
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom polymerase engineering to semi-synthetic life: artificial expansion of the central dogma
Leping
Sun†
,
Xingyun
Ma†
,
Binliang
Zhang
,
Yanjia
Qin
,
Jiezhao
Ma
,
Yuhui
Du
and
Tingjian
Chen
 *
*
MOE International Joint Research Laboratory on Synthetic Biology and Medicines, School of Biology and Biological Engineering, South China University of Technology, 510006, Guangzhou, China. E-mail: chentj@scut.edu.cn
First published on 9th August 2022
Abstract
Nucleic acids have been extensively modified in different moieties to expand the scope of genetic materials in the past few decades. While the development of unnatural base pairs (UBPs) has expanded the genetic information capacity of nucleic acids, the production of synthetic alternatives of DNA and RNA has increased the types of genetic information carriers and introduced novel properties and functionalities into nucleic acids. Moreover, the efforts of tailoring DNA polymerases (DNAPs) and RNA polymerases (RNAPs) to be efficient unnatural nucleic acid polymerases have enabled broad application of these unnatural nucleic acids, ranging from production of stable aptamers to evolution of novel catalysts. The introduction of unnatural nucleic acids into living organisms has also started expanding the central dogma in vivo. In this article, we first summarize the development of unnatural nucleic acids with modifications or alterations in different moieties. The strategies for engineering DNAPs and RNAPs are then extensively reviewed, followed by summarization of predominant polymerase mutants with good activities for synthesizing, reverse transcribing, or even amplifying unnatural nucleic acids. Some recent application examples of unnatural nucleic acids with their polymerases are then introduced. At the end, the approaches of introducing UBPs and synthetic genetic polymers into living organisms for the creation of semi-synthetic organisms are reviewed and discussed.
Introduction
Natural nucleic acids, DNA and RNA, have been employed for storing, retrieving, and transmitting genetic information by all living organisms. Both DNA and RNA are composed of four kinds of nucleotide monomers that are linked by 3′-5′ phosphodiester linkages, and each typical nucleotide unit is composed of nitrogenous base (nucleobase), pentose, and phosphodiester moieties. The Watson–Crick pairing between the nucleobases lays the foundation of storage, replication, retrieval, and transmission of genetic information via nucleic acids and also confers highly programmable structures, properties, and functions to nucleic acids, which lead to broad application of nucleic acids in biotechnology and biomedicine. However, the limited number of natural nucleobases significantly constrains sequence space, genetic information capacity, and chemical diversity of DNA and RNA. Deoxyribose/ribose-phosphate backbones also restrict properties of DNA and RNA and possess relatively poor biological and chemical stability, which severely limits the functions and practical applications of DNA and RNA. Extensive efforts on the design and synthesis of DNA and RNA analogs with unnatural moieties have been made to address these problems in recent years, which greatly expanded the scope and application of genetic materials, and led to the emergence of a thriving field that has been named xenobiology.1 To support the efficient synthesis, replication, and evolution of unnatural nucleic acids, which are crucial for making full use of them, unnatural nucleic acid polymerases are essential. Unfortunately, for many of the exotic unnatural nucleic acids, especially those with modified sugar-phosphate backbones, natural DNA polymerases (DNAPs) and RNA polymerases (RNAPs) are unable to synthesize them efficiently. Approaches of protein engineering have thus been applied for tailoring polymerases for the efficient synthesis of various unnatural nucleic acids, and a number of strategies specifically effective for screening or selecting polymerase mutants have also been developed to facilitate these efforts.2–5 Numerous polymerase mutants with varied activities of synthesizing, reverse transcribing, and amplifying different unnatural nucleic acids have been obtained and broadly applied in the production and evolution of functional unnatural nucleic acids, including aptamers, catalysts, and nanomaterials.6–8 Other than in vitro application of unnatural nucleic acids, efforts have also been made to introduce unnatural nucleic acid components into living organisms, including creation of semi-synthetic organisms (SSOs) that are able to replicate, transcribe and translate unnatural base pairs (UBPs), which greatly expanded the genetic alphabet in vivo.9–11Development of unnatural nucleic acids
Modifications or alterations have been introduced into different moieties of nucleic acids via chemical synthesis of the structural units, which leads to broad expansion of structures, properties, functions, and applications of nucleic acids.12,13 The introduction of chemical modifications into natural nucleobases adds new functionalities to DNA and RNA, while the introduction of UBP pairing orthogonally to natural base pairs increases the genetic information capacity of DNA and RNA.14,15 Modification or replacement of sugar-phosphate backbones with unnatural components usually leads to a significant change of the overall properties of nucleic acids, including an increase of chemical or biological stabilities and a change of electronegativity.16 Combination of modifications on different moieties led to the successful production of nucleic acid analogs with combined properties and functions added.17,18Unnatural nucleic acids with modified nucleobases
Chemical modifications of nucleobases are achieved via direct replacement of atoms in the nucleobases or attachment of functional groups onto the atoms in the nucleobases (Fig. 1). Atoms C5 in pyrimidines and N7 in purines are usually picked as the atoms to be modified, due to their positions in the major groove of the DNA duplex and the correspondingly less steric hindrance for the modifications.19 A broad range of modifications, including hydrophobic groups, side chains of amino acids, fluorophores, and reactive groups for later labelling or coupling, have been introduced into nucleobases to confer desired functionalities, properties, or reactivities to DNA or RNA molecules.4,14 The recognition of nucleoside triphosphates containing modified nucleobases by polymerases is crucial for the practical application of these modified nucleobases, and many of the nucleoside triphosphates containing C5-modified pyrimidines or N7-modified purines have proven to be efficient substrates for polymerases, and broadly used in the field of biotechnology.20 Recently, Herdewijin and co-workers demonstrated efficient polymerase replication of DNA when the natural deoxyribonucleoside triphosphates (dNTPs) were extensively replaced with up to four nucleoside triphosphates containing various non-canonical nucleosides, including 7-deaza-2′-deoxyadenosine, 7-deaza-2′-deoxyguanosine, 5-fluoro-2′-deoxycytidine, and 5-chloro-2′-deoxyuridine.21,22 They termed the DNA with all four nucleobases replaced by 5-substituted pyrimidines and 7-deazapurines DZA, and demonstrated its function for carrying genetic information in living cells and potential use in biotechnology and synthetic biology.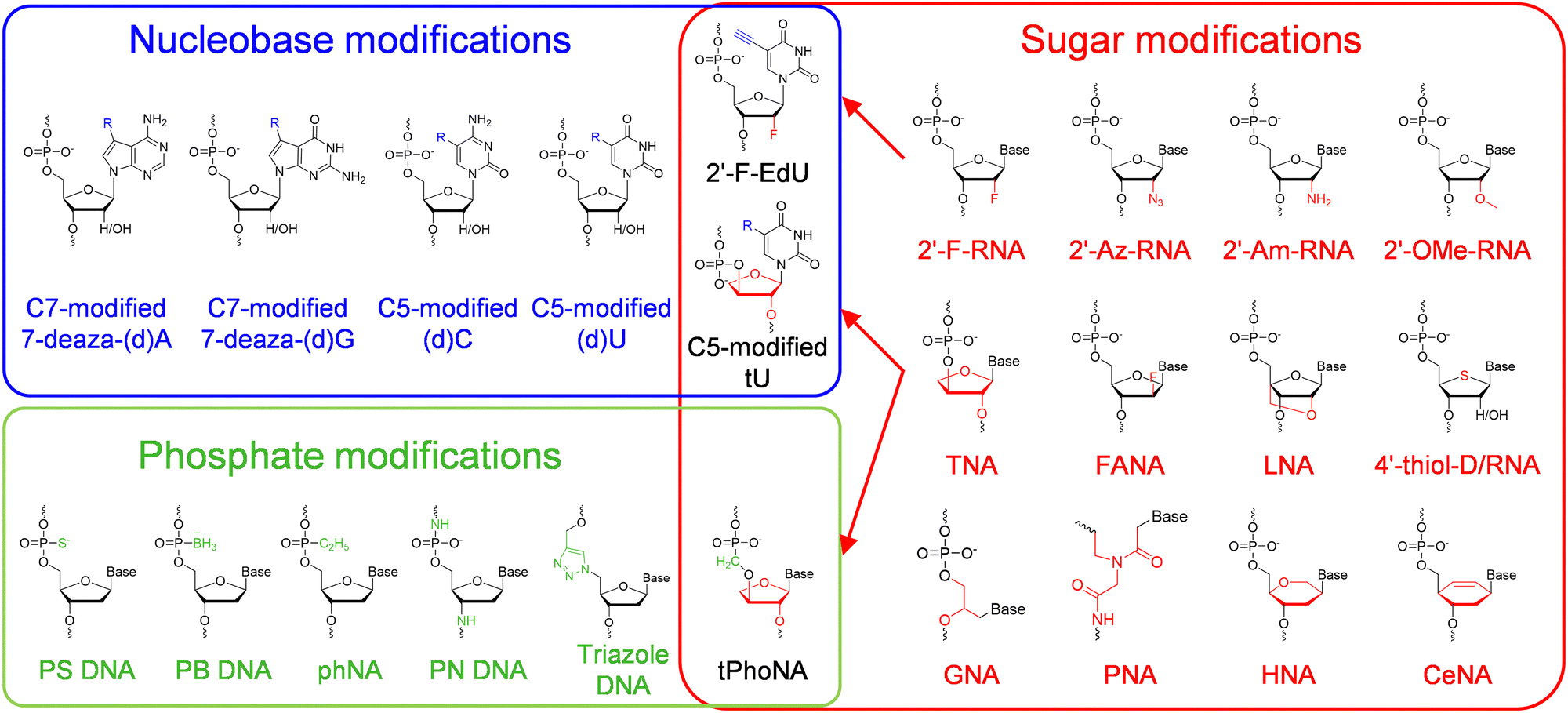 | ||
| Fig. 1 The chemical diversity of unnatural nucleic acids. Modifications or alterations are introduced into the nucleobase (blue block), sugar (red block) or phosphate (green block) moiety of a nucleic acid, and in some cases into multiple moieties. | ||
Unnatural base pairs (UBPs)
Other than modified nucleobases, various UBPs that can pair orthogonally to natural base pairs have been developed in the past few decades, which add not only novel properties and functions, but also a significant increase of the genetic information capacity to DNA and RNA.23,24 Key factors to be considered during the design of UBPs include shape complementarity and forces responsible for base pairing. The most predominant UBPs that can be efficiently replicated similar to natural base pairs have been developed by the Benner, Romesberg, and Hirao groups25,26 (Fig. 2). The pairing of UBPs developed by Benner's group, including isoG–isoC, V–J, K–X, Z–P, and S–B, is mainly based on hydrogen-bonding with rearranged patterns, while the pairing of UBPs developed by Romesberg's group, including MMO2–5SICS, NaM–5SICS, NaM–TPT3, PTMO–TPT3, CNMO–TPT3 and NaM–TAT1, is based on hydrophobic packing and stacking forces.11,27–35 Hirao and co-workers constructed a series of UBP pairing mainly based on hydrophobic interactions, such as Ds–Pa and Ds–Px,36–38 as well as several UBP pairing based on hydrogen bonding with designed steric exclusion, such as x–y, s–y and Q–Pa.39–43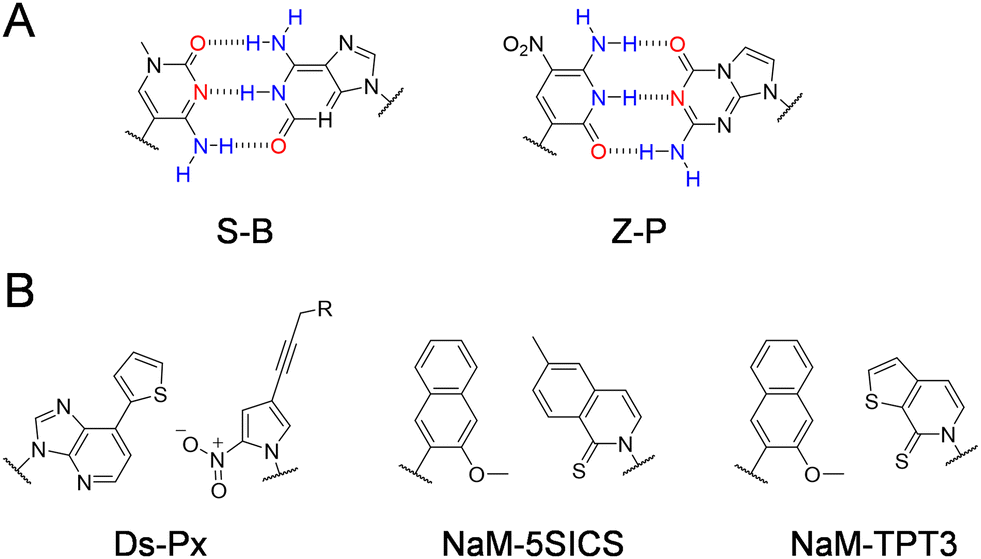 | ||
| Fig. 2 Chemical structures of the representative UBPs. (A) Hydrogen-bonded UBPs. (B) Non-hydrogen-bonded hydrophobic UBPs. | ||
Unnatural nucleic acids with an altered sugar backbone
Modifications or alterations of sugar backbones have also been extensively introduced into nucleic acids, which usually leads to a dramatic change of the overall properties of nucleic acids, including a change of melting temperature, an increase of chemical stability, resistance against nucleases, and a decrease or ablation of electronegativity44 (Fig. 1). Modification on the 2′-position of the ribose or the deoxyribose is one of the most explored modifications, and the hydrogen or hydroxyl group has been replaced with various atoms or groups, including 2′-fluoro (2′-F), 2′-azido (2′-Az), 2′-amino (2′-Am), and 2′-methoxy (2′-OMe). Although only one atom or group in the pentose is substituted, some of these modifications lead to significant changes in the properties of DNA or RNA. For example, 2′-F and 2′-OMe modifications greatly increase the melting temperatures, the duplex stabilities, and nuclease resistances of the nucleic acids.45 Replacement of the oxygen in the sugar ring with sulfur leads to the production of 4′-thiol-modified DNA and RNA, which have been prepared and investigated by Matsuda and co-workers.46,47 They demonstrated that 4′-thiol-modified RNA can be transcribed from a DNA template with 4′-thiol-CTP and 4′-thiol-UTP by T7 RNAP, and 4′-thiol-modified DNA can be efficiently amplified by KOD dash DNAP under appropriate conditions and transcribed by T7 RNAP in vitro and by mammalian RNA polymerases in mammalian cells.48,49 Other than simple substitution of atoms or groups in the pentose, replacement of the entire deoxyribose or ribose with other sugars has also been explored to construct unnatural nucleic acids with varied structures and properties (Fig. 1). For example, replacement of the pentose with arabinose or 2′-fluoro-modified arabinose leads to the production of arabino nucleic acid (ANA) and 2′-fluoro-arabino nucleic acid (FANA). Although the structures of ANA and FANA are similar, the thermal stability of FANA is very different from that of ANA, due to the smaller size and bigger electronegativity of 2′-F, which favors the formation of a pseudo hydrogen bond between 2′-F and purine H8.50 Locked nucleic acid (LNA) was constructed by “locking” the conformation of the ribofuranose with a 2′-O and 4′-C methylene bridge, which increases the stability of the duplex.51 In α-L-threofuranosyl nucleic acid (TNA), the sugar backbone is built with a tetrose instead of a pentose, which does not affect the capability of TNA to hybridize with DNA, RNA, or another strand of TNA, as well as to form G-quadruplexes.52,53 Hexitol nucleic acid (HNA) and cyclohexenyl nucleic acid (CeNA) harbor six-membered rings instead of five-membered rings in their backbones, and retain the ability to hybridize with complementary strands of DNA or RNA.54,55 The sugar rings of different sugar-modified nucleic acids possess varied pucker conformations, which are closely related with the overall helical structures and stabilities of the nucleic acid duplexes.56–58 Replacement of the entire sugar-phosphate backbone of DNA with a peptide backbone produces peptide nucleic acids (PNAs), which can hybridize with DNA or RNA with enhanced melting temperatures, and are highly resistant to nuclease degradation.59,60Unnatural nucleic acids with an altered phosphate moiety
The phosphate moiety links the structural units of DNA and RNA together to form genetic polymers, and is the major contributor to the hydrophilicity and charge of DNA and RNA molecules. Accordingly, chemical modification or substitution of the phosphate moiety in principle will lead to a dramatic change of the physicochemical properties of nucleic acids, and thus has also been extensively explored (Fig. 1). For example, the most straightforward modifications of the phosphate have been done by replacing the non-bridging oxygen with another atom or group, such as a sulfur or a borane, which led to the production of phosphorothioate (PS) DNA or boranophosphate (PB) DNA.61,62 These modifications introduce chirality into the phosphate moieties of the nucleoside triphosphates and the nucleic acid backbones, and the polymerase incorporation efficiencies of the nucleoside triphosphates are affected by the their configurations.63 For example, polymerases, including Escherichia coli (E. coli) DNAP, E. coli RNAP, and T7 RNAP, can polymerize the Sp diastereomers of nucleoside 5′-(1-thiotriphosphates), and inversion of configuration during the polymerization leads to the production of diesters with an Rp configuration.63 Recently, phosphorothioate modification of mRNA in proper patterns was reported to dramatically increase the efficiency of protein synthesis in an E. coli cell-free translation system.61 Holliger and co-workers reported the synthesis of backbone-uncharged phosphonate nucleic acid (phNA) by replacing the non-bridging oxygen with an alkyl group.64 In some other cases, other atoms in the phosphate backbone were replaced to construct nucleic acid analogs. For example, N3′-P5′ phosphoramidate (PN) DNA was synthesized via replacement of the bridging 3′ oxygen with an amino group.65 Specially, triazole DNA was constructed by linking the structural units via click chemistry.66Unnatural nucleic acids with combination of modifications on different moieties
Introduction of two or more modifications into different moieties of one nucleic acid at the same time is supposed to introduce multiple changes in the properties or functions into this nucleic acid, which further expands the chemical diversity of nucleic acids, and thus is attractive. For example, 2′-deoxy-2′-fluoro (5-ethynyl) uridine triphosphate was synthesized and employed to introduce both physiological stability and handles to attach functionalities, such as carbohydrates in this reported case, onto the nucleobases in the same RNA scaffold.67 In another example, Chaput and co-workers reported the synthesis and polymerase recognition of α-L-threofuranosyl uridine nucleoside triphosphate (tUTP) analogs with chemically diverse functional groups attached to the C-5 position, which are very useful for the production or evolution of highly functionalized TNA.68 As to the example for the combination of modifications on both sugar and phosphate moieties, Herdewijn and co-workers synthesized 3′-2′ phosphonomethyl-threosyl nucleic acid (tPhoNA), in which a methylene group was inserted between the phosphorous and the 3′ oxygen of TNA, and demonstrated its potential to be used as a genetic material.69Polymerase engineering for the synthesis, reverse transcription, and amplification of unnatural nucleic acids
Natural nucleic acids are efficiently replicated, transcribed, or reverse transcribed by different natural polymerases, including DNAPs, RNAPs, reverse transcriptases, or RNA-dependent RNAPs, which enables efficient replication and transmission of genetic information among these nucleic acids, and also lays the foundation for broad application of DNA and RNA. Although many unnatural nucleic acids have been designed and synthesized, natural polymerases are unable to synthesize a great number of them efficiently, which has severely limited their potential contribution to the expansion of the central dogma, as well as their immediate use in practical applications. To address this problem, polymerases have to be engineered to be able to recognize and polymerize the building blocks of unnatural nucleic acids efficiently. Among the efforts for engineering polymerases for an expanded substrate spectrum, directed evolution with various strategies has proven to be very effective, and a lot of polymerase mutants with excellent unnatural activities were obtained via directed polymerase evolution.2–5 Directed evolution mimics the natural evolution process in the laboratory, and yet the mutation rate of the target biomolecule is greatly increased and the selection pressure is artificially set to direct the evolution for the desired properties and functions.70 A typical directed polymerase evolution process includes two major steps: diversification of the polymerase gene and screening or selection for the polymerase mutants with desired activities.3Methods for the creation of polymerase libraries
Many strategies for artificial gene diversification have been developed since the early efforts of protein evolution (Table 1), and some of them have been successfully applied, either independently or in combination, in the creation of DNAP or RNAP libraries to be selected for enhanced activities against unnatural substrates.| Method | Advantages | Disadvantages | Application examples in polymerase evolution | Ref. |
|---|---|---|---|---|
| —: no application example in polymerase evolution yet. | ||||
| Error-prone PCR | Simple and easy to implement | Base bias of mutagenesis | Klentaq M1 | 71 and 76–81 |
| Universality | Lack of continuous mutations | Klentaq M2 | ||
| No requirement for structural information of the target protein | Small actual sequence sampling space | SFM4-3 | ||
| SFM4-6 | ||||
| SFM4-9 | ||||
| Taq T8 | ||||
| Taq H15 | ||||
| Taq M1 | ||||
| Taq M4 | ||||
| Tth SΔTthCs12RsEx pol mutants | ||||
| Phi29 DNAP Mut | ||||
| DNA shuffling with fragmentation by DNase I | Simple and easy to implement | Requirement for high sequence homology of the parental proteins | — | 82 |
| No requirement for structural information of the target protein | Hard to control fragmentation | |||
| Low recombination frequency | ||||
| Family shuffling | Parental proteins can be from different species | Requirement for high sequence homology of the parental proteins | Taq/Tth/Tfl 5D4 | 83, 93 and 95 |
| Larger functional sequence space can be sampled | Low recombination frequency | Bst LF/Klentaq v5.9 v7.16 | ||
| No requirement for structural information of the target protein | ||||
| Nucleotide exchange and excision technology (NExT) | Good controllability of the DNA fragment sizes during fragmentation of the parental sequences | Restricted digestion sites during fragmentation of the parental sequences | — | 85 |
| Staggered extension process (StEP) | Simple and easy to implement | Requirement for high sequence homology of the parental proteins | SFM4-3 | 77–79 and 86 |
| SFM4-6 | ||||
| SFM4-9 | ||||
| Taq T8 | ||||
| Taq H15 | ||||
| Taq M1 | ||||
| Taq M4 | ||||
| Synthetic shuffling/assembly of designed oligonucleotides (ADO) | Highly combinatorial DNA library | Requirement for carefully designed oligonucleotides | Pfu DNAP E10 | 80, 88, 89 and 94 |
| Libraries without limits to the length and number of the parental sequences | ||||
| Increased recombination resolution | ||||
| Random chimeragenesis on transient templates (RACHITT) | Lower sequence homology of the parental proteins is required | Requirement for synthesized templates | KlenTaq Mut_ADL | 90 and 96 |
| Higher recombination frequency | KlenTaq Mut_RT | |||
| Incremental truncation for the creation of hybrid enzymes (ITCHY) | No requirement for sequence homology | Numbers of parental sequences and fragments for recombination are limited | — | 91 |
| Site-saturated mutagenesis (SM) | Simple and easy to implement | Requirement for the structural information of the target protein | SFM4-3 | 69, 77, 88, 96, 97, 103, 106, 107 and 163 |
| All possibilities of substituting amino acids at the mutation sites can be sampled | SFM4-6 | |||
| SFM4-9 | ||||
| KlenTaq Mut_ADL | ||||
| KlenTaq Mut_RT | ||||
| 9°N-YRI | ||||
| 9°N-NVA | ||||
| KF I709E E710G | ||||
| T7 RNAP RGVG, E593G V685A | ||||
| SFM19 | ||||
| Combinatorial active-site saturation test (CAST) | Potential synergistic conformational effects can be taken into account | Requirement for the structural information of the target protein | — | 99 |
| Iterative saturation mutation (ISM) | Small but focused high-quality mutant library for each round of evolution | Requirement for the structural information of the target protein | — | 100 |
| Sequence saturation mutagenesis (SeSaM) | Consecutive point mutations | Only one randomized site in each mutant | — | 101 |
| Controllable mutational bias | ||||
| Controllable fragment distribution of the DNA library | ||||
Error-prone PCR introduces random mutations into the genes of target proteins, and is one of the most frequently employed methods for creating protein libraries.71 In a typical error-prone PCR reaction, random mutagenesis is carried out simply by increasing the mutation rate of the gene during PCR amplification, which is achieved by using polymerases of low fidelity, unbalancing the concentrations of the dNTPs, using analogs of some of the dNTPs, increasing PCR cycles, enhancing the concentration of magnesium ions, and adding manganese ions.71–73 It is very important to control the mutation rate of the target gene in an error-prone PCR experiment, since the library size is restricted and only able to cover a small portion of all possible mutants due to limited transformation efficiency and screening or selection throughput, and an excessively high mutation rate usually leads to a rapid loss of protein activity during directed evolution.71,74 Error-prone PCR is especially useful for protein library construction when the structural information of the target protein is not available or sufficient to predict which exact residues are crucial for the desired activity and should be randomized or directly mutated to be certain amino acids. Also, beneficial mutations that are far away from the active site of a protein are frequently revealed from a completely randomized library.75 Error-prone PCR has already been successfully used for constructing libraries of many DNAPs, including Tth DNAP, Klentaq DNAP, Stoffel fragment (SF) of Taq DNAP, full-length Taq DNAP and phi29 DNAP.76–81
The DNA shuffling technique mimics natural hybridization or recombination processes for rapid molecular breeding of proteins by recombining the genes of homologous proteins in vitro.82 In traditional DNA shuffling experiments, the genes of two or more homologous proteins, or mutants of the same protein, are first segmented by DNase I, and then assembled to be recombined full-length genes by PCR to generate the libraries.82 Desired protein mutants with beneficial mutations accumulated and deleterious mutations reduced are then screened or selected from the DNA shuffling libraries. DNA shuffling of a set of homologous genes from different species is called family shuffling.83 The application of family shuffling on genes with relatively low homology may result in less efficient recombination, which could be improved by using restriction endonucleases, instead of DNase I, for DNA fragmentation.84 In another method called nucleotide exchange and excision technology (NExT), DNA fragmentation was achieved by dosing uridine triphosphate (dUTP) into the PCR reaction of the target DNA, excising uracil bases in the PCR product with uracil-DNA-glycosylase, and then cleaving the DNA at the positions where uracil bases were excised with piperidine.85 In this method, the size distribution of the DNA fragments could be easily controlled by the concentration of dosed dUTP. Rather than recombining the target genes by DNA fragmentation and reassembly, Arnold and co-workers developed another strategy called the staggered extension process (StEP) for DNA shuffling.86 In this strategy, the target genes were mixed and subjected to the PCR reaction with a shortened extension time in each PCR cycle, which led to frequent template switching for primer elongation before the elongation reached full-length of the genes every time. In another method for generating recombination libraries, random-priming recombination (RPR), random-priming synthesis is used to generate short gene fragments containing low levels of point mutations to be assembled.87 Synthetic shuffling, in which degenerate oligonucleotides encoding all the variations in the parental genes are used to assemble the mutants, has been demonstrated to be an effective library creation method for evolving highly chimeric enzymes.87,88 In another study, Reetz and co-workers created recombination libraries of proteins by the assembly of designed oligonucleotides (ADO), in which the oligonucleotides for assembly were designed based on sequence information to control the overlapping process and increase the recombination frequency.89 Random chimeragenesis on transient templates (RACHITT) method was developed for creating DNA shuffling libraries with unprecedentedly high recombination frequency.90 In this method, fragments of homologous genes were first annealed onto a transient DNA template, and regions not hybridizing with the template were digested by the nuclease activities of DNAPs. After gap filling, ligation of the nicks, and template destroying, the chimeric library was PCR amplified, cloned, and subjected to screening or selection. Methods for creating homology-independent recombination libraries have also been developed. For example, Benkovic and co-workers developed a method called incremental truncation for the creation of hybrid enzymes (ITCHY), in which the parental genes with low homology were incrementally truncated with exonuclease III first, and then the gene fragments were fused to generate the hybrid library.91 There have already been some successful examples of applying these artificial gene recombination strategies in the evolution of polymerase mutants, including the generation of Taq/Tth/Tfl DNAP variant 5D4, Pfu DNAP variant E10, Bst LF/Klentaq DNAP variants v5.9 and v7.16, Stoffel fragment variants SFM4-3, SFM4-6, and SFM4-9, Taq DNAP variants T5, H8, M1 and M4, Klentaq DNAP variants Mut_ADL and Mut_RT.77–79,92–96
For proteins with more information on structure and structure–activity relationship (SAR) available, semi-rational approaches may be applied for library design and creation to decrease the size of the library to be screened. Site-saturated mutagenesis is broadly used for creating protein libraries in which one or multiple specific amino acid residues that are closely related with desired properties, such as activity, thermal stability, and substrate specificity, of the parental protein are randomized based on structural analysis.97 Oligonucleotides containing randomized degenerate codons, which help further decrease the library size, are used to introduce random mutations into target amino acid residues via overlapping PCR reactions. Recently, Chaput and co-workers demonstrated that the identification of key residues to be mutated could be greatly facilitated by computational analysis of homologous polymerase mutants.98 When there is a synergistic effect of mutations at multiple residues of the parental protein, it is helpful to carry out site-saturation mutagenesis on these residues simultaneously to increase the probability of obtaining protein mutants with desired properties. In the combinatorial active-site saturation test (CAST), protein libraries are generated by simultaneous randomization of groups of two amino acid residues spatially close to each other around the active site, which allows the screening for combinations of side chains on these residues with an optimal synergistic conformational effect.99 To reduce the effort for screening protein libraries with multiple amino acid residues or focused regions to be randomized, the iterative saturation mutation (ISM) method has been developed.100 In this method, rationally chosen sites crucial for the desired properties, each of which consisted of one, two, or three residues, were subjected into iterative cycles of site-saturation mutagenesis and screening. In each cycle, only one site was randomized and screened, which greatly reduced the library size and labor force of screening. A sequence saturation mutation (SeSaM) method was developed to create protein libraries with mutants containing random mutations at every single nucleotide position of the target sequence.101 In this method, the target sequence was segmented to fragments with different lengths first, and the fragments were then 3′ tailed with universal nucleobase using terminal transferase, and elongated to full-length genes. During subsequent PCR amplification of the elongation product, the universal bases were replaced by random standard nucleotides. Some of these semi-rational strategies have been successfully used to obtain polymerases with improved unnatural activities, including variants of Tgo DNAP, KOD DNAP, Deep Vent DNAP, 9°N DNAP, Stoffel fragment of Taq DNAP, full-length Taq DNAP, Klentaq DNAP, Klenow fragment (KF) of E. coli DNAP and T7 RNAP.77,96,98,102–107
Methods for the screening or selection of polymerase mutants
To efficiently identify mutants with desired properties from a polymerase library, a well-designed screening or selection method is essential. The key point of constructing a screening or selection method for mutants of a protein is to establish a linkage between the genotype and the phenotype, i.e., the gene and the activity or other functions, of a protein mutant, which can be achieved by many strategies. For example, this linkage can be built by displaying a protein on a carrier of its gene, such as a cell, a phage particle, a magnetic bead, or an mRNA strand.3,108–113 Some other strategies focus on spatially separating the gene and the expressed protein of each mutant from those of other mutants in a confined space, such as a well of multi-well plates, or a cell-like emulsion compartment, before checking the activity or other properties of each mutant.77,102–104,114,115 Based on these original strategies broadly used in protein evolution and taking advantage of the unique nucleotide polymerization activity of polymerases, various methods have been developed for the screening or selection of polymerase libraries2–4 (Table 2).| Method | Advantages | Disadvantages | Application examples | Ref. |
|---|---|---|---|---|
| Multi-well plate screening | Simple | Time consuming | SFM4-3 | 77, 102, 104, 106 and 115 |
| Direct identification of single active mutants | Limited screening throughput | SFM4-6 | ||
| SFM4-9 | ||||
| Tgo RT-TKK | ||||
| Tgo RT-C8 | ||||
| Tgo Pol6G12 | ||||
| Tgo PolC7 | ||||
| Tgo PolD4K | ||||
| Tgo RT521K | ||||
| Tgo RT521 | ||||
| KF I709E E710G | ||||
| Taq AA40 | ||||
| CSR | High throughput | Target polymerase needs to replicate the full-length of its own gene | Taq T8 | 78, 80, 81, 95, 116 and 167 |
| Simple | High temperature is usually needed to break the emulsified cells | Taq H15 | ||
| Tth SΔTthCs12RsEx pol mutants | ||||
| Phi29 DNAP Mut | ||||
| Bst v5.9 | ||||
| Bst v7.16 | ||||
| KOD RTX | ||||
| KOD RTX-Ome v6 | ||||
| spCSR | High throughput | High temperature is usually needed to break the emulsified cells | Pfu DNAP E10 | 94 and 115 |
| Target polymerase only needs to replicate a part of its own gene | Taq AA40 | |||
| Reduced adaptive burden | ||||
| Tunable selection stringency | ||||
| Improved selection sensitivity and versatility | ||||
| CST | High throughput | High temperature is usually needed to break the emulsified cells | Tgo Pol6G12 | 104 |
| Allows the selection for activities towards difficult nucleoside triphosphate substrates and under challenging conditions | Plasmid DNA has to be used as the extension template for the tagging primer | Tgo PolC7 | ||
| Tgo PolD4K | ||||
| Tgo RT521K | ||||
| Tgo RT521 | ||||
| CPR | High throughput | High temperature is usually needed to break the emulsified cells | T7 RNAP CGG-R7-8 | 117 |
| Expanded scope of proteins to be evolved | Challenging to design genetic circuits | T7 RNAP CGG-R12-KIRV | ||
| Mitigated effect on host fitness | ||||
| CBL | High throughput | High temperature is usually needed to break the emulsified cells | Tgo RT-TKK | 102 |
| Suitable for evolving various reverse transcription activities | Experiment complexity | Tgo RT-C8 | ||
| Phage display | High throughput | The target polymerase needs to be actively displayed on phage | SFM4-3 | 77, 125 and 129 |
| Kinds of the nucleic acid template, primer and nucleoside triphosphates for selection can all be well controlled | SFM4-6 | |||
| Adjustable selection stringency | SFM4-9 | |||
| Rapid reproduction of phage | SFR1 | |||
| SFR2 | ||||
| SFR3 | ||||
| Phi29 DNAP | ||||
| PACE | High throughput | Experiment complexity | T7 RNAP A6-36.4 | 131 |
| Rapid reproduction of phage | Expensive facilities | |||
| Continuous evolution | Challenging to design genetic circuits | |||
| Minimal researcher intervention | ||||
| Rapid evolutionary cycle | ||||
| Cell surface display | High throughput | The target polymerase needs to be actively displayed on cell surface | KF I709E E710G | 106 |
| Expanded scope of polymerases to be displayed for selection |
Multi-well plate screening methods for screening polymerase variants were developed by immobilizing a primer/template complex on the bottom surface of the wells, and extending the primer with certain nucleoside triphosphate substrates using cell lysate of each polymerase mutant in each well.77,104 The success primer extension led to the incorporation of fluorescent, biotinylated, or digoxigenin (DIG)-labelled nucleotides or the annealing of the extension product with labelled oligonucleotides, which could then be detected by reading the fluorescence or by binding with a DIG antibody or streptavidin-coupled enzyme and assaying the activity of this enzyme. Although single clones of active polymerases can be directly identified with these methods, the throughput is limited, which makes these methods more useful for screening pre-enriched or small focused polymerase libraries. For example, variants of Stoffel fragment, Taq DNAP and Tgo DNAP, have been identified with these methods from focused libraries or libraries pre-enriched with other high-throughput selection methods,77,102,104,115 which will be introduced below.
Emulsion or microfluidic system-based compartmentalization technology has been extensively used to develop novel methods for polymerase evolution (Fig. 3). For example, Holliger and co-workers developed a compartmentalized self-replication (CSR) method, in which a water-in-oil emulsion system was employed to confine PCR amplification of the gene of each polymerase mutant by the expressed protein of itself in an emulsion compartment, which led to rapid enrichment of polymerase mutants with good activities78 (Fig. 3A). Using this system, they successfully evolved mutants of Taq DNAP with enhanced thermostability or resistance to inhibitor heparin. Later, they developed a modified version of CSR, short-patch compartmentalized self-replication (spCSR).115 In this method, only a short region of the polymerase gene was diversified and amplified during the evolution, which reduced the requirements for catalytic activity and processivity of polymerases in the early stage of evolution, and thus made this method suitable for the evolution of challenging activities. A variant of Taq DNAP, AA40, which possessed replication, transcription and reverse transcription activities, as well as an expanded substrate spectrum for 2′-modified nucleoside triphosphates, was successfully evolved with this method. Ellington and co-workers developed a modified version of CSR, high-temperature isothermal compartmentalized self-replication (HTI-CSR), in which the self-replication of the polymerase gene was realized via rolling circle amplification (RCA) instead of PCR.95 This method was successfully used to evolve a thermostable strand-displacing polymerase mutant from a shuffled library of Bst LF and Klentaq DNAP. They also developed another modified CSR method, reverse transcription-compartmentalized self-replication (RT-CSR), to evolve reverse transcription activity of a DNAP.116 In the design of this method, to realize self-replication of the polymerase gene, the polymerase mutant had to reverse transcribe several RNA nucleotides in a flank primer, which partially annealed to the polymerase gene, to produce a full-length template that could be PCR amplified with outer primers. A high-fidelity thermostable reverse transcriptase, which they called reverse transcription xenopolymerase (RTX), was then evolved from KOD DNAP with this method. To expand the CSR method for the evolution of more proteins, a compartmentalized partnered replication (CPR) method was developed by the same group117 (Fig. 3B). In the CPR method, the activity of a partner protein that needed to be evolved was linked to the expression of Taq DNAP, which in turn PCR amplified the gene of the partner protein. This method was successfully applied on the evolution of several proteins, including T7 RNAP mutants for the recognition of orthogonal promoters.118
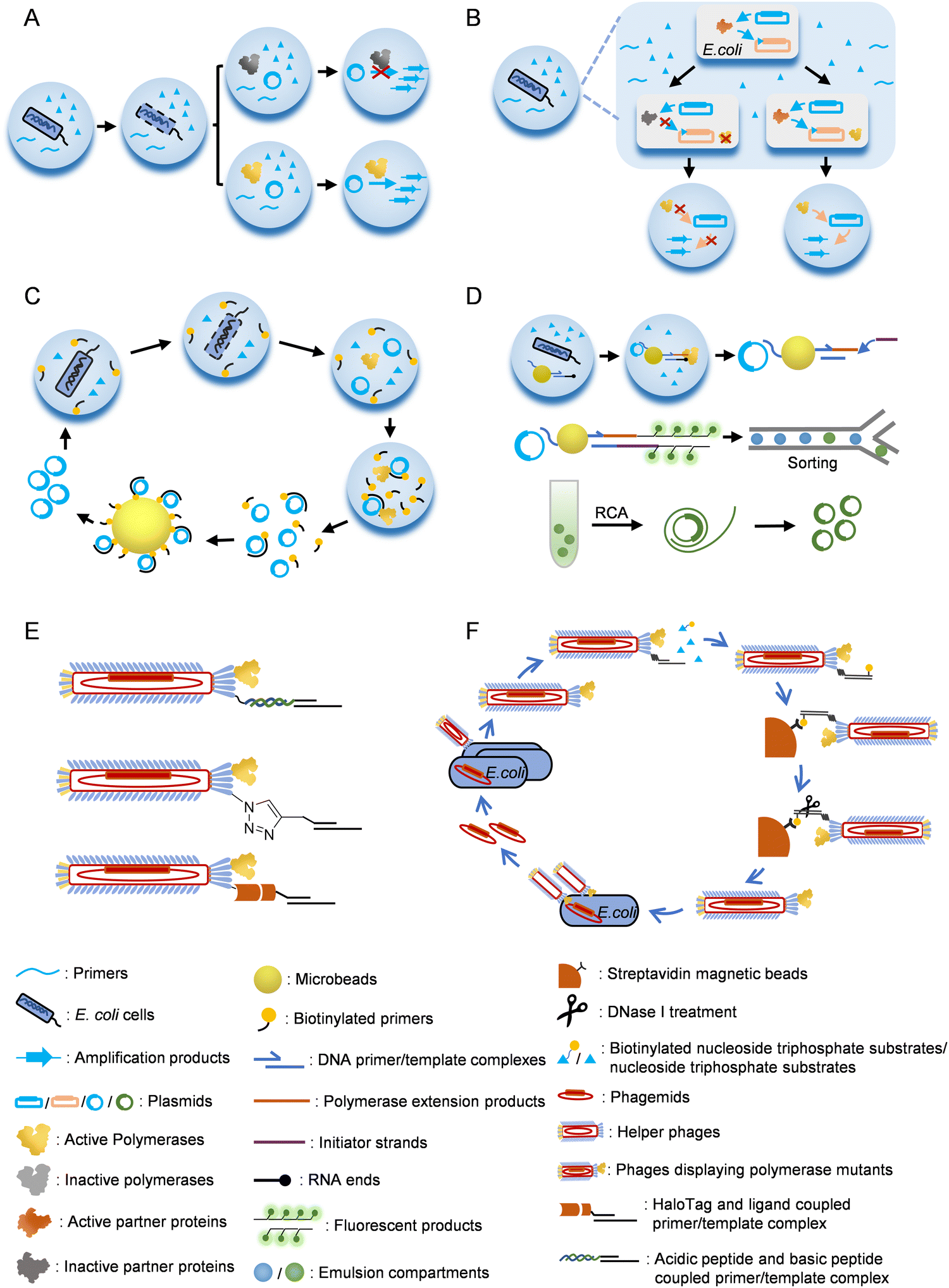 | ||
| Fig. 3 Strategies for the selection/screening of polymerase mutants. (A) Compartmentalized self-replication (CSR). (B) Compartmentalized partnered replication (CPR). (C) Compartmentalized self-tagging (CST). (D) Compartmentalized bead labelling (CBL). (E) Methods for co-displaying polymerases and nucleic acid substrates on phage particles. (F) Procedure for the selection of polymerase mutants with a phage display system. | ||
In CSR or the derivatives of CSR introduced above, full-length or a short region of the polymerase or the partner protein gene needs to be replicated by the polymerase to fulfill the evolution process. However, in some cases, especially when the desired activities are too exotic or challenging to evolve, replication of a gene or part of it during the evolution is unrealistic or hard to be correlated with the desired activities. To address this problem, several other compartmentalization-based strategies for polymerase evolution have been developed. For example, compartmentalized self-tagging (CST) was developed to evolve polymerases for the synthesis of xenobiotic nucleic acids (XNAs)104,119 (Fig. 3C). In this method, the selection of active polymerase mutants did not rely on self-replication in the compartment, but relied on the extension of a short biotinylated primer with unnatural nucleoside triphosphates using the plasmid harboring the polymerase gene as a template. Success extension of the primer resulted in tight binding of the primer and the plasmid, and thus enabled streptavidin bead separation of the active mutants. With this method, TgoT DNAP mutants have been evolved for efficient synthesis and reverse transcription of various XNAs. Recently, Holliger and co-workers developed a compartmentalized bead labelling (CBL) method for the evolution of RNA and XNA reverse transcriptases from a DNAP mutant102 (Fig. 3D). This method employed streptavidin-coated beads to co-display two kinds of oligonucleotides, one of which was responsible for the capture of the plasmid harboring polymerase gene, and another served as the primer for the reverse transcription of an XNA/RNA template. When a polymerase mutant successfully reverse transcribed the XNA/RNA template in a compartment, the reverse transcription product would later trigger a hybridization chain reaction (HCR), resulting in intensive fluorescent labelling of the bead, which then allowed fluorescent-activated bead sorting of the beads carrying plasmids of the active polymerase mutants. Polymerase mutants efficient for the reverse transcription of 2′-OMe-RNA, HNA, D-altritol nucleic acid (AtNA), 2′-methoxyethyl-RNA (2′-MOE-RNA), and P-α-S-phosphorothioate 2′-MOE-RNA (PS 2′-MOE-RNA) were obtained using this method.
In recent years, the rapidly developing microfluidic technology has also been employed in the design of compartmentalization-based methods for polymerase evolution. In these methods, the generation of the compartments was more controllable, and the process of sorting for the active polymerase mutants could also be directly integrated into the system. For example, Chaput and co-workers developed microfluidic-based protein evolution methods, such as droplet-based optical polymerase sorting (DrOPS) and fluorescence-activated droplet sorting (FADS)-based methods, and used them for evolving polymerases with expanded function.103,114 In these methods, polymerase mutants were encapsulated in water-in-oil-in-water or water-in-oil droplets generated by microfluidics. Polymerase-catalyzed primer extension led to the removal of a fluorescent quencher DNA annealed to the fluorophore-labelled template by strand displacement. The generated fluorescence was then used as an optical signal for the sorting of active polymerase droplets.
Phage display technology was initially developed for the evolution of small peptides or proteins, including antibodies, with high affinity towards the targets, and later proved to be a powerful tool for developing methods of polymerase evolution3,111,120–124 (Fig. 3E and F). For example, Romesberg and co-workers developed a phage-display-based method for polymerase evolution, in which a polymerase mutant was displayed on one of the p3 proteins of an M13 phage particle, while the primer/template substrate was attached to other p3 proteins.77,125 The substrate attachment was accomplished either by the coupling of an acidic peptide displayed on a p3 protein with a basic peptide conjugated to the primer, or by click reaction of an unnatural amino acid p-azidophenylalanine (pAzF) displayed on a p3 protein and a cycloalkyne conjugated to the primer. When the primer was extended with unnatural nucleoside triphosphates by the polymerase mutant displayed on the same phage, biotinylated-UTPs were incorporated to the end of the extension product, which allowed subsequent streptavidin bead separation of the active polymerase mutants. Using this method, mutants of SF of Taq DNAP that efficiently synthesize and amplify various 2′-modified nucleic acids have been obtained.77,126–128 Other strategies have also been used to attach the primer/template onto the phage. For example, Delespaul and co-workers co-displayed phi29 DNAP and a modified haloalkane dehalogenase, HaloTag, on M13 phage, which allowed the attachment of a DNA substrate coupled with a haloalkane ligand.129
Other than phage-display-based methods, bacteriophages have also been used to develop other methods for directed protein evolution. For example, Liu and co-workers developed a phage-assisted continuous evolution (PACE) strategy, in which the activity of a protein to be evolved was coupled to the propagation of a bacteriophage, and used it to rapidly evolve a variety of proteins with different traits.130–135 Variants of T7 RNAP with altered promoter specificity were successfully evolved with this method by coupling M13 phage propagation with T7 RNAP-mediated transcription of the phage p3 protein.131
Cell surface display technology has been used for the evolution of numerous proteins for either enhanced affinities against certain targets or increased catalytic activities.112,136–138 Recently, the application of an E. coli cell display system for polymerase evolution was also demonstrated by Schwaneberg and co-workers.106 The Klenow fragment (KF) of E. coli DNAP was displayed on the outer membrane of E. coli cells by fusing with autotransporter proteins, and the polymerase mutant-displaying cells were directly used for screening. The activity of each polymerase mutant was checked by monitoring the fluorescence of a fluorescent dye binding with double-stranded primer-extension product in multi-well plates. With this method, a KF mutant with enhanced activity against 2′-O-methyl nucleoside triphosphates (2′-OMe-NTPs) was successfully evolved.
Polymerases for the synthesis, reverse transcription, and replication of nucleic acids with unnatural moieties
With the library creation methods and screening or selection strategies described above, various polymerases have been engineered to be efficient for synthesizing, reverse transcribing, and even replicating nucleic acids with unnatural moieties. The wild type or engineered polymerases that demonstrated activities with unnatural substrates are summarized in Table 3, and the distribution of predominant mutations in the structures of representative engineered unnatural nucleic acid polymerases is illustrated in Fig. 4.| Polymerase | Mutation sites | Activities | Ref. |
|---|---|---|---|
| DNAP I from E. coli | Incorporation of K–X | 147 | |
| KF of DNAP I from E. coli | Incorporation of MMO2–5SICS, NaM–5SICS, NaM–TPT3, s–z, Q–Pa, Dss–Pa and Ds–Pa | 27, 28, 31, 36, 43, 155 and 156 | |
| Taq | Incorporation of NaM–5SICS, NaM–TPT3, and Z–P | 29, 31 and 148 | |
| TiTaq | Incorporation of isoG–isoC | 146 | |
| Taq DNAP mutant | M444V, P527A, D551E, E832V | Incorporation of Z–P | 105 |
| Taq DNAP mutant | N580S, L628V, E832V | 105 | |
| Taq/Tth/Tfl 5D4 | V62I, Y78H, T88S, P114Q, P264S, E303V, G389V, E424G, E432G, E602G, A608V, I614M, M761T, M775T | Incorporation of 5NI and 5NIC | 93 |
| Taq M1 | G84A, D144G, K314R, E520G, F598L, A608V, E742G | PCR with 7-deaza-dGTP, FITC-12-dATP, Biotin-16-dUTP and αS-dNTPs | 79 |
| SF P2 | F598I, I614F, Q489H | Incorporation of PICS–PICS | 150 |
| OneTaq DNAP | Incorporation of NaM–5SICS, NaM–TPT3 | 31 and 237 | |
| Deep Vent DNAP | Incorporation of NaM–5SICS, MMO2–5SICS, Ds–Px, Dss–Pn and Dss–Px | 29, 157 and 158 | |
| Vent DNAP | Incorporation of Ds–Pa | 36 | |
| Phusion DNAP | Incorporation of NaM–5SICS | 29 | |
| Pfu E10 | V93Q, D141A, E143A, V337I, E399D, N400D, R407I, Y546H | Incorporation of Cy3- or Cy5-modified dCTP | 94 |
| KOD Dash DNAP | Incorporation of dNamTPs | 141 | |
| KOD DNAP mutant | D141A, E143A, A485L | Incorporation of dNamTPs | 142 |
| T7 RNAP | Incorporation of m1Ψ triphosphate | 30, 34, 36, 39, 40, 145, 154–156, 160 and 238 | |
| Transcription of MMO2–5SICS, NaM–5SICS, NaM–TPT3, PTMO–TPT3, CNMO–TPT3, x–y, s–y, s–z, s–Pa, Ds–Pa, Dss–Pa and Ds–Px | |||
| T7 RNAP F | Y639F | Transcription of Ds–Pa and Ds-modified Pa | 159 |
| T7 RNAP F-M5 | Y639F, S430P, N433T, S633P, F849I, F880Y | Transcription of Ds–Pa and Ds-modified Pa | 159 |
| T7 RNAP FA-M5 | Y639F, H784A, S430P, N433T, S633P, F849I, F880Y | Transcription of Ds–Pa and Ds-modified Pa | 159 |
| T7 RNAP VRS-M5 | G542V, H772R, H784S, S430P, N433T, S633P, F849I, F880Y | Transcription of Ds–Pa and Ds-modified Pa | 159 |
| Transcription of 2′-F-C/U modified RNA containing modified Pa | |||
| T7 RNAP FAL | Y639F, H784A, P266L | Transcription of Z–P and S–B | 149 |
| RNAP II from S. cerevisiae | Transcription of NaM–TPT3 | 151 | |
| AMV reverse transcriptase | Reverse transcription of NaM–TPT3 and Q–Pa | 43, 153 and 154 | |
| MMLV reverse transcriptase | Reverse transcription of NaM–TPT3 | 153 | |
| SuperScript II reverse transcriptase | Reverse transcription of NaM–TPT3 | 153 | |
| SuperScript III reverse transcriptase | Reverse transcription of NaM–TPT3 | 154 | |
| SuperScript IV reverse transcriptase | Reverse transcription of NaM–TPT3 | 153 and 154 | |
| Taq Volcano2G | Reverse transcription of NaM–TPT3 | 153 | |
| SFM4-3 | I614E, E615G, V518A, N583S, D655N, E681K, E742Q, M747R | Synthesis or amplification of 2′-OMe, 2′-F, 2′-Az, 2′-Cl, 2′-Am-modified DNA/RNA and ANA | 77 and 127 |
| SFM4-6 | I614E, E615G, D655N, L657M, E681K, E742N, M747R | Synthesis of 2′-F-DNA, 2′-OMe-RNA | 77 |
| SFM4-9 | I614E, E615G, N415Y, V518A, D655N, L657M, E681V, E742N, M747R | Reverse transcription of 2′-F-DNA, 2′-OMe-RNA | 77 |
| Bst DNAP | Reverse transcription of FANA and TNA | 177–179 | |
| Deep Vent DNAP | Synthesis of HNA, ANA and FANA | 177 | |
| Deep Vent-RI | D141A, E143A, A485R, E664I | Synthesis of TNA | 98 |
| Tgo DNAP | Synthesis of HNA, FANA and ANA | 177 and 181 | |
| Incorporation of C8-alkyne-FANA UTP into FANA | |||
| Tgo-RI | D141A, E143A, A485R, E664I | Synthesis of TNA | 98 |
| Tgo Pol6G12 | TgoT: V589A, E609K, I610M, K659Q, E664Q, Q665P, R668K, D669Q, K671H, K674R, T676R, A681S, L704P, E730G | Synthesis of HNA and FANA | 104 and 177 |
| Tgo-6G12-I521L | Pol6G12: I521L | Synthesis of HNA and FANA | 177 |
| Tgo RT521 | TgoT: E429G, I521L, K726R | Synthesis of TNA | 69 and 104 |
| Reverse transcription of HNA, ANA, FANA, TNA and tPhoNA | |||
| Tgo RT521K | RT521: A385V, F445L, E664K | Reverse transcription of LNA and CeNA | 104 |
| Tgo RT-TKK | RT521K: I114T, S383K, N735K | Reverse transcription of 2′-OMe-RNA, AtNA | 102 |
| Tgo RT-C8 | RT-TKK: F493V, Y496N, Y497L, Y499A, A500Q, K501H | Reverse transcription of 2′-OMe, 2′-MOE, PS 2′-MOE-RNA, HNA, and AtNA | 102 |
| Tgo PolC7 | TgoT: E654Q, E658Q, K659Q, V661A, E664Q, Q665P, D669A, K671Q, T676K, R709K | Synthesis of CeNA and LNA | 104 |
| Tgo PolD4K | TgoT: L403P, P657T, E658Q, K659H, Y663H, E664K, D669A, K671N, T676I | Synthesis of FANA, ANA, TNA, HNA, PMT and RNA | 104 and 177 |
| Tgo QGLK | V93Q, D141A, E143A, Y409G, A485L, E664K | Synthesis of RNA, FANA, ANA and HNA | 177 |
| Tgo EPFLH | V93Q, D141A, E143A, H147E, L403P, L408F, A485L, I521L, E664H | Synthesis of PMT, ANA, TNA, RNA, FANA and tPhoNA | 69 and 177 |
| KOD DNAP | Synthesis of FANA | 177 | |
| KOD Dash DNAP | PCR with 4′-Thiol-dTTP and 4′-Thiol-dCTP | 48 | |
| KOD DGLNK | N210D, Y409G, A485L, D614N, E664K | Synthesis of 2′-OMe-RNA and LNA | 176 |
| KOD DLK | N210D, A485L, E664K | Reverse transcription of LNA and 2′-OMe-RNA | 176 |
| Kod RI | D141A, E143A, A485R, E664I | Synthesis of TNA | 98 |
| Kod RS | D141A, E143A, A485R, N491S | Synthesis of TNA | 174 |
| Kod QS | D141A, E143A, L489Q, N491S | Synthesis of TNA | 174 |
| Kod RSGA | D141A, E143A, A485R, N491S, R606G, T723A | Synthesis of FANA, ANA, HNA, TNA, C5-modified TNA, RNA and PMT | 68, 175 and 177 |
| KOD RTX | F38L, R97M, K118I, M137L, R381H, Y384H, V389I, K466R, Y493L, T514I, I521L, F587L, E664K, G711V, N735K, W768R | Reverse transcription of RNA and 2′-OMe-RNA | 116 |
| KOD RTX-Ome v6 | RTX: A40V, E251K, S340P, G350V, V353L, H381R, H384Y, K468N, I488L, G498A, K664R | Reverse transcription of 2′-OMe-RNA | 167 |
| KOD RT521K | V93E, D141A, E143A, A485L, I521L, E664K | Reverse transcription of tPhoNA | 69 |
| 9°N DNAP | Synthesis of FANA, ANA, HNA and TNA | 177 | |
| 9°N-Therminator | D141A, E143A, A485L | Synthesis of TNA | 172 and 173 |
| 9°N-YRI | D141A, E143A, A485R, E664I | Synthesis of TNA | 103 |
| 9°N-NVA | D141A, E143A, Y409N, D432G, A485V, V636A, E664A | Synthesis of TNA | 103 |
| Phi29 DNAP mutant | D12A | Synthesis of HNA, FANA and 2′-F-DNA | 180 |
| Tgo PGV2 | RT521L: D455P, K487G, R606V, R613V | Synthesis of phNA | 64 |
| DNAP from E. coli | Polymerization of the Sp diastereomers of nucleoside 5′-(1-thiotriphosphates) | 63 | |
| RNAP from E. coli | Polymerization of the Sp diastereomers of nucleoside 5′-(1-thiotriphosphates) | 63 | |
| T7 RNAP | Transcription of RNA from 4′-thiol-modified DNA | 46, 63 and 194 | |
| Transcription of 4′-thiol-modified RNA from DNA | |||
| Polymerization of the Sp diastereomers of nucleoside 5′-(1-thiotriphosphates) | |||
| T7 RNAP mutant | Y639F | Transcription of 2′-F, 2′-Am and 2′-F-EdU-modified RNA | 48, 49, 67, 168 and 169 |
| T7 RNAP mutant | Y639F, H784A | Transcription of 2′-OMe and 2′-Az-modified RNA | 170 |
| T7 RNAP RGVG, E593G, V685A | Y639V, H784G, E593G, V685A | Transcription of 2′-OMe-modified RNA | 107 |
| T7 RNAP RGVG-M5 | RGVG: S430P, N433T, S633P, F849I, F880Y | Transcription of 2′-OMe-RNA | 171 |
| T7 RNAP RGVG-M6 | RGVG: P266L, S430P, N433T, S633P, F849I, F880Y | Transcription of 2′-OMe-RNA | 171 |
| RNAPs from mammalian cells | Transcription of RNA from 4′-thiol-modified DNA | 48 and 49 |
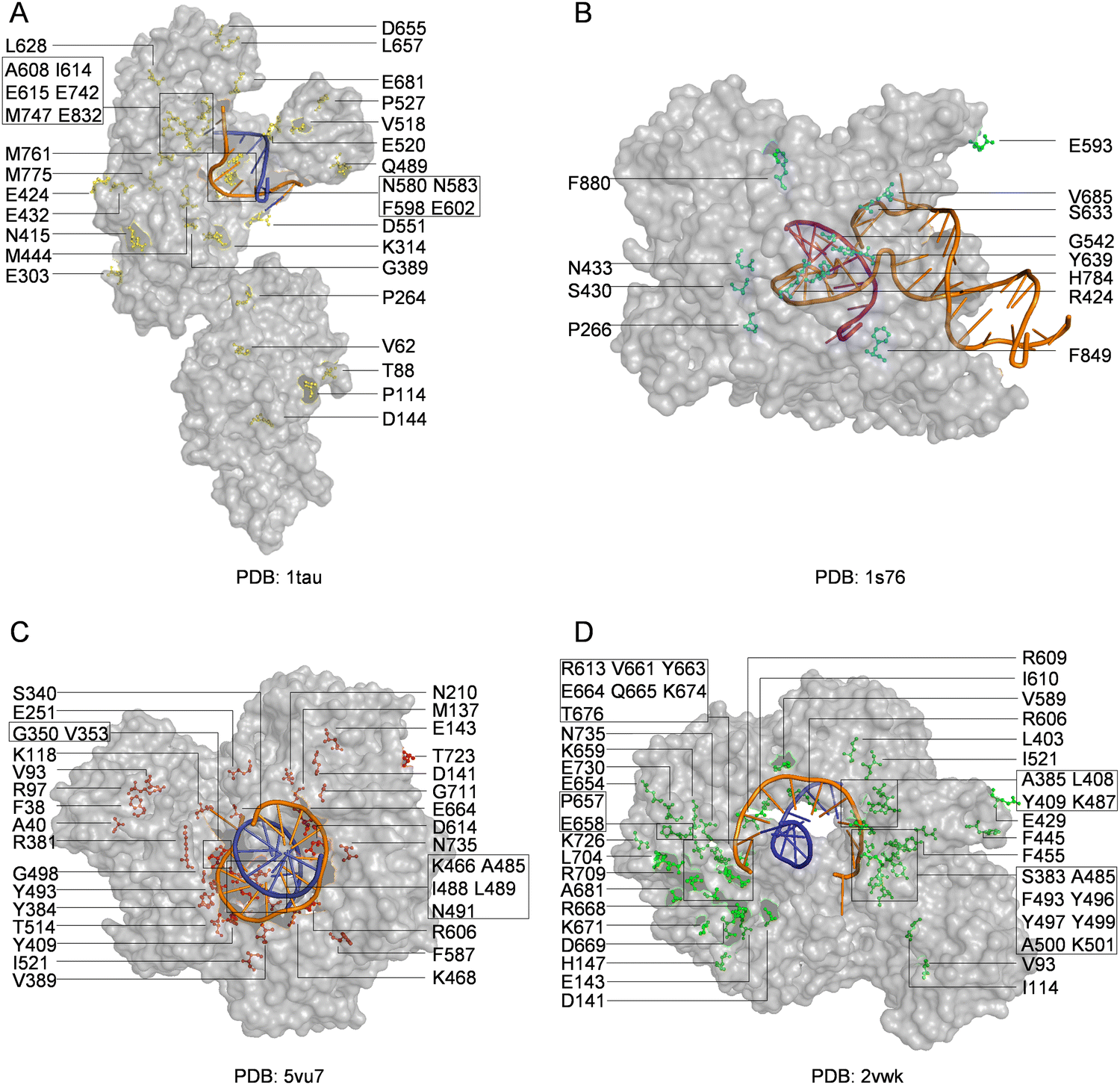 | ||
| Fig. 4 Distribution of mutations in engineered polymerases with synthesis, reverse transcription, or replication activities for unnatural nucleic acids. Key mutations in engineered (A) Taq DNA polymerase (yellow); (B) T7 RNA polymerase (cyan); (C) KOD DNA polymerase (red); and (D) Tgo DNA polymerase (green). The DNA templates and DNA primers are shown in orange and blue, respectively, while RNA product is shown in purple. | ||
Although usual modifications of nucleobases, especially those at the C5 position of pyrimidines and the C7 position of deazapurines, are well tolerated by natural polymerases and broadly used in the labelling and functionalization of DNA and RNA,139,140 engineering of polymerases can help further increase the enzymatic incorporation efficiency of the nucleotides with base modifications, and even achieve efficient PCR amplification of DNA extensively modified on nucleobases. For example, using the CSR method, Holliger and co-workers evolved a mutant of Taq DNAP, M1, that had an expanded substrate spectrum, and could perform efficient PCR amplification of DNAs with 7-deaza-dGTP, FITC-12-dATP, Biotin-16-dUTP or αS-dNTPs replacing the corresponding dNTP(s).79 In another study, they applied the spCSR method on the evolution of a family B DNAP, Pfu, and successfully obtained a mutant, E10, which could PCR amplify a DNA fragment up to 1 kb with dCTP completely substituted by Cy3- or Cy5-modified dCTP.94 Fujita and co-workers reported the enzymatic synthesis of DNA containing high-density amphiphilic functionalities attached to the nucleobases with 7-substituted 7-deazapurine nucleoside triphosphates, dGamTP and dAamTP, and 5-substituted pyrimidine nucleoside triphosphates, dUamTP and dCamTP, using KOD Dash DNAP (KOD XL DNAP).141 Efficient PCR amplification of a 500-bp DNA fragment with the mixture of these nucleobase-modified nucleoside triphosphates and natural dNTPs using the same polymerase was also demonstrated. Later, Hoshino and co-workers demonstrated that a mutant of KOD DNAP, KOD exo−/A485L, could synthesize longer DNA products containing nucleobases modified with these amphiphilic functionalities faithfully and more efficiently.142 T7 RNAP has also been proven to be efficient for incorporating nucleoside triphosphates with various modified nucleobases, including N1-methylpseudouridine (m1Ψ) triphosphate, which allows the in vitro transcription of mRNA vaccines with modified bases against various diseases, such as COVID-19.143–145
UBPs for the expansion of genetic alphabet have been developed and optimized for good recognition by natural polymerases, which is crucial for their in vitro and in vivo applications, and in some cases, the replication or transcription efficiency of the UBPs was increased by engineering the DNAPs or RNAPs employed. Replication and transcription of hydrogen-bonding-based UBPs developed by Benner's group have been demonstrated with various DNAPs and RNAPs or their mutants. For example, DNA containing the isoG–isoC pair was successfully PCR amplified with a truncated mutant of Taq DNAP, TiTaq.146In vitro replication of the K–X pair was also carried out with DNAP I from E. coli.147 Although Taq DNAP can replicate the Z–P pairs,148 for enhanced replication efficiency of the Z–P pair, directed evolution of Taq DNAP was carried out with CSR method, in which oligonucleotides containing multiple P nucleobases were used as the primers for self-replication.105 The evolved Taq DNAP mutants Taq (N580S, L628V, and E832V) and Taq (M444V, P527A, D551E, and E832V) demonstrated a much less pause when incorporating dZTP against P nucleobases in a template. T7 RNAP mutant FAL has been shown to be able to efficiently transcribe DNA containing both the Z–P and S–B pairs (Hachimoji DNA), resulting in the production of RNA containing P, Z, B and S nucleobases (Hachimoji RNA).148,149 The in vitro replication of some of the representative hydrophobic UBPs developed by Romesberg's group, such as MMO2–5SICS, NaM–5SICS and NaM–TPT3, has been shown to be efficient with various family A or B DNAPs, including Klenow fragment of E. coli DNAP I, Taq DNAP, Deep Vent DNAP, Phusion DNAP, and OneTaq DNAP (a mixture of Deep Vent DNAP and Taq DNAP).27–29,31 Directed evolution of polymerases has also proven effective to increase their replication performance for hydrophobic UBPs. For example, a mutant of SF of Taq DNAP, P2, which could synthesize DNA containing PICS self-pair more efficiently than wild type SF, was successfully obtained by directed evolution using the phage-display-based selection method.150 In another approach, with the CSR method, Holliger and co-workers evolved a Taq/Tth/Tfl DNAP mutant, 5D4, for the ability of forming and extending other self-pairs of hydrophobic nucleobase analogs, including 5NI and 5NIC.93 The transcription of some of these UBPs has been demonstrated with several well-studied RNAPs, including T7 RNAP and eukaryotic RNAP II.30,151 Recent in vivo experiments suggested that E. coli RNAP could also transcribe DNA containing some of these UBPs.152 Recently, reverse transcription of RNA containing TPT3 or NaM has been investigated with several reverse transcriptases or DNAP mutants, including avian myeloblastosis virus (AMV) reverse transcriptase, Moloney murine leukemia virus (MMLV) reverse transcriptase, SuperScript II reverse transcriptase, SuperScript III reverse transcriptase, SuperScript IV reverse transcriptase, and an engineered Taq DNAP with reverse transcription activity, Volcano2G (V2G).153,154 It was found that the UBP reverse transcription efficiencies of different reverse transcriptases were sharply different. For UBPs developed by Hirao's group, it has been shown that UBPs Q–Pa, s–z, Ds–Pa and Dss–Pa could be recognized by the Klenow fragment of E. coli DNAP I,36,43,155,156 and remarkably, Ds–Px, Dss–Pn and Dss–Px pairs could be PCR amplified by Deep Vent DNAP efficiently and faithfully.157,158 PCR amplification of DNA containing the Ds–Pa pair has also been carried out with Vent DNAP.36 It has been shown that the x–y, s–y, s–z, s–Pa, Dss–Pa, Ds–Pa, and Ds–Px pairs could be transcribed by T7 RNAP or its mutant VRS-M5,36,39,40,155,156,159–162 and the Q–Pa pair could be reverse transcribed by AMV reverse transcriptase.43
Efficient synthesis of sugar-modified nucleic acids with polymerases is usually more challenging, and thus much effort has been made on engineering polymerases to achieve this goal. Using the phage-display-based method for polymerase evolution, SF of Taq DNAP has been evolved to efficiently synthesize, reverse transcribe, and even amplify nucleic acids with various 2′-modifications, including 2′-OMe, 2′-F, 2′-Cl, 2′-Az, 2′-Am and 2′-arabino-modifications.77,127,128,163–166 Among the evolved SF mutants, SFM4-3 and SFM4-6 demonstrated good activity for the synthesis of 2′-modified nucleic acids, SFM4-9 was more efficient for the reverse transcription of 2′-modified nucleic acids, and SFM4-3 could PCR amplify partially 2′-modified nucleic acids. Recently, Ellington and co-workers employed the RT-CSR method to further evolve a previously evolved mutant of KOD DNAP, RTX, which could reverse transcribe RNA faithfully,116 and obtained mutant RTX-Ome v6 that could reverse transcribe 2′-OMe-RNA efficiently.167 Mutants of T7 RNAP have been extensively investigated for the activity of incorporating 2′-modified nucleotides.159 T7 RNAP mutant Y639F has been found to be able to use various 2′-substituted-NTPs, including dNTPs, 2′-F-dNTPs, and 2′-Am-dNTPs, as substrates during transcription,168,169 and the mutant with one more mutation, T7 RNAP (Y639F, H784A), displayed higher activity against NTPs with bulkier 2′-substitutions, including 2′-OMe and 2′-Az.170 Later, Ellington and co-workers carried out directed evolution of T7 RNAP for enhanced activity towards 2′-modified NTPs by randomizing residues R425, G542, Y639 and H784.107 Active mutants were selected using the autogene selection method, in which the activity of T7 RNAP was coupled with the transcription of an antibiotic resistance gene. The activity towards 2′-modified NTPs of each selected active mutants was then checked. Evolved mutants ‘RGFA’, (‘RGVG’, E593G, and V685A), ‘RGFH’ and ‘RGLH’ showed good activity when 2′-OMe UTP was used as a substrate, and mutants (‘RGVG’, E593G, and V685A) showed the best activity when more kinds of 2′-OMe-NTPs were used as substrates. Further engineering of mutants (‘RGVG’, E593G, and V685A) by introducing more reported mutations responsible for increased activities and thermostability of other T7 RNAP mutants led to the generation of mutants RGVG-M5 and RGVG-M6, which could synthesize 2′-OMe-modified RNA much more efficiently.171 T7 RNAP mutant VRS-M5 has also been demonstrated to be able to efficiently transcribe RNA containing modified unnatural base Pa from a DNA template containing UBP Ds–Px, and allowed the production of functional RNA molecules with both 2′-modification and an expanded genetic alphabet.159
Polymerases for the efficient synthesis of nucleic acids in which the entire pentose is replaced with unnatural sugars have also been developed. For example, a mutant of replicative family B DNAP from Thermococcus gorgonarius, TgoT, has been evolved for the efficient synthesis and reverse transcription of various XNAs with the CST method.104 Among the evolved TgoT mutants, Pol6G12 showed good activity for the synthesis of HNA. PolC7 showed good activities for the syntheses of CeNA and LNA. PolD4K showed good activities for the syntheses of ANA and FANA. RT521 showed good activities for the synthesis of TNA and reverse transcription of HNA, ANA, FANA and TNA. RT521K showed good activities for the reverse transcription of CeNA and LNA. Recently, mutant RT521K was further evolved with the CBL RT selection method and reverse transcription activity screening to be reverse transcriptases for 2′-OMe-RNA, HNA, AtNA, 2′-MOE-RNA and PS 2′-MOE RNA with varied efficiencies.102 Natural 9°N, Deep Vent, and Vent DNAP were shown to be able to synthesize a short stretch of TNA from a DNA template with tNTPs, and several mutants of 9°N DNAP, A485L (Therminator), Y409V, and Y409V, A485L double mutant, demonstrated enhanced activity to extend a primer with tNTPs.172 Among these mutants, Therminator has the highest activity for TNA synthesis, and has been used for the construction of a TNA selection system.173 Using the DrOPS strategy, Chaput and co-workers carried out directed evolution of a mutant of 9°N DNAP, 9n-GLK (Y409G, A485L and E664K), and obtained mutant 9n-YRI harboring mutations A485R and E664I and mutant 9n-NVA harboring mutations Y409N, D432G, A485V, V636A and E664A.103 Both of the mutants could efficiently synthesize TNA in the absence of manganese, and thus increase the fidelity of TNA synthesis. By sampling mutations A485R and E664I in other homologous polymerase scaffolds, efficient TNA polymerases, Kod-RI, Tgo-RI, DV-RI, which are mutants of KOD, Tgo and Deep Vent DNAPs harboring mutations A485R and E664I, have been identified.98 Combining the microfluidic screening method and deep mutational scanning, two other mutants of KOD DNAP with enhanced TNA synthesis activity, Kod-RS and Kod-QS, both of which harbored two epistatic mutations, have been identified.174 Mutant Kod-RS also demonstrated inversed substrate specificity towards tNTPs and dNTPs, compared with wild type KOD DNAP. Further screening of Kod-RS variants with mutations in tiles 6 and 8 of the thumb subdomain led to the discovery of mutant Kod-RSGA, which demonstrated enhanced activity, high fidelity, and low template sequence bias for TNA synthesis.175 KOD DNAP has also been engineered for efficient synthesis of other unnatural nucleic acids. Obika and co-workers developed KOD DNAP mutants KOD DGLNK and KOD DLK, which could efficiently synthesize LNA or 2′-OMe-RNA from DNA templates and reverse transcribe LNA or 2′-OMe-RNA to DNA, respectively.176 Recently, Chaput and co-workers systematically compared the activities of some natural and evolved polymerases for the synthesis and reverse transcription of different XNAs.177 Natural 9°N, Deep Vent, Tgo and KOD DNAPs showed the ability to synthesize full-length FANA and limited activity for the syntheses of other XNAs. Laboratory-evolved polymerases, including Tgo-QGLK, Tgo-6G12, TgoD4K, Tgo-6G12-I521L, Tgo-EPFLH, and Kod-RSGA demonstrated varied activities for the syntheses of RNA, FANA, ANA, HNA, TNA, and 3′-2′ phosphonomethyl-threosyl nucleic acid (PMT). Full-length products of different XNAs could be produced by different polymerase mutants. In another study, Tgo-EPFLH was demonstrated to be a tPhoNA synthase, while Tgo RT521 and KOD RT521K showed efficient ability to reverse transcribe tPhoNA into DNA.69 Bst DNAP displayed good activities for the reverse transcription of FANA and TNA, but much lower activity for the reverse transcription of ANA.177–179 Other than the extensively explored polymerases described above, some other natural or mutated polymerases have also been investigated for the activities towards unnatural substrates. For example, production of HNA, FANA, and 2′-F-DNA with phi29 DNAP mutant D12A has been reported.180
Polymerases can be evolved to be efficient for the synthesis of nucleic acids with bulky modifications on the phosphate moiety as well. For example, Holliger and co-workers further engineered a Tgo DNAP mutant, RT521L, which was previously evolved to be a reverse transcriptase for several XNAs, for efficient synthesis of phNA.64 After screening of a site-saturation mutagenesis library, evolution with the CST method, and reverse introduction of a single point mutation, they successfully obtained a mutant, PGV2, with enhanced activity for the synthesis of fully modified phNAs.
Engineered polymerase mutants have also found application in the synthesis of nucleic acids containing combined modifications on different moieties. For example, KOD DNAP mutant Kod-RSGA, which was evolved for efficient TNA synthesis, also demonstrated good activities against tUTP containing various C5-modifications, and was used to synthesize TNA containing functionalized nucleobases.68 In another example, T7 RNAP mutant Y639F was shown to be efficient for the transcription of RNA with 2′-deoxy-2′-fluoro (5-ethynyl) uridine triphosphate and other natural NTPs, and used for the evolution of 2′-F-modified RNA-scaffolded carbohydrate clusters.67,168 Very recently, Niu and co-workers synthesized C8-alkyne-FANA UTP, and demonstrated its enzymatic incorporation into FANA by Tgo DNAP.181 This work further enriched the XNA toolbox with components containing clickable handles.
Besides proteinaceous polymerases, Z RNA polymerase ribozyme, which is an RNA replicase generated via in vitro evolution, has also been investigated for its activity of incorporating unnatural nucleoside triphosphates.182 It was found that this ribozyme was able to incorporate different sugar or base-modified nucleoside triphosphates with varied efficiencies, as well as efficiently replicate UBP isoG–isoC under appropriate conditions.
In vitro application of unnatural nucleic acids and tailored polymerases
By combining use of the unnatural nucleic acids and their polymerases, either natural or engineered ones, novel aptamers, biocatalysts, and biomaterials can be produced. The acquirement of aptamers or biocatalysts composed of unnatural nucleic acids is usually achieved via evolution of unnatural nucleic acids from a pool of randomized sequences.183 The evolution can be carried out mainly with two procedures when proper polymerases are available.15 In one procedure, a randomized DNA pool is first transcribed into an unnatural nucleic acid pool, and after the selection, the unnatural nucleic acid pool is reverse transcribed back into a DNA pool, amplified, and then subjected into next round of evolution.184 In another procedure, the unnatural nucleic acid pool is directly amplified, and subjected into next round of evolution.128 Novel functionalities and properties can also be incorporated into nucleic-acid-based materials by using nucleic acid components containing unnatural moieties.Selection of improved aptamers
Systematic Evolution of Ligands by Exponential Enrichment (SELEX) technology has been broadly used to evolve aptamers with high affinity and specificity towards specific targets, ranging from small molecules to cells.185 Using unnatural nucleic acid triphosphates and polymerases that can recognize them, unnatural nucleotides can be introduced into the sequences in the pools for selection in SELEX, and serve to integrate novel functionalities and improved properties into the selected aptamers.Modifications on nucleobases can help expand the chemical diversity of and add novel functionalities to aptamers. For example, SELEX experiments have been carried out with DNA containing hydrophobic groups or amino-acid-like modifications attached to uracil or both uracil and cytosine nucleobases, resulting in the generation of protein-targeting high-affinity aptamers, which were called slow off-rate modified aptamers (SOMAmers).183,186 Due to its good acceptance of base-modified triphosphates, KOD (exo−) DNAP has been employed to generate SELEX libraries with 5-modified dC and dU.183 Application of DNA containing UBPs in SELEX significantly expands the sequence diversity of the pools to be selected, and incorporates the properties of the unnatural nucleobases into the selected aptamers, which has proven effective to increase the probability of obtaining aptamers with higher affinities. Hirao and co-workers carried out SELEX with DNA containing Ds, and successfully obtained high-affinity aptamers for vascular endothelial cell growth factor-165 (VEGF-165) and interferon-γ (IFN-γ), with the Kd values in the subnanomole to astonishing subpicomole range.6 Later, DNA aptamers containing Ds or both Ds and Pa with high affinity towards von Willebrand factor A1-domain (vWF) or dengue non-structural protein 1 (DEN-NS1) serotypes were reported.187,188 Hydrophobic UBP Z–P developed by Benner's group has also been extensively applied in the SELEX of aptamers with expanded genetic information for various targets, including different cell lines and proteins.189–192
Development of aptamers with unnatural sugar backbones has drawn even more attention, since modification of the sugar backbone can lead to a dramatic improvement of the overall properties of the aptamers, such as obtaining good chemical or biological stabilities, which are properties that natural DNA and RNA aptamers lack the most for practical applications. By employing evolved SF mutants to transcribe, reverse transcribe, or amplify 2′-modified DNAs, Romesberg and co-workers selected fully 2′-OMe-modified or partially 2′-F-modified aptamers against human neutrophil elastase (HNE), which displayed good biological stability and retained high affinity in a high concentration of salt.126,193 Recently, they reported the selection of HNE and factor IXa aptamers with large hydrophobic groups attached to the 2′-position of the sugar backbone by producing 2′-Az-DNA with SF mutant SFM4-3 and coupling alkyne modified molecules to the 2′-azido group via click chemistry.128 It was found that these 2′-hydrophobic groups significantly increased not only the binding affinity, but also the serum stability of the selected aptamers. With the assistance of T7 RNAP, Matsuda and co-workers successfully selected 4′-thiol-modified RNA aptamers against human α-thrombin, which have not only high binding affinity, but also superior stability toward RNase A.46,194,195 Holliger and co-workers demonstrated the application of Tgo DNAP mutants that they evolved in SELEX experiments for HNA aptamers against different targets, including hen egg lysozyme (HEL) and HIV trans-activating response RNA (TAR).104 Later, using one mutant of Tgo DNAP, D4K, to transcribe and reverse transcribe FANA, DeStefano and co-workers selected FANA aptamers against HIV-1 reverse transcriptase, HIV-1 integrase, and very recently receptor binding domain of SARS-CoV-2 S protein.196–198 TNA aptamers against various targets, including small molecules and proteins, were selected either with a DNA display strategy, in which the polymerase-synthesized TNA was attached to the template DNA annealed with its complementary strand during selection, or through cycles of the transcription–selection–reverse transcription–amplification process.199–201 Different polymerases, including Therminator DNAP and a mutant of KOD DNAP, Kod-RI, have been used for the synthesis/transcription of TNA, and Bst DNAP has been used for the reverse transcription of TNA in these studies. Recently, using TNA polymerase in combination with nucleobase-modified tNTPs, a stable TNA aptamer with functionalized nucleobases has also been selected.202,203 Mirror-image DNA has drawn broad interest in recent years, since it possesses good resistance to nucleases while retaining similar properties and functions of DNA.204 In a very recent study, Zhu and co-workers carried out SELEX experiment with a chemically synthesized mirror-image DNAP D-Dpo4-5m, and successfully obtained biostable L-DNA aptamers against human thrombin.205
Generation of new catalysts
The fact that similar to natural DNA and RNA, nucleic acids with unnatural moieties can fold into structures defined by their sequences makes it possible to build novel ribozyme-like catalysts with these unnatural nucleic acids, and the unnatural moieties can presumably provide these catalysts with much more possibilities for the catalytic activities, properties, and applications compared with natural ribozymes.Nucleobase modification can be used to attach functional groups, including amino acid-like side chains, to nucleic acid catalysts, and thus confer novel activities, such as protein enzyme-like activities, to these catalysts. For example, recently, Perrin and co-workers used dCTP and dUTP modified with arginine and lysine-like side chains for the selection of DNAzymes that could cleave RNA in a divalent metal cation-independent manner.206
Development of nucleic acid catalysts with unnatural sugar backbones not only expands the scope of macromolecular biocatalysts out of DNA, RNA and protein, but also has great potential to provide practically valuable catalysts with superior biostability. Using evolved TgoT DNAP mutants, Holliger and co-workers successfully selected ANA, FANA, HNA, and CeNA enzymes (XNAzymes) that could cleave or ligate RNA substrates, as well as a FANA enzyme with XNA–XNA ligase activity.7 Later, Chaput and co-workers evolved a general RNA-cleaving FANA enzyme with both strong catalytic activity and good nuclease-resistance, which could be further engineered to target different RNA sequences.178 They also reported the introduction of XNA modifications, including FANA and TNA nucleotides, into an existing DNAzyme scaffold for the construction of a novel enzyme, X10–23, with enhanced biological stability and good catalytic activity, and demonstrated the application of X10–23 in gene knockdown and pathogen detection.207–209 Recently, selection of TNA enzymes with RNA cleavage or ligation activity has been reported by Yu and co-workers.210,211
Construction of novel biomaterials
Biomaterials composed of nucleic acids have been extensively developed and used in numerous applications in recent years, due to their fascinating properties, including high programmability and good biocompatibility.45 Integration of unnatural nucleic acid components is helpful to further improve the properties, increase the functions and expand the applications of these materials.14Introduction of nucleobases modified with functional groups into nucleic acid materials immediately enables the coupling of various molecules onto these materials and thus expands the functionalities of these materials. For example, Brown and co-workers employed RCA with base-modified dUTP and dCTP to construct modified DNA nanoflowers, to which various cargos, including fluorophores and functional peptides, could be densely attached, and demonstrated their potential use in diagnostics and therapeutics.8 UBPs can be employed to increase the number of possible DNA or RNA sequences used for the assembly of nucleic acid nanostructures, and also to make these nanostructures uninvadable to natural DNAs or RNAs. For example, Tan and co-workers recently used DNA sequences containing unnatural bases Z and P to construct an aptamer-nanotrain assembly, and demonstrated its application in drug delivery.212
Modifications on the sugar-phosphate backbones of nucleic acid frameworks are valuable for augmentation of nucleic-acid-based materials with enhanced thermal, chemical and biological stabilities. Taylor and co-workers demonstrated the assembly of different nanostructures, including tetrahedron and octahedron, with various XNAs, including 2′-F-DNA, FANA, HNA or CeNA.213 Recently, Li and co-workers constructed FANA-based double crossover nanotiles with increased thermal and biological stability, and demonstrated their potential in cellular delivery of small molecules under physiological conditions.214 Other than improving the properties of nucleic acid materials, modification of the sugar-phosphate backbone can also be used for functionalizing the frameworks of, and even providing new strategies for the construction of, nucleic acid materials. For example, Chen and Romesberg used 2′-Az-DNA produced by SFM4-3 polymerase for the construction of a novel DNA hydrogel, in which the 2′-Az group was coupled with ssDNA primers for PCR crosslinking of the 2′-Az-DNA scaffolds.127
Construction of semi-synthetic organisms (SSOs) with unnatural nucleic acids
With the fast development of unnatural nucleic acids that can efficiently serve as carriers of genetic information, it is possible to apply them for increasing the diversity of or replacing some of the genetic materials in living cells, which leads to the production of SSOs.11 Expansion of the genetic alphabet and even the central dogma in living organisms with unnatural nucleic acid components is attractive, not only because it may help deepen our understanding of life, but also because the development of novel functional biomacromolecules, especially biopharmaceuticals, will greatly benefit from it.4,215 After years of efforts, UBPs developed by different groups have been used to efficiently expand the genetic alphabet to different extents in vitro, and the UBPs developed by Romesberg's group were used to build the first SSO with an expanded genetic alphabet, which was then optimized, and successfully used to express proteins with several unnatural amino acids incorporated at the same time.9–11,216 Meanwhile, several successful preliminary attempts of introducing XNAs into living cells have also been demonstrated. All these efforts have initiated the construction and application of an expanded central dogma.In vivo expansion of the genetic alphabet
Development of UBPs and their introduction into DNA and RNA have significantly expanded the genetic alphabet in vitro. Replication, transcription, and reverse transcription of UBPs developed by different groups have been achieved with various polymerases as described above, and the most representative examples are summarized in Fig. 5. To thoroughly complete the retrieval of increased genetic information encoded by UBPs in DNA, the RNA transcribed from UBP-containing DNA has to be further translated into protein, with the unnatural nucleobase-containing codons properly decoded. In 2002, Hirao and co-workers demonstrated the use of UBP s–y to incorporate unnatural amino acids, exemplified by 3-chlorotyrosine, into proteins in vitro.40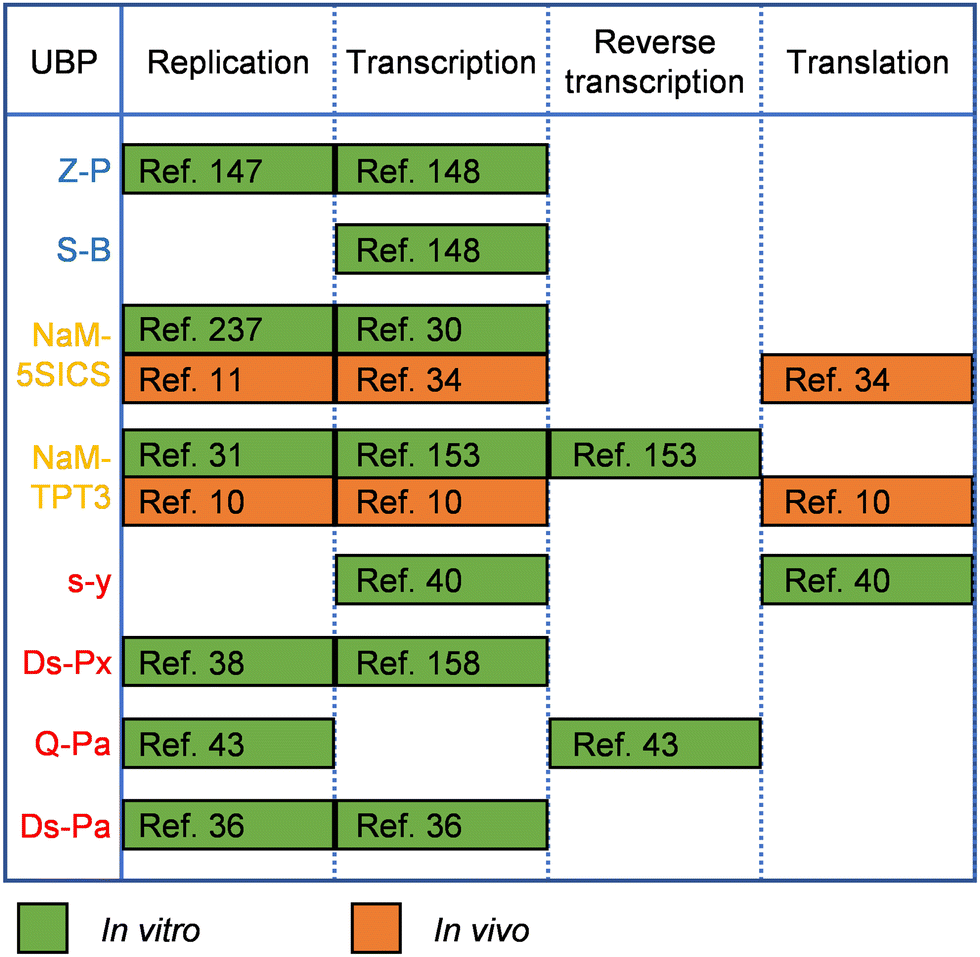 | ||
| Fig. 5 Replication, transcription, reverse transcription, and translation of representative UBPs in vitro (green rectangles) and in vivo (orange rectangles). Blue: UBPs developed by Benner and co-workers; yellow: UBPs developed by Romesberg and co-workers; red: UBPs developed by Hirao and co-workers. | ||
To expand the genetic alphabet with UBPs in vivo is much more challenging, and key issues that need to be addressed include the availability of unnatural nucleoside triphosphates in the cells, recognition of UBPs by endogenous replication, transcription, and translation machineries, and stability of UBPs in the cells during cell growth and propagation. In 2014, Romesberg and co-workers reported the first SSO with an expanded genetic alphabet, in which an initial information plasmid was constructed with UBP NaM–TPT3, and then replicated with dNaMTP and d5SICSTP.11 Nucleoside triphosphate transporter from Phaeodactylum tricornutum (PtNTT2)217 was employed to import dNaMTP and d5SICSTP into the cytoplasm of E. coli cells, allowing the in vivo replication of NaM–5SICS. Later, by using chemically optimized UBP NaM–TPT3 instead of NaM–5SICS for in vivo replication, engineering the PtNTT2 transporter, introducing the CRISPR/Cas system to eliminate DNA sequences that had lost the UBP, the SSO was optimized for robust growth, constitutive unnatural nucleoside triphosphate uptake, and much better UBP retention.9 In 2017, in vivo transcription and translation of UBP to incorporate non-canonical amino acids (ncAAs) into proteins was accomplished with the SSO.10
Since the successful development of SSOs for the storage and retrieval of increased genetic information, lots of efforts have been made to further explore and optimize the SSOs. For example, exploration of the contributions of different endogenous polymerases on UBP replication and the effects of cellular DNA repair mechanisms on UBP retention led to replisome reprogramming of the SSO for increased UBP retention, and subsequently allowed the incorporation of UBP into the chromosome of the SSO.218 Other than chassis cells for SSO construction, UBPs and unnatural triphosphates can also be continuously optimized for higher efficiencies of triphosphate uptake, in vivo replication, transcription, and translation. Early efforts of constructing SSOs used UBPs and unnatural triphosphates that have been screened and optimized based on in vitro SAR analysis, and thus might be less optimal for in vivo performance. The successful construction of SSOs enabled in vivo SAR analysis of UBPs, which led to the identification of more optimal UBPs and unnatural triphosphates for in vivo applications, exemplified by the combination of UBP CNMO–TPT3 and triphosphates NaMTP and TAT1TP, the use of which gave a high yield of a protein with high-fidelity incorporation of an ncAA.34,35
Expansion of the genetic alphabet with UBPs led to a great increase in the number of genetic codons, allowing the incorporation of much more kinds of amino acids into a protein at the same time. However, translation efficiencies of different unnatural base-containing codons can be dramatically different, and selective use of these codons for incorporating ncAAs into proteins is thus important for good protein yields, as well as high translation fidelity. Romesberg and co-workers have systematically analyzed unnatural codons, and identified nine most promising ones for efficient incorporation of ncAAs.216 Using three orthogonal ones of these codons, they successfully constructed an SSO with 67 codons, which includes 64 conventional codons and 3 new codons with unnatural bases. SSOs with additional sense codons containing unnatural bases have immediate application in producing novel protein products, including proteins site-specifically conjugated with other molecules for therapeutic use.23 For example, employing an SSO, human cytokine IL-2 variants, in which a modifiable unnatural amino acid was incorporated by decoding an unnatural base-containing codon, were produced, site-specifically modified with PEG polymers, and screened for altered receptor binding specificities and improved pharmacological properties.215
Expansion of the genetic alphabet in eukaryotes is also attractive, since it will not only allow the incorporation of various ncAAs into proteins that can only be well produced by eukaryotic cells, but also enable the development of molecular tools, including nucleic acid sequences containing unnatural nucleotide derivatives or proteins containing functional ncAAs, for regulating cellular functions or even behaviors of the entire organisms. As an initial effort for constructing eukaryotic SSOs with an expanded genetic alphabet, Romesberg and co-workers carried out translation experiment with unnatural codon–anticodon pairs containing NaM and TPT3 in HEK293 and CHO cells.219 The results suggested that eukaryotic ribosome could decode unnatural codons, and appeared more tolerant to different unnatural codons than prokaryotic ribosomes. Recently, Bornewasser et al. demonstrated the application of functionalized TPT3 for the labeling and visualization of mRNA in living cells.220
Development of methods for the sequencing of UBP-containing DNAs will significantly facilitate the ever-increasing efforts on expanding the genetic alphabet and constructing SSOs. Benner and Hirao groups have developed sequencing methods for their UBPs, respectively, in which the UBPs were first converted into different natural base pairs under different conditions and sequenced, and subsequent alignment and analysis of the resulting sequences revealed the positions of the UBPs.221,222 Hirao's group also developed a method for UBP sequencing, termed Sanger gap sequencing, in which the sequencing processivity was increased and modified Px analogs were used to generate clear gap patterns in the sequencing spectrum, which indicated the UBP positions.223 Recently, Romesberg and co-workers reported the application of nanopore sequencing for the thorough analysis of DNA containing UBP NaM–TPT3.25
Introduction of other unnatural nucleic acid components into living organisms
Other than constructing SSOs with an expanded genetic alphabet, a lot of efforts have also been made on introducing other unnatural nucleic acid components into living cells to expand the scope of genetic materials in vivo. Mutzel and co-workers demonstrated the replacement of a large proportion of thymine with artificial base 5-chlorouracil in the E. coli genome by evolving a thymidylate synthase-deficient E. coli strain with a gradually increased ratio of 5-chlorouracil to thymine in the medium.224 In most of other approaches, the nucleic acids containing unnatural moieties served as the initial replication templates that carried the genetic information, and were converted into natural DNAs after in vivo replication of the carrier plasmids. For example, Herdewijn and co-workers PCR amplified the gene of a dihydrofolate reductase (DHFR) with 5-chloro-2′-deoxyuridine, 7-deaza-2′-deoxyadenosine, 5′-fluoro-2′-deoxycytidine, and 7-deaza-2′-deoxyguanosine triphosphates, cloned and transformed the PCR product into E. coli cells, and found that the fully-modified DNA product could serve as a replication template of E. coli DNAP, and confer the phenotype encoded by the DHFR gene to the cells.21 Kool and co-workers demonstrated that DNA containing single to multiple size-expanded nucleobases (xDNA bases) could be read by E. coli DNAP, and in vivo converted into fully natural DNA that is functional with high fidelity.225 Other than templates containing nucleobase analogs, templates containing unnatural sugar-phosphate backbones have also been explored for encoding genetic information in vivo. For example, Pezo and co-workers found that short G/T sequences of CeNA and I/U sequences of ANA or HNA could guide the faithful biosynthesis of DNA sequences in E. coli cells.226 Liu and co-workers demonstrated that the thyA gene containing a few tPhoNA or dPhoNA oligonucleotides was still able to encode the prototrophic phenotype in thyA-deficient E. coli cells, although DNA propagation was severely diminished due to the addition of these nucleotides.69 Recently, in vivo transliteration of (S)-ZNA, which has an acyclic phosphonate backbone, to DNA was also demonstrated in E. coli cells.227 As an example of introducing unnatural nucleic acid components as genetic information carriers into eukaryotes, Matsuda and co-workers reported the transcription of 4′-thiol-modified DNA into natural RNA in mammalian cells.48,49Conclusion and perspective
With the fast development of nucleic acid chemistry and organic synthesis, various unnatural moieties have been introduced into nucleic acids to expand their sequences, structures, properties, functions, and applications. To make full use of these unnatural nucleic acids, traditional and newly developed approaches for polymerase engineering have been employed to empower DNAPs and RNAPs with the ability to efficiently synthesize, reverse transcribe, and even amplify these unnatural nucleic acids. By combining application of unnatural nucleic acids and their polymerases, a series of novel aptamers, biocatalysts, and materials have been produced. Meanwhile, the UBPs have been successfully introduced into living cells to expand the genetic alphabet in vivo, and successful attempts of introducing unnatural nucleic acids with modified nucleobases or backbones into living cells have also been demonstrated.69,224,227 With the introduction of these artificially synthesized moieties into the genome, E. coli has been made into the first set of SSOs. All these efforts summarized above are gradually and yet rapidly expanding the central dogma in different dimensions.To further expand the central dogma, more genetic polymers with novel modifications or combination of modifications can be designed and synthesized, and their efficient polymerases also need to be discovered or engineered, with the development and employment of novel polymerase evolution strategies, as well as the assistance of computational tools, including novel machine-learning methods.228,229 For existing unnatural nucleic acids, transcription of short stretches of them from a DNA template and reverse transcription of them back into DNA processes are already relatively efficient and sufficient for various in vitro applications, including SELEX for aptamers and XNAzymes, after years of efforts on engineering their polymerases. However, to achieve direct replication and even efficient amplification of the unnatural nucleic acids, the polymerases have to be engineered to be able to synthesize a strand of unnatural nucleic acid from an unnatural nucleic acid template. Although efficient amplification of partially sugar-modified short unnatural nucleic acids has been demonstrated with evolved DNAPs,77,127 further engineering of these polymerases is still needed to achieve efficient replication and amplification of fully sugar-modified long unnatural nucleic acids, which is the prerequisite of actually using these unnatural nucleic acids as full-function and augmented alternatives of DNA for the storage and transmission of genetic information, and will obviously lead to more efficient use of these unnatural nucleic acids, for example, SELEX of unnatural aptamers with less steps. Also, engineering polymerases for efficient transcription of different fully-sugar modified unnatural nucleic acids with big length will enable the full use of these unnatural nucleic acids as RNA alternatives with altered properties and expanded functions, not only for the production of larger biocatalysts or assembled nanomaterials, but also for the transmission of genetic information from the original carrier, such as DNA, to the function performer, say, protein or another genetic polymer. Moreover, in order to translate proteins from an unnatural nucleic acid, efforts have to be made to engineer the translational machinery to well adopt this unnatural nucleic acid, as well as to efficiently decode its genetic information with tRNAs or even other unnatural tRNA alternatives, the efficient charge of which with amino acids again may need extensive engineering of aminoacyl-tRNA synthetases (aaRSs).230,231 In an ideal world, all of the unnatural nucleic acids have efficient polymerases to replicate them, and to transmit genetic information from arbitrary one to another, which will lead to the expansion of the central dogma to higher dimensions (Fig. 6).
 | ||
| Fig. 6 Expansion of the central dogma to higher dimensions with XNAs. XNA-1-n: different XNAs. Solid arrow: processes that have been fully or partially achieved. Dotted arrow: processes that have not been achieved yet. | ||
For expanding the central dogma in vivo, synthesis or replication of unnatural nucleic acids in living cells needs efficient polymerases as well. Moreover, to be used in vivo, unnatural nucleic acid polymerases need to be further engineered for good substrate specificity immediately, since all of these polymerases were derived from natural DNAPs or RNAPs, and may still possess good activities against dNTPs or NTPs, which are abundant in living cells, and will obviously interfere the synthesis of unnatural nucleic acids from unnatural nucleoside triphosphates. To make the mutant polymerases function better in vivo, their optimal working temperatures and ionic strengths may also need to be engineered to adapt to the internal environment of the hosts. Efficient pathways for cellular polymerases to acquire various unnatural nucleoside triphosphates, either direct import from the medium or step-by-step synthesis via metabolic pathways, also need to be further exploited and optimized immediately. For example, kinases for the phosphorylation of nucleosides, nucleoside monophosphates, and nucleoside diphosphates can be engineered for higher activities against the unnatural substrates, and then employed to produce unnatural nucleoside triphosphates in vivo, as well as to regenerate unnatural triphosphates that have been dephosphorylated by endogenous phosphatases. Initial efforts on engineering the phosphorylation pathways to produce unnatural nucleoside triphosphates have already been made by several groups, including Benner's group and Romesberg's group.232–235 Long-term efforts for expanding the central dogma in vivo may include construction of replicable XNA plasmids or chromosomes, establishment of in vivo XNA transcription systems, and engineering of the host cells to balance energy consumption between the pathways for the production of unnatural genetic polymers and natural metabolic pathways, as well as to achieve even distribution of unnatural genetic polymers into divided cells. The orthogonality between unnatural genetic systems and natural genetic systems is also important for not interfering replication and function of endogenous genomes of the hosts,236 and potentially can be achieved by engineering and employing replication or transcription systems with orthogonal replication origins or promoters and corresponding polymerases with good substrate specificity to build the unnatural genetic systems. With all these efforts, organisms with not only an expanded genetic alphabet, but also an increased number of fully functional genetic polymers may be developed to further expand the central dogma in vivo, and find unprecedentedly broad application in the fields of biotechnology and biomedicine in the future (Fig. 7).
 | ||
| Fig. 7 Future expansion of the central dogma in vivo. XNAs can serve as carriers of genetic information, together with DNAs and RNAs, or as functional polymers, together with RNAs and proteins in the cells. UBPs are fully optimized for the expansion of the genetic alphabet, and working in different nucleic acids. Unnatural nucleoside triphosphates for the synthesis of unnatural nucleic acids can be acquired in vivo either via the triphosphate import by nucleoside triphosphate transporters, or through the metabolic pathways for triphosphate synthesis, exemplified by the cascade phosphorylation of unnatural nucleosides. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank National Key R&D Program of China (2019YFA0904102), National Natural Science Foundation of China (21978100), Guangdong Provincial Pearl River Talents Program (2019QN01Y228), and the Program for Guangdong Introducing Innovative and Entrepreneurial Teams (2019ZT08Y318) for the financial support of this work.References
- M. Schmidt, BioEssays, 2010, 32, 322–331 CrossRef CAS.
- A. Nikoomanzar, N. Chim, E. J. Yik and J. C. Chaput, Q. Rev. Biophys., 2020, 53, 1–31 CrossRef PubMed.
- T. Chen and F. E. Romesberg, FEBS Lett., 2014, 588, 219–229 CrossRef CAS PubMed.
- K. Duffy, S. Arangundy-Franklin and P. Holliger, BMC Biol., 2020, 18, 112 CrossRef PubMed.
- Z. Ouaray, S. A. Benner, M. M. Georgiadis and N. G. J. Richards, J. Biol. Chem., 2020, 295, 17046–17059 CrossRef CAS PubMed.
- M. Kimoto, R. Yamashige, K. Matsunaga, S. Yokoyama and I. Hirao, Nat. Biotechnol., 2013, 31, 453 CrossRef CAS PubMed.
- A. I. Taylor, V. B. Pinheiro, M. J. Smola, A. S. Morgunov, S. Peak-Chew, C. Cozens, K. M. Weeks, P. Herdewijn and P. Holliger, Nature, 2015, 518, 427–430 CrossRef CAS.
- Y. R. Baker, L. Yuan, J. Chen, R. Belle, R. Carlisle, A. H. El-Sagheer and T. Brown, Nucleic Acids Res., 2021, 49, 9042–9052 CrossRef CAS PubMed.
- Y. Zhang, B. M. Lamb, A. W. Feldman, A. X. Zhou, T. Lavergne, L. Li and F. E. Romesberg, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1317–1322 CrossRef CAS PubMed.
- Y. Zhang, J. L. Ptacin, E. C. Fischer, H. R. Aerni, C. E. Caffaro, K. S. Jose, A. W. Feldman, C. R. Turner and F. E. Romesberg, Nature, 2017, 551, 644 CrossRef CAS PubMed.
- D. A. Malyshev, K. Dhami, T. Lavergne, T. Chen, N. Dai, J. M. Foster, Jr., I. R. Correa and F. E. Romesberg, Nature, 2014, 509, 385 CrossRef CAS PubMed.
- E. Eremeeva and P. Herdewijn, Curr. Opin. Biotechnol., 2019, 57, 25–33 CrossRef CAS PubMed.
- K. H. Lee, K. Hamashima, M. Kimoto and I. Hirao, Curr. Opin. Biotechnol., 2018, 51, 8–15 CrossRef CAS PubMed.
- V. B. Pinheiro and P. Holliger, Trends Biotechnol., 2014, 32, 321–328 CrossRef CAS PubMed.
- T. Chen, N. Hongdilokkul, Z. Liu, D. Thirunavukarasu and F. E. Romesberg, Curr. Opin. Chem. Biol., 2016, 34, 80–87 CrossRef CAS.
- K. G. Devine and S. Jheeta, Life, 2020, 10, 346 CrossRef CAS.
- M. Kwak and A. Herrmann, Angew. Chem., Int. Ed., 2010, 49, 8574–8587 CrossRef CAS.
- W. B. Wan and P. P. Seth, J. Med. Chem., 2016, 59, 9645–9667 CrossRef CAS PubMed.
- A. Espinasse, H. K. Lembke, A. A. Cao and E. E. Carlson, RSC Chem. Biol., 2020, 1, 333–351 RSC.
- S. Ochoa and V. T. Milam, Molecules, 2020, 25, 4659 CrossRef CAS.
- E. Eremeeva, M. Abramov, L. Margamuljana, J. Rozenski, V. Pezo, P. Marliere and P. Herdewijn, Angew. Chem., Int. Ed., 2016, 55, 7515–7519 CrossRef CAS PubMed.
- E. Eremeeva, M. Abramov, L. Margamuljana and P. Herdewijn, Chem. – Eur. J., 2017, 23, 9560–9576 CrossRef CAS PubMed.
- M. Manandhar, E. Chun and F. E. Romesberg, J. Am. Chem. Soc., 2021, 143, 4859–4878 CrossRef CAS PubMed.
- F. E. Romesberg, J. Mol. Biol., 2021, 434, 167331 CrossRef PubMed.
- M. P. Ledbetter, J. M. Craig, R. J. Karadeema, M. T. Noakes, H. C. Kim, S. J. Abell, J. R. Huang, B. A. Anderson, R. Krishnamurthy, J. H. Gundlach and F. E. Romesberg, J. Am. Chem. Soc., 2020, 142, 2110–2114 CrossRef CAS.
- M. Kimoto and I. Hirao, Chem. Soc. Rev., 2020, 49, 7602–7626 RSC.
- A. M. Leconte, G. T. Hwang, S. Matsuda, P. Capek, Y. Hari and F. E. Romesberg, J. Am. Chem. Soc., 2008, 130, 2336–2343 CrossRef CAS PubMed.
- Y. J. Seo, G. T. Hwang, P. Ordoukhanian and F. E. Romesberg, J. Am. Chem. Soc., 2009, 131, 14596 CrossRef CAS.
- D. A. Malyshev, Y. J. Seo, P. Ordoukhanian and F. E. Romesberg, J. Am. Chem. Soc., 2009, 131, 14620 CrossRef CAS.
- Y. J. Seo, S. Matsuda and F. E. Romesberg, J. Am. Chem. Soc., 2009, 131, 5046 CrossRef CAS PubMed.
- L. Li, M. Degardin, T. Lavergne, D. A. Malyshev, K. Dhami, P. Ordoukhanian and F. E. Romesberg, J. Am. Chem. Soc., 2014, 136, 826–829 CrossRef CAS PubMed.
- V. T. Dien, M. Holcomb, A. W. Feldman, E. C. Fischer, T. J. Dwyer and F. E. Romesberg, J. Am. Chem. Soc., 2018, 140, 16115–16123 CrossRef CAS.
- K. Dhami, D. A. Malyshev, P. Ordoukhanian, T. Kubelka, M. Hocek and F. E. Romesberg, Nucleic Acids Res., 2014, 42, 10235–10244 CrossRef CAS PubMed.
- A. W. Feldman, V. T. Dien, R. J. Karadeema, E. C. Fischer, Y. You, B. A. Anderson, R. Krishnamurthy, J. S. Chen, L. Li and F. E. Romesberg, J. Am. Chem. Soc., 2019, 141, 10644–10653 CrossRef CAS.
- A. W. Feldman and F. E. Romesberg, J. Am. Chem. Soc., 2017, 139, 11427–11433 CrossRef CAS PubMed.
- I. Hirao, M. Kimoto, T. Mitsui, T. Fujiwara, R. Kawai, A. Sato, Y. Harada and S. Yokoyama, Nat. Methods, 2006, 3, 729–735 CrossRef CAS PubMed.
- I. Hirao, T. Mitsui, M. Kimoto and S. Yokoyama, J. Am. Chem. Soc., 2007, 129, 15549–15555 CrossRef CAS PubMed.
- R. Yamashige, M. Kimoto, Y. Takezawa, A. Sato, T. Mitsui, S. Yokoyama and I. Hirao, Nucleic Acids Res., 2012, 40, 2793–2806 CrossRef CAS PubMed.
- T. Ohtsuki, M. Kimoto, M. Ishikawa, T. Mitsui, I. Hirao and S. Yokoyama, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 4922–4925 CrossRef CAS.
- I. Hirao, T. Ohtsuki, T. Fujiwara, T. Mitsui, T. Yokogawa, T. Okuni, H. Nakayama, K. Takio, T. Yabuki, T. Kigawa, K. Kodama, T. Yokogawa, K. Nishikawa and S. Yokoyama, Nat. Biotechnol., 2002, 20, 177–182 CrossRef CAS PubMed.
- T. Fujiwara, M. Kimoto, H. Sugiyama, I. Hirao and S. Yokoyama, Bioorg. Med. Chem. Lett., 2001, 11, 2221–2223 CrossRef CAS PubMed.
- K. Moriyama, M. Kimoto, T. Mitsui, S. Yokoyama and I. Hirao, Nucleic Acids Res., 2005, 33, e129 CrossRef PubMed.
- T. Mitsui, A. Kitamura, M. Kimoto, T. To, A. Sato, I. Hirao and S. Yokoyama, J. Am. Chem. Soc., 2003, 125, 5298–5307 CrossRef CAS.
- L. K. McKenzie, R. El-Khoury, J. D. Thorpe, M. J. Damha and M. Hollenstein, Chem. Soc. Rev., 2021, 50, 5126–5164 RSC.
- G. Zhu, P. Song, J. Wu, M. Luo, Z. Chen and T. Chen, Front. Bioeng. Biotechnol., 2022, 9, 792489 CrossRef.
- Y. Kato, N. Minakawa, Y. Komatsu, H. Kamiya, N. Ogawa, H. Harashima and A. Matsuda, Nucleic Acids Res., 2005, 33, 2942–2951 CrossRef CAS PubMed.
- N. Inoue, N. Minakawa and A. Matsuda, Nucleic Acids Res., 2006, 34, 3476–3483 CrossRef CAS PubMed.
- N. Inoue, A. Shionoya, N. Minakawa, A. Kawakami, N. Ogawa and A. Matsuda, J. Am. Chem. Soc., 2007, 129, 15424 CrossRef CAS PubMed.
- H. Maruyama, K. Furukawa, H. Kamiya, N. Minakawa and A. Matsuda, Chem. Commun., 2015, 51, 7887–7890 RSC.
- J. K. Watts, N. Martin-Pintado, I. Gomez-Pinto, J. Schwartzentruber, G. Portella, M. Orozco, C. Gonzalez and M. J. Damha, Nucleic Acids Res., 2010, 38, 2498–2511 CrossRef CAS PubMed.
- R. N. Veedu and J. Wengel, Chem. Biodiversity, 2010, 7, 536–542 CrossRef CAS PubMed.
- K. U. Schoning, P. Scholz, X. L. Wu, S. Guntha, G. Delgado, R. Krishnamurthy and A. Eschenmoser, Helv. Chim. Acta, 2002, 85, 4111–4153 CrossRef CAS.
- V. Kempeneers, K. Vastmans, J. Rozenski and P. Herdewijn, Nucleic Acids Res., 2003, 31, 6221–6226 CrossRef CAS PubMed.
- I. A. Kozlov, P. K. Politis, A. Van Aerschot, R. Busson, P. Herdewijn and L. E. Orgel, J. Am. Chem. Soc., 1999, 121, 2653–2656 CrossRef CAS PubMed.
- B. Verbeure, E. Lescrinier, J. Wang and P. Herdewijn, Nucleic Acids Res., 2001, 29, 4941–4947 CrossRef CAS.
- J. M. Vargason, K. Henderson and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 7265–7270 CrossRef CAS PubMed.
- C. A. Mattelaer, H. P. Mattelaer, J. Rihon, M. Froeyen and E. Lescrinier, J. Chem. Theory Comput., 2021, 17, 3814–3823 CrossRef CAS PubMed.
- I. Anosova, E. A. Kowal, M. R. Dunn, J. C. Chaput, W. D. Van Horn and M. Egli, Nucleic Acids Res., 2016, 44, 1007–1021 CrossRef CAS.
- M. Egholm, O. Buchardt, L. Christensen, C. Behrens, S. M. Freier, D. A. Driver, R. H. Berg, S. K. Kim, B. Norden and P. E. Nielsen, Nature, 1993, 365, 566–568 CrossRef CAS PubMed.
- N. Kundu, B. E. Young and J. T. Sczepanski, Nucleic Acids Res., 2021, 49, 6114–6127 CrossRef CAS PubMed.
- D. Kawaguchi, A. Kodama, N. Abe, K. Takebuchi, F. Hashiya, F. Tomoike, K. Nakamoto, Y. Kimura, Y. Shimizu and H. Abe, Angew. Chem., Int. Ed., 2020, 59, 17403–17407 CrossRef CAS PubMed.
- A. H. S. Hall, J. Wan, A. Spesock, Z. Sergueeva, B. R. Shaw and K. A. Alexander, Nucleic Acids Res., 2006, 34, 2773–2781 CrossRef CAS PubMed.
- F. Eckstein, Nucleic Acid Ther., 2014, 24, 374–387 CrossRef CAS PubMed.
- S. Arangundy-Franklin, A. I. Taylor, B. T. Porebski, V. Genna, S. Peak-Chew, A. Vaisman, R. Woodgate, M. Orozco and P. Holliger, Nat. Chem., 2019, 11, 533–542 CrossRef CAS.
- S. Gryaznov and J. Chen, J. Am. Chem. Soc., 1994, 7, 3143–3144 CrossRef.
- M. Kukwikila, N. Gale, A. H. El-Sagheer, T. Brown and A. Tavassoli, Nat. Chem., 2017, 9, 1089–1098 CrossRef CAS PubMed.
- R. L. Redman and I. J. Krauss, J. Am. Chem. Soc., 2021, 143, 8565–8571 CrossRef CAS PubMed.
- Q. Li, V. A. Maola, N. Chim, J. Hussain, A. Lozoya-Colinas and J. C. Chaput, J. Am. Chem. Soc., 2021, 143, 17761–17768 CrossRef CAS PubMed.
- C. Liu, C. Cozens, F. Jaziri, J. Rozenski, A. Marechal, S. Dumbre, V. Pezo, P. Marliere, V. B. Pinheiro, E. Groaz and P. Herdewijn, J. Am. Chem. Soc., 2018, 140, 6690–6699 CrossRef CAS PubMed.
- F. H. Arnold, FASEB J., 1997, 11S, A872 Search PubMed.
- P. C. Cirino, K. M. Mayer and D. Umeno, Meth. Mol. Biol., 2003, 231, 3–9 CAS.
- R. C. Cadwell and G. F. Joyce, PCR Methods Appl., 1992, 2, 28–33 CrossRef CAS PubMed.
- M. Zaccolo, D. M. Williams, D. M. Brown and E. Gherardi, J. Mol. Biol., 1996, 255, 589–603 CrossRef CAS PubMed.
- P. A. Romero and F. H. Arnold, Nat. Rev. Mol. Cell Biol., 2009, 10, 866–876 CrossRef CAS PubMed.
- K. L. Morley and R. J. Kazlauskas, Trends Biotechnol., 2005, 23, 231–237 CrossRef CAS PubMed.
- K. B. M. Sauter and A. Marx, Angew. Chem., Int. Ed., 2006, 45, 7633–7635 CrossRef CAS PubMed.
- T. Chen, N. Hongdilokkul, Z. Liu, R. Adhikary, S. S. Tsuen and F. E. Romesberg, Nat. Chem., 2016, 8, 557–563 CrossRef PubMed.
- F. J. Ghadessy, J. L. Ong and P. Holliger, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 4552–4557 CrossRef CAS PubMed.
- F. J. Ghadessy, N. Ramsay, F. Boudsocq, D. Loakes, A. Brown, S. Iwai, A. Vaisman, R. Woodgate and P. Holliger, Nat. Biotechnol., 2004, 22, 755–759 CrossRef CAS PubMed.
- S. L. Aye, K. Fujiwara, A. Ueki and N. Doi, Biochem. Biophys. Res. Commun., 2018, 499, 170–176 CrossRef CAS.
- T. Povilaitis, G. Alzbutas, R. Sukackaite, J. Siurkus and R. Skirgaila, Protein Eng., Des. Sel., 2016, 29, 617–628 CrossRef CAS PubMed.
- W. P. Stemmer, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 10747–10751 CrossRef CAS PubMed.
- A. Crameri, S. A. Raillard, E. Bermudez and W. Stemmer, Nature, 1998, 391, 288–291 CrossRef CAS PubMed.
- M. Kikuchi, K. Ohnishi and S. Harayama, Gene, 1999, 236, 159–167 CrossRef CAS PubMed.
- K. M. Muller, S. C. Stebel, S. Knall, G. Zipf, H. S. Bernauer and K. M. Arndt, Nucleic Acids Res., 2005, 33, e117 CrossRef PubMed.
- H. M. Zhao, L. Giver, Z. X. Shao, J. A. Affholter and F. H. Arnold, Nat. Biotechnol., 1998, 16, 258–261 CrossRef CAS PubMed.
- Z. X. Shao, H. M. Zhao, L. Giver and F. H. Arnold, Nucleic Acids Res., 1998, 26, 681–683 CrossRef CAS PubMed.
- J. E. Ness, S. Kim, A. Gottman, R. Pak, A. Krebber, T. V. Borchert, S. Govindarajan, E. C. Mundorff and J. Minshull, Nat. Biotechnol., 2002, 20, 1251–1255 CrossRef CAS.
- D. X. Zha, A. Eipper and M. T. Reetz, ChemBioChem, 2003, 4, 34–39 CrossRef CAS PubMed.
- W. M. Coco, W. E. Levinson, M. J. Crist, H. J. Hektor, A. Darzins, P. T. Pienkos, C. H. Squires and D. J. Monticello, Nat. Biotechnol., 2001, 19, 354–359 CrossRef CAS PubMed.
- M. Ostermeier, J. H. Shim and S. J. Benkovic, Nat. Biotechnol., 1999, 17, 1205–1209 CrossRef CAS PubMed.
- M. D'Abbadie, M. Hofreiter, A. Vaisman, D. Loakes, D. Gasparutto, J. Cadet, R. Woodgate, S. Paeaebo and P. Holliger, Nat. Biotechnol., 2007, 25, 939–943 CrossRef.
- D. Loakes, J. Gallego, V. B. Pinheiro, E. T. Kool and P. Holliger, J. Am. Chem. Soc., 2009, 131, 14827–14837 CrossRef CAS PubMed.
- N. Ramsay, A. Jemth, A. Brown, N. Crampton, P. Dear and P. Holliger, J. Am. Chem. Soc., 2010, 132, 5096–5104 CrossRef CAS PubMed.
- J. N. Milligan, R. Shroff, D. J. Garry and A. D. Ellington, Biochemistry, 2018, 57, 4607–4619 CrossRef CAS.
- G. Raghunathan and A. Marx, Sci. Rep., 2019, 9, 590 CrossRef.
- M. J. G. Elizabeth, N. C. Janine and A. David, Directed Evolution Library Creation, SpringerLink, 2014 Search PubMed.
- M. R. Dunn, C. Otto, K. E. Fenton and J. C. Chaput, ACS Chem. Biol., 2016, 11, 1210–1219 CrossRef CAS PubMed.
- M. T. Reetz, M. Bocola, J. D. Carballeira, D. X. Zha and A. Vogel, Angew. Chem., Int. Ed., 2005, 44, 4192–4196 CrossRef CAS PubMed.
- M. T. Reetz and J. D. Carballeira, Nat. Protoc., 2007, 2, 891–903 CrossRef CAS PubMed.
- T. S. Wong, K. L. Tee, B. Hauer and U. Schwaneberg, Nucleic Acids Res., 2004, 32, e26 CrossRef.
- G. Houlihan, S. Arangundy-Franklin, B. T. Porebski, N. Subramanian, A. I. Taylor and P. Holliger, Nat. Chem., 2020, 12, 683–690 CrossRef CAS PubMed.
- A. C. Larsen, M. R. Dunn, A. Hatch, S. P. Sau, C. Youngbull and J. C. Chaput, Nat. Commun., 2016, 7, 11235 CrossRef CAS.
- V. B. Pinheiro, A. I. Taylor, C. Cozens, M. Abramov, M. Renders, S. Zhang, J. C. Chaput, J. Wengel, S. Peak-Chew, S. H. McLaughlin, P. Herdewijn and P. Holliger, Science, 2012, 336, 341–344 CrossRef CAS PubMed.
- R. Laos, R. Shaw, N. A. Leal, E. Gaucher and S. Benner, Biochemistry, 2013, 52, 5288–5294 CrossRef CAS PubMed.
- M. Chung, K. Goroncy, A. Kolesnikova, D. Schoenauer and U. Schwaneberg, Biotechnol. Bioeng., 2020, 117, 3699–3711 CrossRef CAS PubMed.
- J. Chelliserrykattil and A. D. Ellington, Nat. Biotechnol., 2004, 22, 1155–1160 CrossRef CAS PubMed.
- J. A. Van Deventer and K. D. Wittrup, Methods Mol. Biol., 2014, 1131, 151–181 CrossRef CAS PubMed.
- J. Hanes and A. Pluckthun, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4937–4942 CrossRef CAS.
- L. Diamante, P. Gatti-Lafranconi, Y. Schaerli and F. Hollfelder, Protein Eng., Des. Sel., 2013, 26, 713–724 CrossRef CAS PubMed.
- I. Benhar, Biotechnol. Adv., 2001, 19, 1–33 CrossRef CAS PubMed.
- S. Y. Lee, J. H. Choi and Z. H. Xu, Trends Biotechnol., 2003, 21, 45–52 CrossRef CAS.
- D. S. Wilson, A. D. Keefe and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 3750–3755 CrossRef CAS.
- D. Vallejo, A. Nikoomanzar, B. M. Paegel and J. C. Chaput, ACS Synth. Biol., 2019, 8, 1430–1440 CrossRef CAS PubMed.
- J. L. Ong, D. Loakes, S. Jaroslawski, K. Too and P. Holliger, J. Mol. Biol., 2006, 361, 537–550 CrossRef CAS PubMed.
- J. W. Ellefson, J. Gollihar, R. Shroff, H. Shivram, V. R. Iyer and A. D. Ellington, Science, 2016, 352, 1590–1593 CrossRef CAS PubMed.
- J. W. Ellefson, A. J. Meyer, R. A. Hughes, J. R. Cannon, J. S. Brodbelt and A. D. Ellington, Nat. Biotechnol., 2014, 32, 97 CrossRef CAS.
- A. J. Meyer, J. W. Ellefson and A. D. Ellington, ACS Synth. Biol., 2015, 4, 1070–1076 CrossRef CAS PubMed.
- V. B. Pinheiro, S. A. Franklin and P. Holliger, Curr. Protoc. Nucleic Acid Chem., 2014, 57, 9 Search PubMed.
- A. Fernandez-Gacio, M. Uguen and J. Fastrez, Trends Biotechnol., 2003, 21, 408–414 CrossRef CAS.
- G. Winter, A. D. Griffiths, R. E. Hawkins and H. R. Hoogenboom, Annu. Rev. Immunol., 1994, 12, 433–455 CrossRef CAS PubMed.
- E. Brunet, C. Chauvin, V. Choumet and J. L. Jestin, Nucleic Acids Res., 2002, 30, e40 CrossRef PubMed.
- L. Rahbarnia, S. Farajnia, H. Babaei, J. Majidi, K. Veisi, V. Ahmadzadeh and B. Akbari, J. Drug Target., 2017, 25, 216–224 CrossRef CAS PubMed.
- J. Pande, M. M. Szewczyk and A. K. Grover, Biotechnol. Adv., 2010, 28, 849–858 CrossRef CAS.
- G. Xia, L. J. Chen, T. Sera, M. Fa, P. G. Schultz and F. E. Romesberg, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 6597–6602 CrossRef CAS.
- D. Thirunavukarasu, T. Chen, Z. Liu, N. Hongdilokkul and F. E. Romesberg, J. Am. Chem. Soc., 2017, 139, 2892–2895 CrossRef CAS PubMed.
- T. Chen and F. E. Romesberg, Angew. Chem., Int. Ed., 2017, 56, 14046–14051 CrossRef CAS PubMed.
- Q. Shao, T. Chen, K. Sheng, Z. Liu, Z. Zhang and F. E. Romesberg, J. Am. Chem. Soc., 2020, 142, 2125–2128 CrossRef CAS.
- W. Delespaul, Y. Peeters, P. Herdewijn and J. Robben, Biochem. Biophys. Res. Commun., 2015, 460, 245–249 CrossRef CAS.
- M. S. Morrison, C. J. Podracky and D. R. Liu, Nat. Chem. Biol., 2020, 16, 610–619 CrossRef CAS PubMed.
- K. M. Esvelt, J. C. Carlson and D. R. Liu, Nature, 2011, 472, 499–550 CrossRef CAS.
- M. S. Packer, H. A. Rees and D. R. Liu, Nat. Commun., 2017, 8, 956 CrossRef.
- M. F. Richter, K. T. Zhao, E. Eton, A. Lapinaite, G. A. Newby, B. W. Thuronyi, C. Wilson, L. W. Koblan, J. Zeng, D. E. Bauer, J. A. Doudna and D. R. Liu, Nat. Biotechnol., 2020, 38, 901 CrossRef CAS.
- B. C. Dickinson, A. M. Leconte, B. Allen, K. M. Esvelt and D. R. Liu, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9007–9012 CrossRef CAS PubMed.
- A. H. Badran and D. R. Liu, Curr. Opin. Chem. Biol., 2015, 24, 1–10 CrossRef CAS PubMed.
- K. D. Wittrup, Curr. Opin. Biotechnol., 2001, 12, 395–399 CrossRef CAS PubMed.
- M. R. Smith, E. Khera and F. Wen, Ind. Eng. Chem. Res., 2015, 54, 4021–4032 CrossRef CAS PubMed.
- Z. Liu, S. Ho, T. Hasunuma, J. Chang, N. Ren and A. Kondo, Bioresour. Technol., 2016, 215, 324–333 CrossRef CAS.
- A. Hottin and A. Marx, Acc. Chem. Res., 2016, 49, 418–427 CrossRef CAS PubMed.
- P. Ghosh, H. M. Kropp, K. Betz, S. Ludmann, K. Diederichs, A. Marx and S. G. Srivatsan, J. Am. Chem. Soc., 2022, 144, 10556–10569 CrossRef CAS PubMed.
- H. Fujita, K. Nakajima, Y. Kasahara, H. Ozaki and M. Kuwahara, Bioorg. Med. Chem. Lett., 2015, 25, 333–336 CrossRef CAS.
- H. Hoshino, Y. Kasahara, H. Fujita, M. Kuwahara, K. Morihiro, S. Tsunoda and S. Obika, Bioorg. Med. Chem. Lett., 2016, 26, 530–533 CrossRef CAS PubMed.
- M. Gao, Q. Zhang, X. Feng and J. Liu, Acta Biomater., 2021, 131, 1–15 CrossRef CAS PubMed.
- J. W. Park, P. Lagniton, Y. Liu and R. H. Xu, Int. J. Biol. Sci., 2021, 17, 1446–1460 CrossRef CAS.
- K. D. Nance and J. L. Meier, ACS Cent. Sci., 2021, 7, 748–756 CrossRef CAS PubMed.
- S. C. Johnson, C. B. Sherrill, D. J. Marshall, M. J. Moser and J. R. Prudent, Nucleic Acids Res., 2004, 32, 1937–1941 CrossRef CAS.
- C. B. Winiger, R. W. Shaw, M. Kim, J. D. Moses, M. F. Matsuura and S. K. Benner, ACS Synth. Biol., 2017, 6, 194–200 CrossRef CAS PubMed.
- Z. Yang, F. Chen, S. G. Chamberlin and S. A. Benner, Angew. Chem., Int. Ed., 2010, 49, 177–180 CrossRef CAS PubMed.
- S. Hoshika, N. A. Leal, M. Kim, M. Kim, N. B. Karalkar, H. Kim, A. M. Bates, Jr., N. E. Watkins, H. A. SantaLucia, A. J. Meyer, S. DasGupta, J. A. Piccirilli, A. D. Ellington, Jr., J. SantaLucia, M. M. Georgiadis and S. A. Benner, Science, 2019, 363, 884 CrossRef CAS PubMed.
- A. M. Leconte, L. J. Chen and F. E. Romesberg, J. Am. Chem. Soc., 2005, 127, 12470–12471 CrossRef CAS.
- J. Oh, J. Shin, I. C. Unarta, W. Wang, A. W. Feldman, R. J. Karadeema, L. Xu, J. Xu, J. Chong, R. Krishnamurthy, X. Huang, F. E. Romesberg and D. Wang, Nat. Chem. Biol., 2021, 17, 906 CrossRef CAS PubMed.
- K. Hashimoto, E. C. Fischer and F. E. Romesberg, J. Am. Chem. Soc., 2021, 143, 8603–8607 CrossRef CAS.
- F. Eggert, K. Kurscheidt, E. Hoffmann and S. Kath Schorr, ChemBioChem, 2019, 20, 1642–1645 CrossRef CAS PubMed.
- A. X. Zhou, X. Dong and F. E. Romesberg, J. Am. Chem. Soc., 2020, 142, 19029–19032 CrossRef CAS PubMed.
- I. Hirao, Y. Harada, M. Kimoto, T. Mitsui, T. Fujiwara and S. Yokoyama, J. Am. Chem. Soc., 2004, 126, 13298–13305 CrossRef CAS PubMed.
- M. Kimoto, T. Mitsui, S. Yokoyama and I. Hirao, J. Am. Chem. Soc., 2010, 132, 4988 CrossRef CAS PubMed.
- M. Kimoto, R. Kawai, T. Mitsui, S. Yokoyama and I. Hirao, Nucleic Acids Res., 2009, 37, e14 CrossRef PubMed.
- M. Kimoto, T. Mitsui, R. Yamashige, A. Sato, S. Yokoyama and I. Hirao, J. Am. Chem. Soc., 2010, 132, 15418–15426 CrossRef CAS PubMed.
- M. Kimoto, A. J. Meyer, I. Hirao and A. D. Ellington, Chem. Commun., 2017, 53, 12309–12312 RSC.
- T. Someya, A. Ando, M. Kimoto and I. Hirao, Nucleic Acids Res., 2015, 43, 6665–6676 CrossRef CAS PubMed.
- M. Kimoto, T. Mitsui, Y. Harada, A. Sato, S. Yokoyama and I. Hirao, Nucleic Acids Res., 2007, 35, 5360–5369 CrossRef CAS.
- K. H. Lee, M. Kimoto, G. Kawai, I. Okamoto, A. Fin and I. Hirao, Chem. – Eur. J., 2022, 28, e202104396 Search PubMed.
- M. Fa, A. Radeghieri, A. A. Henry and F. E. Romesberg, J. Am. Chem. Soc., 2004, 126, 1748–1754 CrossRef CAS PubMed.
- T. Chen and F. E. Romesberg, Biochemistry, 2017, 56, 5227–5228 CrossRef CAS PubMed.
- T. Chen and F. E. Romesberg, J. Am. Chem. Soc., 2017, 139, 9949–9954 CrossRef CAS PubMed.
- P. Song, R. Zhang, C. He and T. Chen, Curr. Protoc., 2021, 1, e188 CAS.
- R. Shroff, J. W. Ellefson, S. S. Wang, A. A. Boulgakov, R. A. Hughes and A. D. Ellington, ACS Synth. Biol., 2022, 11, 554–561 CrossRef CAS PubMed.
- R. Sousa and R. Padilla, EMBO J., 1995, 14, 4609–4621 CrossRef CAS PubMed.
- R. Padilla and R. Sousa, Nucleic Acids Res., 1999, 27, 1561–1563 CrossRef CAS PubMed.
- R. Padilla and R. Sousa, Nucleic Acids Res., 2002, 30, e138 CrossRef PubMed.
- A. J. Meyer, D. J. Garry, B. Hall, M. M. Byrom, H. G. McDonald, X. Yang, Y. W. Yin and A. D. Ellington, Nucleic Acids Res., 2015, 43, 7480–7488 CrossRef CAS PubMed.
- A. Horhota, K. Y. Zou, J. K. Ichida, B. Yu, L. W. McLaughlin, J. W. Szostak and J. C. Chaput, J. Am. Chem. Soc., 2005, 127, 7427–7434 CrossRef CAS PubMed.
- J. K. Ichida, K. Zou, A. Horhota, B. Yu, L. W. McLaughlin and J. W. Szostak, J. Am. Chem. Soc., 2005, 127, 2802–2803 CrossRef CAS PubMed.
- A. Nikoomanzar, D. Vallejo and J. C. Chaput, ACS Synth. Biol., 2019, 8, 1421 CrossRef CAS PubMed.
- A. Nikoomanzar, D. Vallejo, E. J. Yik and J. C. Chaput, ACS Synth. Biol., 2020, 9, 1873–1881 CrossRef CAS PubMed.
- H. Hoshino, Y. Kasahara, M. Kuwahara and S. Obika, J. Am. Chem. Soc., 2020, 142, 21530–21537 CrossRef CAS PubMed.
- E. Medina, E. J. Yik, P. Herdewijn and J. C. Chaput, ACS Synth. Biol., 2021, 10, 1429–1437 CrossRef CAS PubMed.
- Y. Wang, A. K. Ngor, A. Nikoomanzar and J. C. Chaput, Nat. Commun., 2018, 9, 5067 CrossRef PubMed.
- M. R. Dunn and J. C. Chaput, ChemBioChem, 2016, 17, 1804–1808 CrossRef CAS PubMed.
- L. L. Torres and V. B. Pinheiro, Curr. Protoc. Chem. Biol., 2018, 10, e41 CrossRef.
- K. B. Wu, C. Skrodzki, Q. W. Su, J. Lin and J. Niu, Chem. Sci., 2022, 13, 6873–6881 RSC.
- J. Attwater, S. Tagami, M. Kimoto, K. Butler, E. T. Kool, J. Wengel, P. Herdewijn, I. Hirao and P. Holliger, Chem. Sci., 2013, 4, 2804–2814 RSC.
- B. N. Gawande, J. C. Rohloff, J. D. Carter, I. von Carlowitz, C. Zhang, D. J. Schneider and N. Janjic, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2898–2903 CrossRef CAS PubMed.
- A. D. Keefe and S. T. Cload, Curr. Opin. Chem. Biol., 2008, 12, 448–456 CrossRef CAS PubMed.
- S. Qi, N. Duan, I. M. Khan, X. Dong, Y. Zhang, S. Wu and Z. Wang, Biotechnol. Adv., 2022, 55, 107902 CrossRef CAS PubMed.
- D. R. Davies, A. D. Gelinas, C. Zhang, J. C. Rohloff, J. D. Carter, D. O'Connell, S. M. Waugh, S. K. Wolk, W. S. Mayfield, A. B. Burgin, T. E. Edwards, L. J. Stewart, L. Gold, N. Janjic and T. C. Jarvis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19971–19976 CrossRef CAS PubMed.
- K. Matsunaga, M. Kimoto and I. Hirao, J. Am. Chem. Soc., 2017, 139, 324–334 CrossRef CAS PubMed.
- K. Matsunaga, M. Kimoto, V. W. Lim, H. P. Tan, Y. Q. Wong, W. Sun, S. Vasoo, Y. S. Leo and I. Hirao, Nucleic Acids Res., 2021, 49, 11407–11424 CrossRef CAS.
- K. Sefah, Z. Yang, K. M. Bradley, S. Hoshika, E. Jimenez, L. Zhang, G. Zhu, S. Shanker, F. Yu, D. Turek, W. Tan and S. A. Benner, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1449–1454 CrossRef CAS PubMed.
- L. Zhang, Z. Yang, K. Sefah, K. M. Bradley, S. Hoshika, M. Kim, H. Kim, G. Zhu, E. Jiménez, S. Cansiz, I. Teng, C. Champanhac, C. McLendon, C. Liu, W. Zhang, D. L. Gerloff, Z. Huang, W. Tan and S. A. Benner, J. Am. Chem. Soc., 2015, 137, 6734–6737 CrossRef CAS PubMed.
- E. Biondi, J. D. Lane, D. Das, S. Dasgupta, J. A. Piccirilli, S. Hoshika, K. M. Bradley, B. A. Krantz and S. A. Benner, Nucleic Acids Res., 2016, 44, 9565–9577 CAS.
- L. Zhang, Z. Yang, T. Le Trinh, I. Teng, S. Wang, K. M. Bradley, S. Hoshika, Q. Wu, S. Cansiz, D. J. Rowold, C. McLendon, M. Kim, Y. Wu, C. Cui, Y. Liu, W. Hou, K. Stewart, S. Wan, C. Liu, S. A. Benner and W. Tan, Angew. Chem., Int. Ed., 2016, 55, 12372–12375 CrossRef CAS PubMed.
- Z. Liu, T. Chen and F. E. Romesberg, Chem. Sci., 2017, 8, 8179–8182 RSC.
- N. Minakawa, M. Sanji, Y. Kato and A. Matsuda, Bioorg. Med. Chem., 2008, 16, 9450–9456 CrossRef CAS PubMed.
- S. Diafa and M. Hollenstein, Molecules, 2015, 20, 16643–16671 CrossRef CAS PubMed.
- I. A. Ferreira-Bravo, C. Cozens, P. Holliger and J. J. DeStefano, Nucleic Acids Res., 2015, 43, 9587–9599 CAS.
- K. M. Rose, I. A. Ferreira-Bravo, M. Li, R. Craigie, M. A. Ditzler, P. Holliger and J. J. DeStefano, ACS Chem. Biol., 2019, 14, 2166–2175 CAS.
- I. A. Ferreira-Bravo and J. J. DeStefano, Viruses, 2021, 13, 1983 CrossRef PubMed.
- M. R. Dunn, C. M. McCloskey, P. Buckley, K. Rhea and J. C. Chaput, J. Am. Chem. Soc., 2020, 142, 7721–7724 CrossRef CAS.
- H. Yu, S. Zhang and J. C. Chaput, Nat. Chem., 2012, 4, 183–187 CrossRef CAS PubMed.
- A. E. Rangel, Z. Chen, T. M. Ayele and J. M. Heemstra, Nucleic Acids Res., 2018, 46, 8057–8068 CrossRef CAS PubMed.
- H. Mei, J. Liao, R. M. Jimenez, Y. Wang, S. Bala, C. McCloskey, C. Switzer and J. C. Chaput, J. Am. Chem. Soc., 2018, 140, 5706–5713 CrossRef CAS PubMed.
- C. M. McCloskey, Q. Li, E. J. Yik, N. Chim, A. K. Ngor, E. Medina, I. Grubisic, L. Co Ting Keh, R. Poplin and J. C. Chaput, ACS Synth. Biol., 2021, 10, 3190–3199 CrossRef CAS PubMed.
- B. E. Young, N. Kundu and J. T. Sczepanski, Chem. – Eur. J., 2019, 25, 7981–7990 CrossRef CAS PubMed.
- J. Chen, M. Chen and T. F. Zhu, Nat. Biotechnol., 2022 DOI:10.1038/s41587-022-01337-8.
- S. Paul, A. A. W. L. Wong, L. T. Liu and D. M. Perrin, ChemBioChem, 2022, 23, e202100600 CrossRef CAS PubMed.
- Y. Wang, K. Nguyen, R. C. Spitale and J. C. Chaput, Nat. Chem., 2021, 13, 319–326 CrossRef CAS.
- K. Nguyen, Y. Wang, W. E. England, J. C. Chaput and R. C. Spitale, J. Am. Chem. Soc., 2021, 143, 4519–4523 CrossRef CAS.
- K. Yang and J. C. Chaput, J. Am. Chem. Soc., 2021, 143, 8957–8961 CrossRef CAS.
- Y. Wang, Y. Wang, D. Song, X. Sun, Z. Li, J. Chen and H. Yu, Nat. Chem., 2021, 14, 350 CrossRef PubMed.
- Y. Wang, Y. Wang, D. Song, X. Sun, Z. Zhang, X. Li, Z. Li and H. Yu, J. Am. Chem. Soc., 2021, 143, 8154–8163 CrossRef CAS PubMed.
- L. Zhang, S. Wang, Z. Yang, S. Hoshika, S. Xie, J. Li, X. Chen, S. Wan, L. Li, S. A. Benner and W. Tan, Angew. Chem., Int. Ed., 2020, 59, 663–668 CrossRef CAS.
- A. I. Taylor, F. Beuron, S. Peak-Chew, E. P. Morris, P. Herdewijn and P. Holliger, ChemBioChem, 2016, 17, 1107–1110 CrossRef CAS PubMed.
- Q. Wang, X. Chen, X. Li, D. Song, J. Yang, H. Yu and Z. Li, ACS Appl. Mater. Interfaces, 2020, 12, 53592–53597 CrossRef CAS.
- J. L. Ptacin, C. E. Caffaro, L. Ma, K. M. San Jose Gall, H. R. Aerni, N. V. Acuff, R. W. Herman, Y. Pavlova, M. J. Pena, D. B. Chen, L. K. Koriazova, L. K. Shawver, I. B. Joseph and M. E. Milla, Nat. Commun., 2021, 12, 4785 CrossRef CAS.
- E. C. Fischer, K. Hashimoto, Y. Zhang, A. W. Feldman, V. T. Dien, R. J. Karadeema, R. Adhikary, M. P. Ledbetter, R. Krishnamurthy and F. E. Romesberg, Nat. Chem. Biol., 2020, 16, 570 CrossRef CAS.
- M. Ast, A. Gruber, S. Schmitz-Esser, H. E. Neuhaus, P. G. Kroth, M. Horn and I. Haferkamp, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3621–3626 CrossRef CAS PubMed.
- M. P. Ledbetter, R. J. Karadeema and F. E. Romesberg, J. Am. Chem. Soc., 2018, 140, 758–765 CrossRef CAS.
- A. X. Zhou, K. Sheng, A. W. Feldman and F. E. Romesberg, J. Am. Chem. Soc., 2019, 141, 20166–20170 CrossRef CAS PubMed.
- L. Bornewasser, C. Domnick and S. Kath-Schorr, Chem. Sci., 2022, 13, 4753–4761 RSC.
- Z. Yang, F. Chen, J. B. Alvarado and S. A. Benner, J. Am. Chem. Soc., 2011, 133, 15105–15112 CrossRef CAS.
- K. Hamashima, Y. T. Soong, K. Matsunaga, M. Kimoto and I. Hirao, ACS Synth. Biol., 2019, 8, 1401–1410 CrossRef CAS PubMed.
- M. Kimoto, S. H. G. Soh and I. Hirao, ChemBioChem, 2020, 21, 2287–2296 CrossRef CAS.
- P. Marliere, J. Patrouix, V. Doering, P. Herdewijn, S. Tricot, S. Cruveiller, M. Bouzon and R. Mutzel, Angew. Chem., Int. Ed., 2011, 50, 7109–7114 CrossRef CAS PubMed.
- A. T. Krueger, L. W. Peterson, J. Chelliserry, D. J. Kleinbaum and E. T. Kool, J. Am. Chem. Soc., 2011, 133, 18447–18451 CrossRef CAS.
- V. Pezo, F. W. Liu, M. Abramov, M. Froeyen, P. Herdewijn and P. Marliere, Angew. Chem., Int. Ed., 2013, 52, 8139–8143 CrossRef CAS PubMed.
- M. Luo, E. Groaz, M. Froeyen, V. Pezo, F. Jaziri, P. Leonczak, G. Schepers, J. Rozenski, P. Marliere and P. Herdewijn, J. Am. Chem. Soc., 2019, 141, 10844–10851 CrossRef CAS.
- M. Defresne, S. Barbe and T. Schiex, Int. J. Mol. Sci., 2021, 22, 11741 CrossRef CAS PubMed.
- S. Zhou and H. S. Alper, J. Chem. Technol. Biotechnol., 2019, 94, 366–376 CrossRef CAS.
- S. Kim, H. Yi, Y. T. Kim and H. S. Lee, J. Mol. Biol., 2022, 434, 167302 CrossRef CAS PubMed.
- M. Kimoto and I. Hirao, Front. Mol. Biosci., 2022, 9, 851646 CrossRef CAS PubMed.
- M. F. Matsuura, C. B. Winiger, R. W. Shaw, M. Kim, M. Kim, A. B. Daugherty, F. Chen, P. Moussatche, J. D. Moses, S. Lutz and S. A. Benner, ACS Synth. Biol., 2017, 6, 388–394 CrossRef CAS PubMed.
- M. F. Matsuura, R. W. Shaw, J. D. Moses, H. Kim, M. Kim, M. Kim, S. Hoshika, N. Karalkar and S. A. Benner, ACS Synth. Biol., 2016, 5, 234–240 CrossRef CAS PubMed.
- F. Chen, Y. Zhang, A. B. Daugherty, Z. Yang, R. Shaw, M. Dong, S. Lutz and S. A. Benner, PLoS One, 2017, 12, e174163 Search PubMed.
- Y. Wu, M. Fa, E. L. Tae, P. G. Schultz and F. E. Romesberg, J. Am. Chem. Soc., 2002, 124, 14626–14630 CrossRef CAS PubMed.
- J. C. Chaput, P. Herdewijn and M. Hollenstein, ChemBioChem, 2020, 21, 1408–1411 CrossRef CAS.
- D. A. Malyshev, K. Dhami, H. T. Quach, T. Lavergne, P. Ordoukhanian, A. Torkamani and F. E. Romesberg, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12005–12010 CrossRef CAS PubMed.
- V. T. Dien, E. M. Sydney, J. K. Rebekah and E. Floyd Romesberg, Curr. Opin. Chem. Biol., 2018, 46, 196–202 CrossRef CAS.
Footnote |
| † Authors have equal contributions. |
| This journal is © The Royal Society of Chemistry 2022 |