
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceListeria monocytogenes utilizes the ClpP1/2 proteolytic machinery for fine-tuned substrate degradation at elevated temperatures†
Dóra
Balogh‡
 ,
Konstantin
Eckel‡
,
Christian
Fetzer
and
Stephan A.
Sieber
*
,
Konstantin
Eckel‡
,
Christian
Fetzer
and
Stephan A.
Sieber
*
Center for Functional Protein Assemblies (CPA), Department of Chemistry, Chair of Organic Chemistry II, Technische Universität München, 85748, Garching, Germany. E-mail: stephan.sieber@tum.de
First published on 31st May 2022
Abstract
Listeria monocytogenes exhibits two ClpP isoforms (ClpP1/ClpP2) which assemble into a heterooligomeric complex with enhanced proteolytic activity. Herein, we demonstrate that the formation of this complex depends on temperature and reaches a maximum ratio of about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at 30 °C, while almost no complex formation occurred below 4 °C. In order to decipher the role of the two isoforms at elevated temperatures, we constructed L. monocytogenes ClpP1, ClpP2 and ClpP1/2 knockout strains and analyzed their protein regulation in comparison to the wild type (WT) strain via whole proteome mass-spectrometry (MS) at 37 °C and 42 °C. While the ΔclpP1 strain only altered the expression of very few proteins, the ΔclpP2 and ΔclpP1/2 strains revealed the dysregulation of many proteins at both temperatures. These effects were corroborated by crosslinking co-immunoprecipitation MS analysis. Thus, while ClpP1 serves as a mere enhancer of protein degradation in the heterocomplex, ClpP2 is essential for ClpX binding and functions as a gatekeeper for substrate entry. Applying an integrated proteomic approach combining whole proteome and co-immunoprecipitation datasets, several putative ClpP2 substrates were identified in the context of different temperatures and discussed with regards to their function in cellular pathways such as the SOS response.
:1 at 30 °C, while almost no complex formation occurred below 4 °C. In order to decipher the role of the two isoforms at elevated temperatures, we constructed L. monocytogenes ClpP1, ClpP2 and ClpP1/2 knockout strains and analyzed their protein regulation in comparison to the wild type (WT) strain via whole proteome mass-spectrometry (MS) at 37 °C and 42 °C. While the ΔclpP1 strain only altered the expression of very few proteins, the ΔclpP2 and ΔclpP1/2 strains revealed the dysregulation of many proteins at both temperatures. These effects were corroborated by crosslinking co-immunoprecipitation MS analysis. Thus, while ClpP1 serves as a mere enhancer of protein degradation in the heterocomplex, ClpP2 is essential for ClpX binding and functions as a gatekeeper for substrate entry. Applying an integrated proteomic approach combining whole proteome and co-immunoprecipitation datasets, several putative ClpP2 substrates were identified in the context of different temperatures and discussed with regards to their function in cellular pathways such as the SOS response.
Introduction
Listeria monocytogenes is a highly stress resistant pathogenic bacterium that can survive under rapidly changing conditions.1,2 In order to cope with different stresses, the cells must detect environmental changes and promptly adjust protein expression as well as turnover in a strictly regulated manner. One characteristic trait of L. monocytogenes is its growth at various temperatures ranging from −0.4 to +45 °C posing a major challenge for adapting its cellular physiology.2 For example, heat shock induces the SOS response which is initiated by autocleavage of LexA, the repressor of the SOS genes.3,4 N- and C-terminal LexA domains (NTD and CTD, respectively) are further digested by bacterial proteases such as ClpXP (see below) to activate the SOS pathway.5–7 In L. monocytogenes, 28 genes have been identified to be under control of LexA.8 Most of them are DNA polymerases required for DNA repair. Furthermore, the induction of the SOS pathway inhibits bacterial growth, probably in order to prevent cell division after incomplete DNA replication.8,9In addition to gene regulation, heat stress generates damaged proteins, which need to be efficiently removed by the cellular proteolytic machinery. In bacteria, several proteases are capable of this process. These include caseinolytic protease P (ClpP) which, in concert with its cognate chaperones, e.g. ClpC and ClpX, digests misfolded protein substrates. ClpX is a hexameric ATP-dependent chaperone which recognizes protein substrates and directs unfolded peptide chains into the tetradecameric barrel of the ClpP serine protease for degradation.10 Some bacteria such as L. monocytogenes encode two ClpP isoforms (ClpP1 and ClpP2) with yet largely unknown cellular roles.11–15 In L. monocytogenes ClpP1 exhibits low sequence homology to ClpP isoforms from other bacteria and is expressed as an inactive heptamer with an impaired catalytic triad. Co-expression with ClpP2, a close homolog of other ClpP isoforms, yields a heterotetradecamer assembly composed of one ClpP1 and one ClpP2 heptamer ring, ClpP17P27, here referred to as ClpP1/2.12 In association with ClpX, this heterocomplex exhibits enhanced proteolytic activity in comparison to the corresponding uniform ClpX6P214 complex. Structural studies revealed that within this complex ClpP2 serves as a template to force the impaired catalytic triad of ClpP1 into an aligned conformation which enables substrate digestion.12 Moreover, recent cryo-EM data confirmed that ClpX solely docks via ClpP2 to the heterocomplex as ClpP1 lacks cognate chaperone binding sites.16 It is thus assumed that ClpP1 is needed by L. monocytogenes under certain conditions to enhance proteolytic turnover and clearance of misfolded proteins.
It is hitherto unknown why some bacteria have homotetradecameric and others heterotetradecameric ClpPs. In this study, we revealed the thermosensing ability of ClpP1/2 for heterooligomerization and investigated the unique cellular functions of ClpP1 and ClpP2 in L. monocytogenes. To achieve this, the phenotypes of ΔclpP1, ΔclpP2 and double knockout (ΔclpP1/2) strains were examined in an integrative proteomic approach using mass spectrometry-based whole proteome analysis and co-immunoprecipitation. Our data suggest that ClpP2 plays an important role to mediate substrate recognition of e.g. proteins involved in stress response while ClpP1 is a mere enhancer of proteolytic turnover.
Results and discussion
ClpP1 and ClpP2 form a heterocomplex at elevated temperatures
Previous transcription analyses showed that both clpP genes exhibit up to 7-fold higher expression levels under heat stress,4,12 indicating that heterocomplex formation is preferred at high temperatures and might have a specific biological role under these conditions. In line with this observation, heterologous co-expression and purification of L. monocytogenes ClpP1/2 in E. coli revealed that the heterocomplex is unstable at low temperatures (4 °C) and stable tetradecameric ClpP1/2 could only be obtained when the whole purification process after cell lysis was performed at room temperature (∼26 °C, Fig. 1A and B). Interestingly, M. tuberculosis ClpP1 and ClpP2 also heterooligomerize at elevated temperatures,17 which suggests that heat sensing could be a conserved biological function of ClpP. To assess whether the temperature-dependent stabilization is a general feature of ClpP1/2 and not a result of the co-expression and purification conditions, we measured heterooligomerization of separately overexpressed and purified ClpP17 and ClpP214 at different temperatures. For this, equal amounts of both purified enzymes were mixed and incubated at temperatures ranging from 0 °C to 48 °C for 10 or 30 minutes. The samples were subjected to analytical size-exclusion chromatography (SEC), and the protein composition of the tetradecamer peak was analyzed by intact protein mass spectrometry (ip-MS) (Fig. 2A). The ratio of the tetradecamer (ClpP214 and ClpP1/214) and heptamer peaks (ClpP17) differed temperature-dependently with the highest 14-mer amount observed at 30 °C (Fig. 2B). Ip-MS analysis revealed an increasing ClpP1 fraction within the tetradecameric complex up to this temperature with a maximum content of about 47–49% already after an incubation of 10 minutes (Fig. 2C). However, at higher temperatures, the 14-mer:7-mer ratio declined again. Accordingly, the ClpP1 partition decreased. As a control, the ClpP1/2 complex assembled at 30 °C was cooled down to 0 °C which resulted in a decrease of the dominant tetradecamer peak suggesting that heterocomplex formation is reversible (Fig. 2D). In order to rule out the existence of ClpP114 homocomplexes, we incubated ClpP1 at 42 °C. No major shift in the chromatogram compared to 0 °C occurred, which implies that ClpP1 is not able to build homotetradecamers even under elevated temperatures (Fig. 2E).
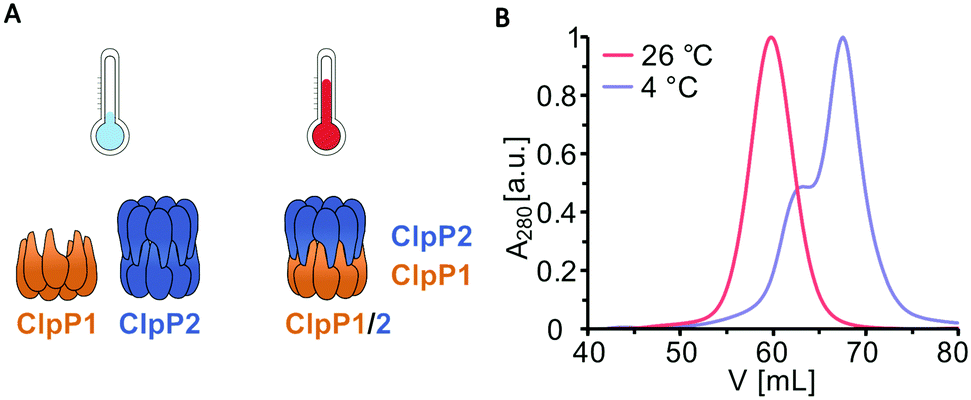 | ||
| Fig. 1 Purification of ClpP1/2 at 4 °C and at room temperature. (A) Schematic representation of ClpP1 (orange) and ClpP2 (blue) compositions at different temperatures according to size-exclusion chromatography. (B) Size-exclusion chromatography was performed on a Superdex 200 pg 16/60 column of co-expressed ClpP1/2 purified at 4 °C and 26 °C. Purifications of L. monocytogenes ClpP1/2 at 4 °C yielded a mixture of heptameric ClpP1 and tetradecameric ClpP2 (blue curve with shoulder), whereas a tetradecameric ClpP1/2 heterocomplex was obtained at room temperature (red curve). | ||
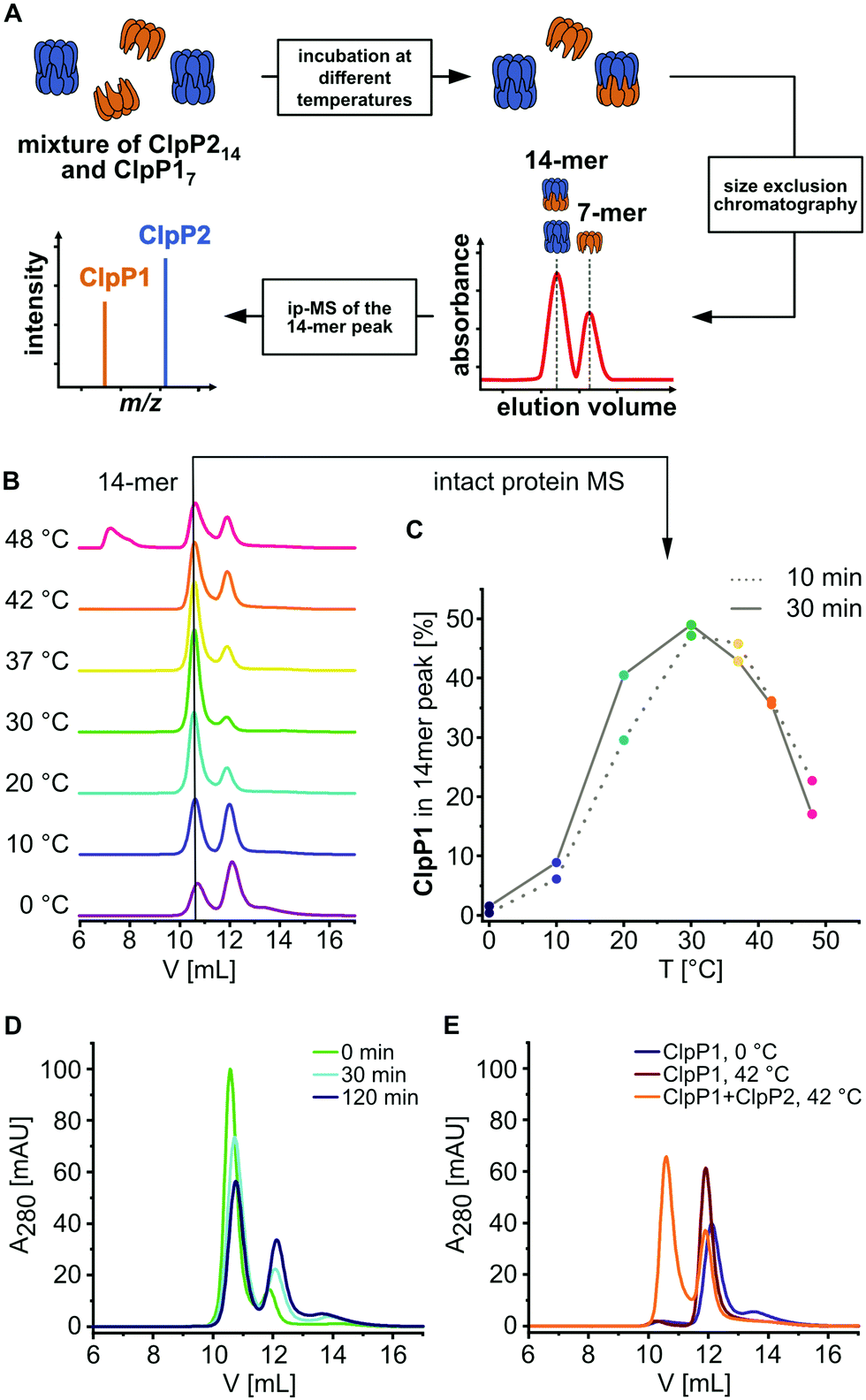 | ||
| Fig. 2 Temperature-dependent formation of the ClpP1/2 heterocomplex. (A) Scheme of the SEC/ip-MS workflow. Orange: ClpP1, blue: ClpP2. (B) Size-exclusion chromatography of ClpP17 and ClpP214 after incubation at the indicated temperatures for 30 min. Black line indicates the tetradecamer (C) Percentage of ClpP1 in the 14-mer peaks after 10 min (dotted line) or 30 min incubation (straight line), measured by intact protein mass spectrometry. (D) Size-exclusion chromatography of ClpP1/2 after incubation at 30 °C for 30 min followed by 0 °C for 0 min (green), 30 min (cyan) and 120 min (dark blue). (E) Size-exclusion chromatography of ClpP17 after incubation at 0 °C for 30 min (dark blue) and at 42 °C for 30 min (dark red) compared to a mixture of ClpP1 and ClpP2 at 42 °C for 30 min (orange). | ||
ClpP1 is not active by itself, however, in association with the heterocomplex it exhibits ten times higher protease activity per subunit compared to the ClpP2 homocomplex.12,18 In order to assess whether the heterocomplex formation translates to increased protease activity at high temperatures, we monitored the degradation of GFP-SsrA by ClpXP in the presence of an ATP regeneration system.19 Using this assay, we compared the protease activity of mixed ClpP17 and ClpP214 to solely ClpP214 at different temperatures with ClpX6 added. While ClpP1 alone is known to be inactive because of its impaired catalytic triad (Ser98, His123, Asn172) and its inability to bind AAA+ chaperones,12,16,18,20 co-incubation with ClpP2 at 37 °C and 42 °C resulted in an elevated proteolytic activity compared to a ClpP2 homocomplex at the same respective temperature (Fig. 3). The overall slower kinetics of the GFP degradation at 42 °C are attributed to the low thermal stability of ClpX and the ATP regenerating enzyme creatine kinase.21
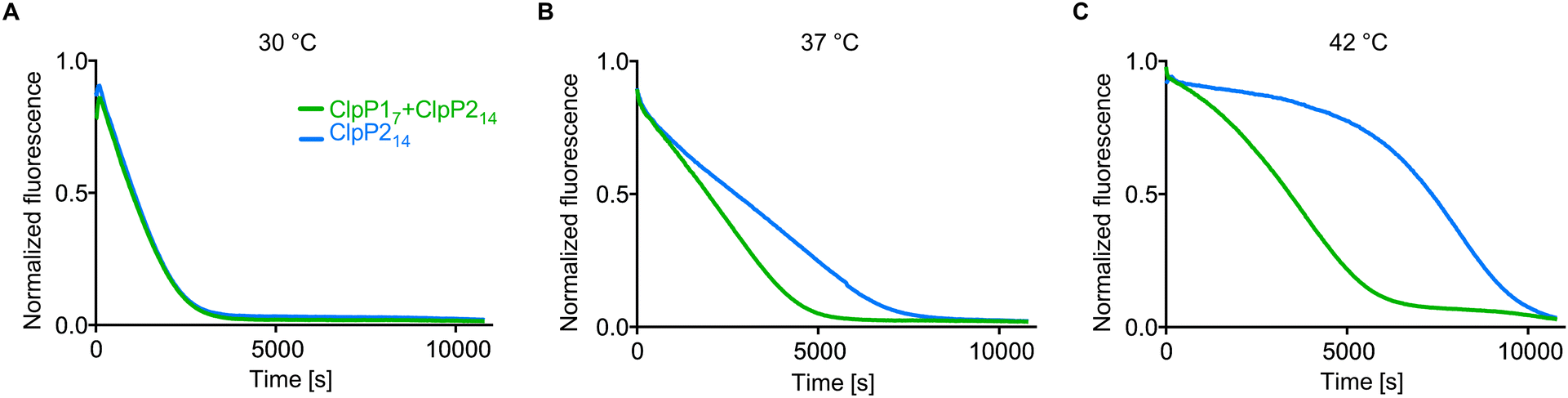 | ||
| Fig. 3 Protease activity of ClpP17 and ClpP214 at different temperatures. ClpP (green line: 0.1 μM ClpP214 and 0.2 μM ClpP17, blue line: 0.1 μM ClpP214) and 0.4 μM ClpX6 were pre-incubated for 30 min at 30 °C (A), 37 °C (B) and 42 °C (C), subsequently the degradation of 0.4 μM GFP-SsrA was measured. Means of triplicates are shown. The experiments were independently repeated with qualitatively identical results (Fig. S3, ESI†). | ||
Intracellular heterooligomerization of ClpP1 and ClpP2 under elevated temperatures
Next, we set out to investigate whether temperature-dependent heterooligomerization also occurs in living L. monocytogenes. For this purpose, we first quantified ClpP1 and ClpP2 levels via western blot at low and high temperatures to investigate if the previously observed increased expression of both clpP genes translates to the protein level.12 In order to detect ClpP isomers selectively, we inserted a 2×myc tag at the end of the endogenous clpP genes. The L. monocytogenes clpP1(191)::2×myc and L. monocytogenes clpP2(199)::2×myc strains constitutively expressed C-terminally myc-tagged ClpP1 (ClpP1-2×myc) and ClpP2 (ClpP2-2×myc) respectively, which can be visualized with an anti-c-Myc antibody in a western blot. In addition, the myc-tag also allows for co-immunoprecipitation (co-IP) experiments to study the interaction of ClpP1 and ClpP2 in situ.Indeed, we observed strongly increased expression of both isoforms at elevated temperatures compared to 10 °C and 20 °C, corroborating previous gene expression studies (Fig. S1, ESI†).12 This increase is especially pronounced for ClpP1, since its expression is lower compared to ClpP2 at temperatures < 42 °C. As both isoforms are highly abundant at elevated temperatures, we investigated a potential role of the proteins under heat stress.
Yet, the extremely weak expression of ClpP1 at low temperatures represents a challenge for co-IP experiments when studying their temperature-dependent interactions in situ, especially when choosing ClpP1 as the bait protein. Despite this limitation, we carried out co-IP experiments at high and low temperatures with an immobilized anti-c-Myc antibody in the presence of disuccinimidyl sulfoxide (DSSO) crosslinker to stabilize transient protein–protein interactions.22 The captured proteins were subjected to a tryptic digest, and the isolated peptides were measured by LC-MS/MS.
As expected, when ClpP1-2×myc was used as bait no difference in ClpP2 enrichment could be observed at 42 °C compared to 20 °C (Fig. S2a and b, ESI†). This result is likely attributed to the low abundance of heptameric ClpP1 at 20 °C which under the huge excess of ClpP2 could form sufficient amounts of heterocomplex. In contrast, as ClpP2 is generally abundant, it represents a more robust reference protein for this study. In fact, when ClpP2-2×myc was used as a bait, analysis of the ClpP1 intensities revealed a 6-times higher enrichment at 42 °C compared to 20 °C (Fig. S2c and d, ESI†). Despite this encouraging result, it is difficult to draw general conclusions due to the lack of a reliable ClpP1 expression at low temperatures.
Phenotypic characterization of L. monocytogenes ΔclpP mutants
To further investigate the cellular role of ClpP1 and ClpP2, we constructed ΔclpP1 and ΔclpP2 single mutants, as well as a ΔclpP1/2 double knockout strain (Fig. 4B, top and Fig. S4, ESI†) in L. monocytogenes EGD-e (WT). Growth curves of the mutants show that the single mutants grow at a similar rate to the wild type strain but ΔclpP2 reaches a higher optical density in the stationary phase (Fig. 4C and Fig. S5, ESI†). The double mutant ΔclpP1/2 grows substantially slower than all other investigated strains, but shows the highest optical density in the stationary phase.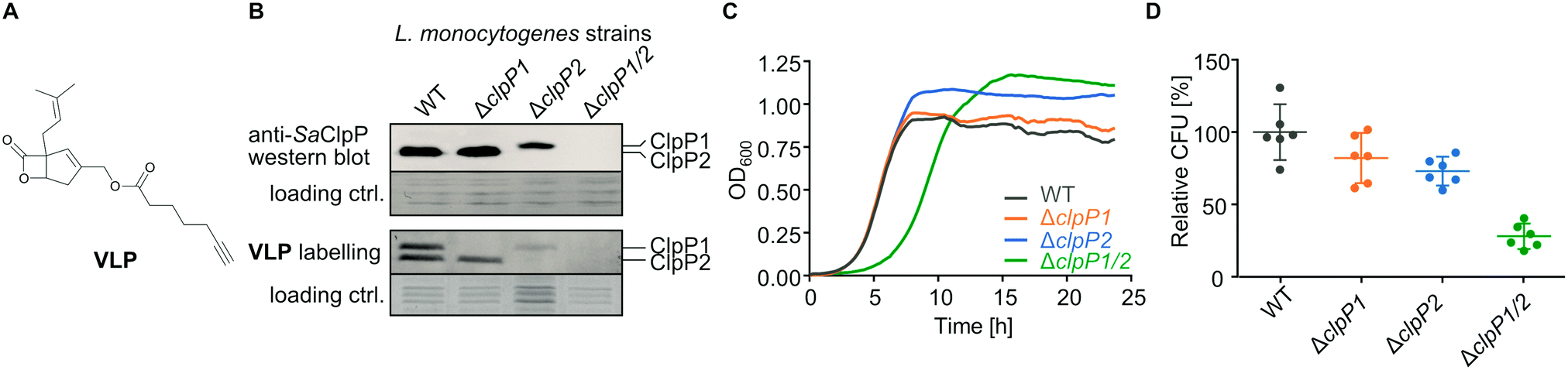 | ||
| Fig. 4 L. monocytogenes ΔclpP mutants. (A) Structure of the vibralactone probe. (B) Validation of the ΔclpP mutants by western blot (top) and by fluorescent labelling with vibralactone probe (bottom). Coomassie-stained gels were used as loading control. Full gels and membranes are depicted in Fig. S8 (ESI†). (C) Growth curves of the ΔclpP mutants in BHI medium at 37 °C. Means of triplicates are shown. The experiment was independently repeated with qualitatively identical results (Fig. S5A, ESI†). (D) Intracellular growth of the ΔclpP mutants in murine macrophages. CFUs were determined after 7 h and normalized to WT as 100% (n = 6, two independent experiments in triplicates were performed, mean ± 95% confidence interval). | ||
ClpP2 is known to be important for intracellular growth in macrophages and we thus investigated the impact of all mutants in this process as well.23 Mouse-derived macrophages were infected with L. monocytogenes EGD-e (WT), ΔclpP1, ΔclpP2 and ΔclpP1/2 and colony forming units (CFUs) determined after 7 hours (Fig. 4D). All mutants were able to replicate inside the cells, with comparable growth behaviors as observed in medium. Contrary to previous findings,23 the intracellular growth of ΔclpP2 was only weakly inhibited which might be attributed to the use of a different strain by Gaillot et al. (L. monocytogenes LO28).
We next assessed the in situ activity of both ClpPs by labelling the whole L. monocytogenes cells with vibralactone probe (VLP) (Fig. 4A). Vibralactone is the only known small molecule, which is able to label both ClpP1 and ClpP2 of L. monocytogenes by binding to their active site serine.11 VLP is equipped with a terminal alkyne tag which enables coupling to an azide-functionalized rhodamine dye via copper-catalyzed click chemistry.24–26 This way, proteins that covalently bind VLP can be visualized by fluorescence on a polyacrylamide gel. As observed previously, VLP is able to label both ClpP2 and ClpP1 in L. monocytogenes EGD-e (Fig. 4B, bottom). In line with the lack of proteolytic activity,12,20 only a weak ClpP1 band is observed in ΔclpP2 which may result from some residual binding to the active site. In addition, as expected a strong ClpP2 signal is detected in ΔclpP1.
Whole proteome analysis of ClpP1 and ClpP2 deletion mutants
ClpP is required for the maintenance and regulation of the proteome by clearing damaged proteins and degrading transcription factors. So far, the specific roles of ClpP1 and ClpP2 in L. monocytogenes are elusive. We analyzed whole proteomes of L. monocytogenes EGD-e (WT), ΔclpP1, ΔclpP2 and ΔclpP1/2 grown to early stationary phase at 37 °C and 42 °C to identify proteomic changes upon deletion of one or both proteins. We have deliberately chosen these two temperatures to ensure that both isomers are expressed, the heterocomplex formed and their function under normal and stress conditions can be compared.At 37 °C, the proteome of ΔclpP1 does not differ markedly from the wild type (Fig. 5A and Table S1, ESI†) but in ΔclpP2 and in ΔclpP1/2 many proteins are dysregulated (Fig. 5B and C). The dysregulated proteins in ΔclpP2 and ΔclpP1/2 are highly overlapping: 89% of the proteins that are upregulated in ΔclpP1/2 compared to the wild type are also upregulated in ΔclpP2 and the same applies for 82% of the downregulated proteins in the double mutant (Fig. 5D and E). However, a notable difference is the exclusive downregulation of 123 proteins solely in ΔclpP2 compared to the double mutant.
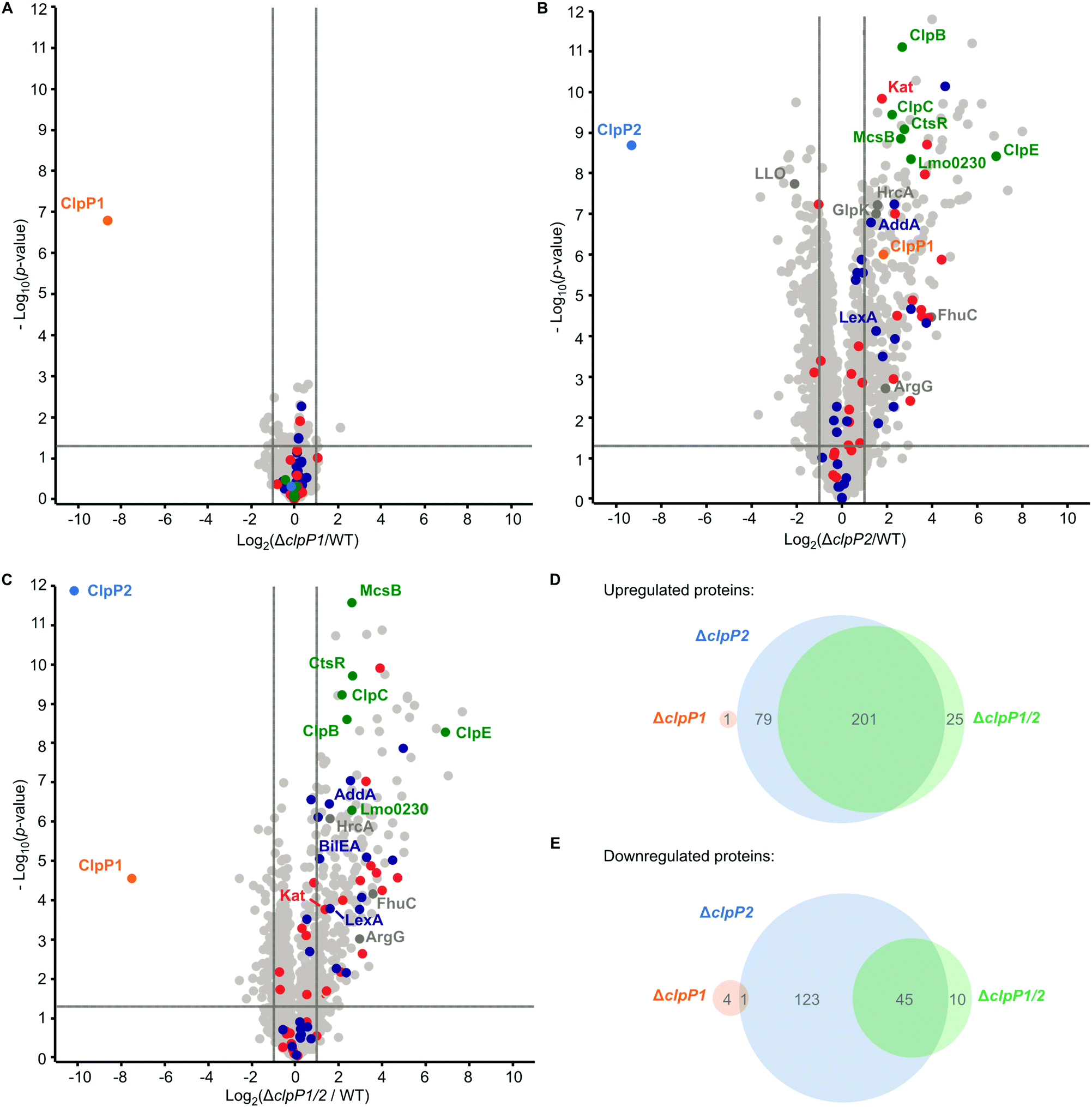 | ||
| Fig. 5 Whole proteome analysis of the L. monocytogenes ΔclpP mutants at 37 °C. (A)–(C) Proteomes of L. monocytogenes ΔclpP1 (A), ΔclpP2 (B) and ΔclpP1/2 (C) compared to the WT. Bacterial cultures were grown to stationary phase at 37 °C. −log10p-values from two-sample Student's t-test are plotted against log2 ratios of LFQ protein intensities. The vertical grey lines show 2-fold enrichment, the horizontal grey lines show −log10t-test p-value = 1.3. Samples were prepared in triplicates in two independent experiments (n = 6). Class III heat shock proteins (green), SOS response proteins (dark blue) and iron-containing proteins (red) are highlighted. Other proteins mentioned in the text are highlighted in dark grey if they are significantly dysregulated in the respective plot. ClpP1 and ClpP2 are shown in orange and blue respectively. (D) and (E) Venn-diagrams showing the up-(D) and downregulated (E) proteins in the proteomes of the ΔclpP mutants compared to the WT (fold enrichment ≥ 2, –log10t-test p-value ≥ 1.3, ClpP1 and ClpP2 excluded). | ||
UniProt keyword and Gene Ontology Biological Process (GOBP) term analyses of the proteomic data were performed with the aGOtool (agotool.org) (Tables S3–S10, ESI†).27 All proteins detected at 37 °C in the whole proteomes of L. monocytogenes EGD-e and all mutants were combined after categorical filtering and used as background. Among the upregulated proteins, the GOBP term “response to stimulus” and “regulation of transcription” was significantly enriched in both ΔclpP2 and in ΔclpP1/2 (Table S3, ESI†). Notably, SOS response-related terms (cellular response to DNA damage stimulus, DNA repair) were specifically enriched only in ΔclpP1/2. This indicates that both ClpPs are needed for full regulation of the SOS response in L. monocytogenes. Activation of the SOS response inhibits cell division in L. monocytogenes4 and in E. coli,28 which rationalizes the observed slower growth of ΔclpP1/2 compared to the wild type.
Additionally, the class III heat shock proteins (CtsR, McsB, ClpB, ClpC, ClpE and the Lmo0230 protein) were upregulated in both ΔclpP2 and ΔclpP1/2. The class I heat shock proteins were not overexpressed, except for their repressor, HrcA. Most of the class II HSPs (except for GlpK and BilEA, which is also an SOS response protein) and their positive regulator σB were also not dysregulated. Of the 28 proteins, which have been found in a genome-wide screen for temperature sensitivity,29 only two (ClpB and AddA) were significantly upregulated in ΔclpP2 and ΔclpP1/2. This, and the fact that the class I and II heat shock proteins were not induced, highlights the differences between the stress caused by clpP2 deletion and heat stress, even though class III heat shock proteins and parts of the SOS response are induced in the mutants lacking clpP2. Iron-containing and iron–sulfur proteins were also significantly upregulated in ΔclpP2 and in ΔclpP1/2. In S. aureus, it has been shown that ClpP degrades damaged iron–sulfur proteins,30,31 which could also be the case in L. monocytogenes. Additionally, ClpP has been connected to iron homeostasis and maintaining the oxidative balance inside the cell.32–34
At 42 °C, in general more proteins are dysregulated than at 37 °C in all whole proteomes. Although there are more proteins dysregulated for ΔclpP1 at 42 °C compared to 37 °C, there is surprisingly little impact of a ClpP1 deletion on the proteome level (Fig. 6A and Table S2, ESI†). Among the upregulated proteins are the two virulence-associated proteins internalin B (InlB) and listeriolysin O (LLO). Internalin B plays a role in receptor-mediated endocytosis of non-phagocytic cells, whereas listeriolysin O is a pore-forming toxin needed for subsequent vacuole opening to enter the cytosol of infected cells.1 In addition, FhuC is upregulated, which is an ABC ATPase involved in the membrane transport of iron(III)hydroxamates in S. aureus.35
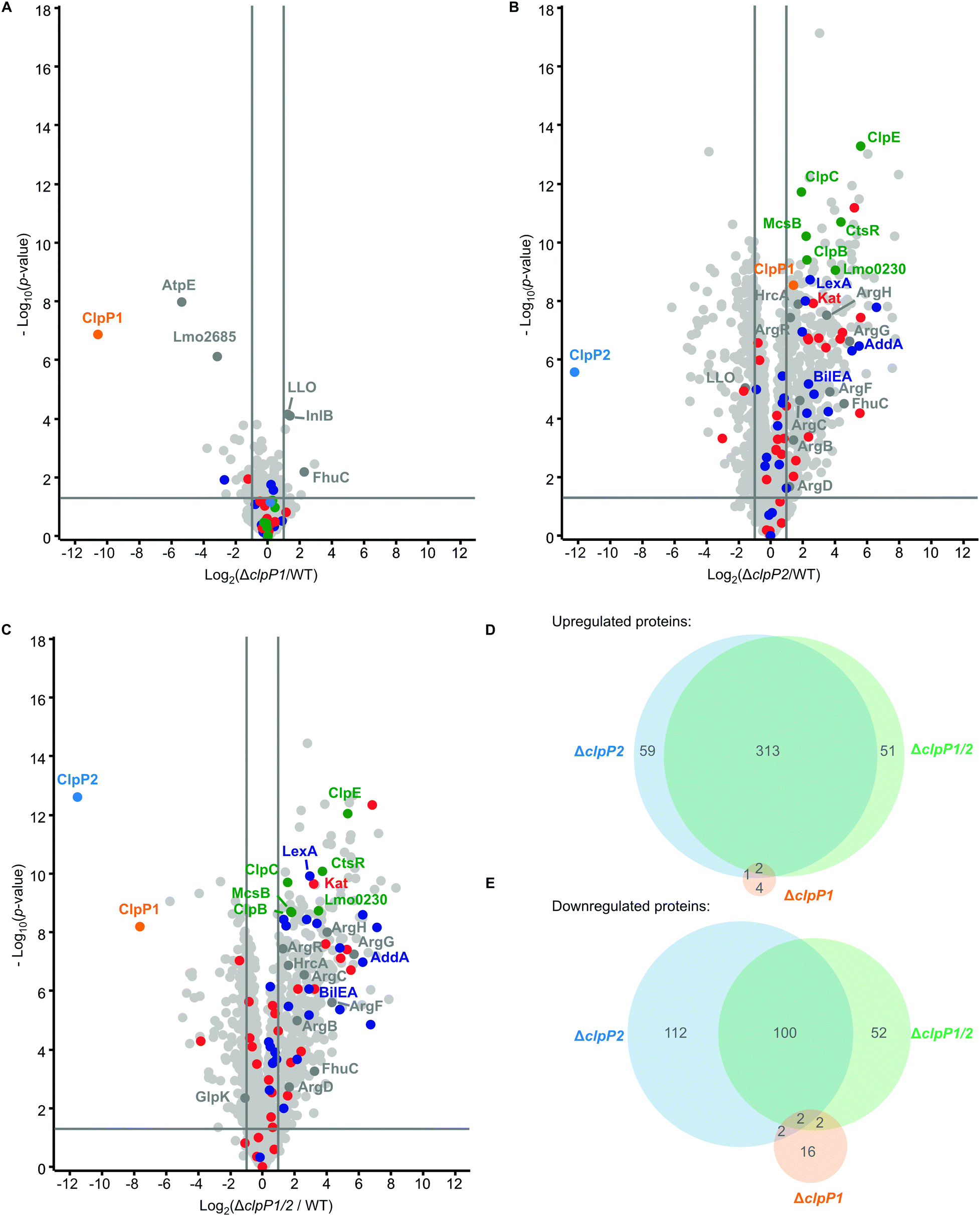 | ||
| Fig. 6 Whole proteome analysis of the L. monocytogenes ΔclpP mutants at 42 °C. (A)–(C) Proteomes of L. monocytogenes ΔclpP1 (A), ΔclpP2 (B) and ΔclpP1/2 (C) compared to the WT. Bacterial cultures were grown to stationary phase at 42 °C. −Log10p-values from two-sample Student's t-test are plotted against log2 ratios of LFQ protein intensities. The vertical grey lines show 2-fold enrichment, the horizontal grey lines show −log10t-test p-value = 1.3. Samples were prepared in triplicates in two independent experiments (n = 6). Class III heat shock proteins (green), SOS response proteins (dark blue) and iron-containing proteins (red) are highlighted. Other proteins mentioned in the text are highlighted in dark grey if they are significantly dysregulated in the respective plot. ClpP1 and ClpP2 are shown in orange and blue respectively. (D) and (E) Venn-diagrams showing the up-(D) and downregulated (E) proteins in the proteomes of the ΔclpP mutants compared to the WT (fold enrichment ≥ 2, –log10t-test p-value ≥ 1.3, ClpP1 and ClpP2 excluded). | ||
The most significantly downregulated proteins for ΔclpP1 at 42 °C are the F-ATPase subunit c (AtpE) and Lmo2685, a component of the phosphotransferase system (PTS). Yet, we identified no protein which is upregulated in ΔclpP1 and ΔclpP1/2, but not in ΔclpP2 at both temperatures. Since ClpX and most likely other chaperones bind solely to ClpP2,16 a deletion of ClpP1 is expected to solely adjust the speed of substrate degradation with little impact on the substrate scope itself. Thus, static proteome analysis may not capture the dynamics of protein digest, as during cell harvest and lysis ClpP2 still retains its activity, which could diminish observed proteome changes between ΔclpP1 and the wild type.
For ΔclpP2 and ΔclpP1/2, again many more proteins are dysregulated (Fig. 6B and C), with 375 and 366 upregulated proteins for either deletion mutant. 216 proteins are downregulated in ΔclpP2 and 156 proteins are downregulated in ΔclpP1/2. Yet, there is still a high overlap between dysregulated proteins of ΔclpP2 and ΔclpP1/2. 86% of proteins upregulated in ΔclpP1/2 are also upregulated in ΔclpP2 and the same applies to 65% of downregulated proteins of ΔclpP1/2 (Fig. 6D and E). Again a notable difference is the downregulation of 112 proteins in ΔclpP2, which are not affected in ΔclpP1/2. In line with whole proteomes at 37 °C, both deletion mutants show an upregulation of iron- or iron–sulfur-containing proteins and class III heat shock proteins. Interestingly, for ΔclpP2 at 42 °C we could identify SOS-related GOBP terms (e.g. DNA repair, base-excision repair, cellular response to DNA damage stimulus) to be upregulated, which distinguishes it from the same deletion mutant at 37 °C (Tables S7 and S8, ESI†). Yet, the actual term “SOS response” is only significantly upregulated for ΔclpP1/2 at 42 °C, further supporting the effect of both ClpPs on the SOS response regulation.
In addition, the pyrimidine and especially the UMP de novo biosynthesis are highly downregulated for ΔclpP2 and ΔclpP1/2 at both temperatures (Tables S9 and S10, ESI†). This downregulation is especially pronounced at 42 °C with nearly every protein of the pyr operon affected. Recently, a downregulation of the UMP biosynthesis was also discovered for ΔclpP of S. aureus, which was subsequently confirmed on the metabolite level.36 In contrast, the purine biosynthesis is not heavily affected in L. monocytogenes, which differs from the S. aureus ΔclpP proteome.
There are also some notable differences to the 37 °C whole proteomes. A majority of arginine biosynthetic proteins (ArgB, ArgC, ArgD, ArgF, ArgG, ArgH) is upregulated only at 42 °C both for ΔclpP2 and ΔclpP1/2, together with their repressor ArgR.
Co-immunoprecipitation of ClpP1 and ClpP2
In case of clpP deletion, substrates are expected to accumulate. However, to distinguish putative ClpP substrates from downstream effects caused by clpP deletion, a more in-depth proteomic investigation was required. As substrates also need to be engaged by chaperones and adaptors bound to ClpP, we conducted co-immunoprecipitation (co-IP) experiments. Lacking an antibody that can distinguish between ClpP1 and ClpP2, we performed the co-IP in ΔclpP1 and ΔclpP2 mutants in order to selectively target one of the two proteins. Cells were grown to stationary phase at 37 °C and 42 °C, respectively, and interacting proteins were covalently crosslinked with DSSO (XL-co-IP). The ClpPs were precipitated with a polyclonal anti-ClpP antibody and binding partners of each ClpP isoform were selectively pulled down. To account for background binding to the antibody, the co-IP was also performed with an isotype control antibody lacking the specific target binding in parallel. The precipitated proteins were digested with trypsin and analyzed by LC-MS/MS. 451 significantly enriched proteins were found for ClpP1 and 468 for ClpP2 at 37 °C as well as 232 and 145 at 42 °C, respectively (Fig. 7 and Fig. S6, ESI†). In order to decipher putative substrates, we searched for common hits between upregulated proteins in ΔclpP strains and the co-IPs (Fig. 8A). Applying these search criteria we identified 26 putative ClpP2 substrate proteins at 37 °C (Table 1). Among these, analogs of four proteins (MecA, LexA, MurC and catalase) are known ClpP substrates.30,37 Six of the 26 classified substrates are oxidoreductases and four other proteins are associated with oxidative stress (LexA, Lmo1515 CymR analog, Lmo2168 putative lactoylglutathione lyase and Lmo2182 ferrichrome ABC transporter) suggesting that ClpP plays a crucial role in redox homeostasis in L. monocytogenes, similar to S. aureus ClpP.33,34 For example, LexA, the repressor of the SOS regulon, is a known ClpP target in E. coli and in S. aureus.5,30 During the activation of the SOS response, LexA undergoes autocleavage and the N- and C-terminal domains are separated.3 Consequently, the ClpX recognition sequence gets exposed and NTD (in some organisms also the CTD) is degraded by ClpXP.5 While we were unable to detect any peptides that stretch across the autocleavage site, the fact that many SOS response proteins were upregulated in both ΔclpP2 and ΔclpP1/2 suggests that cleaved LexA accumulates, which can only weakly bind to the SOS box.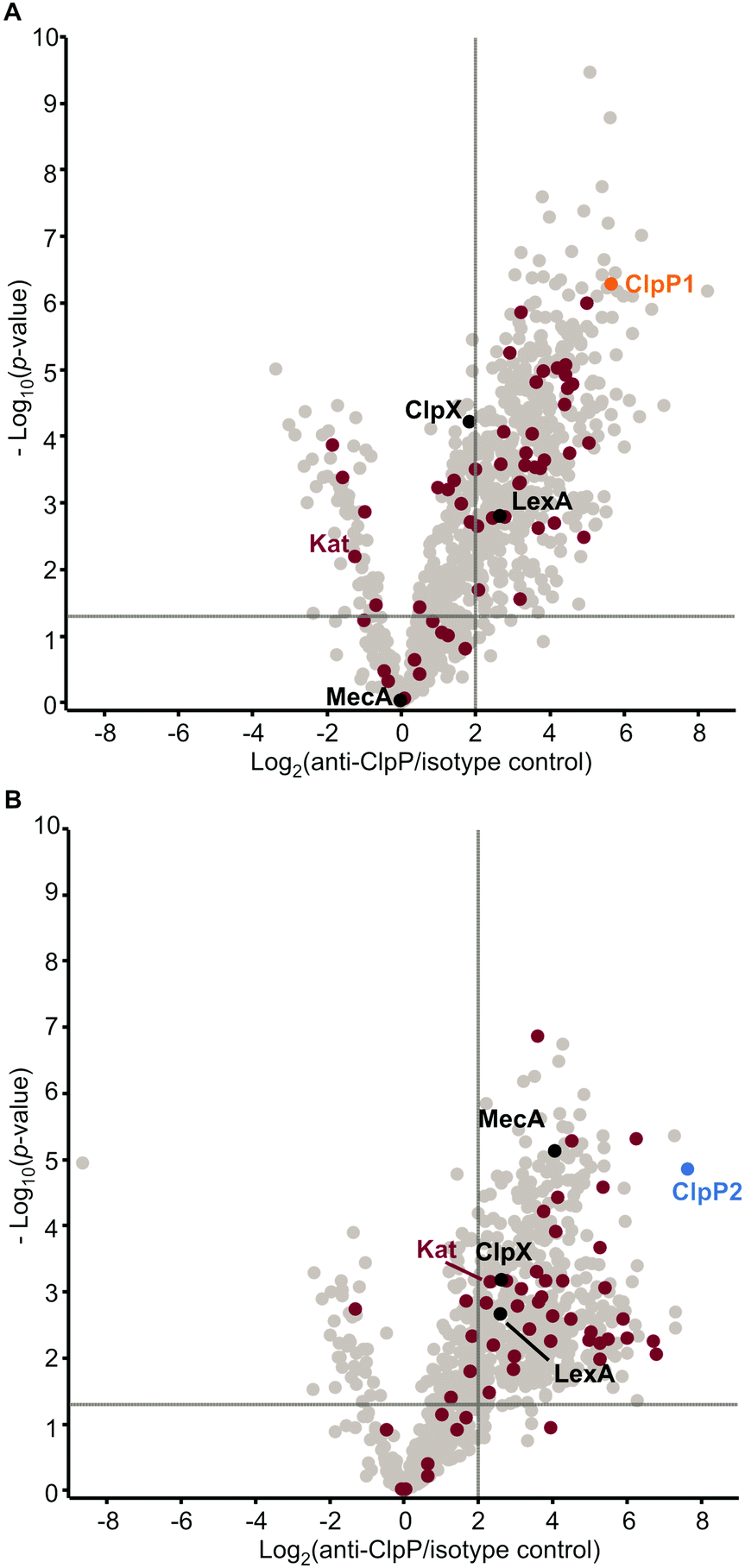 | ||
| Fig. 7 Co-immunoprecipitation of ClpP1 and ClpP2 in L. monocytogenes ΔclpP mutants. Volcano plots of co-IPs with anti-ClpP antibody in L. monocytogenes ΔclpP2 (A) and ΔclpP1 (B) at stationary phase (37 °C). − Log10p-values from two-sample Student's t-test are plotted against log2 ratios of LFQ protein intensities. The vertical grey lines show 4-fold enrichment, the horizontal grey lines show –log10t-test p-value = 1.3 (n = 4). Oxidoreductases are highlighted with purple. ClpP1 and ClpP2 are shown in orange and blue respectively. | ||
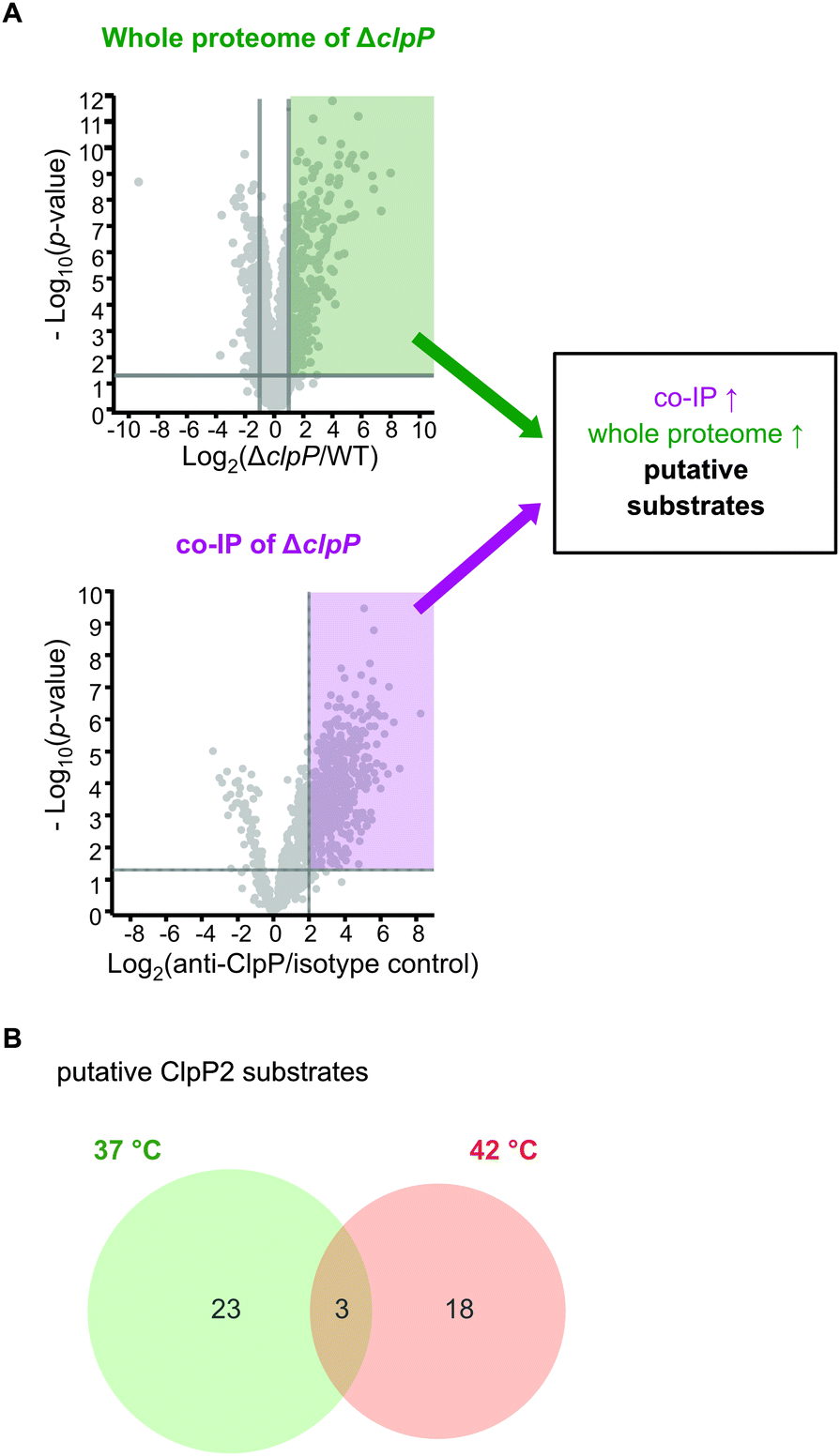 | ||
| Fig. 8 Proteomic analysis of the cellular functions of the ClpP isoforms and identification of putative substrates. (A) Proteins were classified as putative ClpP substrates (see Table 1) if they were significantly enriched both in the whole proteome analysis at 37 °C and/or 42 °C and in the anti-SaClpP co-IP of the respective ΔclpP mutants at the same temperature. Additional proteins that were significantly enriched only in the co-IP are listed in Tables S11 and S12 (ESI†). (B) Venn-diagram showing the putative substrates of ClpP2 at both temperatures. | ||
| Gene | Uniprot ID | Description |
|---|---|---|
| a The functions of not annotated proteins were derived from BLAST searches. | ||
| Putative substrates at 37 °C | ||
| lmo0485 | Q8Y9P0 | Putative oxidoreductase, iron responsea |
| lmo0487 | Q8Y9N8 | Putative hydrolasea |
| lmo0582 (iap) | P21171 | Invasion-associated protein p60 |
| lmo0640 | Q8Y993 | Putative oxidoreductasea |
| lmo0823 | Q8Y8S1 | Putative oxidoreductasea |
| lmo0930 | Q8Y8H4 | Putative lactamasea |
| lmo1320 (polC) | Q8Y7G1 | PolC-type DNA polymerase III |
| lmo1350 (gcvPB) | Q8Y7D3 | Probable glycine dehydrogenase (decarboxylating) subunit 2 |
| lmo1381 (acyP) | Q8Y7A7 | Acylphosphatase (pyruvate metabolism) |
| lmo1406 (pflB) | Q8Y786 | Pyruvate formate–lyase (pyruvate metabolism) |
| lmo1515 | Q8Y711 | Similar to CymR cystein metabolism repressora |
| lmo1538 (glpK) | Q8Y6Z2 | Glycerol kinase (glycerol metabolism) |
| lmo1605 (murC) | Q8Y6S8 | UDP-N-acetylmuramate-L-alanine ligase |
| lmo1921 | Q8Y5Y2 | Unknown function |
| lmo1932 | Q8Y5X2 | Putative heptaprenyl diphosphate synthase (menaquinone biosynthesis)a |
| lmo2168 | Q8Y5A1 | Putative lactoylglutathione lyasea |
| lmo2190 (mecA) | Q9RGW9 | ClpC adapter protein MecA |
| lmo2205 (gpmA) | Q8Y571 | 2,3-Bisphosphoglycerate-dependent phosphoglycerate mutase (glycolysis) |
| lmo2743 (tal1) | Q8Y3T8 | Probable transaldolase 1 (pentose phosphate pathway) |
| lmo2755 | Q8Y3S6 | Putative dipeptidyl-peptidase activitya |
| lmo2759 | Q8Y3S3 | Macro domain-containing protein (putative ADP–ribose binding) |
| lmo2785 (kat) | Q8Y3P9 | Catalase (H2O2 detoxification) |
| lmo2829 | Q8Y3K6 | Putative nitroreductasea |
| Putative substrates at both temperatures | ||
| lmo1302 (lexA) | Q8Y7H7 | LexA SOS response repressor |
| lmo2182 | Q8Y587 | Putative ferrichrome ABC transporter ATP-binding proteina |
| lmo2526 (murA) | Q8Y4C4 | UDP-N-acetylglucosamine 1-carboxyvinyltransferase 1 |
| Putative substrates at 42 °C | ||
| lmo0227 | Q8YAB9 | tRNA–dihydrouridine synthase |
| lmo0229 (ctsR) | Q7AP89 | CtsR (transcription repressor of class III heat shock genes) |
| lmo0231 (mcsB) | Q48759 | Arginine Kinase McsB |
| lmo0454 | Q8Y9R9 | Putative MoxR family ATPasea |
| lmo0608 | Q8Y9C4 | Putative multidrug ABC transportera |
| lmo0785 | Q8Y8V7 | Transcriptional Regulator ManR |
| lmo1293 (glpD) | Q8Y7I4 | Glycerol-3-phosphate dehydrogenase |
| lmo1387 | Q8Y7A2 | Putative pyrrolysine-5-carboxylate reductasea |
| lmo1475 (hrcA) | P0DJM4 | HrcA (heat-inducable transcription repressor A) |
| lmo1631 (trpD) | Q8Y6Q3 | Anthranilate phosphoribosyltransferase |
| lmo1713 (mreB) | Q8Y6H3 | Cell shape-determining protein MreB |
| lmo1813 | Q8Y684 | L-Serine deaminase |
| lmo1881 | Q8Y621 | Putative 5′-3′-exonucleasea |
| lmo2267 (addA) | Q8Y511 | ATP-dependent helicase/nuclease subunit A |
| lmo2352 | Q8Y4T0 | Putative LysR family transcriptional regulatora |
| lmo2489 (uvrB) | Q8Y4F5 | UvrABC system protein B, excision nuclease |
| lmo2552 (murZ) | Q8Y4A2 | UDP-N-acetylglucosamine 1-carboxyvinyltransferase 2 |
| lmo2712 | Q8Y3W7 | Putative gluconate kinase (Pentose phosphate pathway)a |
Interestingly, the number of overall and enriched proteins identified via XL-co-IP largely dropped at 42 °C (Fig. S6, Table S12, ESI†).
In search for ClpP2 substrates at this temperature, we identified 21 putative proteins (Table 1).
Interestingly, only three of those proteins are also substrate candidates at 37 °C (LexA, MurA, Lmo2182 ferrichrome ABC transporter) with 18 additional proteins being substrate candidates solely at 42 °C (Fig. 8B). This indicates that the substrate scope is adapted with changing temperature. There are five additional proteins among those that have been previously described as ClpP substrates in other bacteria, namely the two heat shock protein transcriptional regulators HrcA and CtsR, ClpC adaptor protein McsB, DNA damage repair protein UvrB and glycerol-3-phosphate dehydrogenase GlpD.30,37 HrcA and CtsR regulate the expression of class I and III heat shock proteins, while UvrB is an integral part of the SOS response.8,38–40 In addition, we identified AddAB helicase/nuclease subunit A as putative ClpP2 substrate, which is also an SOS response protein.8 Other identified putative substrates at 42 °C are involved in cell wall synthesis, cell shape (MurA, MurZ, MreB) and metabolic processes (e.g. GlpD, Lmo1387 pyrrolysin-5-carboxylate reductase, TrpD, Lmo1813 L-serine deaminase, Lmo2712 gluconate kinase). Thus, in sum 44 putative ClpP2 substrates were identified in this work. At both temperatures, nearly no putative ClpP1 substrates could be identified with this approach in our datasets (except for FhuC at 42 °C). This lack of substrates was expected as ClpP1 can not bind to AAA+ proteins and therefore does not come into close contact with substrate proteins, which is necessary for co-IP.
With many identified dysregulated proteins and putative substrates being related to oxidative stress, we finally investigated the ability of the ΔclpP mutants to grow in medium supplemented with H2O2 (Fig. 9). Surprisingly, only ΔclpP1/2 could grow in the presence of 100 ppm H2O2. This is in line with the observation that SOS-related GOBP terms were significantly upregulated only in ΔclpP1/2 but not in ΔclpP2 and ΔclpP1 at 37 °C. Thus, this indicates that a constitutively upregulated SOS response system readily protects cells from H2O2 in the ΔclpP1/2 strain.
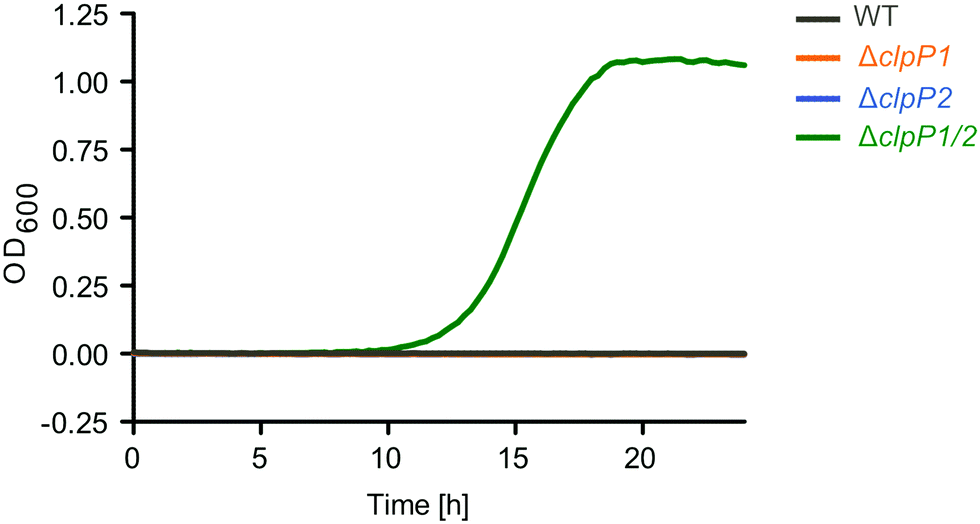 | ||
| Fig. 9 L. monocytogenes ΔclpP1/2 is resistant against oxidative stress. Growth curves of the ΔclpP mutants in the presence of 100 ppm H2O2 (BHI medium, 37 °C). Note that the WT strain and the single clpP knockouts show no growth under these conditions. The experiment was independently repeated with qualitatively identical results (data not shown). | ||
Conclusions
ClpP is a conserved heat shock protein in bacteria and in eukaryotic organelles. Some organisms have more than one clpP gene, but the role of multiple ClpPs in these organisms is not well understood. In bacteria, it is known that two different ClpPs are able to form heterocomplexes to become more active, tune the cleavage specificity or enhance the activity of the homocomplexes.12,13,15,41 Here we examined the biological role of ClpP1/2 heterocomplex formation in L. monocytogenes and the specific physiological functions of both ClpPs.We showed that ClpP1 and ClpP2 do not bind to each other at temperatures below 10 °C, and under these conditions ClpP2 is a homotetradecamer and ClpP1 an inactive heptamer. At higher temperatures, the ClpP1/2 heterocomplex is formed displaying enhanced substrate turnover. We suspected that this trait is important for modulation of ClpP proteolytic activity and is therefore crucial for stress response and virulence regulation. In order to study this effect in intact L. monocytogenes cells, we performed MS-based co-IP experiments at various temperatures. We observed enhanced ClpP1 binding to the bait ClpP2 at 42 °C as compared to 20 °C, which indeed indicates that temperature affects intracellular heterooligomer formation. However, the analysis of ClpP1 as bait was challenged due to its low abundance at 20 °C. Thus, further research is needed to investigate the exact conditions under which heterooligomerization takes place in situ and elucidate whether other factors such as binding partners or post-translational modifications can modulate ClpP1/2 complex formation.
With the aim of dissecting the physiological functions of each ClpP isoforms, we constructed single and double clpP deletion mutants in L. monocytogenes EGD-e. Phenotypic assays showed decreased growth of ΔclpP1/2 in culture medium and in macrophages. MS-based whole proteome analysis demonstrated that the deletion of clpP1 only caused minimal changes in the proteome while ΔclpP2 and ΔclpP1/2 mutants differed greatly from the wild type. These results highlight the predominant role of ClpP1 as an enhancer of catalytic turnover which is unable to recruit substrates by itself. In ΔclpP2 and ΔclpP1/2 mutants, class III heat shock proteins and a subset of the SOS response proteins as well as iron-containing proteins were upregulated. These results suggest that ClpP plays an important role in the regulation of oxidative stress response, which is in line with the results of transcriptomic analysis of the S. aureus ΔclpP mutant.34 Furthermore, the upregulated SOS response predominantly observed in the ΔclpP1/2 mutant led to a strong H2O2 resistance for this strain.
We conducted co-IPs in the single mutants with anti-ClpP antibody in order to identify specific ClpP1 and ClpP2 substrates. Combined analysis of the co-IP and whole proteome data at two temperatures led to the identification of 44 putative ClpP2 substrates. A large fraction of the identified ClpP2 substrates is related to transcriptional regulation, cell wall synthesis, cellular metabolism and oxidative stress, including LexA, corroborating the upregulation of SOS response proteins in the whole proteome.
In summary, we found that the ClpP1/2 heterocomplex in L. monocytogenes assembles at elevated temperatures and revealed ClpP's role in coping with heat-induced stress. Studying ClpP heterocomplex formation in other organisms under varying conditions might reveal that thermosensitivity is a general feature of ClpPs in bacteria carrying more than one clpP genes. This study and initial data from M. tuberculosis,17 showing a temperature-dependent, reversible assembly of a ClpP1/2 heterocomplex without an activator peptide, point in this direction. Based on the phenotypic effect of the ΔclpP1/2 mutant in conjunction with the high overlap in dysregulated whole proteome data between ΔclpP2 and ΔclpP1/2 observed in this study, an additional function of ClpP1 in L. monocytogenes cannot be excluded. It is tempting to speculate that heterocomplex formation could e.g. modulate affinities towards distinct chaperones or adaptor proteins binding to the heterocomplex, which in turn fine-tunes substrate specificity in response to association of ClpP1. However, a more in-depth biophysical investigation is required to elucidate such additional functions of ClpP1 in future studies.
Experimental
Protein overexpression and purification
Full-length ClpP2 was obtained as described previously.20 In short, expression constructs with a C-terminal Strep-tag II were cloned in pET301 plasmids, over-expressed in E. coli BL21(DE3) and purified by affinity chromatography and gel filtration. ClpP1 with a C-terminal Strep-tag II was kindly provided by Dr Maria Dahmen.12 Co-expressed ClpP1/2 was obtained as described previously.16 ClpX was obtained as described previously.16 GFP-SsrA was obtained as described previously.18 Creatine kinase (10 127 566 001) was purchased from Roche (Roche Diagnostics GmbH, Mannheim, Germany).Analytical size-exclusion chromatography followed by intact protein mass spectrometry
544 nmol ClpP17 (1:1 ClpP1:ClpP2 monomeric ratio) and/or 272 nmol ClpP214 were incubated for 10 or 30 min at the indicated temperatures (0–48 °C) in sample buffer (20 mM MOPS, 110 mM KCl, 5% glycerol, pH 7.0) in a final volume of 100 μL. The samples were loaded on a pre-equilibrated Superdex 200 Increase 10/300 gel filtration column (GE Healthcare, Chicago, United States) connected to an ÄKTA Purifier 10 system (GE Healthcare) and eluted with 1 CV ClpP-GF buffer buffer (20 mM MOPS, 100 mM KCl, 5% glycerol, pH 7.0). 200 μL fractions were collected. UV absorption was recorded at 280 nm. The oligomerization state was determined by comparison of the elution volumes to the calibration curve of the column (Gel Filtration Calibration Kit, GE Healthcare). The gel filtration column was equilibrated to room temperature for samples incubated at 20 °C and above, to 10 °C for samples incubated at 10 °C and to 4 °C for samples incubated at below 10 °C. The fraction corresponding to the tetradecamer peak was analyzed by intact protein mass spectrometry. Measurements were carried out on a Dionex Ultimate 3000 HPLC system (Thermo Fisher Scientific, Waltham, United States) coupled to a Thermo LTQ Oribtrap XL mass-spectrometer (Thermo Fisher Scientific) with an electrospray ionization source (H-ESI-II source, spray voltage 4.0 kV, tube lens 110 V, capillary voltage 31 V, capillary temperature 350 °C, sheath gas 30 a.u., aux gas 15 a.u). 1–4 μL were desalted with a MassPREP desalting cartridge (Waters, Milford, United States). The mass spectrometer was operated in positive mode collecting full scans at high resolution (R = 100000) from m/z = 300 to m/z = 2000. Collected data was deconvoluted using the Thermo Xcalibur Xtract algorithm (Thermo Fisher Scientific).
The experiments with a mixture of ClpP17 and ClpP214 at 30 °C and at 42 °C for 30 min were repeated with qualitatively identical results. Plots were made with Microcal OriginPro 2022 (OriginLab Corporation, Northampton, United States).
Protease assay
Protease assays were carried out in flat bottom black 96-well plates in a final volume of 60 μL. 0.1 μM ClpP214 or a mixture of 0.2 μM ClpP17 and 0.1 μM ClpP214 (1:1 ClpP1:ClpP2 monomeric ratio), ClpX6 (0.4 μM) and ATP regeneration mix (4 mM ATP, 16 mM creatine phosphate, 20 U mL−1 creatine kinase) were pre-incubated for 30 min at the indicated temperatures (30 °C, 37 °C and 42 °C) in PZ buffer (25 mM HEPES, 200 mM KCl, 5 mM MgCl2, 1 mM DTT, 10% glycerol, pH 7.6). 0.4 μM eGFP-LmSsrA substrate was added and fluorescence (λex = 485 nm, λem = 535 nm) was measured at the respective temperatures with an infinite M200Pro plate reader (Tecan, Männedorf, Switzerland). Data were recorded in triplicates. The measurements were independently repeated with qualitatively identical results. Protease activity was determined by linear regression using Microsoft Excel and plots were made with GraphPad Prism 6 (GraphPad, San Diego, United States).
Cloning of L. monocytogenes mutants
Construction of pLSV101_clpP-2×myc shuttle vectors. Ca. 1000 base pairs upstream and downstream from the C-terminus of clpP1 were amplified by PCR using the A–B and C–D primer pairs from Table 2 (Phusion polymerase, GC buffer, New England Biolabs, Ipswich, United States). For clpP2, ca. 700 bp upstream and downstream were amplified using the A–B and C–CD primer pairs from Table 2 (Phusion polymerase, GC buffer, New England Biolabs). The 2×myc tag was added to the B primers as overhangs. The PCR products were purified with E.Z.N.A. Cycle Pure Kit (Omega Bio-tek, Norcross, United States). The AB fragments were digested with SalI-HF (New England Biolabs) and BglII (Promega, Madison, United States), the CD fragments were digested with BglII (Promega) and BamHI (New England Biolabs) and the empty pLSV101 vector was digested with SalI-HF and BamHI-HF (New England Biolabs). The digested DNAs were purified with E.Z.N.A. Gel Extraction Kit (Omega Bio-tek) after agarose gel electrophoresis. The AB and CD fragments were ligated with T4 DNA ligase (New England Biolabs) (1
:1 molar ratio, 15 °C, overnight). The ligated fragments were amplified by PCR (Phusion polymerase, HF buffer, New England Biolabs) using the clpP1_A–clpP1D and clpP2_A– clpP2_CD primer pairs (Table 2). The PCR products were purified with E.Z.N.A. Gel Extraction Kit (Omega Bio-tek) after agarose gel electrophoresis. The ABCD fragments were digested with SalI-HF and BamHI-HF (New England Biolabs) and dephosphorylated with Antarctic phosphatase (New England Biolabs). The fragments were purified with E.Z.N.A. Gel Extraction Kit (Omega Bio-tek) after agarose gel electrophoresis. The fragments were ligated into the pLSV101 vector (1:1 and 3:1 molar ratios) with T4 DNA ligase (New England Biolabs) (10 °C for 30 s and 30 °C for 30 s alternating overnight). The ligated vectors were transformed into chemically competent E. coli TOP10. E. coli containing pLSV101 was grown with 200 μg mL−1 erythromycin. Colonies were tested with colony PCR using pLSV101_seq fwd and rev primers (Table 2). The vectors were purified from positive colonies with NucleoSpin Plasmid EasyPure, Mini kit (MACHEREY-NAGEL, Düren, Germany) (elution with ddH2O) and sequenced by Sanger sequencing with A and D primers.
| Primer | Sequence (5′ → 3′) |
|---|---|
| clpP1_A | GTTGCAGTCGACAGGAGGAAACCATGCAAGAG |
| clpP1-Myc_B | TTAGATCTAAATCTTCTTCACTAATTAATTTTTGTTCTAAATCTTCTTCACTAATTAATTTTTGTTCTTTTAAGCCATCGCGATTTTCG |
| clpP1_C | CGGCAGATCTATAAAACCAAAAGGTTCACTTC |
| clpP1_D | CTTTATGGATCCTTGATCCGGTCACTCCAG |
| clpP2_A | GTTGCAGTCGACACAGGAGGAATCTTGATATGAAC |
| clpP2-Myc_B | TTAGATCTAAATCTTCTTCACTAATTAATTTTTGTTCTAAATCTTCTTCACTAATTAATTTTTGTTCGCCTTTTAAGCCAGATTTATTAATG |
| clpP2_C | CGGCAGATCTCTAATAAAAAAAGAGGTTTTGCAC |
| clpP2_CD | CTTTATGGATCCTTCTGCAGTTCTAACAGGAGT |
| pLSV101_seq fwd | AGTACCATTACTTATGAG |
| pLSV101_seq rev | AGGGTTTTCCCAGTCACG |
| clpP1_tag fwd | CGTAATTTCTGGCTTTCTG |
| clpP1_tag rev | GAGTGATAAATGAATTAGGTCAAG |
| clpP2_tag fwd | GCGATACAGATCGTGATAATTTC |
| clpP2_tag rev | GAATACTAGTGTATACATTCTATGGAAG |
Preparation of electrocompetent L. monocytogenes. 200 mL BHI medium (7.5 g L−1 brain infusion, 1 g L−1 peptone, 10 g L−1 heart infusion, 5 g L−1 NaCl, 2.5 g L−1 Na2HPO4, 2 g L−1 glucose, pH 7.4) was inoculated to an initial OD600 of 0.05 with an overnight culture of L. monocytogenes EGD-e. The culture was grown to OD600 = 0.5 at 37 °C, 200 rpm. 5 μg mL−1 penicillin G was added, and the bacteria were incubated at 37 °C, 200 rpm for 15 min and on ice without shaking for 10 min. The cells were harvested (4000 g, 10 min, 4 °C) and washed with 30 mL ice-cold SMHEM medium (952 mM saccharose, 3.5 mM MgCl2, 7 mM HEPES, pH 7.2). The pellet was resuspended in 2 mL cold SMHEM medium. 100 μL aliquots were prepared and shock-frozen in liquid N2 and stored at −80 °C.
Transformation into L. monocytogenes. Electrocompetent L. monocytogenes EGD-e aliquots were thawed on ice and 1 μg plasmid was added. The cells were transferred into ice-cold 2 mm electroporation cuvettes (Bio-Rad, Hercules, United States) and electroporated (2500 V, 200 Ω, 25 μF, exponential decay, time constant <4 ms) using Gene Pulser Xcell (Bio-Rad). 1 mL BHI medium +0.5 mM saccharose was added and the cells were incubated at 30 °C for 4 h and plated on BHI agar plates containing 10 μg mL−1 erythromycin. The plates were incubated at 30 °C for 3 days.
Homologous recombination and colony selection. 2.5 mL BHI medium with 10 μg mL−1 erythromycin was inoculated with single colonies after transformation. 10−2 and 10−6 dilutions were plated on BHI + 10 μg mL−1 erythromycin agar plates and incubated at 42 °C for 2 days. Colony PCR (OneTaq polymerase, New England Biolabs) with the respective primer pairs clp_A–pLSV101_seq rev and pLSV101_seq fwd–clp_D (Table 2) was performed to check the genomic integration of the fragments. Positive colonies were subcultivated several times in 3 mL BHI medium without antibiotic at 30 °C (200 rpm). 10−6 dilutions were plated on BHI agar plates (37 °C, overnight). Single colonies were picked and transferred to BHI agar plates with and without 10 μg mL−1 erythromycin (37 °C, overnight). Erythromycin-sensitive strains were tested with colony PCR (OneTaq DNA polymerase, New England Biolabs) using the clpP_tag fwd and rev primer pair (Table 2) to check for integration of the 2×myc tag into the genome.
Construction of pMAD_ΔclpP1 shuttle vector. A pMAD shuttle vector derivative was used to introduce a deletion of clpP.42 Approx. 1000 bp upstream (clpP1_KO_A and clpP1_KO_B, Table 3) region of clpP1 was amplified by PCR (GC buffer, Phusion polymerase, New England Biolabs) using isolated L. monocytogenes EGD-e DNA as template. The PCR product was purified (Cycle Pure Kit, E.Z.N.A., Omega Bio-tek) and digested with MluI and NcoI (Promega, standard protocol). pMAD plasmid was also digested with MluI and NcoI and dephosphorylated by addition of TSAP (Promega, streamlined restriction digestion protocol) for 20 min. After restriction digest products were purified (MicroElute DNA Clean-Up Kit, E.Z.N.A., Omega Bio-tek). Ligation into pMAD vector was conducted using T4 DNA Ligase (Promega, standard protocol) overnight at 8 °C and a vector:insert ratio of 1
:6. The ligation product (pMAD-AB) was chemically transformed into E. coli TOP10 cells and plated onto LB agar containing ampicillin. Accordingly, a 1000 bp downstream (clpP1_KO_C and clpP1_KO_D, Table 3) region of clpP1 was amplified by PCR (GC buffer, Phusion polymerase, New England Biolabs) using isolated L. monocytogenes EGD-e DNA as template. The PCR product was purified (Cycle Pure Kit, E.Z.N.A., Omega Bio-tek) and digested with MluI and BamHI (Promega, standard protocol). pMAD-AB plasmid was also digested with MluI and BamHI and dephosphorylated by addition of TSAP (Promega, streamlined restriction digestion protocol) for 20 min. After restriction digest products were purified (MicroElute DNA Clean-Up Kit, E.Z.N.A., Omega Bio-tek). Ligation into pMAD-AB vector was conducted using T4 DNA Ligase (Promega, standard protocol) overnight at 8 °C and a vector:insert ratio of 1:6. Insertion of the desired construct was tested after plasmid extraction (Plasmid Mini Kit I, E.Z.N.A., Omega Bio-tek) by analytical restriction digest and sequencing (pMAD-seq-for and pMAD-seq-rev, Table 3).
| Primer | Sequence (5′ → 3′) |
|---|---|
| clpP1_KO_A | GGACCATGGTTTCATCAGCAAACCTCCGCAC |
| clpP1_KO_B | GGAACGCGTGAAAAAATTCCTCCTTAAAAAGCCTTAGTTTATTTG |
| clpP1_KO_C | GGAACGCGTAAGCAAAAGATTACGGCATCG |
| clpP1_KO_D | GGAGGATCCTTGATCCGGTCACTCCAGTA |
| pMAD-seq-for | CCCAATATAATCATTTATCAACTCTTTTACACTTAAATTTCC |
| pMAD-seq-rev | GCAACGCGGGCATCCCGATG |
| clpP2_KO_A | CGAACAGTGTAAGTGTATGCG |
| clpP2_KO_B | AGTTTGAGATCTTACTGTTGGAATTAAGTTCAT |
| clpP2_KO_C | TACGGCAGATCTGATGATATTATCATTAATAAA |
| clpP2_KO_D | TTGCATTTGTAGTGGTTATGG |
| clpP2_AB | GTTGCAGTCGACTCTAACGATGATCTTGTTAGT |
| clpP2_CD | CTTTATGGATCCTTCTGCAGTTCTAACAGGAGT |
:100) from a L. monocytogenes EGD-e overnight culture and incubated at 37 °C until an OD600 of 0.5 was reached. Cells were centrifuged (5000 g, 15 min, 4 °C) and washed three times with cold 10% glycerol (sterile): (1) 100 mL; (2) 50 mL; (3) 25 mL. The pellet was resuspended in 400 μL cold 10% glycerol and 75 μL aliquots were frozen in liquid nitrogen and stored at −80 °C.
:1000 from the overnight culture and incubated 2 h at 30 °C and 6 h at 42 °C. 100 μL diluted cultures (10−2 to 10−6) were plated onto BM agar (containing 1 μg mL−1 erythromycin and 100 μg mL−1 X-gal) and incubated at 42 °C until colonies with blue coloration were visible (enrichment of single crossover). Ten light blue colonies were picked and incubated (together) in 10 mL BM medium at 3 °C for 8 h followed by overnight incubation at 42 °C. 10 mL BM medium were inoculated 1:1000 from this overnight culture and grown for 4 h at 30 °C and additional 4 h at 42 °C. 100 μL of diluted cultures (10−2 to 10−6) were plated onto BM agar containing X-gal and incubated at 4 °C. White colonies were picked and streaked onto BM agar containing erythromycin and X-gal and onto BM agar containing only X-gal. Plates were incubated at 30 °C and erythromycin susceptible colonies further analyzed by colony PCR followed by analytical restriction digest and sequencing. For colony PCR small parts of colonies were resuspended in 50 μL sterile water and 1 μL thereof was used in PCR reactions with an initial denaturation step for 10 min (95 °C).
Construction of pLSV101_ΔclpP2 shuttle vector. A construct derived from the mutagenesis vector pLSV101 was used for clpP2 deletion (pLSV101 was kindly provided by Prof. Dr Thilo M. Fuchs).43Ca. 1000 base pairs upstream and downstream from the clpP2 gene were amplified by PCR using the A–B and C–D primer pairs from Table 3 (Phusion polymerase, GC buffer, New England Biolabs). The PCR products were purified with E.Z.N.A. Gel Extraction Kit (Omega Bio-tek) after agarose gel electrophoresis. The fragments were digested with BglII (Promega) and purified with E.Z.N.A. Cycle Pure Kit (Omega Bio-tek). The AB and CD fragments were ligated with T4 DNA ligase (New England Biolabs) (1
:1 molar ratio, 15 °C, overnight). The ligated fragment was amplified by PCR (Phusion polymerase, HF buffer) using the AB–CD primer pair (Table 3). The PCR product was purified with E.Z.N.A. Cycle Pure Kit (Omega Bio-tek). The insert and the empty pLSV101 vector were digested with SalI-HF and BamHI-HF (New England Biolabs) and purified with E.Z.N.A. Gel Extraction Kit (Omega Bio-tek) after agarose gel electrophoresis. The fragment was ligated into the pLSV101 vector (3:1 molar ratio) with T4 DNA ligase (New England Biolabs) (16 °C overnight). The ligated vector was transformed into chemically competent E. coli TOP10. E. coli containing pLSV101_ΔclpP2 was grown with 300 μg mL−1 erythromycin. Colonies were tested with colony PCR using pLSV101_seq fwd and rev primers (Table 2). The vectors were purified with E.Z.N.A. Plasmid Mini Kit I (Omega Bio-tek) from positive colonies (elution with ddH2O) and sequenced by Sanger sequencing with pLSV101_seq fwd and rev primers.
Transformation into L. monocytogenes. Electrocompetent L. monocytogenes EGD-e and ΔclpP1 cells were prepared as described above. Aliquots of electrocompetent cells were thawed on ice and 2 or 5 μg plasmid was added. The cells were transferred into ice-cold 2 mm electroporation cuvettes (Bio-Rad) and electroporated (2500 V, 200 Ω, 25 μF, exponential decay, time constant ∼ 4 ms) using Gene Pulser Xcell (Bio-Rad). 1 mL warm BHI medium was added and the cells were incubated at 30 °C for 6 h under shaking at 200 rpm and plated on BHI agar plates with 10 μg mL−1 erythromycin. The plates were incubated at 30 °C for 5 days.
Western blot
5 mL BHI medium was inoculated with L. monocytogenes EGD-e, ΔclpP1, ΔclpP2 and ΔclpP1/2 strains. An amount of cells corresponding to 200 μL of OD600 = 20 of each mutant was harvested (4000g, 10 min, 4 °C). The cells were lysed by ultrasonication (3 × 20 s, 75%, cooled on ice during breaks). 2× Laemmli buffer was added and 20 μL sample was separated by SDS–PAGE (12.5% polyacrylamide, 150 V, 2.5 h). The proteins from the polyacrylamide gel were transferred to a methanol–soaked PVDF membrane (Bio-Rad) in a Trans-Blot SD semi-dry western blot cell (Bio-Rad) using blotting buffer (48 mM Trizma, 39 mM glycine, 0.04% SDS, 20% methanol) (10 V, 1 h). The membrane was blocked with 5% milk powder in PBS-T (0.5% Tween-20 in PBS) for 1 h at RT and subsequently incubated with rabbit polyclonal anti-ClpP antibody (custom-made, raised against S. aureus ClpP, 2 mg mL−1, 1:1000 dilution) in PBS-T + 5% milk powder (4 °C, overnight). The membrane was washed three times with PBS-T (15 min, RT) and incubated with Pierce Goat anti-Rabbit poly-HRP secondary antibody (1:10000, Thermo Fisher Scientific) in PBS-T + 5% milk powder (1 h, RT). The membrane was washed three times with PBS-T (15 min, RT) and chemiluminescence was detected after 10 min incubation with freshly prepared Clarity Western ECL Substrate (Bio-Rad) with a LAS-4000 gel scanning station (Fujitsu Life Sciences, Tokyo, Japan).
For detection of myc-tagged ClpP1 and ClpP2, pellets of L. monocytogenes clpP1(191)::2×myc and L. monocytogenes clpP2(199)::2×myc corresponding to 1 mL OD600 = 20 were prepared as described in the MS-based co-immunoprecipitation section without crosslinking. The pellets were resuspended in 200 μL 0.4% SDS–PBS and lyzed by sonication (3 × 20 s, 75%, cooled on ice during breaks). Protein concentration was determined using a BCA assay (Roti-Quant universal, Carl Roth GmbH + Co. KG, Karlsruhe, Germany), all samples were adjusted to 4 mg mL−1 with 2× Laemmli buffer and 20 μL of the samples were separated by SDS–PAGE (12.5% polyacrylamide, 150 V, 2.5 h). Protein transfer and detection was performed as described above, with a 1:5000 dilution of an anti-c-Myc antibody (rabbit polyclonal, ab152146, 1 mg mL−1, Abcam, Cambridge, United Kingdom) in PBS-T + 5% milk powder used as primary antibody and the membrane was stained with Ponceau S.
Fluorescent labelling
25 mL BHI medium was inoculated with L. monocytogenes EGD-e, ΔclpP1, ΔclpP2 and ΔclpP1/2 from a day culture to an initial OD600 of 0.05. The culture was grown to early stationary phase and an amount corresponding to 800 μL OD600 = 20 was harvested (4000g, 4 °C, 10 min). The cells were washed with 1 mL PBS (4000g, 4 °C, 5 min). The pellets were resuspended in 800 μL PBS and aliquots of 250 μL were prepared. 2.5 μL 5 mM vibralactone probe (or 5 mM D3 or DMSO as controls) from a DMSO stock was added to all strains (2 h, RT). The cells were centrifuged (4000g, 5 min, 4 °C), the supernatant was discarded, and the pellets were washed with 1 mL PBS (4000g, 5 min, 4 °C). The pellets were stored at −80 °C until further usage. The cells were resuspended in 250 μL PBS and transferred to 2 mL tubes containing 0.5 mL inlets filled with glass beads of 0.5 mm diameter. The cells were lysed using 2× program #2 in Precellys 24 tissue homogenizer (Bertin Instruments, Montigny-le-Bretonneux, France) coupled to liquid N2-cooled Cryolys (flow rate set to level I during shaking, level 0 during breaks). 200 μL of the lysates were pipetted into microcentrifuge tubes and the insoluble fractions were separated (10000g, 30 min, 4 °C). Click reagents [2 μL 5 mM rhodamine azide, 2 μL 15 mg mL−1 TCEP, 6 μL 1.67 mM tris((1-benzyl-4-triazolyl)methyl)amine ligand and 2 μL 50 mM CuSO4] were added to 88 μL of the supernatant and the reactions were incubated in the dark for 1 h at RT. 2 × Laemmli buffer was added and the samples were stored at −20 °C until further usage. 50 μL of the samples were separated by SDS–PAGE (12.5% polyacrylamide, 150 V, 3 h) and fluorescence was detected with LAS-4000 gel scanning station (Fujitsu Life Sciences).
Intracellular growth assay
J774A.1 murine macrophage-like cells were grown in tissue culture flasks with hydrophobic surface for suspension cells in DMEM/FCS (DMEM high glucose medium (Sigma-Aldrich, St. Louis, United States) supplemented with 2 mM glutamine and 10% heat-deactivated FCS). The flasks were incubated at 37 °C under 5% CO2. The cells were splitted into new flasks every 2–3 days to ca. 5 × 104 cells per cm2. For detachment, cells were washed twice with TEN buffer (40 mM Tris–HCl, 150 mM NaCl, 1 mM EDTA, pH 7.4) and incubated with Accutase solution (Sigma-Aldrich) at 37 °C for 30 min. 105 J774A.1 cells in 100 μL DMEM/FCS were pipetted into the inner wells of a flat-bottom 96-well plate. The outer wells were filled with 150 μL sterile PBS. The plates were incubated overnight at 37 °C under 5% CO2. On the next day, DMEM/FCS was inoculated with L. monocytogenes EDG-e, ΔclpP1, ΔclpP2 and ΔclpP1/2 overnight cultures to 103 CFU μL−1. The J774A.1 cells were washed with 150 μL PBS and 100 μL bacterial suspension was added (multiplicity of infection = 0.5). The plate was incubated on ice for 15 min and at 37 °C for 15 min. The cells were washed three times with 200 μL PBS. 150 μL DMEM/FCS supplemented with 10 μL gentamycin was added to kill extracellular bacteria. The plates were incubated at 37 °C under 5% CO2. After 7 h, the cells were washed three times with 200 μL PBS, and lysed with 2× 100 μL 0.05% Triton X-100 in ddH2O (1 min, RT). Dilution series were prepared from the lysates and plated on BHI agar plates. The agar plates were incubated at 37 °C for 2 days until colonies were counted. Data was recorded in triplicates and two independent experiments were performed. Plots were made with GraphPad Prism 6.Whole proteome analysis
:100 with overnight cultures of L. monocytogenes EGD-e, ΔclpP1, ΔclpP2 and ΔclpP1/2. The first day culture was grown to an OD600 of ca. 0.5 at 37 °C under shaking at 200 rpm. For the second day culture, 3 × 5 mL BHI medium was inoculated with the first day culture to a starting OD600 of 0.05 and incubated at 37 °C or 42 °C under shaking at 200 rpm. After reaching early stationary phase, 1.5 mL of the cultures were harvested (4000g, 10 min, 4 °C). The pellet was washed with 1 mL PBS and stored at −80 °C until further usage. Two biological replicates were generated.
000g at 4 °C for 15 min and the supernatant was discarded. The pellet was resuspended in 500 μL methanol (−80 °C) with ultrasonication (10 s, 10%). After centrifugation at 21000g and at 4 °C for 15 min, the supernatant was discarded and the pellet was air-dried.
000 and an automatic gain control (AGC) ion target value of 2 × 105 in a scan range of 300–1500 m/z with a maximum injection time of 50 ms. Internal calibration was performed using the ion signal of fluoranthene cations (EASY-ETD/IC source). Dynamic exclusion time was set to 60 s with a mass tolerance of 10 ppm (low/high). Precursors with intensities higher than 5 × 103 and charge states 2–7 were selected for fragmentation with HCD (30%). MS2 scans were recorded in the ion trap operating in a rapid mode with an isolation window of 1.6 m/z. The AGC target was set to 1 × 104 with a maximum injection time of 35 ms (100 ms in case of the temperature-dependent co-IP with anti-c-Myc antibody) and the “inject ions for all available parallelizable time” was enabled.
MS-based co-immunoprecipitation
Cultivation of L. monocytogenes with c-Myc-tagged clpP. 30 mL BHI medium were inoculated 1
:100 with overnight cultures of L. monocytogenes clpP1(191)::2×myc and L. monocytogenes clpP2(199)::2×myc. The first day culture was grown to an OD600 of ca. 0.5 at 37 °C under shaking at 200 rpm. For the second day culture, 4 × 100 mL BHI medium was inoculated with the first day cultures to a starting OD600 of 0.05. 2 flasks per condition were incubated at 20 °C and at 42 °C under shaking at 200 rpm. After reaching early stationary phase, an amount of bacteria corresponding to 4 × 1 mL OD600 = 20 per flask was harvested (4000g, 5 min, 4 °C) and washed with 1 mL PBS. The pellets were resuspended in 1 mL PBS and 2 mM DSSO was added (20 μL from a 100 mM DMSO stock). DSSO was kindly provided by Dr Vadim Korotkov and Dr Pavel Kielkowski and synthesized as described previously.22 The bacteria were incubated with the crosslinker for 30 min at 20 °C or 42 °C under shaking at 200 rpm. The reaction was quenched by washing twice with 50 mM Tris–HCl (pH 8.0) and the pellets were stored at −80 °C until further usage.
000g, 30 min, 4 °C) and the supernatant was sterile filtered through a 0.2 μm PTFE filter. Protein concentration was determined using a BCA assay (Roti-Quant universal, Carl Roth GmbH + Co. KG). 30 μL Protein A/G agarose beads (Thermo Fisher Scientific) were transferred to protein low-bind microcentrifuge tubes (Eppendorf) and washed with 1 mL co-IP wash buffer (50 mM Tris–HCl, 150 mM NaCl, 5% glycerol, 0.05% NP-40, pH 7.4) and centrifuged for 1 min at 1000g at 4 °C. 500 μg proteome (in 500 μL) and either 1 μL anti-c-Myc antibody (rabbit polyclonal, ab152146, 1 mg mL−1, Abcam) or 0.4 μL rabbit mAb IgG isotype control (2.5 mg mL−1, Cell Signaling Technology, Danvers, United States) were added. The samples were incubated at 4 °C for 3 h under constant rotation. The supernatant was removed after centrifugation (1000g, 1 min, 4 °C), and the beads were washed twice with 1 mL co-IP wash buffer. The detergent was removed by washing the beads twice with co-IP lysis buffer.
Co-IP with anti-clpP antibody
20 mL BHI medium was inoculated 1:100 with overnight cultures of L. monocytogenes ΔclpP1 and ΔclpP2. The first day culture was grown to an OD600 of ca. 0.5 at 37 °C under shaking at 200 rpm. For the second day culture, 50 mL BHI medium was inoculated with the first day cultures to a starting OD600 of 0.05 and incubated at 37 °C or 42 °C under shaking at 200 rpm. After reaching early stationary phase, an amount of bacteria corresponding to 2 × 1 mL OD600 = 20 per replicate was harvested (4000g, 5 min, 4 °C) and washed with 1 mL PBS. The pellets were resuspended in 1 mL PBS and 2 mM DSSO was added (20 μL from a 100 mM DMSO stock). The bacteria were incubated with the crosslinker for 30 min at 37 °C or 42 °C and under shaking at 200 rpm. The reaction was quenched by washing twice with 50 mM Tris–HCl (pH 8.0) and the pellets were stored at −80 °C until further usage. Four replicates from independent overnight cultures were generated for each experiment.
Cell lysis, co-IP and sample preparation were conducted as described for the temperature-dependent co-IP with anti-c-Myc antibody, except that either 5 μL anti-ClpP antibody (custom-made, polyclonal, raised against S. aureus ClpP in rabbit, 2 mg mL−1) or 4 μL rabbit mAb IgG isotype control (2.5 mg mL−1, Cell Signaling Technology) were used. 300 μg proteome was used in case of the 42 °C XL-co-IP. LC-MS/MS measurements and data analysis was done as described for the temperature-dependent co-IP with anti-c-Myc antibody. Oxidoreductases were searched for with the UniProt Keyword “Oxidoreductase”.
Author contributions
S. A. S. and D. B. conceived the project. D. B. and K. E. planned and performed the experimental work and analysed obtained data. C. F. constructed a mutant strain. S. A. S., D. B. and K. E. wrote the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was performed within the framework of SFB 1035 (German Research Foundation DFG, Sonderforschungsbereich 1035, Projektnummer 201302640, project A09). We thank Mona Wolff and Katja Bäuml for technical support as well as Dr Stuart Ruddell and Dr Thomas Gronauer for critical proofreading of the manuscript. We are grateful to Prof. Dr Thilo M. Fuchs and Dr Jakob Schardt for their help with cloning in L. monocytogenes. We thank Dr David Lyon (UZH, Institute of Molecular Life Sciences, Switzerland) for support concerning the aGOtool.References
- L. Radoshevich and P. Cossart, Nat. Rev. Microbiol., 2018, 16, 32–46 CrossRef CAS PubMed.
- F. I. Bucur, L. Grigore-Gurgu, P. Crauwels, C. U. Riedel and A. I. Nicolau, Front. Microbiol., 2018, 9, 2700 CrossRef PubMed.
- B. Michel, PLoS Biol., 2005, 3, e255 CrossRef.
- S. van der Veen, T. Hain, J. A. Wouters, H. Hossain, W. M. de Vos, T. Abee, T. Chakraborty and M. H. J. Wells-Bennik, Microbiology, 2007, 153, 3593–3607 CrossRef CAS.
- M. T. Cohn, P. Kjelgaard, D. Frees, J. R. Penadés and H. Ingmer, Microbiology, 2011, 157, 677–684 CrossRef CAS.
- J. W. Little and M. Gellert, J. Mol. Biol., 1983, 167, 791–808 CrossRef CAS.
- S. B. Neher, J. M. Flynn, R. T. Sauer and T. A. Baker, Genes Dev., 2003, 17, 1084–1089 CrossRef CAS PubMed.
- S. van der Veen, S. van Schalkwijk, D. Molenaar, W. M. De Vos, T. Abee and M. H. J. Wells-Bennik, Microbiology, 2010, 156, 374–384 CrossRef CAS.
- Y. Kawai, S. Moriya and N. Ogasawara, Mol. Microbiol., 2003, 47, 1113–1122 CrossRef CAS PubMed.
- T. A. Baker and R. T. Sauer, Biochim. Biophys. Acta, Mol. Cell Res., 2012, 1823, 15–28 CrossRef CAS.
- E. Zeiler, N. Braun, T. Böttcher, A. Kastenmüller, S. Weinkauf and S. A. Sieber, Angew. Chem., Int. Ed., 2011, 50, 11001–11004 CrossRef CAS PubMed.
- M. Dahmen, M. T. Vielberg, M. Groll and S. A. Sieber, Angew. Chem., Int. Ed., 2015, 54, 3598–3602 CrossRef CAS PubMed.
- S. Pan, I. T. Malik, D. Thomy, B. Henrichfreise and P. Sass, Sci. Rep., 2019, 9, 14129 CrossRef PubMed.
- B. M. Hall, E. B. M. Breidenstein, C. de la Fuente-Núñez, F. Reffuveille, G. D. Mawla, R. E. W. Hancock and T. A. Baker, J. Bacteriol., 2017, 199, e00568-16 CrossRef PubMed.
- G. D. Mawla, B. M. Hall, G. Cárcamo-Oyarce, R. A. Grant, J. J. Zhang, J. R. Kardon, K. Ribbeck, R. T. Sauer and T. A. Baker, Mol. Microbiol., 2021, 115, 1094–1109 CrossRef CAS PubMed.
- C. Gatsogiannis, D. Balogh, F. Merino, S. A. Sieber and S. Raunser, Nat. Struct. Mol. Biol., 2019, 26, 946–954 CrossRef CAS.
- J. Leodolter, J. Warweg and E. Weber-Ban, PLoS One, 2015, 10, e0131132 CrossRef PubMed.
- D. Balogh, M. Dahmen, M. Stahl, M. Poreba, M. Gersch, M. Drag and S. A. Sieber, Chem. Sci., 2017, 8, 1592–1600 RSC.
- Y. I. Kim, R. E. Burton, B. M. Burton, R. T. Sauer and T. A. Baker, Mol. Cell, 2000, 5, 639–648 CrossRef CAS PubMed.
- E. Zeiler, A. List, F. Alte, M. Gersch, R. Wachtel, M. Poreba, M. Drag, M. Groll and S. A. Sieber, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11302–11307 CrossRef CAS.
- C.-L. Wu, Y.-H. Li, H.-C. Lin, Y.-H. Yeh, H.-Y. Yan, C.-D. Hsiao, C.-F. Hui and J.-L. Wu, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 2011, 158, 189–198 CrossRef PubMed.
- A. Fux, V. S. Korotkov, M. Schneider, I. Antes and S. A. Sieber, Cell Chem. Biol., 2019, 26, 48–59 CrossRef CAS PubMed.
- O. Gaillot, E. Pellegrini, S. Bregenholt, S. Nair and P. Berche, Mol. Microbiol., 2000, 35, 1286–1294 CrossRef CAS.
- R. Huisgen, Proc. Chem. Soc., London, 1961, 357–396 Search PubMed.
- V. V. Rostovtsev, J. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- C. Schölz, D. Lyon, J. C. Refsgaard, L. J. Jensen, C. Choudhary and B. T. Weinert, Nat. Methods, 2015, 12, 1003–1004 CrossRef PubMed.
- C. Miller, L. E. Thomsen, C. Gaggero, R. Mosseri, H. Ingmer and S. N. Cohen, Science, 2004, 305, 1629–1631 CrossRef CAS PubMed.
- S. Van Der Veen, T. Abee, W. M. De Vos and M. H. J. Wells-Bennik, FEMS Microbiol. Lett., 2009, 295, 195–203 CrossRef CAS.
- J. M. Flynn, S. B. Neher, Y. I. Kim, R. T. Sauer and T. A. Baker, Mol. Cell, 2003, 11, 671–683 CrossRef CAS.
- B. Guillon, A.-L. Bulteau, M. Wattenhofer-Donzé, S. Schmucker, B. Friguet, H. Puccio, J.-C. Drapier and C. Bouton, FEBS J., 2009, 276, 1036–1047 CrossRef CAS PubMed.
- D. Frees, S. N. Qazi, P. J. Hill and H. Ingmer, Mol. Microbiol., 2003, 48, 1565–1578 CrossRef CAS PubMed.
- A. J. Farrand, D. B. Friedman, M. L. Reniere, H. Ingmer, D. Frees and E. P. Skaar, Pathog. Dis., 2015, 73, ftv004 Search PubMed.
- A. Michel, F. Agerer, C. R. Hauck, M. Herrmann, J. Ullrich, J. Hacker and K. Ohlsen, J. Bacteriol., 2006, 188, 5783–5796 CrossRef CAS.
- C. D. Speziali, S. E. Dale, J. A. Henderson, E. D. Vinés and D. E. Heinrichs, J. Bacteriol., 2006, 188, 2048–2055 CrossRef CAS.
- V. C. Kirsch, C. Fetzer and S. A. Sieber, J. Proteome Res., 2021, 20, 867–879 CrossRef CAS PubMed.
- J. Feng, S. Michalik, A. N. Varming, J. H. Andersen, D. Albrecht, L. Jelsbak, S. Krieger, K. Ohlsen, M. Hecker, U. Gerth, H. Ingmer and D. Frees, J. Proteome Res., 2013, 12, 547–558 CrossRef CAS PubMed.
- C. Kisker, J. Kuper and B. Van Houten, Cold Spring Harbor Perspect. Biol., 2013, 5, a012591 Search PubMed.
- S. Nair, I. Derré, T. Msadek, O. Gaillot and P. Berche, Mol. Microbiol., 2000, 35, 800–811 CrossRef CAS PubMed.
- A. Schulz and W. Schumann, J. Bacteriol., 1996, 178, 1088–1093 CrossRef CAS.
- M. Li, O. Kandror, T. Akopian, P. Dharkar, A. Wlodawer, M. R. Maurizi and A. L. Goldberg, J. Biol. Chem., 2016, 291, 7465–7476 CrossRef CAS.
- M. Arnaud, A. Chastanet and M. Débarbouillé, Appl. Environ. Microbiol., 2004, 70, 6887–6891 CrossRef CAS PubMed.
- B. Joseph, K. Przybilla, C. Stühler, K. Schauer, J. Slaghuis, T. M. Fuchs and W. Goebel, J. Bacteriol., 2006, 188, 556–568 CrossRef CAS PubMed.
- J. Cox and M. Mann, Nat. Biotechnol., 2008, 26, 1367–1372 CrossRef CAS PubMed.
- S. Tyanova, T. Temu, P. Sinitcyn, A. Carlson, M. Y. Hein, T. Geiger, M. Mann and J. Cox, Nat. Methods, 2016, 13(9), 731–740 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cb00077f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |