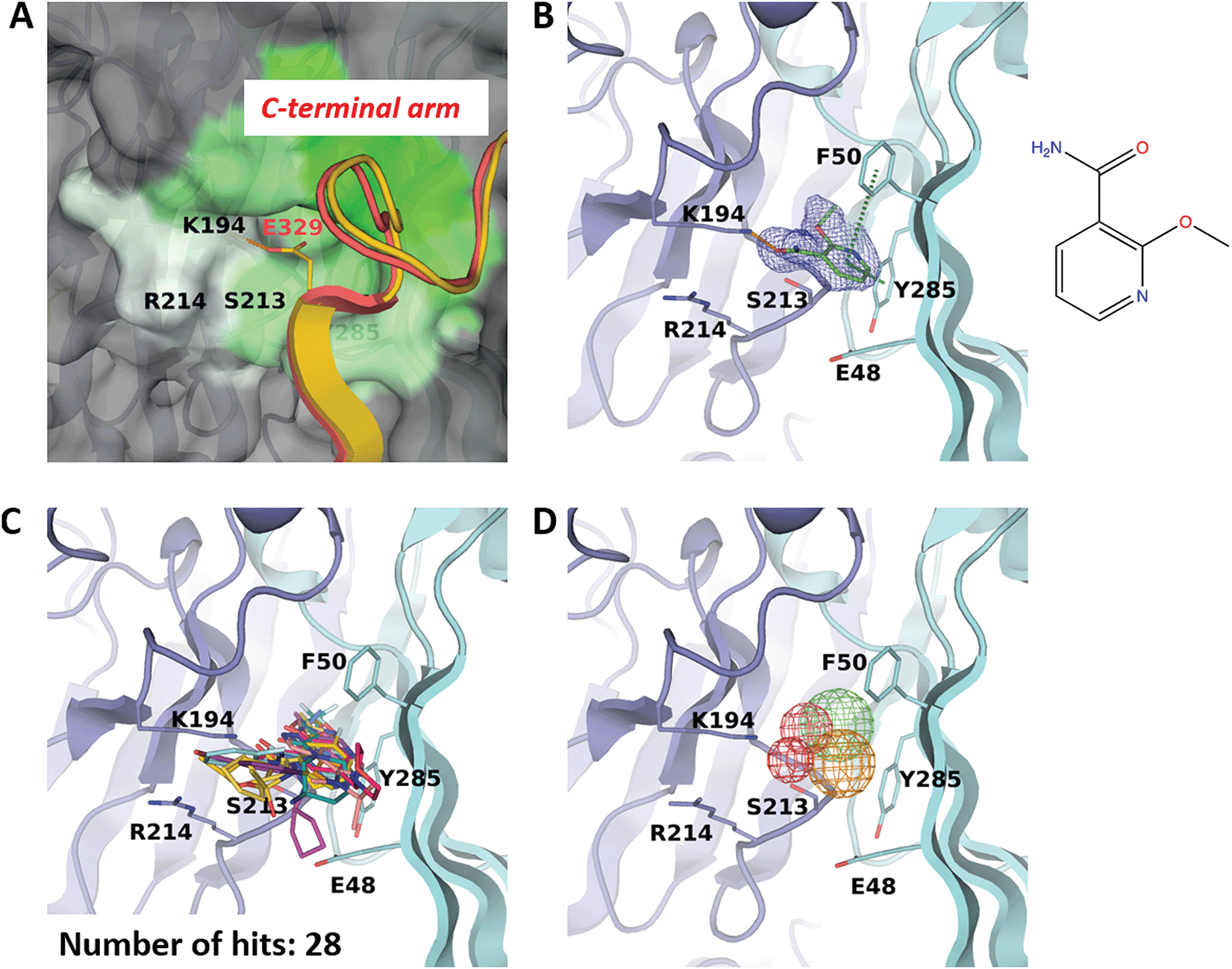 | ||
| Fig. 3 F50 site. (A) Close-up view of the pocket coloured by hydrophobicity, with the most hydrophobic regions shown in green and hydrophilic regions in white. The rest of the pentamer surface is coloured grey. The superimposed invading C-terminal arm is rendered as appearing in the crystal structure of the BKVP1.Cfusionlong construct (yellow ribbon) and in the cryo-EM structure of the VLP9 (red ribbon). (B) Binding of 2-ethoxypyridine-3-carboxamide (ZINC ID Z272156568) in the pocket. The chemical structure is shown on the right. Polder difference map44 at 3σ level is shown as blue mesh. The H-bond with the K194 side chain and the edge-to-face π–π interaction with F50 are depicted as orange and green dashed lines, respectively. (C) Superposition of all drug-like fragments binding in the F50 pocket. (D) Consensus pharmacophores derived from all binders. Colours of the pharmacophore elements (mesh) are defined as follows: red for hydrogen bond acceptors, blue (absent on this figure) for hydrogen bond donors, green for hydrophobic groups and orange for aromatics (capable of π–π stacking). | ||
Of note, only the open pocket could be detected by typical binding site prediction algorithms. In our BKVP1 structures, this situation drastically changes after drug-like fragment binding. Fig. 3B shows an example of a well-resolved fragment which binds to all five chains of the pentamer, whereby all F50 side chains flip to the open position. These observations allow us to designate the F50 site as ‘inducible’ or ‘cryptic’.
Importantly, the F50 pocket forms part of the core pentamer interface which accommodates the ‘invading’ C-terminal arm upon capsid assembly (Fig. 3A). Since at the start of our work no atomic resolution structure of BKPyV capsid was available, we created a novel fusion protein construct to analyse the C-terminal arm interactions using crystallography. To this end, we produced two chimeric constructs (designated BKVP1.Cfusionlong and BKVP1.Cfusionshort, see Fig. 1C, Fig. S1C and Table S1, ESI†) which contain the residues 315–335 or 325–335 respectively fused to the N-terminus of the BKVP1 core. These designs were based on the observation that, in the full-length SV40 VP1 structure,12 the invading C-terminal arm revealed a β-strand (starting with residue 328) which aligned in an antiparallel fashion with the first β-strand of the VP1 core. The designs were therefore aimed at creating an antiparallel β-hairpin at the fusion point. This arrangement could indeed be observed in the X-ray structure of the BKVP1.Cfusionlong construct (Fig. S1B and Table S2, ESI†). The structure revealed opening of the F50 pocket and insertion of residue E329 of the C-terminal arm, so that a salt bridge formed with the residue K194 located deep in the pocket. Of note, our structure is in good agreement (root mean-square deviation of 0.4 Å for Cα positions of the C-terminal arm portion) with the full-length VLP structure as determined using cryo-EM9 (Fig. 3A). Here our X-ray structure provides a greater molecular detail (2.5 Å resolution) compared to the cryo-EM structure (3.4 Å resolution). The crystal structure of BKVP1.Cfusionshort revealed deviations in the C-terminal arm part (Fig. S1B, ESI†), apparently due to its insufficient length.
Biological significance of the interactions made by the invading C-terminal arm with the F50 pocket and proximal residues was previously documented through mutagenesis studies in the SV40 polyomavirus (Table S3, ESI†). This virus is closely related to BKPyV with a sequence identity of 81% and a direct correspondence of residue numbering throughout the sequence. In particular, the importance of the C-terminal arm residues E329 and E330 was established, as the E330K single mutant and E329A-E330K/R double mutants of SV40 were non-viable (Table S3, ESI†).14 The first residue forms a salt bridge with K194 within the F50 pocket. The second residue contributes to a Ca2+ binding site which is situated next to the F50 pocket and involved in capsid assembly. In addition, point mutations E48A and E216K were shown to decrease the viral viability by several orders of magnitude. These two glutamine residues also form part of the Ca2+ binding site.14
Over the course of multiple screening rounds (Table 1), a total of 28 distinct drug-like fragment binders in the F50 pocket could be established (Fig. 3C). This rich collection allows us to formulate the pharmacophore features of this pocket (Fig. 3D). First, all hit compounds form a hydrogen bond with residue K194 in the position equivalent to the salt bridge with E329 observed in the assembled capsid. Second, these fragments typically include single or double aromatic systems that are engaged in edge-to-face π–π interactions with F50. Finally, the hydrophobic inner part of the pocket is optimally filled by aliphatic groups and occasionally halogens.
R92 site binds a conservative tetrapeptide from the C-terminal arm and can accommodate a known antipsychotic drug
While the F50 pocket is deep and relatively small, the R92 site can be best described as a larger groove located on the outer side of one VP1 monomer (Fig. 4A). The site is highly conserved in BKPyV and JCPyV with only one difference in residue P158 which is replaced by an alanine in the latter virus (Fig. 2C). Our X-ray structure of the BKVP1.Cfusionlong construct and the cryo-EM structure of the virion9 reveal that the R92 site binds the tetrapeptide 316-VDGQ-319 of the C-terminal arm. This tetrapeptide is conserved across polyomaviruses, with the residue D317 sometimes replaced with a threonine, residue Q319 being strictly conserved, and a lesser conservation of the two middle residues (Fig. 2C). These residues are involved in the interactions between the C-terminal arm and the core pentamer, as the sidechain of V316 sits deep within the R92 site and the sidechain Q319 forms two hydrogen bonds with the protein backbone in the region between F50 and R92 sites. In line with that, mutation of the equivalent or BKVP1 residue G318 to alanine in hamster polyomavirus resulted in a formation of aberrant particles.16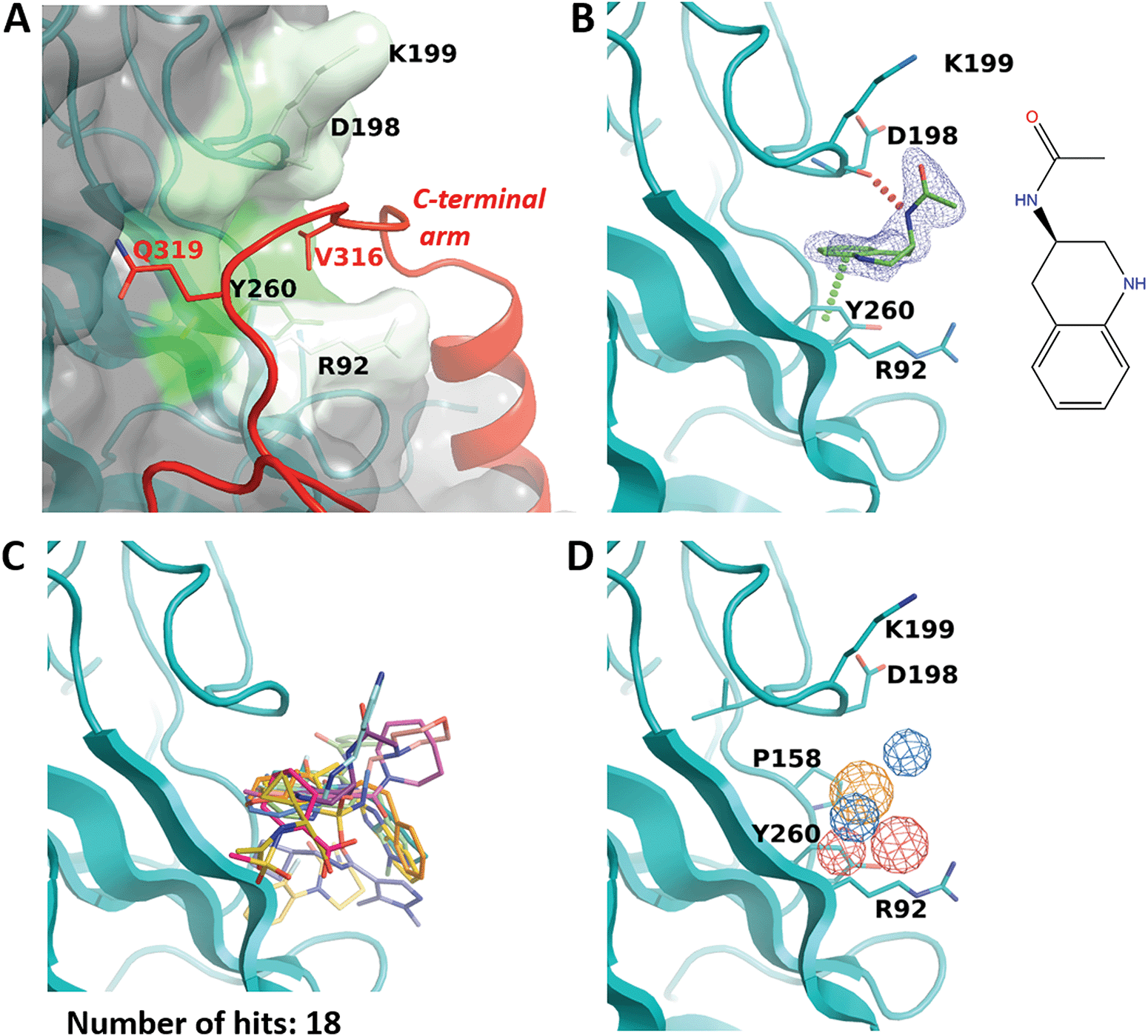 | ||
| Fig. 4 R92 site. (A) Close-up view of the pocket coloured by hydrophobicity. The invading C-terminal arm (red ribbon) from the cryo-EM structure of the VLP9 is superimposed. Residue labels for C-terminal arm are coloured red. (B) Binding of N-(1,2,3,4-tetrahydroquinolin-3-yl)acetamide (ZINC ID Z1492796719) in the R92 site. Polder difference map44 at 3σ level is shown as blue mesh. The H-bond with the main-chain carbonyl of D298 and the edge-to-face π–π interaction with Y260 are depicted by dashed lines. (C) Superposition of all fragments binding in the R92 site. (D) Consensus pharmacophore for the site. Colour coding is given in the legend to Fig. 3. | ||
Eighteen fragments were observed to bind in this pocket (Fig. 4B and C). Most typically such fragments include an aromatic ring which makes an edge-to-face π–π interaction with Y260, and engage in further hydrophobic interactions with P158, D198 and K199. The binding induces a small rotation of the Y260 side chain which allows the fragment to bind deeper in the groove. In a few cases, the fragment binding also affects the conformation of the R92 side chain.
Interestingly, we noticed that a family of antipsychotic drugs related to promazine produce a match to our derived pharmacophore in the R92 site. In the past, activity of chlorpromazine against both JCPyV and SV40 was demonstrated.56 Other drugs from this family were also shown to suppress pathogenic polyomaviruses.57 Indeed, our experiments indicated that the R92 pocket can accommodate trifluoperazine (TFP) which is chemically closely related to chlorpromazine. When BKVP1.26-299 crystals were soaked with TFP, rapid cracking and dissolution of the crystals was observed at compound concentrations above 1 mM. Thereafter we switched to using crystals of the equivalent fragment of JC polyomavirus (JCVP1.18-291). These crystals diffracted to 2.5 Å resolution and revealed a strong electron density for TFP in the equivalent pocket centred around residue R84 (Fig. 5). Here, the edge-to-face interaction of residue Y252 (Y260 in BKVP1) with one of the aromatic rings of the drug corresponds to the common pharmacophore (Fig. 4D). In addition, there is a π–cation interaction involving residue R84 (R92 in BKVP1), and finally a salt bridge between D190 (D198 in BKVP1) and the positive charge on the piperazine moiety. Using surface plasmon resonance (SPR), we measured a Kd of 0.68 mM for binding of the drug to the BKVP1.26-299 construct (Fig. S5, ESI†). The more efficient binding of TFP in the JC pocket may be due to a distinct, smaller residue (A150 in JC vs. P158 in BK) located deep in the pocket (Fig. 5 and 4D). Our discovery of a direct interaction between polyomavirus VP1 and TFP is unexpected, given the currently assumed function of this drug as endocytosis inhibitor,57 and calls for further investigation of its action mechanism. Alongside with the smaller fragments, the TFP molecule can provide a further starting point for rational inhibitor design.
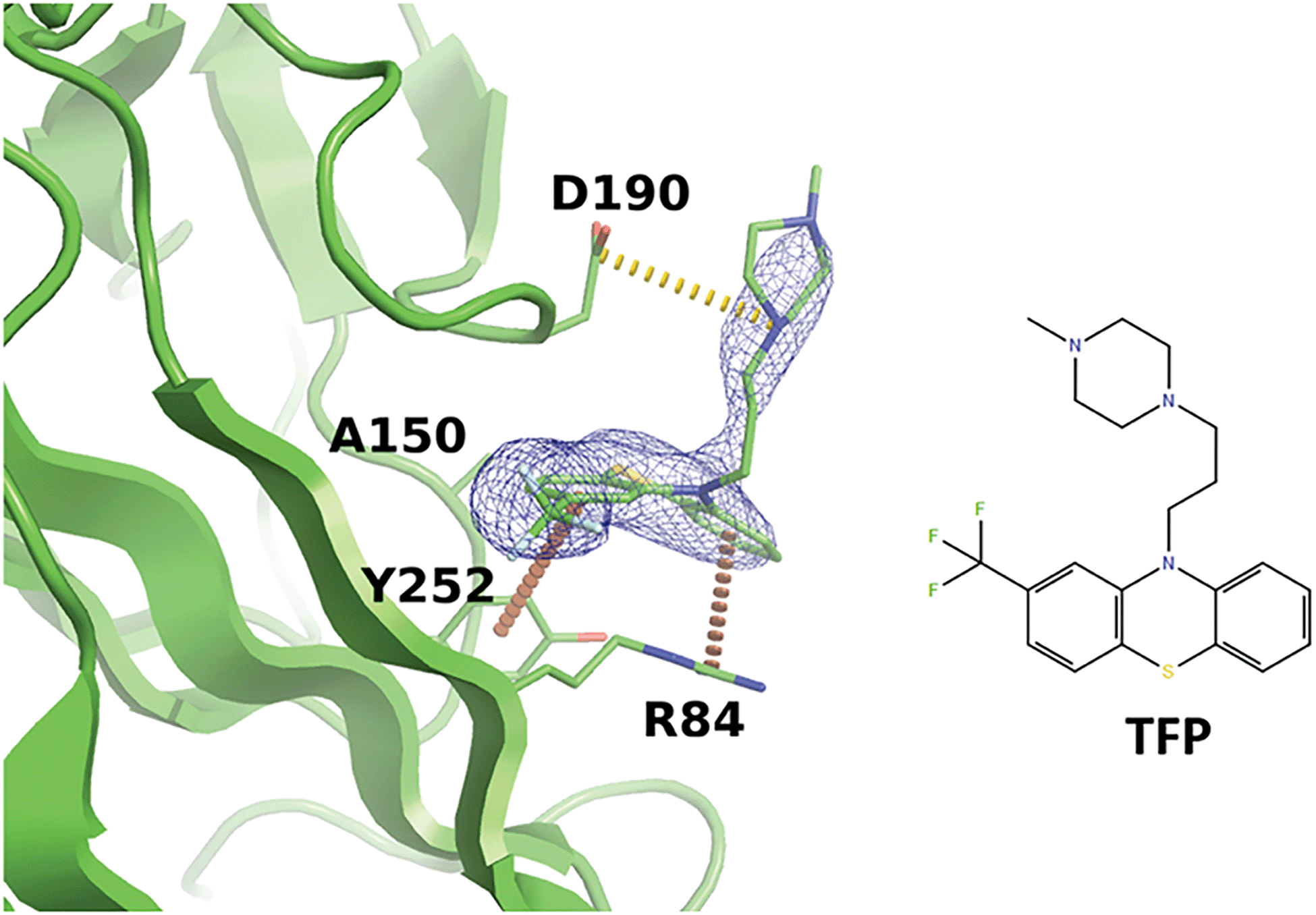 | ||
| Fig. 5 Binding of trifluoperazine in the R92 site equivalent of JCPyV VP1. Polder difference map44 at 3σ is shown as a blue mesh. Π-stacking interactions and a salt bridge are depicted by orange and yellow dashed lines, respectively. | ||
Finally, we note that the sites R92 and F50 are located at a fairly short distance (20 Å between their geometrical centres) (Fig. 2A). Within the invading C-terminal arm, the last residue of the tetrapeptide patching the R92 site, Q319, is followed by a compact outward loop (Fig. 3B). This loop continues into a β-strand starting with residue Glu329 which inserts into the F50 site. Combining pairs of fragments binding in the R92 and F50 sites respectively may be possible, provided the resulting compound includes a relatively long connector.
M109 site accommodates an α-helix from the invading C-terminal arm
The third site identified by fragment screening is M109 (Fig. 6). This site is situated at each of the five ‘feet’ of the pentamer. At this location, multiple interpentamer interactions are taking place within the assembled capsid (Fig. 1B). A major part of site M109 is made up by the outermost loop CD (residues 98–106). In addition, one side of the binding pocket is made by the first three residues of the ‘connector‘ between the core domain and the C-terminal extension (residues 296-KNPYP-300).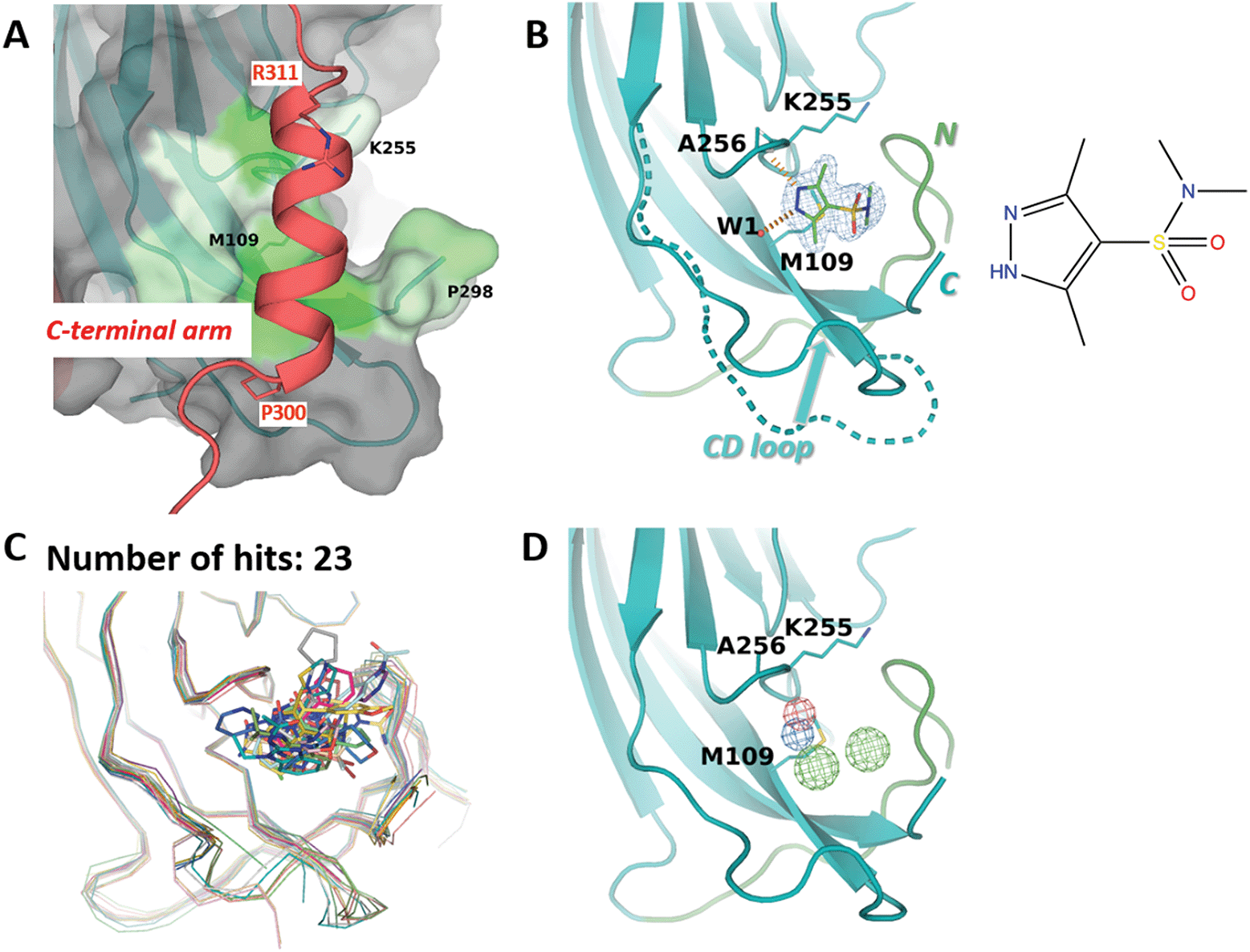 | ||
| Fig. 6 M109 site. (A) Close-up view of the binding site (surface, coloured by hydrophobicity) as observed in the cryo-EM structure.8 The invading C-terminal arm is shown as red ribbon. Both ends of C-terminal arm α-helix are depicted by sticks. (B) Binding of 3,5-dimethyl-1H-pyrazole-4-sulfonamide (ZINC ID Z94597856) to BKVP1.26-299.C104S crystals. Polder difference map44 at 3 σ level is shown (blue mesh). H-bonds with the main-chain carbonyl of A256 and a buried water molecule are depicted by red dashed lines. N-terminal residues (26–40) are shown in green. The distinct conformation of the loop CD (residues 98 to 106) in the full-length cryo-EM structure is superimposed in a dashed line. (C) Superposition of all drug-like fragments (stick representation) binding in the M109 site, including the Cα-traces of the protein in respective colours. (D) Consensus pharmacophore for the site. Colour coding is given in the legend to Fig. 3. | ||
Importantly, in the assembled capsid, the M109 site accommodates an α-helix (residues 300–311) from the invading C-terminal arm (Fig. 6A), even though there is no pronounced surface complementarity between this pocket and the helix. Notably, the M109 site has the highest predicted druggability score of the three sites (Table 2), which appears to be linked to its larger volume and a significant number of hydrophobic residues. In line with that, we could identify 23 fragments binding at this site, which represent a variety of poses and interactions with the target pocket (Fig. 6C). Most of these fragments reveal a hydrogen bond formation with the main-chain amide of residue Ala256 and hydrophobic interactions with the side chain of M109. As seen in our ligand-bound crystal structures but also observed previously41 both the CD loop and the connector (residues 296–300) between the core domain and the C-terminal extension can accommodate a multitude of conformations (Fig. 6B and C). This flexibility appears to contribute to an increased repertoire of fragments binding in the M109 site and poses thereof, opening a perspective of fragment merging towards advanced compounds.
The importance of interactions made by the C-terminal arm in the M109 site is clearly supported by mutagenesis data. In particular, mutations of residues Y299 and P300 located in the connector region of SV40 have been reported to compromise viral assembly.15 Moreover, multiple mutations within the α-helix (residues 300–311) of the C-terminal arm were shown to either completely abolish capsid assembly or yield aberrant capsids17 (Table S3, ESI†).
Conclusions and outlook
In this work we report the first expansive mapping of putative drug binding sites on the major capsid protein VP1 of a polyomavirus. Until today, identification of specific drugs targeting pathogenic polyomaviruses has proven challenging. Here we show that direct crystallographic screening with fragment libraries is a highly efficient approach to initiate rational drug design against BKVP1. As many as 144 distinct drug-like fragments were observed to bind to specific pockets on the VP1 pentamer surface, in line with previous observations of a higher sensitivity of FBS-X compared to traditional fragment screening.30Our study highlights the importance of establishing a highly performant crystallographic system as a prerequisite for a successful FBS-X study. Previously, VP1 core pentamers could be crystallized and resolved for a number of polyomaviruses.24,50,53,54,58 However, in stark contrast to mainstream crystallographic studies where one or several good crystals are typically sufficient,59 FBS-X is only possible if hundreds of isomorphous and well-diffracting crystals36,45 can be obtained. In the past, meticulous efforts to refine the crystallographic system were necessary towards a successful FBS-X with the Nsp3 macrodomain of SARS-CoV-2.40 Here we improved the reproducibility of crystallization by adjusting the length of the truncated BKVP1 construct and removing a surface-exposed cysteine residue. As a result, for the final screening round with the DSiP library43 using the XChem facility at Diamond, we have used 768 soaked crystals and obtained 634 diffraction datasets with better than 2.5 Å resolution and Rfree below 0.4. This corresponded to a remarkably successful progression (83% of mounted crystals) through several consecutive stages of the pipeline i.e. crystal mounting, data collection, phasing and refinement (Fig. S2, ESI†).
Importantly, processing of the large number of collected datasets with the latest developmental software PanDDA2 included clustering of the electron density maps, which was necessary for the efficient detection of the binding events. We suggest that such processing should become standard for future large-scale FBS-X experiments, since obtaining hundreds of perfectly isomorphous crystals is rarely feasible. In addition, improved resolution of the crystals enabled a more accurate determination of the fragment poses (Fig. S4, ESI†).
As a result, during the final FBS-X round, we detected fragment binding in as many as 19.5% of the crystals that retained diffraction quality upon soaking. In comparison, a recent FBS-X effort with SARS-CoV-2 helicase Nsp13 yielded 52 structures with bound drug-like fragments after screening 648 compounds, corresponding to a hit rate of 8%.38 A 12% hit rate has been reported by Astex for FBS-X against five protein targets.34 Most recently, screening of SARS-CoV-2 protease involved a collection of 5953 repurposed drugs.60 As expected for full-sized drugs compared to fragments, here the hit rate was much lower (0.6%), but nevertheless several compounds with cell culture activity were established.
Inhibition of capsid assembly and prevention of host receptor recognition are the two main strategies currently being considered to combat pathogenic polyomaviruses.23–26 The three novel fragment binding sites described here have a distinct relevance towards a rational design of capsid assembly inhibitors. Indeed, these sites locate on the outside of the pentameric core and map on the regions of the C-terminal arm attachment which is essential for correct capsid assembly (Fig. 2A). The functional significance of these regions is evident from previous mutagenesis studies.14–17 These data highlighted the role of interactions made by the invading C-terminal arm in each of the sites F50, R92 and M109, with the evidence for the first and the third sites being extensive (Table S3, ESI†). In addition, progressive deletions of up to 34 residues at the C-terminus of VP1 in SV40 were shown to lead to the assembly of capsids of altered size such as T = 1 particles, while larger deletions abolished the assembly completely.17
For each of the three sites we established a rich collection of chemically diverse drug-like fragment binders (28, 18 and 23 fragments for sites F50, R92 and M109 respectively, Table 2). Importantly, our bioinformatics analysis suggests that all three sites are well suited towards a rational design of drug leads. Furthermore, the obtained hits offer multiple direct possibilities towards fragment merging and growth using the established routes.61 In addition, we consolidated the recurring fragment interaction patterns seen in each site to establish the corresponding pharmacophore models (Fig. 3, 4 and 6). At the same time, the vast array of fragment binders in each of the three sites is directly providing numerous ideas on scaffold diversification beyond the core pharmacophore. Moreover, we show that the R92 site can accommodate a known antipsychotic drug, offering a further starting point of inhibitor design. As the next step, the designed inhibitors will be synthesized, tested in vitro for their affinity to VP1 pentamers using several biophysical techniques, and assessed for their antiviral activity using established cell-based assays.62 Indeed, a key strength of FBS-X is its sensitivity, while the affinity of the initial drug-like fragment hits like reported here is typically weak.27,28 In turn, the downstream designed compounds should furnish sufficient affinity and ultimately biological activity.
While we have focused on BK polyomavirus because of its clinical importance, our results are applicable for other polyomaviruses. Indeed, analysis of VP1 sequences (Fig. 2C) indicates a high conservation of the residues forming each of the F50, R92 and M109 sites. In particular, between BKPyV and JCPyV, the F50 site is highly conserved except for a single S → T substitution, the R92 is conserved with a single P → A substitution and the M109 site is conserved except for two substitutions L → I and A → G. Such high conservation opens up a prospect of developing antivirals that would be active against both BKPyV and JCPyV, and possibly other pathogenic polyomaviruses.
Experimental section
Recombinant protein production and purification
Truncated variants BKVP1.30-299 and BKVP1.26-299 were subcloned from the full-length VP1 of BKPyV (Uniprot entry P03088) using standard techniques. We used the residue numbering which differs by −1 compared to the numbering of the Uniprot entry, in line with most other BKVP1 and SV40 VP1 structures already present in the PDB. In a similar fashion, a truncated variant JCVP1.18-291 was subcloned from the full-length VP1 of JCPyV (Uniprot entry P03039).The constructs were placed into the pETSUK vector which includes a cleavable N-terminal His6-SUMO tag.63 For all constructs except BKVP1.30-299, an extra glycine was added in between of the tag and the construct, to increase the effectiveness of SUMO hydrolase. The construct BKVP1.30-299 starts with a glycine residue already. The additional C104S mutation was introduced using the QuickChange Site-Directed Mutagenesis kit (Agilent Technologies). The chimeras BKVP1.Cfusionlong and BKVP1.Cfusionshort were expressed with the same His6-SUMO tag, followed by an extra glycine, residues 315–335 and 325–335 respectively, and finally residues 39–299 of BKVP1.
Overexpression was carried out in Rosetta pLysS E. coli strain grown on ZYP-5052 autoinduction media at 24 °C until OD600 reached 4.64 Thereafter the temperature was decreased to 18 °C and the incubation was continued for additional 24 hours. Cells were pelleted by centrifugation at 4000 rpm for 45 min at 4 °C and lysed by sonication. Homogenate was clarified by centrifugation at 13000 rpm for 45 min at 4 °C. Supernatant was applied onto a Ni-chelating column equilibrated with 12.5 mM imidazole, 10 mM Tris pH 7.4, 150 mM NaCl and 5 mM β-mercaptoethanol. The column was washed by elution of 10 column volumes of IMAC buffer supplemented with 0.1% Triton X-100. The target protein was eluted with 250 mM imidazole, 10 mM Tris pH 7.4, 150 mM NaCl and 5 mM β-mercaptoethanol. His6-SUMO tag was cleaved off by adding His6-tagged SUMO hydrolase (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 ratio) during overnight dialysis against 20 mM Tris pH 7.4, 10% glycerol and 5 mM DTT. Dialyzed sample was applied on the Ni chelating column again to capture the uncleaved material, the cleaved tag and the hydrolase. Unbound fraction containing the protein of interest was dialyzed overnight against 20 mM Tris pH 7.4 with 10% glycerol, and applied on 2 × 5 mL HiTrapQ HP anion-exchange column equilibrated with 20 mM Tris pH 7.4, 5 mM DTT. After elution with a linear gradient of 0–0.6 M NaCl, the fractions containing protein were pooled together, concentrated by ultrafiltration on YM-30 (Merck Millipore) to 2 mL and applied onto a Superdex S200 10/300 GL gel-filtration column equilibrated with 20 mM Tris pH7.4, 150 mM NaCl and 5 mM DTT (for BKVP1.26-299.C104S no reducing agent was added). Fractions containing protein (>95% pure according the Coomassie-stained SDS-PAGE) were pooled together, concentrated by ultrafiltration on YM-30 to 20 mg mL−1 and stored at −80 °C until further use.
:1000 ratio) during overnight dialysis against 20 mM Tris pH 7.4, 10% glycerol and 5 mM DTT. Dialyzed sample was applied on the Ni chelating column again to capture the uncleaved material, the cleaved tag and the hydrolase. Unbound fraction containing the protein of interest was dialyzed overnight against 20 mM Tris pH 7.4 with 10% glycerol, and applied on 2 × 5 mL HiTrapQ HP anion-exchange column equilibrated with 20 mM Tris pH 7.4, 5 mM DTT. After elution with a linear gradient of 0–0.6 M NaCl, the fractions containing protein were pooled together, concentrated by ultrafiltration on YM-30 (Merck Millipore) to 2 mL and applied onto a Superdex S200 10/300 GL gel-filtration column equilibrated with 20 mM Tris pH7.4, 150 mM NaCl and 5 mM DTT (for BKVP1.26-299.C104S no reducing agent was added). Fractions containing protein (>95% pure according the Coomassie-stained SDS-PAGE) were pooled together, concentrated by ultrafiltration on YM-30 to 20 mg mL−1 and stored at −80 °C until further use.
Crystallization
Initial crystallization conditions for all constructs were found after screening with commercial crystallization kits (Hampton Research, Molecular Dimensions, Qiagen and Rigaku), followed by systematic optimization. Most of the time, sitting crystallization drops in 96-well plates were used, consisting of 0.2 μL protein solution and 0.2 μL crystallization solution. Protein stock solution was at 10 mg mL−1, supplemented with 20 mM of fresh DTT. For some crystallizations, hanging drops in 24-well plates were also made, consisting of 1 μL protein solution, 1 μL crystallization solution and 0.2 μL seed stock. The latter was prepared from one week old crystals which were crushed, diluted with crystallization solution at 1:500 ratio and stored at −20 °C. Growth time of optimized crystals was several days to one week. Optimized crystallization conditions are provided in Table S1 (ESI†).
Fragment soaking
For high-throughput soaking at XChem facility (Diamond Light Source, Oxford, UK), two-day old crystals of BKVP1.30-299 (preliminary experiments) or one-week-old BKVP1.26-299.C104S (final round) were used. Fragment soaking was done with the Echo robot.37 To this end, drug-like fragments from library stock in 100% DMSO were added to a plate with crystals to a final fragment concentration of 50 mM, while additional DMSO was added to a final concentration of 20%. Crystals were soaked for 1–2 hours, harvested and flash-cooled in liquid nitrogen.Manual soaking of the obtained crystals using a subselection of the FragLites library was done with 60 mM fragment in the presence of 10% DMSO, produced by the addition of 0.6 M fragment stock in DMSO to crystallization solution (Table S1, ESI†). During this procedure, precipitation was seen for several compounds, implying that the effective fragment concentration was lower than indicated. After 1 hour soaking, crystals were briefly dipped in a drop made of 2 μL of crystallization solution and 1 μL of 0.6 M fragment stock (final DMSO concentration 33%), which served as cryoprotectant, and flash-cooled in liquid nitrogen.
X-ray data collection and structure solution
X-ray datasets were collected using the beamlines I04-1 of the Diamond Light Source (UK), ID30B of the ESRF (France), P13 of Petra III/DESY (Germany) and F1 of CHESS (USA). For the crystals soaked with the Fraglites library, λ = 0.91 Å (13.6 keV) was used to collect anomalous scattering from Br or I atoms. For other data collections, the wavelength was close to 1 Å. Data processing was performed with autoPROC.65 High-resolution cut-offs for datasets utilized in PanDDA44 analysis and for the final PDB deposition were defined by CC1/2 > 0.366 and CC1/2 > 0.6 respectively. The apo structure of BKVP1.30-299 was solved by molecular replacement in MolRep67 using the VP1 pentamer (PDB code 4mj0) as a search model. Data for soaked crystals were processed using the DIMPLE pipeline.68 Anomalous difference maps were generated using ANODE69 as implemented in DIMPLE.After the main screening at the XChem facility, datasets containing bound fragments were identified using the PanDDA algorithm.36 Early on, PanDDA version 0.2.14 failed to capture a number of hits that were apparent in the Fo-Fc maps. It was realized that this suboptimal performance had been caused by heterogeneity of the diffraction data. Next, clustering of electron maps using the developmental PanDDA2 software (https://github.com/ConorFWild/pandda_2_gemmi) was performed. To this end, the individual datasets were scaled, phased and sharpened70 with respect to the reference ground state model, and subjected to dimensionality reduction with UMAP71 and clustering using HDBSCAN72 with default parameters. This procedure resulted in three clusters (Fig. S3, ESI†). Across the clusters, some heterogeneity was observed in the crystal cell parameters (Fig. S3A, ESI†). The most notable difference between the clusters is in the packing of the two VP1 pentamers within asymmetric unit, as one of the VP1 pentamers in cluster 3 is shifted by 4 Å along the 5-fold axis compared to two other clusters (Fig. S3B, ESI†). Finally, there is variability in the CD loop across the clusters (Fig. S3C, ESI†). We were not able to link the differences of the three clusters to fragment binding, suggesting that the observed heterogeneity originated either at crystal growth or during flash cooling. Placement of the ligands into PanDDA event maps was performed separately for each cluster.
Structure refinement was carried out in REFMAC573 using TLS parameters with automatic group definition and automatically generated local NCS restraints. Ligand restraints were generated using the Grade server (https://grade.globalphasing.org). Final crystallographic statistics for unliganded BKVP1 structures is provided in Table S2 (ESI†). Polder type electron density maps were generated using PHENIX.polder.44
Additionally, crystals of the JCVP1.18-291 construct were obtained (Table S1, ESI†). These crystals belonged to space group P21 with cell parameters a = 127.4 Å, b = 83.9 Å, c = 142.1 Å and β = 90.2°, contained two pentamers per ASU and were thus distinct from the known crystals of JCVP1 core pentamer (PDB entry 3NXG), which have space group C2 and contain one pentamer per ASU. After soaking with 25 mM TFP, our JCVP1.18-291 crystals could be used to collect diffraction data to 2.5 Å, followed by phasing by molecular replacement.
Bioinformatics
Multiple sequence alignment for polyomavirus VP1 was calculated using ClustalO74 using protein sequences downloaded from Uniprot (https://www.uniprot.org). The numbering of BKVP1 which excludes the N-terminal methionine and thus differs by −1 relative to the Uniprot numbering was used. Sequence identity and similarity were calculated in Multiple sequence viewer in Maestro Suite version 2020-1 (Schrodinger). Sequence visualizations were prepared in Jalview.75Specific binding pockets were defined through all protein residues that interacted with at least one bound fragment in our FBS-X experiments (within 4.5 Å cut-off). Druggability prediction for the binding sites was performed using Sitemap46 from the Maestro suite. Initially, searching for druggable pockets on the surface of VP1 pentamer was performed. This search was limited by the vicinity of all fragment binders (6 Å radius).
Structure renderings were done in Pymol 2.4 (Schrödinger LLC). Drug-binding pockets were coloured by hydrophobicity according to the scale proposed by Eisenber et al.76
Affinity measurements
SPR measurements were performed at the Core Facility of Biomolecular Interactions and Crystallization (CF-BIC) hosted by the Central European Institute of Technology (CEITEC) in Brno, Czech Republic, using a Bioacore T200 instrument and CM5 sensor chips (Cytiva). BKVP1.30-299 sample was immobilized via surface cysteine residues. The binding of small-molecule ligands was recorded in 20 mM Tris buffer, pH7.4, 150 mM NaCl, 5% DMSO, and analysed assuming steady-state affinity.Author contributions
A. H. M. and S. V. S. designed the research, E. M. O., A. H. M. and S. B. contributed to various stages of experimental work, D. F. and A. D. contributed to FBS-X, D. F. and C. W. developed the PanDDA2 software. E. M. O. and S. V. S. wrote the manuscript, A. H. M., A. V. A., F. V. D. and S. V. S. secured funding, all authors contributed to data analysis, manuscript editing and approved the manuscript before submission.Conflicts of interest
A. H. M. and S. D. W. are currently employed by and are shareholders in Orthogon Therapeutics. The remaining authors declare no competing interests.Acknowledgements
The authors thank Dr Vipul Chitalia (Boston University Medical Center) for discussions and support, and the staff of synchrotron beamlines I04-1 (Diamond), F1 (MacCHESS), ID30B (ESRF) and P13 (DESY) for assistance with X-ray data collection. We are also grateful to Dr Josef Houser and the CF-BIC Brno (CEITEC) team for SPR measurements. This research was supported by grants G0A5718N from the Research Foundation Flanders and C24/18/081 from Internal Fonds of KU Leuven (both to S. V. S. and A. V. A.) A. H. M. was further supported by Orthogon Therapeutics LLC. The authors acknowledge the Diamond Light Source for access to the XChem facility under proposals LB16816 and LB25586, and Instruct-ERIC for providing funding towards SPR measurements at CEITEC under project ID 16717. Fragment screening at XChem was supported by iNEXT-Discovery, grant number 871037 by the Horizon 2020 program of the European Commission.References
- Z. Wu, F. E. Graf and H. H. Hirsch, Rev. Med. Virol., 2021, 31, e2220 Search PubMed.
- F. Helle, E. Brochot, L. Handala, E. Martin, S. Castelain, C. Francois and G. Duverlie, Viruses, 2017, 9, 327 CrossRef PubMed.
- A. Cohen-Bucay, S. E. Ramirez-Andrade, C. E. Gordon, J. M. Francis and V. C. Chitalia, Kidney Med., 2020, 2, 771–786 CrossRef PubMed.
- M. Jiang, J. R. Abend, S. F. Johnson and M. J. Imperiale, Virology, 2009, 384, 266–273 CrossRef CAS PubMed.
- J. A. DeCaprio, Philos. Trans. R. Soc., B, 2017, 372, 20160276 CrossRef PubMed.
- G. R. Ambalathingal, R. S. Francis, M. J. Smyth, C. Smith and R. Khanna, Clin. Microbiol. Rev., 2017, 30, 503–528 CrossRef CAS PubMed.
- M. Philippe, F. Ranchon, L. Gilis, V. Schwiertz, N. Vantard, F. Ader, H. Labussiere-Wallet, X. Thomas, F.-E. Nicolini, E. Wattel, S. Ducastelle-Leprêtre, F. Barraco, L. Lebras, G. Salles, M. Michallet and C. Rioufol, Biol. Blood Marrow Transplant., 2016, 22, 723–730 CrossRef CAS PubMed.
- D. Sawinski and S. Goral, Nephrol., Dial., Transplant., 2015, 30, 209–217 CrossRef CAS PubMed.
- D. L. Hurdiss, M. Frank, J. S. Snowden, A. Macdonald and N. A. Ranson, Structure, 2018, 26, 839–847 CrossRef CAS PubMed.
- T. Stehle and S. C. Harrison, EMBO J., 1997, 16, 5139–5148 CrossRef CAS PubMed.
- C. D. S. Nelson, L. J. Ströh, G. V. Gee, B. A. O'Hara, T. Stehle and W. J. Atwood, J. Virol., 2015, 89, 3910–3921 CrossRef CAS PubMed.
- T. Stehle, S. J. Gamblin, Y. Yan and S. C. Harrison, Structure, 1996, 4, 165–182 CrossRef CAS PubMed.
- D. L. Hurdiss, E. L. Morgan, R. F. Thompson, E. L. Prescott, M. M. Panou, A. Macdonald and N. A. Ranson, Structure, 2016, 24, 528–536 CrossRef CAS PubMed.
- P. P. Li, A. Naknanishi, M. A. Tran, K.-I. Ishizu, M. Kawano, M. Phillips, H. Handa, R. C. Liddington and H. Kasamatsu, J. Virol., 2003, 77, 7527–7538 CrossRef CAS PubMed.
- O. Ben-nun-Shaul, H. Bronfeld, D. Reshef, O. Schueler-Furman and A. Oppenheim, J. Mol. Biol., 2009, 386, 1382–1391 CrossRef CAS PubMed.
- A. Gedvilaite, E. Aleksaite, J. Staniulis, R. Ulrich and K. Sasnauskas, Arch. Virol., 2006, 151, 1811–1825 CrossRef CAS PubMed.
- N. Yokoyama, M.-a Kawano, H. Tsukamoto, T. Enomoto, T. Inoue, R.-u Takahashi, A. Nakanishi, T. Imai, T. Wada and H. Handa, J. Biochem., 2007, 141, 279–286 CrossRef CAS PubMed.
- D. M. Salunke, D. L. Caspar and R. L. Garcea, Biophys. J., 1989, 56, 887–900 CrossRef CAS PubMed.
- J. Nilsson, N. Miyazaki, L. Xing, B. Wu, L. Hammar, T. C. Li, N. Takeda, T. Miyamura and R. H. Cheng, J. Virol., 2005, 79, 5337–5345 CrossRef CAS PubMed.
- A. Nakanishi, N. Itoh, P. P. Li, H. Handa, R. C. Liddington and H. Kasamatsu, J. Virol., 2007, 81, 3778–3785 CrossRef CAS PubMed.
- S. M. Bennett, L. Zhao, C. Bosard and M. J. Imperiale, Virology, 2015, 474, 110–116 CrossRef CAS PubMed.
- R. Sahli, R. Freund, T. Dubensky, R. Garcea, R. Bronson and T. Benjamin, Virology, 1993, 192, 142–153 CrossRef CAS PubMed.
- U. Neu, K. Woellner, G. Gauglitz and T. Stehle, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5219–5224 CrossRef CAS PubMed.
- L. J. Ströh, N. H. Rustmeier, B. S. Blaum, J. Botsch, P. Rößler, F. Wedekink, W. I. Lipkin, N. Mishra and T. Stehle, mBio, 2020, 11, e00745 CrossRef PubMed.
- M. Baier, N. H. Rustmeier, J. Harr, N. Cyrus, G. J. Reiss, A. Grafmüller, B. S. Blaum, T. Stehle and L. Hartmann, Macromol. Biosci., 2019, 19, 1800426 CrossRef PubMed.
- J. R. Kane, S. Fong, J. Shaul, A. Frommlet, A. O. Frank, M. Knapp, D. E. Bussiere, P. Kim, E. Ornelas, C. Cuellar, A. Hyrina, J. R. Abend and C. A. Wartchow, eLife, 2020, 9, e50722 CrossRef CAS PubMed.
- D. E. Scott, A. R. Bayly, C. Abell and J. Skidmore, Nat. Rev. Drug Discovery, 2016, 15, 533–550 CrossRef CAS PubMed.
- Q. Li, Front. Mol. Biosci., 2020, 7, 1–13 CrossRef PubMed.
- J. Schiebel, S. G. Krimmer, K. Röwer, A. Knörlein, X. Wang, A. Y. Park, M. Stieler, F. R. Ehrmann, K. Fu, N. Radeva, M. Krug, F. U. Huschmann, S. Glöckner, M. S. Weiss, U. Mueller, G. Klebe and A. Heine, Structure, 2016, 24, 1398–1409 CrossRef CAS PubMed.
- J. Schiebel, N. Radeva, S. G. Krimmer, X. Wang, M. Stieler, F. R. Ehrmann, K. Fu, A. Metz, F. U. Huschmann, M. S. Weiss, U. Mueller, A. Heine and G. Klebe, ACS Chem. Biol., 2016, 11, 1693–1701 CrossRef CAS PubMed.
- M. J. Hartshorn, C. W. Murray, A. Cleasby, M. Frederickson, I. J. Tickle and H. Jhoti, J. Med. Chem., 2005, 48, 403–413 CrossRef CAS PubMed.
- R. Wilcken, X. Liu, M. O. Zimmermann, T. J. Rutherford, A. R. Fersht, A. C. Joerger and F. M. Boeckler, J. Am. Chem. Soc., 2012, 134, 6810–6818 CrossRef CAS PubMed.
- D. J. Wood, J. D. Lopez-Fernandez, L. E. Knight, I. Al-Khawaldeh, C. Gai, S. Lin, M. P. Martin, D. C. Miller, C. Cano, J. A. Endicott, I. R. Hardcastle, M. E. M. Noble and M. J. Waring, J. Med. Chem., 2019, 62, 3741–3752 CrossRef CAS PubMed.
- M. O’Reilly, A. Cleasby, T. G. Davies, R. J. Hall, R. F. Ludlow, C. W. Murray, D. Tisi and H. Jhoti, Drug Discovery Today, 2019, 24, 1081–1086 CrossRef PubMed.
- N. S. Troelsen and M. H. Clausen, Chem. – Eur. J., 2020, 26, 11391–11403 CrossRef CAS PubMed.
- N. M. Pearce, T. Krojer, A. R. Bradley, P. Collins, R. P. Nowak, R. Talon, B. D. Marsden, S. Kelm, J. Shi, C. M. Deane and F. von Delft, Nat. Commun., 2017, 8, 15123 CrossRef PubMed.
- P. M. Collins, J. T. Ng, R. Talon, K. Nekrosiute, T. Krojer, A. Douangamath, J. Brandao-Neto, N. Wright, N. M. Pearce and F. von Delft, Acta Crystallogr., Sect. D: Struct. Biol., 2017, 73, 246–255 CrossRef CAS PubMed.
- J. A. Newman, A. Douangamath, S. Yadzani, Y. Yosaatmadja, A. Aimon, J. Brandão-Neto, L. Dunnett, T. Gorrie-stone, R. Skyner, D. Fearon, M. Schapira, F. von Delft and O. Gileadi, Nat. Commun., 2021, 12, 1–11 CrossRef PubMed.
- A. Douangamath, D. Fearon, P. Gehrtz, T. Krojer, P. Lukacik, C. D. Owen, E. Resnick, C. Strain-Damerell, A. Aimon, P. Ábrányi-Balogh, J. Brandão-Neto, A. Carbery, G. Davison, A. Dias, T. D. Downes, L. Dunnett, M. Fairhead, J. D. Firth, S. P. Jones, A. Keeley, G. M. Keserü, H. F. Klein, M. P. Martin, M. E. M. Noble, P. O’Brien, A. Powell, R. N. Reddi, R. Skyner, M. Snee, M. J. Waring, C. Wild, N. London, F. von Delft and M. A. Walsh, Nat. Commun., 2020, 11, 1–11 CrossRef PubMed.
- M. Schuller, G. J. Correy, S. Gahbauer, D. Fearon, T. Wu, R. E. Díaz, I. D. Young, L. C. Martins, D. H. Smith, U. Schulze-Gahmen, T. W. Owens, I. Deshpande, G. E. Merz, A. C. Thwin, J. T. Biel, J. K. Peters, M. Moritz, N. Herrera, H. T. Kratochvil, A. Aimon, J. M. Bennett, J. B. Neto, A. E. Cohen, A. Dias, A. Douangamath, L. Dunnett, O. Fedorov, M. P. Ferla, M. R. Fuchs, T. J. Gorrie-Stone, J. M. Holton, M. G. Johnson, T. Krojer, G. Meigs, A. J. Powell, J. G. M. Rack, V. L. Rangel, S. Russi, R. E. Skyner, C. A. Smith, A. S. Soares, J. L. Wierman, K. Zhu, P. O. Brien, N. Jura, A. Ashworth, J. J. Irwin, M. C. Thompson, J. E. Gestwicki, F. Von Delft, B. K. Shoichet, J. S. Fraser and I. Ahel, Sci. Adv., 2021, 7, eabf8711 CrossRef PubMed.
- U. Neu, S.-a A. Allen, B. S. Blaum, Y. Liu, M. Frank, A. S. Palma, L. J. Ströh, T. Feizi, T. Peters, W. J. Atwood and T. Stehle, PLoS Pathog., 2013, 9, e1003688 CrossRef PubMed.
- T. Krojer, R. Talon, N. Pearce, P. Collins, A. Douangamath, J. Brandao-Neto, A. Dias, B. Marsden and F. von Delft, Acta Crystallogr., Sect. D: Struct. Biol., 2017, 73, 267–278 CrossRef CAS PubMed.
- O. B. Cox, T. Krojer, P. Collins, O. Monteiro, R. Talon, A. Bradley, O. Fedorov, J. Amin, B. D. Marsden, J. Spencer, F. von Delft and P. E. Brennan, Chem. Sci., 2016, 7, 2322–2330 RSC.
- D. Liebschner, P. V. Afonine, N. W. Moriarty, B. K. Poon, O. V. Sobolev, T. C. Terwilliger and P. D. Adams, Acta Crystallogr., Sect. D: Struct. Biol., 2017, 73, 148–157 CrossRef CAS PubMed.
- P. M. Collins, A. Douangamath, R. Talon, A. Dias, J. Brandao-Neto, T. Krojer and F. von Delft, Methods Enzymol., 2018, 610, 251–264 CAS.
- T. A. Halgren, J. Chem. Inf. Model., 2009, 49, 377–389 CrossRef CAS PubMed.
- J. Wanner, D. C. Fry, Z. Peng and J. Roberts, Future Med. Chem., 2011, 3, 2021–2038 CrossRef CAS PubMed.
- D. Bowkett, R. Talon, C. Tallant, C. Schofield, F. von Delft, S. Knapp, G. Bruton and P. E. Brennan, ChemMedChem, 2018, 13, 1051–1057 CrossRef CAS PubMed.
- N. J. Bayer, D. Januliene, G. Zocher, T. Stehle, A. Moeller and B. S. Blaum, J. Virol., 2020, 94, e01664 CrossRef CAS PubMed.
- X. S. Chen, EMBO J., 1998, 17, 3233–3240 CrossRef CAS PubMed.
- A. S. Dugan, S. Eash and W. J. Atwood, J. Virol., 2005, 79, 14442–14445 CrossRef CAS PubMed.
- H.-Y. Zheng, H. Ikegaya, T. Takasaka, T. Matsushima-Ohno, M. Sakurai, I. Kanazawa, S. Kishida, K. Nagashima, T. Kitamura and Y. Yogo, Biochem. Biophys. Res. Commun., 2005, 333, 996–1002 CrossRef CAS PubMed.
- U. Neu, J. Wang, D. Macejak, R. L. Garcea and T. Stehle, J. Virol., 2011, 85, 7384–7392 CrossRef CAS PubMed.
- L. J. Ströh, M. S. Maginnis, B. S. Blaum, C. D. S. Nelson, U. Neu, G. V. Gee, B. A. O'Hara, N. Motamedi, D. DiMaio, W. J. Atwood and T. Stehle, J. Virol., 2015, 89, 6364–6375 CrossRef PubMed.
- A. C. Cheng, R. G. Coleman, K. T. Smyth, Q. Cao, P. Soulard, D. R. Caffrey, A. C. Salzberg and E. S. Huang, Nat. Biotechnol., 2007, 25, 71–75 CrossRef CAS PubMed.
- M. T. Pho, A. Ashok and W. J. Atwood, J. Virol., 2000, 74, 2288–2292 CrossRef CAS PubMed.
- S. R. Golden, D. L. Rosenstein, T. Belhorn and J. Blatt, Assay Drug Dev. Technol., 2021, 19, 373–385 CrossRef CAS PubMed.
- U. Neu, Z. M. Khan, B. Schuch, A. S. Palma, Y. Liu, M. Pawlita, T. Feizi and T. Stehle, PLoS Pathog., 2013, 9, e1003714 CrossRef PubMed.
- A. Ducruix and R. Giegé, Crystallization of Nucleic Acids and Proteins: A Practical Approach, Oxford University Press, 2nd edn, 1999 Search PubMed.
- S. Günther, P. Y. A. Reinke, Y. Fernández-Garciá, J. Lieske, T. J. Lane, H. M. Ginn, F. H. M. Koua, C. Ehrt, W. Ewert, D. Oberthuer, O. Yefanov, S. Meier, K. Lorenzen, B. Krichel, J. D. Kopicki, L. Gelisio, W. Brehm, I. Dunkel, B. Seychell, H. Gieseler, B. Norton-Baker, B. Escudero-Pérez, M. Domaracky, S. Saouane, A. Tolstikova, T. A. White, A. Hänle, M. Groessler, H. Fleckenstein, F. Trost, M. Galchenkova, Y. Gevorkov, C. Li, S. Awel, A. Peck, M. Barthelmess, F. Schlünzen, P. L. Xavier, N. Werner, H. Andaleeb, N. Ullah, S. Falke, V. Srinivasan, B. A. Francą, M. Schwinzer, H. Brognaro, C. Rogers, D. Melo, J. J. Zaitseva-Doyle, J. Knoska, G. E. Penã-Murillo, A. R. Mashhour, V. Hennicke, P. Fischer, J. Hakanpa, J. Meyer, P. Gribbon, B. Ellinger, M. Kuzikov, M. Wolf, A. R. Beccari, G. Bourenkov, D. V. Stetten, G. Pompidor, I. Bento, S. Panneerselvam, I. Karpics, T. R. Schneider, M. M. Garcia-Alai, S. Niebling, C. Günther, C. Schmidt, R. Schubert, H. Han, J. Boger, D. C. F. Monteiro, L. Zhang, X. Sun, J. Pletzer-Zelgert, J. Wollenhaupt, C. G. Feiler, M. S. Weiss, E. C. Schulz, P. Mehrabi, K. Karnĭcar, A. Usenik, J. Loboda, H. Tidow, A. Chari, R. Hilgenfeld, C. Uetrech, R. Cox, A. Zaliani, T. Beck, M. Rarey, S. Günther, D. Turk, W. Hinrichs, H. N. Chapman, A. R. Pearson, C. Betzel and A. Meents, Science, 2021, 372, 642–646 CrossRef PubMed.
- L. R. de Souza Neto, J. T. Moreira-Filho, B. J. Neves, R. L. B. R. Maidana, A. C. R. Guimarães, N. Furnham, C. H. Andrade and F. P. Silva, Jr., Front. Chem., 2020, 8, 1–18 CrossRef PubMed.
- D. Topalis, I. Lebeau, M. Krečmerová, G. Andrei and R. Snoeck, Antimicrob. Agents Chemother., 2011, 55, 1961–1967 CrossRef CAS PubMed.
- S. D. Weeks, M. Drinker and P. J. Loll, Protein Expression Purif., 2007, 53, 40–50 CrossRef CAS PubMed.
- F. W. Studier, Protein Expression Purif., 2005, 41, 207–234 CrossRef CAS PubMed.
- C. Vonrhein, C. Flensburg, P. Keller, A. Sharff, O. Smart, W. Paciorek, T. Womack and G. Bricogne, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 293–302 CrossRef CAS PubMed.
- J. Beilsten-Edmands, G. Winter, R. Gildea, J. Parkhurst, D. Waterman and G. Evans, Acta Crystallogr., Sect. D: Struct. Biol., 2020, 76, 385–399 CrossRef CAS PubMed.
- A. Vagin and A. Teplyakov, J. Appl. Crystallogr., 1997, 30, 1022–1025 CrossRef CAS.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- A. Thorn and G. M. Sheldrick, J. Appl. Crystallogr., 2011, 44, 1285–1287 CrossRef CAS PubMed.
- C. Liu and Y. Xiong, J. Mol. Biol., 2014, 426, 980–993 CrossRef CAS PubMed.
- L. McInnes, J. Healy and J. Melville, 2020, arXiv:1802.03426.
- L. McInnes, J. Healy and S. Astels, J. Open Source Softw., 2017, 2, 205 CrossRef.
- G. N. Murshudov, P. Skubák, A. A. Lebedev, N. S. Pannu, R. A. Steiner, R. A. Nicholls, M. D. Winn, F. Long and A. A. Vagin, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 355–367 CrossRef CAS PubMed.
- F. Madeira, Y. M. Park, J. Lee, N. Buso, T. Gur, N. Madhusoodanan, P. Basutkar, A. R. N. Tivey, S. C. Potter, R. D. Finn and R. Lopez, Nucleic Acids Res., 2019, 47, 636–641 CrossRef PubMed.
- A. M. Waterhouse, J. B. Procter, D. M. A. Martin, M. Clamp and G. J. Barton, Bioinformatics, 2009, 25, 1189–1191 CrossRef CAS PubMed.
- D. Eisenberg, E. Schwarz, M. Komaromy and R. Wall, J. Mol. Biol., 1984, 179, 125–142 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cb00052k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |