
Si-containing polycyclic aromatic hydrocarbons: synthesis and opto-electronic properties†
Thomas
Delouche
 a,
Ghizlene
Taifour
a,
Marie
Cordier
a,
Thierry
Roisnel
a,
Denis
Tondelier
b,
Payal
Manzhi
bc,
Bernard
Geffroy
bc,
Boris
Le Guennic
a,
Denis
Jacquemin
*d,
Muriel
Hissler
a and
Pierre-Antoine
Bouit
*a
a,
Ghizlene
Taifour
a,
Marie
Cordier
a,
Thierry
Roisnel
a,
Denis
Tondelier
b,
Payal
Manzhi
bc,
Bernard
Geffroy
bc,
Boris
Le Guennic
a,
Denis
Jacquemin
*d,
Muriel
Hissler
a and
Pierre-Antoine
Bouit
*a
aUniv Rennes, CNRS, ISCR – UMR 6226, Rennes 35000, France. E-mail: pierre-antoine.bouit@univ-rennes1.fr
bLaboratoire de Physique des Interfaces et des Couches Minces (LPICM), CNRS, Ecole Polytechnique, IP Paris, Palaiseau Cedex, France
cUniversité Paris-Saclay, CEA, CNRS, NIMBE, LICSEN, Gif-sur-Yvette, 91191, France
dCEISAM UMR CNRS 6230, University of Nantes, Nantes 44322, France. E-mail: denis.jacquemin@univ-nantes.fr
First published on 23rd November 2021
Abstract
We report a straightforward synthesis of Si-containing Polycyclic Aromatic Hydrocarbons (PAHs). The impact of π-extension and exocyclic modifications on both the optical and redox properties is investigated using a joint experimental/theoretical approach. By taking advantage of the solid-state luminescence of these derivatives, electroluminescent devices are prepared. Such preliminary opto-electronic results highlight that these heteroatom-containing PAHs are promising building blocks for organic electronics.
Polycyclic aromatic hydrocarbons (PAHs) are organic compounds consisting of ortho- and peri-fused aromatic rings.1 These derivatives appeared during the last few years as efficient semiconductors in a wide range of applications linked with plastic electronics (organic solar cells (OSCs), field-effect transistors, organic batteries, etc.). Various strategies have been used to tune their molecular properties and consequently the device performance. One of them is to modify the C-sp2 skeleton by introducing various ring-sizes (5, 6 or 7-membered rings) or edge configurations (armchair, zigzag, cove, etc.).2 Another approach is to introduce heteroatoms into the C-scaffold, a strategy successfully applied with second row heteroatoms such as B, N, and O.3 While the insertion of third row elements, such as P, also proved to be a valuable molecular engineering strategy,4 there are surprisingly few examples of Si-containing PAHs. This is rather surprising considering the important role played by silole (5-membered unsaturated Si-heterocycles) oligomers and polymers in opto-electronic applications, and notably OSCs.5 Such devices benefit from the low lying LUMO of the silole.6 Moreover, the possibility to prepare stable derivatives with unusual coordination numbers (hyper-7 or hypo-coordinated8) or displaying π-single bonding9 makes organosilicon derivatives very original scaffolds for π-conjugated system engineering. In order to take advantage of the presence of heteroatoms in PAHs, the main challenge is to find efficient synthetic pathways compatible with both the preparation of large C-sp2 frameworks and the presence of the heteroatom(s). Until now only a few “Si-compatible” approaches toward Si-doped PAHs A–D (Fig. 1) have been developed including lithiation, sila-Friedel–Crafts reaction, transition-metal catalysis and dehydro-Diels–Alder.10 In some cases like C,11 the compounds are mainly built as precursors for the corresponding B-doped PAHs, as it is the case for many group-14 containing acenes.12 For these reasons, the study of the opto-electronic properties of Si-containing PAHs is clearly underexplored.13
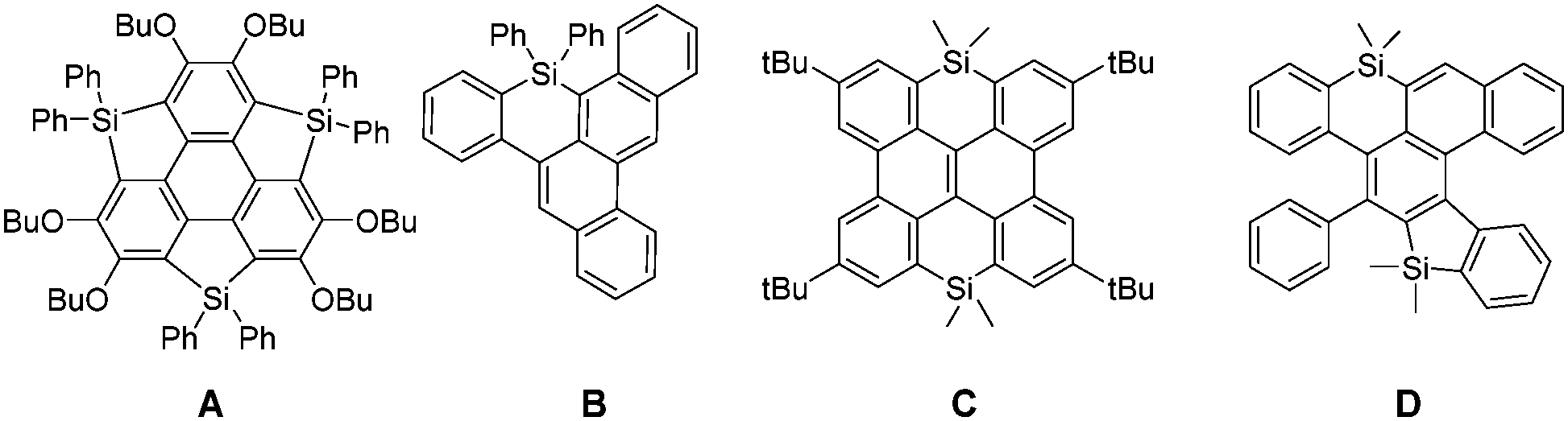 | ||
| Fig. 1 Examples of Si-containing PAHs A–D reported in the literature. | ||
To develop novel Si-containing PAHs, we decided to adapt the Ru-catalyzed annulation described by Fukuzawa et al.14 toward the preparation of PAHs featuring two Si-atoms and investigate the optical/redox properties and device incorporation of the novel derivatives generated in that way. Using commercial starting materials that we previously used to prepare polyaromatic bisphosphoniums,15 we synthesized in one step the corresponding dimethylsilane 1a (Scheme 1). 1a was then engaged in the Ru-catalyzed cyclization with diphenylacetylene.18 The resulting disilapyrene 2a was obtained in a moderate isolated yield, because of purification difficulties. 2a was fully characterized (see ESI†). To illustrate that this strategy allows for easy molecular engineering, various polyaromatic bis-silanes were converted into the corresponding disilapyrene 3 (isomer of 1a) and disilanthanthrene 4 in good yields (Scheme 1). One should note that this approach is considerably simpler than the previous route toward 3.16 The exocyclic Si-substituent could also be modified (from Me to Ph) for the preparation of 2b. In contrast to the reports based on the annulation of only one Si-cycle, the use of electron-rich or electron-poor alkynes led to incomplete cyclization products only (see Scheme S1 and Fig S12, ESI†). However, this approach allowed us to prepare a novel family of Si-doped PAHs 2–4 with variable π-extension and Si-substituents. All compounds are perfectly air and moisture stable and display good solubility in classical organic solvents.
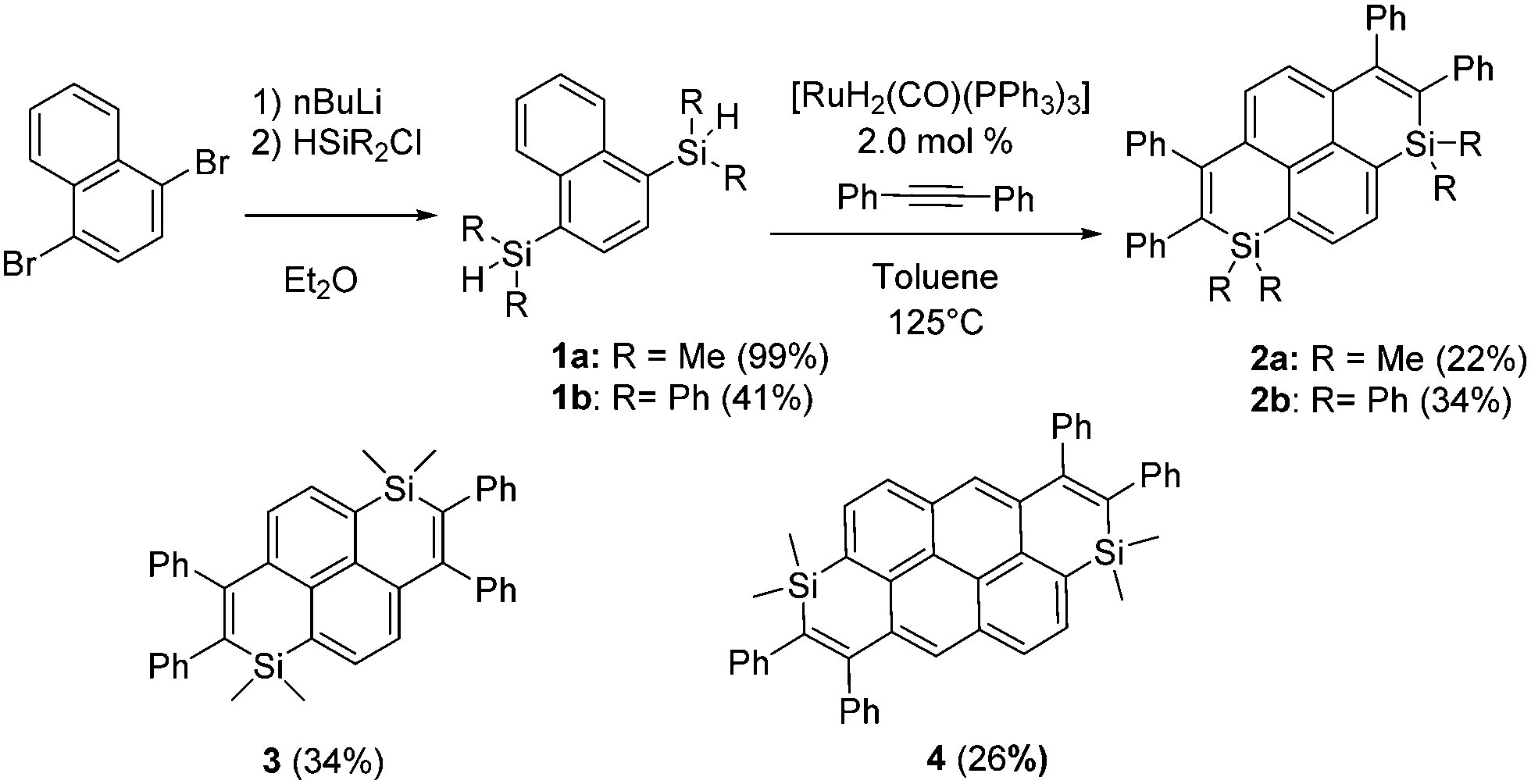 | ||
| Scheme 1 Synthetic access to 2a–b and chemical structures of compounds 3–4. | ||
The structures of 2a, 2b and 4 were confirmed by single-crystal X-ray diffraction (Fig. 2).17 In all compounds, the Si-atom shows a classical tetrahedral shape with usual valence angles (103° < C–Si–C < 104° for the intracyclic bonds, 108° < C–Si–C < 113° for the exocyclic bonds,) and bond lengths (1.85 Å <dSi–C< 1.86 Å, Table S2, ESI†). The non-equalized C–C bond lengths in the Si-heterocycle account for its non-aromaticity (1.35 Å < dC–C < 1.50 Å, Table S2, ESI†). The C-sp2 skeleton including the heterocycles is mainly planar (maximum deviation from the mean C-sp2 plane between 0.08 Å for 2a and 0.19 Å for 2b, Fig. 2), and the peripheral phenyls lie perpendicular to this plane. These structural characteristics are conserved in solution according to DFT calculations (Fig. S15, ESI†). No intermolecular interactions were observed in the crystal packing, likely due to the presence of tetrahedral Si-atoms and the steric hindrance of the peripheral phenyls. Such a property is beneficial for the design of solid-state emitters.
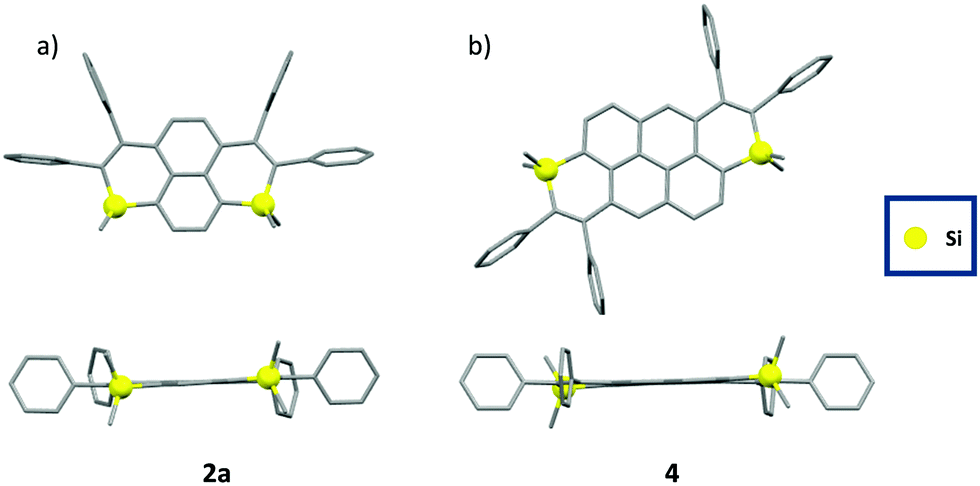 | ||
| Fig. 2 X-Ray crystallographic structures of 2a and 4 in top view (top) and lateral view (bottom). | ||
The optical properties (absorption/emission) of 2–4 have been investigated in dilute DCM solutions (c = 5.10−6 mol L−1, Fig. 3 and Table 1).17 They all display structured absorption bands in the 340–440 nm range, classical for PAH derivatives. This absorption is attributed to a delocalized π–π* transition with limited Si contributions according to TD-DFT calculations (Fig. 3 and Fig. S16, ESI†). While the absorption of disilapyrenes 2–3 markedly differs from that of pristine pyrene (red-shifted absorption), the properties of 4 are somehow similar to their C-sp2-hydrocarbon analogue (Table S3, ESI†).18 Both endo- and exo-skeletal modifications have minor impact on the absorption properties. However, as observed in the silole series, a small red-shift occurs when Me substituents of 2a are replaced by Ph in 2b, due to negative hyperconjugation within the Si ring (Fig. S16, ESI†).19 Moreover, one notices in Fig. S16 (ESI†) that the central carbon atoms are not involved in the transition in 2, contrary to 3 and 4. In addition in 4, we notice that the excitation is mostly centred on the four central phenyl rings, whereas the Si-containing rings play significantly stronger roles in 2 and 3.
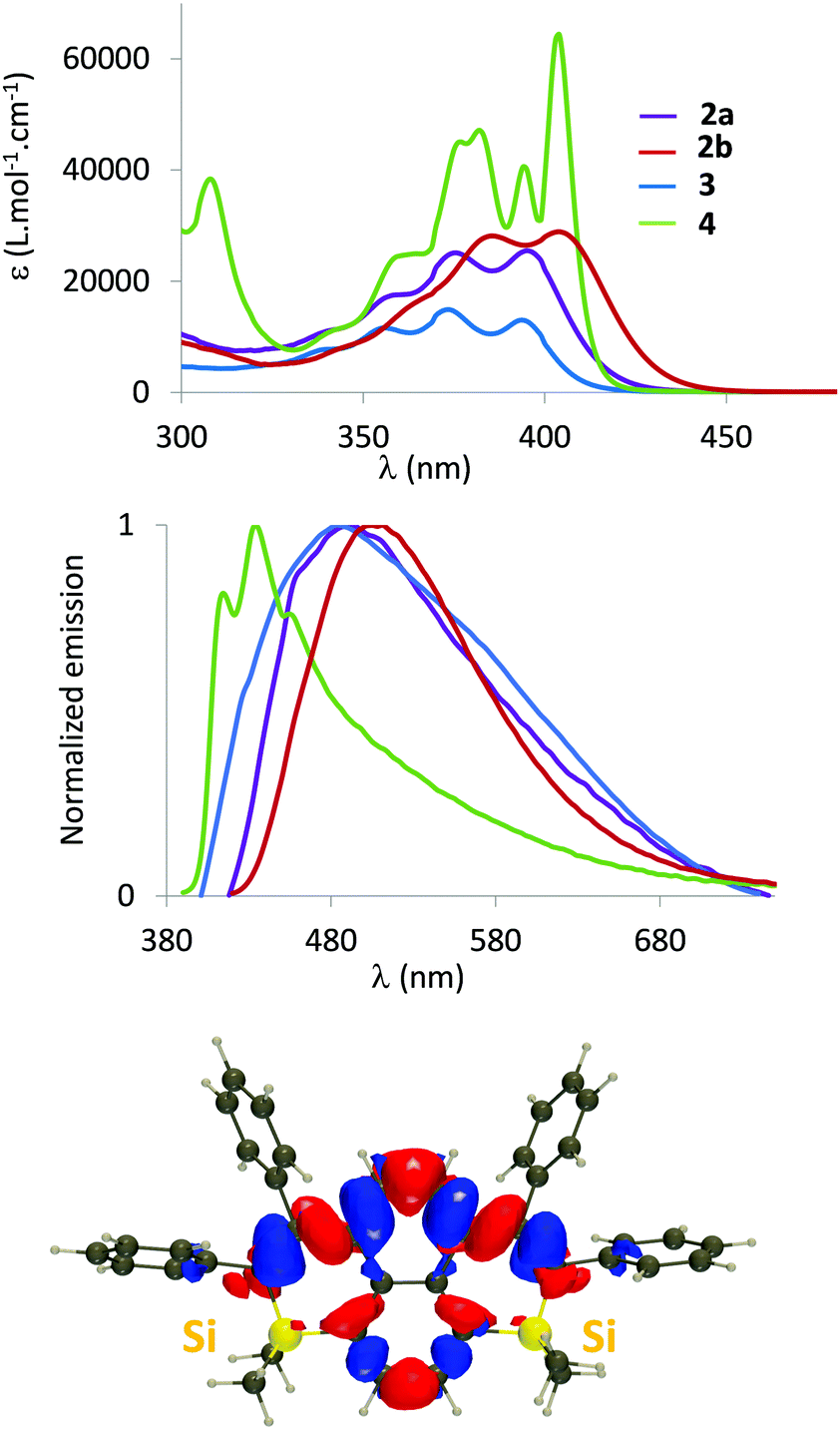 | ||
| Fig. 3 UV-Vis absorption (top) and normalized emission spectra (middle) of 2–4 in DCM (c = 5.10−6 M). Electron density difference computed for 2a (bottom). The blue and red regions, respectively, indicate the decrease and increase of electron density upon photon absorption (contour threshold: 8 10−4). | ||
| λ abs (nm) | ε (L Mol−1 cm−1) | λ em (nm) | ϕ (%) | σ (cm−1) | λ em (nm) | ϕ (%) | E ox (V vs. Fc) | |
|---|---|---|---|---|---|---|---|---|
| a In CH2Cl2 (10−5 M). b Measured using a calibrated integration sphere. c Measured in powder. d (c = 10−3 M) recorded in CH2Cl2 with Bu4N+PF6−(0.2 M) at a scan rate of 200 mV s−1. Potentials vs. Fc+/Fc. | ||||||||
| 2a | 395 | 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 500 |
480 | 6 | 4483 | 446 | 19 | +0.88 |
| 2b | 404 | 28900 |
499 | 25 | 4712 | 467 | 35 | +0.95 |
| 3 | 396 | 14900 |
485 | 2 | 4634 | 446 | 29 | +0.97 |
| 4 | 404 | 64200 |
435 | 10 | 1764 | 437 | 27 | +0.83 |
2–4 are also emissive in dilute solution in the 400–600 nm range. As can be seen in Table S4 (ESI†), there is an excellent agreement between the experimental and theoretical 0–0 energies. While the optical properties of 2a and 3 are very similar, we notice a slight red-shift of the emission for 2b, and more importantly, a blue-shifted emission for 4 (Table 1). This is somewhat surprising as 4 is the most π-extended derivative and as the position of the absorption peak is not strongly displaced as compared to the other systems. Interestingly, the theory accurately reproduces these trends (Table S4, ESI†), including the much smaller Stokes shift of 4 as compared to the other derivatives. Fig. S17 (ESI†) allows this behaviour to be rationalized: the ground and excited state geometries are extremely similar in 4, but significantly different in, e.g., 2a, consistent with the experimental trends in Stokes shifts. All compounds display low to moderate photoluminescence quantum yields in solution (2% < ϕ < 25%). As expected, 2–4 are fluorescent in powder form (Fig. S14, ESI† and Table 1) and the trend for the emission wavelengths in powders parallels the one found in solution. The luminescence quantum yields are considerably increased (19–35%, with the maximum value for 2b) compared to those of the dilute solutions. Such behaviour was previously observed for our related polyaromatic bisphosphoniums15 and can be rationalized on the basis of the rotation restriction of the peripheral phenyl rotors. Importantly, disilapyrenes 2 and 3 markedly differ from the pristine pyrene as no excimeric emission could be observed. The tetrahedral Si atom and the perpendicularly oriented exocyclic phenyl rings thus efficiently limit intermolecular interactions, as already observed in the crystals.
The redox properties of 2–4 were investigated by cyclic voltammetry in DCM (Fig. S15, ESI†). All derivatives display one quasi-reversible oxidation wave (for example, Eox(2a) = +0.88 V vs. Fc, Table 1). No reduction is observed under these experimental conditions. As expected, the use of more π-extended structure in 4 (Eox(4) = +0.83 V vs. Fc, Table 1) or richer groups on the Si-atom Me in 2avs. Ph in 2b (Eox(2b) = +0.95 V vs. Fc, Table 1) allows decreasing the oxidation potential. We also underline that the redox behavior of these compounds markedly differs compared with that of previously reported B- and P-analogs featuring similar carbon domains (Table S3, ESI†),19,20 which were strong electron acceptors. This nicely illustrates how one can tune the redox properties by modifying the main group element of the heteroatom-doped PAHs. The overall physico-chemical study further confirms that Si-containing PAHs are electron rich π-systems showing bright fluorescence in solution but also in the solid state, thus nicely completing the study of the data reported for A–D (Fig. 1).5b,14–16
Considering its thermal stability (Td10 = 411 °C, Fig. S18, ESI†) associated with favourable electronic properties (vide supra), 2b was used as an emitter in a multilayer organic light-emitting diode (OLED) architecture (see the ESI† for device details). Device performance is reported in Table S4 and Fig. S19–S21 (ESI†). When 2b is used as a dopant in a mCP (1,3-bis(N-carbazolyl)benzene) host matrix, the electroluminescence resembles the one of the dilute solution (λEL = 460 nm), with a turn on voltage Von of 4.2 V and a moderate external quantum efficiency (EQE) for a fluorescent emitter of 1.1% (Table S4, ESI†). However, non-quantitative energy transfer from the matrix is evidenced by the presence of residual mCP emission at high energy (Fig. S19, ESI†), independent of the doping rate (1–10%). Changing the mCP to CBP (4,4′-bis(N-carbazolyl)-1,1′-biphenyl) favours the total energy transfer when the doping rate is superior to 2% (Table S4 and Fig. S20, ESI†). In this case the EQE increases to 1.5%, the turn on voltage is 3.5 V and the maximal brightness is 2054 cd m−2 at 250 mA cm2. Interestingly, total energy transfer and EQE of 1% is also achieved upon solution processing of 2b using a PVK (poly(9-vinylcarbazole):oxadiazole (2:1)) matrix (Table S4 and Fig. S21, ESI†). These preliminary experiments illustrate that Si-containing PAHs such as 2b can be used in electroluminescent devices and more generally they fulfil stability requirements of opto-electronic devices.
In this communication, the straightforward synthesis of Si-containing PAHs 2–4 using a Ru-catalyzed synthetic approach was reported. The impact of π-extension and exocyclic modifications on the optical (absorption and emission) and redox properties has been investigated and rationalized using a joint experimental/theoretical approach. Finally, one compound with satisfying luminescence properties and thermal stability was selected to prepare an OLED device. This work highlights the potential of Si-doped PAHs for opto-electronic applications and paves the way toward the preparation of functional Si-containing nanographenes.
This work is supported by the Ministère de la Recherche et de l’Enseignement Supérieur, the CNRS, the Région Bretagne, the French National Research Agency (ANR Heterographene ANR-16-CE05-0003-01). Y. Molard and G. Taupier (Scanmat-UMS 2001) are thanked for PLQY measurements. This work used the computational resources of the CCIPL supercomputing center installed in Nantes.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. D. Watson, A. Fechtenkotter and K. Müllen, Chem. Rev., 2001, 101, 1267–1300 CrossRef CAS PubMed; (b) J. S. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS.
- (a) K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739–744 CrossRef CAS PubMed; (b) W. Zeng, T. Y. Gopalakrishna, H. Phan, T. Tanaka, T. S. Herng, J. Ding, A. Osuka and J. Wu, J. Am. Chem. Soc., 2018, 140, 14054–14058 CrossRef CAS.
- (a) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2016, 117, 3479–3716 CrossRef; (b) A. Narita, X.-Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC; (c) X.-Y. Wang, X. Yao, A. Narita and K. Müllen, Acc. Chem. Res., 2019, 52, 2491–2505 CrossRef CAS; (d) E. von Grotthuss, A. John, T. Kaese and M. Wagner, Asian J. Org. Chem., 2018, 7, 37–53 CrossRef CAS; (e) J. Liu and X. Feng, Synlett, 2020, 211–222 Search PubMed.
- R. Szűcs, P.-A. Bouit, L. Nyulászi and M. Hissler, ChemPhysChem, 2017, 18, 2618–2630 CrossRef PubMed.
- (a) T. Baumgartner and F. Jaekle, Main Group Strategies toward Functional Hybrid Materials, John Wiley & Sons, UK, 2018 Search PubMed; (b) E. Hey-Hawkins and M. Hissler, Smart Inorganic Polymers, John Wiley & Sons, UK, 2019 CrossRef.
- (a) M. Hissler, P. W. Dyer and R. Réau, Coord. Chem. Rev., 2003, 244, 1–44 CrossRef CAS; (b) X. Zhan, S. Barlow and S. R. Marder, Chem. Commun., 2009, 1948–1955 RSC.
- S. Yamaguchi, S. Akiyama and K. Tamao, J. Am. Chem. Soc., 2000, 122, 6793–6794 CrossRef CAS.
- B. Su, A. Kostenko, S. Yao and M. Driess, J. Am. Chem. Soc., 2020, 142, 16935–16941 CrossRef CAS.
- T. Nukazawa and T. Iwamoto, J. Am. Chem. Soc., 2020, 142, 9920–9924 CrossRef CAS PubMed.
- (a) S. Furakawa, K. Hayashi, K. Yamagishi and M. Saito, Mater. Chem. Front., 2018, 2, 929–934 RSC; (b) S. Furukawa, J. Kobayashi and T. Kawashima, J. Am. Chem. Soc., 2009, 131(40), 14192–14193 CrossRef CAS; (c) T. Tsuda, Y. Kawakami, S.-M. Choi and R. Shintani, Angew. Chem., Int. Ed., 2020, 59, 8057–8061 CrossRef CAS; (d) Y. Tokoro and T. Oyama, Chem. Lett., 2018, 47, 130–133 CrossRef CAS; (e) A. Mitake, R. Nagai, A. Sekine, H. Takano, N. Sugimura, K. S. Kanyiva and T. Shibata, Chem. Sci., 2019, 10, 6715–6720 RSC.
- (a) V. M. Hertz, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2015, 54, 8800–8804 CrossRef CAS; (b) V. M. Hertz, J. G. Massoth, M. Bolte, H. W. Lerner and M. Wagner, Chem. – Eur. J., 2016, 22, 13181–13188 CrossRef CAS PubMed.
- L. G. Mercier, W. E. Piers and M. Parvez, Angew. Chem., Int. Ed., 2009, 48, 6108–6111 CrossRef CAS PubMed.
- Z. Ma, C. Xiao, C. Liu, D. Meng, W. Jiang and Z. Wang, Org. Lett., 2017, 19, 4331–4334 CrossRef CAS PubMed.
- Y. Tokoro, K. Sugita and S. Fukuzawa, Chem. – Eur. J., 2015, 21, 13229–13232 CrossRef CAS.
- (a) T. Delouche, A. Vacher, E. Caytan, T. Roisnel, B. Le Guennic, D. Jacquemin, M. Hissler and P.-A. Bouit, Chem. – Eur. J., 2020, 26, 8226–8229 CrossRef CAS; (b) T. Delouche, A. Vacher, T. Roisnel, M. Cordier, J.-F. Audibert, B. Le Guennic, F. Miomandre, D. Jacquemin, M. Hissler and P.-A. Bouit, Mater. Adv., 2020, 1, 3369–3377 RSC.
- T. Saeki, A. Toshimitsu and K. Tamao, J. Organomet. Chem., 2003, 686, 215–222 CrossRef CAS.
- The X-ray structure and optical properties in solution of 3 have been described in ref. 16.
- (a) A. G. Crawford, A. D. Dwyer, Z. Liu, A. Steffen, A. Beeby, L.-O. Pålsson, D. J. Tozer and T. B. Marder, J. Am. Chem. Soc., 2011, 133, 13349–13362 CrossRef CAS; (b) B. K. Shah, D. C. Neckers, J. Shi, E. Forsythe and D. Morton, J. Phys. Chem. A, 2005, 109, 7677–7681 CrossRef CAS PubMed.
- H. Chen, M. Denis, P.-A. Bouit, Y. Zhang, X. Wei, D. Tondelier, B. Geffroy, Z. Duan and M. Hissler, Appl. Sci., 2018, 8, 812 CrossRef.
- J. M. Farrell, C. Mützel, D. Bialas, M. Rudolf, K. Menekse, A.-M. Krause, M. Stolte and F. Würthner, J. Am. Chem. Soc., 2019, 141, 9096–9104 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1982459, 1982458, 1982457 and 2001027. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc06309j |
| This journal is © The Royal Society of Chemistry 2022 |