
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonitoring GAPDH activity and inhibition with cysteine-reactive chemical probes†
Sarah E.
Canarelli
a,
Brooke M.
Swalm
b,
Eric T.
Larson
 *b,
Michael J.
Morrison‡
*b and
Eranthie
Weerapana
*a
*b,
Michael J.
Morrison‡
*b and
Eranthie
Weerapana
*a
aDepartment of Chemistry, Boston College, Chestnut Hill, Massachusetts 02467, USA. E-mail: eranthie@bc.edu
bRheos Medicines, Inc, Cambridge, Massachusetts 02142, USA. E-mail: elarson@rheosrx.com; mmorrison57@gmail.com
First published on 9th June 2022
Abstract
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) is a central enzyme in glycolysis that regulates the Warburg effect in cancer cells. In addition to its role in metabolism, GAPDH is also implicated in diverse cellular processes, including transcription and apoptosis. Dysregulated GAPDH activity is associated with a variety of pathologies, and GAPDH inhibitors have demonstrated therapeutic potential as anticancer and immunomodulatory agents. Given the critical role of GAPDH in pathophysiology, it is important to have access to tools that enable rapid monitoring of GAPDH activity and inhibition within a complex biological system. Here, we report an electrophilic peptide-based probe, SEC1, which covalently modifies the active-site cysteine, C152, of GAPDH to directly report on GAPDH activity within a proteome. We demonstrate the utility of SEC1 to assess changes in GAPDH activity in response to oncogenic transformation, reactive oxygen species (ROS) and small-molecule GAPDH inhibitors, including Koningic acid (KA). We then further evaluated KA, to determine the detailed mechanism of inhibition. Our mechanistic studies confirm that KA is a highly effective irreversible inhibitor of GAPDH, which acts through a NAD+-uncompetitive and G3P-competitive mechanism. Proteome-wide evaluation of the cysteine targets of KA demonstrated high selectivity for the active-site cysteine of GAPDH over other reactive cysteines within the proteome. Lastly, the therapeutic potential of KA was investigated in an autoimmune model, where treatment with KA resulted in decreased cytokine production by Th1 effector cells. Together, these studies describe methods to evaluate GAPDH activity and inhibition within a proteome, and report on the high potency and selectivity of KA as an irreversible inhibitor of GAPDH.
Introduction
Dysregulation of glycolysis is a hallmark of cancer and other diseases. In order to support the biosynthetic demands of rapidly dividing cells, energy metabolism is reprogrammed to increase glucose uptake and lactic-acid fermentation, independent of oxygen supply. This metabolic reprogramming is known as the Warburg effect.1,2 Compared to normal cells, cancer cells and activated immune cells are more dependent on glycolysis than oxidative phosphorylation for ATP production. As a result, glycolytic enzymes have emerged as promising therapeutic targets for cancer and immunomodulation. Recently, D-mannoheptulose, an inhibitor of hexokinase, was shown to enhance anti-tumor effects and reduce growth of breast cancer cells by inducing apoptosis.3 Another hexokinase inhibitor, 2-deoxyglucose (2-DG), reduced joint inflammation and immune-cell activation in an autoimmune model.4 Therapeutic strategies for targeting Warburg metabolism have largely focused on targeting hexokinase and phosphofructokinase, enzymes that traditionally regulate glycolytic flux. However, flux control analysis identified glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as a rate-limiting step in Warburg metabolism.5 Furthermore, overexpression of GAPDH has been reported in a variety of cancers and is associated with a poor survival rate.6,7 As a result, GAPDH inhibition has been identified as a promising strategy for the treatment of cancer and autoimmune diseases.8GAPDH catalyzes the conversion of glyceraldehyde 3-phosphate (G3P) to 1,3-bisphosphoglycerate (1,3 BPG) in the presence of inorganic phosphate and nicotinamide adenine dinucleotide (NAD+). The mechanism of the GAPDH-catalyzed reaction involves nucleophilic attack of the active-site cysteine, C152, on the aldehyde group of G3P, followed by subsequent hydride transfer to NAD+, resulting in the formation of a covalent enzyme-substrate complex. The thioester linkage in this enzyme-substrate complex is then cleaved by inorganic phosphate to generate the 1,3 BPG product (Fig. 1(A)). The glycolytic functions of GAPDH rely on the homo-tetrameric form of GAPDH that is localized in the cytosol. However, distinct pools of GAPDH have also been observed in membranes, mitochondria, and the nucleus.9 In these subcellular compartments, GAPDH participates in a variety of cellular functions, including ER to golgi vesicle transport, cell signaling, extracellular vesicle biogenesis, DNA replication and repair, and regulation of cell death pathways.10–12 This functional diversity of GAPDH is mediated, in part, by post-translational modifications and protein–protein interactions of GAPDH.13 For example, S-nitrosated GAPDH has been shown to interact with the ubiquitin ligase Siah1, resulting in translocation to the nucleus and subsequent induction of apoptosis.14 Protein–protein interactions of GAPDH can govern both physiological and pathological processes. For example, in the case of Alzheimer's disease, GAPDH binds amyloid-β to form stable neurotoxic aggregates.15,16
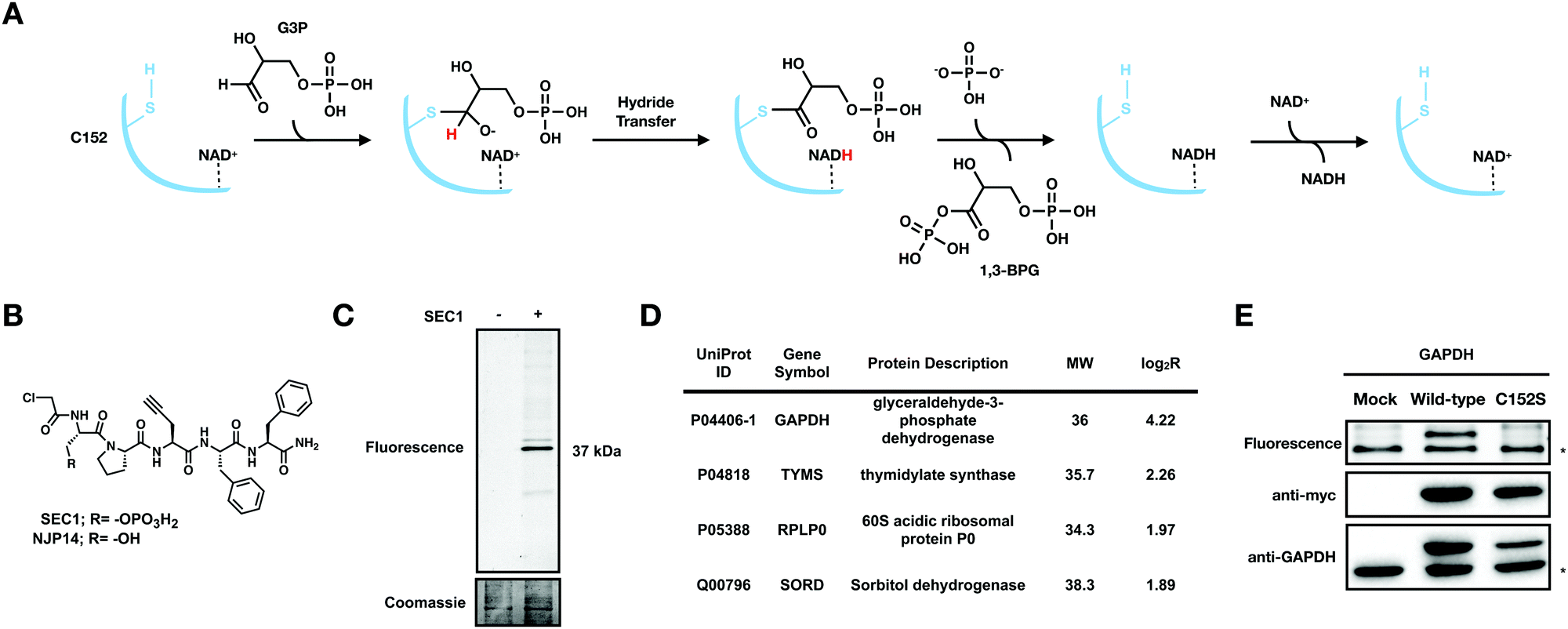 | ||
Fig. 1 SEC1 is a phosphopeptide probe that covalently modifies GAPDH (A) Scheme for the GAPDH catalyzed conversion of glyceraldehyde 3-phosphate (G3P) to 1,3 bisphosphoglycerate (1,3 BPG). (B) Structure of chloroacetamide-bearing peptide probes, SEC1 and NJP14. (C) In-gel fluorescence of HeLa lysates labeled with SEC1 (100 μM, 1 h) shows labeling of one major protein at ∼37 kDa. (D) Protein targets of SEC1 identified in Jurkat cells with a MW between 30 and 45 kDa (light:heavy log2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) R > 1). Unfiltered MS data is provided in Table S1 (ESI†). (E) HEK293T cells were transiently transfected with a pcDNA3.1 plasmid encoding GAPDH to identify C152 as the site of modification. In-gel fluorescence (top panel) of mock transfected, GAPDH WT, and GAPDH C152S mutant-expressing cells labeled with SEC1. Anti-myc (middle panel) and anti-GAPDH (bottom panel) western blots to confirm overexpression. The lower band * represents endogenous protein and the upper band represents the overexpressed myc/His-tagged GAPDH. Western blotting with an anti-myc antibody verified equal protein expression for mutant and WT proteins. R > 1). Unfiltered MS data is provided in Table S1 (ESI†). (E) HEK293T cells were transiently transfected with a pcDNA3.1 plasmid encoding GAPDH to identify C152 as the site of modification. In-gel fluorescence (top panel) of mock transfected, GAPDH WT, and GAPDH C152S mutant-expressing cells labeled with SEC1. Anti-myc (middle panel) and anti-GAPDH (bottom panel) western blots to confirm overexpression. The lower band * represents endogenous protein and the upper band represents the overexpressed myc/His-tagged GAPDH. Western blotting with an anti-myc antibody verified equal protein expression for mutant and WT proteins. | ||
Given the key role of GAPDH in a variety of pathophysiological processes, it is important to have access to methods that allow for rapid interrogation of GAPDH activity within a complex proteome. Current methods for evaluating GAPDH activity involve spectrophotometric assays that monitor NADH levels.17,18 These assays are highly effective at quantitatively assessing the activity of purified GAPDH in vitro, however, the sensitivity and accuracy is reduced when applied within a complex biological sample. As an alternative to traditional activity assays, activity-based protein profiling (ABPP) is a technology that applies chemical probes to report on the activity state of enzymes within a native and complex proteome. The development of an activity-based probe (ABP) for a protein of interest relies on the presence of a highly nucleophilic active-site residue that can be targeted by a complementary electrophile on the ABP. Here, we report on a phospho-peptide ABP, SEC1, which bears a chloroacetamide electrophile that covalently modifies the active-site cysteine, C152, of GAPDH. SEC1 has sufficient selectivity to enable visualization of GAPDH labeling within a proteome using in-gel fluorescence analysis. We demonstrate that SEC1 can report on relative GAPDH activity levels across two biological systems, with increased GAPDH activity observed upon oncogenic transformation of the MCF10A cell line. Oxidation of C152, or inhibition of GAPDH by covalent inhibitors, will result in diminished SEC1 labeling. Therefore, the extent of SEC1 labeling provides a measure of the pool of GAPDH that is present in the reduced and active form. Importantly, traditional abundance-based methods that monitor GAPDH protein levels are unable to inform on inhibition or oxidation of GAPDH. Upon screening a panel of GAPDH inhibitors using SEC1, we identified Koningic acid (KA) as highly potent and selective, and performed detailed in vitro evaluation of the mechanism of inhibition by this epoxide-containing natural product. The selectivity of KA for C152 of GAPDH over other reactive cysteines in the proteome was evaluated using isotopic tandem orthogonal proteolysis-activity-based protein profiling (isoTOP-ABPP).19 Lastly, we investigated the effects of KA in a cytokine production assay, providing extensive evaluation of the mechanism, selectivity, and cellular effects of KA. Together, these studies provide tools and insight into the activity, inhibition, and therapeutic potential of GAPDH.
Results and discussion
SEC1 is a phospho-peptide probe that covalently modifies GAPDH
We previously generated a library of peptide-based probes that contained the following functionalities: (1) a variable peptide region comprised of ∼5 amino acids; (2) a cysteine-reactive electrophile such as a chloroacetamide or acrylamide group; and, (3) a propargyl glycine residue to provide an alkyne handle for biorthogonal conjugation. Initial evaluation of the peptide library identified NJP14 as a peptide probe that labeled a unique subset of reactive cysteine residues in the human proteome, determined to be zinc-chelating residues.20 Here, we report on the proteome-wide reactivity of another member of this peptide-based probe library, SEC1. SEC1 is a penta-peptide with the sequence pSer-Pro-Pra-Phe-Phe (pSer: phosphoserine, Pra: propargylglycine), bearing an N-terminal chloroacetamide moiety as the cysteine-targeting electrophile (Fig. 1(B)). The Pra residue enables conjugation of a fluorescent or biotin tag using copper-catalyzed azide-alkyne cycloaddition (CuAAC) chemistry.To evaluate the proteome-wide reactivity of SEC1, SEC1-labeled proteins from cell lysates were conjugated to rhodamine-azide (Rh-N3) and visualized by in-gel fluorescence. The predominant labeling event was a protein at ∼37 kDa (Fig. 1(C)). To determine the identity of this 37 kDa band, cell lysates were treated with SEC1 or DMSO, enriched on streptavidin beads following conjugation of biotin azide using CuAAC and subjected to on-bead trypsin digestion. The resulting tryptic peptides were then isotopically labeled with light (SEC1) and heavy (DMSO) formaldehyde, combined, and analyzed by LC/LC-MS/MS. The relative abundance of each identified peptide in the SEC1 and DMSO-treated samples was quantified to generate light: heavy ratios (R). Proteins with a log2R > 1 were designated as putative SEC1 targets. Filtering the resulting target proteins by molecular weight identified GAPDH as the ∼37 kDa predominant protein target of SEC1 (Fig. 1(D) and Table S1, ESI†).
To confirm covalent targeting of GAPDH by SEC1, and to identify the cysteine residue on GAPDH that is modified by SEC1, HEK293T cells were transiently transfected with C-terminal myc/His-tagged wild-type (WT) and active-site cysteine mutant (C152S) GAPDH. Anti-myc and anti-GAPDH blots were used to confirm expression of the myc/His-tagged isoforms of GAPDH. Upon treatment of these cell lysates with SEC1, labeling of bands corresponding to endogenous GAPDH (lower band) and overexpressed GAPDH (upper band) were observed for the WT (Fig. 1(E)). No labeling of the overexpressed GAPDH was observed for the C152S mutant, confirming that SEC1 covalently modifies the active-site cysteine, C152, in GAPDH.
To further explore the features of SEC1 that were essential for driving GAPDH reactivity, several structural analogs of SEC1 were generated. These analogs included the non-phosphorylated derivative of SEC1, NJP14, as well as analogs where each amino acid (except Pra) was replaced with alanine (Ala) (SEC2-5, Fig. 2(A)). The proteome-wide reactivity of these analogs was determined by in-gel fluorescence (Fig. 2(B)). GAPDH reactivity was completely diminished for NJP14 and SEC5, confirming that the pSer is a critical recognition element in driving GAPDH selectivity. Of the remaining amino acids, replacement of either of the two Phe residues (SEC2 and SEC3) resulted in an increase in GAPDH labeling, and replacement of Pro (SEC4) did not appear to have any effect on GAPDH labeling. Together, these studies indicate that there is significant flexibility to further modulate the structure of SEC1 to improve potency and selectivity for GAPDH. As expected, the pSer residue was essential for GAPDH recognition, since the phosphate moiety of pSer likely mimics the phosphate group of the G3P substrate for GAPDH (Fig. 2(C)). With this knowledge in hand, we generated an analog of SEC1 that was pre-conjugated to rhodamine (Fig. S1a, ESI†), to circumvent the need to perform CuAAC to conjugate a reporter tag. This fluorescent version, SEC6, maintained GAPDH labeling (Fig. S1b, ESI†), and therefore offers a more versatile analog of the GAPDH activity based probe for users without expertise in CuAAC.
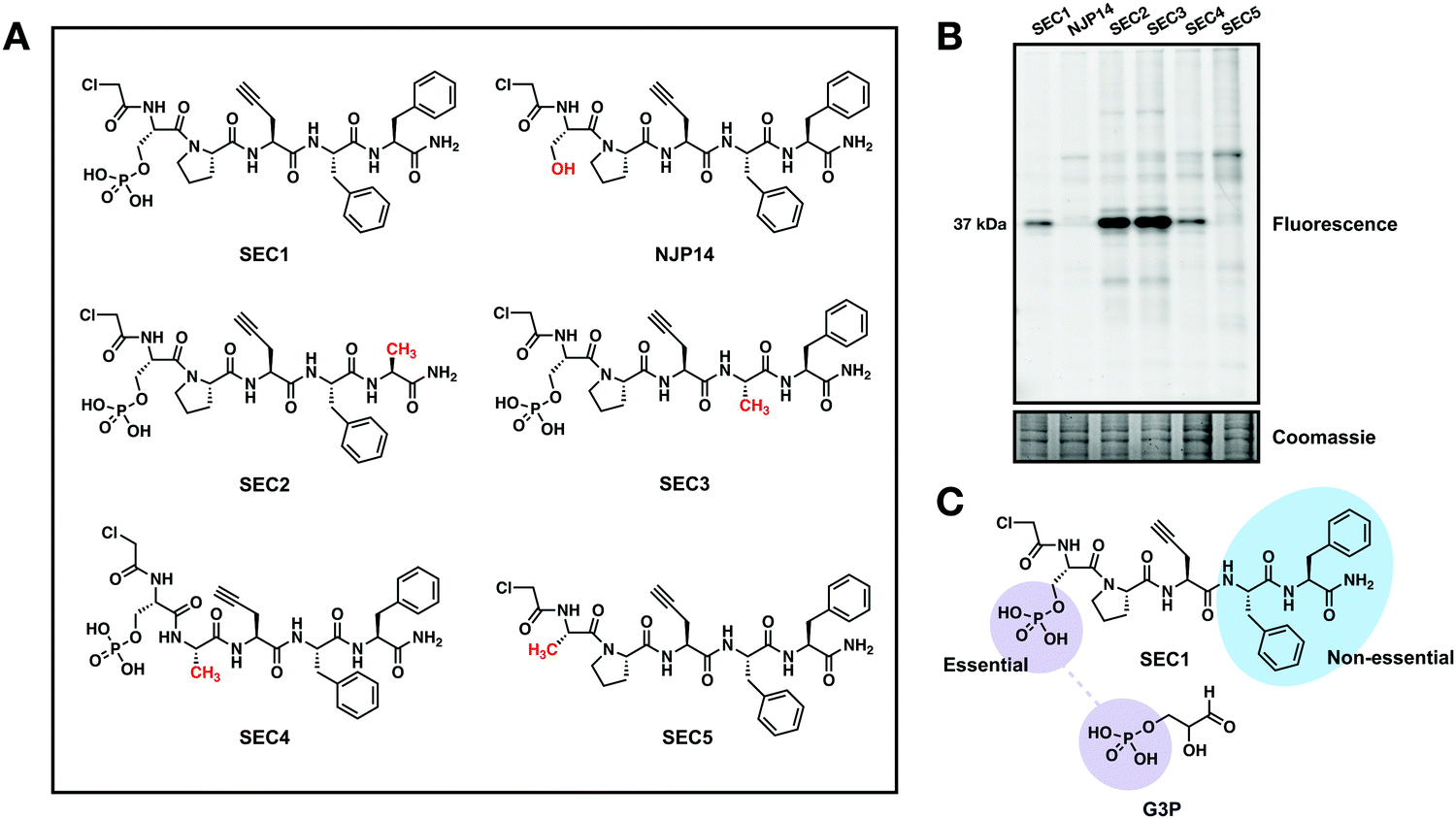 | ||
| Fig. 2 The phosphoserine of SEC1 is essential for GAPDH binding (A) Library of chloroacetamide bearing peptide probe analogs used to investigate critical residues essential for GAPDH binding. (B) In-gel fluorescence of HeLa lysates labeled with library members (100 μM, 1 h). The Coomassie-stained gel confirms equal protein loading in all lanes. (C) Chemical structures of SEC1 and glyceraldehyde 3-phosphate (G3P), the substrate of GAPDH. | ||
GAPDH activity increases with oncogenic transformation
It is known that GAPDH expression is increased in human cancers, and overexpression of GAPDH results in increased cancer cell proliferation.6,21 To highlight the ability of SEC1 to report on GAPDH activity in different cell lines, we utilized the benign MCF10A epithelial cell line, and the malignant counterpart, MCF10CA1a, which overexpresses the HRAS oncogene.22 Lysates from each cell line were labeled with SEC1, and GAPDH labeling was visualized by in-gel fluorescence (Fig. 3(A)). Anti-GAPDH western blots were used to demonstrate that GAPDH levels are significantly higher in MCF10CA1a cells relative to MCF10A cells. Similarly, SEC1 labeling reflected a ∼7-fold increase in GAPDH activity in MCF10CA1a cells. These studies confirm that GAPDH levels increase with oncogenic transformation. Additionally, we demonstrate that SEC1 can assess the activity-state of GAPDH, thereby complementing abundance-based western blot measurements. Treatment of MCF10CA1a cells with the GAPDH inhibitor, KA, resulted in a loss in SEC1 labeling, confirming that the highly intense protein band at ∼37 kDa is predominantly GAPDH.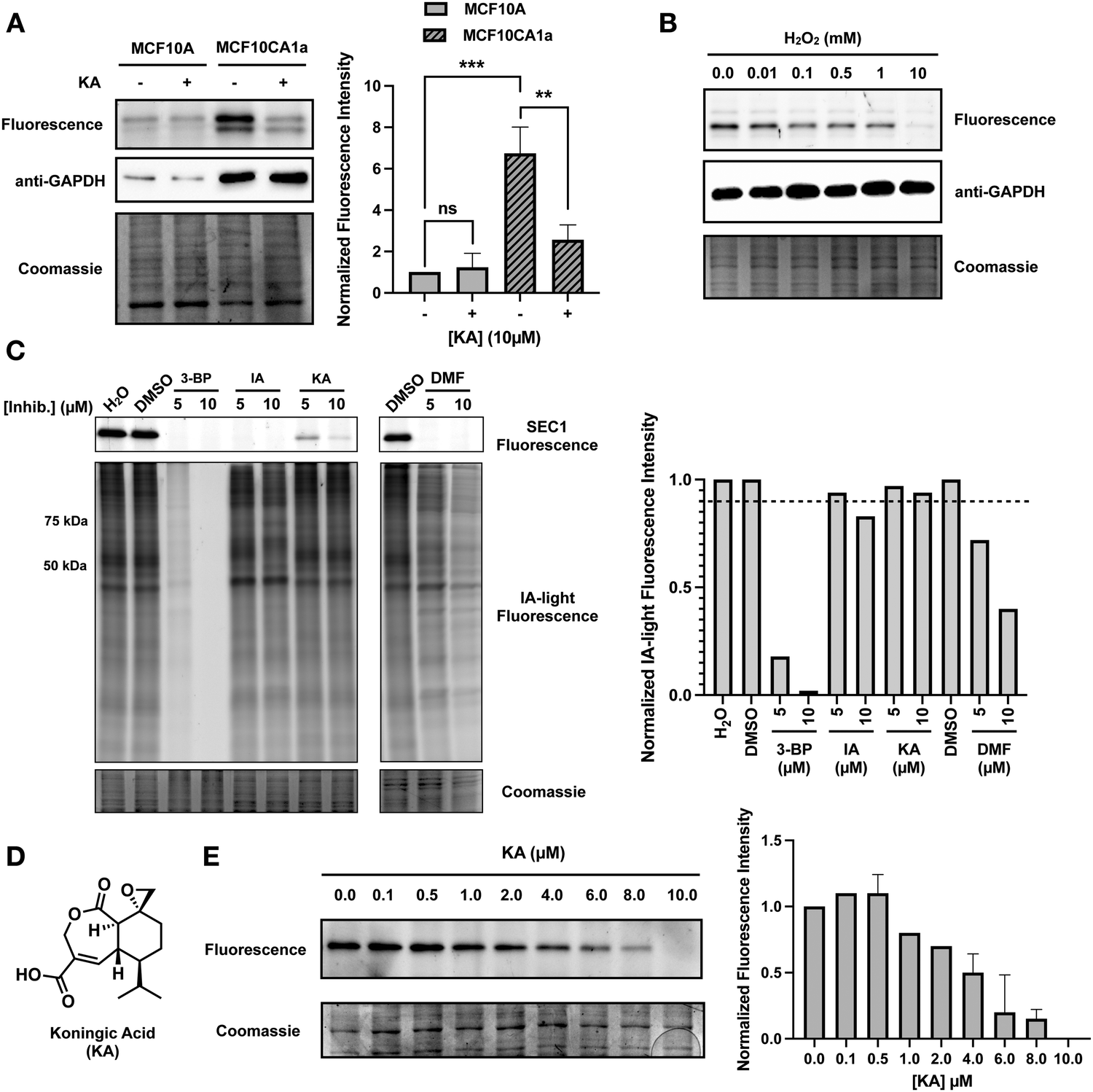 | ||
| Fig. 3 SEC1 reports on changes in GAPDH activity associated with oncogenic transformation, reactive oxygen species and covalent inhibitors. (A) MCF10A and MCF10CA1a cells were treated with vehicle or KA (10 μM, 1 h) and cell lysates were labeled with SEC1 (100 μM, 1 h). Protein labeling was evaluated by in-gel fluorescence following CuACC with Rh-N3, and protein loading was evaluated by Coomassie stain. Western blot analysis with an anti-GAPDH antibody was used to evaluate GAPDH expression. Band intensities were analyzed and compared using ImageJ software and GraphPad Prism version 9. All values are means ± S.E. (error bars) from three replicates (n = 3). *Significantly different p < 0.05, **p < 0.01, ***p < 0.001. ns., not significant. (B) Jurkat cell lysates were treated with increasing concentrations of H2O2 prior to labeling with SEC1, and protein labeling was visualized by in-gel fluorescence. Protein loading was evaluated by Coomassie stain and western blot analysis with an anti-GAPDH antibody was used to evaluate GAPDH expression. (C) Jurkat cell lysates were incubated with two different concentrations (5 and 10 μM) of GAPDH inhibitors: 3-bromopyruvate (3-BP), iodoacetate (IA), KA, and dimethyl fumarate (DMF). Treated lysates were labeled with SEC1 (100 μM, 1 h) and IA-light (100 μM, 1 h) and labeled proteins were visualized by in-gel fluorescence. Band intensities were analyzed and compared using ImageJ software and GraphPad Prism version 9 for IA-light. (D) The chemical structure of KA, a previously reported GAPDH inhibitor. (E) Jurkat cells were incubated with increasing concentrations of KA and lysates were labeled with SEC1 (100 μM, 1 h). Protein labeling was evaluated by in-gel fluorescence and protein loading was evaluated by Coomassie stain. Band intensities were analyzed and compared using ImageJ software and GraphPad Prism version 9. All values are means ± S.E. (error bars) from two replicates (n = 2). | ||
GAPDH is susceptible to oxidation by reactive oxygen species
Reactive oxygen species (ROS) are highly reactive byproducts of metabolism that can act in a signaling role to regulate cellular pathways.23 Elevated levels of ROS can result in both reversible and irreversible cysteine oxidation. GAPDH is a known target of ROS, and oxidation of the active-site cysteine, C152, leads to enzyme inactivation, and formation of GAPDH aggregates, which are associated with cell death.24,25 Oxidation of C152 would lead to a loss of covalent SEC1 labeling. To evaluate the ability of SEC1 to report on the oxidation state of GAPDH, HeLa cell lysates were treated with increasing concentrations of hydrogen peroxide. Following 1 h of peroxide treatment, lysates were labeled with SEC1 and GAPDH labeling was visualized by in-gel fluorescence (Fig. 3(B)). A concentration-dependent decrease in SEC1 labeling of GAPDH was observed in the presence of peroxide. While a low concentration (10 μM) of peroxide had little effect on SEC1 labeling, high concentrations (10 mM) completely abolished GAPDH reactivity. These observations support the use of SEC1 as a chemical probe to provide a rapid readout of the oxidation state of GAPDH upon exposure of a proteome to diverse sources of ROS.KA is a potent inhibitor of GAPDH
Due to the involvement of GAPDH in diverse pathologies, many GAPDH inhibitors have been reported to date.26 We screened a subset of these reported GAPDH inhibitors for potency and selectivity of GAPDH inhibition, including 3-bromopyruvate (3-BP), iodoacetate (IA), KA, and dimethyl fumarate (DMF). Cell lysates were treated with two different concentrations of each inhibitor (5 μM and 10 μM) and subsequently treated with SEC1 for in-gel fluorescence visualization (Fig. 3(C)). Inhibition of GAPDH would result in a decrease in SEC1 labeling. At the highest inhibitor concentration tested (10 μM), all of the inhibitors displayed at least a partial decrease in SEC1 labeling, however, 3-BP, IA and DMF showed complete loss of SEC1 labeling even at the lower concentration tested (5 μM). To assess selectivity across the proteome, SEC1 was replaced with a general cysteine-reactive iodoacetamide-alkyne probe (IA-light), which covalently modifies a large subset of highly reactive cysteine-containing proteins within a proteome (Fig. 3(C)).27 This gel-based evaluation of global cysteine reactivity demonstrated that 3-BP and DMF are broadly cysteine reactive, resulting in diminished IA-light labeling across a wide array of proteins. IA was less promiscuous, but loss in labeling of proteins around 50 and 75 kDa was observed. In contrast, KA appeared to be the most selective (albeit least potent) of the panel of inhibitors tested, and was thereby selected for further evaluation of potency and selectivity as described below.KA (Fig. 3(D)) is a sesquiterpene natural product isolated from the fungus Trichoderma koningi, and inhibits GAPDH through covalent modification of the catalytic cysteine.28,29 To further evaluate the potency of KA, a gel-based assessment of GAPDH labeling by SEC1 was performed over a range of KA concentrations (Fig. 3(E)). Jurkat cells were treated with increasing concentrations of KA (0–10 μM) for one hour prior to cell lysis. KA-treated cell lysates were labeled with SEC1, and GAPDH activity was visualized by in-gel fluorescence. A clear concentration-dependent loss in GAPDH activity was observed with increasing KA treatment, with ∼4 μM KA resulting in 50% inhibition. The potency of KA for GAPDH was also evaluated in HeLa cells (Fig. S2a, ESI†) and demonstrated a similar concentration-dependent decrease in GAPDH activity with increasing KA. Again, these studies serve to highlight the utility of SEC1, which enables facile screening of different inhibitors and concentrations for follow up biological investigations.
Mechanistic characterization of the GAPDH inhibitor KA
To further investigate the mechanism of GAPDH inhibition by KA in the context of the above cellular results, several in vitro studies were performed. First, a luminescent GAPDH activity assay based on detection of the reaction product NADH was developed. This assay was utilized to determine the mechanism of inhibition of KA in the presence of the physiological substrates of GAPDH (NAD+ and G3P). Increasing NAD+ concentrations lead to the enhancement of GAPDH inhibition by KA (Fig. 4(A)), whereas increasing G3P concentration resulted in a decrease in KA potency (Fig. 4(B)). Fitting each data set to the mixed model inhibition equation resulted in an alpha value much greater than one for G3P (α ∼ 280) and much less than one for NAD+ (α ∼ 0.0003). KA is therefore an NAD+-uncompetitive, G3P-competitive GAPDH inhibitor.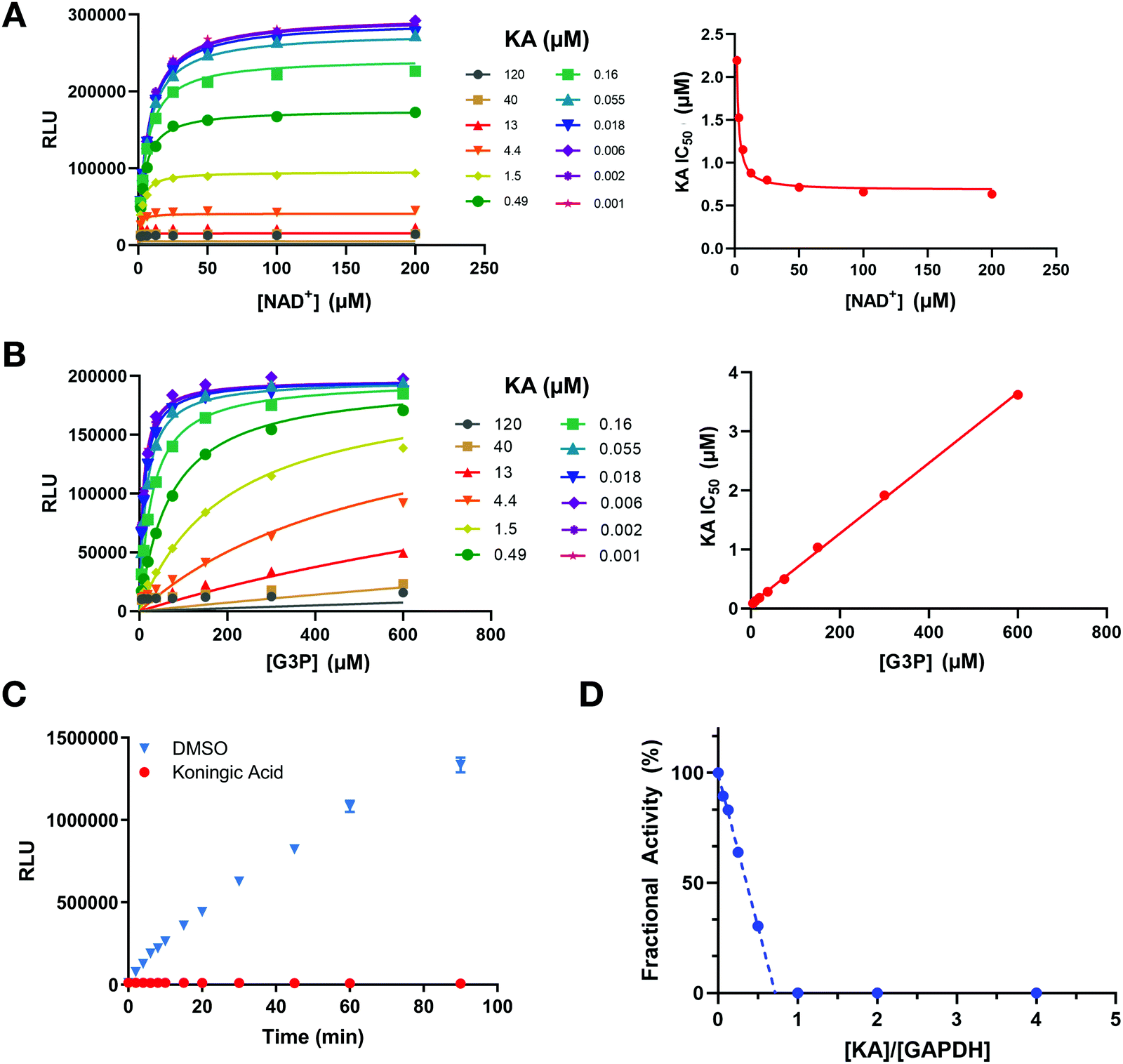 | ||
| Fig. 4 KA is a potent, irreversible, NAD+ uncompetitive, and G3P competitive inhibitor of GAPDH. (A) Global (left) and local (right) fits of varying concentrations of NAD+ (8-point, 2-fold dilution) and KA (12-point, 3-fold dilution). αKi = 0.70 ± 0.02 μM (global) and αKi = 0.68 ± 0.02 μM (local). (B) Global (left) and local (right) fits of varying concentrations of G3P (8-point, 2-fold dilution) and KA (12-point, 3-fold dilution). Ki = 0.06 ± 0.003 μM (global) and Ki = 0.08 ± 0.02 μM (local). Each data point is an average of 3 replicates. (C) Recovery of GAPDH biochemical activity after rapid dilution of the enzyme-inhibitor complex; (blue) DMSO and (red) KA. GAPDH and KA were pre-incubated for 30 minutes followed by dilution to a negligible concentration of free compound. Enzymatic activity was quantified by generation of NADH in a bioluminescent readout. Error bars for each point represent the standard deviation from two replicates. (D) KA is an extremely effective inactivator of GAPDH. An excess of enzyme was preincubated with varying concentrations of KA, diluted 100×, and the remaining catalytic activity measured. The partition ratio was calculated to be approximately 0 (−0.27 ± 0.01) resulting in completely efficient inactivation of GAPDH (KA is sequestered by the enzyme upon each binding event forming the covalent complex). Standard deviation was calculated based on two independent measurements of the partition ratio. | ||
To evaluate the reversibility of the interaction between KA and GAPDH, GAPDH activity was assessed after rapid dilution of the enzyme-inhibitor complex. GAPDH activity was not recovered upon dilution after KA treatment (Fig. 4(C)), confirming the irreversible nature of the inhibition. The binding affinity of KA was also evaluated by SPR (Fig. S3, ESI†), and confirmed that KA binds irreversibly to WT GAPDH. In contrast, binding to the C152S active-site mutant is rapid and reversible. Interestingly, KA potency almost exclusively relies on its ability to inactivate the enzyme (kinact) as opposed to its binding capacity (Ki) as exhibited through biophysical studies with the C152S mutant protein. This was confirmed biochemically by measuring the rate of GAPDH inactivation (kobs) as a function of KA concentration (kinact = 1.44 min−1, Ki = 6.3 μM; Fig. S4, ESI†).
GAPDH activity following KA treatment was further evaluated in a fractional activity assay to determine the efficiency of inactivation that is better known as the partition ratio (r).30 In this assay, GAPDH activity was measured after excess enzyme was incubated with increasing concentrations of KA prior to dilution and measurement of activity. GAPDH activity was found to be significantly reduced even when the enzyme is in excess, where a partition ratio was calculated to be ∼−0.27 (Fig. 4(D)). The x-intercept ([KA]/[GAPDH]) approaches a value of one indicating that 1 mol of KA is required to inactivate 1 mol of GAPDH. Therefore, KA is a highly efficient inhibitor that generates an inactivated enzyme with each binding event. Together, these data confirm that KA is a highly potent irreversible inhibitor of GAPDH that is competitive with the G3P substrate and relies on the ability to quickly and efficiently inactivate the enzyme as its principal source of inhibition.
Proteome-wide selectivity of KA
Proteome-wide selectivity has been reported for only a few GAPDH inhibitors.31,32 Evaluation of 3-BP targets using a chemoproteomic method revealed 62 significant protein targets, including GAPDH.31 A similar proteome-wide reactivity profile is not available for KA. Metabolomics data suggests that KA selectively perturbs glycolytic metabolites, however a global picture of the protein targets of KA has not been reported.5 Here, we focused on assessing the cellular cysteine targets of KA. To achieve this, we performed an isoTOP-ABPP analysis, which allows for monitoring changes in cysteine reactivity between two proteomes.27 In this workflow, Jurkat cells were treated with KA or DMSO for 1 h, and then the resulting cell lysates were labeled with IA-light or IA-heavy, respectively (Fig. 5(A)). A cleavable biotin tag was appended to probe-labeled proteins using CuAAC, allowing for streptavidin enrichment, on-bead trypsin digestion, and selective elution of probe-labeled peptides. The resulting isotopically tagged peptides were analyzed by LC/LC-MS/MS, to identify and quantify cysteine-labeled peptides from the DMSO and KA samples. This competitive isoTOP-ABPP strategy was used to evaluate three different KA concentrations (1 μM, 5 μM, and 10 μM). For each identified peptide, a heavy:light isotopic ratio (R) was calculated, and provides a measure of the extent of probe labeling in the KA versus DMSO-treated samples. A cysteine with log2R > 0 indicates a decrease in cysteine reactivity upon treatment with KA, whereas a log2R = 0, denotes no change in cysteine reactivity upon treatment. Over 1000 cysteine sites were quantified from triplicate data for each condition (Table S2, ESI†). At 10 μM KA treatment, the majority of cysteines (98%) displayed log2R = 0, signifying negligible covalent modification of these cysteines by KA. In contrast, C152 of GAPDH displayed a significant decrease in cysteine reactivity with a high log2R value of ∼4, indicating that close to ∼100% of this cysteine is modified at 10 μM KA (Fig. 5(B)). As expected, the log2R values for C152 display a clear dependence on KA concentration (Fig. 5(C)). Additionally, this competitive isoTOP-ABPP workflow was also used to evaluate KA targets in HeLa cells (Fig. S2b and Table S3, ESI†). Similar to Jurkat cells, C152 of GAPDH displayed a significant decrease in cysteine reactivity in HeLa cells in the presence of 10 μM KA (log2R of ∼2). These data demonstrate the high selectivity of KA for C152 of GAPDH over other highly reactive cellular cysteine residues.
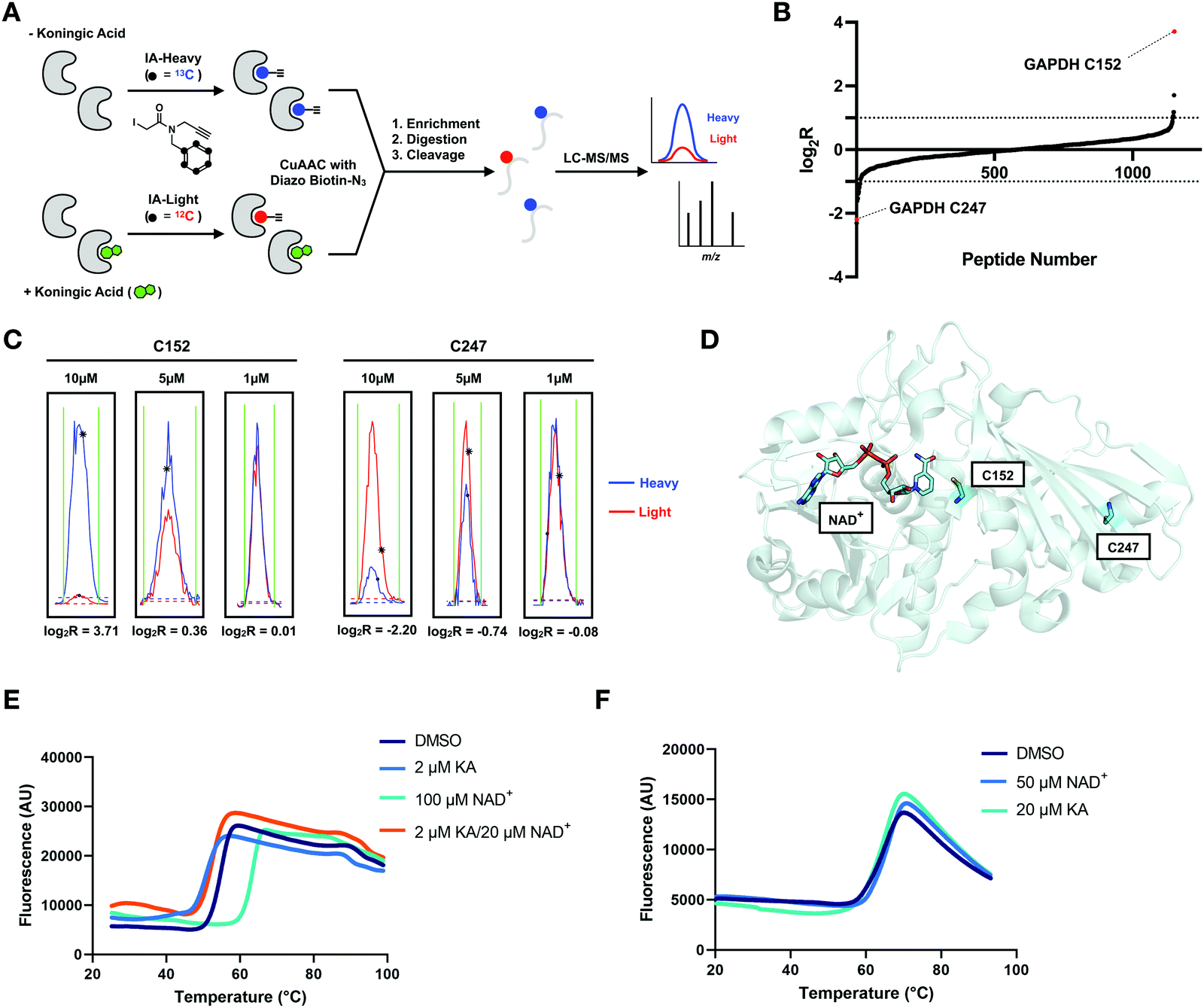 | ||
| Fig. 5 KA is a highly selective GAPDH inhibitor. (A) isoTOP-ABPP workflow used to study KA-induced inhibition of GAPDH. The proteomes of vehicle and KA treated (1, 5, 10 μM) cells were labeled with cysteine-reactive IA-light and IA-heavy probes, respectively, and compared to identify cellular cysteine targets of KA. (B) Heavy:light log2R for all identified cysteines in vehicle (heavy) and 10 μM KA (light) treated Jurkat cells (n = 3, s.d. <25%). (C) Representative traces for the C152-containing peptide and C247 containing peptide of GAPDH from the Jurkat reactivity data. (D) Crystal structure of GAPDH with NAD+ bound (adapted from PDB:4wnc). (E) Thermal shift assay data for WT GAPDH showing stabilization by the addition of its physiological substrate NAD+, and destabilization upon KA binding. Protein stability in the presence of ligand was assessed using SYPRO Orange with increasing temperature (20 to 95 °C). (F) Thermal shift assay data for the C152S mutant GAPDH showing no change in stability upon addition of NAD+ and KA. | ||
Interestingly, we identified a second cysteine on GAPDH in our isoTOP-ABPP analyses, C247, which is located distal to the GAPDH active site (Fig. 5(D)). In contrast to C152, which displayed a decrease in cysteine reactivity, C247 showed an increase in cysteine reactivity with a log2R value of ∼−2 (Fig. 5(B)). This increase in cysteine reactivity is dependent on KA concentration (Fig. 5(C)), and could be indicative of a conformational change upon KA binding that makes C247 more accessible for IA-light labeling. To explore this hypothesis, a thermal shift assay was performed with Sypro Orange. GAPDH was stabilized in a concentration dependent manner, by its physiological substrate NAD+ (Fig. 5(E) and Table S4, ESI†). In contrast, KA binding was shown to have a destabilizing effect on GAPDH (Fig. 5(E) and Table S4, ESI†). Upon mutation of the active site cysteine to an alanine or serine, neither NAD+ nor KA had an effect on protein thermal stability (Fig. 5(F) and Table S4, ESI†). Interestingly, mutation of C152 to either serine or alanine had minimal impact of NAD+ binding to GAPDH by SPR (data not shown). Considering KA is a weak binder to GAPDH C152S, the thermal shift assay results are not surprising. The thermal shift assay results reported herein contradict a previous report in the literature, where KA was shown to have a very minimal stabilizing effect on GAPDH, however those studies were performed in the absence of NAD+, which likely impacted the extent of KA binding.33 Further investigation using size exclusion chromatography suggests that this destabilization is not a result of dissociation of the biological tetramer (Table S5, ESI†). Nevertheless, the observed thermal destabilization may explain localized structural changes that result in the observed increase in C247 labeling.
Inhibition of GAPDH reduces cytokine production
Autoimmune and inflammatory diseases are caused by uncontrolled long-term activation of immune cells. Similar to cancer cells, these activated immune cells undergo metabolic reprogramming to prioritize efficient and rapid biosynthesis over energy production.34 GAPDH inhibition has emerged as a therapeutic strategy to modulate immunity. In fact, DMF has been applied in the treatment of psoriasis and multiple sclerosis.35 To investigate the clinical potential of KA, GAPDH activity and inhibition were evaluated upon activation of Jurkat cells. Jurkat cells were activated by stimulating the Raf-MEK-MAPK pathway that promotes cell proliferation and cell survival using phorbol 12-myristate 13-acetate (PMA) for 10 and 20 minutes prior to cell lysis. Jurkat activation was confirmed by western blotting with an anti-pMAPK antibody, where increased pMAPK was observed after 10 and 20 minutes of PMA treatment (Fig. 6(A)). To evaluate changes in GAPDH activity upon Jurkat activation, cell lysates were treated with SEC1 and GAPDH labeling was visualized by in-gel fluorescence. Changes in GAPDH protein levels were simultaneously evaluated by western blot with an anti-GAPDH antibody. A slight increase in SEC1-labeling of GAPDH was observed in PMA-treated cells compared to the control, with the highest labeling observed at the 20 minute time point. Based on the anti-GAPDH western blot, no significant change in protein levels was observed.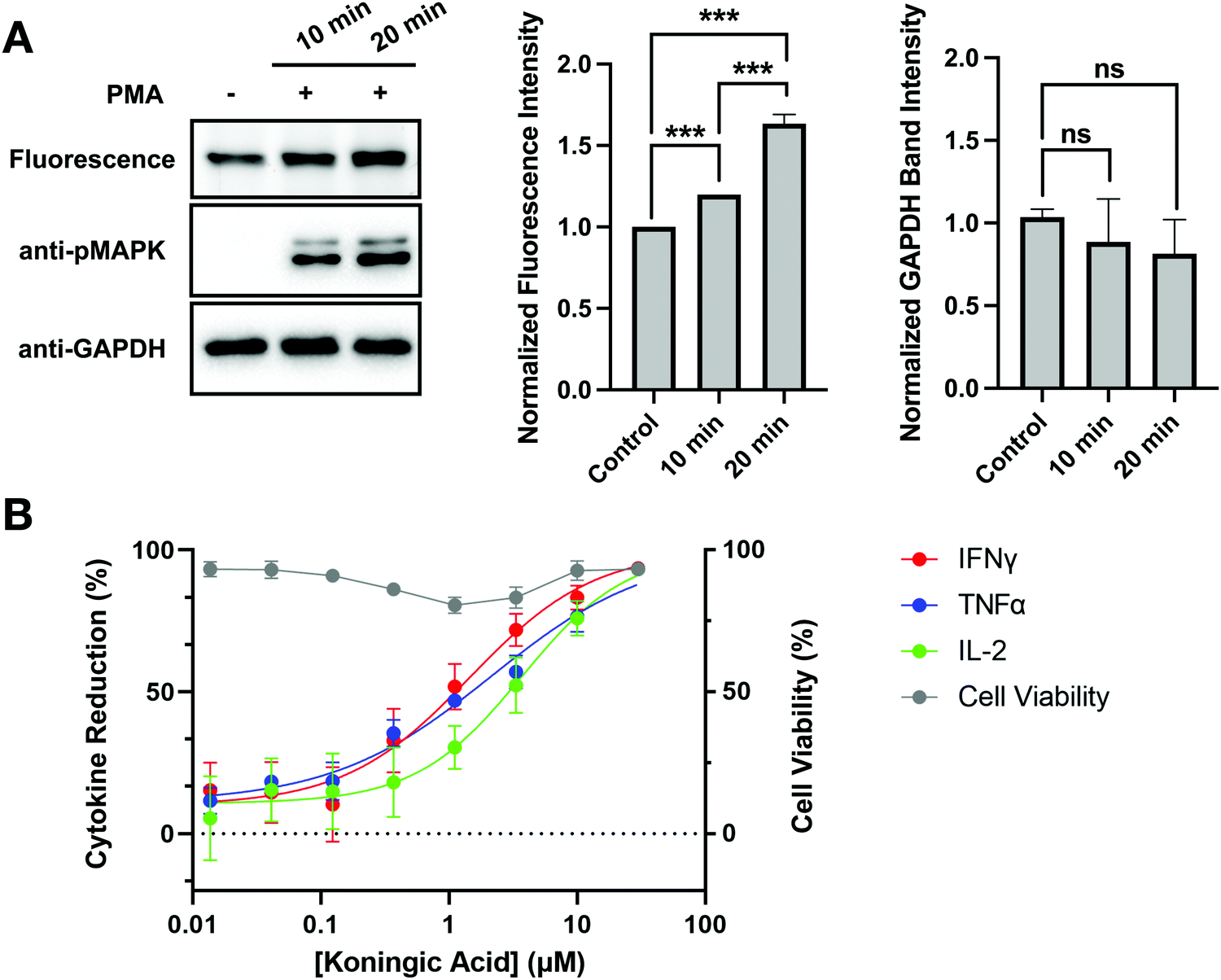 | ||
| Fig. 6 GAPDH activity and inhibition in activated immune cells. (A) Jurkat cells were activated with PMA (20 ng mL−1) for the listed time. In gel fluorescence (top panel) evaluation of activated and un-activated Jurkat lysates labeled with SEC1 (100 μM, 1 h). Anti-pMAPK (middle panel) was used to verify activation of Jurkat cells and anti-GAPDH was used to evaluate protein expression. Band intensities were analyzed and compared using ImageJ software and GraphPad Prism version 9. All values are means ± S.E. (error bars) from three replicates (n = 3) for fluorescence data and two replicates (n = 2) for western blots. *Significantly different p < 0.05, **p < 0.01, ***p < 0.001. ns., not significant. (B) Percentage reduction in cytokine production of Th1 effector cells following addition of KA with no loss in cellular viability. Representative IC50 curves measuring cytokine reduction with varying concentrations of KA following stimulation by CD3/CD28/CD2 after 24 h. Mean IC50 values were calculated by a three-parameter fit constraining the top to 100% inhibition; IFNγ (red) IC50 = 1.4 ± 0.3 μM, TNFα (blue) IC50 = 2.1 ± 0.4 μM, IL-2 (green) IC50 = 3.7 ± 0.7 μM. All values are means ± S.E. (error bars) from three replicates (n = 3). | ||
To determine the effects of GAPDH inhibition on immune-cell activation, cytokine production was measured in KA treated human Th1 effector cells following stimulation by CD3/CD28/CD2. Cellular viability was maintained in activated Th1 effector cells following KA treatment, however production of IFNγ, TNFα, and IL-2 decreased with increasing concentrations of KA (Fig. 6(B)). These data agree with previous studies, which suggest that GAPDH inhibition can be used as a strategy to modulate the immune response.34,35 To determine if the observed reduction in cytokine production was mediated specifically by changes in GAPDH, a GAPDH knockout cell line was generated using CRISPR in human CD4+ T cells. Evaluation of IFNγ in the CRISPR knockout cells showed significant inhibition in cytokine production upon GAPDH knockout (Fig. S5, ESI†). This inhibition in IFNγ production was comparable to the reduction observed following KA treatment in activated Th1 effector cells. A CRISPR knockout of the tyrosine kinase ZAP70 was also generated in CD4+ T cells. Given the importance of ZAP70 activity in T-cell activation, knockout of this protein was expected to modulate cytokine activity and thus be a positive control for a reduction in IFNγ production. The reduction in IFNγ production observed in the ZAP70 knockout was comparable to that observed in the GAPDH knockout (Fig. S5, ESI†). Together, these data suggest that KA-mediated inhibition of GAPDH effectively modulates cytokine production in T cells. Furthermore, these data highlight the clinical potential that KA has in the treatment of autoimmune diseases.
Conclusion
In summary, we report SEC1, a peptide-based probe for rapid assessment of GAPDH activity in proteomes using an in-gel fluorescence readout. SEC1 can be used to rapidly assess changes in GAPDH activity in response to oxidation by ROS or inhibition by small-molecule GAPDH inhibitors. Information regarding GAPDH oxidation or covalent inhibition cannot be obtained with traditional western blotting with an anti-GAPDH antibody. With rising interest in GAPDH inhibition as a therapeutic strategy to modulate Warburg metabolism, SEC1 allows for facile identification and characterization of GAPDH inhibitors in vitro. Our structure-activity relationship studies on SEC1 indicate that the phosphate modification on serine is a critical component for GAPDH binding, likely mimicking the phosphate of the G3P substrate. Other components of SEC1 were less critical, and future studies will focus on further optimizing this probe for increased potency and selectivity for GAPDH. Additionally, a limitation of SEC1 is the inability to apply this probe to label GAPDH directly in cells due to the poor cell permeability of the peptide-like structure, and the rapid de-phosphorylation of the phosphoserine. Future work will explore the incorporation of peptidomimetic and phosphomimetic groups to improve the cell permeability and stability, thereby enabling direct monitoring of GAPDH in cells.Of the previously reported GAPDH inhibitors that were tested, KA displayed a promising combination of potency for GAPDH with reduced cross-reactivity with other cysteine-containing proteins within the proteome. Use of a competitive isoTOP-ABPP proteomics study corroborated the observed selectivity of this inhibitor and identified the GAPDH active-site cysteine as the primary target of KA. Using purified GAPDH and a variety of in vitro characterization tools, KA was shown to be a highly potent, irreversible, and G3P-competitive inhibitor for GAPDH. To demonstrate the therapeutic potential of GAPDH inhibitors in T-cell activation, treatment of T-cells with KA decreased cytokine production without compromising cellular viability. Together, these studies provide tools and insight into the activity and inhibition of GAPDH and the therapeutic potential of KA as a potent and selective GAPDH inhibitor.
Cell lines
HeLa, HEK293T, Jurkat and MCF10a cells were purchased from ATCC. Th1 cells were generated from peripheral blood mononuclear cells (PBMCs) from a healthy adult donor. Informed consent was obtained for any experimentation with human subjects.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was funded by NIH grant R35GM134964 to E. W. and a sponsored research award from Rheos Medicines Inc. to E. W. We would like to thank Dr. Brian Albrecht and Dr. Guillaume Barbe for project conception, Dr. Brian DeChristopher for KA synthesis, Marshall Zingg for providing technical expertise, Yanlong Zhao and Fei Xue for protein production and thermal shift experiments, Qiyang Jiang for SPR experiments, and Dr. Samantha Hiemer for KA cell viability experiments. We also thank members of the Weerapana Lab for helpful discussions and critical reading of the manuscript.References
- O. Warburg, F. Wind and E. Negelein, The metabolism of tumors in the body., J. Gen. Physiol., 1927, 8, 519–530 CrossRef CAS PubMed.
- C. A. Cori and G. T. Cori, The carbohydrate metabolism of tumours, J. Biol. Chem., 1925, 65, 397–405 CrossRef CAS.
- A. G. Al-Ziaydi, A. M. Al-Shammari, M. I. Hamzah, H. S. Kadhim and M. S. Jabir, Hexokinase inhibition using D-mannoheptulose enhances oncolytic newcastle disease virus-mediated killing of breast cancer cells, Cancer Cell Int., 2020, 20, 420 CrossRef CAS PubMed.
- G. Abboud, S. Choi, N. Kanda, L. Zeumer-Spataro, D. C. Roopenian and L. Morel, Inhibition of glycolysis reduces disease severity in an autoimmune model of rheumatoid arthritis, Front. Immunol., 2018, 9, 1973 CrossRef PubMed.
- M. V. Liberti, Z. Dai, S. E. Wardell, J. A. Baccile, X. Liu, X. Gao, R. Baldi, M. Mehrmohamadi, M. O. Johnson, N. S. Madhukar, A. A. Shestov, I. Chio, O. Elemento, J. C. Rathmell, F. C. Schroeder, D. P. McDonnell and J. W. Locasale, A Predictive model for selective targeting of the Warburg effect through GAPDH inhibition with a natural product., Cell Metab., 2017, 26(4), 648–659 CrossRef CAS PubMed.
- B. Altenberg and K. O. Greulich, Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes., Genomics, 2004, 84, 1014–1020 CrossRef CAS PubMed.
- C. Guo, S. Liu and M. Z. Sun, Novel insight into the role of GAPDH playing in tumor., Clin. Transl. Oncol., 2013, 15, 167–172 CrossRef CAS PubMed.
- V. F. Lazarev, I. V. Guzhova and B. A. Margulis, Glyceraldehyde-3-phosphate dehydrogenase is a multifaceted therapeutic target., Pharmaceutics, 2020, 12(5), 416 CrossRef CAS PubMed.
- C. Tristana, N. Shahania, T. W. Sedlaka and A. Sawaa, The diverse functions of GAPDH:views from different subcellular compartments., Cell. Signalling, 2011, 23(2), 317–323 CrossRef PubMed.
- M. A. Sirover, Role of the glycolytic protein, glyceraldehyde-3-phosphate dehydrogenase, in normal cell function and in cell pathology., J. Cell. Biochem., 1997, 66, 133–140 CrossRef CAS PubMed.
- M. A. Sirover, New insights into an old protein: The functional diversity of mammalian glyceraldehyde-3-phosphate dehydrogenase., Biochim. Biophys. Acta, 1999, 1432(2), 159–184 CrossRef CAS.
- M. A. Sirover, New nuclear functions of the glycolytic protein, glyceraldehyde-3-phosphate dehydrogenase, in mammalian cells., J. Cell. Biochem., 2005, 95, 45–52 CrossRef CAS PubMed.
- M. A. Sirorver, Subcellular dynamics of multifunctional protein regulation: Mechanisms of GAPDH intracellular translocation, J. Cell. Biochem., 2012, 113, 2193–2200 CrossRef PubMed.
- M. R. Hara, N. Agrawal, S. F. Kim, M. B. Cascio, M. Fujimuro, Y. Ozeki, M. Takahashi, J. H. Cheah, S. K. Tankou, L. D. Hester, C. D. Ferris, S. D. Hayward, S. H. Snyder and A. Sawa, S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding., Nat. Cell Biol., 2005, 7(7), 665–674 CrossRef CAS PubMed.
- V. F. Lazarev, M. Tsolaki, E. R. Mikhaylova, K. A. Benken, M. A. Shevtsov, A. D. Nikotina, M. Lechpammer, V. A. Mitkevich, A. A. Makarov, A. A. Moskalev, S. A. Kozin, B. A. Margulis, I. V. Guzhova and E. Nudler, Extracellular GAPDH promotes alzheimer disease progression by enhancing amyloid-β aggregation and cytotoxicity, Aging Dis., 2021, 12(5), 1223–1237 CrossRef PubMed.
- D. A. Butterfield, S. S. Hardas and M. L. Lange, Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer's disease: Many pathways to neurodegeneration, J. Alzheimer's Dis., 2010, 20(2), 369–393 CAS.
- S. Ganapathy-Kanniappan, Analysis of GAPDH enzyme activity: A quantitative and qualitative approach, Advances in GAPDH Protein Analysis: A Functional and Biochemical Approach, Springer, 2017, pp. 5–15, ISBN-13: 9789811073403.
- S. Ganapathy-Kanniappan, Analysis of GAPDH by mass spectrometry, Advances in GAPDH Protein Analysis: A Functional and Biochemical Approach, Springer, 2017, pp. 95–98, ISBN-13: 9789811073403.
- E. Weerapana, A. E. Speers and B. F. Cravatt, Tandem orthogonal proteolysis-activity-based protein profiling (TOP-ABPP) – a general method for mapping sites of probe modification in proteomes., Nat. Protoc., 2007, 2, 1414–1425 CrossRef CAS PubMed.
- N. J. Pace and E. Weerapana, A competitive chemical-proteomic platform to identify zinc-binding cysteines., ACS Chem. Biol., 2013, 9, 258–265 CrossRef PubMed.
- F. Revillion, V. Pawlowski, L. Hornez and J. P. Peyrat, Glyceraldehyde-3-phosphate dehydrogenase gene expression in human breast cancer., Eur. J. Cancer, 2000, 36(8), 1038–1042 CrossRef CAS PubMed.
- S. J. Santner, P. J. Dawson, L. Tait, H. D. Soule, J. Eliason, A. N. Mohamed, S. R. Wolman, G. H. Heppner and F. R. Miller, Malignant MCF10CA1 cell lines derived from premalignant human breast epithelial MCF10AT cells., Breast Cancer Res. Treat., 2001, 65(2), 101–110 CrossRef CAS PubMed.
- H. Sies and D. P. Jones, Reactive oxygen species (ROS) as pleitropic physiological signaling agents., Nat. Rev. Mol. Cell Biol., 2020, 21, 363–383 CrossRef CAS PubMed.
- H. Nakajima, M. Itakura, T. Kubo, A. Kaneshige, N. Harada, T. Izawa, Y.-T. Azuma, M. Kuwamura, R. Yamaji and T. Takeuchi, Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) aggregation causes mitochondrial dysfunction during oxidative stress-induced cell death., J. Biol. Chem., 2017, 292(11), 4727–4742 CrossRef CAS PubMed.
- T. Kubo, H. Nakajima, M. Nakatsuji, M. Itakura, A. Kaneshige, Y. T. Azuma, T. Inui and T. Takeuchi, Active site cysteine-null glyceraldehyde-3-phosphate dehydrogenase (GAPDH) rescues nitric oxide-induced cell death., Nitric oxide, 2016, 53, 13–21 CrossRef CAS PubMed.
- A. Galbiati, A. Zana and P. Conti, Covalent inhibitors of GAPDH: From unspecific warheads to selective compounds, Eur. J. Med. Chem., 2020, 207, 112740 CrossRef CAS PubMed.
- M. Abo, C. Li and E. Weerapana, Isotopically-labeled iodoacetamide-alkyne probes for quantitative cysteine-reactivity profiling, Mol. Pharmacol., 2018, 15, 743–749 CrossRef CAS PubMed.
- A. Endo, K. Hasumi, K. Sakai and T. Kanbe, Specific inhibition of glyceraldehyde-3-phosphate dehydrogenase by koningic acid (heptelidic acid)., J. Antibiot., 1985, 38, 920–925 CrossRef CAS PubMed.
- K. Sakai, K. Hasumi and A. Endo, Identification of koningic acid (heptelidic acid)-modified site in rabbit muscle glyceraldehyde-3-phosphate dehydrogenase., Biochim. Biophys. Acta, 1991, 1077, 192–196 CrossRef CAS.
- R. A. Copeland, Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, 2nd edn, Wiley, 2013, pp. 366–367 Search PubMed.
- N. Darabedian, T. C. Chen, H. Molina, M. R. Pratt and A. H. Schönthal, Bioorthogonal profiling of a cancer cell proteome identifies a large set of 3-bromopyruvate targets beyond glycolysis., ACS Chem. Biol., 2018, 13(11), 3054–3058 CrossRef CAS PubMed.
- M. M. Blewett, J. Xie, B. W. Zaro, K. M. Backus, A. Altman, J. R. Teijaro and B. F. Cravatt, Chemical proteomic map of dimethyl fumarate-sensitive cysteines in primary human T cells, Sci. Signaling, 2016, 9, 445 CrossRef PubMed.
- T. Li, X. Tan, R. Yang, Y. Miao, M. Zhang, Y. Xi, R. Guo, M. Zheng and B. Li, Discovery of novel glyceraldehyde-3-phosphate dehydrogenase inhibitor via docking-based virtual screening., Bioorg. Chem., 2020, 96 Search PubMed.
- M. D. Kornberg, The immunologic Warburg effect: Evidence and therapeutic opportunities in autoimmunity, Wiley Interdiscip. Rev.: Syst. Biol. Med., 2020, 12, 5 Search PubMed.
- M. D. Kornberg, P. Bhargava, P. M. Kim, V. Putluri, A. M. Snowman, N. Putluri, P. A. Calabresi and S. H. Snyder, Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity, Science, 2018, 360(6387), 449–453 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, Supplemental Figures, and Supplemental Tables. See DOI: https://doi.org/10.1039/d2cb00091a |
| ‡ Former address: Rheos Medicines, Inc, Cambridge, Massachusetts 02142, USA. |
| This journal is © The Royal Society of Chemistry 2022 |