
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free C–Se cross-coupling enabled by photoinduced inter-molecular charge transfer†
Chen
Zhu‡
 ,
Serik
Zhumagazy‡
,
Huifeng
Yue
* and
Magnus
Rueping
*
,
Serik
Zhumagazy‡
,
Huifeng
Yue
* and
Magnus
Rueping
*
KAUST Catalysis Center, KCC, King Abdullah University of Science and Technology, KAUST, Thuwal 23955-6900, Saudi Arabia. E-mail: huifeng.yue@kaust.edu.sa; magnus.rueping@kaust.edu.sa
First published on 29th November 2021
Abstract
Metal-free C–Se cross-couplings via the formation of electron-donor–acceptor (EDA) complexes have been developed. The visible-light induced reactions can be applied for the synthesis of a series of unsymmetrical diaryl selenides employing aryl bromides, aryl iodides as well as aryl chlorides under mild reaction conditions. The scale-up was readily achieved. UV-Vis spectroscopy measurements provide insight into the reaction mechanism.
Aryl and heteroaryl selenides represent an important class of organic compounds due to their application in the fields of pharmaceuticals, agrochemicals and materials (Fig. 1).1–7 As a consequence, the development of methods to access such compounds is highly sought-after. Among the developed strategies, palladium-8–10 or copper-11–13 catalyzed selenylation reactions of aryl halides play an important role in the construction of unsymmetrical diaryl selenides due to their high efficiency. However, the necessity of a transition-metal catalysts, strong bases, or high temperature limits the application of these protocols to a certain extent (Scheme 1a).
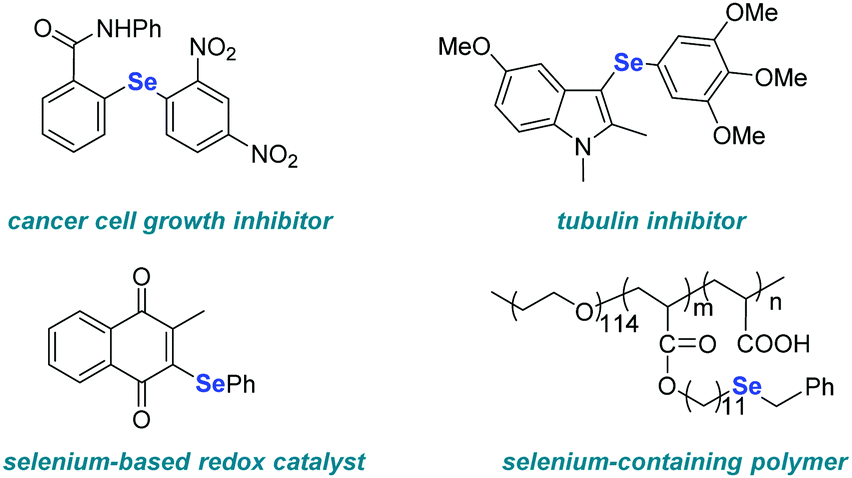 | ||
| Fig. 1 Examples of selenium-containing bioactive molecules, catalysts, and polymers. | ||
In recent years the use of the electron donor–acceptor (EDA) complex concept has led to the development of efficient cross-coupling reactions.14–16 In these cases, the stoichiometric electron-rich substrates (donors) and electron-deficient substrate (acceptors) form complexes in the ground state and these complexes can undergo intermolecular single-electron transfer (SET) upon irradiation with light.17–21 For instance, hydroimination cyclization products have been prepared via the EDA complex of dinitro-substituted O-aryl oximes and trimethylamine as demonstrated by Leonori and coworkers.22 Also, Miyake and coworkers realized the C–S bond formation via the EDA complex between electron-poor aryl halide and electron-rich thiolate anion.18 A C–N bond cross-coupling was realized by König and coworkers via the generation of an EDA complex between electron-poor (hetero)aryl halides and anilines.23 In addition, by employing the EDA strategy, Aggarwal and coworkers realized deoxygenative borylation reaction of 2-iodophenyl-thionocarbonates; thus converting efficiently aliphatic alcohols into boronic esters.24 Furthermore, a decarbonylative arylation of redox-active esters as well as the trifluoromethylthiolation of aldehydes have been achieved by Molander and coworkers via photoactive EDA complexes.25–27
In addition to these advances using stoichiometric reagents, new developments are being made in EDA catalysis. Pioneering work has been performed by Fu, Shang, and coworkers who realized the alkylation of silyl enol ethers, N-heteroarenes, and alkenes via EDA complex formation employing catalytic amount of sodium iodide and triphenylphosphine.28 Furthermore, catalytic amounts of B(C6F5)3 were applied in the oxidative C–S cross-coupling reaction of thiophenol with indoles.29 Considering the advantages of the EDA concept and the demand for general methods to construct diaryl selenides, we decided to investigate a metal-free C–Se bond cross-coupling enabled by photoinduced intermolecular charge transfer (Scheme 1b). We here describe the development of such metal-free C–Se bond forming reaction, the scale-up and mechanistic investigations to gain insight into the reaction mechanism.
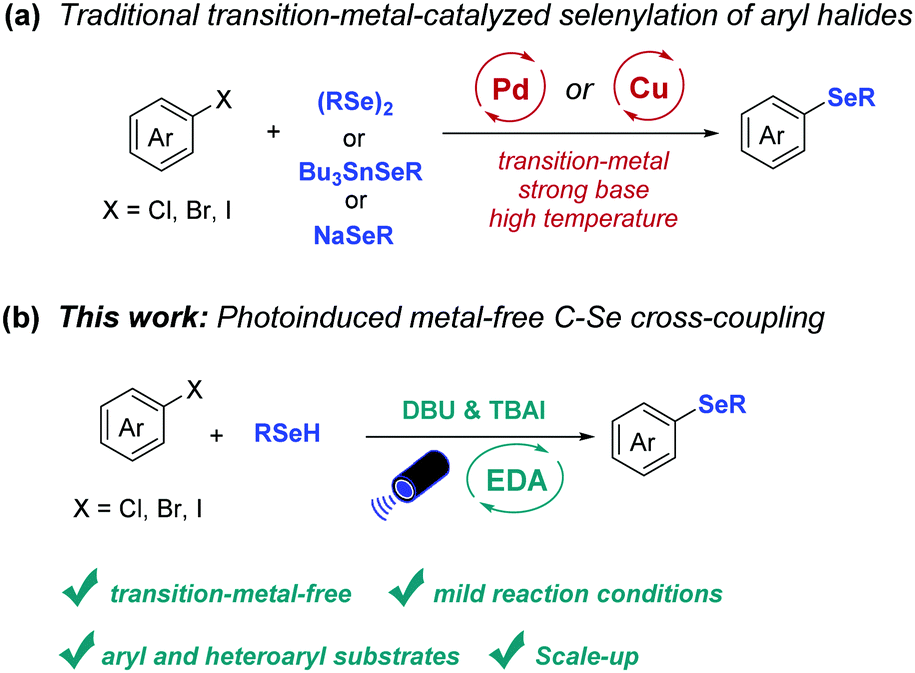 | ||
| Scheme 1 Currently available selenylation processes and the newly developed C–Se cross-coupling. | ||
We started to explore the photoinduced selenylation reaction of aryl halides by choosing 4-bromo-acetophenone 1a as a model substrate in reaction with benzeneselenol 2a (Table 1). After evaluation of the reaction parameters promising results were obtained by applying two equivalents of DBU as the base, DMF as the solvent, and 440 nm blue LED as light source (entry 1). Further optimization showed that the use of 390 nm purple LED resulted in lower yield (entry 2). Application of bases including K3PO4 and Cs2CO3 gave slightly lower yields (entries 3 and 4). Solvents such as DMA and DMSO resulted in decreased yield, while the use of MeCN improved the yield to 82% (entries 5–7). Moreover, the addition of 10 mol% of TBAI as an additive is beneficial for the transformation (entry 8).30,31
|
|
||
|---|---|---|
| Entry | Variables | Yieldb (%) |
| a Reaction conditions: 1a (0.20 mmol), 2a (0.40 mmol), DBU (2 equiv.) in DMF (1 mL) was irradiated under 440 nm blue LED at r.t. for 12 h. b GC Yields using dodecane as internal standard. c Reaction conditions as entry 8. | ||
| 1 | None | 75 |
| 2 | 390 nm purple LED | 65 |
| 3 | K3PO4 as base | 70 |
| 4 | Cs2CO3 as base | 72 |
| 5 | DMA as solvent | 68 |
| 6 | DMSO as solvent | 30 |
| 7 | MeCN as solvent | 82 |
| 8 | 10 mol% TBAI, MeCN as solvent | 90 |
| 9c | 0.5 mL MeCN | 70 |
| 10c | 2 mL MeCN | 82 |
| 11c | 20% light intensity | 65 |
| 12c | 75 °C | 73 |
| 13c | 1% V(H2O)/V(MeCN) | 76 |
| 14c | Under air | 0 |
| 15c | w/o DBU | 0 |
| 16c | w/o Blue LED | 0 |
Adjusting concentration, decreasing light intensity, elevating the temperature or adding water as additive led to lower yields (entries 9–13). The reaction gave no product when performed under air (entry 14). Control experiments showed that both, light and base are essential for the success of the reaction (entries 15 and 16).
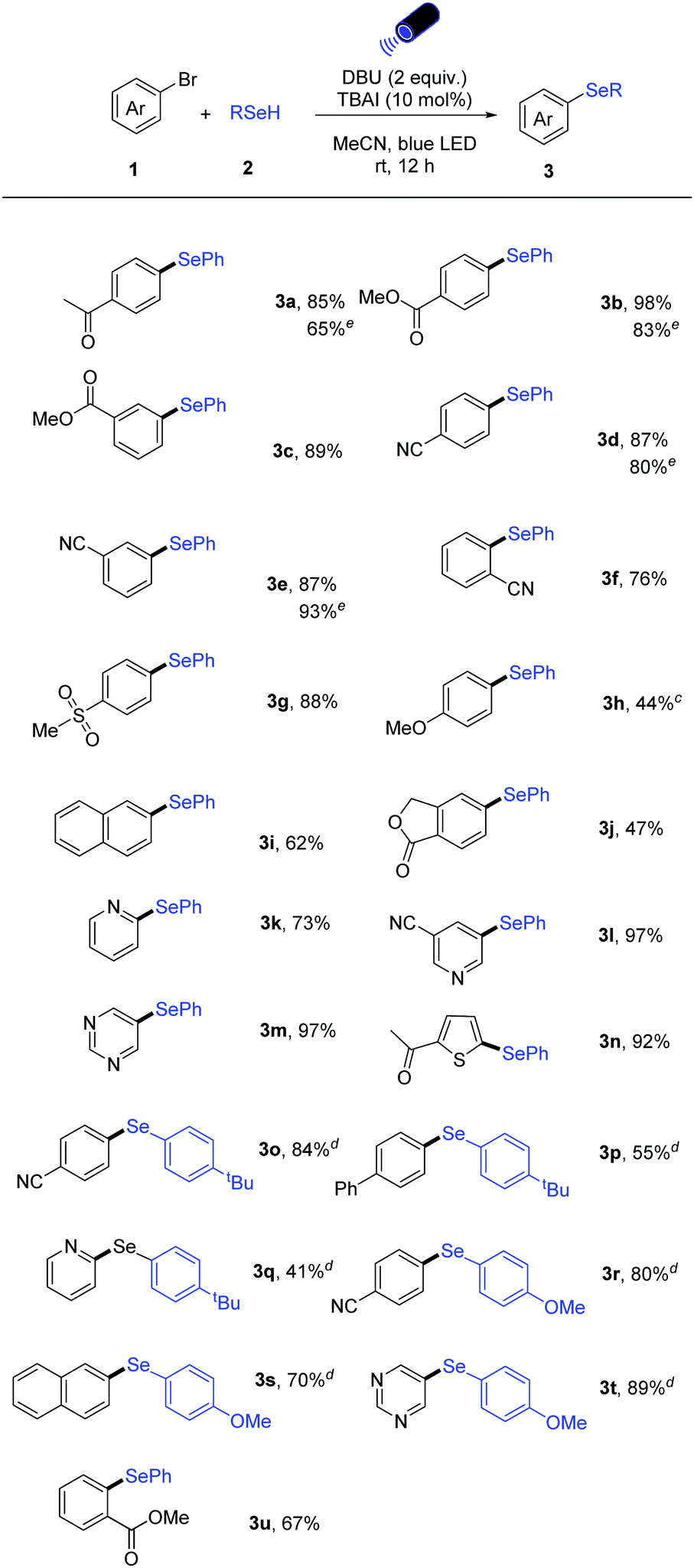
With the optimized reaction conditions in hand, the scope with respect to the aryl bromides was first explored (Table 2). A series of aryl halides bearing electron-withdrawing and electron-donating groups could be converted into the corresponding diarylselenide products. Functional groups including ketone, ester, cyano, sulfone as well as ether moieties are all tolerated in this photoinduced selenylation reaction (3a–3h). It is noteworthy that ortho-, meta-, and para-substituted aryl bromides all gave the corresponding products in high yields (3b–3f, and 3u). Also, π-extended naphthyl bromide and the bicyclic aryl bromide underwent this transformation effectively (3i and 3j). For electron-rich substrates (1h), the use of iodo-surrogates and 390 nm light source was found to be beneficial.
| a Reaction conditions: reaction conditions: 1 (0.20 mmol), 2 (0.40 mmol), DBU (2 equiv.), TBAI (10 mol%) in MeCN (1 mL) was irradiated under 440 nm blue LED at r.t. for 12 h. b Yield after purification. c 4-Iodoanisole, 390 nm LED and 2 equiv. NatOBu was used. d The reaction was performed on 0.10 mmol scale using the corresponding selenolate without the addition of DBU. e Aryl iodides were used. |
|---|
|
Importantly, pharmaceutically relevant heteroaryl bromides bearing pyridine, pyrimidine, thiophene reacted well in this catalytic system, affording the heteroaryl selenides in good to excellent yields (3k–3n). In addition, other sodium aryl selenolates could be applied in the reaction with both aryl- and heteroaryl bromides (3o–3t).
Moreover, our catalytic protocol can also be applied to aryl chlorides. Para, ortho ester- and para cyano-substituted aryl chlorides could be converted to the corresponding unsymmetrical diaryl selenides in high yields (Scheme 2).
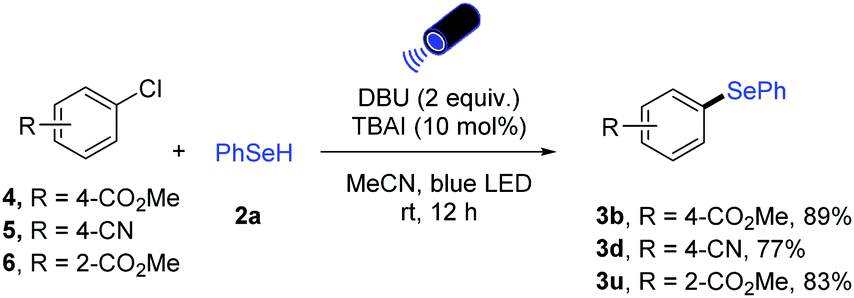 | ||
| Scheme 2 Selenylation of aryl chlorides. | ||
Furthermore, the effectiveness of this newly developed methodology is illustrated by the transformation of α-D-galactopyranose derivative 7 to the corresponding selenide 8 (82% yield, Scheme 3a). Furthermore, a gram-scale reaction with aryl bromide 1d was successfully performed in 73% yield, demonstrating the practicality of this metal-free selenylation protocol (Scheme 3b).
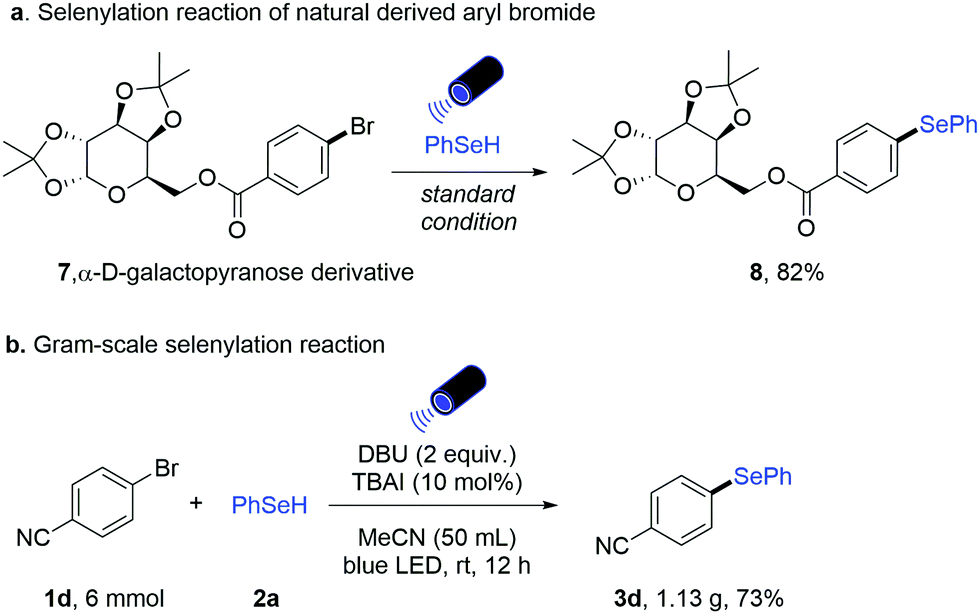 | ||
| Scheme 3 Selenylation of the nature-derived substrate and gram-scale reaction. | ||
To further understand the C–Se cross-coupling mechanism, UV-vis absorption spectra of mixtures 1a, 2a, and DBU in MeCN (path length = 1 cm) were measured at concentrations of 0.1 M (Scheme 4a). The absorption of selenol 2a increased considerably in the 400–475 nm region upon the addition of DBU, and the colour of the mixture of 2a and DBU turned to light yellow. Also, the subsequent addition of 4-bromo-acetophenone 1a to the mixture resulted in a stronger absorption, and the colour become orange, indicating an EDA complex is formed between 1a, and deprotonated 2a. The addition of 10% TBAI slightly enhanced the absorption of the mixture. Based on these results, the mechanism was proposed as follows (Scheme 4b): 1a and deprotonated 2a first forms the EDA complex. Upon the visible light irradiation, the intermolecular charge transfer event happened between the EDA complex from selenolate anion to the aryl halide, generating selenyl radical, aryl radical, and halide anion. The diaryl selenide forms via radical–radical cross-coupling with the release of an ammonium salt.
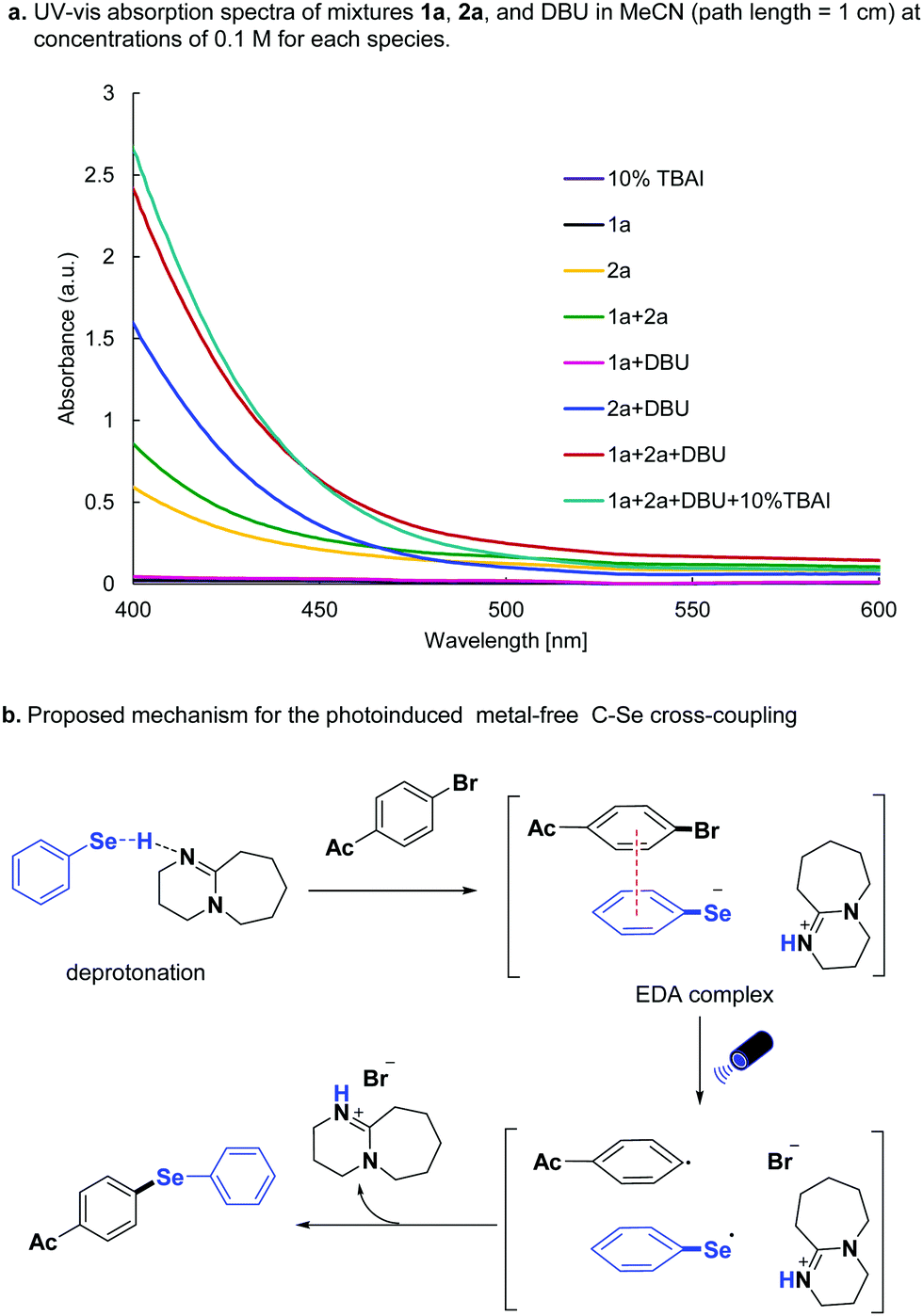 | ||
| Scheme 4 UV-Vis absorption study and proposed mechanism. | ||
In conclusion, a mild and efficient visible-light method for C–Se bond formation has been developed. The new protocol proceeds via a photoinduced intermolecular charge transfer between the EDA complex of aryl halide, selenol, and base. The practicality of this metal-free selenylation methodology is illustrated by the broad substrate scope, the late-stage functionalization of more complex molecules, and the successful gram-scale reaction. UV-Vis absorption spectroscopy measurements reveal that EDA complexes are involved in this process. The catalytic protocol is a general, metal-free, and low-cost alternative to transition-metal-catalyzed C–Se bond cross-coupling reactions and, thus, should find use in applications in which metal impurities are detrimental.
This work was financially supported by the King Abdullah University of Science and Technology (KAUST), Saudi Arabia, Office of Sponsored Research (URF/1/4384).
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. Kundu, RSC Adv., 2021, 11, 6682–6698 RSC.
- C. W. Nogueira, G. Zeni and J. B. Rocha, Chem. Rev., 2004, 104, 6255–6286 CrossRef CAS.
- B. K. Sarma, D. Manna, M. Minoura and G. Mugesh, J. Am. Chem. Soc., 2010, 132, 5364–5374 CrossRef CAS.
- G. Mugesh, W.-W. du Mont and H. Sies, Chem. Rev., 2001, 101, 2125–2180 CrossRef CAS PubMed.
- J. Trenner, C. Depken, T. Weber and A. Breder, Angew. Chem., Int. Ed., 2013, 52, 8952–8956 CrossRef CAS PubMed.
- H. Xu, W. Cao and X. Zhang, Acc. Chem. Res., 2013, 46, 1647–1658 CrossRef CAS.
- G. I. Giles, N. M. Giles, C. A. Collins, K. Holt, F. H. Fry, P. A. Lowden, N. J. Gutowski and C. Jacob, Chem. Commun., 2003, 2030–2031 RSC.
- E. Senol, T. Scattolin and F. Schoenebeck, Chem. – Eur. J., 2019, 25, 9419–9422 CrossRef CAS.
- I. Beletskaya, A. Sigeev, A. Peregudov and P. Petrovskii, Russ. J. Org. Chem., 2001, 37, 1463–1475 CrossRef CAS.
- Y. Nishiyama, K. Tokunaga and N. Sonoda, Org. Lett., 1999, 1, 1725–1727 CrossRef CAS.
- V. P. Reddy, A. V. Kumar, K. Swapna and K. R. Rao, Org. Lett., 2009, 11, 951–953 CrossRef CAS PubMed.
- D. Singh, E. E. Alberto, O. E. D. Rodrigues and A. L. Braga, Green Chem., 2009, 11, 1521–1524 RSC.
- N. Taniguchi and T. Onami, J. Org. Chem., 2004, 69, 915–920 CrossRef CAS PubMed.
- G. E. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed.
- C. G. Lima, T. de, M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixao, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS.
- S. V. Rosokha and J. K. Kochi, Acc. Chem. Res., 2008, 41, 641–653 CrossRef CAS.
- L. Candish, M. Teders and F. Glorius, J. Am. Chem. Soc., 2017, 139, 7440–7443 CrossRef CAS.
- B. Liu, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 13616–13619 CrossRef CAS.
- Y. Cheng and S. Yu, Org. Lett., 2016, 18, 2962–2965 CrossRef CAS.
- E. Arceo, I. D. Jurberg, A. Alvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750–756 CrossRef CAS.
- Y. Cheng, C. Mück-Lichtenfeld and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 16832–16836 CrossRef CAS PubMed.
- J. Davies, S. G. Booth, S. Essafi, R. A. Dryfe and D. Leonori, Angew. Chem., Int. Ed., 2015, 54, 14017–14021 CrossRef CAS.
- L. Marzo, S. Wang and B. König, Org. Lett., 2017, 19, 5976–5979 CrossRef CAS PubMed.
- J. Wu, R. M. Bär, L. Guo, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2019, 58, 18830–18834 CrossRef CAS PubMed.
- V. C. Polites, S. O. Badir, S. Keess, A. Jolit and G. A. Molander, Org. Lett., 2021, 23, 4828–4833 CrossRef CAS PubMed.
- L. M. Kammer, S. O. Badir, R.-M. Hu and G. A. Molander, Chem. Sci., 2021, 12, 5450–5457 RSC.
- A. Lipp, S. Badir, R. Dykstra, O. Gutierrez and G. Molander, Adv. Synth. Catal., 2021, 363, 3507–3520 CrossRef CAS.
- M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429–1434 CrossRef CAS PubMed.
- W. Yuan, J. Huang, X. Xu, L. Wang and X.-Y. Tang, Org. Lett., 2021, 23, 7139–7143 CrossRef CAS PubMed.
- G.-Z. Wang, M.-C. Fu, B. Zhao and R. Shang, Sci. China: Chem., 2021, 64, 439–444 CrossRef CAS.
- C. Liu, N. Shen and R. Shang, Org. Chem. Front., 2021, 8, 4166–4170 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc06152f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |