
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereochemical course of cobalamin-dependent radical SAM methylation by TokK and ThnK†
Michael S.
Lichstrahl‡
 ,
Craig A.
Townsend
* and
Erica K.
Sinner‡
*
,
Craig A.
Townsend
* and
Erica K.
Sinner‡
*
Department of Chemistry, The Johns Hopkins University, 3400 N Charles St, Baltimore, Maryland, USA. E-mail: ctownsend@jhu.edu
First published on 6th June 2022
Abstract
Complex carbapenems are important clinical antibiotics for difficult-to-treat infections. An essential step in the biosyntheses of these natural products is stereospecific methylation at C6 and subsequent alkylations by cobalamin-dependent radical SAM methylases such as TokK and ThnK. We have prepared isotopically labeled substrates in a stereospecific manner and found that both homologous enzymes selectively abstract the 6-pro-S hydrogen, followed by methyl transfer to the opposite face to give the (6R)-methyl carbapenam product proceeding, therefore, by inversion of absolute configuration at C6. These data clarify an unexpected ambiguity in the recently solved substrate-bound crystal structure of TokK and have led to a stereochemically complete mechanistic proposal for both TokK and ThnK.
Introduction
TokK and ThnK are cobalamin (Cbl)-dependent radical S-adenosylmethionine (rSAM) methylases that catalyze sequential methylations in complex carbapenem antibiotic biosynthesis. These enzymes share 79% sequence identity and a common substrate, (2R,3R,5R)-pantetheinylated carbapenam (PCPM, 1). However, TokK and ThnK differ in the number of methylations they catalyze in the biosyntheses of asparenomycin (6) and thienamycin (5), respectively (Scheme 1).1–4 The differences in the kinetic behavior of these two enzymes have been previously discussed,1 and the recent structural characterization of TokK greatly improved our mechanistic understanding of Cbl-dependent rSAM methyl transfer.5 Despite these advances, however, the stereochemical course of methylation by TokK could not be determined directly from the structure.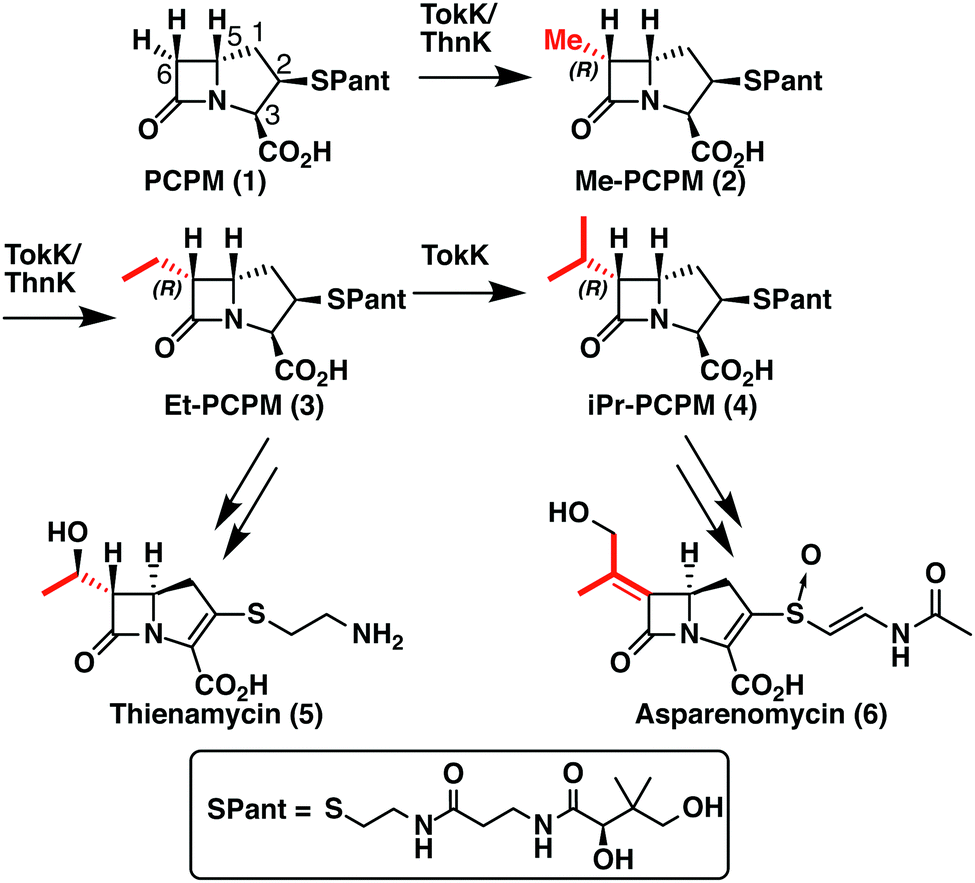 | ||
| Scheme 1 Reactions catalyzed by ThnK and TokK in the biosyntheses of Thienamycin and Asparenomycin. | ||
TokK uses a [4Fe–4S] cluster to reductively cleave SAM, generating a 5′-deoxyadenosyl radical (5′-dA˙), which abstracts a hydrogen atom from C6 of PCPM (1) to form 5′-deoxyadenosine (5′-dAH). The substrate radical is methylated by the homolytic cleavage of methylcobalamin (MeCbl), which is formed by the nucleophilic attack of cob(I)alamin on a second equivalent of SAM.6–8 A similar mechanistic strategy is employed by the related sequential methylase CysS, which, unlike TokK and ThnK, acts on a carrier protein-bound substrate.9 The first methylation by both TokK and ThnK yields Me-PCPM (2) with the R configuration at C61,2 (Scheme 1). Early studies with the thienamycin producer showed that the methyl groups are methionine-derived.3 This result has been confirmed by using isotopically labeled SAM in assays with ThnK, which leads to a corresponding mass shift in the methylated product.2 Additionally, experiments with chiral-methyl methionine gave overall retention of absolute configuration in the transferred methyl group.10 Because this result was inconsistent with direct transfer from SAM, Floss and colleagues correctly predicted the involvement of a MeCbl intermediate.
These insights notwithstanding, the identity of the hydrogen atom abstracted by the 5′-dA˙ generated by ThnK and TokK is unknown. Detailed in vitro stereochemical analysis has only been done with a small number of Cbl-dependent rSAM methylases, and has not always been straightforward. Fom3, which methylates a fosfomycin precursor, was first thought to be a nonstereoselective catalyst,11 but upon further inspection was found to proceed with inversion of configuration.12–14 Conversely, GenK, which is a methylase in gentamycin biosynthesis, proceeds with retention of configuration.15 The stereochemical relationship between H-atom abstraction and methylation is likely determined by the relative placement of the substrate to 5′-dA˙ and MeCbl in the active site. Therefore, it was thought that structural characterization of Cbl-dependent rSAM enzymes would reveal the stereochemistry of each step of the reaction. However, due to the practical limitations of working with these enzymes, progress in this area has been slow.16 There are currently four structurally characterized Cbl-dependent rSAM enzymes: OxsB,17 TsrM,18 Mmp10,19 and TokK.5 The first two structures solved in this small set of enzymes are mechanistic outliers, limiting their utility in connecting active site architecture and catalytic function. OxsB is thought to perform a radical ring contraction,17,20 and TsrM is a methylase that uses an unconventional non-radical mechanism.18,21,22 Moreover, OxsB was not co-crystallized with its substrate,17 and the position of the substrate bound to TsrM is incompetent for catalysis.18 Mmp10 was crystallized with its substrate,19 but has a non-canonical domain architecture and does not cluster with the Cbl-dependent subfamily in a sequence similarity network of rSAM enzymes.23 Therefore, the recently solved 1.9 Å X-ray structure of TokK co-crystallized with substrate, hydroxycobalamin, methionine, and 5′-deoxyadenosine (5′-dAH)5 is an advance likely to be the most representative structure of the Cbl-dependent rSAM subfamily. The TokK structure clearly shows the Cbl cofactor oriented to methylate from the bottom face of the β-lactam ring to give the (6R)-Me-PCPM (2) product.5 However, to our surprise, the structure did not clearly reveal which C6 hydrogen is abstracted by 5′-dA˙. As shown in Fig. 1, 5′-dAH is situated approximately halfway between the two modeled C6 hydrogen atoms. It was therefore unclear if hydrogen atom abstraction would be stereorandom or stereoselective, and if selective, which hydrogen would be preferentially removed by TokK. We set out to address this question by using the unambiguous chemical approach of examining the fate of stereospecifically deuterated substrates. Together with the structural insight into TokK, we now have a more complete picture of the mechanism of methylation by the Cbl-dependent rSAM subfamily.
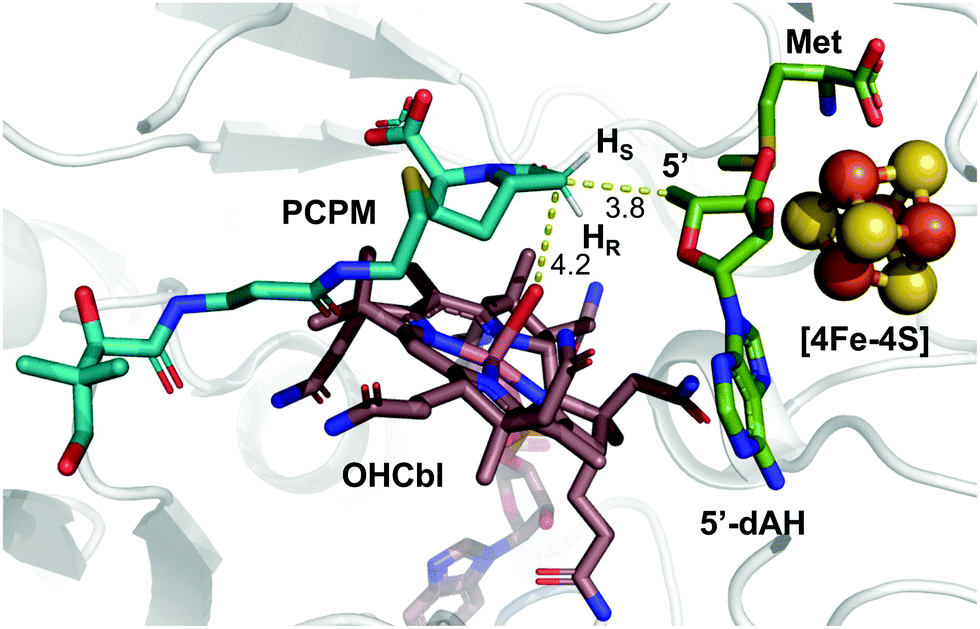 | ||
| Fig. 1 Substrate-bound TokK active site with pro-S and pro-R hydrogens modeled. (PDB ID: 7KDY). | ||
Results and discussion
Synthesis of deuterated probes
To achieve highly stereoselective labeling at C6 of the carbapenam PCPM (1) we elected to set this stereocenter prior to formation of the β-lactam ring. The instability of the carbapenam nucleus and the functionally indistinguishable reactivity of the two C6 hydrogens precludes late-stage labeling. We therefore assessed our previous syntheses of carbapenams1,24 to ensure both scalability and high deuterium incorporation at an earlier step. Monocyclic β-lactams 14a/14b were identified as key intermediates known to be accessible from dibenzyl D-aspartate.25 We envisioned, therefore, the preparation of diastereomeric [3-2H] D-aspartates 11a and 11b.Isotopic labelling of amino acids is well precedented and many deuterated amino acid derivatives are commercially available. While the asymmetric deuteration of numerous natural and non-canonical amino acids at the α-carbon (C2) has been described,26–28 stereoselective generation of β-carbon (C3) labeled substrates is far less common. A chemoenzymatic synthesis of both diastereomers of [3-2H] L-aspartic acid on large scale and with high incorporation of deuterium has been reported.29 Unfortunately, the reliance on biocatalysts precludes the ability to generate unnatural D-amino acids, necessitating a fully chemical approach.
Retrosynthetic analysis of aspartic acid led us to propose tartrate as a chiral-pool starting material. While the mono-reduction of tartrate to malate has been extensively explored in the literature,30,31 most processes rely upon single-electron mechanisms unsuitable for stereoselective introduction of deuterium at C3. We initially explored the direct reduction (D2) of a symmetrical aziridine dicarboxylate readily accessible from tartrate,32 in the hope that deuterium uptake from the catalyst surface would be stereospecific along one of the symmetry-equivalent edges of the aziridine. Instructively, reduction was observed to be stereorandom, which has precedence in the literature.33 Attempts to accomplish the same transformation using alternative reducing agents were either unsuccessful or incompatible with other functional groups. Therefore, we pursued an alternative approach using a similar symmetrical heterocycle that would serve as a chiral synthon for stereospecific deuteration.
Sharpless et al. have previously described the reductive opening of tartrate-derived cyclic sulfates using hydride reagents, including NaBH4.34,35 This reduction occurs by an SN2 mechanism and, therefore, should occur with complete inversion at the C3 stereocenter. We repeated this reduction on cyclic sulfates 8a and 8b derived from dibenzyl D- and L-tartrate with NaBD4 (Fig. 2(A) and (B)). Following hydrolysis of the intermediate alkyl sulfate and purification, 1H-NMR analysis of the product malates showed a simplified coupling pattern consistent with excellent deuterium incorporation (96–99%, over several preparations, confirmed by HRMS) and the expected stereoinversion. This procedure could be used to prepare both the (2S,3R)- and (2R,3S)-enantiomers of dibenzyl malate (9a and 9b).
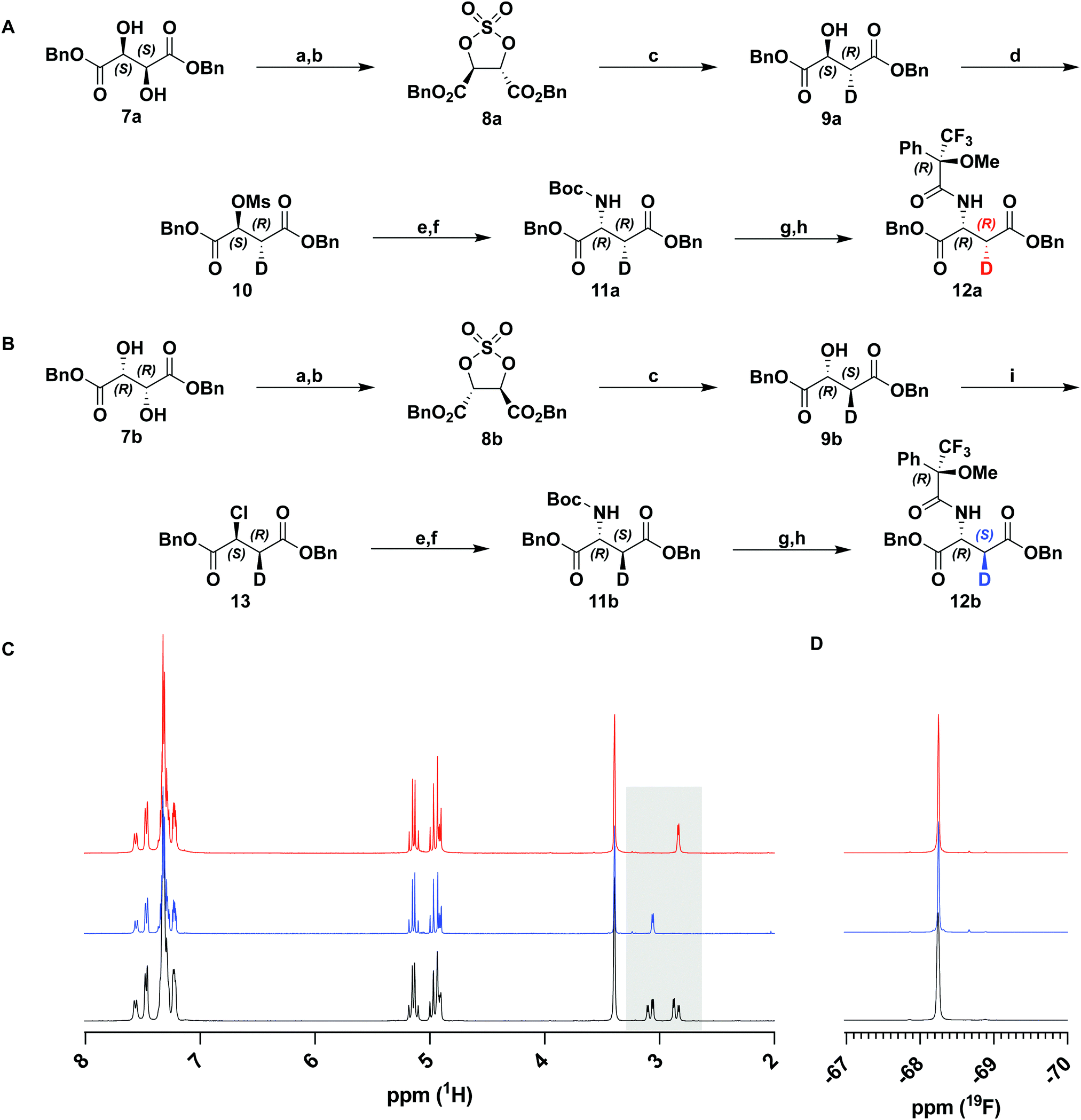 | ||
| Fig. 2 (A) and (B) Preparation of deuterated D-aspartates and their respective Mosher amides. Reagents and conditions: (a) SOCl2, DMF (cat.), DCM, 0 to 50 °C, 30 min; (b) NaIO4, RuCl3·3H2O (cat.), CH3CN, H2O, rt, 1 h; (c) NaBD4, DMAc, rt, 1 h, then, (NH4)2SO4, H2SO4, Et2O, rt, 24 h; (d) MsCl, DIPEA, DCM, 0 °C, 1.5 h; (e) NaN3, DMSO, rt, 18 h; (f) SnCl2, dioxane, H2O, 0 °C to rt, 2 h, then Boc2O, NaHCO3, 0 °C to rt, 18 h; (g) TFA, DCM, rt, 2 h; (h) (S)-(+)-MTP-Cl, NEt3, DMAP, DCM, rt, 2 h; (i) SOCl2, pyridine, CHCl3, 0 to 60 °C, 4 h. (C) 1H NMR spectra of 12a (red), 12b (blue), and unlabeled control (black). (D) 19F NMR spectra of 12a (red), 12b (blue), and unlabeled control (black). | ||
With deuterium installed stereospecifically and at a high level, we proceeded to prepare the desired D-aspartate, and further the β-lactam. Against expectation, we found very few examples of the conversion of malate to aspartate in the literature. Most precedence involves the inversion of triflates with hydroxylamine36 or hydrazine37 derivatives. Although these transformations proceeded in high yields and optical purity, the reduction of the product derivatives would not be compatible with our ester protecting groups. Instead, we chose to use azide as a nucleophilic source of nitrogen in analogous substitution reactions. Activation of 9a as mesylate 10 was accomplished in high yields with minimal elimination and was followed by displacement by NaN3. Substitution by azide gave an inseparable ca. 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of substitution and elimination products. Rather than attempt to purify the reaction product, the crude material was subjected to a one-pot tin(II) chloride-mediated reduction and subsequent Boc protection. The protected D-aspartate 11a was easily purified and isolated in 55% yield over two steps (Fig. 2(A)).
:1 mixture of substitution and elimination products. Rather than attempt to purify the reaction product, the crude material was subjected to a one-pot tin(II) chloride-mediated reduction and subsequent Boc protection. The protected D-aspartate 11a was easily purified and isolated in 55% yield over two steps (Fig. 2(A)).
Having prepared the D-aspartate from D-tartrate derived 9a, we sought to achieve the same using the L-tartrate derived 9b (Fig. 2(B)). To achieve the desired stereochemistry at C2, it was necessary to perform a double-inversion at this center. SOCl2/pyridine simultaneously accomplished the first inversion and generation of a leaving group in 13. Appel bromination was initially explored to effect a similar inversion and generate a better leaving group, but resulted in significant racemization at C2, a known phenomenon owing to repeated bromide displacements in such activated systems.36 Using an identical procedure as above, chloride 13 was displaced with azide and telescoped to 11b. A reduced yield was obtained in this step due to substantially greater elimination (>60%) in the azidation reaction. This observation suggests that both leaving group identity and stereochemistry of the adjacent deuterium significantly affect the ratio of substitution and elimination products.
To verify the desired stereochemistry and optical purity at C2 in 11a and 11b, we converted each to its respective (R)-Mosher amide 12a and 12b. Comparison of their 1H NMR spectra as shown in Fig. 2(C) demonstrated that the compounds were diastereomeric at C3. Further comparison of the corresponding 19F NMR spectra (Fig. 2(D)) to the Mosher amide prepared from unlabeled dibenzyl D-aspartate confirmed the stereochemistry at C2. Following deprotection and deprotonation to the free amine, an established procedure25 was slightly modified to efficiently cyclize both substrates to the diastereomeric [3-2H]-β-lactams 14a and 14b (Fig. 3(A)). Direct comparison of their 1H-NMR spectra, as well as the unlabeled reference compound, further secured the assigned stereochemistries (Fig. 3(B)). A simple doublet was observed at C4 of the azetidinone with J = 6.0 or 2.8 Hz for 14a and 14b, respectively, in accord with Karplus dihedral angle dependence.38 These key intermediates were separately elaborated to the correspondingly labeled PCPM isotopologues using a strategy similar to that described previously (Scheme 2).1,24 Briefly, after removal of the benzyl ester by hydrogenolysis to afford 15a and 15b, each carboxylic acid was subjected to an Arndt-Eistert homologation.39 The resultant 16a and 16b were activated by carbonyldiimidazole (CDI) and extended by the decarboxylative addition of Mg(mono-PNB malonate)2.40 Subsequent removal of the silane resulted in the formation of 17a and 17b, which were diazotized to 18a and 18b. This transformation enabled rhodium(II) catalyzed carbene insertion to form the 5-membered ring as a 2-oxocarbapenam, which was reduced, mesylated, and eliminated in each case to give carbapenems 19a and 19b. Pantetheine 1,4 addition was followed by HPLC separation of the C2 diastereomers to yield 20a and 20b. Hydrogenolysis of the PNB esters gave PCPMs 21a and 21b in 21 steps from dibenzyl tartrate.
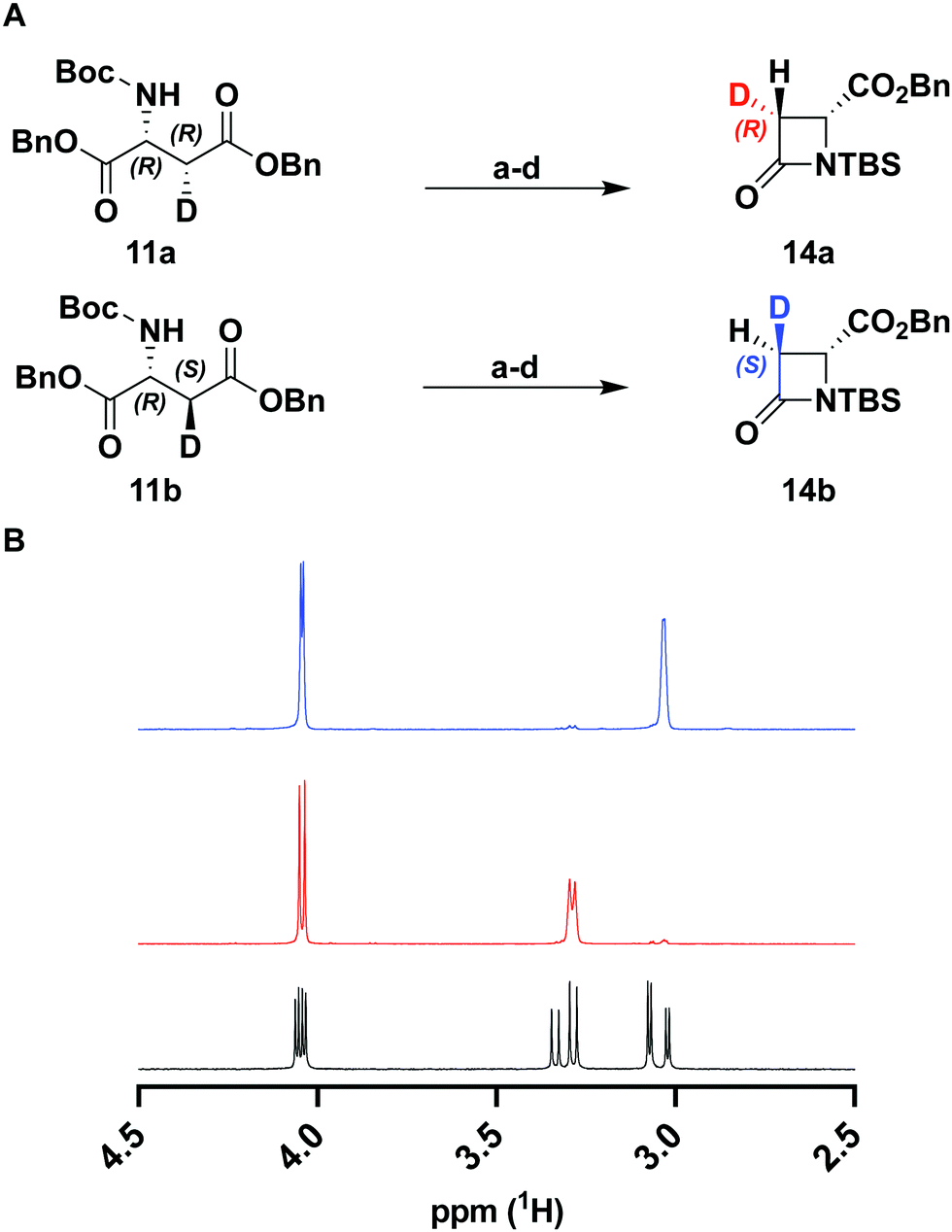 | ||
| Fig. 3 (A) synthesis of β-lactams from D-aspartates. Reagents and conditions: (a) TFA, DCM, rt, 2 h; (b) 1 M K2CO3, DCM, rt, 45 min; (c) TBSOTf, NEt3, DCM, rt, 4 h; (d) tBuMgCl, Et2O, 0 °C to rt, 16 h. (B) 1H NMR spectra of 14a (red), 14b (blue), and unlabeled control (black). | ||
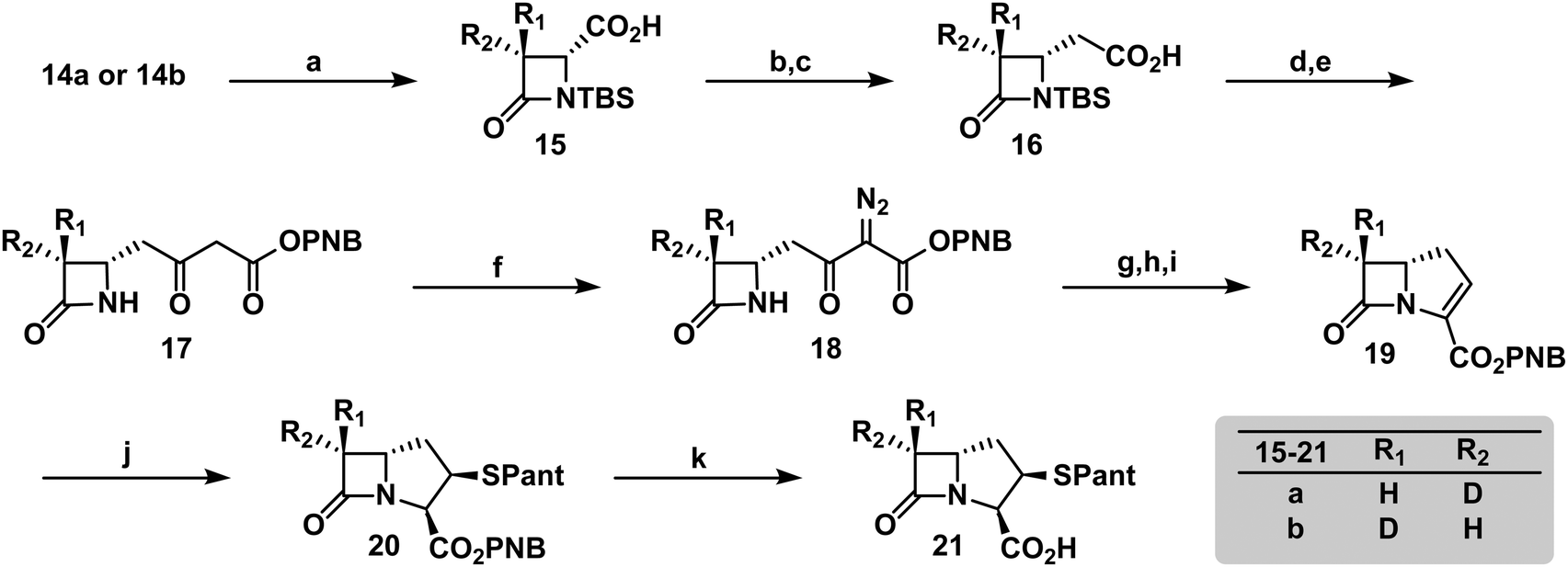 | ||
| Scheme 2 Elaboration of β-lactams 14a and 14b to the corresponding PCPMs 21a and 21b. Reagents and conditions: (a) H2 (1 atm), Pd/C, EtOAc, rt, 3 h; (b) iBuCO2Cl, NEt3, THF, 0 °C, 30 min, then CH2N2, Et2O, 0 °C to rt, 3.5 h; (c) hν, THF, H2O, rt, 18 h; (d) CDI, CH3CN, rt, 1 h, then Mg(mono-PNB malonate)2, 60 °C, 18 h; (e) TBAF, AcOH, THF, 0 °C, 30 min; (f) MsN3, DIPEA, CH3CN, 0 °C to rt, 1 h; (g) Rh2(OAc)4, PhH, reflux, 30 min; (h) NaBH4, MeOH/THF, −78 °C, 30 min; (i) MsCl, NEt3, CH2Cl2, rt, 1 h; (j) pantetheine, NEt3, CH3CN, rt, 1 h, then HPLC separation; (k) H2 (30 psi), Pd/C, THF, KHPO4 (aq), pH 7, rt, 1 h. | ||
ThnK and TokK are stereoselective
TokK and ThnK were expressed and purified as reported previously.41 Each enzyme (100 μM) was incubated with each substrate (1 mM) for 90 min and the formation of products was monitored by UPLC-HRMS. By using a substrate concentration 10-fold higher than the enzyme concentration, the first methylation in the sequence is the dominant reaction for each enzyme, giving Me-PCPM (2) as the major product. A minor amount of Et-PCPM (3) is also formed in reactions with both ThnK and TokK (Fig. S1, ESI†). When (S)-[6-2H]PCPM (21b) was incubated with each enzyme (Fig. 4 blue traces), the major product was unlabeled Me-PCPM (2), which closely matched the product formed in control reactions with unlabeled PCPM (1, Fig. 4 black traces). The formation of unlabeled product indicates that the pro-S deuterium is abstracted by 5′-dA˙ in both enzymes. The production of 5′-dA2H was also observed in reactions with (S)-[6-2H]PCPM (21b), confirming this result. Conversely, when (R)-[6-2H]PCPM (21a) was used (Fig. 4 red traces), the major product was [6-2H]Me-PCPM, showing that deuterium was retained in the product of the reaction. These reactions also produced unlabeled 5′-dAH in accord with pro-S hydrogen abstraction by 5′-dA˙. The fact that reactions with TokK and ThnK gave virtually identical results reveals that the overall architecture of their active sites is highly conserved despite the differences in their kinetic profiles.1 These results establish that both ThnK and TokK proceed with inversion of configuration at C6. The high specificity of both enzymes for the pro-S hydrogen indicates that the position of PCPM in the substrate-bound structure of TokK is not precisely poised for hydrogen abstraction. Rather, C6 of PCPM must move slightly in the active site to facilitate alignment of the pro-S hydrogen with 5′-dA˙. This motion could also place the resulting C6 radical at a more ideal distance for methyl transfer from MeCbl. Although characterized examples are still quite limited, inversion of configuration seems to be more commonly the case for Cbl-dependent rSAM methylases, as this stereochemical process is shared by Fom3,12–14 as well as GenD1,42 and MoeK.43 The latter two enzymes have been assigned a function using traditional knock-out experiments rather than through in vitro study, but as they both catalyze methylation of chiral centers, their stereochemical course can be directly inferred. In contrast, as mentioned above, GenK44 has been shown to proceed with overall retention of configuration,15 suggesting that the active site of a Cbl-dependent rSAM methylase can accommodate the cofactors in varying orientations. Interestingly, the specificity of GenK appears to be weakened when the preferred hydrogen is replaced with deuterium. In this reaction, approximately 20% of the accumulated product is deuterated, potentially due to a primary kinetic isotope effect in the initial H-abstraction step.15 This phenomenon is observed to a lesser degree in both TokK and ThnK, which accumulate ca. 7% and 4% of [6-2H]Me-PCPM, respectively, when (S)-[6-2H]PCPM (21b) is used.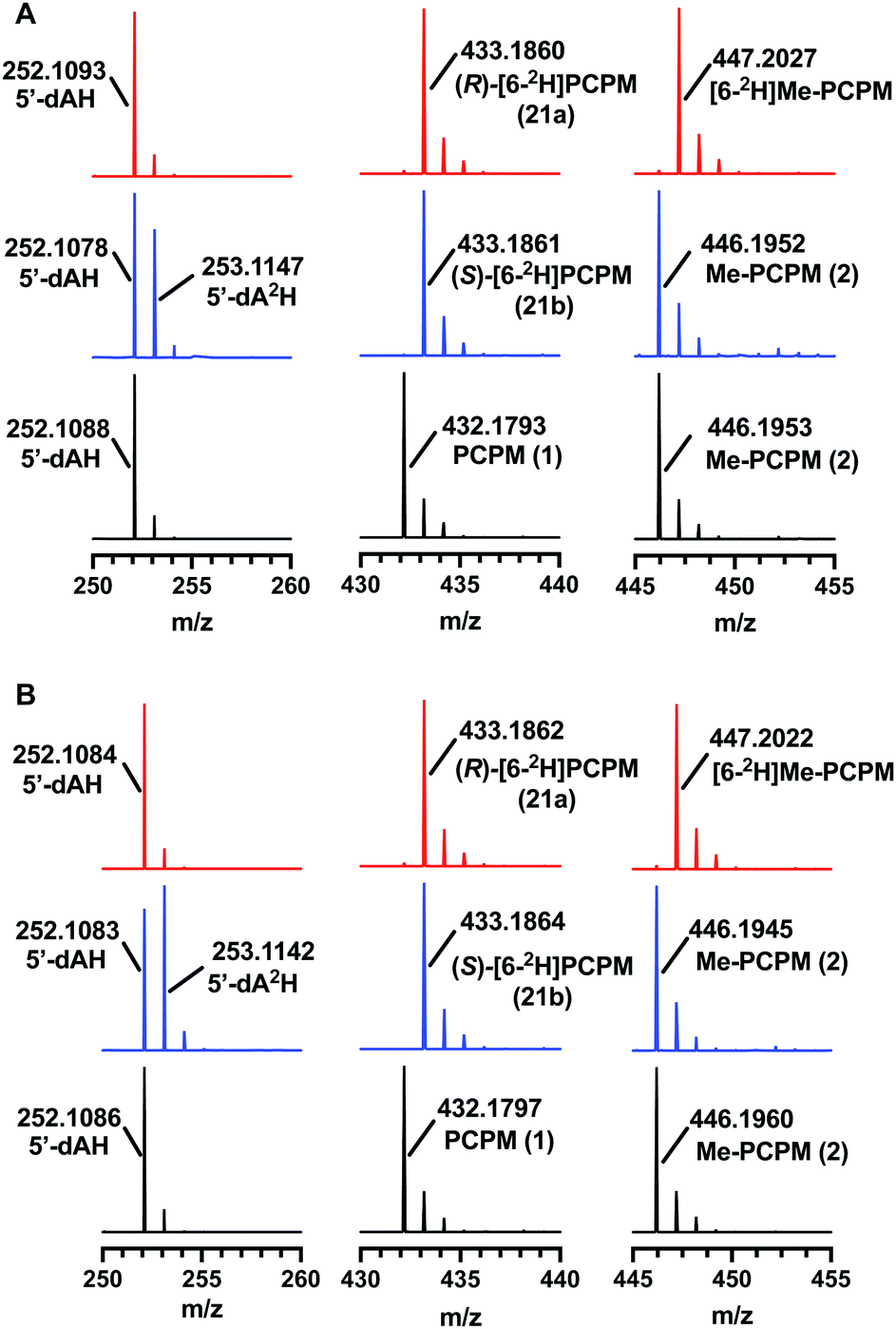 | ||
| Fig. 4 Electrospray ionization (ESI) mass data from reactions with (A) TokK and (B) ThnK. Red traces are from reactions with (R)-[6-2H]PCPM (21a), blue traces are from reactions with (S)-[6-2H]PCPM (21b), black traces are from reactions with unlabeled PCPM (1). Calculated masses are as follows: 5′-dAH, 252.1091; 5′dA2H, 253.1154; PCPM, 432.1799; [6-2H1]PCPM, 433.1862; Me-PCPM, 446.1955; [6-2H1]Me-PCPM, 447.2018. | ||
Conclusions
Here we present the synthesis of deuterated PCPMs of high isotopic and stereochemical purity from diastereomeric [3-2H]-aspartates and their subsequent enzymatic analysis with both TokK and ThnK. These experiments lead to the mechanism proposed in Scheme 3, wherein the reductive cleavage of SAM generates 5′-dA˙, which stereospecifically abstracts the pro-S hydrogen at C6 of PCPM. The resulting planar PCPM radical is methylated from the opposite face by MeCbl to give (6R)-Me-PCPM (2), affording inversion of absolute configuration at C6. Single-electron reduction of Cob(II)alamin and subsequent methylation by SAM regenerates MeCbl for the next round of methylation. Comparison of the structures of TokK solved with and without bound substrate shows that only a few residues shift upon substrate binding, while the positions of the metallocofactors remain unchanged. Moreover, a single active site must accommodate the growing alkyl chain constructed by TokK and ThnK. While our stereochemical experiments only provide insight into the first methylation by both enzymes, they complement the structural characterization of TokK to give a more complete understanding of the interactions between reacting groups. PCPMs are anchored by the binding of the C3-carboxylate to Arg280, but the stereospecificity of hydrogen abstraction points to allowable motion of PCPM in the active site, and could suggest that the PCPM crystallized in the TokK active site is not precisely poised for methyl transfer. Potential PCPM mobility could also help to explain how a single enzyme can catalyze multiple methylations both at C6 and the newly introduced carbon. The kinetic differences between TokK and ThnK and the catalysis of three vs. two methyl transfers have been partially attributed to three amino acid substitutions proximal to the active site.5 Taken together with previous kinetic and structural characterization of TokK, our stereochemical exploration of ThnK and TokK expands the limited understanding of methylation by the large and growing subfamily of Cbl-dependent rSAM enzymes and provides further insight into the even smaller subset of sequential methylases.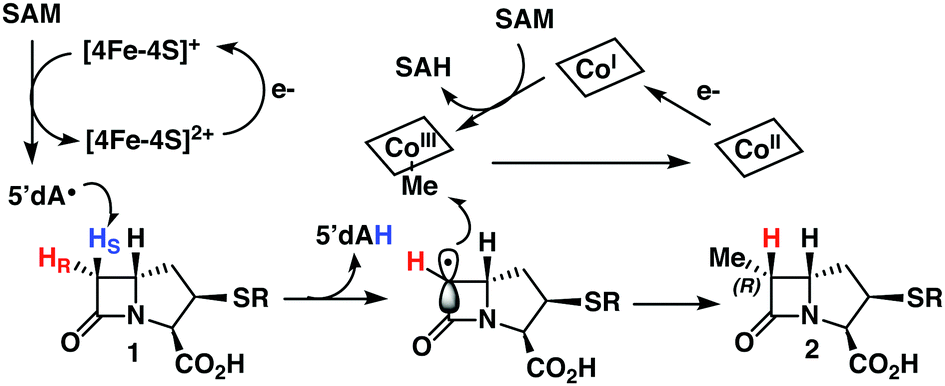 | ||
| Scheme 3 Proposed mechanism of Cbl-dependent rSAM methylation by TokK and ThnK. | ||
Author contributions
All authors were involved in conceptualizing experiments, which were carried out by M. S. L. and E. K. S. All authors were involved in data analysis, as well as writing and editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by NIH AI121072 to CAT and GM080189 to EKS. We would also like to thank Dr. I. P. Mortimer and J. Catazaro for their help with ESI-MS and NMR experiments, respectively.Notes and references
- E. K. Sinner, M. S. Lichstrahl, R. Li, D. R. Marous and C. A. Townsend, Chem. Commun., 2019, 55, 14934–14937 RSC.
- D. R. Marous, E. P. Lloyd, A. R. Buller, K. A. Moshos, T. L. Grove, A. J. Blaszczyk, S. J. Booker and C. A. Townsend, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10354 CrossRef CAS PubMed.
- J. M. Williamson, E. Inamine, K. E. Wilson, A. W. Douglas, J. M. Liesch and G. Albers-Schonberg, J. Biol. Chem., 1985, 260, 4637–4647 CrossRef CAS PubMed.
- J. Shoji, H. Hinoo, R. Sakazaki, N. Tsuji, K. Nagashima, K. Matsumoto, Y. Takahashi, S. Kozuki, T. Hattori, E. Kondo and K. Tanaka, J. Antibiot., 1982, 35, 15–23 CrossRef CAS PubMed.
- H. L. Knox, E. K. Sinner, C. A. Townsend, A. K. Boal and S. J. Booker, Nature, 2022, 602, 343–348 CrossRef CAS PubMed.
- M. R. Bauerle, E. L. Schwalm and S. J. Booker, J. Biol. Chem., 2015, 290, 3995–4002 CrossRef CAS PubMed.
- E. K. Sinner, D. R. Marous and C. A. Townsend, ACS Bio Med Chem Au, 2022, 2, 4–10 CrossRef CAS PubMed.
- S. C. Wang, Nat. Prod. Rep., 2018, 35, 707–720 RSC.
- Y. Wang, B. Schnell, S. Baumann, R. Müller and T. P. Begley, J. Am. Chem. Soc., 2017, 139, 1742–1745 CrossRef CAS PubMed.
- D. R. Houck, K. Kobayashi, J. M. Williamson and H. G. Floss, J. Am. Chem. Soc., 1986, 108, 5365–5366 CrossRef CAS.
- S. Sato, F. Kudo, S.-Y. Kim, T. Kuzuyama and T. Eguchi, Biochemistry, 2017, 56, 3519–3522 CrossRef CAS PubMed.
- M. I. McLaughlin and W. A. van der Donk, Biochemistry, 2018, 57, 4967–4971 CrossRef CAS PubMed.
- S. Sato, F. Kudo, T. Kuzuyama, F. Hammerschmidt and T. Eguchi, Biochemistry, 2018, 57, 4963–4966 CrossRef CAS PubMed.
- B. Wang, A. J. Blaszczyk, H. L. Knox, S. Zhou, E. J. Blaesi, C. Krebs, R. X. Wang and S. J. Booker, Biochemistry, 2018, 57, 4972–4984 CrossRef CAS PubMed.
- H. J. Kim, Y. Liu, R. M. McCarty and H. Liu, J. Am. Chem. Soc., 2017, 139, 16084–16087 CrossRef CAS PubMed.
- J. Bridwell-Rabb, B. Li and C. L. Drennan, ACS Bio Med Chem Au, 2022 DOI:10.1021/acsbiomedchemau.1c00051.
- J. Bridwell-Rabb, A. Zhong, H. G. Sun, C. L. Drennan and H. Liu, Nature, 2017, 544, 322–326 CrossRef CAS PubMed.
- H. L. Knox, P. Y.-T. Chen, A. J. Blaszczyk, A. Mukherjee, T. L. Grove, E. L. Schwalm, B. Wang, C. L. Drennan and S. J. Booker, Nat. Chem. Biol., 2021, 17, 485–491 CrossRef CAS PubMed.
- C. D. Fyfe, N. Bernardo-García, L. Fradale, S. Grimaldi, A. Guillot, C. Brewee, L. M. G. Chavas, P. Legrand, A. Benjdia and O. Berteau, Nature, 2022, 602, 336–342 CrossRef CAS PubMed.
- A. Zhong, Y.-H. Lee, Y. Liu and H. Liu, Biochemistry, 2021, 60, 537–546 CrossRef CAS PubMed.
- S. Pierre, A. Guillot, A. Benjdia, C. Sandström, P. Langella and O. Berteau, Nat. Chem. Biol., 2012, 8, 957–959 CrossRef CAS PubMed.
- A. J. Blaszczyk, A. Silakov, B. Zhang, S. J. Maiocco, N. D. Lanz, W. L. Kelly, S. J. Elliott, C. Krebs and S. J. Booker, J. Am. Chem. Soc., 2016, 138, 3416–3426 CrossRef CAS PubMed.
- M. I. Radle, D. V. Miller, T. N. Laremore and S. J. Booker, J. Biol. Chem., 2019, 294, 11712–11725 CrossRef CAS PubMed.
- M. J. Bodner, R. M. Phelan and C. A. Townsend, Org. Lett., 2009, 11, 3606–3609 CrossRef CAS PubMed.
- A. Soicke, C. Reuter, M. Winter, J.-M. Neudörfl, N. Schlörer, R. Kühne and H.-G. Schmalz, Eur. J. Org. Chem., 2014, 6467–6480 CrossRef CAS.
- R. Takeda, H. Abe, N. Shibata, H. Moriwaki, K. Izawa and V. A. Soloshonok, Org. Biomol. Chem., 2017, 15, 6978–6983 RSC.
- P. Ji, Y. Zhang, Y. Dong, H. Huang, Y. Wei and W. Wang, Org. Lett., 2020, 22, 1557–1562 CrossRef CAS PubMed.
- S. W. Chun and A. R. H. Narayan, ACS Catal., 2020, 10, 7413–7418 CrossRef CAS PubMed.
- K. M. Lee, K. Ramalingam, J. K. Son and R. W. Woodard, J. Org. Chem., 1989, 54, 3195–3198 CrossRef CAS.
- K. Kusuda, J. Inanaga and M. Yamaguchi, Tetrahedron Lett., 1989, 30, 2945–2948 CrossRef CAS.
- Y. Gao and C. M. Zepp, Tetrahedron Lett., 1991, 32, 3155–3158 CrossRef CAS.
- R. Hili, V. Rai and A. K. Yudin, J. Am. Chem. Soc., 2010, 132, 2889–2891 CrossRef CAS PubMed.
- F. A. Davis, C. H. Liang and H. Liu, J. Org. Chem., 1997, 62, 3796–3797 CrossRef CAS.
- Y. Gao and K. B. Sharpless, J. Am. Chem. Soc., 1988, 110, 7538–7539 CrossRef CAS.
- K. B. Sharpless and Y. Gao, US Pat., US5321143A, 1994 Search PubMed.
- R. W. Feenstra, E. H. M. Stokkingreef, R. J. F. Nivard and H. C. J. Ottenheijm, Tetrahedron, 1988, 44, 5583–5595 CrossRef CAS.
- R. V. Hoffman and K. Hwa-Ok, Tetrahedron Lett., 1990, 31, 2953–2956 CrossRef CAS.
- L. Decuyper, J. Franceus, S. Dhaene, M. Debruyne, K. Vandoorne, N. Piens, G. Dewitte, T. Desmet and M. D’hooghe, ACS Omega, 2018, 3, 15235–15245 CrossRef CAS PubMed.
- J. Fetter, K. Lempert, T. Gizur, J. Nyitrai, M. Kajtár-Peredy, G. Simig, G. Hornyák and G. Doleschall, J. Chem. Soc., Perkin Trans. 1, 1986, 221–227 RSC.
- D. W. Brooks, L. D.-L. Lu and S. Masamune, Angew. Chem., Int. Ed. Engl., 1979, 18, 72–74 CrossRef.
- E. K. Sinner and C. A. Townsend, Methods Enzymol., 2021, 669, 29–44 Search PubMed.
- C. Huang, F. Huang, E. Moison, J. Guo, X. Jian, X. Duan, Z. Deng, P. F. Leadlay and Y. Sun, Chem. Biol., 2015, 22, 251–261 CrossRef CAS PubMed.
- B. Ostash, E. H. Doud, C. Lin, I. Ostash, D. L. Perlstein, S. Fuse, M. Wolpert, D. Kahne and S. Walker, Biochemistry, 2009, 48, 8830–8841 CrossRef CAS PubMed.
- H. J. Kim, R. M. McCarty, Y. Ogasawara, Y. N. Liu, S. O. Mansoorabadi, J. LeVieux and H. W. Liu, J. Am. Chem. Soc., 2013, 135, 8093–8096 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cb00113f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |