
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn intramodular thioesterase domain catalyses chain release in the biosynthesis of a cytotoxic virulence factor†
Rory
Little
 ,
Felix
Trottmann
,
Miriam
Preissler
and
Christian
Hertweck
*
,
Felix
Trottmann
,
Miriam
Preissler
and
Christian
Hertweck
*
Department of Biomolecular Chemistry, Leibniz Institute for Natural Product Research and Infection Biology HKI. Beutenbergstr. 11a, 07745 Jena, Germany. E-mail: christian.hertweck@hki-jena.de
First published on 25th July 2022
Abstract
An essential step in the biosynthesis of polyketide and non-ribosomal peptide natural products is cleavage of the thioester bond that tethers the acyl/peptidyl chain to its biosynthetic enzyme. In modular polyketide synthases (PKS) and non-ribosomal peptide synthetases (NRPS) chain release is typically catalysed by a single C-terminal thioesterase domain. A clear exception is the bimodular PKS-NRPS BurA that produces gonyol—an intermediate in the biosynthesis of the cytotoxic Burkholderia virulence factor malleicyprol. While BurA lacks a C-terminal thioesterase domain, making the mechanism by which gonyol is released unclear, it contains two uncommon non-C-terminal thioesterase domains: one at the N-terminus of module one (BurA TE-A) and one within module two (BurA TE-B). Here we show using a sequence similarity network and site-directed mutagenesis that BurA TE-A resembles proofreading type II thioesterases and is not essential for gonyol biosynthesis, indicating a hydrolytic proofreading role. In contrast, the intramodular BurA TE-B is essential and catalyses the hydrolytic release of gonyol. Furthermore, unlike typical type I thioesterase domains, BurA TE-B accepts its acyl substrate from a downstream carrier-protein domain as opposed to an upstream one. Our findings clarify an important step in malleicyprol biosynthesis, reveal the flexibility of thioesterase domain positioning, and will serve as a basis for understanding other intramodular thioesterase domains.
Introduction
Polyketides and non-ribosomal peptides are both large families of natural products whose members often possess pharmacologically relevant bioactivities, including antibiotic, anti-inflammatory, or anticancer.1,2 A key feature of many polyketides and non-ribosomal peptides is that during biosynthesis they are covalently attached via a thioester bond to their biosynthetic enzymes. In modular polyketide synthases (PKSs) and non-ribosomal peptide synthetases (NRPSs) this covalent attachment is mediated by dedicated carrier protein domains via phosphopantetheine prosthetic groups. The thiol of the ca. 18 Å long flexible phosphopantetheine moiety forms a thioester bond to a growing acyl/peptidyl chain and delivers it to each of the different catalytic domains within a given module, before passing it to the carrier protein of the downstream module via a chain extension reaction.1,2 Once all of the chain extension reactions have completed, however, the covalent attachment must be broken in order to produce a free and functional polyketide or peptide natural product. A wide range of different enzymes that catalyse chain release have been characterised, with type I thioesterase (TE) domains being one of the most common.3 Typically found on the C-terminus of the final PKS or NRPS in a pathway, TE domains possess a serine protease-like α/β hydrolase fold and catalyse acyl/peptidyl chain release using a conserved Ser-Asp-His catalytic triad.3 The Ser forms an ester linkage with the C1 carbonyl of the nascent acyl/peptidyl chain, while the catalytic His (stabilised by the Asp) activates a nucleophile via deprotonation to attack and cleave the ester bond. If an exogenous nucleophile (typically water) is selected then the product released is a linear carboxylic acid, while if an endogenous nucleophile is used (such as a hydroxyl or amino group) a cyclic product is released.3 Aside from type I TE domains, many polyketide and non-ribosomal peptide biosynthetic gene clusters also encode a standalone α/β hydrolase fold thioesterase (called a type II thioesterase). In contrast to type I TE domains, type II TEs are typically not responsible for catalysing product release.4,5 Instead, their function is more akin to “proofreading” through catalysing the hydrolytic removal of aberrant residues attached to phosphopantetheine groups, such as unreactive acetyl groups that prohibit subsequent chain extension reactions.5 While the activities of C-terminal type I TE domains and type II thioesterases are well defined, not all thioesterases in polyketide and non-ribosomal peptide pathways fall clearly into these two groups. In particular, some PKS and NRPS enzymes contain unusual non C-terminal TE domains (Table S1, ESI†).3,6–10 Such TE domains are poorly understood and may catalyse reactions unlike canonical type I or type II TEs, exemplified by the intramodular TE domain of the PKS Fr9C that catalyses cis double bond formation in the biosynthesis of the polyketide FR901464.7 A standout among PKS/NRPS enzymes with unusually placed TE domains is the bimodular PKS-NRPS hybrid BurA from the bacterium Burkholderia thailandensis. BurA contains two non-C-terminal TE domains: an N-terminal TE domain in module 1 (BurA TE-A) and an intramodular TE domain in module 2 (BurA TE-B) (Fig. 1A).11 BurA catalyses the condensation of the L-methionine (1)-derived sulfonium acid dimethylsulfoniopropionate (DMSP) (2) with malonyl-CoA, followed by a ketoreductase (KR) domain-catalysed β-ketoreduction, to produce gonyol (3S-5-dimethylsulfonio-3-hydroxylpentanoate) (3).11–13 The burA gene is found in the bur biosynthetic gene cluster—which is also present in the human pathogenic B. mallei and B. pseudomallei—responsible for the production of the polyketide malleicyprol (4) and its downstream metabolite burkholderic acid (5) (syn. malleilactone).11,14,15 The cyclopropanol moiety of malleicyprol is the main difference to burkholderic acid and is responsible for the ca. 110 fold higher cytotoxicity of malleicyprol.11,14 The gonyol created by BurA is converted, via gonydiol (6), into trigonic acid (7), which becomes the virulence-conferring cyclopropanol moiety of malleicyprol.14–16 Given that BurA lacks an orthodox C-terminal TE domain, and contains two unusual non-C-terminal ones, how gonyol is released was unclear. One possibility was that akin to the biosynthesis of polyketide ionophores like monensin,3,4 a separate type II TE catalyses gonyol release. However, the bur gene cluster does not encode an obvious candidate to catalyse this reaction. In addition, gonyol was still produced when, in a previous study, our group heterologously expressed burA in E. coli, suggesting that BurA is capable of catalysing gonyol release on its own.13 In this study, we therefore turned our attention to the two non-C-terminal TE domains of BurA to determine whether, despite their unusual placement, one or both is involved in gonyol release. Here we show that the intramodular BurA TE-B is responsible for catalysing the hydrolytic release of gonyol. Furthermore, BurA TE-B is unique in being the first characterised example of a TE domain that receives its substrate from a downstream carrier protein domain as opposed to an upstream domain.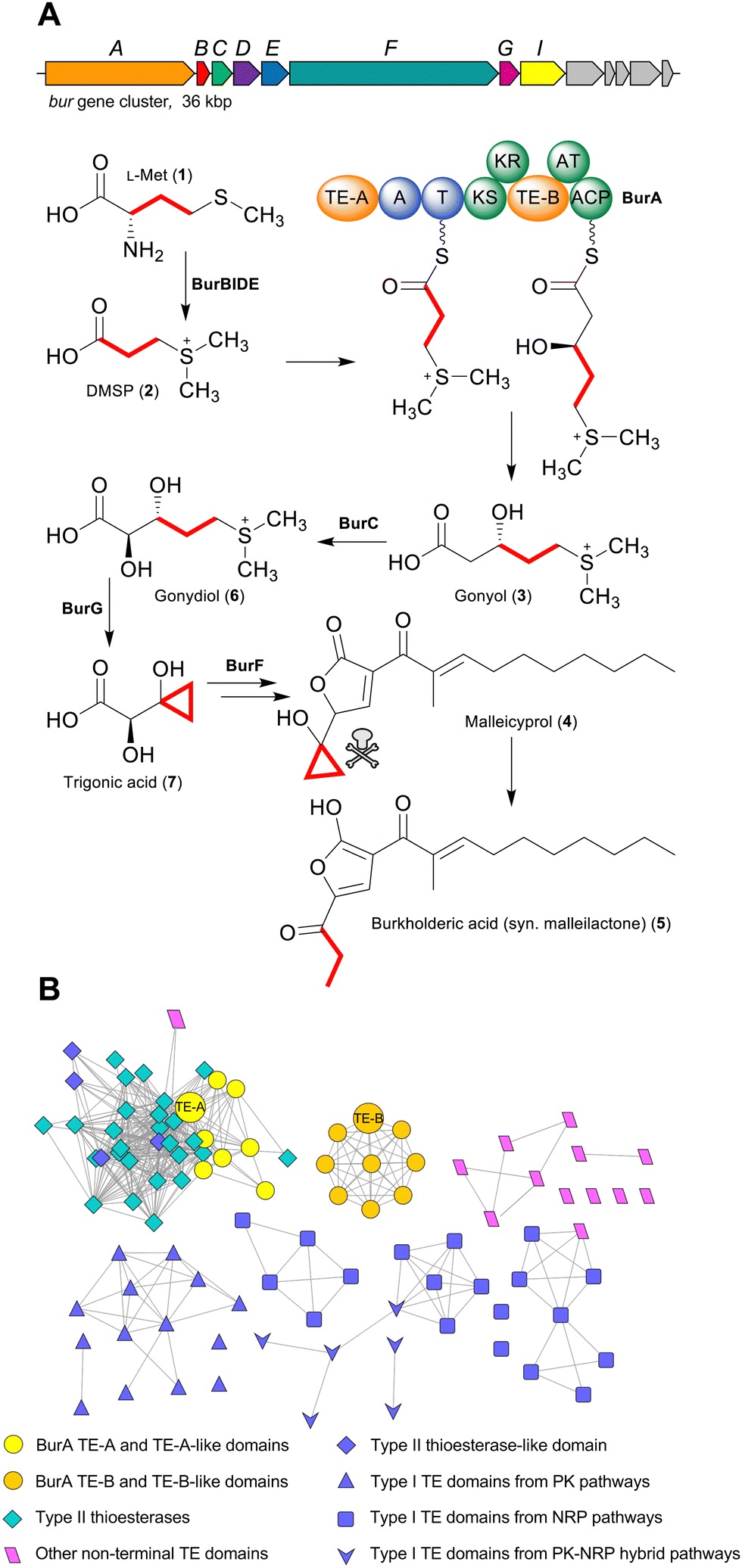 | ||
| Fig. 1 (A) The bur gene cluster and the biosynthesis pathway to malleicyprol in Burkholderia thailandensis. Dimethylsulfoniopropionate (DMSP) produced from L-methionine is converted into gonyol by the bimodular PKS-NRPS hybrid BurA. Gonyol is then converted into the cyclopropanol-containing trigonic acid, which becomes the virulence-conferring cyclopropanol moiety of malleicyprol. (B) An α/β hydrolase fold thioesterase sequence similarity network. BurA TE-A and BurA TE-B from B. thailandensis are labelled and both depicted as large circular nodes. | ||
Results and discussion
As a starting point for investigating the functions of BurA TE-A and TE-B, we first compared their amino acid sequences to other type I TE- and type II TE-domains in a sequence similarity network (Fig. 1B). In addition to BurA TE-A and TE-B, we included the equivalent TE domains from BurA homologues encoded in several non-malleicyprol-producing bacteria (named TE-A-like and TE-B-like domains),16 and other non-C-terminal TE domains from other PKS and NRPS enzymes (Table S1, ESI†). BurA TE-A and BurA TE-B both contain a conserved catalytic serine (within a GxSxG motif) and histidine typical of α/β hydrolases fold thioesterases, while the catalytic Asp residue, typically located ca. 20 amino acids upstream of the catalytic Ser, is absent in both (though is not always essential for catalytic activity) (Fig. S1, ESI†).17 A previous study using Bayesian analysis had found that both BurA TE-A and BurA TE-B resemble type II thioesterases more than type I.18 Here, our sequence similarity network analysis revealed that, of the two, BurA TE-A and the TE-A-like domains are more similar to type II thioesterases (and also to four type I TE domains that resemble type II TEs)18–21 than BurA TE-B or any of the TE-B-like domains (Fig. 1B). In contrast, BurA TE-B and the TE-B-like domains clustered together, separate from the type II thioesterases, type I TE domains, or the other non-C-terminal TE domains included in the network. Taken together, the network analysis suggested that, while BurA TE-A may have a type II TE-like “proofreading” function, the function of TE-B is difficult to predict based on sequence data alone. We therefore next turned to a functional investigation of both domains.To determine if BurA TE-A and BurA TE-B have a role in gonyol biosynthesis, the catalytic serine residues of each were mutated to alanine, producing burA_TE-A S90A and burA_TE-B S2023A. Each mutant gene was then independently expressed in E. coli and the corresponding cultures supplied with DMSP (allowing them to synthesise gonyol using endogenous malonyl-CoA), followed by LC-HRMS analysis to search for gonyol (m/z = 179.0736 [M+H]+, M as zwitterionic species) (Fig. 2A). Mutating the catalytic serine of BurA TE-A did not affect gonyol production (Fig. 2B(a, d and e)). Following on from this result, we then made the same burA_TE-A S90A point mutation in the genome of B. thailandensis to determine if, when examined in vivo on the level of output of the whole bur gene cluster (rather than just gonyol), the mutation had an effect. To assess this, we chose to monitor the level of burkholderic acid (5), the downstream metabolite of malleicyprol, due to its strong UV-Vis absorbance readily facilitating quantification. We found no difference in burkholderic acid production in B. thailandensis burA_TE-A S90A compared to the wild type (Fig. S2 and S3, ESI†). The experiments therefore demonstrated that BurA TE-A is not essential for gonyol biosynthesis or measurably affects the production of the late-stage bur product burkholderic acid.
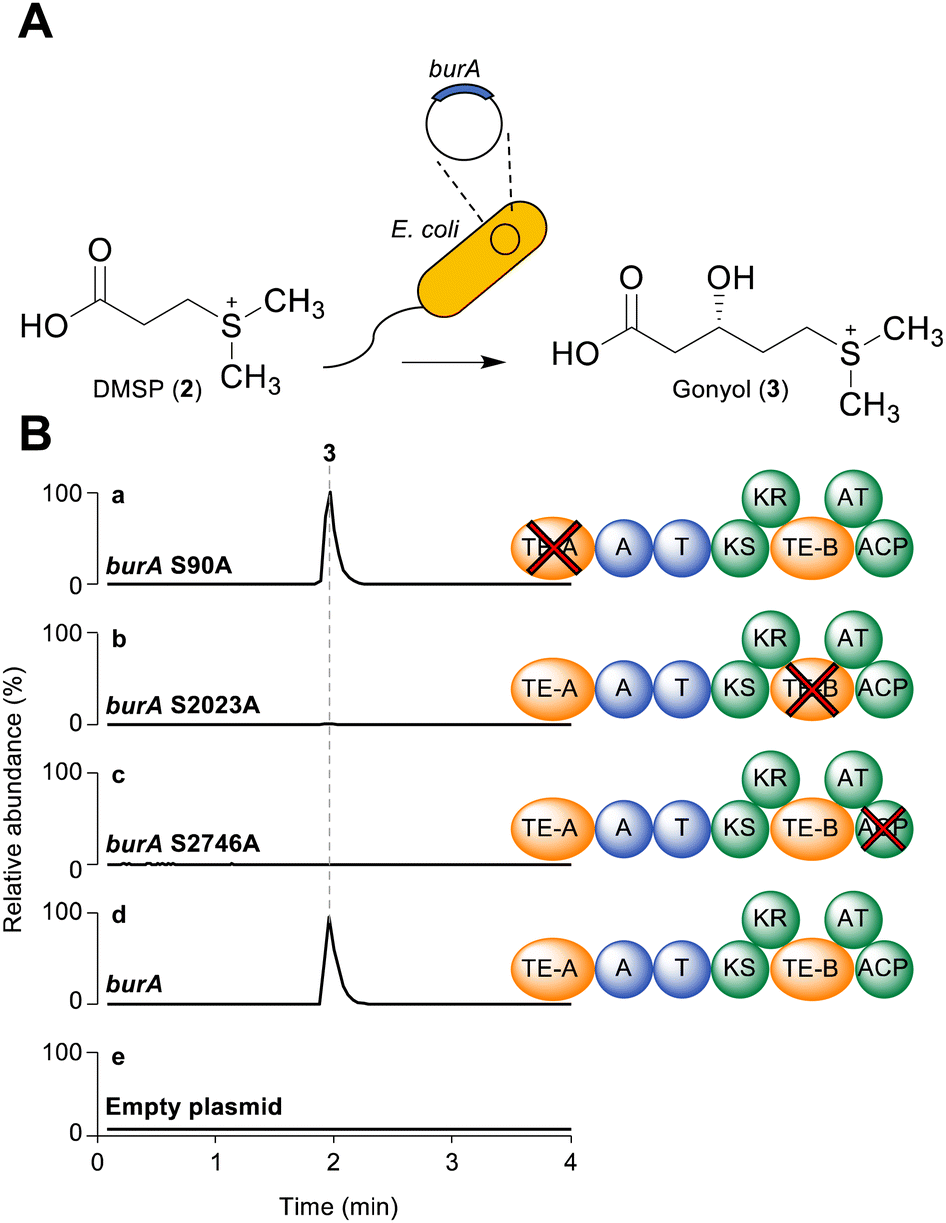 | ||
| Fig. 2 (A) Schematic of assay used to determine the effect of BurA TE-A and BurA TE-B inactivation on gonyol production. E. coli Rosetta cells expressed the full length burA (or a point mutant) from a plasmid and were grown with added DMSP. The production of gonyol was assessed using LC-HRMS. (B) LC-HRMS extracted ion chromatogram traces of organic extracts isolated from E. coli Rosetta expressing (a) burA S90A (TE-A mutant); (b) burA S2023A; (c) burA S2746A; d. burA wild type (positive control); e. empty plasmid (negative control). All data representative of three independent experiments. The scale of all plots is identical (NL: 7.60 × 106). The m/z of gonyol ion extracted for the chromatogram was 179.0736 in positive ion mode. | ||
In contrast to the burA_TE-A S90A mutant, gonyol production was abolished in the burA TE-B S2023A mutant (Fig. 2B(b, d and e)). BurA TE-B is therefore essential for gonyol biosynthesis, strongly implicating it as the TE domain responsible for catalysing the gonyol release. To validate this, we sought to demonstrate the hydrolytic activity of BurA TE-B in vitro. To achieve this an N-acetyl cysteamine (SNAC) (8) version of gonyol (9) was synthesised to serve as a surrogate for the natural phosphopantetheine-tethered gonyol substrate (Fig S4, S8–S13, ESI†). Next, the gene region corresponding to TE-B of BurA was expressed and the protein fragment purified to homogeneity using metal-affinity chromatography (Fig. S5 and S6, ESI†). Following purification, the TE-B protein and SNAC-gonyol were immediately incubated together (due to the instability of the purified excised TE-B) in the presence of 5,5′-dithiobis-(2-nitrobenzoic acid) (Ellman's reagent), allowing thiol formation to be monitored spectroscopically (Fig. 3A, B and Fig. S7, ESI†).22 The in vitro assay clearly demonstrated that TE-B is able to effectively hydrolyse SNAC-gonyol compared to a TE-B S2023A negative control, consistent with the abolished gonyol production in the burA S2023A mutant. TE-B therefore possesses the hydrolytic activity necessary for releasing gonyol (3) from BurA.
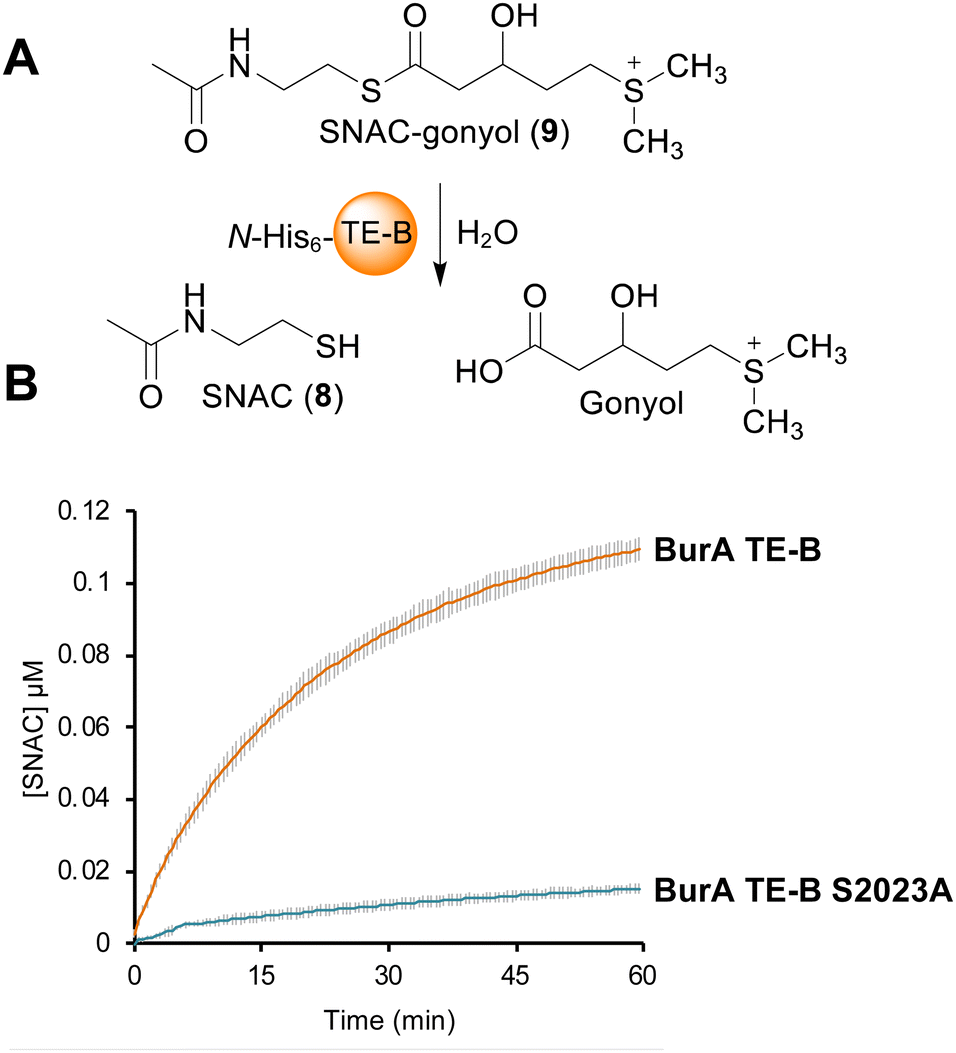 | ||
| Fig. 3 (A) Schematic of assay used to measure the hydrolytic activity of BurA TE-B with racemic SNAC-gonyol. The free thiol of the liberated SNAC was detected using Ellman's reagent. (B) in vitro activity assay using SNAC gonyol and purified BurA TE-B or BurA TE-B S2023A (negative control). The graph depicts the change in 412 nm absorbance (indicative of liberated SNAC) over time. The graph in the figure is representative of three independent experiments, each with three technical repeats. Error bars are ± the standard deviation. | ||
In addition to the unusual placement of TE-B, BurA also contains a C-terminal ACP domain in module 2. While final module C-terminal ACP domains are found in PKS enzymes responsible for polyketide tetronate biosynthesis (where chain release is catalysed by a standalone FabH-like enzyme), they are an uncommon feature when a TE domain is present.3,23 The presence of both the intramodular TE-B and a C-terminal ACP domain within the same module indicated that, for gonyol to enter the active site of TE-B, it must be passed upstream from the ACP, as opposed to the typical forward pass that occurs when the TE domain is located on the C-terminus (Fig. 4B).3,24 To verify this, we mutated the conserved serine residue in the ACP domain (part of a DSL motif)25 that serves as the attachment point for a phosphopantetheine group, generating a burA_ACP S2746A mutant. Gonyol production was completely abolished in this mutant, confirming that it must serve as the attachment point for gonyol prior to release by BurA TE-B (Fig. 2B(c, d and e)). In addition to being a rare example of a non-terminal hydrolytic thioesterase, BurA TE-B therefore also receives its substrate from the opposite direction of canonically placed C-terminal thioesterase domains (Fig. 4A and B).
 | ||
| Fig. 4 (A) The typical mechanism where the TE receives its substrate from an upstream carrier protein domain via a downstream pass. (B) The unusual “upstream pass” mechanism discovered here in BurA-catalysed gonyol biosynthesis. Here, the intramodular TE receives its substrate from a downstream carrier protein domain rather than an upstream one. | ||
Aside from BurA, PKS and NRPS enzymes with multiple TE domains and/or a non-C-terminal thioesterase domain have only been identified in a few other biosynthesis pathways.3 NRPS enzymes containing tandem TE domains on their C-terminus are present in the biosynthesis pathways of several cyclic peptides including teixobactin,26 arthrofactin27 and lysobactin.21,25,28,29 In such cases, the first TE domain of the pair is typically responsible for catalysing peptide release/cyclisation, while the role of the second is variable. In the case of Txo2 from teixobactin biosynthesis, the second TE of the tandem pair cooperates with the first in catalysing chain release.26 In contrast, inactivating the second TE domain in LybB from lysobactin28 biosynthesis had no detectable effect on activity.28 This second TE domain in lysobactin biosynthesis possesses deacetylase activity, consistent with a type II thioesterase-like role in hydrolysing mis-primed PCPs.28 Such a C-terminal type II thioesterase-like domain has also been identified in other pathways, including the PKS CylH from cylindrocyclophane biosynthesis.18–21 In this study we found that BurA TE-A is not essential for gonyol biosynthesis, nor does mutating it affect titres of the bur product burkholderic acid. Such a result indicates that BurA TE-A also has a proofreading function, as the genetic inactivation of type II TEs does not necessarily affect product yields.5 Type II thioesterase-like activity for BurA TE-A also agrees with our sequence similarity network analysis that revealed that BurA TE-A is more similar to type II thioesterases (and type I TE domains that resemble type II thioesterases) than to type I thioesterases. While our results indicate that BurA TE-A is a non-essential type II thioesterase-like domain, the conservation of a TE-A-like domain across all known BurA homologues suggests an important role, and that its activity and function are not yet fully understood. While purified TE-B is shown to hydrolyse SNAC-gonyol, the excised domain clearly lost activity over the course of the assay and is notably slower than other excised TE domains.30 The reasons for this are unclear, but could be due to the intramodular nature of TE-B making it more sensitive to being expressed in isolation compared to the typical C-terminal TE domains. The future characterisation of additional hydrolytic intramodular TE domains will be valuable for assessing whether these observations are specific for BurA TE-B or are more general. Another intramodular TE domain that, while lacking experimental verification, likely catalyses chain release is present in ObiF, an NRPS responsible for the release/lactonisation of the tripeptide obafluorin.31 The ObiF TE domain is located between a T domain and a C-terminal A domain.31 The critical difference with BurA TE-B and ObiF TE (and tandem C-terminal thioesterases) is that the terminal domain of BurA is an ACP, meaning that gonyol is delivered to TE-B from a downstream ACP domain (via an upstream pass) as opposed to the typical downstream pass from an upstream carrier protein domain. While BurA is the first characterised example of this, it also likely occurs in the biosynthesis of the non-ribosomal peptide JBIR-34, where the final NRPS module of FmoA5 contains a C-terminal T domain and two upstream intramodular TE domains (Table S1, ESI†).6 The evolutionary advantages of an intramodular TE domain are unclear at this point, but one possibility is that it helps to shield the active site of the TE from unwanted substrates that could impede metabolic flux.
Conclusions
Here we report that the sulfonium acid gonyol is hydrolytically released from BurA by the intramodular thioesterase domain TE-B. This finding clarifies an important step in the biosynthesis of the virulence-conferring cyclopropanol ring of malleicyprol. In addition, TE-B is the first characterised example of a thioesterase that receives its substrate from a downstream carrier protein domain rather than an upstream one. It is currently unknown if other TE domains can tolerate this alternative method of substrate delivery, or whether BurA TE-B contains special features to facilitate such a mechanism. Regardless, the finding illuminates the previously unappreciated flexibility of TE domain placement within modular PKS/NRPS enzymes. Furthermore, the new knowledge that at least some TE domain can accept their substrate from a downstream ACP offers flexibility in future experiments that aim to create engineered PKS/NRPS assembly lines. Finally, the characterisation of BurA TE-B will serve as the basis for understanding other PKS/NRPS enzymes that contain an intramodular TE domain and a C-terminal carrier protein domain.Experimental
General molecular biology techniques
All PCR amplifications were performed using Q5 high-fidelity DNA polymerase (New England Biolabs). Restriction digestions were performed using enzymes purchased from New England Biolabs according to the manufacturer's instructions. All chemicals were purchased from Merck KGaA (Germany) or TCI (Japan). All DNA sequencing was performed by Genewiz (Germany). The sequences of all oligonucleotide primers used are listed in Table S2 (ESI†). All plasmids used are listed in Table S3 (ESI†). All bacteria are listed in Table S4 (ESI†).LC-HRMS/HPLC analysis
LC-HRMS measurements to detect gonyol were carried out on an UltiMate 3000 UHPLC (Thermo Fisher Scientific) coupled to a Thermo Fisher Scientific QExactive HF-X Hybrid Quadrupole-Orbitrap equipped with an electrospray ion source using a Nucleodur 100-2 C18 column (100 × 2 mm, Macherey-Nagel) and an elution gradient [solvent A: H2O + 0.1% HCOOH, solvent B: CH3CN, 5% B for 0.5 min, from 5% to 100% B in 6.5 min, 100% B for 3 min, 100% B to 5% B in 0.01 min, 5% B for 2.9 min; flow rate: 0.4 mL min−1, injection volume: 2 μL] for chromatographic separation.Sequence similarity network generation
The FASTA sequences of the 100 α/β hydrolase fold thioesterases (including both domains and standalone proteins) (Table S1, ESI†) were collected from GenBank. The domain boundaries of TE domains were determined using the online PKS/NRPS predictor tool.29 A sequence similarity network of the 100 sequences was created using the ESI-EFT online tool.32 The alignment score filter value used was 30. The network was visualised using Cytoscape.33Site-directed mutagenesis of burA
To create the burA S90A mutation (corresponding to the TE-A gene region) the full-length burA gene was mutated using overlap-extension PCR. The upstream fragment was amplified using the PCR primers S90A_OL1_Fw and S90A_OL1_Rv. The downstream fragment was amplified using the primers S90A_OL2_Fw and S90A_OL2_Rv. The two fragments were then seamlessly joined together using overlap extension PCR. The fragment was then inserted into the pHIS8_burA vector between the XbaI and EcoRI cut sites.To create the burA S2023A mutation (corresponding to the TE-B gene region) an upstream fragment was amplified using the PCR primers S2023A_OL1_Fw and S2023A_OL1_Rv while the downstream fragment was amplified using the PCR primers S2023A_OL2_Fw and S2023A_OL2_Rv. The two fragments were ligated between the EcoRI/HindIII sites of pHIS8_burA in a Gibson assembly34 reaction.
To create the burA S2749A mutation (corresponding to the ACP gene region of burA) an upstream fragment was amplified using the PCR primers S2749A_OL1_Fw and S2749A_OL1_Rv while the downstream fragment was amplified using the primers S2749A_OL2_Fw and S2749A_OL2_Rv. The two fragments were then seamlessly joined using overlap extension PCR. The product was then digested using the restriction enzymes HindIII and PaqCI and cloned into pHIS8_burA between the HindIII and PaqCI sites. All cloning reactions were confirmed by Sanger DNA sequencing (Genewiz).
Heterologous expression of burA and burA mutants
E. coli Rosetta cells transformed with a pHIS8_burA plasmid were selected on LB agar supplemented with kanamycin (50 μg mL−1) and chloramphenicol (25 μg mL−1). Overnight cultures derived from a single transformant were inoculated into 50 mL of LB containing kanamycin (50 μg mL−1) and chloramphenicol (25 μg mL−1) and grown to an OD600 of ca. 0.5. The culture was then chilled on ice for 10 minutes, after which DMSP (0.1 mg mL−1) and IPTG (0.5 mM) were added. The culture was then grown at 16 °C with shaking at 120 rpm overnight. The following day the cells were collected by centrifugation and the pellet extracted using methanol. The methanol was evaporated under reduced pressure. The organic residue was redissolved in 1 mL of methanol, filtered, and analysed by LC-HRMS.Creation and analysis of the S90A mutation in B. thailandensis
To create the S90A mutation in the genome of B. thailandensis, the S90A mutation was first introduced into the pGEM_pbur plasmid created in (11). Using the pGEM_pbur plasmid as the DNA template, the upstream fragment was amplified using the primers pGEM_pbur_S90A_Up_Fw and pGEM_pbur_S90A_Up_Rv, and the downstream fragment was amplified using the primers pGEM_pbur_S90A_Dn_Fw and pGEM_pbur_S90A_Dn_Rv. The fragments were joined and cloned into pGEM_pbur at the KpnI and NdeI sites using Gibson assembly,34 creating pGEM_pbur_S90A. B. thailandensis was transformed with pGEM_pbur_S90A using the same method described in (11). The desired integration of the pGEM_pbur_S90A plasmid into the genome of B. thailandensis was confirmed using the primers Tet_rv and II2087rvConf.To assess the effect of the S90A mutation in B. thailandensis, overnight cultures of B. thailandensis Pbur and B. thailandensis Pbur S90A were independently inoculated into 50 mL of LB and grown at 37 °C with shaking at 120 rpm for 16 h. A caffeine internal standard was then added to each culture at a final concentration of 5 mg mL−1. The cultures were then extracted using ethyl acetate, which was then collected and evaporated under reduced pressure. HPLC analysis was then performed to measure the absorbance peak area of burkholderic acid (370 nm) relative to peak area of the caffeine internal standard (270 nm). Peak integration was performed using the software Xcalibur (Thermo Fisher).
Purification of N-terminal His6-tagged BurA TE-B and BurA TE-B S2023A
The domain boundaries of TE-B were predicted using the online PKS/NRPS analysis tool.29 PCR primers were then designed to specifically amplify the gene region corresponding to the TE-B region. To amplify wildtype TE-B, pHIS8_burA was used as the template DNA. To amplify TE-B S90A, pHIS8_burA S90A was used a template. In either case the primers pET28a_TE-B_Fw and pET28a_TE-B_Fw were used to amplify the TE-B gene fragment and introduce a 5′ NdeI site and a downstream HindIII site. The amplified products, along with expression vector pET-28a(+), were independently digested using NdeI and HindIII. The vector and an insert were then joined using DNA ligase (New England Biolabs), making the gene fragments in frame with the N-terminal His6 tag of pET-28a(+) (MGSSHHHHHHSSGLVPRGSH). For the heterologous expression, a pET-28a(+) construct containing a target gene cloned in-frame to the N-terminal His6 tag was transformed into chemically competent E. coli BL21 (λDE3). A single transformant was selected to inoculate a 3 mL overnight culture of LBK (LB containing 50 μg mL−1 kanamycin) medium. The following day the overnight culture was added to 400-1000 mL of LBK medium and grown at 37 °C 200 rpm to an OD600 of 0.5–0.6 before being placed on ice for 10 min. Isopropyl β-D-1-thiogalactopyranoside (IPTG) was then added to a final concentration of 0.5 mM and the cells were incubated at 16 °C with shaking at 200 rpm overnight. The cells were then spun down and frozen at −80 °C until needed. A frozen aliquot of E. coli BL21 (λDE3) cells in which protein had been induced was resuspended in 30–40 mL of protein binding buffer (20 mM TrisCl, 0.5 M NaCl, pH 7.9). For small-scale protein purifications (<400 mL) cell lysis was performed using sonication. The lysed cells were centrifuged at 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ×g, 4 °C for 20 min. The supernatant was run through a column containing 1 mL of charged cobalt-NTA resin (Takara). After the supernatant had been applied to the column, non-specifically bound proteins were washed off the resin using 10 mL of protein binding buffer followed by 6 mL of protein wash buffer (20 mM TrisCl pH 7.9, 0.5 M NaCl, 50 mM imidazole, 10% glycerol). The fusion-protein was eluted from the column in binding buffer containing escalating imidazole concentrations from 100–200 mM. All elution fractions were run on an SDS-PAGE gel and samples containing highly enriched TE-B (the 200 mM and 400 mM fractions) were pooled and concentrated using an Amicon Ultra-15 Centrifugal Filter Unit (Merck Millipore, USA). The buffer was then exchanged to 100 mM potassium phosphate pH 8.0, 1 mM EDTA buffer using a Minitrap G-25 column. Protein concentration was determined Pierce™ 660 nm Protein Assay Reagent (Thermo Scientific) with a BSA standard curve according to instructions. Activity assays were performed on the same day.
000 ×g, 4 °C for 20 min. The supernatant was run through a column containing 1 mL of charged cobalt-NTA resin (Takara). After the supernatant had been applied to the column, non-specifically bound proteins were washed off the resin using 10 mL of protein binding buffer followed by 6 mL of protein wash buffer (20 mM TrisCl pH 7.9, 0.5 M NaCl, 50 mM imidazole, 10% glycerol). The fusion-protein was eluted from the column in binding buffer containing escalating imidazole concentrations from 100–200 mM. All elution fractions were run on an SDS-PAGE gel and samples containing highly enriched TE-B (the 200 mM and 400 mM fractions) were pooled and concentrated using an Amicon Ultra-15 Centrifugal Filter Unit (Merck Millipore, USA). The buffer was then exchanged to 100 mM potassium phosphate pH 8.0, 1 mM EDTA buffer using a Minitrap G-25 column. Protein concentration was determined Pierce™ 660 nm Protein Assay Reagent (Thermo Scientific) with a BSA standard curve according to instructions. Activity assays were performed on the same day.
Protein melting point analysis
The thermal stability of purified TE-B and TE-B S2023A was determined by using a Tycho NT.6 (NanoTemper Technologies) monitoring the fluorescence of each protein preparation at 330 nm and 350 nm during thermal escalation. Inflection temperatures were obtained at a concentration of 20 μM enzyme in 100 mM potassium phosphate pH 8.0, 1 mM EDTA buffer.In vitro activity of purified TE-B using Ellman's reagent
Ellman's reagent (5,5′-dithio-bis-[2-nitrobenzoic acid) was used to assay the activity of the purified TE-B domains. Reactions were performed in 100 μL volumes in a 96 well plate in triplicate. For each reaction, 1 μM of purified TE-B or TE-B S2023A was incubated with 500 μM of SNAC-gonyol and 10 μM Ellman's reagent in 100 mM potassium phosphate pH 8.0, 1 mM EDTA buffer. Enzyme was added last to start the reaction. The reaction was monitored using a Varioskan Lux plate reader (Thermo Fisher Scientific). The formation of free thiol groups produced by SNAC-gonyol hydrolysis was monitored by measuring the absorbance at 412 nm. A SNAC standard curve was used to convert the absorbance values recoded during the assay into SNAC concentration (μM). Data from three independent protein preparations of was BurA TE-B and BurA TE-B S2023A was collected.Creation and analysis of the S90A mutation in B. thailandensis
To create the S90A mutation in the genome of B. thailandensis, the S90A mutation was first introduced into the pGEM_pbur plasmid created in (11). Using the pGEM_pbur plasmid as the DNA template, the upstream fragment was amplified using the primers pGEM_pbur_S90A_Up_Fw and pGEM_pbur_S90A_Up_Rv, and the downstream fragment was amplified using the primers pGEM_pbur_S90A_Dn_Fw and pGEM_pbur_S90A_Dn_Rv. The fragments were joined and cloned into pGEM_pbur at the KpnI and NdeI sites using Gibson assembly,34 creating pGEM_pbur_S90A. B. thailandensis was transformed with pGEM_pbur_S90A using the same method described in (11). The desired integration of the pGEM_pbur_S90A plasmid into the genome of B. thailandensis was confirmed using the primers Tet_rv and II2087rvConf.To assess the effect of the S90A mutation in B. thailandensis, overnight cultures of B. thailandensis Pbur and B. thailandensis Pbur S90A were independently inoculated into 50 mL of LB and grown at 37 °C with shaking at 120 rpm for 16 h. A caffeine internal standard was then added to each culture at a final concentration of 5 mg mL−1. The cultures were then extracted using ethyl acetate, which was then collected and evaporated under reduced pressure. HPLC analysis was then performed to measure the absorbance peak area of burkholderic acid (370 nm) relative to the absorbance peak area of the caffeine internal standard (270 nm). Peak integration was performed using the software Xcalibur (Thermo Fisher).
Synthetic schemes
Synthesis of racemic gonyol SNAC thioester
The racemic (2-acetamidoethyl) 3-hydroxy-5-(methylthio)pentanethioate (1.9 mg; 7.2 μmol; 1 eq.) was dissolved in 200 μL of acetone. Next, 48 μL of a 160.6 mM solution of iodomethane in acetone (7.7 μmol; 1.1 eq.) was added and the resulting mixture stirred at ambient temperature. A reaction control performed via LC-HRMS after 16 h indicated incomplete turnover. Hence, a further 2.2 eq. of MeI were added. After a second period of stirring for 16 h at ambient temperature complete turnover was still not observed. Consequently, 8 μL of pure MeI (18.3 mg; 153 μmol; 21.4 eq.) was added, the mixture was heated to 40 °C and stirred for an additional 20 h, after which 15 μL of pure MeI (34.2 mg; 289 μmol; 40.4 eq.) was added, followed by stirring at 40 °C for 16 h. Following a final addition of 10 μL of pure MeI (22.8 mg; 191 μmol; 25 eq.) and stirring for 16 h at 40 °C, the reaction mixture was concentrated under reduced pressure and subjected to purification via preparative HPLC using a Nucleodur C18 PolarTec column (5 μm, 250 × 10 mm, Macherey-Nagel) [solvent A: H2O + 0.1% TFA, solvent B: acetonitrile 83%, isocratic condition with 100% A, flow rate: 4 mL min−1]. Through this, 1.93 mg (69%) of the title compound (as a racemic TFA salt) was obtained in the form of a colourless oil. During NMR analysis, we noted unusually broad and split signals in the 13C NMR spectrum (see Fig. S12, ESI†), which we attributed to different rotomers; split signals are denoted with an asterisk (*) in the list below. 1H NMR (600 MHz, CD3CN): δ 6.76 (br. s, 1H), 4.16 (m, 1H), 3.34 (m, 2H), 3.31 (m, 2H), 2.96 (m, 2H), 2.80 (s, 3H), 2.78 (s, 3H), 2.74 (m, 2H), 2.01 (m, 1H), 1.89 (m, 1H), 1.84 (s, 3H). 13C NMR (151 MHz, CD3CN): δ 198.0, 171.2, 67.7*, 51.9*, 41.9*, 39.1*, 31.4*, 29.8*, 26.1, 25.7, 23.0. HRMS: Calculated for C11H22NO3S2, ([M]+): 280.1036; found, 280.1031. See Fig. S4, ESI† for synthetic scheme.NMR Spectroscopy
NMR spectra were measured on a Bruker Avance III 500 MHz or a Bruker Avance III 600 MHz spectrometer (600 MHz with cryo probe) in CD3CN or CD2Cl2. The spectra were referenced relative to the residual solvent peak (CD3CN: δH = 1.94, δC = 1.3; 118.3 ppm; CD2Cl2: δH = 5.32, δC = 53.5 ppm).Author contributions
Rory Little: conceptualization, formal analysis, investigation, methodology, project administration, visualisation, supervision, validation, writing – original draft, writing – review & editing; Felix Trottmann: investigation, formal analysis, visualisation, writing – original draft, writing – review & editing; Miriam Preissler: investigation, formal analysis, visualisation, writing – review & editing; Christian Hertweck: conceptualization, supervision, resources, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Leon Katzengruber for his help with cloning BurA TE-B. Many thanks to the Alexander von Humboldt foundation for Research Fellowship for Postdoctoral Researchers awarded to R. Little. We thank F. Kloss for provision of a Tycho NT.6 for protein melting point measurements and Andrea Perner for mass spectrometer operation.References
- J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380–416 RSC.
- R. D. Süssmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56, 3770–3821 CrossRef PubMed.
- R. F. Little and C. Hertweck, Nat. Prod. Rep., 2022, 39, 163–205 RSC.
- B. M. Harvey, H. Hong, M. A. Jones, Z. A. Hughes-Thomas, R. M. Goss, M. L. Heathcote, V. M. Bolanos-Garcia, W. Kroutil, J. Staunton, P. F. Leadlay and J. B. Spencer, ChemBioChem, 2006, 7, 1435–1442 CrossRef CAS PubMed.
- M. Kotowska and K. Pawlik, Appl. Environ. Microbiol., 2014, 98, 7735–7746 CAS.
- A. Muliandi, Y. Katsuyama, K. Sone, M. Izumikawa, T. Moriya, J. Hashimoto, I. Kozone, M. Takagi, K. Shin-ya and Y. Ohnishi, Chem. Biol., 2014, 21, 923–934 CrossRef CAS PubMed.
- H.-Y. He, M.-C. Tang, F. Zhang and G.-L. Tang, J. Am. Chem. Soc., 2014, 136, 4488–4491 CrossRef CAS PubMed.
- X. Liu, S. Biswas, M. G. Berg, C. M. Antapli, F. Xie, Q. Wang, M. C. Tang, G. L. Tang, L. Zhang, G. Dreyfuss and Y. Q. Cheng, J. Nat. Prod., 2013, 76, 685–693 CrossRef CAS PubMed.
- M. Rust, E. J. N. Helfrich, M. F. Freeman, P. Nanudorn, C. M. Field, C. Rückert, T. Kündig, M. J. Page, V. L. Webb, J. Kalinowski, S. Sunagawa and J. Piel, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 9508–9518 CrossRef CAS PubMed.
- M. A. Storey, S. K. Andreassend, J. Bracegirdle, A. Brown, R. A. Keyzers, D. F. Ackerley, P. T. Northcote and J. G. Owen, mBio, 2020, 11 Search PubMed.
- J. Franke, K. Ishida and C. Hertweck, Angew. Chem., Int. Ed., 2012, 51, 11611–11615 CrossRef CAS PubMed.
- C. Liao and F. P. Seebeck, Angew. Chem., Int. Ed., 2019, 58, 3553–3556 CrossRef CAS PubMed.
- F. Trottmann, K. Ishida, J. Franke, A. Stanišić, M. Ishida-Ito, H. Kries, G. Pohnert and C. Hertweck, Angew. Chem., Int. Ed., 2020, 59, 13511–13515 CrossRef CAS PubMed.
- F. Trottmann, J. Franke, I. Richter, K. Ishida, M. Cyrulies, H.-M. Dahse, L. Regestein and C. Hertweck, Angew. Chem., Int. Ed., 2019, 58, 14129–14133 CrossRef CAS PubMed.
- J. B. Biggins, M. A. Ternei and S. F. Brady, J. Am. Chem. Soc., 2012, 134, 13192–13195 CrossRef CAS PubMed.
- F. Trottmann, K. Ishida, M. Ishida-Ito, H. Kries, M. Groll and C. Hertweck, Nat. Chem., 2022 DOI:10.1038/s41557-022-01005-z.
- C. A. Shaw-Reid, N. L. Kelleher, H. C. Losey, A. M. Gehring, C. Berg and C. T. Walsh, Chem. Biol., 1999, 6, 385–400 CrossRef CAS PubMed.
- H. Nakamura, J. X. Wang and E. P. Balskus, Chem. Sci., 2015, 6, 3816–3822 RSC.
- B. Julien, Z.-Q. Tian, R. Reid and C. D. Reeves, Chem. Biol., 2006, 13, 1277–1286 CrossRef CAS PubMed.
- T. Ahrendt, M. Miltenberger, I. Haneburger, F. Kirchner, M. Kronenwerth, A. O. Brachmann, H. Hilbi and H. B. Bode, ChemBioChem, 2013, 14, 1415–1418 CrossRef CAS PubMed.
- K. Buntin, K. J. Weissman and R. Müller, ChemBioChem, 2010, 11, 1137–1146 CrossRef CAS PubMed.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS PubMed.
- L. Vieweg, S. Reichau, R. Schobert, P. F. Leadlay and R. D. Süssmuth, Nat. Prod. Rep., 2014, 31, 1554–1584 RSC.
- M. E. Horsman, T. P. A. Hari and C. N. Boddy, Nat. Prod. Rep., 2016, 33, 183–202 RSC.
- A. T. Keatinge-Clay, Nat. Prod. Rep., 2012, 29, 1050–1073 RSC.
- D. Mandalapu, X. Ji, J. Chen, C. Guo, W.-Q. Liu, W. Ding, J. Zhou and Q. Zhang, J. Org. Chem., 2018, 83, 7271–7275 CrossRef CAS PubMed.
- N. Roongsawang, K. Washio and M. Morikawa, ChemBioChem, 2007, 8, 501–512 CrossRef CAS PubMed.
- J. Hou, L. Robbel and M. A. Marahiel, Chem. Biol., 2011, 18, 655–664 CrossRef CAS PubMed.
- B. O. Bachmann and J. Ravel, Methods in Enzymology, Academic Press, 2009, vol. 458, pp. 181–217 Search PubMed.
- R. S. Gokhale, D. Hunziker, D. E. Cane and C. Khosla, Chem. Biol., 1999, 6, 117–125 CrossRef CAS PubMed.
- J. E. Schaffer, M. R. Reck, N. K. Prasad and T. A. Wencewicz, Nat. Chem. Biol., 2017, 13, 737–744 CrossRef CAS PubMed.
- R. Zallot, N. Oberg and J. A. Gerlt, Biochemistry, 2019, 58, 4169–4182 CrossRef CAS PubMed.
- P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, N. Amin, B. Schwikowski and T. Ideker, Genome Res., 2003, 13, 2498–2504 CrossRef CAS PubMed.
- D. G. Gibson, L. Young, R. Y. Chuang, J. C. Venter, C. A. Hutchison, 3rd and H. O. Smith, Nat. Methods, 2009, 6, 343–345 CrossRef CAS PubMed.
- B. Gebser and G. Pohnert, Mar. Drugs, 2013, 11 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cb00121g |
| This journal is © The Royal Society of Chemistry 2022 |