
Recent advances on non-precious metal-catalyzed C–H functionalization of N-heteroarenes
Atanu
Bera†
,
Lalit Mohan
Kabadwal†
,
Sourajit
Bera
and
Debasis
Banerjee
 *
*
Department of Chemistry, Laboratory of Catalysis and Organic Synthesis, Indian Institute of Technology Roorkee, Roorkee-247667, Uttarakhand, India. E-mail: debasis.banerjee@cy.iitr.ac.in
First published on 18th November 2021
Abstract
N-Heteroarenes are widely used for numerous medicinal applications, lifesaving drugs and show utmost importance as intermediates in chemical synthesis. This feature article highlights the recent advances, from 2015 to August 2021, on sp2 and sp3 C–H bond functionalization reactions of various N-heteroarenes catalyzed by non-precious transition metals (Mn, Co, Fe, Ni, etc.). The salient features of the report are: (i) the development of newer catalysis for Csp2–H activation of N-heteroarenes and categorized into alkylation, alkenylation, borylation, cyanation, and annulation reactions, (ii) recent advances on Csp3–H bond functionalization of N-heteroarenes considering newer approaches for alkylation as well as alkenylation processes, and (iii) synthetic applications and practical utility of the catalytic protocols utilized for late-stage drug development; (iv) scope for the development of newer catalytic protocols along with mechanistic studies and detail mechanistic findings of various important processes.
 Atanu Bera | Atanu Bera was born in West Bengal, Kolkata, in 1993. He obtained his BSc degree in Chemistry (Hons.) from St. Paul's Cathedral Mission College (under University of Calcutta) in 2015, followed by MSc degree in Chemical Science from Pondicherry University in 2017. Currently, he is a PhD student at the Department of Chemistry, Indian Institute of Technology, Roorkee, under the supervision of Prof. Debasis Banerjee. His current research work focuses on non-precious 3d-transition metal-catalyzed based activation of small molecules and photo-redox catalysis for sustainable organic transformations. |
 Lalit Mohan Kabadwal | Lalit Mohan Kabadwal was born in Uttarakhand, India. He obtained his BSc in Chemistry from S. S. Jeena Campus Almora, Kumaun University, Nainital, in 2013, followed by MSc in Chemistry from the same university in 2015. Currently, he is a PhD student at the Department of Chemistry, Indian Institute of Technology, Roorkee, under the supervision of Prof. Debasis Banerjee. His current research work focuses on sustainable organic transformation for C–X bonds (X = C, N, O, etc.) using transition metal catalysis. |
 Sourajit Bera | Sourajit Bera was born in West Bengal, India. He obtained his BSc in Chemistry from Ramakrishna Mission Vidyamandira (affiliated to the University of Calcutta), Belur Math, Howrah, in 2015, followed by MSc from West Bengal State University, Barasat in 2017. Currently, he is a PhD student at the Department of Chemistry, Indian Institute of Technology, Roorkee, under the supervision of Prof. Debasis Banerjee. His current research work focuses on sustainable approaches for the synthesis of small molecules using transition metal catalysis. |
 Debasis Banerjee | Debasis Banerjee graduated with an MSc degree in Organic Chemistry from Banaras Hindu University and obtained PhD in organic chemistry from the Indian Institute of Technology Kanpur in 2011. Thereafter he moved to Leibniz Institute for Catalysis (LIKAT), Germany as Leibniz postdoctoral fellow with Prof. Matthias Beller (2011–2014) and subsequently held another postdoctoral position (2014–2015) at Stockholm University, Sweden with Prof. Jan-Erling Bäckvall. Currently, he is an Associate Professor at the Indian Institute of Technology Roorkee. His research interests include sustainable catalysis, C–H bond functionalization, and enantioselective catalysis. He is a recipient of the DAE-Young Scientist Research Award (2016) and the Thieme Chemistry Journals Award (2020). Presently, he is serving as a Guest Editor in Tetrahedron and Tetrahedron Letters. |
1. Introduction
Transition metal-catalyzed C–H functionalization has emerged as an alternative strategy to the traditional organic transformations. Though conventional organic synthesis is highly powerful, it often relies on the pre-functionalization of substrates and generates stoichiometric waste. Thus, C–H bond functionalization has been recognized as a powerful tool to streamline organic synthesis due to its inherent merits of the atom- and step-economy and is widely employed in the synthesis of drugs, natural products, functional materials, and fine industrial chemicals.1,2Nitrogen-based heterocyclic compounds are privileged structure motifs and have received significant attention due to their existence in numerous naturally occurring bioactive molecules, drugs, natural products (alkaloids, flavonoids), vitamins, and co-enzyme, etc. (Fig. 1A).1–17 Therefore, transition metal catalyzed C–H bond activation of N-heteroarenes has become one of the most popular strategies due to its atom and resource economy;3,18–28 However, their inertness renders their functionalization very difficult.5
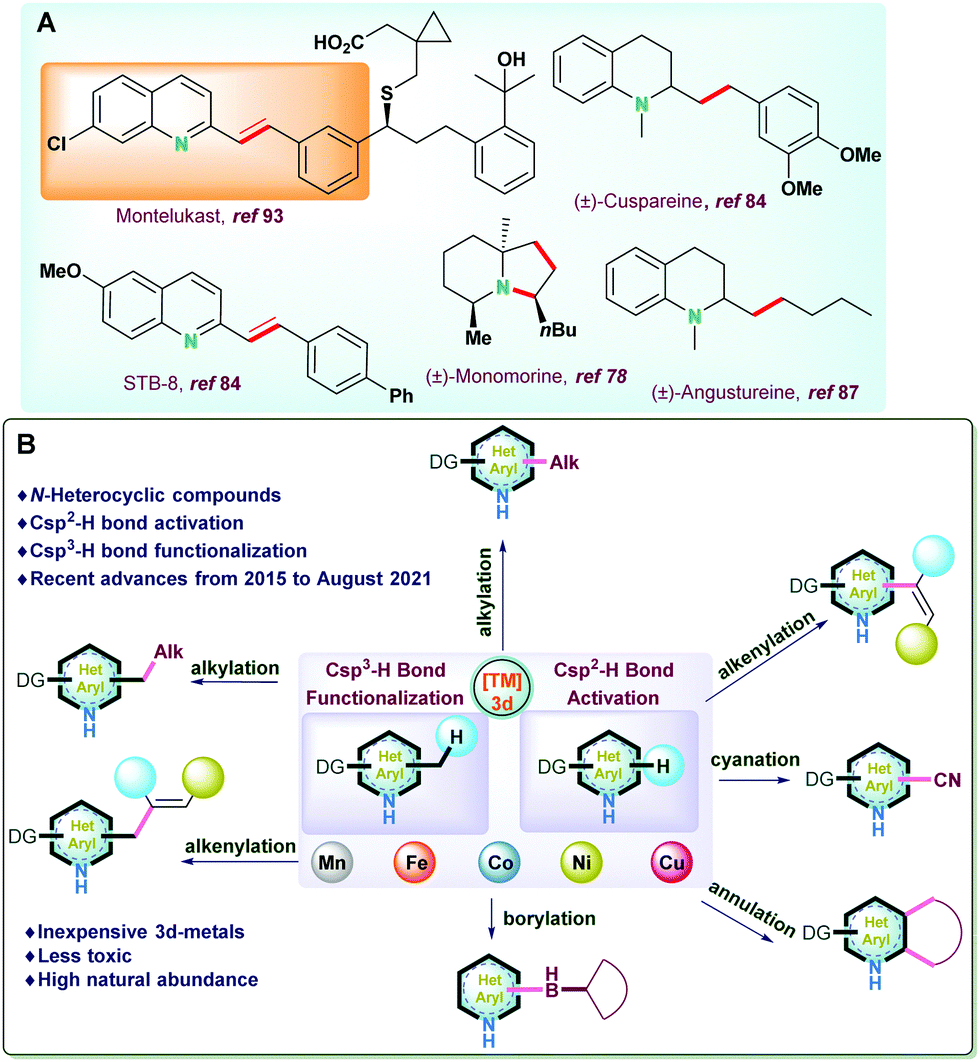 | ||
| Fig. 1 (A) Selected examples of natural products and biologically active molecules synthesized via C–H bond functionalization. (B) Scope of the present review. | ||
Predominant accomplishments have been reported in this direction, mostly by noble metal based catalysts.5,29,30 Due to its poor natural abundance, toxicity issue, and expensive nature raise severe concern. Therefore, the application of non-precious 3d transition metal catalysts encourage significant attention due to high natural abundance, low cost, and have the potential to explore many important chemical transformations.8,9
Despite the recent advances in C–H bond functionalization using 3d-metal catalysts, there are some challenges.10,11 Such 3d-metal complexes have low reactivity and moderate stability in any metal–organic intermediate, which inherently controls the selectivity and undesired side products.12 Although 3d transition metals have some drawbacks, they can be easily overcome by using suitable ligand and additive combinations.13
Over the past few decades, the regioselective C–H bond activation or functionalization of N-heteroarenes using 3d metal catalysts has significantly increased because of their synthetic and industrial importance. Several important reviews have been reported in this direction, mainly based on 4d and 5d transition metals.5,29,30 A few reviews were also dedicated to specific 3d-metals, namely Mn,3 Fe,18 Ni,24–27 and Co.19–23 For instance, in 2019, Cramer and co-workers, reported enantioselective C–H bond functionalization by 3d transition-metal catalysts.28 In 2020, Ackermann and co-workers documented recent progress in Co-catalyzed oxidative C–H bond activation reactions.20 Recently, Anilkumar and co-workers reported C–H bond activation reactions through Mn-based catalysts.3 However, all these above mentioned reviews are mainly based on C–H activation by precious metal catalysts or are either very specific to a particular metal-based catalyst among Mn, Co, Fe, or Ni. To the best of our knowledge to date, there is no such comprehensive review known for the 3d-transition metal-catalyzed functionalization of sp2 and sp3 C–H bonds of N-heteroarenes.
In the current review, we have tried to accommodate the most recent advances (from 2015 to August 2021) related to sp2 and sp3 C–H bond activation and functionalization reactions of various N-heteroarenes catalyzed by non-precious transition metals (Mn, Co, Fe, Ni, etc.). The salient features of the report are (i) catalytic development based on Csp2–H activation of N-heteroarenes and categorized into alkylation, alkenylation, borylation, cyanation, and annulation reactions; (ii) recent advances on Csp3–H bond activation and functionalization of N-heteroarenes considering newer approaches for alkylation as well as alkenylation processes; (iii) synthetic applications and practical utility of the catalytic protocols utilized for late-stage drug development; (iv) scope for the development newer catalytic protocols along with mechanistic studies and detailed mechanistic findings of various important protocols (Fig. 1B).
2. Non-precious metal-catalyzed sp2 C–H bond functionalization for N-heteroarenes
Selective functionalization of C–H bonds of N-heteroarenes attracted significant attention in organic chemistry to install appended functionalities having potential importance. Though noble metal catalyzed sp2 C–H functionalization has been well explored since last decades, the toxic nature and expensive catalyst system are often the associated issues for bulk scale organic transformations. Therefore, the application of cost-efficient and earth abundant metal catalysts for such applications with comparable efficiency and selectivity are the future focus. Hence, in this feature article, we have summarized the recent advances on the various 3d-transition metal catalyzed approaches for selective functionalization of the sp2 C–H bond of N-heteroarenes.2.1 Alkylation via C(sp2)–H bond activation
This part of the review highlights the recent advances in the alkylation of various types of sp2 C–H bond activation of N-heterocycles. Detailed mechanistic aspects have been considered along with different prospects of activation modes for such transformations.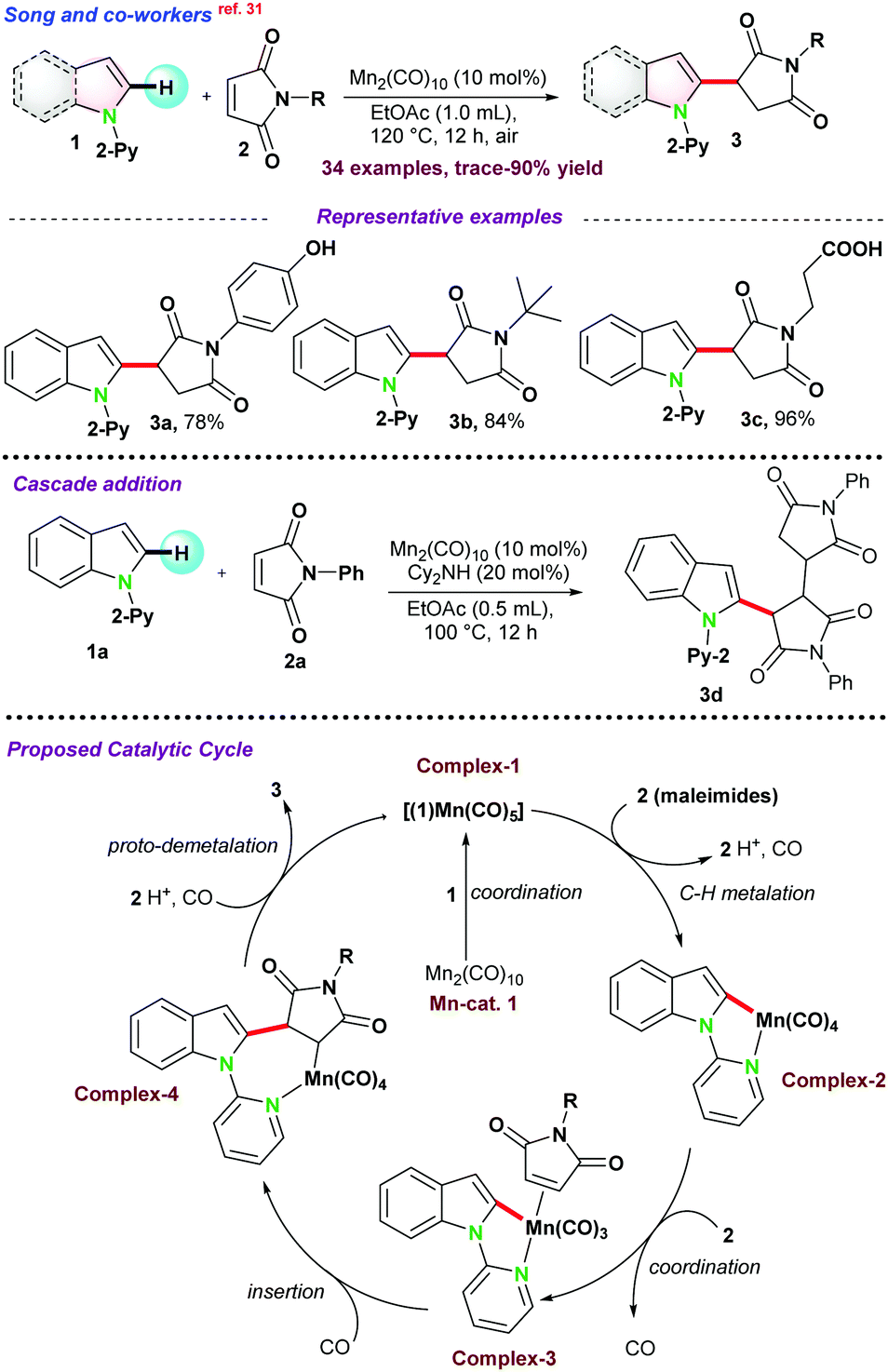 | ||
| Scheme 1 Mn-catalyzed C(sp2)–H activation reaction of 1-(pyridine-2-yl)-1H-indole. | ||
In the same year, Glorious and co-workers reported C-2 alkylation of 1via activation of C(sp2)–H bond using manganese pre-catalysts in assistance with pyridine as the directing group. The catalytic protocol resulted in a series of allyl alcohols, allylated arenes, functionalized cyclopentenes, and dienes.32 Integrated C–H, C–C, or C–N bond functionalizations of vinyl-1,3-dioxolan-2-one 4, vinyl-cyclopropane 5, and di-azabicycle 6 with indole 1 gave access to 7–9 in good to excellent yield, respectively (Scheme 2a). It was proposed that the solvent played a pivotal role in successful transformations. For instance, control experiments using DMF as the solvent had a negative E/Z selectivity, and when the reaction was carried out in diethyl ether, an improved E/Z selectivity was observed. These results revealed that the product selectivity might have occurred due to the solvent effect and not by the formation of π-allylic manganese intermediate.
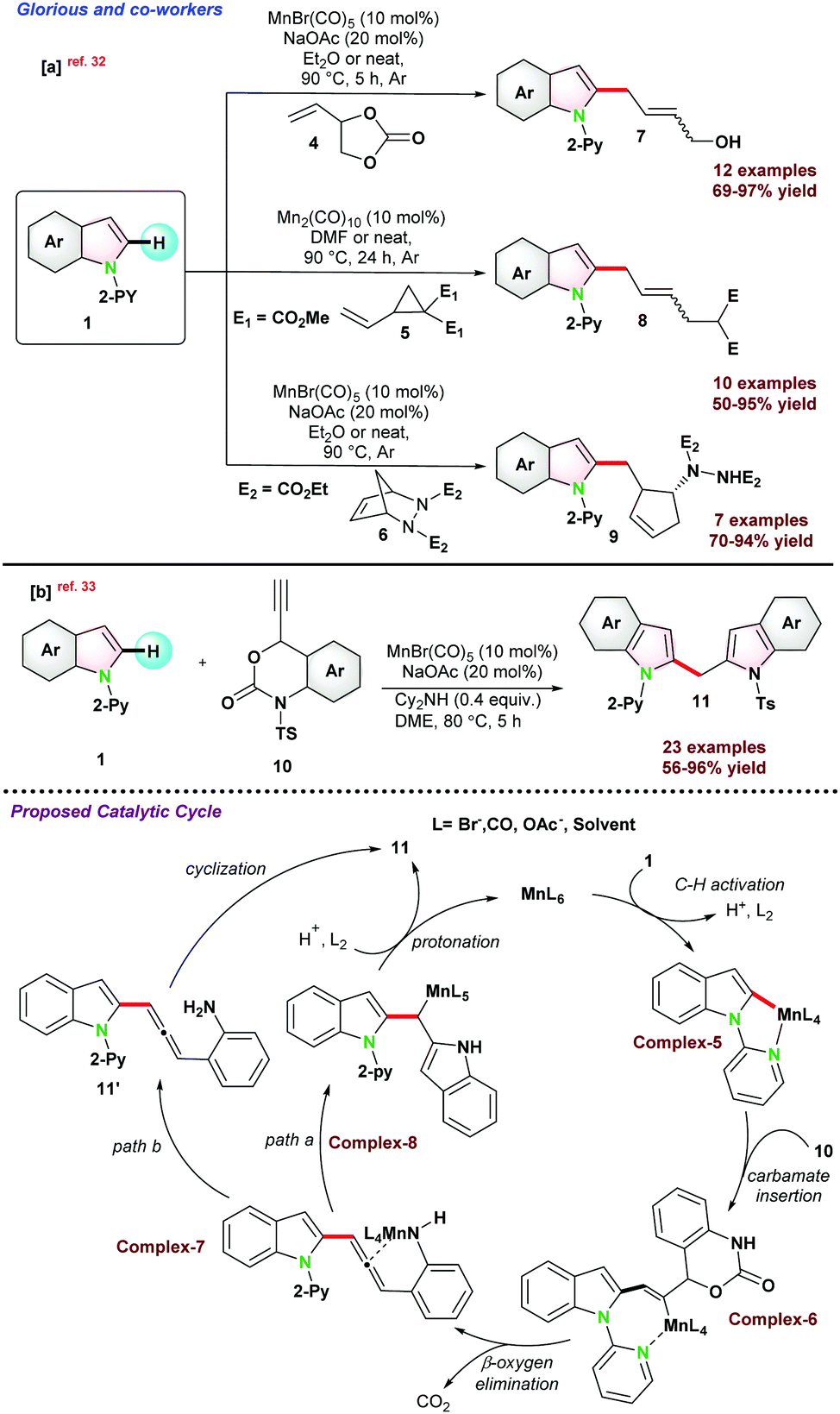 | ||
| Scheme 2 (a) Mn-catalyzed C–H arylation of picolinamide via C(sp2)–H activation in continuous flow medium. (b) Mn(I)-catalyzed synthesis of di-heteroaryl methanes. | ||
In 2018, the same group,33 reported Mn(I) catalyzed C–H bond functionalization to install (2-indolyl)methyl group on heteroarenes to access di-heteroarylmethanes. Ethynyl benzoxazinanone 10 was chosen as the coupling partner with 1 for the synthesis of bis(2-indolyl)methane 11 (Scheme 2b). The catalytic pathways went through a cascade strategy, where multiple bonds were reorganized in a one-pot operation and cyclized into unsymmetrical heteroarenes. Thereafter, to get a detailed insight into the reaction pathways, they performed some compatible reactions using 1,1-diphenylethylene and BHT as the radical scavengers, resulting in the desired product with a satisfactory yield. These control experiments provide evidence for the reaction and suggest following the organometallic C–H bond activation instead of the radical mechanism. They proposed a catalytic cycle where base-assisted coordination of 1 with metal generated a five-membered complex-5, which underwent addition with carbamate 10 to complex-6. Next, β-oxygen elimination from complex-6 generates intermediate complex-7, which undergoes regio-selective allene insertion and is transformed into complex-8. Thereafter, Complex-8 was protonated to raise the target product 11 and the active Mn(I) complex. Alternatively, intermediate complex-7 might be protonated to intermediate 11′, which was converted into the product through highly selective intramolecular cyclization (Scheme 2b).
In 2017, Ackermann and co-workers disclosed an Mn-catalyzed site-selective C–H/C–C functionalization of indoles assisted by the 2-pyridyl group for the synthesis of allylated alkenes with excellent diastereo-selectivity.34 HNCy2 was used as a co-catalyst along with MnBr(CO)5 for successful conversions (Scheme 3a). C–H/C–C functionalization in the presence of deuterated methanol CD3OD resulted in no deuterium scrambled product, suggesting that the reaction follows an organometallic C–H activation pathway. Observed kH/kD = 1.5 suggested that electron-rich substituents on 1 were more reactive and facilitated base-assisted electrophilic type substitution. Next, In 2018, the same group unrevealed the C–H arylations of N-substituted picolinamide 12 using Mn-catalyst through continuous flow technology (100 min of continuous flow).35 The catalytic protocol consisted of MnCl2, neocuproine, TMEDA, and DCIB (1,2-dichloro-2-methylpropane) and a variety of aromatic motifs easily coupled with diversely decorated picolinamides with high positional selectivity (Scheme 3b). Interestingly, iso-nicotinamide also reacted well, and gram scale synthesis, including late-stage derivatization of 16a, showed the practical utility of the protocol. Control experiments in the presence of manganese-neocuproine complex resulted in an 86% yield. Computational studies based on PW6B95-D3BJ/def2-TZVP + SMD(THF)//TPSS-D3BJ/def2-SVP theory revealed a Sigma-bond metathesis type ligand to ligand hydrogen transfer (LLHT) during the reaction (Scheme 3b).
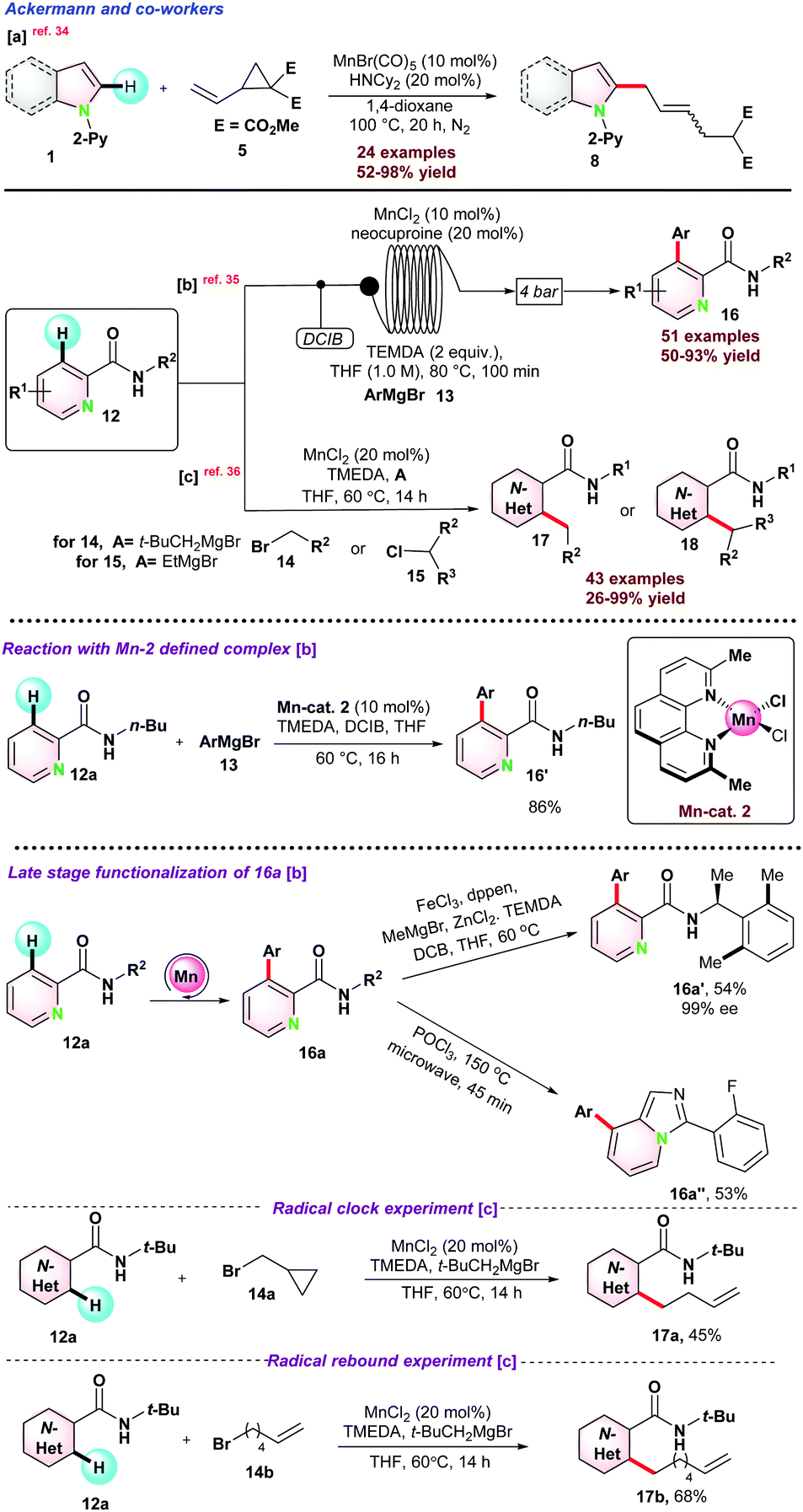 | ||
| Scheme 3 MnCl2-catalyzed C(sp2)–H alkylation of picolinamide with alkyl halides. | ||
In 2019, they again reported a new strategy for catalytic C–H alkylation of N-substituted picolinamide 12 with primary and more challenging secondary alkyl halides (14–15) using low valent and non-toxic MnCl2 (Scheme 3c).36 EtMgBr and t-BuCH2MgBr, respectively, were observed as the most effective bases for the transformations. Mechanistic investigation using classical radical clock agent bromo–methyl cyclopropane 14a resulted in the ring-opening butenylated product 17a, which signified the homolytic cleavage of the C–Br bond (Scheme 3). 6-Bromohex-1-ene 14b reacted with 12a to 17d, which emphasized the possibilities of a quick radical pick-up within the radical period i.e., 1.3 × 108 s−1 to 1.0 × 105 s−1. Further, as proposed, the resulting kinetic isotopic effect (kH/kD = 2.6) might reveal the possibilities of C–H bond breaking during or before the rate determining step (rds).
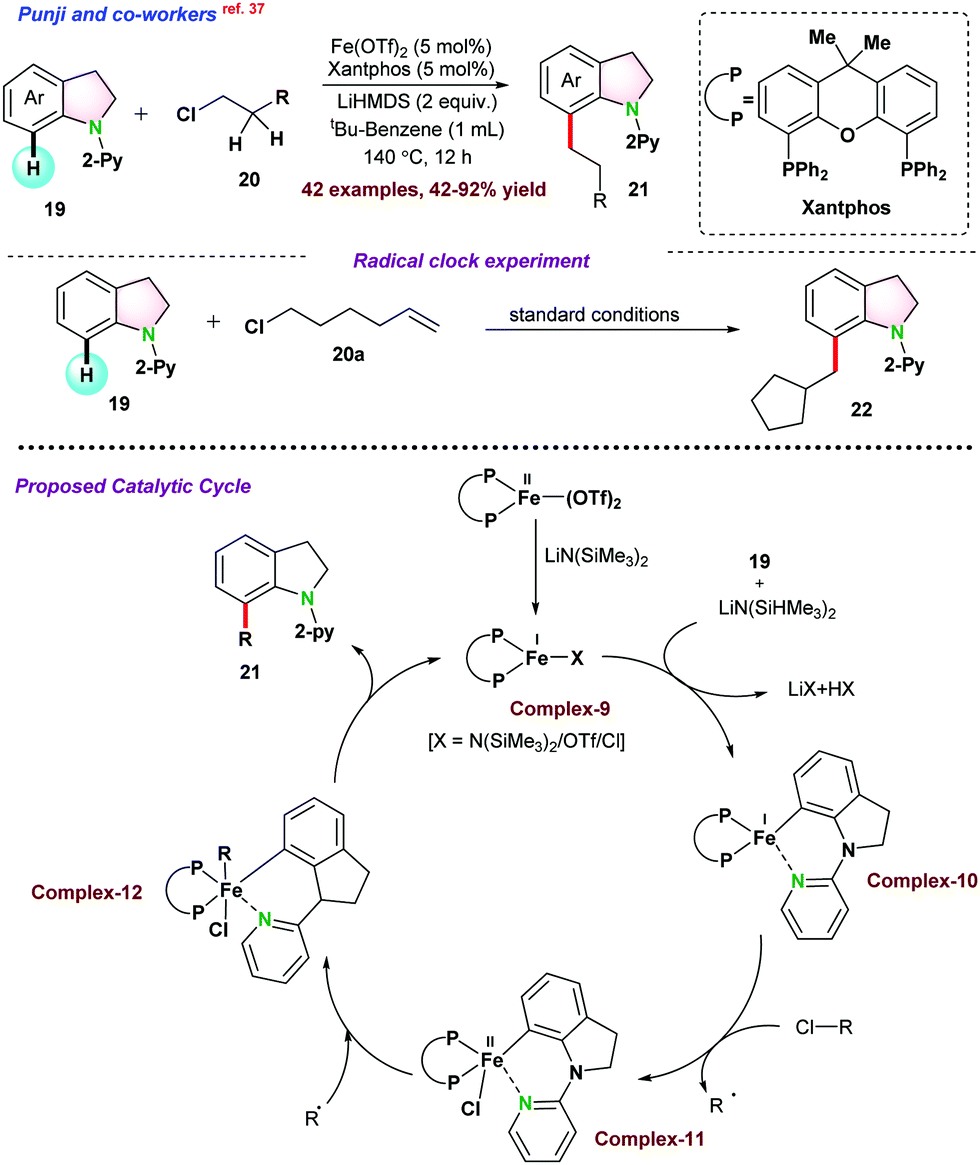 | ||
| Scheme 4 Fe-catalyzed cross-coupling of indoline C(7)–H with alkyl chlorides. | ||
Initial mechanistic investigations in the presence of radical scavengers (TEMPO, galvinoxyl, and BHT), were performed. It was observed that in the presence of TEMPO and galvinoxyl, the alkylation reaction did not progress, whereas BHT significantly suppressed the product yield. Radical clock experiment using 6-chlorohex-1-ene 20a ended up in the cyclized product 22, which proved the possibilities of an alkyl radical intermediate. EPR spectroscopy analysis of the incomplete reaction mixture showed a sharp peak at g = 1.98, clearly indicating the existence of the carbon-centric organic radical. The reaction followed the first-order kinetics with respect to indoline. Based on the experimental results and DFT studies, it was proposed that Fe(II)-complex was reduced into Fe(I)-active species complex-9 in the presence of LiHMDS. Subsequently, indoline directly coordinated with Fe(I)-complex leading to the electron-rich iron complex-10, which was the rate-limiting step. Thereafter, iron complex-10 coordinated with halogen extracted from the electrophilic alkyl chloride via a single electron transfer pathway and transforms into complex-11 and generates the alkyl radical. In the final step, complex-12 furnished the desired product 21via reductive elimination and regenerated complex-9 (Scheme 4).
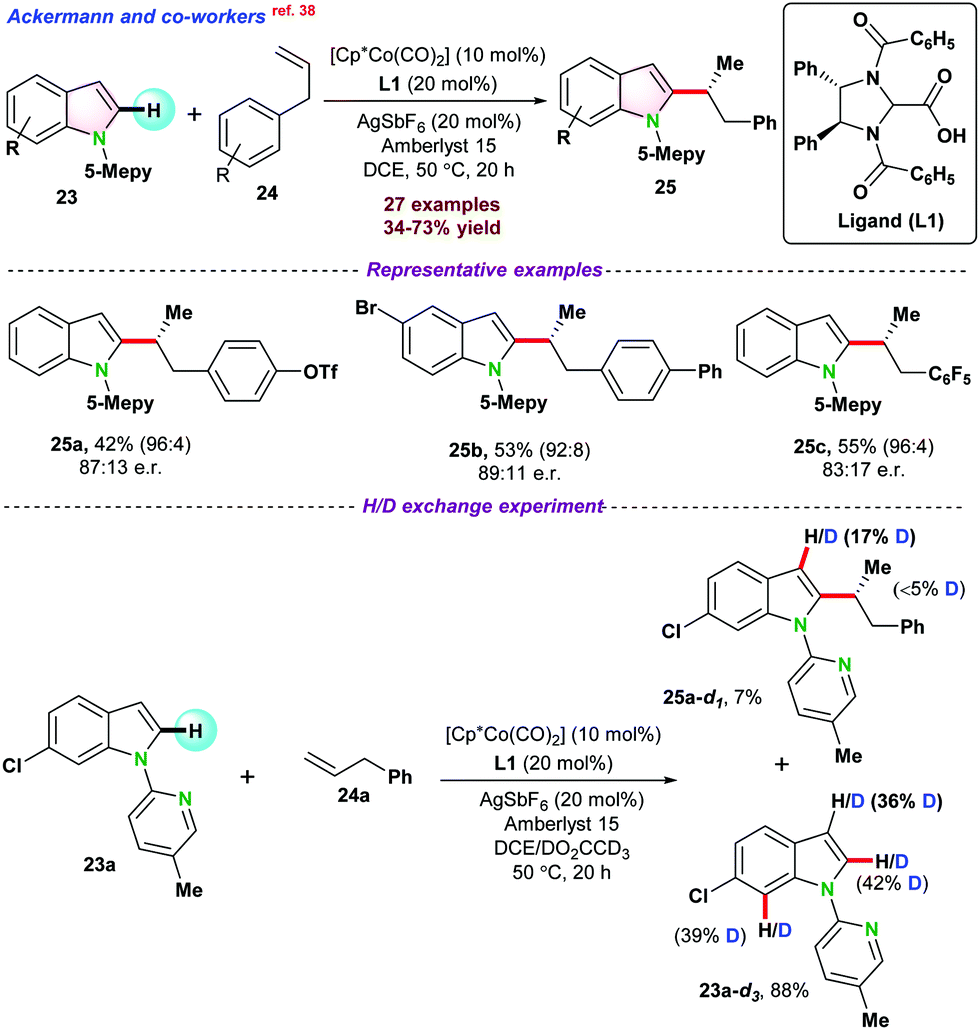 | ||
| Scheme 5 Enantioselective Co(III)-catalyzed C–H functionalization of 1-(5-methyl pyridine-2-yl)-1H-indole. | ||
Later on, Zeng and co-workers,39 demonstrated the cross-coupling of pyridines 26 with diazoesters 27 for selective carbenoid insertion at the C3 positions of pyridines under Co(II) catalytic system (Scheme 6). Commercially available CoBr2 in the presence of Lewis acid Cu(OAc)2 gave maximum product yield. A series of control experiments, such as H/D exchange and kinetic isotopic effect (KIE) reactions, were performed to get a deeper insight into the reaction mechanism. H/D exchange reactions pointed towards the electrophilic addition for the formation of cobalt carbenoid and indicated that the reaction followed irreversible C–H activation pathways. Next, the coupling reaction between 26a and 27 using standard conditions with 2 equiv. of CH3OD gave 28a-d1 with 26% deuterium incorporation. However, by the reaction of 28a with CH3OD, no 28a-d1 product was obtained. These two experiments exposed that enol isomerization ruled the deuterium exchange of 28a in lieu of metal protonation (Scheme 6).
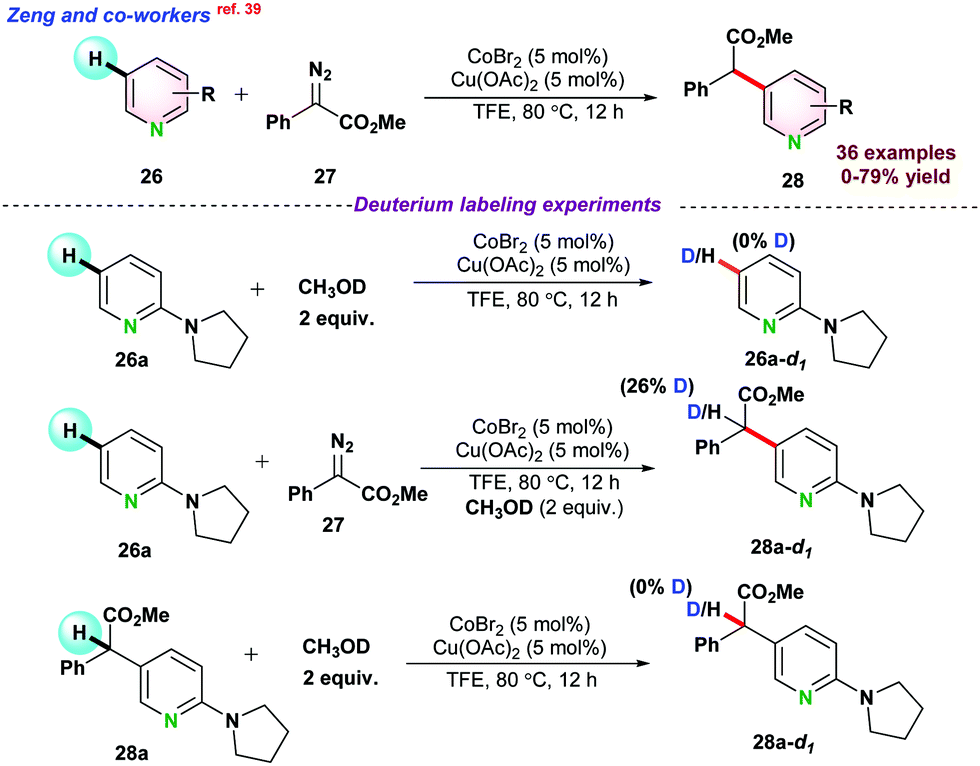 | ||
| Scheme 6 Co(II)-catalyzed regioselective C–H alkylation of pyridine with diazoacetates through C(sp2)–H activation. | ||
Recently, in 2020, Lautens and co-workers40 disclosed cobalt catalyzed asymmetric hydroarylative cyclization of 1,6-enynes. A series of chiral bi-phosphine ligands were used for such studies, and (R,R)-QuinoxP* furnished the desired product in up to 90% yield with a single diastereomer and regioisomer (95.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4.5 er). It was proposed that the catalytic cycle might follow either oxidative cyclization (depicted in Scheme 7) or oxidative addition pathway. In the case of oxidative cyclization mechanism, initially bi-phosphine-ligated Co(I) cationic species complex-13 is generated in the presence of Zn and NaBARF, which further coordinated with enynes 29 and transformed into the intermediate cobalt-cycle complex-15 following enantioselective oxidative cyclization. In the next step, 1 directly coordinated with complex-15 and subsequently underwent a series of intermediate Co-complexes and resulted in product 30 following σ bond metathesis and reductive elimination.
:4.5 er). It was proposed that the catalytic cycle might follow either oxidative cyclization (depicted in Scheme 7) or oxidative addition pathway. In the case of oxidative cyclization mechanism, initially bi-phosphine-ligated Co(I) cationic species complex-13 is generated in the presence of Zn and NaBARF, which further coordinated with enynes 29 and transformed into the intermediate cobalt-cycle complex-15 following enantioselective oxidative cyclization. In the next step, 1 directly coordinated with complex-15 and subsequently underwent a series of intermediate Co-complexes and resulted in product 30 following σ bond metathesis and reductive elimination.
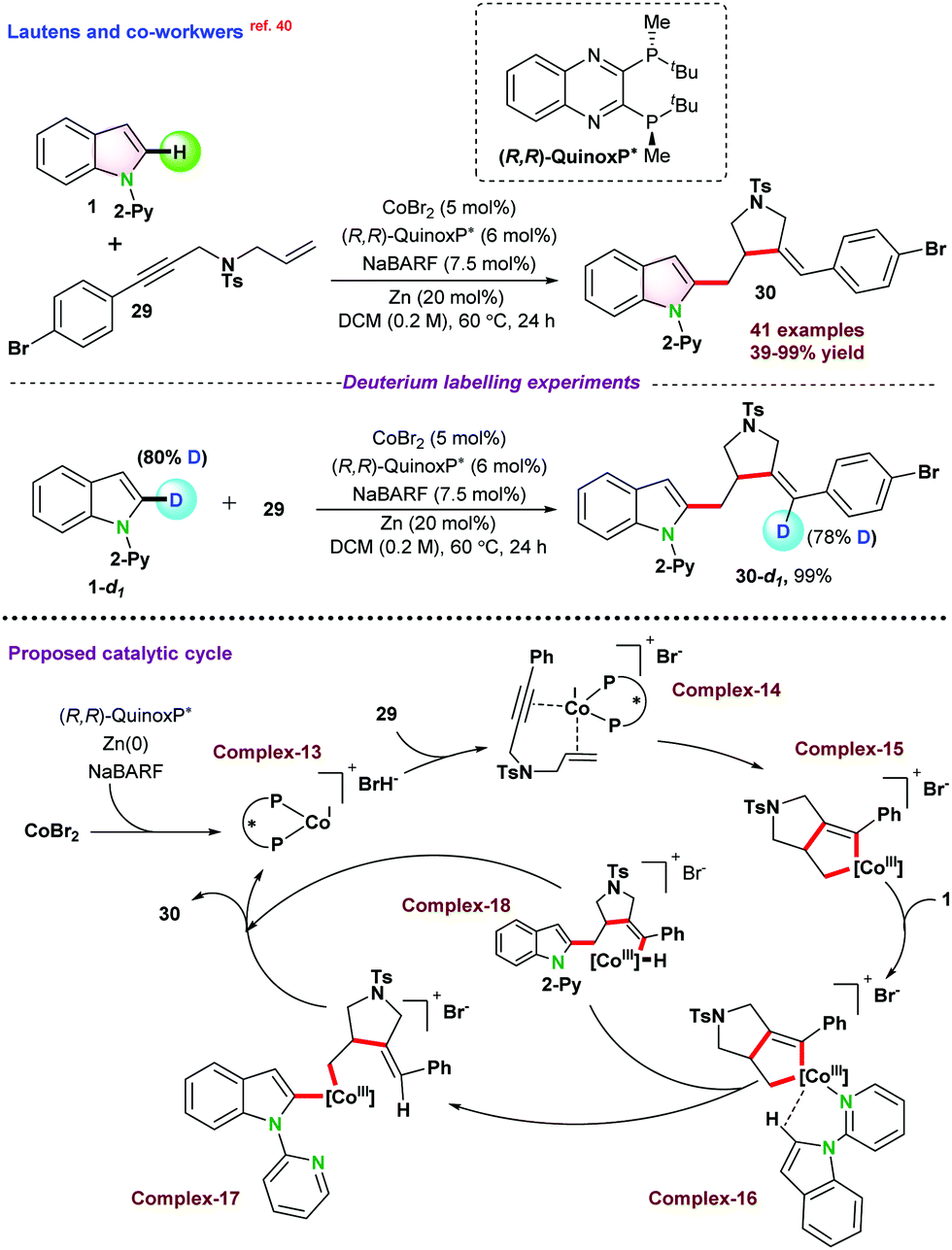 | ||
| Scheme 7 Co(II)-catalyzed enantioselective hydroarylation of 1-(pyridine-2-yl)-1H-indole with 1,6-enyens. | ||
It was proposed that the reaction pathways involved two catalytic cycles. Cycle A allowed the alkene isomerization following chain walking strategy, and Cycle B followed a slow C–H functionalization mechanism. Initially, allyl benzene isomerized to give thermodynamically more stable conformation 24′ through η3-allyl Ni–H intermediate via 1,3-hydride shift. Thereafter, 24′ participated in Cycle B, and functionalization was succeeded by oxidative addition, migratory insertion, and reductive elimination steps to the final product 32a and re-generated the pre-catalyst (Scheme 8). In 2020, Surawatanawong and co-workers revealed the density functional theory (DFT) study for the above reaction.42 Ong and co-workers,41 proposed that C–H oxidative addition of pyridine-AlMe3 species occurred before the migratory insertion of allyl benzene. However, the density functional theory calculations by Surawatanawong and co-workers suggested that for the migratory insertion, a higher amount of energy barrier had to be overcome, which was not feasible under the catalytic conditions reported.
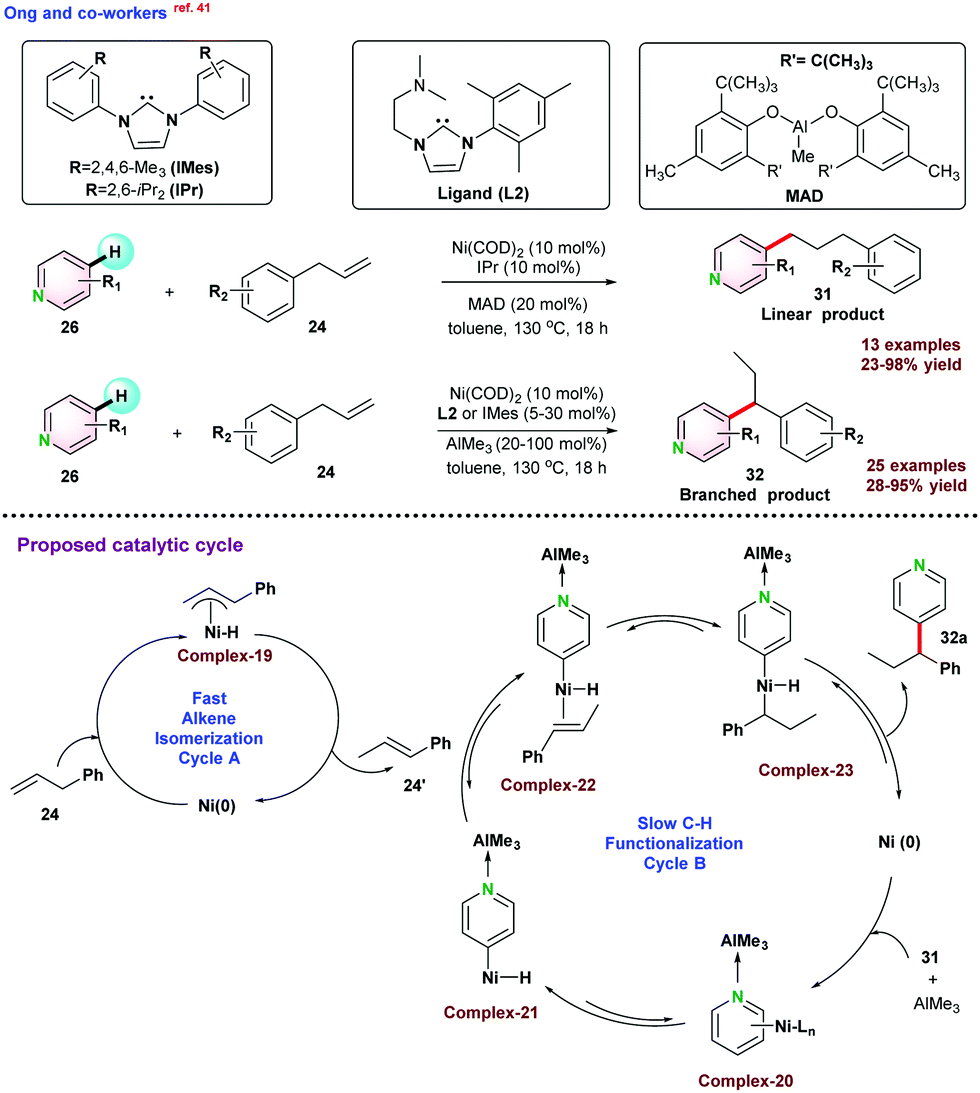 | ||
| Scheme 8 Ni–NHC catalytic C–H alkylation of pyridine with allyl benzene. | ||
 | ||
| Scheme 9 Cu-catalyzed (a) regio-selective oxidative homo-coupling of imidazo[1,2-a]pyridines, (b) regio-selective di-carbonylation of imidazo[1,2-a]pyridines with N,N-di-substituted acetamide, (c) Cu-catalyzed carbonylation of imidazo[1,2-a]pyridines with methyl heteroarenes through C–H bond activation. | ||
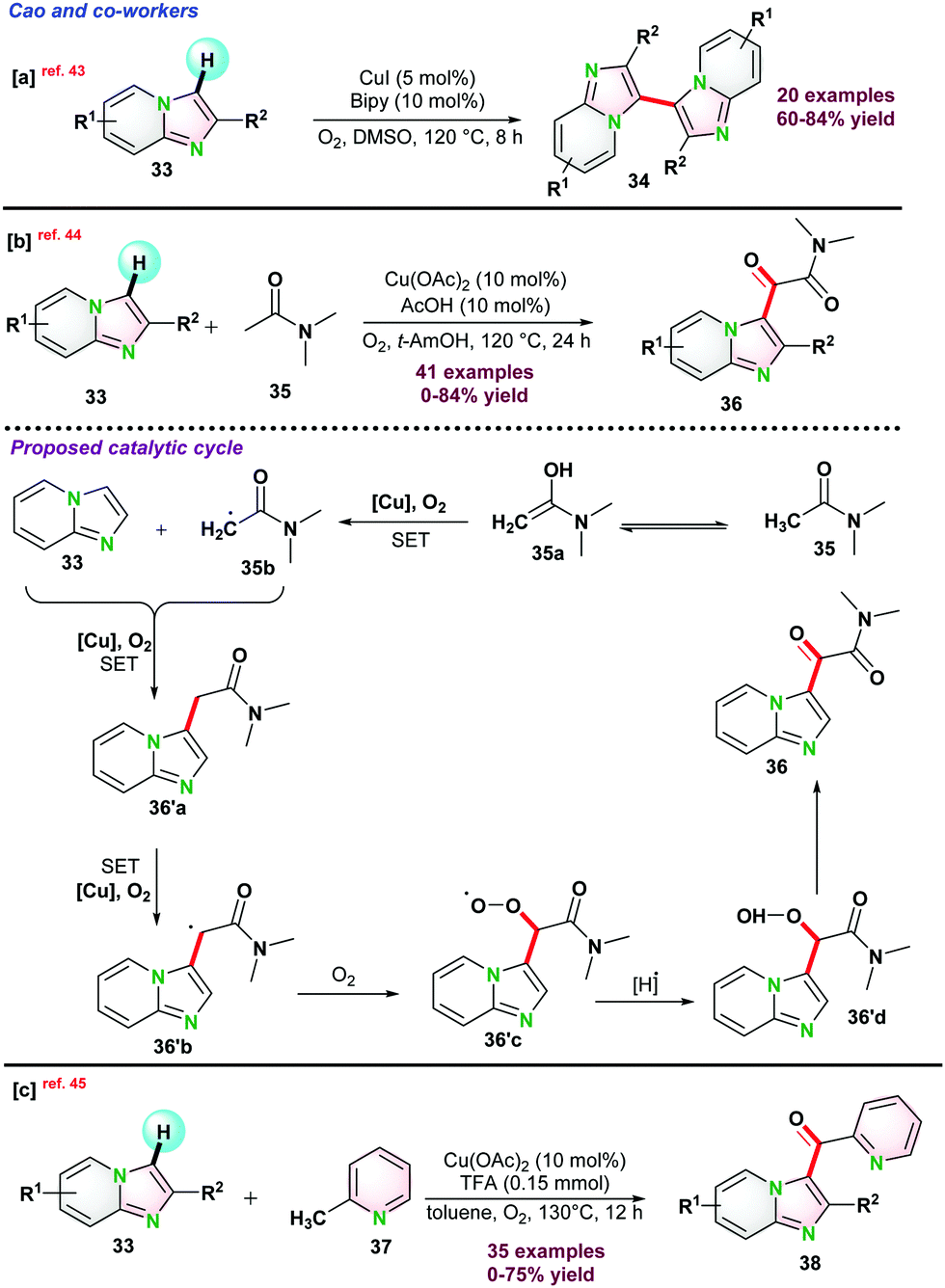
Recently, Miura and co-workers disclosed Cu-catalyzed dehydrative biaryl-coupling between azine-N-oxides 39 and 5-phenyloxazoles 40. Interestingly, the reaction that occurred in the presence of pyridine and acid was essential for the successful transformations. It was observed that bis-oxazole was formed as a major by-product via homo-coupling of oxazoles. However, N-oxide product (39-O) was not detected (Scheme 10a).46 Very recently, in 2021, they again reported de-hydrogenative bi-aryl coupling between o-cresol 42 and 5-phenyloxazole 43. It was proposed that the introduction of 4,4′-di(tert-butyl)-2,2′-bipyridine(dtbpy) group into the phenolic oxygen was the key for high selectivity with broad functional group tolerance under comparatively milder reaction conditions (Scheme 10b).47
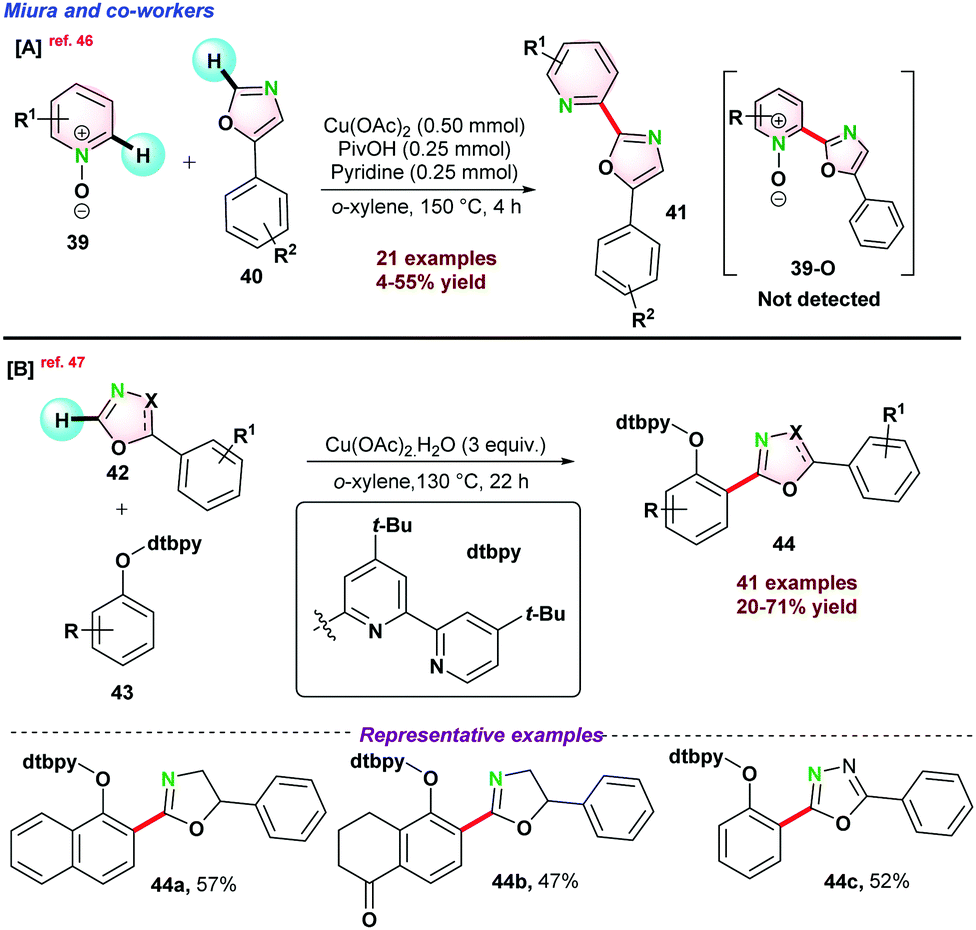 | ||
| Scheme 10 Cu-catalyzed process for de-hydrative biaryl-coupling. | ||
In 2016, Sawamura and co-workers reported enantioselective allylic alkylation of azoles 44dvia Csp2–H bond activation. They used γ,γ-di-substituted primary allylic phosphate 44e as a coupling partner with azoles and generated quaternary carbon centre 44f at the α-position of the azole ring. Application of the chiral NHC-ligand allowed access to high enantio-selectivity (Scheme 11a).48a In 2017, Jain and co-workers studied the oxidative homo-coupling of 2-phenylpyridine N-oxide 45 using Cu(OAc)2. The catalytic process was initiated by the abstraction of a C-2 proton of 45a by LiOtBu and subsequently resulted in intermediate Complex-24 via transmetallation. Next, the oxidative coupling of complex-24 in the presence of air gave the desired product 46 (Scheme 11b).48b
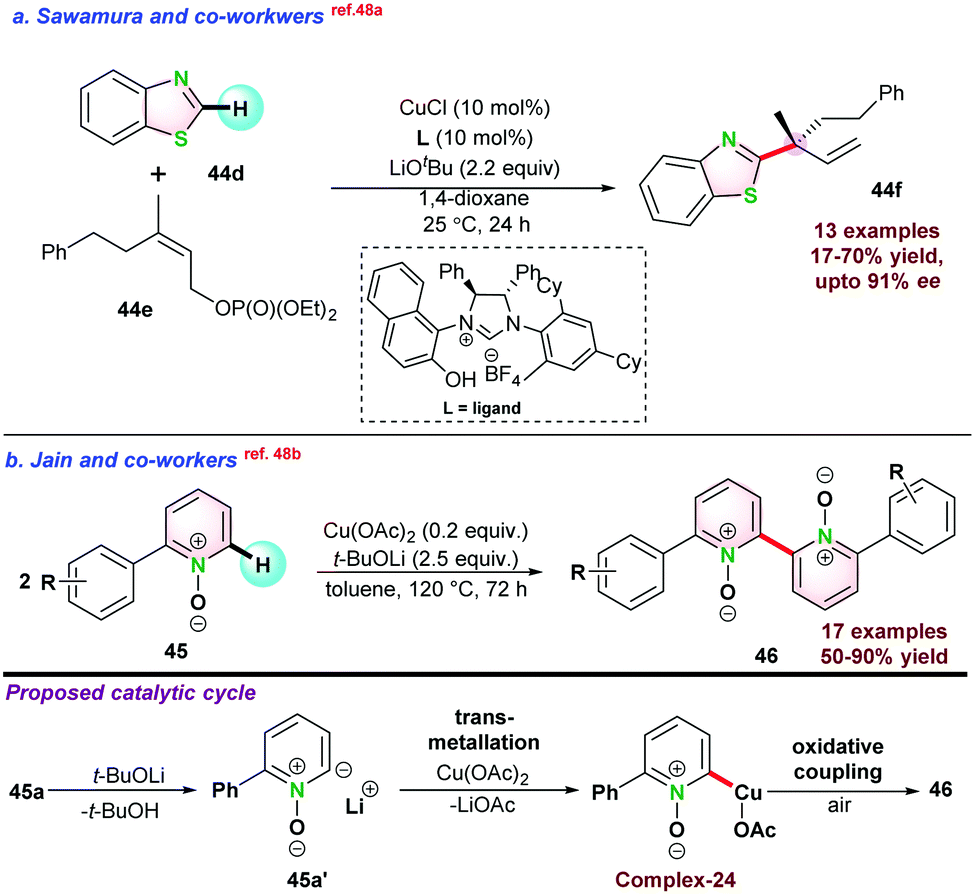 | ||
| Scheme 11 (a) Cu-catalyzed enantioselective synthesis of quaternary carbon center. (b) Cu-catalyzed homo-coupling of 2-azine-N-oxide. | ||
In 2019, Li and co-workers49 demonstrated copper and cobalt co-catalyzed dehydrogenative cross-coupling (CDC) between benzothiazole 47 and benzoxazole 40 (Scheme 12). 20 mol% Co(NO3)2.6H2O and 1.5 equivalent Cu(OAc)2 were required in 1-fluoro-2-trifluoromethyl-benzene as the solvent. NaOAc was used to increase acetate ion level in the reaction medium, and air acted as a terminal oxidant. Catalytic studies showed that initially Cu(OAc)2 reacted with benzoxazole 40 following metalation-deprotonation process with the assistance of NaOAc and gave intermediate complex-25, which further reacted with another molecule of Cu(OAc)2 to form Cu(III) intermediate complex-26. Next, benzothiazole 47 coupled with the intermediate complex complex-26 through second concerted metalation-deprotonation to the key hetero-coupling intermediate species complex-27, which underwent reductive elimination to the targeted cross-coupling product 48. It was proposed that CuOAc was finally oxidised by Co(III) species and NaOAc facilitated to regenerate Cu(OAc)2 for the next cycle. The active Co(III) species was formed from the oxidation of Co(II) in the presence of air.
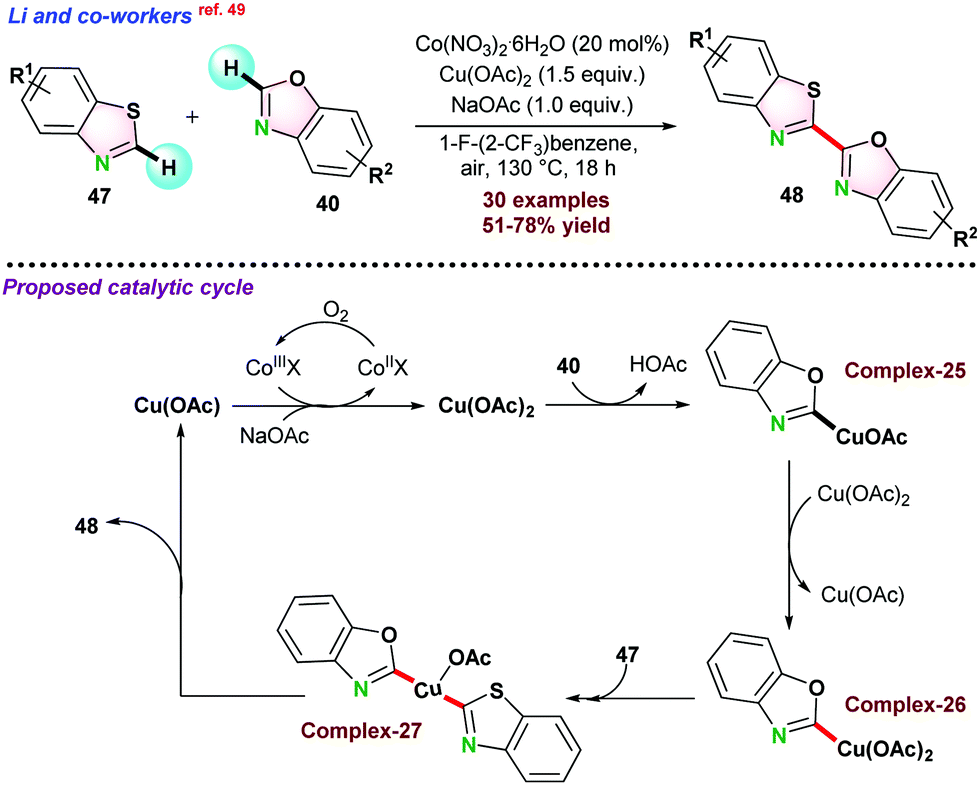 | ||
| Scheme 12 Cu-based and Co-co-catalyzed oxidative cross-dehydrogenative coupling of benzothiazole with benzoxazole. | ||
Peng and co-workers,50a reported CuCl2-catalyzed cross-de-hydrogenative coupling of quinazoline-3-oxides 49 and N-methylindole 50 to 4-(indol-3-yl)quinazoline 51 under air atmosphere. A series of bi-heteroaryl products were synthesized in moderate to good yields. Based on a series of control experiments, a catalytic mechanism is proposed, and it was observed that initially CuCl2 reacted with 49 to give intermediate complex-28via C–H bond activation. Next, complex-28 was inserted at the C-3 position of the indole 50 through another C–H bond activation. Cu(II) intermediate complex-29 further transformed into Cu(III) complex-30 in the presence of another equivalent of CuCl2, and CuCl2 converted into CuCl. Finally, intermediate complex-30 underwent reductive elimination to product 51. It is to be noted that CuCl generated through the process is oxidized into CuCl2 by oxygen and continues the catalytic cycle (Scheme 13a).
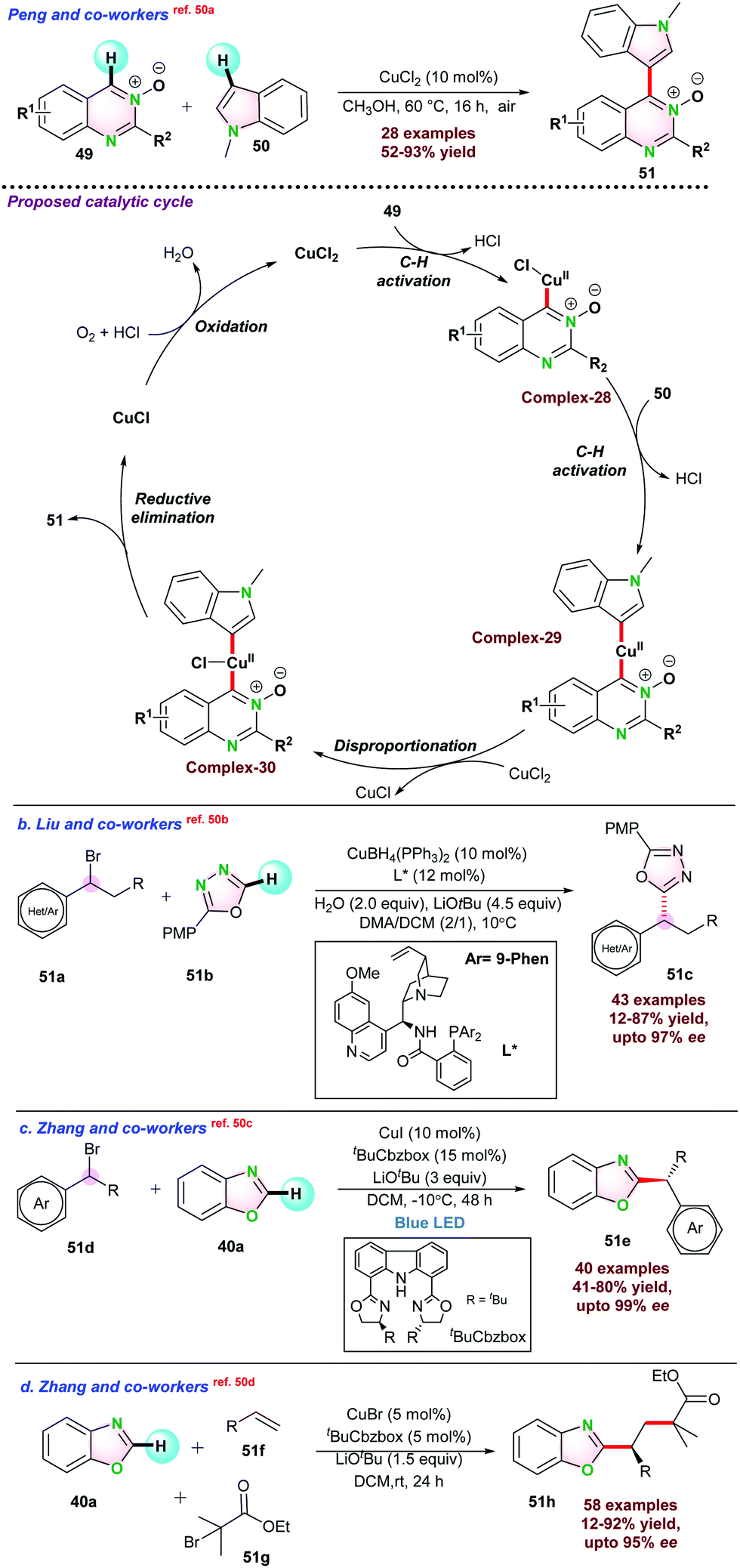 | ||
| Scheme 13 Cu-catalyzed processes for: (a) cross-dehydrogenative coupling of quinazoline-3-oxides and N-methyl-indole; (b) cross-coupling of azoles with racemic alkyl bromide; (c) light promoted asymmetric alkylation of azoles with secondary aryl/alkyl bromide; and (d) asymmetric alkylation of azoles with olefins. | ||
In 2021, Liu and co-workers demonstrated Cu(I)-catalyzed asymmetric cross-coupling reaction between racemic alkyl bromide 51a and 51bvia Csp3–H and Csp2–H bond activation. They prepared a series of cinchona–alkaloid-derived N,N,P-type chiral ligands, and the Cu(I)/cinchona–alkaloid systems were efficient for oxidative addition of alkyl bromides under mild conditions. 10 mol% CuBH4(PPh3)2, 12 mol% cinchona–alkaloid ligand, 2.0 equiv. of H2O and 4.5 equiv. of LiOtBu in DMA/DCM was required for a-alkylated product 51c with optimal yield and 90% ee. A series of control experiments were performed and established that the reaction went through a radical pathway (Scheme 13b).50b
In the same year, Zhang and co-workers reported the photocatalytic asymmetric C–H functionalization of azoles 40a with secondary aryl/alkyl bromides 51d to α-alkylated azoles 51e. The catalytic protocol required CuI/Cbzbox (carbazole based bisoxazoline ligand) under blue light. In the absence of blue light, the product yield was decreased significantly. This result indicated that blue light promoted the reaction with a higher ee value (Scheme 13c).50c Again, they have also established CuBr-catalyzed highly enantioselective three-component coupling of 51f and 51g with azole 40a through Csp2–H bond activation, which resulted in the product 51h. Chiral carbazole-based bisoxazoline (Cbzbox) ligand played a critical role in such transformations (Scheme 13d).50d
2.2 Alkenylation reaction via C(sp2)–H bond activation
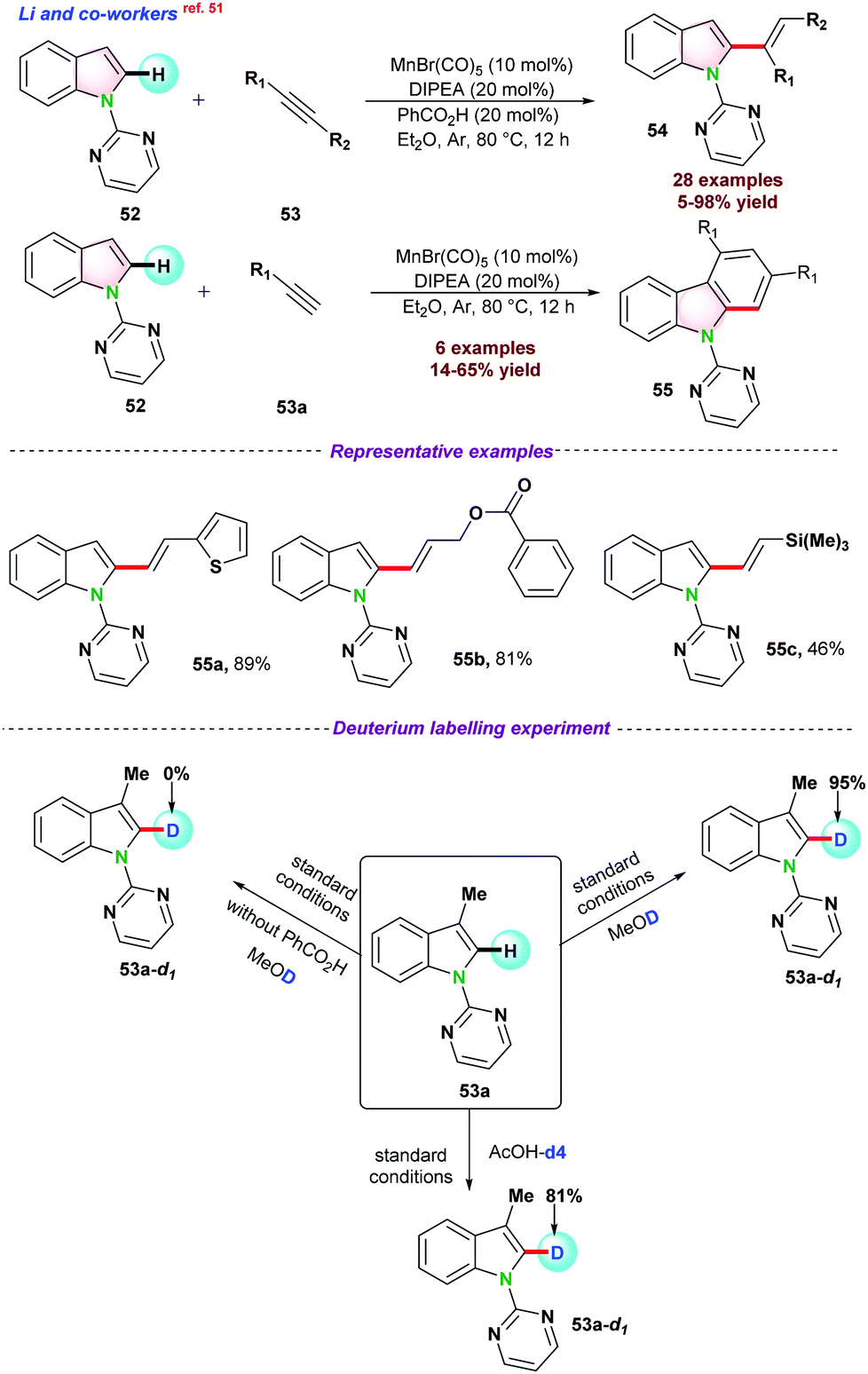 | ||
| Scheme 14 Mn-catalyzed C-2-alkenylation of indoles. | ||
In 2017, Ackermann and co-workers,52 reported chemo-selective hydroarylation of indole to C-2-alkenylated indoles under a continuous flow of current using a combination of MnBr(CO)5-BrØnsted acid catalysts. The catalytic protocol follows the C–H bond activation strategy without β–O elimination. A series of mechanistic studies were performed, and complex-31 was independently prepared. The stoichiometric amount of complex-31 was used for two sets of independent reactions. In the presence of an acid additive, alkenylated product 57 was obtained, whereas allene 58 was formed in the absence of acid additive (Scheme 15a).
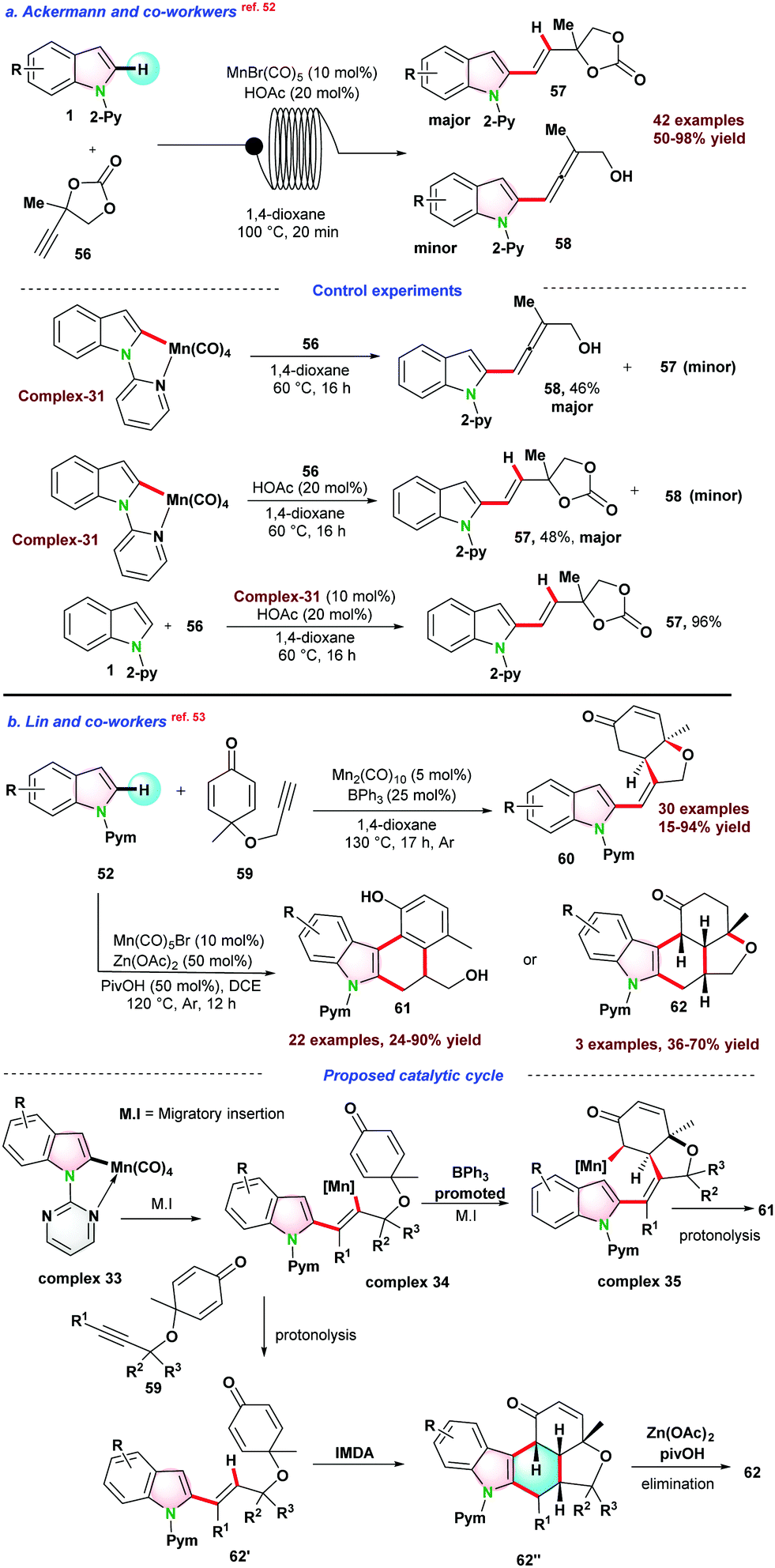 | ||
| Scheme 15 (a) Mn(I)-catalyzed chemo-selective hydroarylation via C–H bond activation in a continuous flow; (b) Mn-catalyzed C–H activation for annulation C–C coupling. | ||
In 2018, Lin and co-workers53 demonstrated Mn-catalyzed selective synthesis of two divergent cyclic products 61 or 62 using Csp2-H bond activation strategy. The catalytic reaction of 52 with 3,3-dimethyl-substituted 1,6-enyne 59 yielded alkenylated cyclic product 60 generated through C–H alkyl insertion or Michael addition sequences. Annulation products 61 or 62 were formed following sequential C–H insertion of alkyne/intramolecular Diels–Alder reaction/aromatization pathways. For the Michael addition, a Lewis acid was required to simplify the activation of enone. It was observed that BPh3 facilitates the positive consequences for the reaction (Scheme 15b).
Recently, in 2020, Banerjee and co-workers54 reported C-2 alkenylation of 1 with substituted ethynylbenzene 63via Mn-catalysed mechanochemical C–H bond activation pathway. They used mild acidic media, and silica was utilized as a milling media for the reaction. This catalytic protocol worked nicely for both electron-rich and electron-deficient substituents (Scheme 16).
 | ||
| Scheme 16 Mn(I)-catalyzed mechanochemical process for C-2 alkenylated indole. | ||
The catalytic system was successfully employed to activate C4 and C6 position of 66 and rendered di-alkylated product 67 using an excess (3.0 equiv.) of alkyne 53. Next, they studied the effectiveness of the catalytic conditions using imidazo[1,2-a]pyridines 33a. AlMe3 was used as a Lewis acid, which coordinated with the N-centre of 33a and subsequently reduced the electron density at the C-3, C-4, and C-5 positions, thereby increasing the reactivity toward the C–H bond activation. Benzothiazole 47a also followed the same catalytic conditions as used for imidazo[1,2-a]pyridines derivatives. But in the case of benzoxazole 40a, a combination of CoI2, Zn, and Ti(OiPr)4 was used to achieve successful product transformations. Mechanistic studies along with DFT calculation suggested that the C–H activation proceeds via ligand to ligand charge transfer (LLHT) (Scheme 17).
 | ||
| Scheme 17 Cobalt-catalyzed functionalization of alkynes through C–H activation. | ||
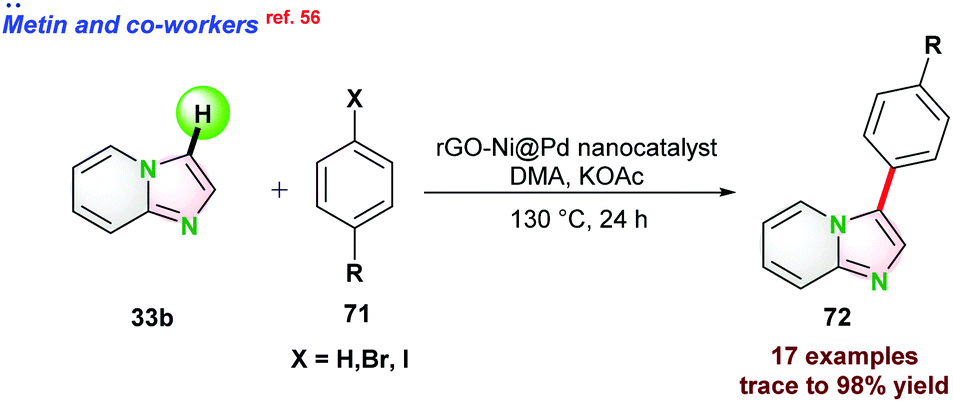 | ||
| Scheme 18 Monodisperse Ni@Pd core@shell NP's based direct C–H arylation of imidazo[1,2-a]pyridines. | ||
2.3 Borylation and cyanation reaction via C(sp2)–H bond activation
In 2017, Driess and co-workers57 reported Co–SiNSi based pincer catalyst for the highly regioselective C–H borylation of 2,6-lutidine 73. Bis-(pinacolato)diborane (B2Pin2) 74 and NaBHEt3 were used as the borylating agent and reductant, respectively. In the presence of B2Pin2, 2,6-lutidine 73 could not completely convert to the desired borylated product 75 even at 100 °C after prolonged reaction time due to the formation of HBpin as a side product, which suppressed the product yield. To overcome such issues, 1 equiv. of cyclohexene was used, which reacted with HBpin and in situ generated C6H11BPin to render complete conversion (Scheme 19).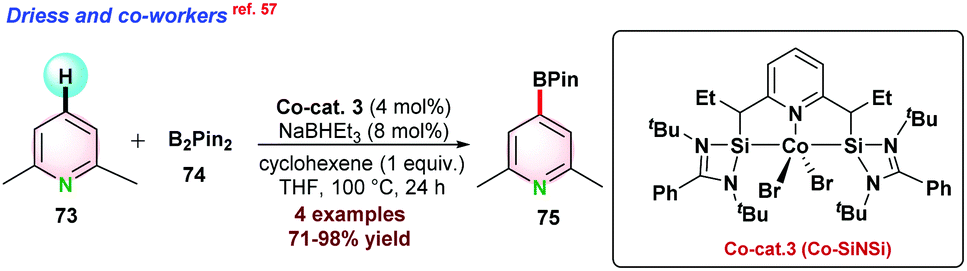 | ||
| Scheme 19 Co–SiNSi pincer catalyst for regio-selective borylation of arenes. | ||
In 2015, Ackermann and co-workers58 reported cobalt(III) catalyzed cyanation of N-heteroarene 53 by using 4-cyano-N-phenyl-p-toluenesulfonamide 76 as a cyanating reagent (Scheme 20a). Catalyst Cp*Co(CO)I2 proved to be effective in the presence of a catalytic amount of AgSbF6 and KOAc. The catalytic protocol was well tolerated for various functional groups and also assisted in removing the directing groups attached to the substrates.
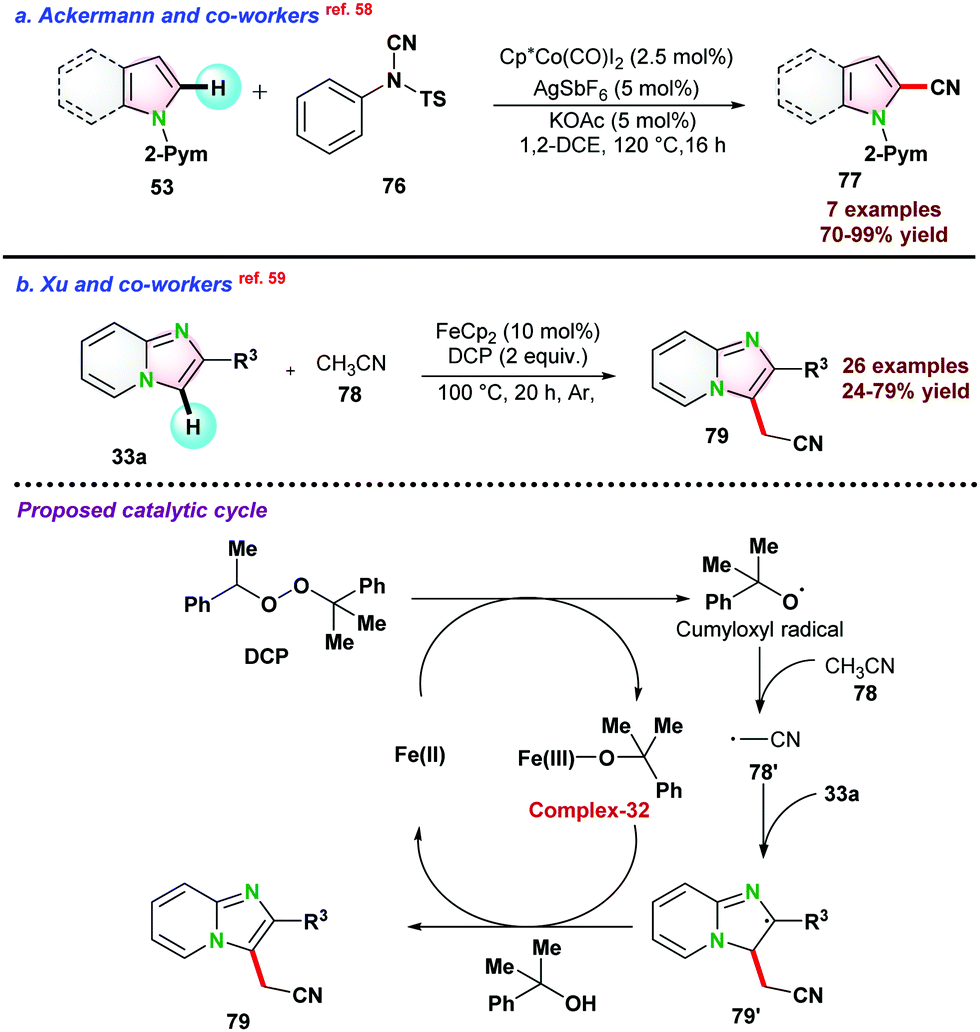 | ||
| Scheme 20 Co-catalyzed cyanation of N-heteroarene. | ||
In 2017, Xu and co-workers59 demonstrated Fe-catalyzed Csp2–H/Csp3–H cross-coupling between 33a and acetonitrile following oxidative C–H bond activation pathway. A series of oxidants were examined; however, DCP showed the best result for the transformations. Mechanistic studies revealed that single electron transfer pathways favour catalytic reactions. Based on control experiments, a catalytic cycle was proposed initially, and DCP was reduced in the presence of FeCp2 and transformed into complex-32 as well as cumyloxyl radical. Next, cumyloxyl radical abstracted a proton from acetonitrile to the radical intermediate 78′, which further reacted with 33a to give the intermediate 79′. Finally, intermediate 79′ was oxidized by complex-32, and deprotonation led to the final product 79 and subsequently regenerating the Fe(II)-species and continuing the catalytic cycle (Scheme 20b).
2.4 Annulation reaction
 | ||
| Scheme 21 Mn-catalyzed annulation reaction with allenes. | ||
In 2018, Lin and co-workers53 also demonstrated the Mn-catalyzed annulation reaction as presented in Scheme 15b. Mn(CO)5Br was used as pre-catalyst, and a fused 6-membered product 62 was obtained via C–H activation followed by intramolecular Diels–Alder (IMDA) reaction. The selectivity of this catalytic protocol was different from other reported catalytic systems (Rh(III), Co(III)), where IMDA metal adduct delivered the final product. Based on control experiments and previous reports, they proposed a reaction pathway for the C–H activation of indole in the presence of Mn(CO)5Br. Initially, metalacyclic complex 33 was formed, and regio-selective migratory insertion of metalacyclic complex 33 into alkyne leads to intermediate complex 34. In the presence of additive BPh3, complex 34 transformed into complex 35via diastereoselective addition, which further underwent protonolysis to form product 61. Contrary to this pathway, when Zn(OAc)2 and PivOH were used as an additive, complex 34 transformed into intermediate 62′ and finally, 62 was obtained through IMDA reaction and Zn(OAc)2-mediated intramolecular elimination (Scheme 15b).
Next, in 2018 Cui and co-workers61 reported three-component one-pot synthesis of fused hetero-tricyclic ring system from N-substituted-3-substituted pyridin-2-amine 83 and di-substituted acetylene di-carboxylate 84via cascade N–H and C–H activation reactions (Scheme 22). The catalytic process proceeds via intramolecular Diels–Alder/retro-Diels–Alder reactions to form the fused N-heterocycle moiety. Pyridine having a substitution at C4 or C5-position and un-symmetrical alkynes limit the catalytic transformations.
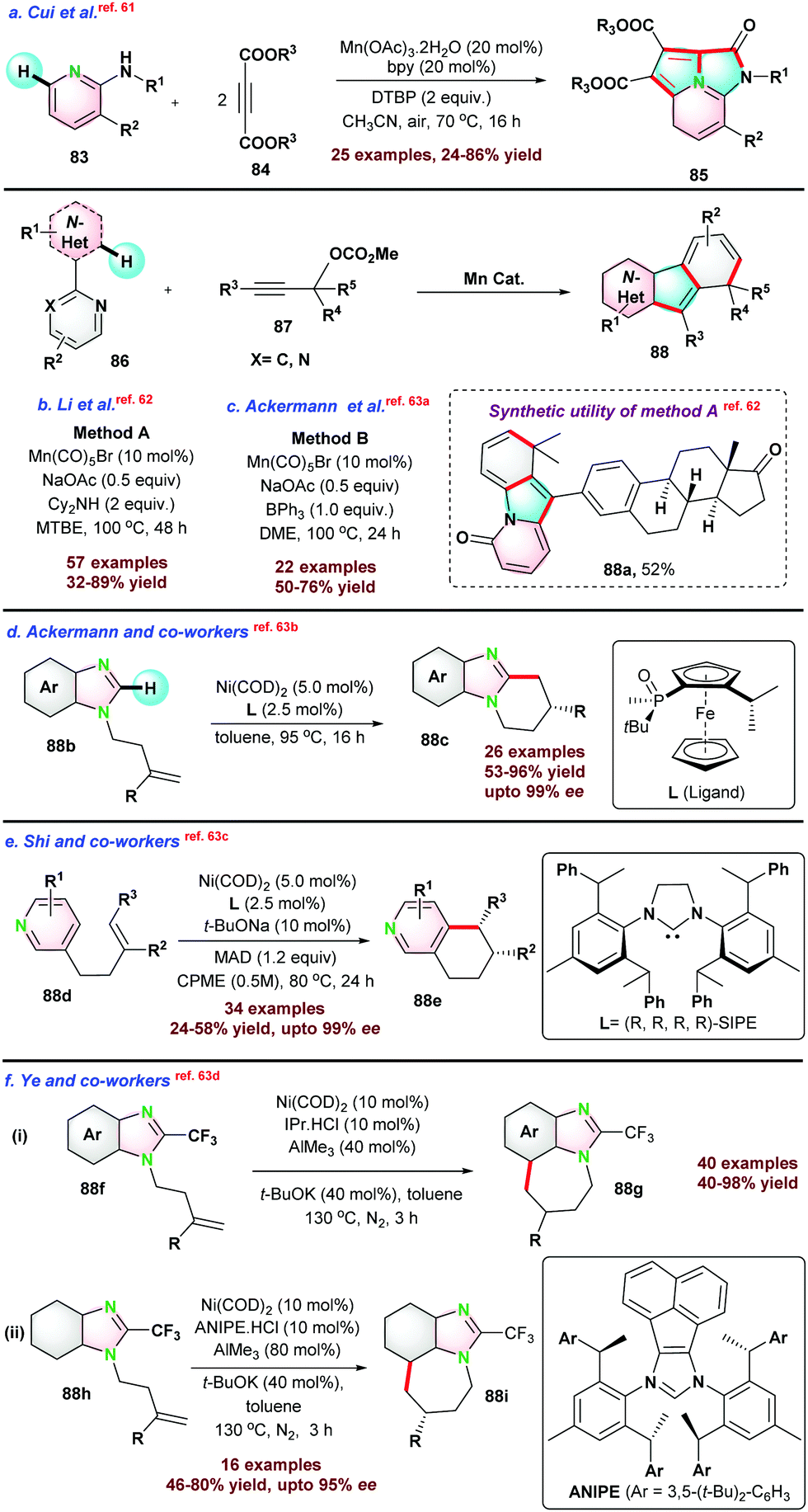 | ||
| Scheme 22 Mn-catalyzed annulation reactions via C–H functionalization. | ||
Thereafter, Li et al.,62 and Ackermann et al.,63a employed similar catalytic systems for the de-hydrocyanative trans-annulation reaction between N-heteroarene and propargyl carbonates assisted by the directing group. The key difference between the reaction conditions used by the respective groups was the use of additives. Li's group used 2 equiv. of Cy2NH with MTBE solvent system, whereas Ackermann's group used 1 equiv. of BPh3 with DME solvent system (Scheme 22b and c).
Recently, Ye et al.63d demonstrated the cyclization of alkene-tethered imidazole 88f and 88h to tri-cyclic imidazoles 88g and 88i (Scheme 22f). In the same report, the authors extended the protocol towards the enantioselective synthesis of substituted tri-cyclic imidazoles using chiral ANIPE·HCl.
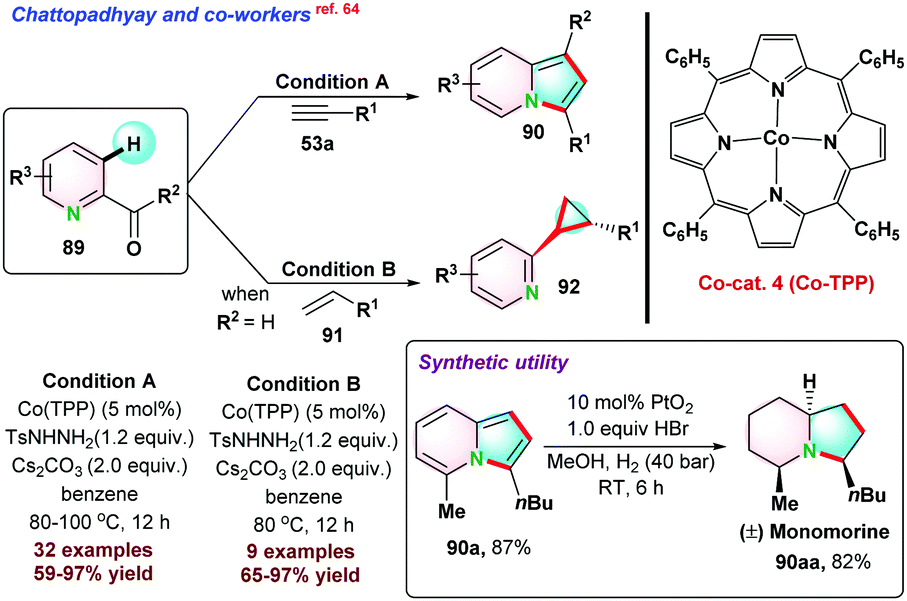 | ||
| Scheme 23 Co-porphyrin-based catalyst for annulation via C–H functionalization. | ||
In 2020, Ackermann and co-workers66 used Cp*Co(CO)I2 system for sp2 C–H activation directed by pyridine moiety. In the overall process, two new C–C bonds and a new C–N bond were formed with the migration of pyridine moiety through C–N bond cleavage (Scheme 24). Furthermore, to understand the electronic effect on reactants, a series of competitive reactions between pyridines 94a–d and alkynes were performed, suggesting that electronically rich pyridones or alkynes were more reactive for the process. Next, deuterium labelling experiments concluded that deuterium exchange at the C–H position strongly supports the reversible nature of C-H activation. DFT studies were conducted to understand the mechanical aspects of the reaction.
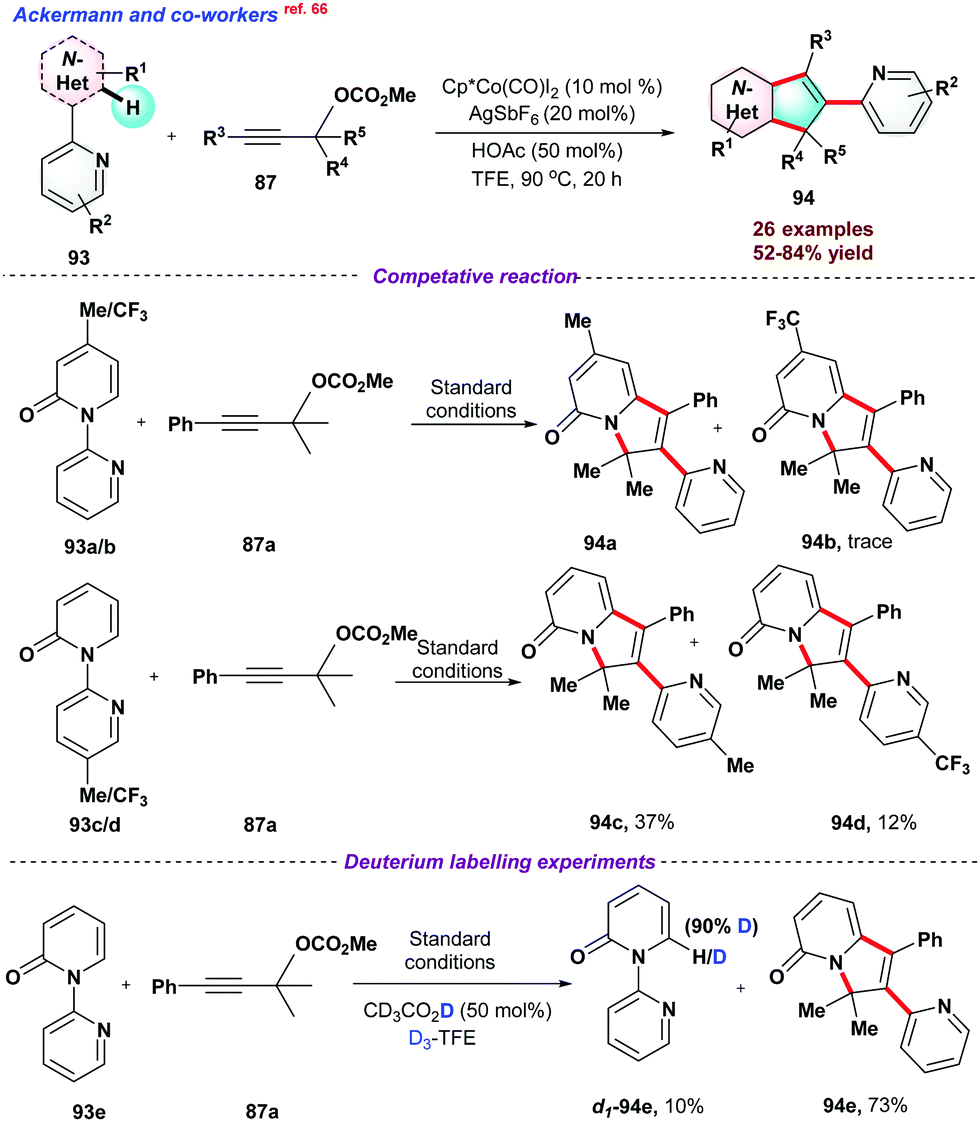 | ||
| Scheme 24 Co-catalyzed annulation reaction via C–H activation/functionalization. | ||
3. sp3 C–H functionalization/activation of azaarenes
Heteroarenes and their derivatives are omnipresent in bioactive natural products and widely used for medicinal applications, material chemistry, and dye industries. Often, functionalization of sp3 C–H bonds in heteroarenes is quite challenging compared to the sp2 C–H bond. Due to weak acidity, sp3 C–H bonds are associated with high bond dissociation energy. Classical approaches required pre-functionalization of one or both the coupling partners, and thus the development of newer technologies for functionalization of sp3 C–H bond has attracted significant attention. In this section, we have tried to compile recent advances on sp3 C–H functionalization of azaarenes via base metal catalysis. Further, we have sub-categorized this section into different 3d-transition metal catalysed alkylation and alkenylation reactions.3.1. Alkylation reaction
In this section, we have incorporated C-alkylation reactions of azaarenes via 3d metal catalyzed functionalization of sp3 C–H bonds.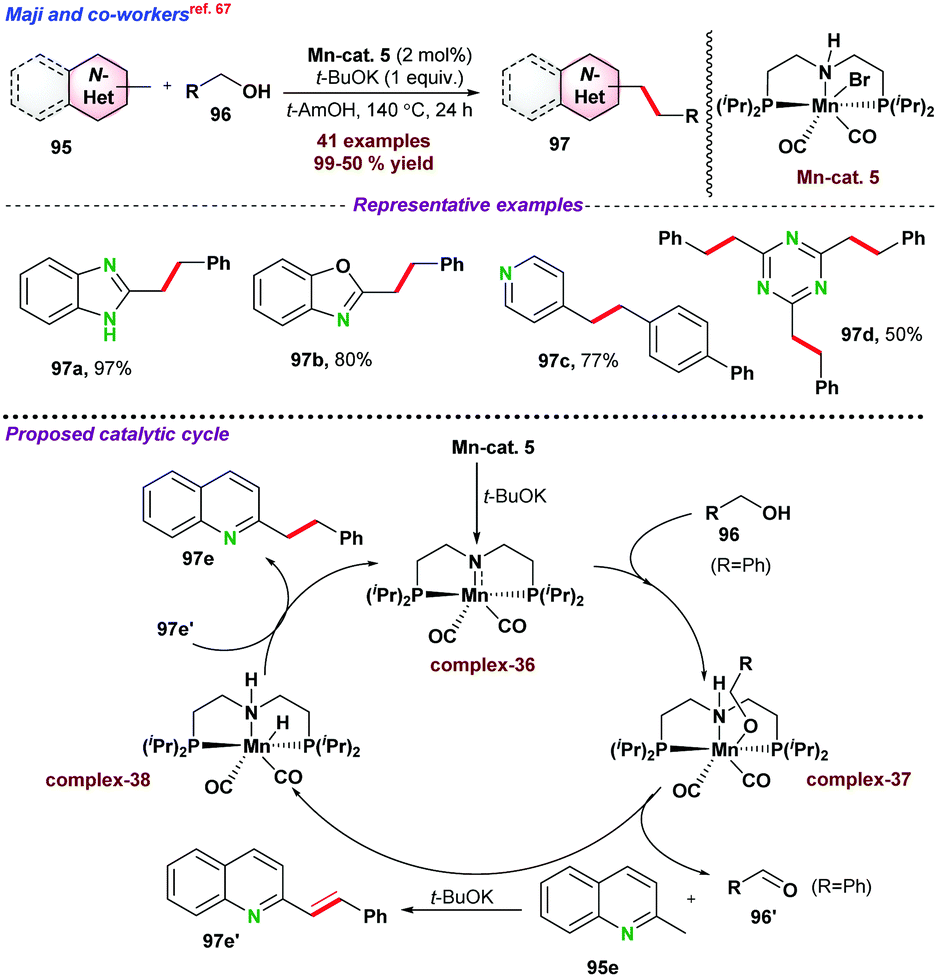 | ||
| Scheme 25 Mn catalyzed alkylation of N-heteroaromatics. | ||
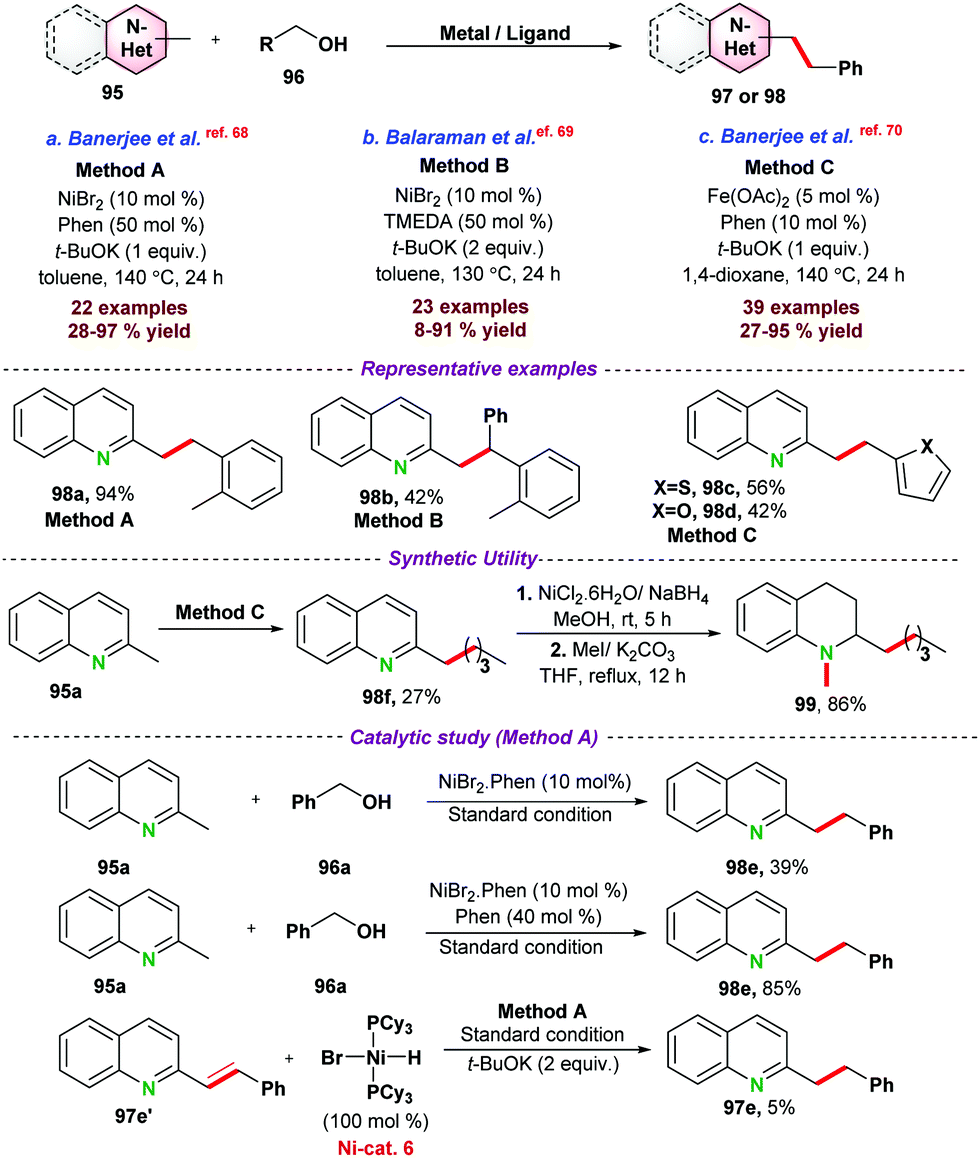 | ||
| Scheme 26 Ni-and Fe-catalyzed alkylation of N-heteroaromatics. | ||
 | ||
| Scheme 27 Co-catalyzed alkylation of N-heteroaromatics. | ||
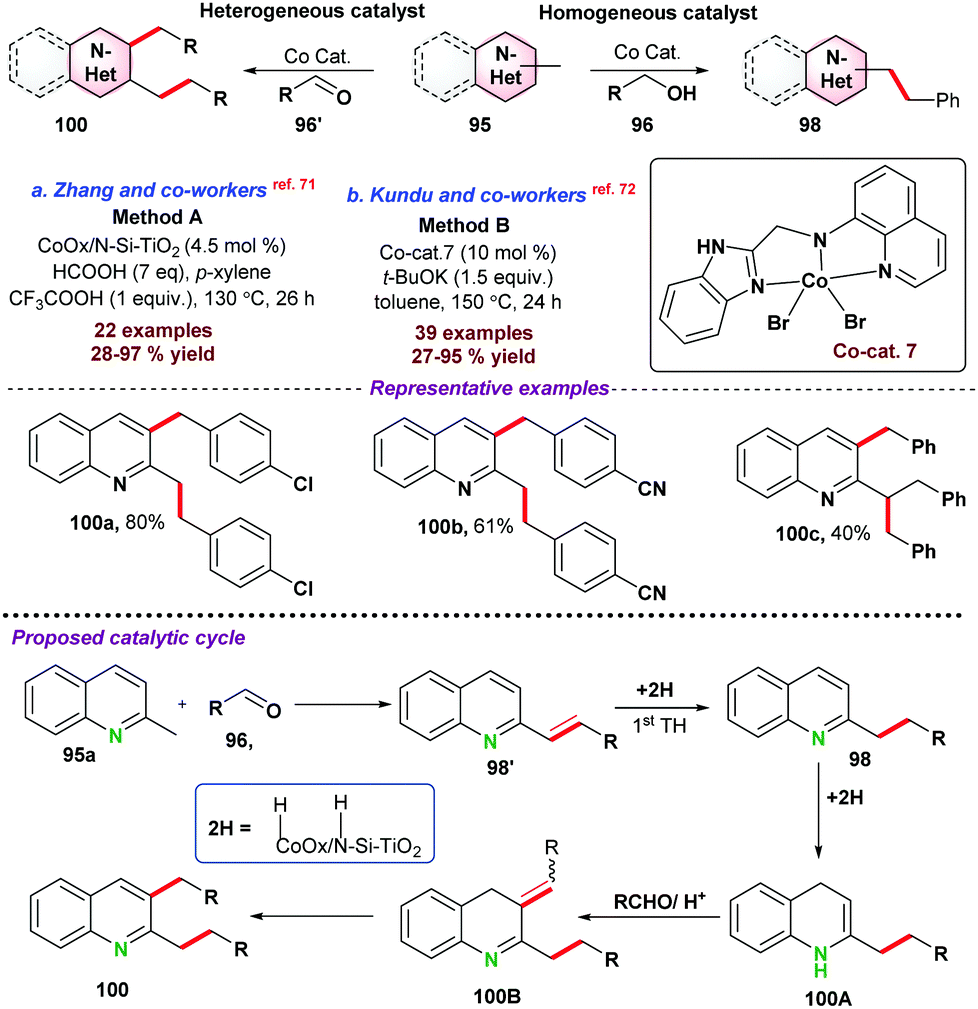
In 2016, Sundararaju and co-workers73 demonstrated cobalt catalyzed amidation of 8-methyl quinoline 101 through Csp3–H bond functionalization (Scheme 28a). The coupling of 8-methyl quinoline with oxazolone (as an amidating reagent) leads to the corresponding product 103 after the elimination of CO2 from oxazolone. During catalytic optimization studies, bis-amidation product was observed as a minor side product. For instance, in the case of aryl oxazolones, product yields significantly decreased due to the generation of the bis-amidation product. Again, due to steric hindrance, functionalization of more challenging secondary Csp3–H bond did not result in any product. Initial mechanistic studies and control experiments were performed to understand the individual role of the catalyst, additive, and AgSbF6.
 | ||
| Scheme 28 Co-catalyzed alkylation of N-heteroaromatics. | ||
Next, in 2018, Sharma and co-workers74 also reported the cobalt-catalyzed C-alkylation of 8-methyl quinoline 101 with maleimides 104 (Scheme 28b). A variety of functional groups were well tolerated under the optimized reaction conditions and N-alkyl or N-arylmaleimides were effectively utilized. Notably, alkylation of the more challenging secondary Csp3–H bond was obtained with moderate product yield.
Fan and co-workers75 employed a similar cobalt-catalyst system for the ring-opening addition of azabenzonorborna dienes with 8-methyl quinoline 101via Csp3–H bond activation (Scheme 28c). X-ray crystallography structure proved that the product was in cis-configuration. Fe(OAc)2 and AgSbF6 were used as an additive and Lewis acid, respectively, for the transformation. Deuterium labelling and kinetic experiments suggested that C–H bond cleavage was a reversible pathway, and the C–H bond cleavage step was the slowest step for the reaction. It was proposed that the reaction mechanism started with the formation of active species Cp*CoX2, formed by the reaction between [Cp*CoI2(CO)], AgSbF6, and Fe(OAc)2. In the next step, Cp*CoX2 coordinated with 101via the formation of a 5-membered cyclic intermediate complex-39, followed by coordination with an alkene to form complex-40. Subsequently, insertion of olefin resulted in a seven-membered ring intermediate species, which underwent β-nitrogen elimination to give ring-open intermediate complex-42, which on protonation provided the desired product 107a with the regeneration of active catalyst Cp*CoX2.
3.2. Alkenylation reaction
Olefins and E-di-substituted alkenes have attracted potential attention in chemical research as alkenes are widely utilized as precursors for fine and bulk chemicals, pharmaceuticals, and biologically active molecules. Though, chemical synthesis of E-olefins are well known following Witting reaction, Julia olefination, etc., which requires multistep manipulation of functional groups. Another important route involving transition metal catalyzed transformations provides a promising alternative for E-olefination in terms of selectivity. In this section, we have tried to summarize the recent advances for C-alkenylation of azaarenes via base-metal catalysis following functionalization of the Csp3–H bond. | ||
| Scheme 29 Mn-catalyzed alkenylation of N-heteroaromatics. | ||
At the same time, Kempe and co-workers77 introduced a phosphine ligand-based Mn-PNP-pincer catalyst for E-olefination of methyl heteroarenes 95 (Scheme 29b). Apart from excellent activity toward various substituted heteroarenes, the catalytic protocol was efficiently utilized towards the functionalization of the secondary Csp3–H bond. As proposed, initially complex-43 is formed by the reaction of the base and Mn-PNP catalyst. Next, dehydrogenation of alcohol into aldehyde resulted in the formation of Mn–H species (complex-44). In the next step, desired E-olefination products were obtained as presented in Scheme 29. Further, MnO2 was also used as a heterogeneous catalyst for the above transformations using air as a mild oxidant (Scheme 29c).78 The catalytic system could be recycled up to five times. However, the product yield significantly dropped during recycling experiments.
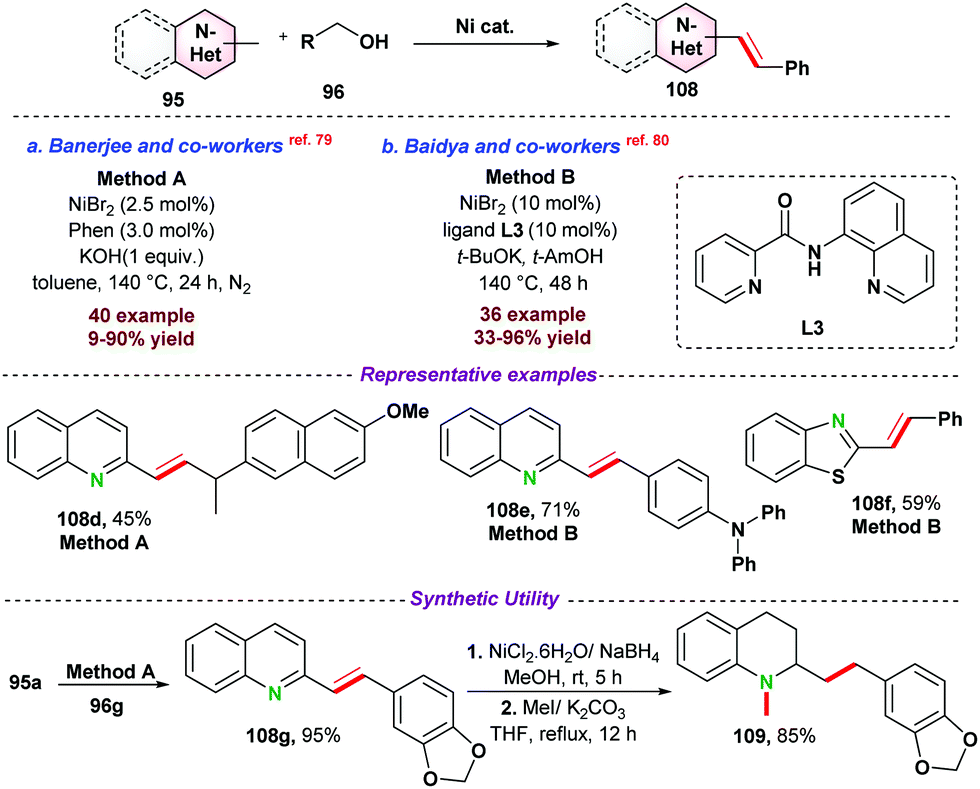 | ||
| Scheme 30 Ni-catalyzed alkenylation of N-heteroaromatics. | ||
 | ||
| Scheme 31 Fe-catalysed alkenylation of N-heteroaromatics. | ||
In 2020, Li and co-workers,82 explored the catalytic application of Fe(III)–nitrate–nonahydrate in combination with TEMPO and O2 as a terminal oxidant for olefination of N-heteroarenes under relatively milder reaction conditions (Scheme 31b). Drug molecule 108h (used for the treatment of Alzheimer's disease) was obtained in up to 70% isolated yield.
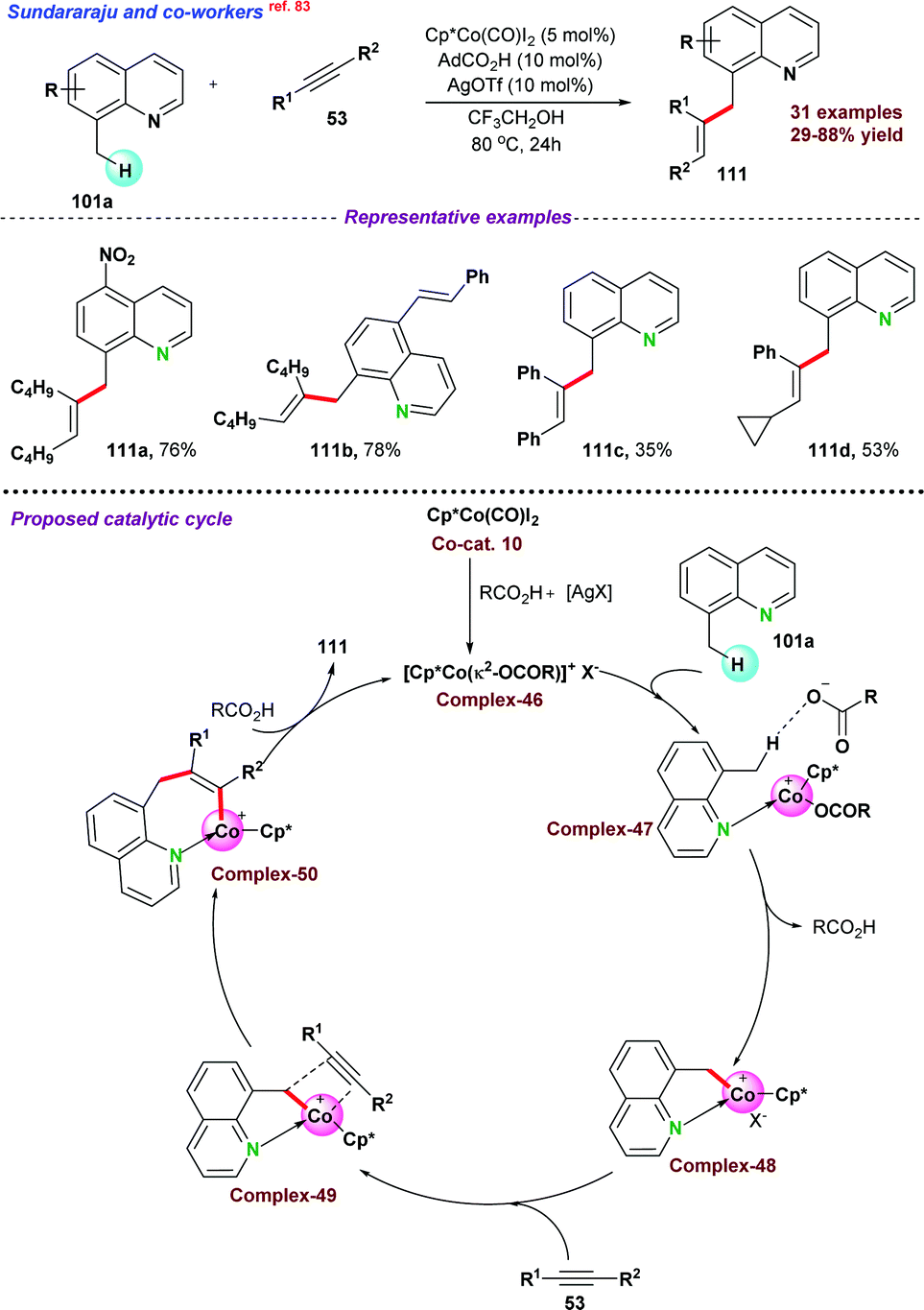 | ||
| Scheme 32 Co-catalyzed alkenylation of N-heteroaromatics. | ||
4. Conclusions
Non-precious 3d-transition metal-based C–H bond activation and functionalization of N-heterocycles is undoubtedly a fast-growing field, which has found an exciting possibility in synthetic organic chemistry beyond 4d and 5d-transitional metals. Since the last decade, earth-abundant metal-based catalysts have been involved in many challenging and important coupling reactions involving N-heteroarenes with alkyl halides, di-oxolane, allyl benzene, enynes, alcohols, alkyne, B2Pin2 as well as cyanating agents. These newly developed and evolving methodologies have widespread applications in the pharmaceutical industry, natural product chemistry, and dye industry. Most of the reported C–H activations proceed via de-protonative metalation, oxidative addition, insertion and reductive elimination for Csp2–H bonds, and radical abstraction as well as single electron transfer (SET) for Csp3–H bonds. Nevertheless, the use of this methodology in industrial processes is not very encouraging. Therefore, there is still scope for improvement in these areas for the development of higher industrial applications. Importantly, the synthesis of chiral molecules through C–H bond activation has not been explored thoroughly. We believe that further advances in the direction of enantioselective C–H functionalization by earth abundant 3d-metals will enrich the synthetic tools for the synthesis of chiral molecules. We further hope that this review will be a potential guide for future synthetic chemists working in academia and industry towards the utilization of 3d-metals for C–H functionalization of N-heterocycles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank CSIR-India (02(0373)/19/EMR-II) for financial support. DAE-BRNS, India (Young Scientist Research Award to D. B., 37(2)/20/33/2016-BRNS), IIT Roorkee (SMILE-32) and FIST-DST are gratefully acknowledged for instrument facilities. A. B. thank IIT Roorkee, L. M. K. thank UGC and S. B. thank INSPIRE (DST) New Delhi for financial support.Notes and references
- D. S. Latha and S. Yaragorla, Eur. J. Org. Chem., 2020, 2155–2179 CrossRef CAS.
- Y. Hu, B. Zhou and C. Wang, Acc. Chem. Res., 2018, 51, 816–827 CrossRef CAS PubMed.
- T. Aneeja, M. Neetha, C. M. A. Afsina and G. Anilkumar, Catal. Sci. Technol., 2021, 11, 444–458 RSC.
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS.
- I. Hussain and T. Singh, Adv. Synth. Catal., 2014, 356, 1661–1696 CrossRef CAS.
- J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1469 CrossRef CAS.
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS.
- B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308–1318 CrossRef CAS.
- (a) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef CAS; (b) S. Bera, L. M. Kabadwal and D. Banerjee, Chem. Commun., 2021, 57, 9807–9819 RSC; (c) L. M. Kabadwal, S. Bera and D. Banerjee, Org. Chem. Front., 2021 10.1039/D1QO01412A; (d) A. Bera, S. Bera and D. Banerjee, Chem. Commun., 2021 10.1039/D1CC04919D.
- A. H. Sandtorv, Adv. Synth. Catal., 2015, 357, 2403–2435 CrossRef CAS.
- J. A. Leitch, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 5618–5627 CrossRef CAS.
- V. P. Ananikov, ACS Catal., 2015, 5, 1964–1971 CrossRef CAS.
- R. A. Jagtap and B. Punji, Asian J. Org. Chem., 2020, 9, 326–342 CrossRef CAS.
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, Molecules, 2020, 25, 1909 CrossRef CAS PubMed.
- Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS.
- I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173–1193 RSC.
- L. Ackermann, Chem. Commun., 2010, 46, 4866–4877 RSC.
- R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086–9139 CrossRef CAS PubMed.
- L. Ackermann, J. Org. Chem., 2014, 79, 8948–8954 CrossRef CAS.
- R. Mei, U. Dhawa, R. C. Samanta, W. Ma, J. Wencel-Delord and L. Ackermann, ChemSusChem, 2020, 13, 3306–3356 CrossRef CAS PubMed.
- D. Wei, X. Zhu, J. L. Niu and M. P. Song, ChemCatChem, 2016, 8, 1242–1263 CrossRef CAS.
- Y. Kommagalla and N. Chatani, Coord. Chem. Rev., 2017, 350, 117–135 CrossRef CAS.
- S. Prakash, R. Kuppusamy and C. H. Cheng, ChemCatChem, 2018, 10, 683–705 CrossRef CAS.
- A. Thakur and J. Louie, Acc. Chem. Res., 2015, 48, 2354–2365 CrossRef CAS.
- X. H. Cai and B. Xie, ARKIVOC, 2015, i, 184–211 Search PubMed.
- N. A. Harry, S. Saranya, S. M. Ujwaldev and G. Anilkumar, Catal. Sci. Technol., 2019, 9, 1726–1743 RSC.
- S. M. Khake and N. Chatani, Chem, 2020, 6, 1056–1081 CAS.
- Ł. Woźniak and N. Cramer, Trends Chem., 2019, 1, 471–484 CrossRef.
- L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed.
- (a) G. Bertuzzi, L. Bernardi and M. Fochi, Catalysts, 2018, 8, 632 CrossRef; (b) X.-L. Liu, L.-B. Jiang, M.-P. Luo, Z. Ren and S.-G. Wang, Org. Chem. Front., 2021 10.1039/d1qo01223a.
- S. L. Liu, Y. Li, J. R. Guo, G. C. Yang, X. H. Li, J. F. Gong and M. P. Song, Org. Lett., 2017, 19, 4042–4045 CrossRef CAS PubMed.
- Q. Lu, F. J. R. Klauck and F. Glorius, Chem. Sci., 2017, 8, 3379–3383 RSC.
- Q. Lu, S. Cembellín, S. Greßies, S. Singha, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 1399–1403 CrossRef CAS.
- T. H. Meyer, W. Liu, M. Feldt, A. Wuttke, R. A. Mata and L. Ackermann, Chem. – Eur. J., 2017, 23, 5443–5447 CrossRef CAS.
- C. Zhu, J. C. A. Oliveira, Z. Shen, H. Huang and L. Ackermann, ACS Catal., 2018, 8, 4402–4407 CrossRef CAS.
- Z. Shen, H. Huang, C. Zhu, S. Warratz and L. Ackermann, Org. Lett., 2019, 21, 571–574 CrossRef CAS.
- R. A. Jagtap, P. P. Samal, C. P. Vinod, S. Krishnamurty and B. Punji, ACS Catal., 2020, 10, 7312–7321 CrossRef CAS.
- F. Pesciaioli, U. Dhawa, J. C. A. Oliveira, R. Yin, M. John and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 15425–15429 CrossRef CAS.
- H. Xie, Y. Shao, J. Gui, J. Lan, Z. Liu, Z. Ke, Y. Deng, H. Jiang and W. Zeng, Org. Lett., 2019, 21, 3427–3430 CrossRef CAS PubMed.
- A. Whyte, A. Torelli, B. Mirabi, L. Prieto, J. F. Rodríguez and M. Lautens, J. Am. Chem. Soc., 2020, 142, 9510–9517 CrossRef CAS PubMed.
- W. C. Lee, C. H. Chen, C. Y. Liu, M. S. Yu, Y. H. Lin and T. G. Ong, Chem. Commun., 2015, 51, 17104–17107 RSC.
- P. Naweephattana, B. Sawatlon and P. Surawatanawong, J. Org. Chem., 2020, 85, 11340–11349 CrossRef CAS.
- S. Lei, H. Cao, L. Chen, J. Liu, H. Cai and J. Tan, Adv. Synth. Catal., 2015, 357, 3109–3114 CrossRef CAS.
- C. Wang, S. Lei, H. Cao, S. Qiu, J. Liu, H. Deng and C. Yan, J. Org. Chem., 2015, 80, 12725–12732 CrossRef CAS.
- S. Lei, Y. Mai, C. Yan, J. Mao and H. Cao, Org. Lett., 2016, 18, 3582–3585 CrossRef CAS.
- R. Odani, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2015, 80, 2384–2391 CrossRef CAS.
- R. Kajiwara, S. Xu, K. Hirano and M. Miura, Org. Lett., 2021, 23, 5405–5409 CrossRef CAS PubMed.
- (a) H. Ohmiya, H. Zhang, S. Shibata, A. Harada and M. Sawamura, Angew. Chem., Int. Ed., 2016, 55, 4777–4780 CrossRef CAS; (b) A. K. Jha and N. Jain, Eur. J. Inorg. Chem., 2017, 4765–4772 CrossRef CAS.
- Y. Li, F. Qian, X. Ge, T. Liu, H. B. Jalani, H. Lu and G. Li, Green Chem., 2019, 21, 5797–5802 RSC.
- (a) Q. Yang, Z. Yin, L. Zheng, J. Yuan, S. Wei, Q. Ding and Y. Peng, RSC Adv., 2019, 9, 5870–5877 RSC; (b) X. L. Su, L. Ye, J. J. Chen, X. D. Liu, S. P. Jiang, F. L. Wang, L. Liu, C. J. Yang, X. Y. Chang, Z. L. Li, Q. S. Gu and X. Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 380–384 CrossRef CAS; (c) C. Li, B. Chen, X. Ma, X. Mo and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 2130–2134 CrossRef CAS; (d) X. Ma and G. Zhang, ACS Catal., 2021, 11, 5108–5118 CrossRef CAS.
- L. Shi, X. Zhong, H. She, Z. Lei and F. Li, Chem. Commun., 2015, 51, 7136–7139 RSC.
- H. Wang, F. Pesciaioli, J. C. A. Oliveira, S. Warratz and L. Ackermann, Angew. Chem., 2017, 129, 15259–15263 CrossRef.
- Y.-X. Tan, X.-Y. Liu, Y.-S. Zhao, P. Tin and G.-Q. Lin, Org. Lett., 2019, 21, 5–9 CrossRef.
- D. Das, A. A. Bhosle, P. C. Panjikar, A. Chatterjee and M. Banerjee, ACS Sustainable Chem. Eng., 2020, 8, 19105–19116 CrossRef CAS.
- C. S. Wang, S. Di Monaco, A. N. Thai, M. S. Rahman, B. P. Pang, C. Wang and N. Yoshikai, J. Am. Chem. Soc., 2020, 142, 12878–12889 CrossRef CAS.
- H. Kilic, M. Turgut, M. S. Yilmaz, O. Dalkilic and Ö. Metin, ACS Sustainable Chem. Eng., 2018, 6, 11433–11440 CrossRef CAS.
- H. Ren, Y. Zhou, Y. Bai, C. Cui and M. Driess, Chem. – Eur. J., 2017, 23, 5663–5667 CrossRef CAS.
- J. Li and L. Ackermann, Angew. Chem., 2015, 127, 3706–3709 CrossRef.
- H. Su, L. Wang, H. Rao and H. Xu, Org. Lett., 2017, 19, 2226–2229 CrossRef CAS.
- S.-Y. Chen, X.-L. Han, J.-Q. Wu, Q. Li, Y. Chen and H. Wang, Angew. Chem., Int. Ed., 2017, 56, 9939–9943 CrossRef CAS PubMed.
- Y. Yu, Y. Feng, R. Chauvin, S. Ma, L. Wang and X. Cui, Org. Lett., 2018, 20, 4209–4212 CrossRef CAS PubMed.
- G. Zheng, J. Sun, Y. Xu, S. Zhai and X. Li, Angew. Chem., Int. Ed., 2019, 58, 5090–5094 CrossRef CAS.
- (a) C. Zhu, R. Kuniyil and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 5338–5342 CrossRef CAS; (b) W.-B. Zhang, X.-T. Yang, J.-B. Ma, Z.-M. Su and S.-L. Shi, J. Am. Chem. Soc., 2019, 141, 5628–5634 CrossRef CAS; (c) J. Loup, V. Muller, D. Ghorai and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 1749–1753 CrossRef CAS; (d) J.-F. Li, W.-W. Xu, R.-H. Wang, Y. Li, G. Yin and M. Ye, Nat. Commun., 2021, 12, 3070–3078 CrossRef CAS.
- S. Roy, S. K. Das and B. Chattopadhyay, Angew. Chem., Int. Ed., 2018, 57, 2238–2243 CrossRef CAS.
- D. A. Babar and H. B. Rode, Eur. J. Org. Chem., 2020, 1823–1827 CrossRef CAS.
- C. Zhu, R. Kuniyil, B. B. Jei and L. Ackermann, ACS Catal., 2020, 10, 4444–4450 CrossRef CAS.
- A. Jana, A. Kumar and B. Maji, Chem. Commun., 2021, 57, 3026–3029 RSC.
- M. Vellakkaran, J. Das, S. Bera and D. Banerjee, Chem. Commun., 2018, 54, 12369–12372 RSC.
- J. Rana, R. Babu, M. Subaramanian and E. Balaraman, Org. Chem. Front., 2018, 5, 3250–3255 RSC.
- L. M. Kabadwal, S. Bera and D. Banerjee, Chem. Commun., 2020, 56, 4777–4780 RSC.
- R. Xie, F. Xie, C. J. Zhou, H. F. Jiang and M. Zhang, J. Catal., 2019, 377, 449–454 CrossRef CAS.
- A. Mishra, A. D. Dwivedi, S. Shee and S. Kundu, Chem. Commun., 2019, 56, 249–252 RSC.
- N. Barsu, M. A. Rahman, M. Sen and B. Sundararaju, Chem. – Eur. J., 2016, 22, 9135–9138 CrossRef CAS.
- R. Kumar, R. Kumar, D. Chandra and U. Sharma, J. Org. Chem., 2019, 84, 1542–1552 CrossRef CAS.
- H. Tan, R. Khan, D. Xu, Y. Zhou, X. Zhang, G. Shi and B. Fan, Chem. Commun., 2020, 56, 12570–12573 RSC.
- M. K. Barman, S. Waiba and B. Maji, Angew. Chem., Int. Ed., 2018, 57, 9126–9130 CrossRef CAS.
- G. Zhang, T. Irrgang, T. Dietel, F. Kallmeier and R. Kempe, Angew. Chem., Int. Ed., 2018, 57, 9131–9135 CrossRef CAS PubMed.
- C. Zhang, Z. Li, Y. Fang, S. Jiang, M. Wang and G. Zhang, Tetrahedron, 2020, 76, 130968 CrossRef CAS.
- J. Das, M. Vellakkaran and D. Banerjee, Chem. Commun., 2019, 55, 7530–7533 RSC.
- B. M. Ramalingam, I. Ramakrishna and M. Baidya, J. Org. Chem., 2019, 84, 9819–9825 CrossRef CAS.
- J. Das, M. Vellakkaran, M. Sk and D. Banerjee, Org. Lett., 2019, 21, 7514–7518 CrossRef CAS PubMed.
- Z. Zhang, Y. Ma, S. Dai, L. Li, Y. Zhang and H. Li, Tetrahedron Lett., 2020, 61, 151885–151888 CrossRef CAS.
- M. Sen, B. Emayavaramban, N. Barsu, J. R. Premkumar and B. Sundararaju, ACS Catal., 2016, 6, 2792–2796 CrossRef CAS.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |