
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Long-lived, near-IR emission from Cr(III) under ambient conditions†
Natalia
Sawicka
a,
Chloe J.
Craze
a,
Peter N.
Horton
b,
Simon J.
Coles
 b,
Emma
Richards
*a and
Simon J. A.
Pope
*a
b,
Emma
Richards
*a and
Simon J. A.
Pope
*a
aSchool of Chemistry, Main Building, Cardiff University, Cardiff CF10 3AT, Cymru/Wales, UK. E-mail: Richardse10@cardiff.ac.uk; popesj@cardiff.ac.uk
bUK National Crystallographic Service, Chemistry, Faculty of Natural and Environmental Sciences, University of Southampton, Highfield, Southampton, SO17 1BJ, England, UK
First published on 19th April 2022
Abstract
Bis-terdentate (N^N^N) ligands coordinated to Cr(III) yield complexes that display near-IR emission under aerated solvent conditions at room temperature.
Cr(III) complexes have attracted interest due to their optical,1 (photo)redox and magnetic properties.2 Cr(III) photoluminescence,3 from a discreet coordination complex was first reported in the 1960s.4 The photophysics of Cr(III) complexes is generally dominated by metal-centred excited states.5–7 A strong, pseudo-octahedral ligand field at Cr(III) yields the possibility for populating doublet excited states (2E and 2T), while simultaneously preventing 4T2/2E back intersystem crossing. As relaxation from these excited states to the 4A2 ground state is spin forbidden, long-lived phosphorescence can be observed. However, Cr(III) species are often hampered by poor emissivity. Deoxygenated, deuterated solvents and deuteriation of ligands has been employed to help minimise quenching from non-radiative multiphonon relaxation pathways.8
In contrast to well-known precious metal lumophores, earth-abundant Cr(III) complexes of archetypal diimine ligands typically show Cr-centred phosphorescence in the 650–760 nm range. Related heteroleptic complexes provide some rational tuning of emission within similar wavelength ranges.9
Scheme 1 describes some important luminescent Cr(III) complexes, most of which have been reported in the last few years. Consideration of the N–Cr–N bite angles of the coordinated ligands is key. Thus, larger six-membered chelate rings are well suited in optimizing N–Cr–N bite angles towards 90° and this in turn reduces non-radiative deactivation.7 Recent studies have clearly demonstrated the use of ligands such as N,N′-dimethyl-N-N′-dipyridine-2-yl-pyridine-2,6-diamine (ddpd),10 2,6-bis(2-pyridylmethyl)pyridine (bpmp),11 and 1,1,1-tris(pyrid-2-yl)ethane (tpe)12 that yield Cr(III) complexes with more intense emission properties and longer lifetimes.
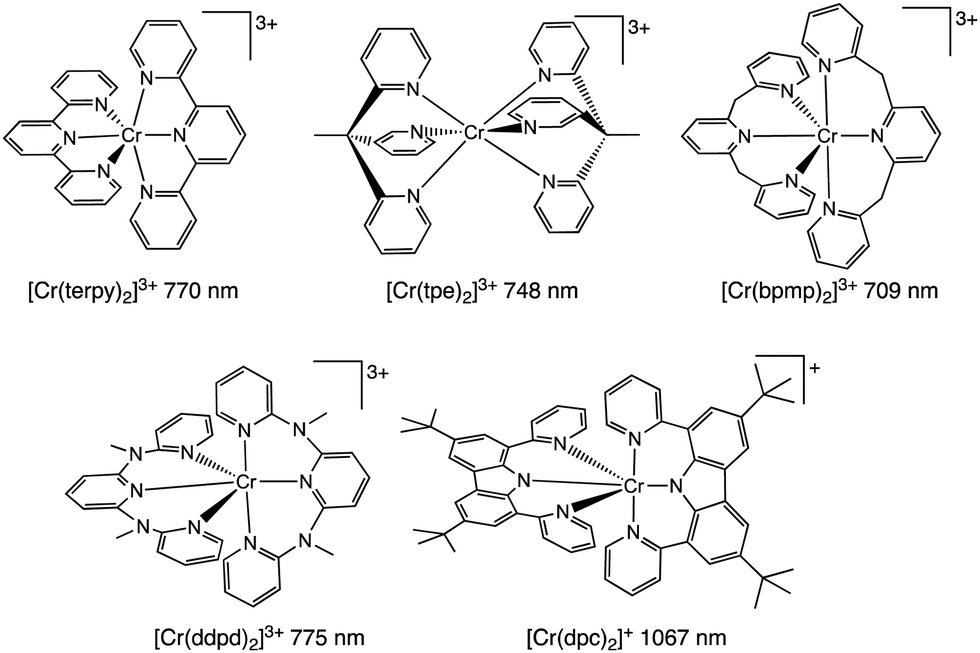 | ||
| Scheme 1 Molecular structures of emissive Cr(III) complexes (with indicative luminescence wavelengths) using terdentate (N^N^N) ligands. | ||
During our current studies, Wenger, Piguet and co-workers described the first report of a Cr(III) complex with emission in the near-IR-II region (ca. 1067 nm) using ligand fields that combine optimised octahedral geometries with both σ- and π-donor/π-acceptor characteristics that potentially increase metal–ligand covalency.13 This was achieved using a 3,6-di-tert-butyl-1,8-di(-pyridine-2-yl)-carbazole (dpc) ligand (Scheme 1) which imparts trans N–Cr–N bond angles of around 174° for [Cr(dpc)2]+. Excitation at 450 nm yielded an emission peaking at 1067 nm (with dual lifetimes of 1.4 and 6.3 μs) assigned to a possible admixture of 2LMCT/2E excited states. Unfortunately, the complex was only emissive at 77 K in a frozen matrix suggesting that further advances are necessary to identify near-IR luminescent Cr(III) species with broader potential applicability and utility.
We now report the development of luminescent Cr(III) complexes using 1,3-bis(2′-pyridylimino)-isoindoline (bpi) derivatives as terdentate N^N^N ligands.
The two Hbpi pro-ligands (Scheme 2) were synthesized in a similar manner to previous reports on closely related complexes.14 Reaction of the phthalonitrile reagent with a 2-aminopyridine species in the presence of CaCl2 resulted15 in the formation of the Hbpi ligands (see ESI† for details and characterisation data).
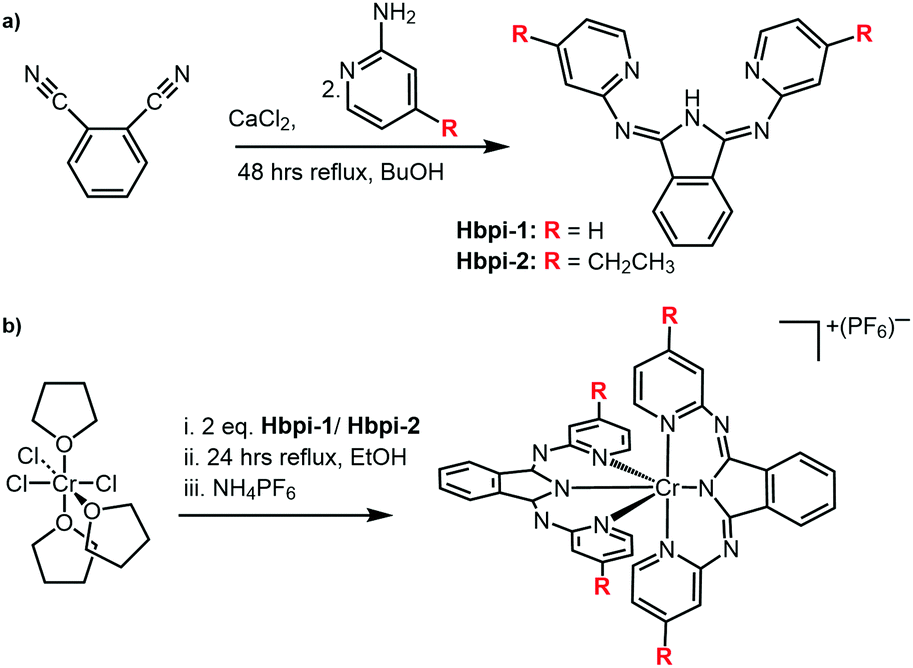 | ||
| Scheme 2 (a) The synthetic route towards Hbpi ligands, and (b) coordination of the ligands to the Cr(III) centre. Full reagents and conditions are detailed in the ESI.† | ||
For the coordination chemistry, mer-CrCl3(THF)3 was reacted with two equivalents of ligand in EtOH to yield [Cr(bpi)2]PF6 (see ESI† for details). The complexes were purified using HPLC (using a water/MeCN solvent gradient) and corresponding high resolution mass spectra were obtained for both complexes (Fig. S1, ESI†) confirming the proposed formulation where each ligand is deprotonated and thus the complexes are monocationic.
Single, red needle crystals of [Cr(bpi-1)2]PF6 suitable for X-ray diffraction16 were obtained from slow evaporation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 diethyl ether/toluene/acetonitrile solution of complex (Table S1, ESI†) over 7 days. The structure revealed the complex crystallised as the diethyl ether solvate.
:1:1 diethyl ether/toluene/acetonitrile solution of complex (Table S1, ESI†) over 7 days. The structure revealed the complex crystallised as the diethyl ether solvate.
The bond angles that describe the coordination sphere can have a fundamental influence upon the photophysics of Cr(III): the N–Cr–N bite angles reveal the extent of distortion away from Oh geometry. For [Cr(bpi-1)2]PF6 the structure (Fig. 1) shows three trans N–Cr–N angles of 172.8(2)°, 175.6(2)° and 173.5(2)° (average = 174.0°). The average Cr–N bond length is 2.0615 Å with the Cr–N(isoindoline) bonds giving the two shortest distances. In comparison the N–Cr–N angles for [Cr(dpc)2]PF6 are 173.69(8)°, 173.66(7)°, 173.16(7)°, which are clearly comparable, and the Cr–N bond lengths also show a similar trend and average value. Therefore the bpi-1 ligand imparts (Tables S2 and S3 ESI†) a favourable geometry at Cr(III).
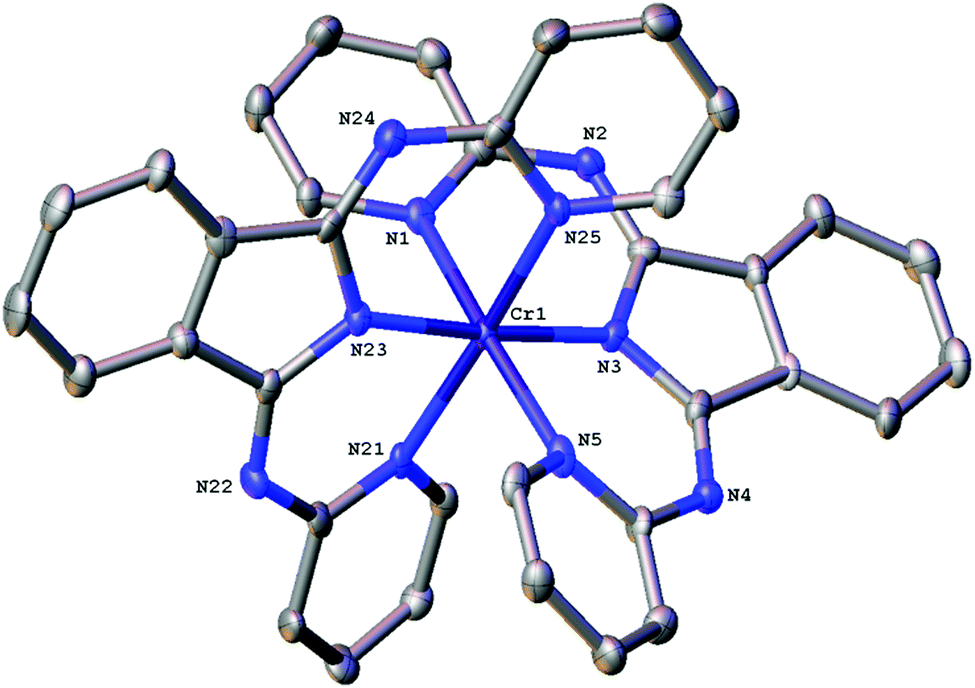 | ||
| Fig. 1 X-Ray structure of [Cr(bpi-1)2]PF6. (H atoms, the counter ion and solvent molecules are omitted for clarity, ellipsoids are shown at 50% probability level.) Selected bond lengths (Å) and angles (°): N1–Cr 2.097(6), N5–Cr 2.088(6), N3–Cr 1.993(6), N21–Cr 2.107(6), N25–Cr 2.098(6), N23–Cr 1.986(6); N1–Cr–N5 172.8(2), N3–Cr–N23 175.6(2), N21–Cr–N25 173.5(2), N1–Cr–N3 86.2(2), N1–Cr–N21 92.1(2), N1–Cr–N23 91.0(2), N1–Cr–N25 90.4(2), N5–Cr–N3 87.4(2), N5–Cr–N21 85.3(2), N5–Cr–N23 95.6(2), N5–Cr–N25 92.9(2). | ||
To probe the electronic properties, a series of X-band EPR spectra were recorded for the complexes as frozen solutions in MeCN (140 K) and revealed broad signals at ∼340 mT (g ∼2), confirming the paramagnetic nature of the high-spin S = 3/2 centre of the ground state Cr3+ (3d3) cation. Due to the poor resolution of the frozen solution spectra, both [Cr(bpi-1)2]PF6 and [Cr(bpi-2)2]PF6 were prepared as 1% doped solids in the corresponding [Co(bpi)2]PF6 matrix, a methodology previously indicated by Di Bilio et al.17 to have good success in characterisation of Cr(III) systems and commonly utilised in EPR studies of first-row transition metal complexes.18,19 This approach resulted in some improvements although the spectra are still complicated by some anisotropy within the material, presumably arising from uptake of the dopant cation in several sites within the host cobalt matrix (see Fig. 2; the cobalt matrices yielded no resolvable signals in their EPR spectra, confirming the diamagnetic nature of the host lattice). The Cr(III) spectra can be fully described by the spin Hamiltonian parameters for S >1/2 systems:
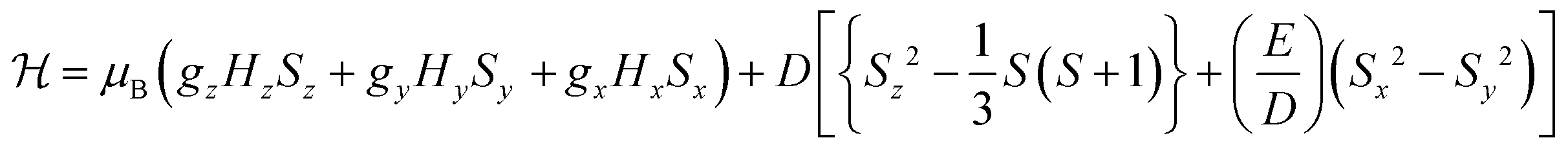
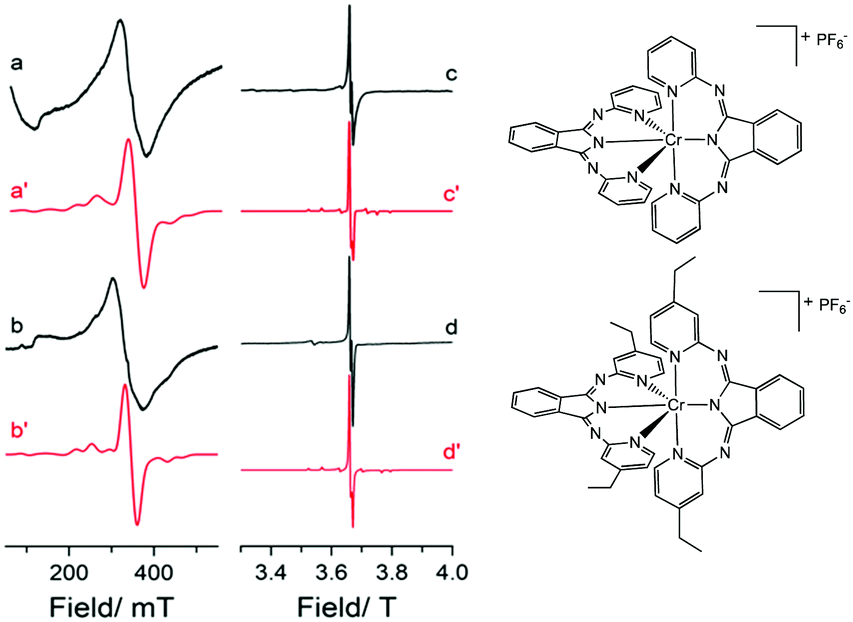 | ||
| Fig. 2 Experimental and simulated EPR spectra of (a and c) [Cr(bpi-1)2]PF6 and (b and d) [Cr(bpi-2)2]PF6 recorded as 1% doped solids in their corresponding diamagnetic cobalt matrix. Experimental data were recorded at (a and b) X-band frequency, 140 K, and (c and d) W-band frequency, 20 K. | ||
The small negative shift from ge ≈ 2.0023 for the two complexes is to be expected as a result of the less than half-filled d-orbitals, and the relatively small spin–orbit coupling parameter of the central chromium ion (ζCr3+ = 275 cm−1).21 The experimentally determined ZFS parameters (Table S4, ESI†) are noticeably smaller than other mononuclear Cr(III) complexes for which D is typically <0.6 cm−1. This is justified (see Table S2) given that a purely cubic field would lead to D = 0.22 Furthermore, there is good agreement with the complex [CrLCl3] that displays similar geometry, incorporating a tridentate ligand (L = (4′-2-pyridyl)-2,2′:6′,2′′-terpyridine; similar Cr–N bond lengths) and a π-donor (Cl−) in the axial coordinate site (known to increase the value of D; see ESI†).23
The redox characteristics of [Cr(bpi-1)2]PF6 and [Cr(bpi-2)2]PF6 were obtained through cyclic voltammetry. Firstly, the resultant voltammograms (Fig. S5, ESI†) did not show any features in the oxidative cathodic region. This is in general agreement with previous reports24 on the redox features of Cr(III) complexes, including [Cr(ddpd)2]3+, but Wenger and co-workers report a reversible feature for [Cr(dpc)2]+ca. +0.46 V vs. Fc0/+ which was ascribed to a Cr3+/4+ redox couple.13 Polypyridyl Cr(III) complexes such as [Cr(bipy)2]3+ (ref. 25) and [Cr(terpy)2]3+ (ref. 26) are well known to show many ligand-based, reversible reductions.27 For [Cr(bpi-1)2]PF6 we observed four reversible or quasi-reversible features in the anodic window (E1/2 at −0.55, −1.19, −1.49 and −1.72 V) which are mainly assigned to ligand-based processes. The voltammogram for [Cr(bpi-2)2]PF6 was slightly different with reversible and irreversible features at −0.75, −1.05 and −1.38 V suggesting poorer electrochemical stability. It was notable that the first reversible reduction was subtly shifted to a more negative potential versus [Cr(bpi-1)2]PF6 and thus consistent with a ligand-based process. The second, irreversible, feature at around −1.05 V may therefore be due to the Cr3+/2+ couple as noted in both [Cr(ddpd)2]3+ and [Cr(dpc)2]+ca. −1.1 V.
The UV-vis absorption properties of the complexes were ascertained using MeCN solutions. For reference, the ligands (Fig. S6, ESI†) absorb similarly between ca. 250–425 nm which is attributed to the different π → π* transitions (>10000 M−1 cm−1) within the aromatic units. At 10−5 M the spectra of the complexes reveal features that are attributable to different ligand-based transitions (ε >5000 M−1 cm−1), with overlapping spin-allowed charge transfer bands also likely to contribute (supported by DFT, Fig. S7, ESI†). A moderately intense band at 450–525 nm is ascribed to an intraligand charge transfer (ILCT) arising from the amido donor of the isoindoline moiety and the conjugated iminopyridine groups; the subtle hypsochromic shift observed for ethyl-functionalised [Cr(bpi-2)2]PF6 is consistent with this assignment. These absorption features and the tail of the ILCT band appear to obscure the spin-allowed (4A2 → 4T2, 4A2 → 4T1(F), 4A2 → 4T1(P)) d–d transitions expected for Cr(III). At higher concentrations and longer wavelengths both complexes displayed a band ca. 750 nm (∼10 M−1 cm−1) and another even weaker feature ∼850 nm. It is likely that one of these bands is due to the spin forbidden transition 4A2 → 2E/2T1. Previous work on related [Cr(dpc)2]+ has proposed, through computational studies, that a 2LMCT band may also contribute in this region.13
For the luminescence studies, the complexes were prepared in MeCN and excited using 430 nm irradiation. As shown in Fig. 3, room temperature emission spectra were obtained under ambient, aerated sample conditions. [Cr(bpi-1)2]PF6 has a main feature at approximately 950 nm with a shoulder to lower energy ca. 1050 nm with a tail extending to 1200 nm; the peak ca. 1270 nm is assigned to the 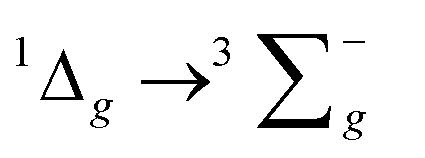 transition of oxygen. In a frozen matrix (77 K, MeCN) the emission maximum is hypsochromically shifted by ∼20 nm, the shoulder is somewhat more pronounced and photogeneration of 1O2 is inhibited. For [Cr(bpi-2)2]PF6 a relative hypsochromic shift in emission maximum was observed at λem = 929 nm (Table 1), which is attributed to the effect of the ethyl substituents and modulation of the ligand field strength at Cr(III).
transition of oxygen. In a frozen matrix (77 K, MeCN) the emission maximum is hypsochromically shifted by ∼20 nm, the shoulder is somewhat more pronounced and photogeneration of 1O2 is inhibited. For [Cr(bpi-2)2]PF6 a relative hypsochromic shift in emission maximum was observed at λem = 929 nm (Table 1), which is attributed to the effect of the ethyl substituents and modulation of the ligand field strength at Cr(III).
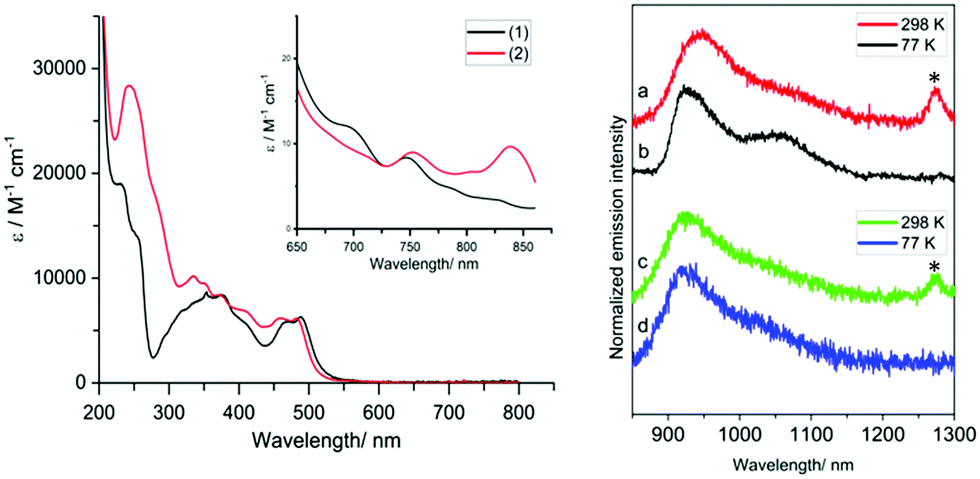 | ||
Fig. 3 Left: UV-vis absorption data for [Cr(bpi-1)2]PF6 (1) and [Cr(bpi-2)2]PF6 (2) including expansion of the long wavelength region. Emission data (λex = 430 nm; 850 nm cut-on longpass filter) for [Cr(bpi-1)2]PF6 (red, black) and [Cr(bpi-2)2]PF6 (green, blue) recorded in MeCN. 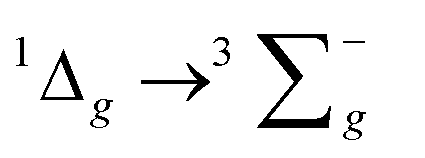 emission of oxygen highlighted by an asterisk. emission of oxygen highlighted by an asterisk. | ||
| Complex | λ abs (nm) | λ em (nm) | τ obs (μs) |
|---|---|---|---|
| a λ ex = 430 nm. b Values in parenthesis recorded at 77 K. c λ ex = 355 nm. d Values in parentheses recorded in deoxygenated solvent. | |||
| [Cr(bpi-1)2]PF6 | 234, 256, 293 sh, 320 sh, 354 sh, 380, 412 sh, 464, 490, 607, 699, 750, 825 sh | 948 (930)b | 4.5 (8.0)d |
| [Cr(bpi-2)2]PF6 | 242, 282 sh, 335, 353, 377, 411 sh, 459, 483, 752, 840 | 929 (924)b | 8.1 (25.0)d |
The lifetimes of the emitting complexes were measured under aerated and degassed conditions (Table 1). In all cases the kinetic traces were fitted (Fig. S8, ESI†) to a single exponential decay yielding lifetimes in the μs domain under ambient conditions, which extended to around 8 μs and 25 μs, for [Cr(bpi-1)2]PF6 and [Cr(bpi-2)2]PF6 respectively, when deoxygenated, implying that oxygen acts as a quencher of these species. These lifetimes are consistent with a phosphorescent emission, which is likely to contain significant spin forbidden 2E → 4A2 character. The longer lifetime noted for [Cr(bpi-2)2]PF6 is consistent with the ligand-induced blue-shift in the emission maximum. Despite appearing in the near-IR range, the deoxygenated lifetimes for [Cr(bpi)2]PF6 are significantly longer than for [Cr(terpy)2]3+ (0.14 μs, de-aerated MeCN),24 and [Cr(dpc)2]+ (see earlier discussion), but appear an order(s) of magnitude shorter than species such as [Cr(ddpd)2]3+ (898 μs, de-aerated H2O) and [Cr(tpe)2]3+ (2800 μs, de-aerated H2O/HClO4) both of which emit ca. 750 nm.
The near-IR photophysical properties of [Cr(bpi)2]PF6 also invite comparison with Yb(III) species which display Yb(III)-centred luminescence at 980 nm (also typically with μs lifetimes) (Fig. S9, ESI†). However, unlike Yb(III), where the emission wavelength is generally insensitive to the ligand environment, the case of [Cr(bpi-2)2]PF6 demonstrates that substituents on the pyridyl donors can modulate and thus tune Cr-centred emission. This paves the way for future ligand iterations allowing the rational tuning of the Cr(III) emission within the attractive near-IR window. Further, the promising emission properties under oxygenated conditions at room temperature (and without recourse to deuterated solvents or ligands) imply that a range of applications could be considered for these complexes, including bioimaging and energy upconversion studies.
We thank Cardiff University and EPSRC (EP/R513003/1) for funding.
Conflicts of interest
There are no conflicts to declare.Notes and references
- L. A. Büldt and O. S. Wenger, Chem. Sci., 2017, 8, 7359 RSC.
- L. A. Büldt and O. S. Wenger, Dalton Trans., 2017, 46, 15175 RSC.
- L. S. Förster, Chem. Rev., 1990, 90, 331 CrossRef.
- K. De Armond and L. S. Förster, J. Chem. Phys., 1961, 55, 2193 Search PubMed.
- J.-R. Jimenez, B. Doistau, M. Poncet and C. Piguet, Coord. Chem. Rev., 2021, 434, 213750 CrossRef CAS.
- P. A. Scattergood, Organomet. Chem., 2021, 43, 1 CAS.
- S. Otto, M. Dorn, C. Förster, M. Bauer, M. Seitz and K. Heinze, Coord. Chem. Rev., 2018, 359, 102 CrossRef CAS.
- J.-C. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048 RSC; A. J. Amoroso and S. J. A. Pope, Chem. Soc. Rev., 2015, 44, 4723 RSC.
- B. Doistau, G. Collet, E. A. Bolomey, V. Sadat-Noorbakhsh, C. Besnard and C. Piguet, Inorg. Chem., 2018, 57, 14362 CrossRef CAS PubMed; J.-R. Jimenez, B. Doistau, C. Besnard and C. Piguet, Chem. Commun., 2018, 54, 13228 RSC; J.-R. Jimenez, M. Poncet, B. Doistau, C. Besnard and C. Piguet, Dalton Trans., 2020, 49, 13528 RSC.
- S. Otto, M. Grabolle, C. Förster, C. Kreitner, U. Resch-Genger and K. Heinze, Angew. Chem., Int. Ed., 2015, 54, 11572 CrossRef CAS PubMed.
- F. Reichenauer, C. Wang, C. Förster, P. Boden, N. Ugur, R. Baez-Cruz, J. Kalmbach, L. M. Carrella, E. Rentschler, C. Ramanan, G. Niedner-Schatteburg, M. Gerhards, M. Seitz, U. Resch-Genger and K. Heinze, J. Am. Chem. Soc., 2021, 143, 11843 CrossRef CAS PubMed.
- S. Treiling, C. Wang, C. Förster, F. Reichenauer, J. Kalmbach, P. Boden, J. P. Harris, L. Carrella, E. Rentschler, U. Resch-Genger, C. Reber, M. Seitz, M. Gerhards and K. Heinze, Angew. Chem., Int. Ed., 2019, 58, 18075 CrossRef CAS PubMed.
- N. Sinha, J.-R. Jimenez, B. Pfund, A. Prescimone, C. Piguet and O. S. Wenger, Angew. Chem., Int. Ed., 2021, 133, 2 CrossRef.
- K.-N. T. Tseng, J. W. Kampf and N. K. Szymczak, Organometallics, 2013, 32, 2046 CrossRef CAS; E. Balogh-Hergovich, G. Speier, M. Reglier, M. Giorgi, E. Kuzmann and A. Vertes, Inorg. Chem. Commun., 2005, 8, 457 CrossRef.
- W. O. J. Siegl, Org. Chem., 1977, 42, 1872 CrossRef CAS.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683 RSC.
- R. P. Bonomo and A. J. Di Bilio, Chem. Phys., 1991, 151, 323 CrossRef CAS.
- R. S. Drago, Physical Methods for Chemists, Surfside Scientific Publishers, Florida, USA, 2nd edn, 1992 Search PubMed.
- B. R. McGarvey, J. Chem. Phys., 1964, 40, 809 CrossRef CAS.
- J. Telser and J. Braz, Chem. Soc., 2006, 17, 1501 CAS.
- G. M. Cole Jr. and B. B. Garrett, Inorg. Chem., 1970, 9, 1898 CrossRef.
- J. M. López Plá, A. K. Boudalis, J. Telser and R. G. Raptis, Inorg. Chim. Acta, 2020, 502, 119299 CrossRef.
- T. Goswami and A. Misra, J. Phys. Chem. A, 2012, 116, 5207 CrossRef CAS PubMed.
- J. C. Barbour, A. J. I. Kim, E. deVries, S. E. Shaner and B. M. Lovaasen, Inorg. Chem., 2017, 56, 8212 CrossRef CAS PubMed.
- Y. Sato and N. Tanaka, Bull. Chem. Soc. Jpn., 1969, 42, 1021 CrossRef CAS; M. C. Hughes, J. M. Rao and D. J. Macero, Inorg. Chim. Acta, 1979, 35, L321 CrossRef.
- M. C. Hughes and D. J. Macero, Inorg. Chem., 1976, 15, 2040 CrossRef CAS.
- C. C. Scarborough, K. M. Lancaster, S. DeBeer, T. Weyhermuller, S. Sproules and K. Wieghardt, Inorg. Chem., 2012, 51, 3718 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, additional spectra and DFT details. CCDC 2155916. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc01434c |
| This journal is © The Royal Society of Chemistry 2022 |