
Deep-blue-emitting nanoaggregates from carbazole-based dyes in water†
Xiaohui
Li‡
,
Qi
Zhang‡
and
Xin
Zhang
 *
*
School of Chemical Engineering and Technology, Collaborative Innovation Center of Chemistry Science and Engineering, Tianjin University, Tianjin, 300072, China. E-mail: xzhangchem@tju.edu.cn
First published on 29th November 2021
Abstract
New amphiphilic carbazole-based dyes assemble in water into deep-blue-emitting, highly fluorescent helical aggregates as observed by transmission electron microscopy and atomic force microscopy. Single crystal X-ray diffraction and NMR spectroscopy reveal that self-complementary, antiparallel H-bonding and π–π stacking interactions are the driving forces for the formation of these dye aggregates.
Fluorescent dye aggregates have recently attracted much interest due to their great application potential in fluorescent probes and sensors,1 artificial photosynthesis systems,2 light-emitting diodes,3 and optoelectrical devices.4 The optical properties of dye aggregates are closely related to the dye chromophore arrangements within the aggregates. In natural photosynthesis systems, the bacteriochlorophyll dyes organize into a tubular aggregate consisting of 22 circularly arranged dye stacks, which demonstrates highly efficient light-harvesting properties.5 These miraculously sophisticated nanoaggregates in Nature inspire chemists to create synthetic dye aggregates with unique optical properties. Deep-blue-emitting dye materials are most valuable in practical applications.6–8 Carbazole chromophores are strongly hydrophobic; thus, studies on carbazole-based dyes in water are considerably scarce. In this work, we reported the synthesis of two new amphiphilic carbazole-based dyes by Michael addition “click” reactions, and investigated their optical and assembly behaviors in aqueous solution. In particular, we demonstrate unprecedented, deep-blue-emitting dye aggregates with exceptional nanostructures in water.
Two new hydrophilic chain-modified, carbazole-based dyes 1 and 2 were synthesized by a 7-step reaction (Fig. 1). Hydrophilic chains were synthesized according to the literature9 with some modifications. In the last synthetic step, N-(4-phenylcarbazole) maleimide was reacted with a hydrophilic amine chain at 0 °C, which is a Michael addition “click” reaction. The precursor N-(4-phenylcarbazole) maleimide is nonfluorescent due to the quenching effect of maleimide double bonds, and the adduct product is highly fluorescent. Thus, this “click” reaction is an interesting fluorescence “turn on” reaction. The chemical structures of 1 and 2 were confirmed and fully characterized by 1H and 13C NMR spectroscopy, and high resolution mass spectrometry (Fig. S1–S4, ESI†).
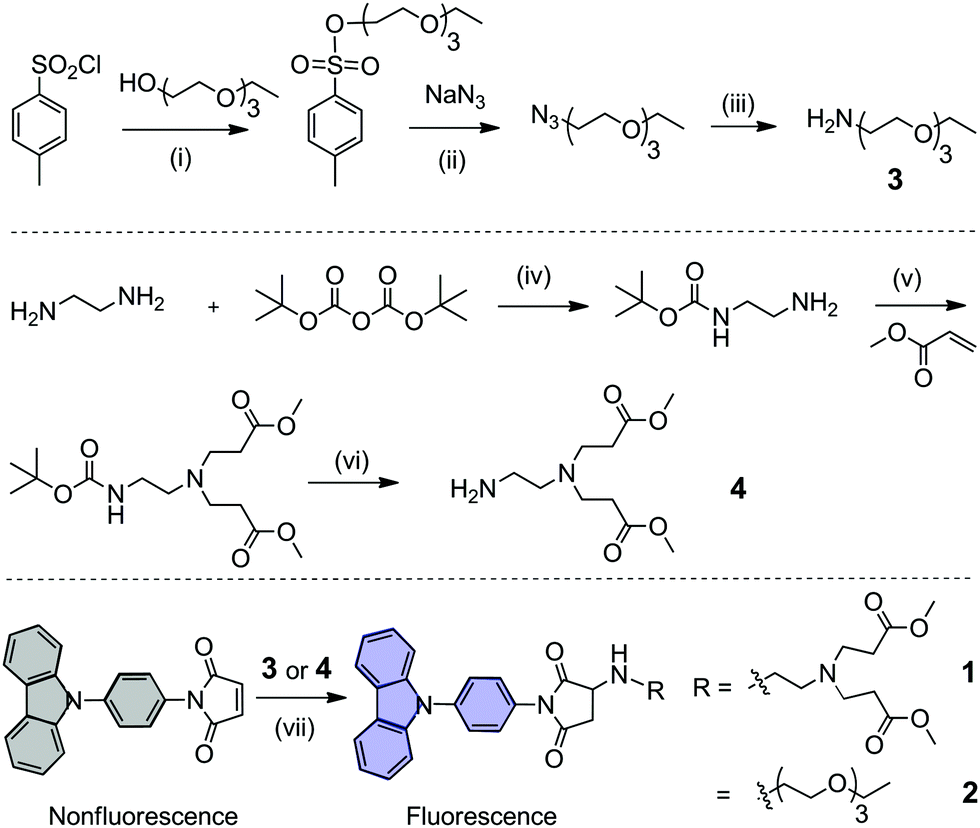 | ||
| Fig. 1 Synthetic routes towards carbazole-based dyes 1 and 2. Reagents and conditions, yield: (i) 0 °C, triethylamine, 25 °C, and 5 h, 85.1%; (ii) methanol, H2O, 80 °C, and 12 h, 60.1%; (iii) triphenylphosphine, THF, 25 °C, 5 h, H2O, 80 °C, and 12 h, 99.1%; (iv) N2, 0 °C, DCM, 25 °C, and 12 h, 65.1%; (v) N2, methanol, 25 °C, and 48 h, 50.1%; (vi) N2, trifluoroacetate, DCM, 25 °C, and 2 h, 35.5%; (vii) N2, 0 °C, DCM, triethylamine, and 1 h, 43.9% for dye 1; N2, 0 °C, DCM, and 2 h, 54.4% for dye 2. | ||
In good solvents such as tetrahydrofuran (THF) and dichloromethane (DCM), dyes 1 and 2 are molecularly dissolved in the monomer state, and they show approximate absorption spectra with the maximum absorption wavelength at 293 nm (Fig. 2), which is attributed to the π–π* electronic transition of the carbazole chromophore.6d Dyes 1 and 2 show high fluorescence at 330–420 nm in tetrahydrofuran (THF) or dichloromethane (DCM). The monomer emission bands of 1 and 2 are narrow, and finely structural with two well-resolved peaks at 346 nm and 360–361 nm (Table 1). After adding deionized water to their THF solutions, 1 and 2 assemble into highly ordered aggregates. The emisssion bands of these aggregates lose their fine structures (Fig. 2), and their emission maximum wavelengths show a slight red shift of 10–16 nm compared with their monomers (Fig. 3). These aggregates in aqueous solution display narrow emission bands and high fluorescence quantum yields of 0.43–0.44 as their monomers (Fig. 3). Generally, the fluorescence quantum yields of aggregates are much lower than those of monomers due to concentration self-quenching, energy migration or other reasons.11,12 In our case, our aggregate fluorescence does not display a large red shift relative to monomer emission, and the emission bands of the aggregates do not become significantly broad. Thus, almost no fluorescence quenching was observed for our dye aggregates in aqueous solution.
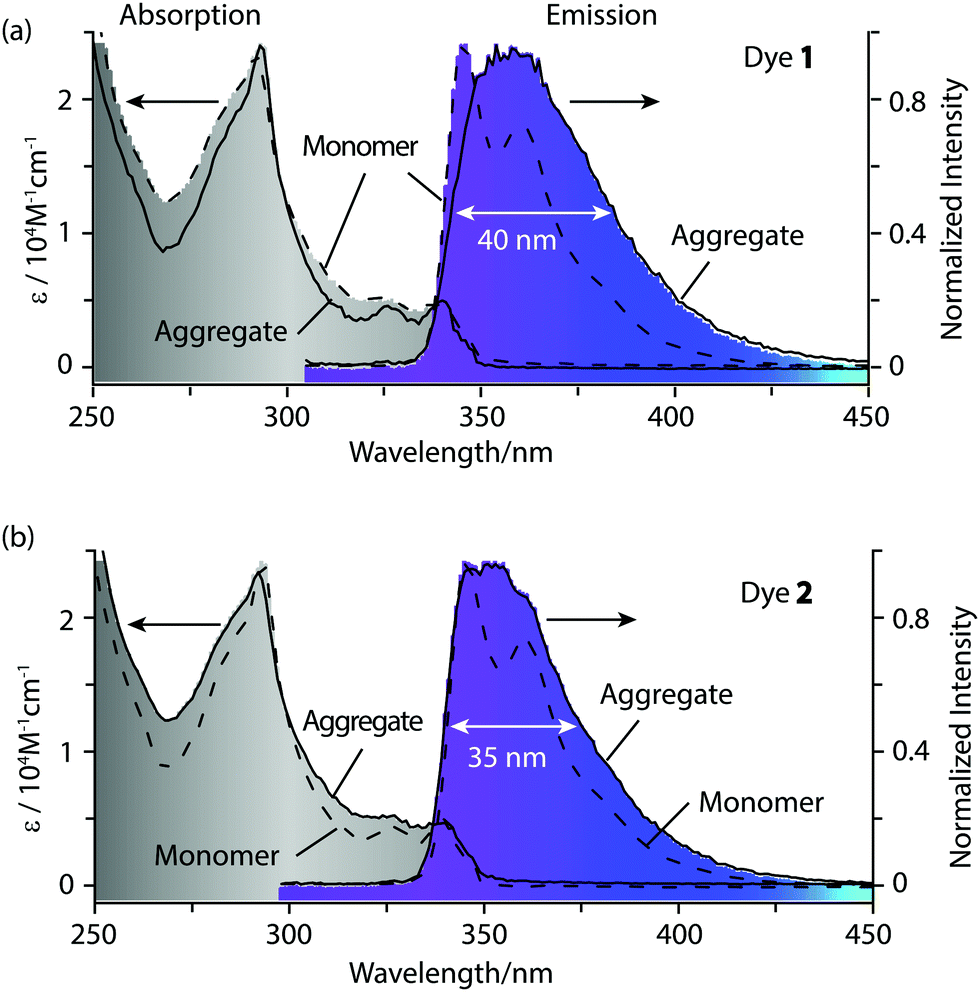 | ||
Fig. 2 UV-vis absorption and fluorescence spectra of monomers (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) -) of 1 (a) and 2 (b) in tetrahydrofuran (THF), and their aggregates (—) in aqueous solution (H2O/THF = 9/1), [1] = [2] =4 × 10−5 M, λex = 295 nm. -) of 1 (a) and 2 (b) in tetrahydrofuran (THF), and their aggregates (—) in aqueous solution (H2O/THF = 9/1), [1] = [2] =4 × 10−5 M, λex = 295 nm. | ||
| Dyes | Absorption | Fluorescence | |||||
|---|---|---|---|---|---|---|---|
| λ A /nm | ε/104 M−1 cm−1 | λ F/nm | τ/ns | Φ | Stokes shift/nm | FWHM/nm | |
| a Relative fluorescence quantum yields were determined by using 9,10-diphenylanthracene as a reference (Φ = 0.90 in cyclohexane).10 | |||||||
| 1 | 292 | 2.34 | 346 | 5.7 | 0.48 | 54 | 27 |
| 2 | 294 | 2.40 | 345 | 5.8 | 0.44 | 51 | 28 |
| Agg. of 1 | 295 | 3.09 | 362 | 6.1 | 0.43 | 67 | 40 |
| Agg. of 2 | 292 | 2.0 | 356 | 5.6 | 0.44 | 64 | 35 |
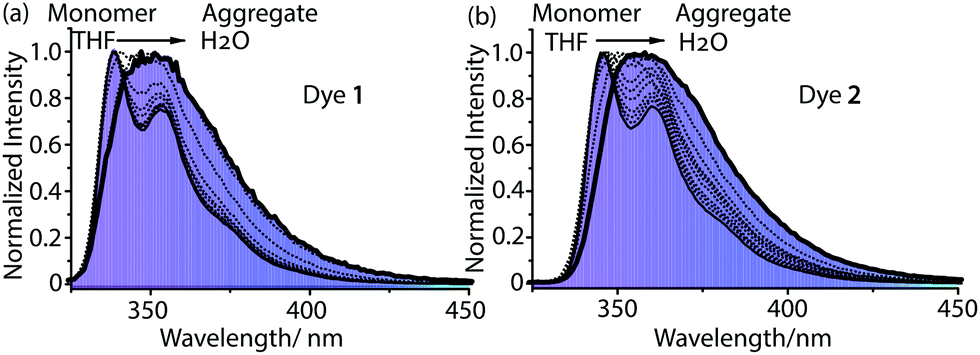 | ||
| Fig. 3 Fluorescence spectra of 1 (a) and 2 (b) in different THF/H2O ratios (THF/H2O = 100/0, 90/10, 80/20, 70/30, 60/40, 50/50, 40/60, 30/70, 20/80, 10/90, and 5/95) from monomers to aggregates, λex = 295 nm; [1] = [2] = 4 × 10−5 M. | ||
We further investigated the formation of these aggregates in aqueous solution by fluorescence spectroscopy. We measured the fluorescence intensity of these aggregates in aqueous solution (H2O/THF = 9/1) at different concentrations, and plotted the fluorescence intensity versus concentration (Fig. S11, ESI†). At low concentrations, the fluorescence intensity of 1 or 2 increases with increasing concentration in aqueous solution, which shows a linear relationship (Fig. S11, ESI† inset). At high concentrations, the plot of fluorescence intensity versus concentration does not display the same slope as at low concentrations due to aggregate formation. The transition points were observed at 1.08 × 10−7 M for 1 and 3.53 × 10−7 M for 2, indicating that dye aggregates start to form at this concentration in H2O/THF (9/1, v/v). The transition points are called the critical aggregation concentrations (CACs).11
The formation process and mechanism of these aggregates were further investigated by NMR spectroscopy. We measured the NMR spectra of the aggregate formation process of dye 1 at various D2O/THF-d8 ratios (Fig. 4). When adding D2O to the THF-d8 solution of 1, the aggregates form gradually. Compared with alkyl protons (Hn), the NMR peak of the active hydrogen (Hi) at the –HN– group shows a relatively large shift from 3.11 ppm to 3.53 ppm (Fig. 4 inset), suggesting that H-bond interaction occurs. The NMR peaks of protons (Ha−f) from aromatic carbazole groups become broad and shift to lower fields, indicating that π–π interaction occurs between carbazole rings. In pure D2O, the NMR peaks of aromatic protons disappear (Fig. 4). These aromatic signals are shielded from magnetic fields due to aggregate formation, where hydrophobic aromatic groups were embedded and surrounded peripherally by hydrophilic chains. These NMR results revealed that the driving forces of aggregate formation in water are H-bonding and π–π stacking interactions (Fig. 4 top).
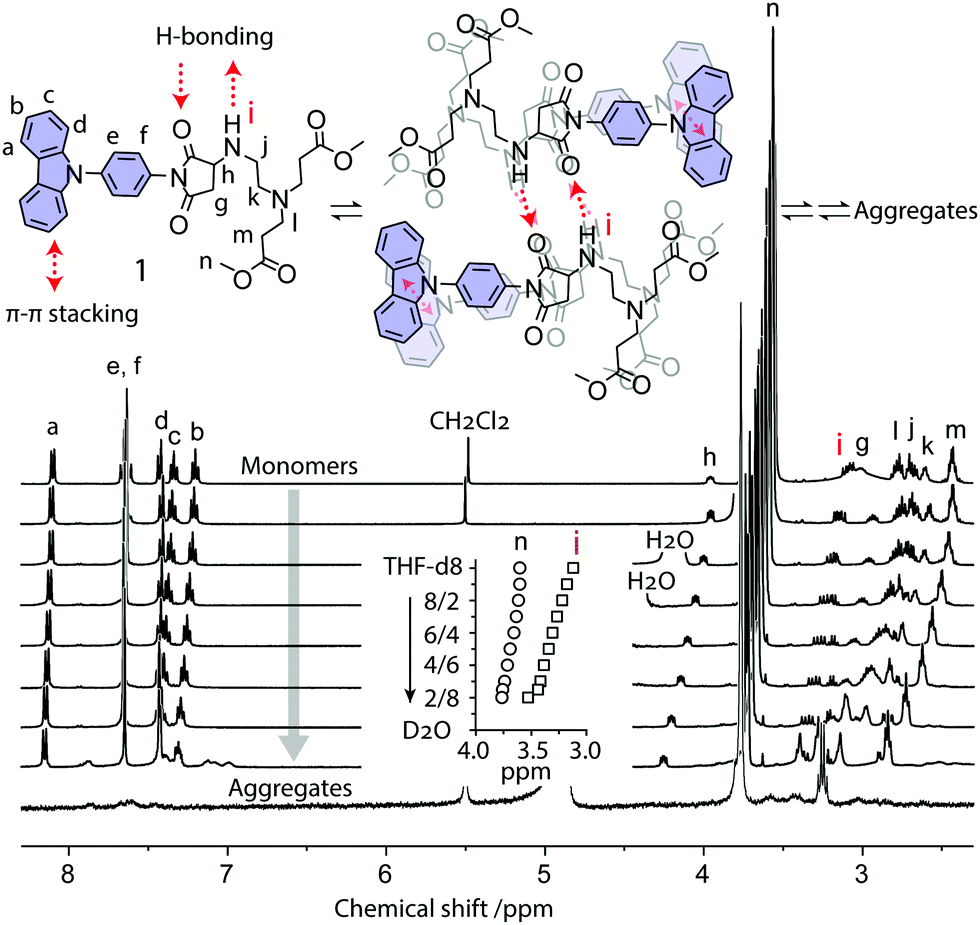 | ||
| Fig. 4 1H NMR spectra of aggregate formation in THF-d8/D2O (100/0, 90/10, 80/20, 70/30, 60/40, 50/50, 40/60, 30/70, and 10/90 from top to bottom), and chemical structures with corresponding NMR signal assignments. Inset shows the changes in chemical shifts of the proton (Hi) of the –NH– group and alkyl protons (Hn) during aggregate formation. [1] = 0.01 M. | ||
The aggregate morphologies in water were directly observed by transmission electron microscopy (Fig. 5a and b) and atomic force microscopy (AFM, Fig. 5c and Fig. S15, ESI†). Dye 1 was first dissolved in THF (0.5 mL). Then, the deionized water (4.5 mL) was added to the above THF solution to induce aggregate formation. These aggregates were then further purified by dialysis against deionized water using a dialysis membrane (MWCO 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) to remove THF and small molecular monomers. The TEM results show that these aggregates are very long, up to several tens of micrometers. Interestingly, highly ordered, helical aggregates were observed with a diameter of 620 nm and helical pitch of 1260 nm (Fig. 5b). The tilted angle of the helix is 35–40° relative to the long axis of the helical aggregates (Fig. 5b). In theory, a small tilt angle cannot form a single-stranded helix, because one helical strand cannot provide sufficient space-filling within one helical pitch.13 This small tilt angle (35–40°) implies that these helical aggregates are not single-stranded, but multi-stranded. Surprisingly, the multi-stranded, helical structures were directly observed by TEM. One magnified TEM image (Fig. 5b) clearly shows that our helical structures are composed of two strands. The double helix can be well interpreted from the formation mechanism of aggregates. Two molecules interact via two complementary H-bonds in an antiparallel manner (Fig. 6b), and the two molecules further form two strands by π–π stacking interactions, which entangle each other to finally form higher-ordered superstructures, i.e., double helical structures. The interesting helical structure was further confirmed by AFM. AFM shows that the helical aggregates have a diameter of 670 nm and helical pitch of 1300 nm, which basically agrees with the TEM results. The TEM and AFM measurements gave direct evidence for helical morphologies. These helical aggregates in water are light-emitting with colour coordinates (0.16, 0.08) (Fig. 5d), which falls into the range of deep-blue (CIE (x, y) < (0.17, 0.09)).6d It should be noted that the main contribution to color coordinates comes from the emission at the wavelengths longer than 360 nm, although the aggregate emission maximum at 350–360 nm. The emission at the wavelengths shorter than 350 nm falls out of visible region, which almost does not contribute to color coordinates.
000) to remove THF and small molecular monomers. The TEM results show that these aggregates are very long, up to several tens of micrometers. Interestingly, highly ordered, helical aggregates were observed with a diameter of 620 nm and helical pitch of 1260 nm (Fig. 5b). The tilted angle of the helix is 35–40° relative to the long axis of the helical aggregates (Fig. 5b). In theory, a small tilt angle cannot form a single-stranded helix, because one helical strand cannot provide sufficient space-filling within one helical pitch.13 This small tilt angle (35–40°) implies that these helical aggregates are not single-stranded, but multi-stranded. Surprisingly, the multi-stranded, helical structures were directly observed by TEM. One magnified TEM image (Fig. 5b) clearly shows that our helical structures are composed of two strands. The double helix can be well interpreted from the formation mechanism of aggregates. Two molecules interact via two complementary H-bonds in an antiparallel manner (Fig. 6b), and the two molecules further form two strands by π–π stacking interactions, which entangle each other to finally form higher-ordered superstructures, i.e., double helical structures. The interesting helical structure was further confirmed by AFM. AFM shows that the helical aggregates have a diameter of 670 nm and helical pitch of 1300 nm, which basically agrees with the TEM results. The TEM and AFM measurements gave direct evidence for helical morphologies. These helical aggregates in water are light-emitting with colour coordinates (0.16, 0.08) (Fig. 5d), which falls into the range of deep-blue (CIE (x, y) < (0.17, 0.09)).6d It should be noted that the main contribution to color coordinates comes from the emission at the wavelengths longer than 360 nm, although the aggregate emission maximum at 350–360 nm. The emission at the wavelengths shorter than 350 nm falls out of visible region, which almost does not contribute to color coordinates.
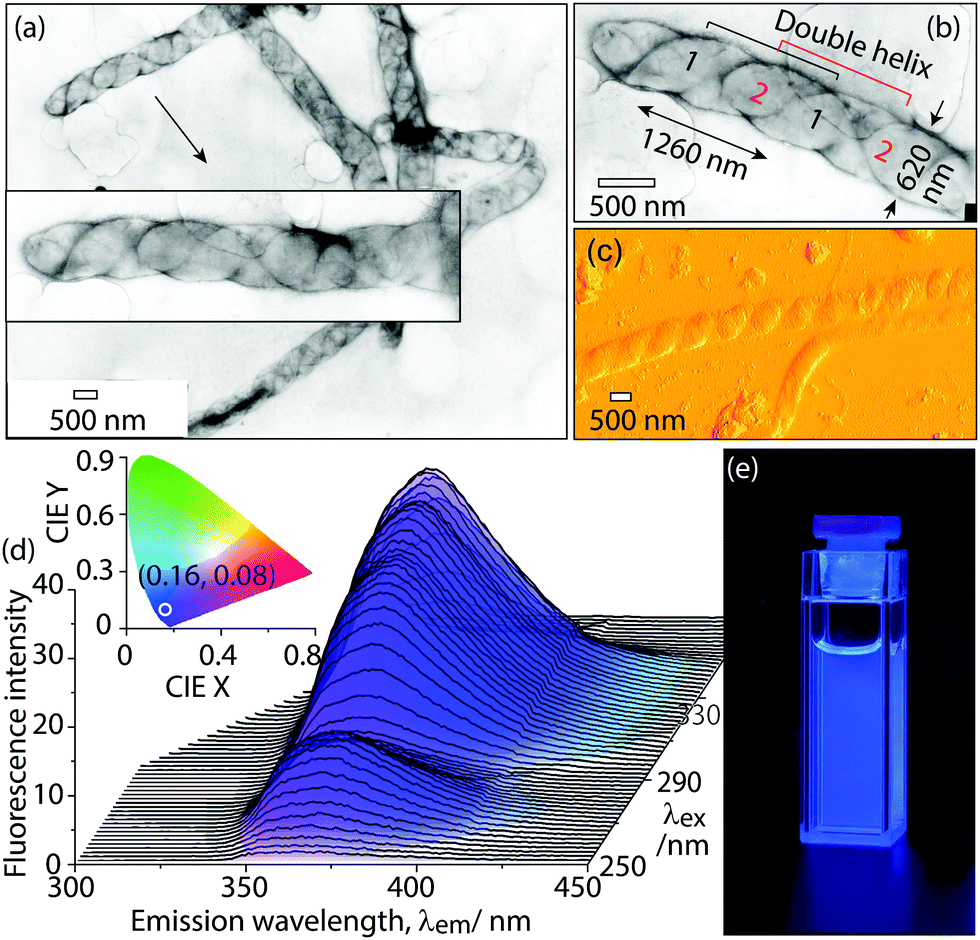 | ||
| Fig. 5 (a) TEM image of aggregates of dye 1 prepared from deionized water, [1] = 1.7 × 10−4 M. (b) Magnified TEM image of a representative double-stranded helical aggregate. [1] = 1.7 × 10−4 M. (c) AFM image of aggregates of 1 prepared from deionized water, [1] = 1.7 × 10−4 M. (d) Excitation-dependent 3D fluorescence spectrum of aggregates of 1 in aqueous solution (H2O/THF = 9/1). Inset: CIE 1931 chromaticity diagram, the circle point is emission colour coordinates (0.16, 0.08) of aggregates, [1] = 2.0 × 10−4 M. (e) Photograph of the aggregate suspension under 365 nm UV lamp. [1] = 2.0 × 10−4 M. | ||
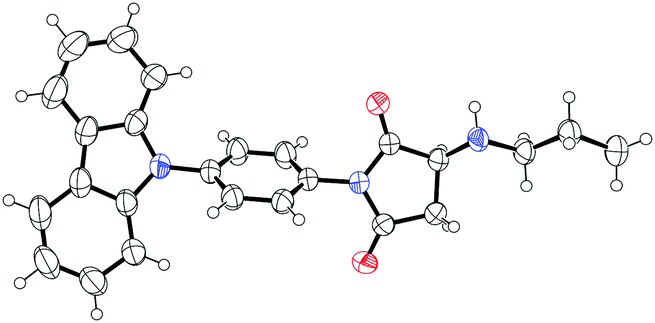
To further confirm the formation mechanism of the helical aggregates, we synthesized a model compound M and prepared its single crystals from a mixed solution (Fig. 6a).14 The single-crystal X-ray diffraction reveals that π–π interaction occurs between carbazole rings with a distance of 3.81 Å, which is slightly longer than general π–π stacking distance (3.5 Å). From the single crystal structures, we observed self-complementary, antiparallel, intermolecular H-bonding interactions between two molecules (Fig. 6a). The first H-bond forms between the H atom of the –HN– group of molecule I, and the carbonyl O atom of the imide group of molecule II. The second H-bond forms in the opposite direction between the carbonyl O atom of the imide group of molecule I, and the H atom of the –HN– group of molecule II. The H-bond length is 2.29 Å, which is shorter than normal H-bond length (2.6–3.3 Å), suggesting that the H-bond interaction is stronger than general H-bonds. From these single crystal X-ray diffraction analyses combined with NMR results (Fig. 4), we concluded that H-bonding and π–π interactions were the driving forces of aggregate formation (Fig. 6b).
 | ||
| Fig. 6 (a) Chemical structures and crystal packing structures of model compound M determined by single-crystal X-ray diffraction. Thermal ellipsoids are drawn at the 50% probability level. (b) Chemical structures and packing structures of dye 1. The red arrows indicate driving forces of assembly: self-complementary antiparallel H-bonding and π–π interactions. | ||
In summary, we synthesized two new carbazole-based dyes by a fluorescence turn on amino-ene “click” reaction. These amphiphilic dyes assemble into deep-blue emitting, helical aggregates with relatively high fluorescence quantum yields of 0.43–0.44 in water. The single-crystal X-ray diffraction and NMR spectroscopy reveal the formation mechanism of these fluorescent dye aggregates. Our results may encourage the development of high performance, optical dye aggregates in environment-friendly water systems.
X. Z. conceived and designed the experiments. X. L. and Q. Z. synthesized the molecules and performed the experiments and wrote the manuscript. X. Z. revised the manuscript.
This research was financially supported by the National Natural Science Foundation of China (Grant No. 21975177 and No. 21674079).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) P. Rajdev and S. Ghosh, J. Phys. Chem. B, 2019, 123, 327–342 CrossRef CAS; (b) A. Sikder, S. Chakraborty, P. Rajdev, P. Dey and S. Ghosh, Acc. Chem. Res., 2021, 54, 2670–2682 CrossRef CAS; (c) A. Mukherjee, D. Sankar Pal, H. Kar and S. Ghosh, Polym. Chem., 2020, 11, 7481–7486 RSC; (d) S. Dou, Y. Wang and X. Zhang, Chem. – Eur. J., 2020, 26, 11503–11510 CrossRef CAS.
- (a) M. A. Fox, Acc. Chem. Res., 2012, 45, 1875–1886 CrossRef CAS; (b) T. Aoki, H. Sakai, K. Ohkubo, T. Sakanoue, T. Takenobu, S. Fukuzumi and T. Hasobe, Chem. Sci., 2015, 6, 1498–1509 RSC; (c) N. R. Reddy, M. Aubin, A. Kushima and J. Fang, J. Phys. Chem. B, 2021, 125, 7911–7918 CrossRef CAS.
- Q. Liang, J. Xu, J. Xue and J. Qiao, Chem. Commun., 2020, 56, 8988–8991 RSC.
- (a) R. Zhao, J. Liu and L. Wang, Acc. Chem. Res., 2020, 53, 1557–1567 CrossRef CAS PubMed; (b) J. Yan, L. X. Zhao, C. Li, Z. Hu, G. F. Zhang, Z. Q. Chen, T. Chen, Z. L. Huang, J. Zhu and M. Q. Zhu, J. Am. Chem. Soc., 2015, 137, 2436–2439 CrossRef CAS PubMed.
- R. Blankenship, Molecular mechanisms of photosynthesis, Wiley, 2002 Search PubMed.
- (a) R. K. Konidena, K. H. Lee and J. Y. Lee, Chem. Commun., 2019, 55, 8178–8181 RSC; (b) R. K. Konidena, W. J. Chung and J. Y. Lee, Org. Lett., 2020, 22, 2786–2790 CrossRef CAS; (c) S. Oda, W. Kumano, T. Hama, R. Kawasumi, K. Yoshiura and T. Hatakeyama, Angew. Chem., Int. Ed., 2021, 60, 2882–2886 CrossRef CAS; (d) D. M. Mayder, C. M. Tonge, G. D. Nguyen, M. V. Tran, G. Tom, G. H. Darwish, R. Gupta, K. Lix, S. Kamal, W. R. Algar, S. A. Burke and Z. M. Hudson, J. Am. Chem. Soc., 2021, 143, 16976–16992 CrossRef CAS PubMed.
- (a) J. V. Grazulevicius, P. Strohriegl, J. Pielichowski and K. Pielichowski, Prog. Polym. Sci., 2003, 28, 1297–1353 CrossRef CAS; (b) X. H. Li, Y. Wang, F. Li and X. Zhang, Dyes Pigm., 2020, 182, 108474 CrossRef CAS; (c) Y. Wang, Q. Zhang, J. B. Gong and X. Zhang, Dyes Pigm., 2020, 172, 107823 CrossRef.
- (a) J. Lia and A. C. Grimsdale, Chem. Soc. Rev., 2010, 39, 2399–2410 RSC; (b) M. Yang, S. Sheykhi, Y. Zhang, C. Milsmann and F. N. Castellano, Chem. Sci., 2021, 12, 9069–9077 RSC.
- (a) J. R. Harjani, C. Liang and P. G. Jessop, J. Org. Chem., 2011, 76, 1683–1691 CrossRef CAS; (b) A. Alliband, F. A. Meece, C. Jayasinghe and D. H. Burns, J. Org. Chem., 2013, 78, 356–362 CrossRef CAS PubMed.
- D. F. Eaton, Pure Appl. Chem., 1988, 60, 1107–1114 CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 2006 Search PubMed.
- N. J. Hestand and F. C. Spano, Acc. Chem. Res., 2017, 50, 341–350 CrossRef CAS PubMed.
- (a) F. Li, X. H. Li, Y. Wang and X. Zhang, Angew. Chem., Int. Ed., 2019, 58, 17994–18002 CrossRef CAS PubMed; (b) S. Sakurai, S. Ohsawa, K. Nagai, K. Okoshi, J. Kumaki and E. Yashima, Angew. Chem., Int. Ed., 2007, 46, 7605–7608 CrossRef CAS PubMed.
- Crystallographic data for dye 1: 0.19 × 0.15 × 0.13 mm, triclinic, a = 4.40 Å, b = 13.5 Å, c = 18.3 Å, α = 71.7, β = 90, γ =80.6°, V = 1015.4 Å, space group P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, ρcalcd = 1.30 g cm−3, λ(MoKα) = 0.71073, T = 273 K, 13347 reflections, 7055 unique [R(int) = 0.084], R1 = 0.0648, wR2 = 0.1983, for 422 parameters and 0 restraints. The X-ray single crystal data were deposited with (CCDC 2111609)†
, Z = 2, ρcalcd = 1.30 g cm−3, λ(MoKα) = 0.71073, T = 273 K, 13347 reflections, 7055 unique [R(int) = 0.084], R1 = 0.0648, wR2 = 0.1983, for 422 parameters and 0 restraints. The X-ray single crystal data were deposited with (CCDC 2111609)†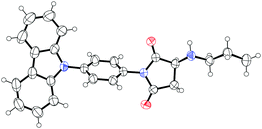 .
.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2111609. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc05441d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |