
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmall molecule activation with bimetallic systems: a landscape of cooperative reactivity
Miquel
Navarro
 ,
Juan José
Moreno
,
Marina
Pérez-Jiménez
and
Jesús
Campos
*
,
Juan José
Moreno
,
Marina
Pérez-Jiménez
and
Jesús
Campos
*
Instituto de Investigaciones Químicas (IIQ), Departamento de Química Inorgánica and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Consejo Superior de Investigaciones Científicas (CSIC) and University of Sevilla, Avenida Américo Vespucio 49, 41092 Sevilla, Spain. E-mail: jesus.campos@iiq.csic.es; Web: https://jcamposgroup.iiq.us-csic.es/
First published on 6th September 2022
Abstract
There is growing interest in the design of bimetallic cooperative complexes, which have emerged due to their potential for bond activation and catalysis, a feature widely exploited by nature in metalloenzymes, and also in the field of heterogeneous catalysis. Herein, we discuss the widespread opportunities derived from combining two metals in close proximity, ranging from systems containing multiple M–M bonds to others in which bimetallic cooperation occurs even in the absence of M⋯M interactions. The choice of metal pairs is crucial for the reactivity of the resulting complexes. In this context, we describe the prospects of combining not only transition metals but also those of the main group series, which offer additional avenues for cooperative pathways and reaction discovery. Emphasis is given to mechanisms by which bond activation occurs across bimetallic structures, which is ascribed to the precise synergy between the two metal atoms. The results discussed herein indicate a future landscape full of possibilities within our reach, where we anticipate that bimetallic synergism will have an important impact in the design of more efficient catalytic processes and the discovery of new catalytic transformations.
 Miquel Navarro | Dr Miquel Navarro studied Chemistry at Universitat de Barcelona. He obtained his PhD in organometallic chemistry from Universität Bern under the supervision of Prof. Martin Albrecht in 2017. Then, he joined the group of Dr Didier Bourissou at Laboratoire Hétérochemie Fondamentale et Appliquée (CNRS – Université Paul Sabatier, Toulouse) as a Postdoctoral Fellow thanks to an Early Postdoc Mobility Fellowship from the Swiss National Science Foundation. In 2020, he joined the group of Dr Jesus Campos (CSIC – Universidad de Sevilla) as a Juan de la Cierva Fellow. His current research is focused on the design of bulky phosphine ligands for metal-containing cooperative systems. |
 Juan José Moreno | Juan José Moreno completed his BSc and MSc in Chemistry at the University of Sevilla in 2014, after which he joined the group of Professor Ernesto Carmona to pursue a PhD in organometallic chemistry. In 2019, he moved to the University of Virginia to perform electrocatalytic CO2 reduction under the guidance of Professor Charles Machan. In 2021, he secured a Junta de Andalucía Postdoctoral Fellowship to join the Campos group, where he is currently exploring the reactivity of metalloradical species in bond activation processes. |
 Marina Pérez-Jiménez | Marina Perez-Jimenez obtained her PhD in Organometallic Chemistry in 2021 at the University of Sevilla under the supervision of Professor E. Carmona and Dr J. Campos. Her PhD work focused on the synthesis and reactivity of dimolybdenum complexes containing quadruple metal–metal bonds. During her PhD, in 2019, she had a Predoctoral stay at the University of Berkeley, California, with the group of Professor T. Don Tilley. In 2022, she was awarded a Postdoctoral Margarita Salas Fellowship to join the group of Professor Mark R. Crimmin at Imperial College London, where she is developing her research at the moment. |
 Jesús Campos | Jesús Campos obtained his PhD (2012) in Organometallic Chemistry at the University of Sevilla (E. Carmona). He developed his Postdoctoral research career at Yale (R. Crabtree) and Oxford University (S. Aldridge). In 2017, he became a CSIC tenured researcher at the Institute for Chemical Research and was awarded an ERC Starting Grant on molecular cooperative systems. In 2020, he was appointed Fellow of the Spanish Young Academy. His interests include all aspects of organometallic chemistry, particularly on the study of cooperative mechanisms for bond activation and catalysis. |
Introduction
The field of organometallic chemistry, especially its application in homogeneous catalysis, has been largely dominated by mononuclear transition metal complexes. However, a vivid interest in bimetallic and polymetallic compounds has recently emerged.1 Nonetheless, the historical development of organometallic chemistry contradicts the potentially misleading notion of genuine novelty based on the concept bimetallic design. In fact, the study of M–M bonds has sparked interest from organometallic chemists since the early beginnings of this discipline. Seminal discoveries at that time included, for instance, the first unambiguous recognition of a M–M bond in Mn2(CO)10,2 or shortly after the disclosure of multiple bonding in [Re2Cl8]−2,3 which shattered the assumption of a maximum bond order of three. However, the evolution of bimetallic complexes has been discontinuous since then,4 where it was only in the last decade that renewed efforts have been devoted to the design and use of bimetallic organometallic architectures in a plethora of applications.This re-emergence is substantiated by several facts. Firstly, in the search for more efficient catalysts, chemists have gained inspiration from nature. In metalloenzymes, it is common to find active sites that rely on the cooperative action of two or more metals to effect catalysis with unparalleled efficiency,5 especially in transformations that remain among the most challenging for modern synthetic chemistry.6 Thus far, bioinspired and biomimetic bimetallic approaches have shown great prospects for future developments.7 The area of heterogeneous catalysis also serves as inspiration for molecular chemists seeking innovative bimetallic designs.8 This is not surprising given that metal–metal cooperation is crucial in many mixed-metal heterogeneous catalysts and nanoparticles.9 Moreover, the advancement of operando techniques and more sophisticated computational approaches evidence increasing cases where catalytic transformations that were believed to be mediated by genuine monometallic species involve the key participation of bimetallic intermediates and transition states.10
Continuous research on bimetallic complexes demonstrates that the presence of a second metal provides multiple tunable features that are unattainable for mononuclear species (Fig. 1). In the latter case, organometallic chemists have traditionally focused on tuning the stereoelectronic properties at the metal center by ligand modification. Beyond this approach, the presence of a second metal enables the polarity, distance and order of the M–M bonds to be tuned, which are features exhibiting a strong impact on their reactivity (activity and selectivity) and other intrinsic properties such as photoluminescence, stability, solubility, magnetism and structural conformation. For instance, the polarity of the M–M bond is associated with the mechanism by which small molecules are activated, usually following homolytic pathways in non-polarized bonds and heterolytic pathways for polar M–M bonds. Besides, bond activation can proceed either by single-site or multi-site pathways, thus offering mechanistic possibilities that are absent in mononuclear complexes, which may lead to the discovery of new transformations.
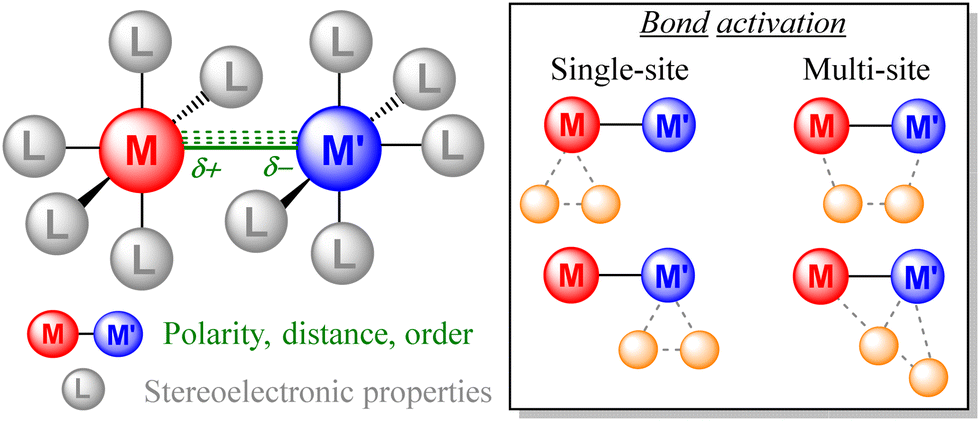 | ||
| Fig. 1 Tunable features of bimetallic complexes and some examples of representative pathways for single vs. multi-site bond activation. | ||
The above-mentioned features have been elegantly exploited by many research groups for a variety of purposes (Fig. 2). For instance, the pseudo-C3-symmetric Zr/Co complex developed by Thomas is versatile, exhibiting either single-site or multi-site activation depending on the added substrate.11 An additional advantage of bimetallic species is the ability to circumvent the often undesirable one-electron pathways, thus providing nobility to first-row transition metals. This concept has been exploited by Mankad in highly polarized group 8/group 11 complexes, which exhibit rich bond activation reactivity that can be implemented in, for instance, borylation catalysis.12 Alternatively, the non-polarized naphthyridine-diimine dinickel complex developed by the Uyeda group is a highly active catalyst for unusual vinylidene transfer reactions.13 Moreover, the M–M bond polarity can be tuned in such a way that the intrinsic polarity of a certain atom can be even reversed, as demonstrated by Aldridge and Goicoechea in a Auδ−/Alδ+ complex, where the gold center behaves as a nucleophile towards small molecules.14 This example demonstrates that besides transition metals, main group-based combinations also hold great potential. In a related example, Camp reported a strongly polarized Irδ−/Alδ+ system, which promotes the unusual reductive cleavage of carbon dioxide, relying on the synergistic cooperation between the two metals.15 Whittlesey and Macgregor examined the effects of incorporating several main group metal fragments in the [Ru(PPh3)3HCl] compound, revealing the remarkable acceleration of dihydrogen activation due to the formation of the very uncommon Ru/Zn bimetallic pair.16 The partnership between transition and main group metals, although in its infancy, offers a landscape of possibilities, even for the apparently simple s-block metals,. A classic example is the formation of the inverse crown complex [Na4Mg2(TMP)6(nBu)2] (TMP = 2,2,6,6-tetramethylpiperidine) reported by Mulvey and O’Hara, which promotes dimetalation reactions with meta–meta’ regioselectivity.17 In fact, the combination of alkali metals with less polar organometallics has proven to be a highly prolific strategy for altering selectivity during C–H bond activation.18 Take into account that this is just a small collection of selected examples that demonstrate the unlimited possibilities within our grasp.
 | ||
| Fig. 2 Selected examples of bimetallic systems that exhibit remarkable cooperative reactivity. | ||
In this work, we describe our efforts to better understand the synergy and cooperative mechanisms evolving from bimetallic complexes of different nature. In the last few years, our group focused on the study of bimetallic species that contain not only multiple and single M–M bonds, but also the extreme case of those that cooperate without exhibiting a direct bimetallic interaction. Our designs include systems in which the two active sites are transition metals and also those incorporating a metal of the main group series. Herein, we discuss our results pertaining to their synthesis and especially their reactivity towards small molecules, focusing on key mechanistic aspects. The discussion is organized in the following sections:
– Frustration versus interaction in bimetallic systems.
○ Frustrated Lewis pairs based on transition metals.
○ Bimetallic frustrated Lewis pairs.
○ M–M-polarized single bonds.
– Multiple metal–metal bonds in X–Y bond activation.
– Transition metal–main group bimetallic cooperativity.
Frustration versus interaction in bimetallic systems
Frustrated Lewis pairs based on transition metals
It was long believed that despite the well-known ability of Lewis acids and bases to promote chemical transformations, their cooperative involvement in bond activation and catalysis was inhibited by the irreversible formation of Lewis adducts. In 1942, Brown first recognized that steric effects precluded the formation of a Lewis adduct between BMe3 and 2,6-lutidine.19 In the following years, various combinations of sterically demanding Lewis acids and bases were found to promote unforeseen stoichiometric reactions,20 but it was not until 2006 that Stephan and coworkers reported the reversible cleavage of H2 by a phosphino–borane (Fig. 3(a)).21 This reactivity, considered at the time exclusive to transition and f-block metals, revolutionized main group chemistry and laid the foundation for the development of frustrated Lewis pairs (FLPs).22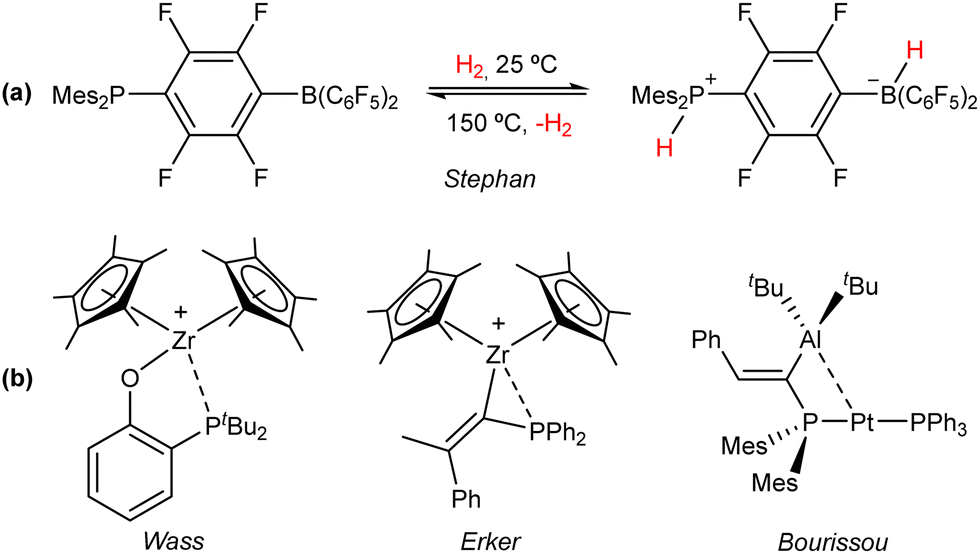 | ||
| Fig. 3 (a) Seminal discovery of reversible dihydrogen activation at a phosphino–borane pair.21 (b) Representative examples of the first transition metal FLPs developed based on Zr(IV)23–25 and first example of a bimetallic FLP based on the Pt(0)/Al(III) pair.26 | ||
Although performing metal-free bond activation and catalysis is one of the preeminent features of FLPs, the reluctance of main group elements to partake in elemental reactions, such as oxidative addition, reductive elimination and migratory insertion encouraged the incorporation of Lewis acidic and basic transition metal sites as FLP constituents, aiming to expand the limited scope of traditional main-group FLPs by exploiting the rich reactivity of the d orbital shell.27 The pioneering work by the group of Wass leveraged the Lewis acidity of Zr(IV) to perform stoichiometric23 and catalytic24 FLP reactivity in conjunction with a pending phosphane base (Fig. 3(b)). Soon after, Erker and coworkers expanded the scope of Zr/P systems by employing a geminal pair in FLP addition reactions and hydrogenation catalysis (Fig. 3(b)).25 Recently, Bourissou et al. developed a bimetallic Pt(0)/Al(III) complex, which displayed thermally induced FLP behaviour (Fig. 3(b)).26
Bimetallic frustrated Lewis pairs
Attempts to synthesize FLPs solely constructed around transition metal centres remained unfruitful until 2017,28 when the use of bulky phosphine ligands allowed us to report the first example of a transition metal-only frustrated Lewis pair (TMOFLP).29 The combination of a sufficiently congested Lewis acidic Au(I) fragment and a Lewis basic Pt(0) centre enabled the activation of dihydrogen and acetylene in a way that resembled main group FLP systems (Scheme 1), whereas the individual fragments were unreactive. In the case of dihydrogen, the heterolytic bond cleavage was followed by the formation of a heterobimetallic Au(I)/Pt(II) complex featuring a bridging and a terminal hydride. In turn, two competitive reaction pathways existed in the activation of acetylene, i.e., alkyne deprotonation, giving rise to a bridged acetylide π-bonded to the cationic Au(I) centre and σ-bonded to a formal Pt(II) hydride, and the formation of two metal–carbon σ bonds (1,2-addition), establishing a bridging vinylene linker between the two metallic fragments. These two isomers, which were obtained in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, highly resemble the products typically obtained during alkyne activation by traditional phosphine/borane FLPs.30
:1 ratio, highly resemble the products typically obtained during alkyne activation by traditional phosphine/borane FLPs.30
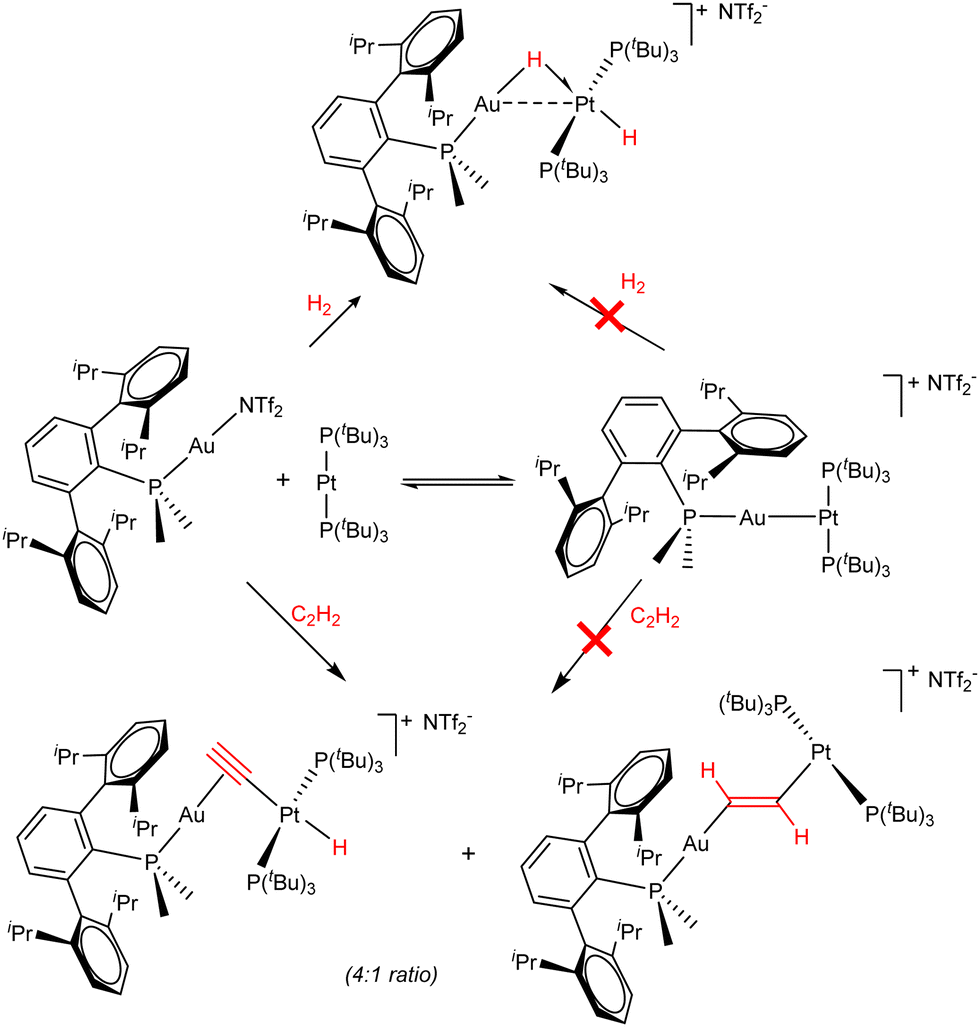 | ||
| Scheme 1 Lewis adduct formation equilibrium and FLP-type bimetallic activation of dihydrogen and acetylene by Au(I)/Pt(0) system. | ||
Following this seminal report, our group studied a family of Au(I)/Pt(0) systems to gain insight into the mechanism of the reaction between dihydrogen and the Au(I)/Pt(0) TMOFLP by experimental and computational means.31 By only varying the steric profile of the terphenyl phosphine of the Au fragment, it became evident that, similar to conventional FLPs, the degree of frustration had a strong impact on the reaction rate. Fig. 4 presents a summary of our findings in this regard. Employing the smaller terphenyl phosphine PMe2ArXyl2 (ArXyl2 = C6H3-2,6-(C6H3-2,6-Me2)2) led to the observation of a heterobimetallic Lewis adduct, whereas the bulkier one, PCyp2ArXyl2 (Cyp = cyclopentyl), ensured complete frustration even in more polar solvents, which favoured the formation of the ionic Lewis adduct. The intermediate phosphine, PMe2ArDipp2 (ArDipp2 = C6H3-2,6-(C6H3-2,6-iPr2)2), as presented in Scheme 1, displayed solvent-dependent behaviour, where in apolar solvents such as benzene, frustration was retained. In dichloromethane, the broadening of the NMR resonances was consistent with the dynamic equilibrium between the independent fragments and the bimetallic Lewis adduct, which could be shifted to the latter by increasing the polarity of the medium by adding methanol. Density functional theory (DFT) calculations displayed excellent agreement with the thermodynamics of these solution equilibria.
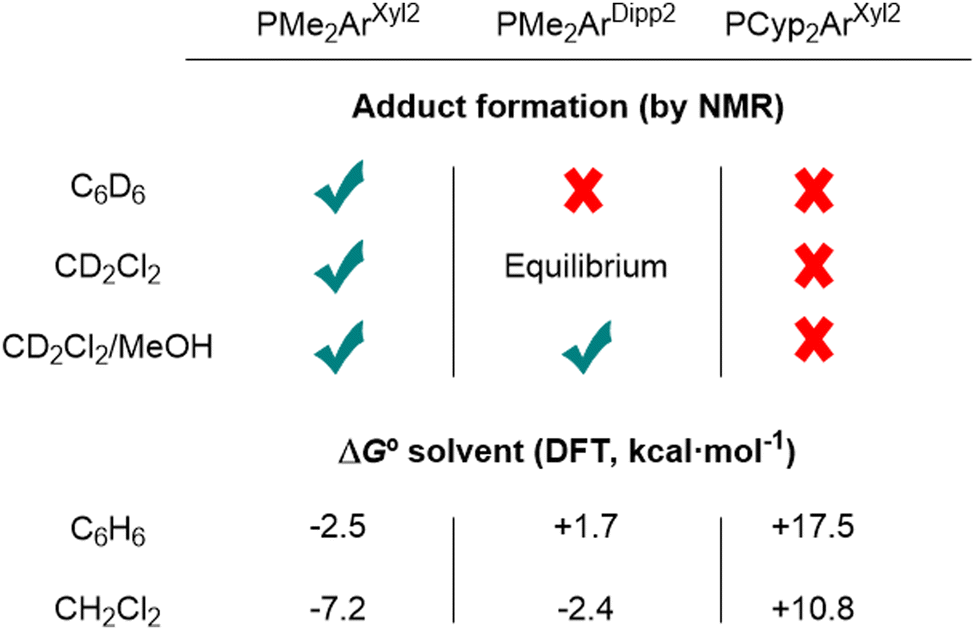 | ||
| Fig. 4 Solvent-dependent thermodynamics of the FLP equilibrium. | ||
Importantly, while bulkier systems effected fast dihydrogen activation at room temperature, that based on the PMe2AXyl2 phosphine, for which the resting state was the heterobimetallic adduct, displayed sluggish reaction kinetics. This was consistent with an FLP-type mechanism, which requires prior dissociation of the bimetallic adduct, and thus an additional barrier to overcome. Furthermore, the addition of either excess Au or Pt precursor to the latter system had a detrimental effect on the reaction rate, supporting that access to both independent metallic fragments was paramount to the observed reactivity. The system constructed around PMe2ArDipp2 displayed greater activity than that based on PCyp2ArXyl2, where similar to traditional FLPs, there is an optimal degree of bulkiness, above which reactions become slower or even do not occur. The product speciation was also affected by the steric profile of the phosphine ligands bound to the Au centre, where in the case of PMe2ArXyl2, the only discernible product was the bridged heterobimetallic dihydride, whereas for PMe2ArDipp2, the rapid formation of cationic digold and platinum hydride complexes eventually evolved to the thermodynamic product, i.e., the heterobimetallic dihydride. However, while for PCyp2ArXyl2 the digold hydride was observed initially, the formation of the bridged heterobimetallic dihydride did not occur, which allowed us to disclose the gold-catalyzed formation of the trans Pt(II) dihydride.
One of our main goals is the precise determination of the mechanism for dihydrogen splitting. This transformation has served as the benchmark to gauge FLP reactivity, although it still remains under debate for main-group FLPs.32 For this, we examined all the potential relevant pathways by computational means (see representation of key transition states in Fig. 5), including activation of the H–H bond at the Pt centre followed by Au-assisted cis–trans isomerization (orthogonal activation), across the Pt–Au bond in the Lewis adduct (bimetallic activation), and at the Au centre followed by deprotonation by the NTf2 anion, which were all found to be unfeasible. In turn, the only energetically accessible route encompassed a true FLP-type transition state. The intermediate preceding this TS is an encounter complex in which the H2 molecule interacts with both metal centres in an end-on fashion. Interestingly, despite its weak coordinating ability, a stabilizing interaction between the NTf2 anion and the Au centre was found to be essential, shifting the Au–H–H angle from 79.3° to 136.5°. A strong inverse kinetic isotope effect (KIE) was found for the systems containing PMe2ArXyl2 (0.46) and PMe2ArDipp2 (0.50, carried out at −20 °C), suggesting that similar reaction pathways were operative for both. The calculated zero-point energy differences (ΔΔZPE) for the PMe2ArXyl2 system gave an inverse primary KIE kH/kD = 0.40, in excellent agreement with the experimentally determined values, thus further supporting our genuine FLP-type mechanism for a bimetallic system. We also found the same feature in a more recent study based on related Au(I)/Pt(0) pairs, in which the platinum species also contains a terphenyl phosphine.33 In contrast to the majority of examples of inverse KIEs which involve H–H bond cleavage, we attributed its origin to the collective isotopically sensitive vibrational modes at the rate-determining transition state and not due to a preequilibrium involving an inverse equilibrium isotope effect (EIE).
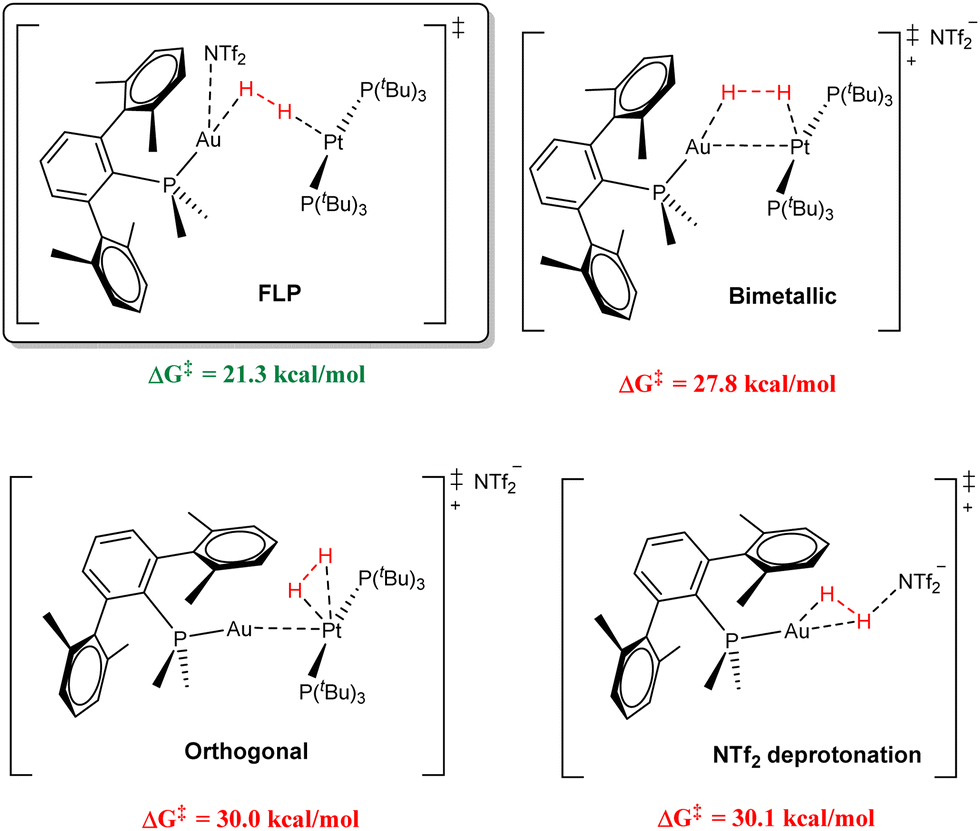 | ||
| Fig. 5 Representation of the key transition states for the most relevant modelled mechanistic pathways and their associated overall energy barriers for H2 splitting. | ||
Another aspect that has attracted our attention in this area is controlling the selectivity. In the case of acetylene activation, the ratio between the isomers described in Fig. 6 was highly sensitive to the steric profile of the phosphines employed.34 As mentioned above, the system based on PMe2ArDipp2 gave an 80:20 mixture of a σ,π-acetylide and a vinylene, respectively. For the least sterically demanding Au complex, (PMe2ArXyl2)Au(NTf2), the regioselectivity shifted towards the formation of the bridging acetylide isomer (95:5 ratio), whereas for the bulkier complex, (PCyp2ArXyl2)Au(NTf2), a drastic change was observed, as the heterobimetallic vinylene was quantitatively formed (Fig. 6). Low-temperature NMR spectroscopy and DFT calculations indicated alkyne coordination to the electrophilic Au centre takes place first, where the attack of the Pt atom towards the C or H atoms of the activated alkyne determines the selectivity of the process. Calculations indicated that the transmetalation of the acetylide ligand following deprotonation is a facile process. Interestingly, the divergence in the reaction outcome was achieved by modifying the steric hindrance around the Lewis acid and not the strength of the Lewis base alike in main-group FLPs. This is particularly advantageous considering that modifying the structure of the Lewis acid in traditional FLPs (typically fluorinated boranes) may be synthetically challenging,35 while subtle changes on the ligand attached to the electrophilic gold site is straightforward.36
 | ||
| Fig. 6 Selectivity during acetylene activation by Au(I)/Pt(0) TMOFLPs. | ||
Heavier tetrylenes, which are species containing divalent group 14 elements (from Si to Pb), display ambiphilic character, given that they present both a vacant p orbital and lone pair with s character on the same atom, which can cause them to behave as Lewis acids and/or bases. This single-site ambiphilicity resembles the concept of frustration, given that the aforesaid empty and filled orbitals cannot form a dative bond. However, this aspect has been exploited by some authors to stabilize highly reactive tetrylene fragments by push–pull interactions in the presence of a donor and an acceptor.37 In fact, Rivard used metallic Lewis acidic W(CO)5 and basic (C5H5)Rh(PMe2Ph)2 and Pt(PCy3)2 moieties to trap simple forms of divalent group 14 compounds.38 These results prompted us to study the reactivity of tetrel dihalides (GeCl2 and SnCl2) towards the Au/Pt TMOFLP presented above.39 The addition of GeCl2·dioxane to a solution of the frustrated Lewis pair based on PMe2ArDipp2 shifted the aforesaid equilibrium towards the formation of the Lewis adduct, likely by sequestration of the triflimide anion (Scheme 2(a)). In turn, SnCl2 promoted the transfer of a phosphine ligand from the Pt complex to the Au centre (Scheme 2(a)). In fact, the major Pt-containing species in this process is a Pt/Sn cluster with a 1:1 phosphine : Pt ratio, for which computational studies indicate that all but one metal atom display ambiphilic donor–acceptor character (Scheme 2(b)). In the presence of added phosphine ligands, SnCl2 was also found to be an effective promoter of phosphine exchange reactions between Pt(0) compounds of formula Pt(PR3)2 and free phosphine, offering a route towards unusual heteroleptic Pt(PR3)(PR’3) complexes.
 | ||
| Scheme 2 Reactivity of Au(I)/Pt(0) TMOFLP towards tetrylene dihalides. | ||
Subtle changes on the steric profile of the electrophilic Au(I) fragment proved impactful when combined with the Rh(I) Lewis base (C5Me5)Rh(PMe3)2, where intriguing divergent reactivity was observed (Scheme 3).40 The system bearing PMe2ArXyl2, the least sterically demanding ligand, quantitatively formed the corresponding bimetallic Lewis adduct, which is a Rh–Au heterobimetallic complex. In turn, the use of the bulkiest PCyp2ArXyl2 promoted an unforeseen C–H activation event at the pentamethylcyclopentadienyl (C5Me5) ligand, which led to the formation of new Au–C and Rh–H bonds. The direct migration of a hydride from a methyl group of the C5Me5 unit to the metal is well known for early transition metals,41 but unprecedented for late ones. In the case of the intermediate PMe2ArTripp2 (ArTripp2 = C6H3-2,6-(C6H2-2,4,6-iPr3)2), the product distribution was a 70:30 ratio between the Lewis adduct and the C–H activation product. The latter fully converted into the Lewis adduct after 24 h at room temperature, showcasing the reversibility of the hydride migration process and indicating that the Lewis adduct was the thermodynamic product. Interestingly, while the Lewis adducts displayed no reactivity towards small molecule activation, the Rh hydrides derived from C–H activation rapidly carried out X–H bond activation reactions (X = N and O). Due to their coordinative saturation and the reversibility of their formation, we proposed that these complexes behave as unusual thermally induced TMOFLPs in the activation of ammonia, water and methanol. The cleavage of N–H bonds in ammonia is a reaction of particular interest, given that it is often challenging to achieve with monometallic transition metal complexes due to the undesirable formation of unreactive Werner-type adducts. The treatment of independently prepared [(PCyp2ArXyl2)Au(NH3)](NTf2) with the Rh base gave the gold amide and rhodium hydride species stemming from N–H bond activation, supporting our hypothesis. Isotopic labelling further confirmed the reversibility of hydride migration and the heterolytic FLP-type mechanism by which the X–H bonds are cleaved by the cooperative action of gold and rhodium.
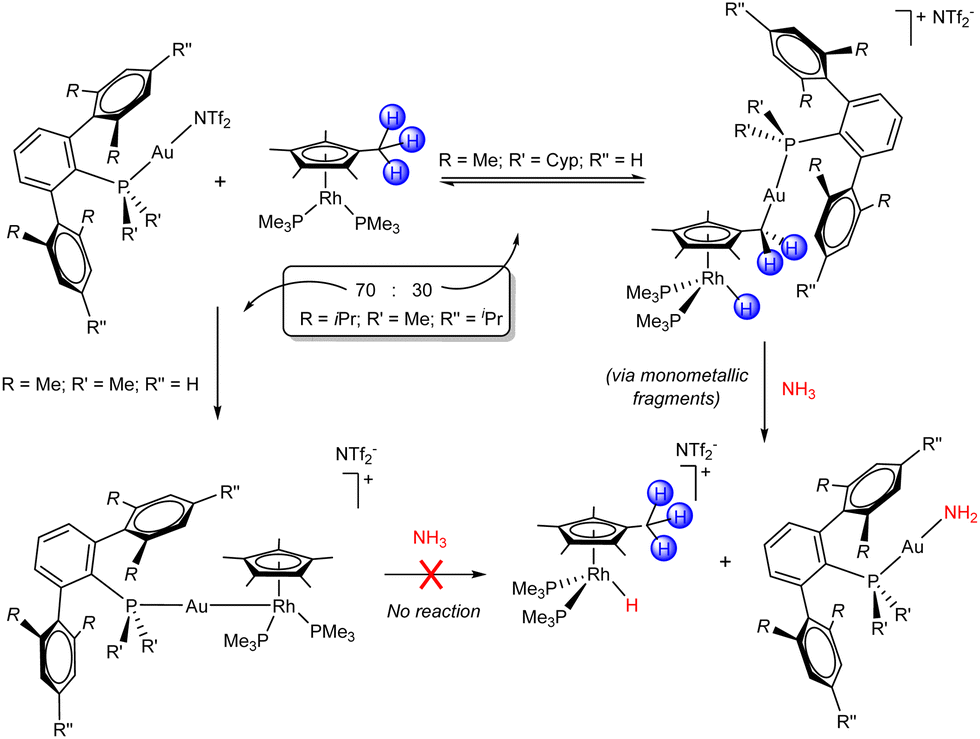 | ||
| Scheme 3 Bimetallic FLPs based on Au(I)/Rh(I) combinations. Intramolecular reactivity and N–H bond activation. | ||
M–M polarized single bonds
Heterobimetallic complexes, in contrast to their homobimetallic counterparts, present intrinsic polarization at the M–M bond, and therefore they often exhibit cooperative reactivity towards different substrates in a way that may resemble FLPs. In this regard, bimetallic complexes without bridging ligand frameworks that are only stabilized through a M–M dative bond between a Lewis basic and a Lewis acidic metal are known as metal-only Lewis pairs (MOLPs).42 In fact, some of the complexes discussed in the previous section fall in this category, in particular those in which the steric constrains are reduced to the extent that the formation of a M–M dative bond is allowed. This type of complex has gathered growing attention in recent years, in no little part due to the key role of metal–metal interactions in a broad variety of catalytic transformations.43 Braunschweig and co-workers explored the formation of a wide variety of unsupported dative bonds between the transition metal base Pt(PCy3)2 and s- and p-block metal acidic fragments (Scheme 4(a)).44 Exchange reactions served as a useful tool for experimentally studying the basicity of different metallic fragments and showed the lability and dynamic behaviour of the M–M bond, forecasting the great potential of this type of system to act as thermally induced FLPs,45 as shortly after demonstrated for the Au(I)/Pt(0) pairs described in the prior section. Despite their great potential, the ability of MOLPs to activate small molecules has been rarely investigated. In a pioneering work, Cutler described the activation of carbon dioxide through a M → Zr bond (M = Ru or Fe), leading to metallocarboxylates stabilized by push–pull interactions (Scheme 4(b)).46 Recently, Mankad investigated the capability of unbridged polarized heterobimetallic systems based on earth–abundant transition metals to activate different small molecules such as carbon disulfide, iodomethane, benzyl chloride and dihydrogen (Scheme 4(c)).47 In addition, these heterobimetallic complexes have shown high activity in a variety of applications in catalysis.48 | ||
| Scheme 4 Representative examples of MOLPs for the activation of small molecules. | ||
Our group also investigated the ability of Pt(PtBu3)2 as a basic metal fragment to form different MOLP systems. The reaction of Pt(PtBu3)2 with the acidic fragment AgX (X = NTf2 or OTf) in either benzene or dichloromethane in the absence of light readily formed the [(PtBu3)2Pt → AgX] adducts (Scheme 5).49 In addition to crystallographic characterization, 31P{1H} NMR spectroscopy revealed a slight change in the chemical shift together with a pronounced decrease in the 1JPPt coupling constant, where 1JPPt = 3244 Hz vs.1JPPt = 4410 Hz in the Pt(PtBu3)2 precursor. This decrease in the 1JPPt value indicates the reduced s character of the P–Pt bond as a consequence of the new Pt → Ag interaction. The use of coordinating anions such as NTf2− or OTf− was shown to be crucial for the stability of these Pt(0)/Ag(I) adducts, given that the analogous MOLPs presenting less coordinating counteranions (i.e., BF4− and PF6−) display limited stability. The reactivity of the [(PtBu3)2Pt → AgNTf2] adduct towards the activation of small molecules was examined. For instance, the MOLP readily reacted with dihydrogen and phenylacetylene under mild conditions to form the corresponding heterobimetallic dihydride and an uncommon trimetallic dibridged bisacetylide, respectively (Scheme 5). It is worth noting that none of the metal precursors exhibited any reactivity towards dihydrogen or phenylacetylene even under harsher conditions, demonstrating the cooperative cleavage of the H–H and C–H bonds. The [(PtBu3)2Pt → AgNTf2] MOLP could also activate X–H bonds in water and ammonia, generating the oxidized platinum(II) hydrides and the corresponding precipitation of the silver hydroxide or amide salts. As discussed earlier, the activation of the N–H bond in ammonia, although less efficient than that for the above-mentioned Au(I)/Rh(I) pair, constitutes a relevant result given its typically challenging nature.
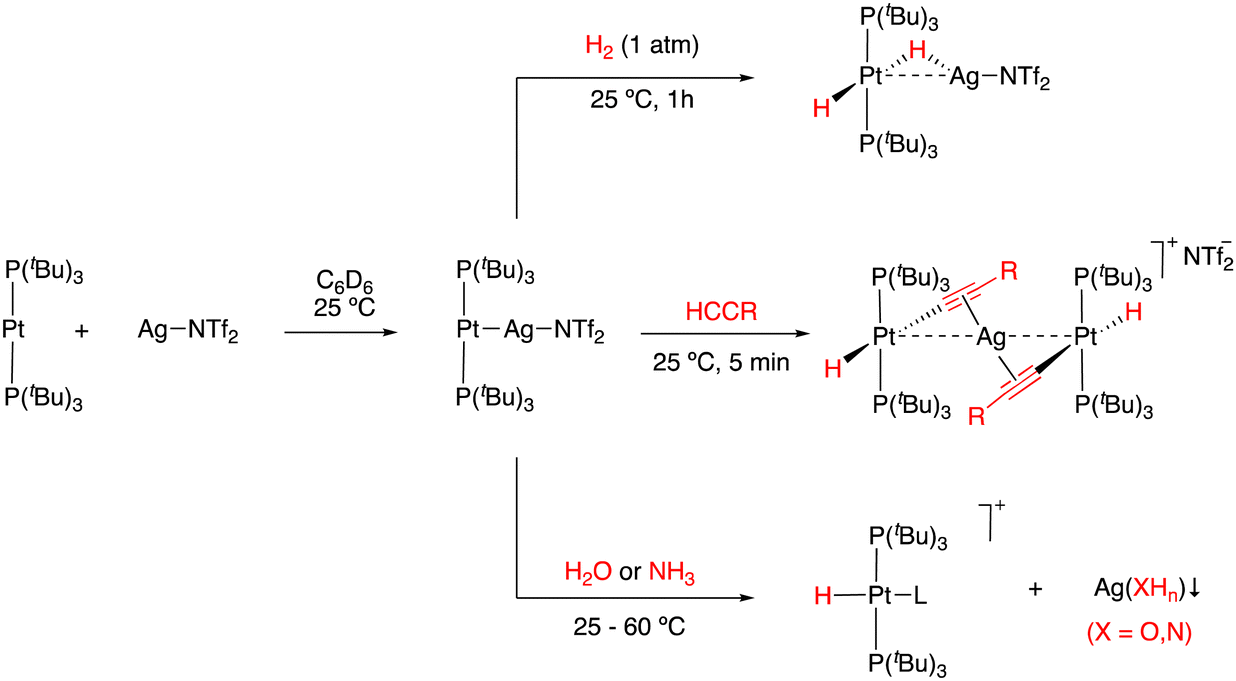 | ||
| Scheme 5 Activation of dihydrogen, alkynes, water and ammonia by a metal-only Lewis pair based on Pt and Ag. | ||
We also investigated the formation and reactivity of zinc-containing MOLPs based on the basic metal fragment Pt(PtBu3)2.50 Treatment of Pt(PtBu3)2 with zinc halides and pseudohalides in several solvents did not result in the expected adduct formation, in contrast to the readily accessible [(PCy3)2Pt → ZnBr2].51 We attributed this reluctance towards adduct formation to steric reasons. However, the reaction of Pt(PtBu3)2 with 1 equivalent of Zn(C6F5)2 in benzene afforded the bimetallic adduct [(PtBu3)2Pt → Zn(C6F5)2], which represents the first example of a Pt(0)/organozinc MOLP (Scheme 6(a)). Similarly to the Pt(0)/Ag(I) adduct, a pronounced decrease in the 1JPPt coupling constant to 3328 Hz (1JPPt = 4410 Hz in Pt(PtBu3)2) was detected, indicating the formation of an adduct. In contrast, the less acidic ZnR2 (R = Me, Et, Ph and η5-C5Me5) did not react with Pt(PtBu3)2 even under harsher conditions, highlighting the necessity for a highly electrophilic zinc centre to overcome the distortion of the linear Pt(0) precursor to accommodate the bimetallic dative bond. The reaction of Pt(PtBu3)2 with the more exotic Zn(I) dimer Zn2(C5Me5)2 resulted in the release of two equivalents of free phosphine and the precipitation of bright orange crystals of the Pt(ZnC5Me5)6 complex (Scheme 6(a)). This adduct is formed through the insertion of the Pt centre into the Zn–Zn bonds of three molecules of Zn2(C5Me5)2. It represents an unusual 16-electron octahedral complex in which each vertex is occupied by a neutral 1-electron Zn(C5Me5) ligand providing steric crowding, which stabilizes the encapsulated electron-rich platinum centre. This is different from all the prior Zn-rich polymetallic compounds of late transition metals, which consistently fulfil the 18-valence electron rule. Computational exploration of the topology of the Pt(ZnH)6 model system showed bond critical points (BCPs) together with their associated bond paths (BPs) between the zinc and platinum centres. In addition, no BCPs or BPs were located between the zinc atoms, indicating the absence of Zn⋯Zn interactions in the Pt(ZnC5Me5)6 complex.
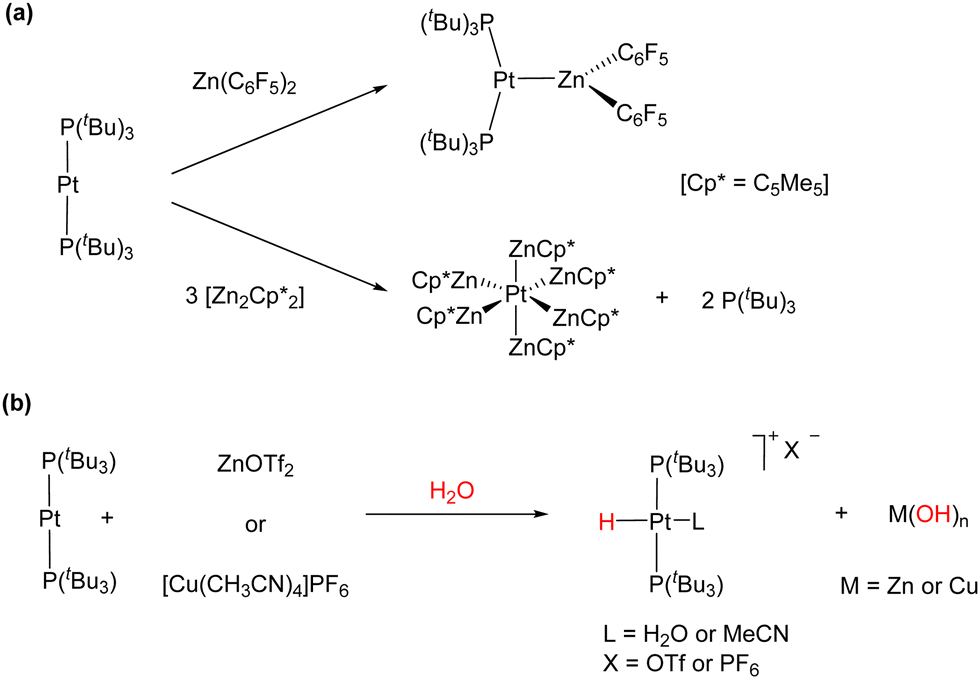 | ||
| Scheme 6 (a) Reaction of Pt(PtBu3)2 with Lewis acidic organozinc compounds. (b) Activation of polar O–H bonds by Pt(0)/Zn(II) and Pt(0)/Cu(I) cooperative bimetallic species. | ||
The [(PtBu3)2Pt → Zn(C6F5)2] and Pt(ZnC5Me5)6 complexes, which are inactive towards H2, rapidly hydrolyse in the presence of water. Similarly, a mixture of Pt(PtBu3)2 and ZnX2 (X = Cl, Br, I and OTf) readily activated the O–H bond of H2O, affording platinum hydride complexes of the formula [PtHX(PtBu3)2], accompanied by the precipitation of zinc hydroxide salts (Scheme 6(b)). This reactivity is analogous to the activation of polar bonds by the combination of Pt(PtBu3)2 with the transition metal Lewis acid [Cu(CH3CN)4]PF6 previously reported by Jamali and co-workers,52 as well as the reactivity with AgNTf2 described in Scheme 5.49 The Lewis basic Pt(PtBu3)2 complex could also activate dihydrogen in the presence of catalytic amounts of Zn(OTf)2, generating the trans Pt(II) dihydride under mild conditions. This process was found to be reversible and presented a strong inverse kinetic isotopic effect (KIE), which suggests that an FLP-type dihydrogen activation similar to the one previously detailed for the Au(I)/Pt(0) pair may be operative.
As with the Lewis base Pt(PtBu3)2, we also capitalized on the ability of [(η5-C5Me5)Rh(PMe3)2] as a transition metal Lewis base to design MOLPs, where the highly constrained Au(I) electrophiles described in Scheme 3 were substituted by smaller Lewis acidic fragments (Scheme 7).53 Treatment of [(η5-C5Me5)Rh(PMe3)2] with lithium and sodium salts of the weakly-coordinating tetrakis(3,5-bis(trifluoromethyl)phenyl)borate anion (BArF−) in the non-coordinating solvent bromobenzene generated the corresponding Rh → M (M = Li and Na) MOLPs. Isolation of these Lewis adducts was not possible due to rapid hydrolysis, which generated the corresponding [(η5-C5Me5)Rh(PMe3)2H][BArF] and alkali hydroxides, illustrating the weakness of the Rh → Li/Na interaction. However, reaction of [(η5-C5Me5)Rh(PMe3)2] with the Grignard reagent MgMeBr affords the dimeric MOLP [(η5-C5Me5)Rh → Mg(MexBr1−x)(μ-Br)]2, in which the methyl group bound to magnesium is mostly exchanged by a bromide (Me:Br with 15:85 occupancies). The Rh → Mg bond length in complex [(η5-C5Me5)Rh → Mg(MexBr1−x)(μ-Br)]2 accounts for 2.651(3) Å, which is shorter than the sum of the covalent radii of these metals and represents the first unambiguous example of an unsupported Rh → Mg bond. The [(η5-C5Me5)Rh(PMe3)2] Lewis base is also capable of forming MOLPs with p-block acids. The reaction of the heavier tetrylenes, GeCl2 and SnCl2, and AlMe3 with the Rh(I) base afforded the corresponding Rh(I) → E Lewis adducts (E = Ge, Sn and Al). The formation of the bimetallic adducts was illustrated by a marked decrease in the 1JPRh coupling constant of ca. 40 Hz and by the corresponding upfield 103Rh{1H} NMR resonances, shifted by ∼400 ppm compared to the Rh(I) precursor. The solid-state structure of these MOLPs showed unsupported Rh → M bonds, with distances shorter than the sum of the respective covalent radii. Interestingly, the germanium and tin MOLPs represent the first examples of rhodium-bound germylene and stannylene not stabilized by the coordination of a base. Finally, [(η5-C5Me5)Rh(PMe3)2] also reacts with the d-block Lewis acids ZnMe2, Zn(C6F5)2 and CuCl, forming the corresponding Rh(I) MOLPs. Overall, the [(η5-C5Me5)Rh(PMe3)2] transition metal Lewis base has proven to be useful to construct a family of MOLPs with a wide range of s-, p- and d-block Lewis acids with unsupported Rh → M bonds. Curiously, DFT calculations demonstrated that the dative Rh → M bond is dominated by electron donation from the Rh–P σ-bonds to the electrophilic metal, rather than from a filled Rh d-orbital to the acidic site, as we originally anticipated. This finding is consistent with the decrease in the M–P coupling constants upon the formation of an adduct.
 | ||
| Scheme 7 Synthesis of Rh(I) MOLPs with s-, p- and d-block Lewis acids. | ||
Multiple metal–metal bonds in X–Y bond activation
Highly polarized M–M bonds hold intrinsic reactivity, as evidenced by the selected examples discussed in the section above. However, the electron density contained in multiply bonded homobinuclear complexes also offers fertile ground for bond activation and catalysis. Since the discovery of the first quadruple metal–metal bond between Re atoms (Scheme 8(a)),3 the vast majority of research in the field of multiple metal–metal bonding has focused on the isolation of complexes exhibiting high bond orders between different metal centres. This has led to important landmarks such as the synthesis of the first compound with a Cr–Cr quintuple bond reported by Power's group in 2005 (Scheme 8(a)).54 After this discovery, diverse homobimetallic55 and some heterobimetallic56 complexes were synthesised, including those based on first-row transition metals.57 However, the exploitation of the close proximity of the two metals (ca. 2 Å)58 for the activation of small molecules has not been explored to a great extent and its true potential still remains mostly undisclosed. Nonetheless, promising reactivity studies regarding compounds containing multiple metal–metal bonds with high bond orders (more than three) have been accomplished by the groups of Tsai and Kempe, among others. Some instances include the carboalumination reactions towards Cr–Cr quintuply bonded units reported by Kempe's group,59 as well as the activation of small molecules such as CO2 or SO2,60 white phosphorous, yellow arsenic,61 alkynes, ketones and allenes.62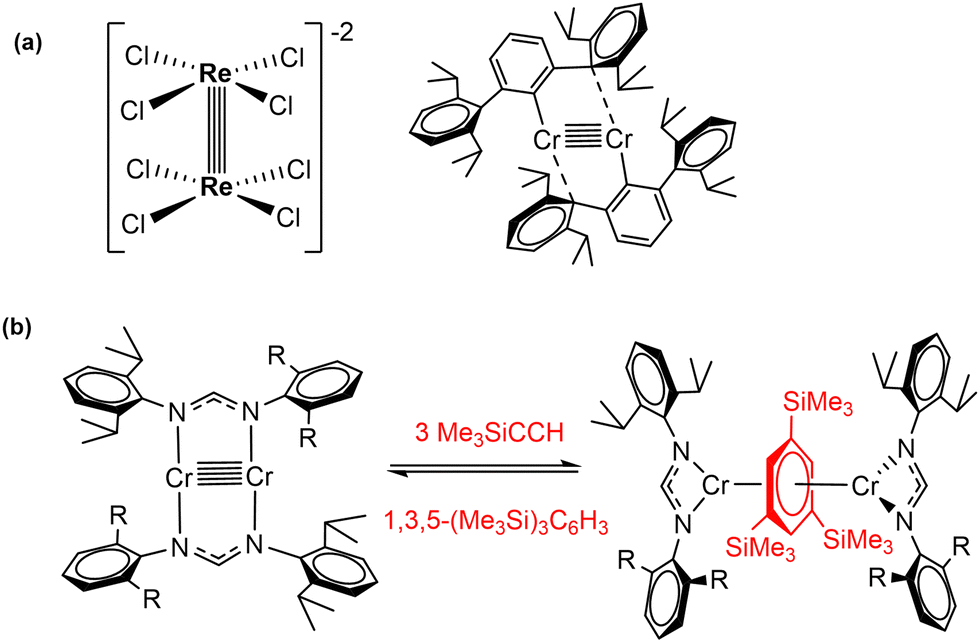 | ||
| Scheme 8 (a) Representative examples of quadruple (Cotton) and quintuple (Power) bonds in bimetallic complexes. (b) [2+2+2] alkyne cyclotrimerization mediated by a quintuply bonded Cr2 complex (Tsai). | ||
The reactivity of quintuply bonded Cr2 species towards terminal alkynes revealed the reversible cleavage of a Cr–Cr quintuple bond to yield an inverted arene sandwich dichromium complex.63 These species, as well as similar quintuply bonded Mo2 complexes,64 can act as catalysts in [2+2+2] cyclotrimerization reactions of terminal alkynes, forming 1,3,5-trisubstituted benzenes (Scheme 8(b)). The catalytic cycle for this transformation was studied theoretically by Sakaki and co-workers.65 However, the development of useful catalytic transformations mediated by metal–metal multiple bonds is still in its infancy.66 Although paddlewheel compounds with metal–metal bonds, mainly single bonds, have been reported as active species in catalytic processes, where dirhodium(II) carboxylates are the classic example,67 only a limited number of multiply bonded bimetallic species behaves as effective catalysts.68 Remarkably, heterobimetallic Zr/Co complexes containing triple metal–metal bonds exhibit good catalytic activity for the hydrogenation of unsaturated hydrocarbons, which is partially attributed to the presence of a metal–metal bond, contributing to lowering the energy barrier required for the H2 cleavage.69
In the last few years, our group contributed to this field by investigating the reactivity of quadruple Mo–Mo bonds. We studied the activation of unsaturated molecules such as alkenes, alkynes and heterocumulenes (CO2 and CS2) via a quadruply bonded dimolybdenum system, stabilized by the coordination of two amidinate AdDipp2 (AdDipp2 = HC(NDipp)2; Dipp = 2,6-iPr2C6H3) ligands in a trans position to each other, featuring, in addition, two trans hydride donor groups.70 Structure A in Scheme 9 illustrates the ambivalent Lewis acid/Lewis base character of this unique molecule, which presents polarized Moδ+–Hδ− bonds next to empty or readily available coordination sites. The short Mo–Mo distance in these complexes of around 2.10 Å, a typical value for quadruple dimolybdenum bonds,58 allows cooperative effects between the two Mo–H units. The reactivity towards alkenes demonstrated the participation of the trans-[H–Mo![[quadruple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e003.gif) Mo–H] central unit in elementary organometallic reactions as reversible migratory insertion, β-H elimination and alkane reductive elimination, which are well-known transformations for mononuclear compounds but have been less studied in bimetallic complexes.71
Mo–H] central unit in elementary organometallic reactions as reversible migratory insertion, β-H elimination and alkane reductive elimination, which are well-known transformations for mononuclear compounds but have been less studied in bimetallic complexes.71
 | ||
| Scheme 9 Bimetallic elementary reactions for the trans-[H–MoMo–H] fragment. | ||
The sequential and reversible bimetallic migratory insertion of ethylene formed an unsaturated bis(hydrocarbyl) species. The latter evolved under an ethylene atmosphere to an ethyl-vinyl complex (Scheme 9(a)) via irreversible reductive elimination of ethane and subsequent oxidative addition of an olefinic C(sp2)–H bond.70 Isomerization of the resulting cis-[H–MoMo–(CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)] moiety72 and migratory insertion of C2H4 into the Mo–H bond led to the final ethyl-vinyl product (Scheme 9(a)). The DFT calculations performed for the first migratory insertion step disclosed that the hydride attack on the coordinated olefin molecule yields a mixed valence Mo(I)Mo(III) species, which reorganizes via barrierless hydride transfer assisted by the THF molecules present in the reaction mixture to form the more stable Mo(II)Mo(II) intermediate. In contrast, the reaction of the trans-[H–MoMo–H] fragment towards phenylacetylene involves hydride abstraction and generation of H2 from the weakly acidic C–H bond of PhC
CH2)] moiety72 and migratory insertion of C2H4 into the Mo–H bond led to the final ethyl-vinyl product (Scheme 9(a)). The DFT calculations performed for the first migratory insertion step disclosed that the hydride attack on the coordinated olefin molecule yields a mixed valence Mo(I)Mo(III) species, which reorganizes via barrierless hydride transfer assisted by the THF molecules present in the reaction mixture to form the more stable Mo(II)Mo(II) intermediate. In contrast, the reaction of the trans-[H–MoMo–H] fragment towards phenylacetylene involves hydride abstraction and generation of H2 from the weakly acidic C–H bond of PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH, leading to a bis(phenylacetylide) dimolybdenum complex (Scheme 9(b)). Treatment with CS2 and CO2 formed the corresponding bis(dithioformate) and bis(formate)-bis(amidinate) paddlewheel complexes by attack of the electrophilic carbon atom of the heterocumulene to the Mo–H bond, respectively. This process was assisted by the coordination of the heteroatom to the adjacent Mo atom, evidencing the cooperative effects between the two metals (Scheme 9(c)).70b
CH, leading to a bis(phenylacetylide) dimolybdenum complex (Scheme 9(b)). Treatment with CS2 and CO2 formed the corresponding bis(dithioformate) and bis(formate)-bis(amidinate) paddlewheel complexes by attack of the electrophilic carbon atom of the heterocumulene to the Mo–H bond, respectively. This process was assisted by the coordination of the heteroatom to the adjacent Mo atom, evidencing the cooperative effects between the two metals (Scheme 9(c)).70b
After studying the activation of C–H, C–O and C–S bonds, we focused on the more polar E–H and E–C bonds, where E is an electropositive main group element (i.e., Li, Mg, Al, Ga and Zn). The interest in the coordination mode of these bonds towards transition metals has rapidly grown in the last decade, in part due to their relevance to a variety of catalytic processes.73 However, little information is available regarding the coordination chemistry of these bonds to bimetallic structures. Thus, we investigated their reactivity towards the quadruply bonded dimolybdenum complex discussed herein. The interaction between the main group element-hydrogen and -carbon bonds with the Mo2 fragment led to the formation of five-membered rings stabilized by 3c–2e interactions, stemming from the coordination of the E–C or E–H bond to one Mo centre, assisted by the electron donation of the adjacent Mo–H bonds. The synergistic effects between both Mo atoms, strongly bonded by a quadruple metal–metal bond, are once again responsible for the stabilization of peculiar and unique molecules. Besides, bimetallic compounds containing M–C–E links have been postulated as key intermediates in cross-coupling reactions,74 but structural information on these species is scarce.75
In this context, we studied the coordination of Zn–C bonds in organozinc molecules such as ZnEt2, ZnMe2 and ZnPh2 (Scheme 10(a)) towards the trans-[H–MoMo–H] core. The Zn–C bonds are weakened upon coordination due to the interaction with the bimetallic core, and the resulting species can be envisioned as snapshots along the reaction coordinate of a transmetalation process, where the Zn–C bond is not completely broken and a new M–C is formed.76 The behaviour towards the Zn(C5Me5)2 and Zn2(C5Me5)2 zincocenes is different and leads to the formation of new compounds in which H–Zn(C5Me5) and H–ZnZn(C5Me5) units bind to the dimolybdenum core through zinc hydride bonds (Scheme 10(b)). The uniqueness of the trans-[H–MoMo–H] core is again responsible for the stabilization of these reactive zinc hydride species, which have not been isolated as free molecules.
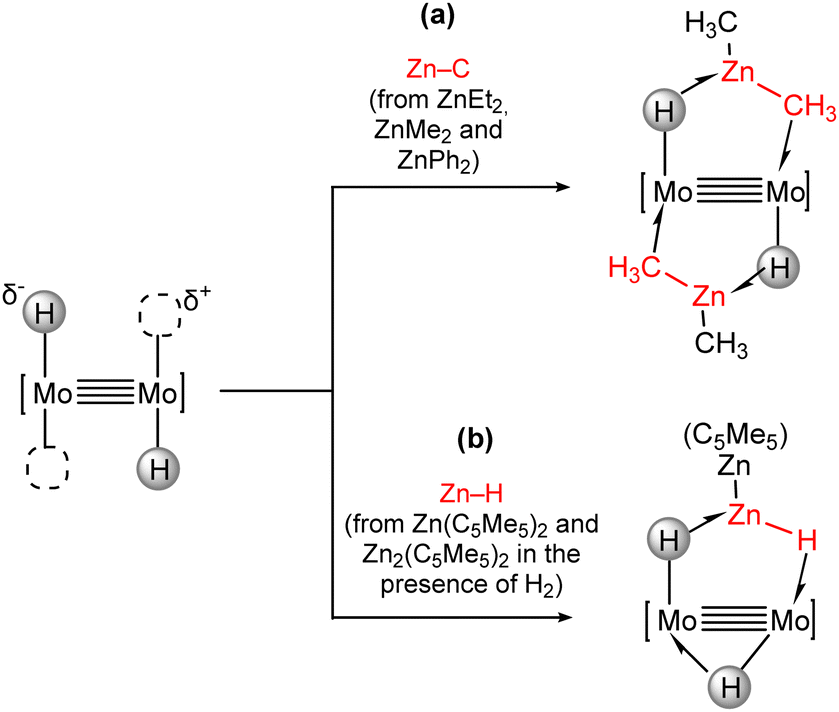 | ||
| Scheme 10 Incorporation of Zn–C and Zn–H bonds in the coordination sphere of quadruple metal–metal bonds. | ||
The unusual coordination of organozinc species to the Mo2 core resembles the even more unusual coordination of Li–H bonds. In fact, a single molecule of lithium hydride can be incorporated to the Mo2 core. This coordination is facilitated by the interaction with a Mo-hydride ligand, which compensates the unsaturation of the lithium atom and increases the σ-donor strength of the polar Li–H bond (Scheme 11(a)). We were able to build molecular structures containing one, two and three LiH monomers per Mo2 unit.77 In the case of three LiH entities, spontaneous trimerization occurs, forming an unexpected Mo6Li9H18 cluster whose structure was determined by X-ray crystallography. The detection of 1J(7Li–1H) coupling constants with values of ca. 17 Hz in the 1H and 7Li NMR spectra confirmed the non-negligible covalent contribution in the bonding of the LiH to the Mo2 entity. A computational analysis was also performed, showing the donor–acceptor interactions between the Mo–H bonds and the Li atoms, and between the electron rich multiple M–M bond and empty orbitals with major contribution of the main group element. All these interactions contribute to the stabilization of the metalacyclic five-membered rings.
 | ||
| Scheme 11 Coordination of Li–H (a) and Li–C (b) bonds to a bimetallic dimolybdenum fragment. Dinickel complex with C6H4 bridging ligand from the reactivity of Ni(COD)2 and LiPh (c). | ||
Encouraged by these studies, the coordination of organolithium reagents became an obvious fundamental target. It is important to note that the coordination of Li–C bonds to transition metal centres is even rarer, although pioneering work was performed by Wilke and coworkers while studying the nickel effect.78 After the analysis on Li–H bonds, we envisioned that a similar situation could occur for Li–C bonds. Therefore, we explored the reactivity of the trans-[H–MoMo–H] core towards commonly used organolithium reagents such as methyl, ethyl and phenyl lithium.79 These studies demonstrated that the dimolybdenum platform has adequate electronic features and coordination environment to incorporate highly reactive monomers of MeLi, EtLi and PhLi, preventing their natural tendency to aggregate and form oligomeric and polymeric structures through Li–C–Li linkages.80 X-ray diffraction data and multinuclear NMR spectroscopic analysis supported the presence of covalent bonding interactions between the Li–C moieties and the Mo2 core, that is, the Li–C fragment behaves as an exotic σ-donor ligand. A short distance of 2.12 Å for the bis(methyllithium) adduct (Scheme 11(b)) was recorded in the solid state. This distance is longer compared to free methyllithium in the gas phase (ca. 1.9 Å).81 This elongation, together with an acute Mo–C–Li angle of 80° and a short Mo–Li distance of 2.84 Å (slightly above the sum of the covalent radii of the atoms82), supports its formulation as σ-complexes of MeLi, assisted by intramolecular donation of electron density from the adjacent Mo–H bond. Moreover, large 1J(7Li–13C) and 1J(6Li–13C) coupling constants of 16 and 6 Hz, respectively, for the bis(methyllithium) complex, (Scheme 11(b)) were measured in solution by 13C and 7Li NMR spectroscopy of the 13C-enriched samples, which further support this rationalization.
Our interest in Li–C bonds acting as σ-donor ligands led us to investigate their reactivity with other transition metal precursors. A proposed “Li3NiPh3(solv)3” structure was reported decades ago by Taube and coworkers,83 which was described as a planar hexacoordinated compound but without authentication by X-ray crystallographic studies. The recent discovery of Pd(0) hexagonal complexes by Crimmin's group84 together with our curiosity towards the LiPh bonding to the Ni atoms led us to examine the reactivity of Ni(COD)2 with LiPh in collaboration with Hevia's group. Instead of the reported molecule, what we found was the formation of a unique dinickel complex, [{Li3(solv)2Ph3Ni}2(μ-η2:η2-C6H4)], which contains a C6H4 bridging ligand (Scheme 11(c)). Theoretical analysis showed strong backdonation from the Ni d orbitals to the antibonding C–C molecular orbital of the C6H4 moiety. The phenyl lithium molecules are linked to the Ni atoms, forming strong Ni–C bonds with a small participation of the Li atoms in a weak σ(C–Li) → s(Ni) interaction.85 The homobimetallic compound does not exhibit a Ni–Ni bond (Ni⋯Ni distance of ca. 2.71 Å), which was characterized by NMR spectroscopic techniques as a diamagnetic complex, in agreement with the computational analysis (NBO, QTAIM and ELI techniques). Although the mechanism involved in the formation of this structure was not investigated in detail, the participation of the two Ni centres in a bimetallic mechanism is likely given that both Ni atoms are engaged in this unusual activation of the LiPh molecule through the formation of a benzyne-type unit, finally leading to a C6H4 bridging ligand with strong dimetallabicyclobutane character. The implications of these species in Ni-catalyzed transformations employing organolithium reagents remain unexplored, although we demonstrated that this complex multi-metallic aggregate behaves as a catalyst in several cross-coupling processes.
Transition metal–main group bimetallic cooperativity
Under certain conditions, main group metals can behave in many ways (reactivity towards small molecules, redox chemistry and structural conformations) as transition metals.86 Therefore, the design of mixed transition/main group bimetallic compounds has also attracted great attention in recent years. In particular, the combination of d-block elements with main group metals, which exhibit amphiphilic character, provides complementary reactivity, and thus has emerged as a powerful frontier research area in the broader field of cooperative chemistry.87 In transition metal-main group metal complexes, there is the possibility that elementary reactions take place at any one of the two active sites, which can then further interact through 1,2-migration events of the activated fragment. Besides, bond activation across the TM–E bond is also possible and provides additional routes for cooperativity reactivity. As mentioned before, combining tetrylenes (:ER2; E = Si, Ge, Sn and Pb) with transition metals represents an appealing research avenue in this field, which offers unique opportunities because the tetrel atom acts as a single-site ambiphile (σ-donating lone pair and empty p orbital), and thus exhibits unusual coordination modes and reactivity.88 Compared to their lighter and widely used carbene analogues, the coordination chemistry and reactivity of tetrylenes remains considerably less explored, in part due to their reduced stability.Recently, our research group focused on the coordination chemistry of germylene [(ArMes2)2Ge:] (ArMes = C6H3-2,6-(C6H2-2,4,6-Me3)2) to late transition metals, with emphasis on the potential cooperative reactivity of the resulting metal/metalloid compounds. For instance, the reaction of [(ArMes2)2Ge:] with [RhCl(COD)2]2 (COD = 1,4-cyclooctadiene) afforded the formation of a neutral germyl rhodium complex. In this species, the rhodium centre exhibits both η6- and η2-coordination to two mesityl rings as a result of the unusual pincer-type structure obtained after the release of the COD ligand (Scheme 12).89 Chloride abstraction was first attempted using AgNTf2 or the trityl salt [CPh3][B(C6F5)4], but surprisingly the withdrawal of a hydride from a benzylic position readily generated a pseudoallylic germyl-rhodium complex. Alternatively, treatment of the neutral germyl rhodium complex with NaBArF successfully effected the targeted chloride. However, the resulting cationic germylene species could not be detected given that it rapidly evolved by benzylic C–H bond activation to form a similar pseudoallylic complex as that generated with AgNTf2 but with a Ge–H moiety.
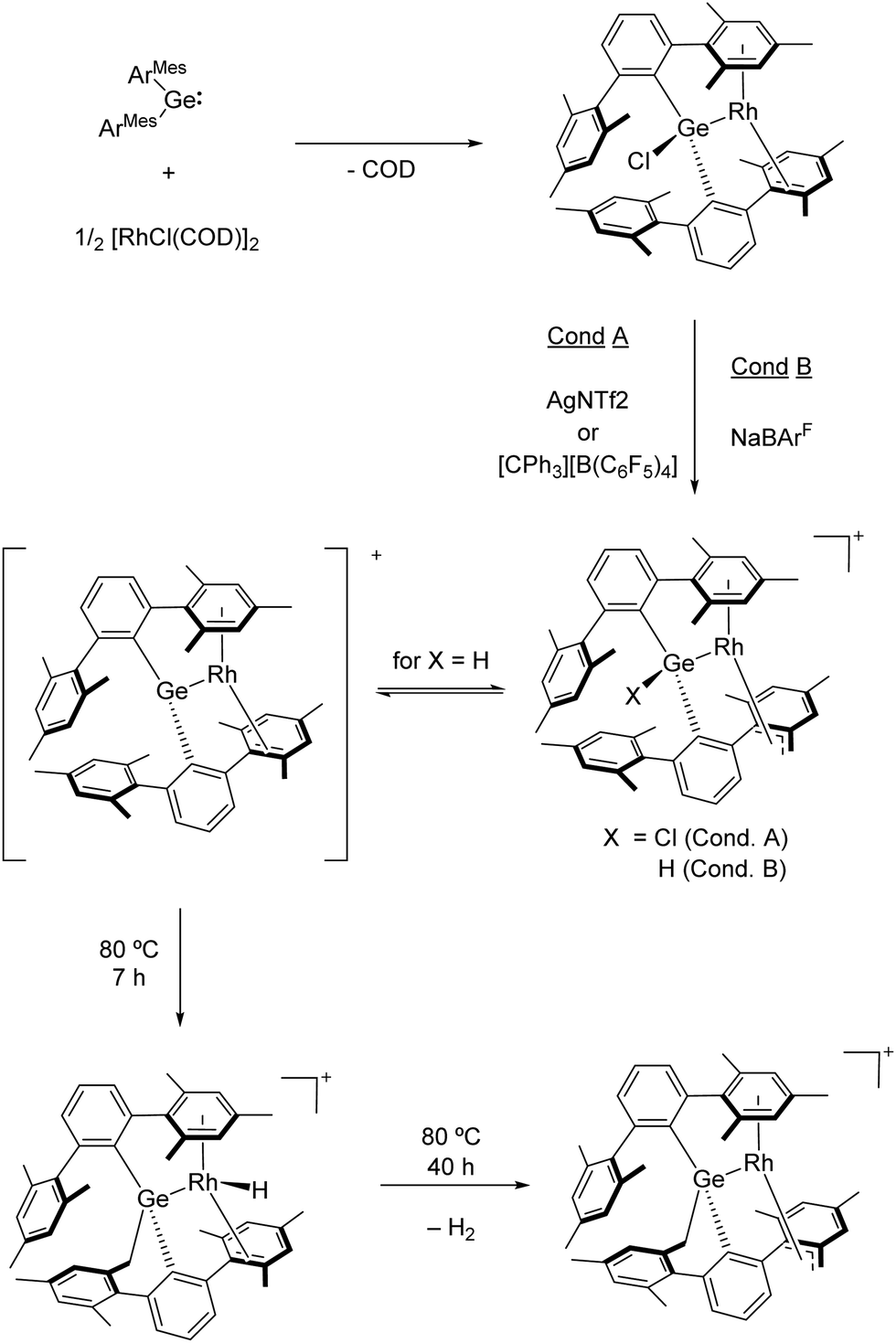 | ||
| Scheme 12 Synthesis and reactivity of germyl-rhodium complexes. | ||
Mechanistic investigations support the formation of a transient cationic germanium site prior to the activation of the C–H bond, also demonstrating that the cooperative participation of both the Ge and Rh sites is crucial. This complex quantitatively evolved when heating in benzene for 7 h, yielding a new species with a distinctive Rh–H bond through a formal hydride/methylene exchange between Rh and Ge sites. Nonetheless, this exchange is proposed to proceed through the formation of the parent reactive germylene intermediate, from which a competence between the activation of a benzylic C–H bond at either Rh (kinetic product) or Ge (thermodynamic product) is operative. Moreover, further heating for 40 h resulted in clean and complete conversion into a dehydrogenated complex after an additional C–H bond activation event. Thus, the overall process involves up to three C–H bond cleavage steps, as well as reversible hydride migration and formal hydrocarbyl migration between germanium and rhodium.
This rich intramolecular reactivity prompted us to examine the catalytic potential of the Rh/Ge system. In these ongoing studies in our group, we found that the Rh/Ge system is a competent catalyst for the trans-semi-hydrogenation of internal alkynes (Scheme 13).90 Kinetic mechanistic investigations disclosed two independent cycles for cis-semi-hydrogenation and trans-isomerization, which together account for the overall trans-semi-hydrogenation reaction. In addition, while direct participation of the germanium centre could not be demonstrated, the presence of the germyl fragment as the only embracing ligand for rhodium is essential for the observed selectivity, which is most likely due to a combination of its strong σ-donation character and the high steric profile of the substituents on germanium. In terms of selectivity towards E-isomers, this was increased by a tandem catalytic one-pot/two-step procedure combining Lindlar catalysts and the Rh/Ge complex. Besides, unusual selectivity towards α-isomers during the hydrosilylation of terminal alkynes was also disclosed, which again is different to the other rhodium catalysts tested.
 | ||
| Scheme 13 Cis-semi-hydrogenation of alkynes followed by the trans-isomerisation of alkenes of the overall Rh/Ge-catalysed trans-semi-hydrogenation of alkynes. | ||
The intramolecular reactivity of the germylene towards the C–H bonds in this system likely stems from its electrophilic character after chloride abstraction. In fact, germylenium cations exhibit remarkable reactivity, which have not only been exploited in mononuclear main group systems, but also in bimetallic designs containing transition metals.91 Based on this, we have investigated the formal combination of half of the related germylene dimer [(ArDipp2)GeCl]2 with the cationic gold fragment [(PMe2ArDipp2)Au]+. This allowed us to access different systems with π-type interactions between the lateral ring of a terphenyl substituent and the highly electrophilic germanium centre, which is crucial for its stabilization.92 Precisely, we described the trajectory for the reversible formation of π-arene bonding to germanium by a series of solid-state structures, which represent frozen snapshots of dynamic behaviour for the same metallogermylene cation.
For these studies, several chloride abstractors were examined to generate an electrophilic germanium centre from a gold-germyl compound formed by the insertion of the above-mentioned germylene into the Au–Cl bond of the (PMe2ArDipp2)AuCl precursor. Surprisingly, the reaction with AgNTf2 did not lead to the cationic complex, but instead led to an unexpected trimetallic complex (Scheme 14). Conversely, the cationic gold–germanium complex was successfully obtained when using either NaBArF or GaCl3 as chloride abstractors. However, diverse solid-state structures were observed with different π-arene germanium contacts (from 2.492(6) to 2.959(8) Å) depending on the counteranion. These structures account for the dynamic structural rearrangement that defines the reversible formation of π-arene interactions in group 14 cations. DFT calculations revealed that a small overall energy of 2.2 kcal·mol−1 is associated with this interaction. It is likely that this small energy difference is responsible for this type of bonding being overlooked in main group compounds, in contrast to the chemistry of transition metals, where it is rather common. It also represents an example of the advantages of exploring unusual bimetallic designs, which tend to offer unforeseen structural features and reactivity.
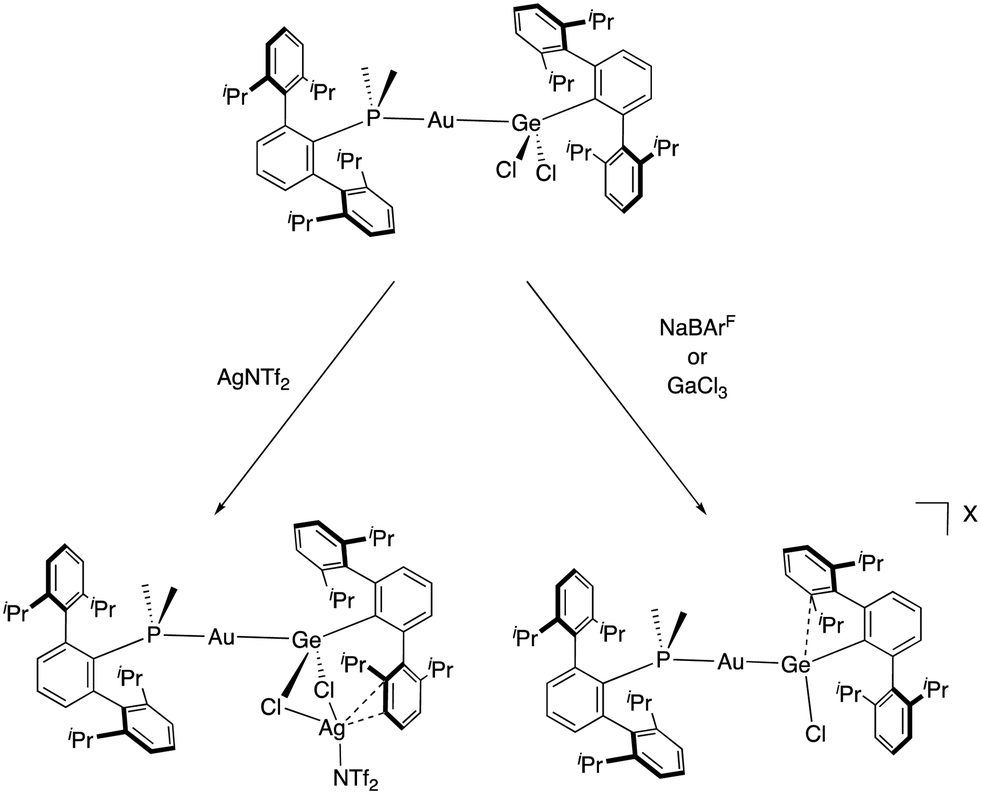 | ||
| Scheme 14 Synthesis of a cationic gold germylene compound stabilized by a Ge-based π-interaction. | ||
Conclusions
Despite occupying a prominent position at the origin of modern organometallic chemistry, the evolution of bimetallic chemistry has followed a discontinuous development over the last six decades. This curiosity-driven intermittent progress has recently given rise to continuously growing efforts to design and investigate bimetallic architectures of highly diverse nature. This re-emergence has been mostly driven by the realization that bimetallic complexes hold great prospects for reaction development and to accomplish some of the most challenging catalytic transformations faced by contemporary chemists. In the natural world metalloenzymes, in numerous cases containing more than one metal, rely on highly sophisticated synergistic mechanisms to succeed in many of the latter chemical transformations. As synthetic chemists, we have the capacity to escape from the constraints imposed by nature, but at the same time get inspiration from its cooperative strategies. The development of innovative bimetallic complexes feed to great extent from this inspiration, as well as from the important knowledge gained from heterogeneous catalytic systems. Overall, the development of bimetallic compounds has become a frontier research area, which complements traditional mononuclear catalysis and other innovative catalytic approaches.Herein, we discussed our group's efforts in the area of bimetallic bond activation and catalysis. Along this journey, we have investigated bimetallic species, ranging from those containing a multiply bonded bimetallic core to others with single and highly polarized dative M → M bonds to others in which there is virtually no interaction between the metals. We investigated in detail the nature of these bonds and interactions and how they influence the resulting reactivity towards a series of small molecules. Tuning the stereoelectronic properties in the ligands that stabilize the two metals has been shown to be extremely important in sterically constrained heterobimetallic designs. Subtle modifications on ligand substituents permits, for instance, accessing bimetallic frustrated Lewis pairs, whose reactivity is equivalent to that found in their main group counterparts. These species display particular advantages derived from easy ligand-tuning and from the available partly-filled d-orbitals, which offer opportunities for further reactivity. Beyond transition metals, we also investigated heterobimetallic complexes that contain a low-valent main group metal or metalloid, which are single-site ambiphilic fragments. We demonstrated bimetallic cooperation for bond activation in these systems, which have been proven to be active in several catalytic transformations.
These still exploratory results are just a hint of what we believe will come in the future. With the increasing number of research groups across the globe joining this collective effort of invigorating the field of bimetallic chemistry, we are confident that the best is still to come. We anticipate the discovery of many new catalytic transformations, relying on synergies that are just out of reach for mononuclear complexes, the isolation of structures with unprecedent physicochemical properties and the permeation of all this knowledge into other areas, which will benefit from bimetallic designs.
Author contributions
All authors have discussed the organization of the Feature Article and have collaboratively written the original draft and directly participated on the review and editing process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Research Council (ERC Starting Grant, CoopCat, Project 756575), the Spanish Ministry of Science and Innovation (Grants PID2019-110856GAI00) and Junta de Andalucía (P18-FR-4688 and US-1380849). M. N. and J. J. M. also acknowledge Junta de Andalucía for postdoctoral fellowships (DOC_00149 and DOC_00153). This contribution discusses work carried out by many students and postdoctoral researchers whose effort and dedication are enormously acknowledged, especially M. G. Alférez, Dr S. Bajo, Dr N. Hidalgo, Dr R. Somerville and Dr. C. Theulier.References
- (a) L. H. Gade, Angew. Chem., Int. Ed., 2000, 39, 2658 CrossRef CAS; (b) J. P. Krogman and C. M. Thomas, Chem. Commun., 2014, 50, 5115 RSC; (c) J. F. Berry and C. M. Thomas, Dalton Trans., 2017, 46, 5472 RSC; (d) J. F. Berry and C. C. Lu, Inorg. Chem., 2017, 56, 7577 CrossRef CAS PubMed; (e) R. C. Cammarota, L. J. Clouston and C. C. Lu, Coord. Chem. Rev., 2017, 334, 100 CrossRef CAS; (f) N. P. Mankad, Chem. Commun., 2018, 54, 1291 RSC; (g) J. Campos, Nat. Rev. Chem., 2020, 4, 696 CrossRef CAS; (h) B. Chatterjee, W.-C. Chang, S. Jena and C. Werlé, ACS Catal., 2020, 10, 14024 CrossRef CAS.
- L. F. Dahl, E. Ishishi and R. E. Rundle, J. Chem. Phys., 1957, 26, 1750 CrossRef CAS.
- F. A. Cotton, N. F. Curtis, C. B. Harris, B. F. G. Johnson, S. J. Lippard, J. T. Mague, W. R. Robinson and J. S. Wood, Science, 1964, 145, 1305 CrossRef CAS.
- S. T. Liddle, Molecular Metal–Metal Bonds: Compounds, Synthesis, Properties, 1st edn, Wiley, 2015 Search PubMed.
- P. A. Lindahl, J. Inorg. Biochem., 2012, 106, 172 CrossRef CAS PubMed.
- (a) J. Clausen and W. Junge, Nature, 2004, 430, 480 CrossRef CAS; (b) S. J. Lee, M. S. McCormick, S. J. Lippard and U.-S. Cho, Nature, 2013, 494, 380 CrossRef CAS.
- (a) Bioinspired Catalysis, ed. W. Weigand and P. Schollhammer, Wiley, 2015 Search PubMed; (b) S. Ogo, K. Ichikawa, T. Kishima, T. Matsumoto, H. Nakai, K. Kusaka and T. Ohhara, Science, 2013, 339, 682 CrossRef CAS PubMed; (c) A. C. Ghosh, C. Duboc and M. Gennari, Coord. Chem. Rev., 2021, 428, 213606 CrossRef CAS; (d) Q.-F. Chen, Y.-H. Guo, Y.-H. Yu and M.-T. Zhang, Coord. Chem. Rev., 2021, 448, 214164 CrossRef CAS; (e) R. M. Bullock, J. G. Chen, L. Gagliardi, P. J. Chirik, O. K. Farha, C. H. Hendon, C. W. Jones, J. A. Keith, J. Klosin, S. D. Minteer, R. H. Morris, A. T. Radosevich, T. B. Rauchfuss, N. A. Strotman, A. Vojvodic, T. R. Ward, J. Y. Yang and Y. Surendranath, Science, 2020, 369, eabc3183 CrossRef CAS.
- (a) M. K. Samantaray, S. Kavitake, N. Morlanés, E. Abou-Hamad, A. Hamieh, R. Dey and J.-M. Basset, J. Am. Chem. Soc., 2017, 139, 3522 CrossRef CAS PubMed; (b) M. K. Samantaray, R. Dey, S. Kavitake, E. Abou-Hamad, A. Bendjeriou-Sedjerari, A. Hamieh and J.-M. Basset, J. Am. Chem. Soc., 2016, 138, 8595 CrossRef CAS PubMed; (c) S. Lassalle, R. Jabbour, P. Schiltz, P. Berruyer, T. K. Todorova, L. Veyre, D. Gajan, A. Lesage, C. Thieuleux and C. Camp, J. Am. Chem. Soc., 2019, 141, 19321 CrossRef CAS PubMed; (d) S. Lassallea, R. Jabbour, I. del Rosal, L. Maron, E. Fonda, L. Veyre, D. Gajan, A. Lesage, C. Thieuleux and C. Camp, J. Catal., 2020, 392, 287 CrossRef.
- See for example: (a) S. Zhang, L. Nguyen, J.-X. Liang, J. Shan, J. Liu, A. I. Frenkel, A. Patlolla, W. Huang, J. Li and F. Tao, Nat. Commun., 2015, 6, 7938 CrossRef CAS PubMed; (b) T. Chen and V. O. Rodionov, ACS Catal., 2016, 6, 4025 CrossRef CAS; (c) M. Filez, Angew. Chem., Int. Ed., 2019, 58, 13220 CrossRef CAS; (d) A. K. Goulas, S. Sreekumar, Y. Song, P. Kharidehal, G. Gunbas, P. J. Dietrich, G. R. Johnson, Y. C. Wang, A. M. Grippo, L. C. Grabow, A. A. Gokhale and F. D. Toste, J. Am. Chem. Soc., 2016, 138, 6805 CrossRef PubMed.
- See for example: (a) M. H. Pérez- Temprano, J. A. Casares, A. R. de Lera, R. Alvarez and P. Espinet, Angew. Chem., Int. Ed., 2012, 51, 4917 CrossRef; (b) R. J. Oeschger and P. Chen, J. Am. Chem. Soc., 2017, 139, 1069 CrossRef CAS PubMed; (c) C. Chen, C. Hou, Y. Wang, T. S. A. Hor and Z. Weng, Org. Lett., 2014, 16, 524 CrossRef CAS PubMed; (d) L. Jin, D. R. Tolentino, M. Melaimi and G. Bertrand, Sci. Adv., 2015, 1, e1500304 CrossRef; (e) D. Campillo, D. Escudero, M. Baya and A. Martín, Chem. – Eur. J., 2022, 28, e202104538 CAS.
- (a) H. Zhang, B. Wu, S. L. Marquard, E. D. Litle, D. A. Dickie, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Organometallics, 2017, 36, 3498 CrossRef CAS; (b) H. Zhang, G. P. Hatzis, C. E. Moore, D. A. Dickie, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2019, 141, 9516 CrossRef CAS PubMed.
- (a) S. Bagherzadeh and N. P. Mankad, J. Am. Chem. Soc., 2015, 137, 10898 CrossRef CAS PubMed; (b) T. J. Mazzacano and N. P. Mankad, J. Am. Chem. Soc., 2013, 135, 17258 CrossRef CAS PubMed.
- (a) Y.-Y. Zhou and C. Uyeda, Angew. Chem., Int. Ed., 2016, 55, 3171 CrossRef CAS PubMed; (b) S. Pal, Y.-Y. Zhou and C. Uyeda, J. Am. Chem. Soc., 2017, 139, 11686 CrossRef CAS PubMed; (c) Y.-Y. Zhou and C. Uyeda, Science, 2019, 363, 857 CrossRef CAS PubMed.
- J. Hicks, A. Mansikkamäki, P. Vasko, J. M. Goicoechea and S. Aldridge, Nat. Chem., 2019, 11, 237 CrossRef CAS PubMed.
- L. Escomel, I. Del Rosal, L. Maron, E. Jeanneau, L. Veyre, C. Thieuleux and C. Camp, J. Am. Chem. Soc., 2021, 143, 4844 CrossRef CAS PubMed.
- F. M. Miloserdo, N. A. Rajabi, J. P. Lowe, M. F. Mahon, S. A. Macgregor and M. K. Whittlesey, J. Am. Chem. Soc., 2020, 142, 6340 CrossRef PubMed.
- A. J. Martínez-Martínez, A. R. Kennedy, R. E. Mulvey and C. T. O’Hara, Science, 2014, 346, 834 CrossRef PubMed.
- (a) S. D. Robertson, M. Uzelac and R. E. Mulvey, Chem. Rev., 2019, 119, 8332 CrossRef CAS PubMed; (b) L. J. Bole and E. Hevia, Nat. Synth., 2022, 1, 195 CrossRef; (c) J. M. Gil-Negrete and E. Hevia, Chem. Sci., 2021, 12, 1982 RSC.
- (a) H. C. Brown, H. I. Schlesinger and S. Z. Cardon, J. Am. Chem. Soc., 1942, 64, 325 CrossRef CAS; (b) H. C. Brown and B. Kanner, J. Am. Chem. Soc., 1966, 88, 986 CrossRef CAS.
- (a) G. Wittig and E. Benz, Chem. Ber., 1959, 92, 1999 CrossRef CAS; (b) W. Tochtermann, Angew. Chem., Int. Ed. Engl., 1966, 5, 351 CrossRef CAS; (c) G. Wittig and A. Rückert, Liebigs Ann. Chem., 1950, 566, 101 CrossRef CAS; (d) S. Döring, G. Erker, R. Fröhlich, O. Meyer and K. Bergander, Organometallics, 1998, 17, 2183 CrossRef.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124 CrossRef CAS PubMed.
- See for example: (a) D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed; (b) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400 CrossRef CAS; (c) A. R. Jupp and D. W. Stephan, Trends Chem., 2019, 1, 35 CrossRef CAS; (d) J. C. Slootweg and A. R. Jupp. Frustrated Lewis Pairs, Molecular Catalysis, Sprinter, Cham, 2021 CrossRef; (e) M. Navarro, J. J. Moreno and J. Campos in Comprehensive Organometallic Chemistry IV (COMC-IV), ed. K. Meyer, D. O’Hare and G. Parkin, Elsevier, Amsterdam, 2022 Search PubMed.
- A. M. Chapman, M. F. Haddow and D. F. Wass, J. Am. Chem. Soc., 2011, 133, 18463 CrossRef CAS PubMed.
- A. M. Chapman, M. F. Haddow and D. F. Wass, J. Am. Chem. Soc., 2013, 52, 4256 Search PubMed.
- X. Xu, G. Kehr, C. G. Daniluc and G. Erker, J. Am. Chem. Soc., 2013, 135, 6465 CrossRef CAS PubMed.
- M. Dellivard, R. Declercq, E. Nicolas, A. W. Ehlers, J. Backs, N. Saffon-Merceron, G. Bouhadir, J. C. Slootweg, W. Uhl and D. Bourissou, J. Am. Chem. Soc., 2016, 138, 4917 CrossRef PubMed.
- (a) S. R. Flynn and D. F. Wass, ACS Catal., 2013, 3, 2574 CrossRef CAS; (b) M. Navarro and J. Campos, J. Organomet. Chem., 2021, 75, 95 Search PubMed.
- A. M. Chapman, S. R. Flynn and D. F. Wass, Inorg. Chem., 2016, 55, 1017 CrossRef CAS PubMed.
- J. Campos, J. Am. Chem. Soc., 2017, 139, 2944 CrossRef CAS PubMed.
- (a) M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 8396 CrossRef CAS PubMed; (b) S. J. Geier, M. A. Dureen, E. Y. Ouyang and D. W. Stephan, Chem. – Eur. J., 2010, 16, 988 CrossRef CAS.
- N. Hidalgo, J. J. Moreno, M. Pérez-Jiménez, C. Maya, J. López-Serrano and J. Campos, Chem. – Eur. J., 2020, 26, 5982 CrossRef CAS PubMed.
- See for example: (a) G. Skara, F. De Vleeschouwer, P. Geerlings, F. De Proft and B. Pinter, Sci. Rep., 2017, 7, 16024 CrossRef PubMed; (b) J. Daru, I. Bakj, A. Stirling and I. Papai, ACS Catal., 2019, 9, 6049 CrossRef CAS; (c) S. Grimme, H. Kruse, L. Goerigk and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 1402 CrossRef CAS PubMed; (d) D. Yepes, P. Jaque and I. Fernández, Chem. – Eur. J., 2016, 22, 18801 CrossRef CAS PubMed; (e) L. Liu, L. L. Cao, Y. Shao, G. Ménard and D. W. Stephan, Chem, 2017, 3, 259 CrossRef CAS; (f) H. B. Hamilton and D. F. Wass, Chem, 2017, 3, 198 CrossRef CAS.
- N. Hidalgo, F. de la Cruz-Martínez, M. T. Martín, M. C. Nicasio and J. Campos, Chem. Commun., 2022, 58, 9144 RSC.
- N. Hidalgo, J. J. Moreno, M. Pérez-Jiménez, C. Maya, J. López-Serrano and J. Campos, Organometallics, 2020, 39, 2534 CrossRef CAS.
- Z. Lu, H. Ye and H. Wang, New Organoboranes in “Frustrated Lewis Pair” Chemistry, Top. Curr. Chem., 2012, 334, 59 CrossRef PubMed.
- (a) L. Ortega-Moreno, M. Fernández-Espada, J. J. Moreno, C. Navarro-Gilabert, J. Campos, S. Conejero, J. López-Serrano, C. Maya, R. Peloso and E. Carmona, Polyhedron, 2016, 116, 170 CrossRef CAS; (b) M. Marín, J. J. Moreno, C. Navarro-Gilabert, E. Álvarez, C. Maya, R. Peloso, M. C. Nicasio and E. Carmona, Chem. – Eur. J., 2019, 25, 260 CrossRef; (c) M. Marín, J. J. Moreno, M. M. Alcaide, E. Álvarez, J. López-Serrano, J. Campos, M. C. Nicasio and E. Carmona, J. Organomet. Chem., 2019, 896, 120 CrossRef.
- (a) A. K. Swarnakar, S. M. McDonald, K. C. Deutsch, P. Choi, M. J. Ferguson, R. McDonald and E. Rivard, Inorg. Chem., 2014, 53, 8662 CrossRef CAS; (b) S. M. I. Al-Rafia, A. C. Malcolm, R. McDonald, M. J. Ferguson and E. Rivard, Chem. Commun., 2012, 48, 1308 RSC; (c) M. Y. Abraham, Y. Wang, Y. Xie, P. Wei, H. F. Schaefer III, P. v R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2011, 133, 8874 CrossRef CAS PubMed.
- A. K. Swarnakar, M. J. Ferguson, R. McDonald and E. Rivard, Dalton Trans., 2016, 45, 6071 RSC.
- N. Hidalgo, S. Bajo, J. J. Moreno, C. Navarro-Gilabert, B. Mercado and J. Campos, Dalton Trans., 2019, 48, 9127 RSC.
- M. G. Alférez, N. Hidalgo, J. J. Moreno and J. Campos, Angew. Chem., Int. Ed., 2020, 59, 20863 CrossRef PubMed.
- (a) J. E. Bercaw, J. Am. Chem. Soc., 1974, 96, 5087 CrossRef CAS; (b) C. McDade, J. C. Green and J. E. Bercaw, Organometallics, 1982, 1, 1629 CrossRef CAS.
- J. Bauer, H. Braunschweig and R. D. Dewhurst, Chem. Rev., 2012, 112, 4329 CrossRef CAS PubMed.
- (a) C. M. Farley and C. Uyeda, Trends Chem., 2019, 1, 497 CrossRef CAS; (b) P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2015, 115, 28 CrossRef CAS PubMed; (c) D. R. Pye and N. P. Mankad, Chem. Sci., 2017, 8, 1705 RSC; (d) I. G. Powers and C. Uyeda, ACS Catal., 2017, 7, 936 CrossRef CAS; (e) A. M. Borys and E. Hevia, Trends Chem., 2021, 3, 803 CrossRef CAS; (f) J. Park and S. Hong, Chem. Soc. Rev., 2012, 41, 6931 RSC; (g) B. G. Cooper, J. W. Napoline and C. M. Thomas, Catal. Rev., 2012, 54, 1 CrossRef CAS; (h) G. Sciortino and F. Maseras, Top. Catal., 2022, 65, 105 CrossRef CAS.
- (a) H. Braunschweig, K. Gruss and K. Radacki, Angew. Chem., Int. Ed., 2007, 46, 7782 CrossRef CAS; (b) H. Braunschweig, K. Gruss and K. Radacki, Angew. Chem., Int. Ed., 2009, 48, 4239 CrossRef CAS PubMed; (c) H. Braunschweig, K. Gruss and K. Radacki, Inorg. Chem., 2008, 47, 8595 CrossRef CAS PubMed; (d) H. Braunschweig, A. Damme, R. D. Dewhurst, F. Hupp, J. O. C. Jimenez-Halla and K. Radacki, Chem. Commun., 2012, 48, 10410 RSC.
- (a) H. Braunschweig, R. D. Dewhurst, F. Hupp, C. Kaufmann, A. K. Phukan, C. Schneider and Q. Ye, Chem. Sci., 2014, 5, 4099 RSC; (b) R. Bissert, H. Braunschweig, R. D. Dewhurst and C. Schneider, Organometallics, 2016, 35, 2567 CrossRef CAS.
- J. R. Pinkes, B. D. Steffy, J. C. Vites and A. R. Cutler, Organometallics, 1994, 13, 21 CrossRef CAS.
- (a) U. Jayarathne, S. R. Parmelee and N. P. Mankad, Inorg. Chem., 2014, 53, 7730 CrossRef CAS PubMed; (b) M. K. Karunananda, F. X. Vázquez, E. E. Alp, W. Bi, S. Chattopadhyay, T. Shibata and N. P. Mankad, Dalton Trans., 2014, 43, 13661 RSC; (c) M. K. Karunananda, S. R. Parmelee, G. W. Waldhart and N. P. Mankad, Organometallics, 2015, 34, 3857 CrossRef CAS; (d) N. P. Mankad, Chem. Commun., 2018, 54, 1291 RSC.
- (a) T. J. Mazzacano and N. P. Mankad, J. Am. Chem. Soc., 2013, 135, 17258 CrossRef CAS PubMed; (b) M. K. Karunananda and N. P. Mankad, J. Am. Chem. Soc., 2015, 137, 14598 CrossRef CAS; (c) L.-J. Cheng and N. P. Mankad, J. Am. Chem. Soc., 2019, 141, 3710 CrossRef CAS PubMed; (d) H.-C. Yu, S. M. Islam and N. P. Mankad, ACS Catal., 2020, 10, 3670 CrossRef CAS; (e) N. J. Leon, H.-C. Yu, T. J. Mazzacano and N. P. Mankad, Synlett, 2020, 125 CAS; (f) H.-C. Yu and N. P. Mankad, Synthesis, 2021, 1409 CAS.
- N. Hidalgo, C. Maya and J. Campos, Chem. Commun., 2019, 55, 8812 RSC.
- N. Hidalgo, C. Romero-Pérez, C. Maya, I. Fernández and J. Campos, Organometallics, 2021, 40, 1113 CrossRef CAS.
- M. Ma, A. Sidiropoulos, L. Ralte, A. Stasch and C. Jones, Chem. Commun., 2013, 49, 48 RSC.
- S. Jamali, S. Abedanzadeh, N. K. Khaledi, H. Samouei, Z. Hendi, S. Zacchini, R. Kia and H. R. Shahsavari, Dalton Trans., 2016, 45, 17644 RSC.
- S. Bajo, M. G. Alférez, M. M. Alcaide, J. López-Serrano and J. Campos, Chem. – Eur. J., 2020, 26, 16833 CrossRef CAS PubMed.
- T. Nguyen, A. D. Sutton, M. Brynda, J. C. Fettinger, G. J. Long and P. P. Power, Science, 2005, 310, 844 CrossRef CAS PubMed.
- N. V. S. Harisomayajula, A. K. Nair and Y.-C. Tsai, Chem. Commun., 2014, 50, 3391 RSC.
- L. J. Clouston, R. B. Siedschlag, P. A. Rudd, N. Planas, S. Hu, A. D. Miller, L. Gagliardi and C. C. Lu, J. Am. Chem. Soc., 2013, 135, 13142 CrossRef CAS PubMed.
- P. A. Rudd, S. Liu, N. Planas, E. Bill, L. Gagliardi and C. C. Lu, Angew. Chem., Int. Ed., 2013, 52, 4449 CrossRef CAS PubMed.
- F. A. Cotton, L. A. Murillo and R. A. Walton, Multiple Bonds Between Metal Atoms, 3rd edn, Springer, New York, 2005, [F. A. Cotton, R. A. Walton, in 1st (1981) and 2nd (1992) ed.] Search PubMed.
- A. Noor, G. Glatz, R. Müller, M. Kaupp, S. Demeshko and R. Kempe, Nat. Chem., 2009, 1, 322 CrossRef CAS PubMed.
- A. Noor, S. Qayyum, T. Bauer, S. Schwarz, B. Weber and R. Kempe, Chem. Commun., 2014, 50, 13127 RSC.
- C. Schwarzmaier, A. Noor, G. Glatz, M. Zabel, A. Y. Timoshkin, B. M. Cossairt, C. C. Cummins, R. Kempe and M. Scheer, Angew. Chem., Int. Ed., 2011, 50, 7283 CrossRef CAS PubMed.
- J. Shen, G. P. A. Yap and K. H. Theopold, Chem. Commun., 2014, 50, 2579 RSC.
- Y.-S. Huang, G.-T. Huang, Y.-L. Liu, J.-S. K. Yu and Y.-C. Tsai, Angew. Chem., Int. Ed., 2017, 56, 15427 CrossRef CAS PubMed.
- H. Z. Chen, S.-C. Liu, C.-H. Yen, J.-S. K. Yu, Y.-J. Shieh, T.-S. Kuo and Y.-C. Tsai, Angew. Chem., Int. Ed., 2012, 51, 10342 CrossRef CAS.
- Y. Chen and S. Sakaki, Dalton Trans., 2014, 43, 11478 RSC.
- J. F. Berry and C. C. Lu, Inorg. Chem., 2017, 56, 7577 CrossRef CAS PubMed.
- K. Liao, T. C. Pickel, V. Boyarskikh, J. Bacsa, D. G. Musaev and H. M. Davies, Nature, 2017, 551, 609 CrossRef CAS PubMed.
- (a) H. Tsurugi, A. Hayakawa, S. Kando, Y. Sugino and K. Mashima, Chem. Sci., 2015, 6, 3434 RSC; (b) S. Rej, M. Majumdar, S. Kando, Y. Sugino, H. Tsurugi and K. Mashima, Inorg. Chem., 2017, 56, 634 CrossRef CAS PubMed.
- K. M. Gramigna, D. A. Dickie, B. M. Foxman and C. M. Thomas, ACS Catal., 2019, 9, 3153 CrossRef CAS.
- (a) M. Pérez-Jiménez, J. Campos, J. López-Serrano and E. Carmona, Chem. Commun., 2018, 54, 9186 RSC; (b) M. Pérez-Jiménez, N. Curado, C. Maya, J. Campos, E. Ruiz, S. Álvarez and E. Carmona, Chem. – Eur. J., 2021, 27, 6569 CrossRef PubMed.
- T. J. Mazzacano, N. J. Leon, G. W. Waldhart and N. P. Mankad, Dalton Trans., 2017, 46, 5518 RSC.
- Y. Chen and S. Sakaki, Inorg. Chem., 2017, 56, 4011 CrossRef CAS PubMed.
- (a) M. J. Butler and M. R. Crimmin, Chem. Commun., 2017, 53, 1348 RSC; (b) I. M. Riddlestone, J. A. B. Abdalla and S. Aldridge, Adv. Organomet. Chem., 2015, 63, 1 CrossRef CAS.
- J. Pozo, G. Salas, R. Álvarez, J. A. Casares and P. Espinet, Organometallics, 2016, 35, 3604 CrossRef.
- (a) A. L. Liberman-Martin, D. S. Levine, M. S. Ziegler, R. G. Bergman and T. D. Tilley, Chem. Commun., 2016, 52, 7039 RSC; (b) R. J. Oeschger and P. Chen, Organometallics, 2017, 36, 1465 CrossRef CAS.
- M. Pérez-Jiménez, J. Campos, J. Jover, S. Álvarez and E. Carmona, Organometallics, 2022 DOI:10.1021/acs.organomet.2c00216.
- M. Pérez-Jiménez, N. Curado, C. Maya, J. Campos, J. Jover, S. Álvarez and E. Carmona, J. Am. Chem. Soc., 2021, 143, 5222 CrossRef PubMed.
- K. Fischer, K. Jonas, P. Misbach, R. Stabba and G. Wilke, Angew. Chem., Int. Ed. Engl., 1973, 12, 943 CrossRef.
- M. Pérez-Jiménez, J. Campos, J. Jover, S. Álvarez and E. Carmona, Angew. Chem., Int. Ed., 2022, 61, e202116009 CrossRef PubMed.
- V. Gessner, C. Däschlein and C. Strohmann, Chem. – Eur. J., 2009, 15, 3320 CrossRef CAS PubMed.
- D. B. Grotjahn, T. C. Pesch, J. Xin and L. M. Ziurys, J. Am. Chem. Soc., 1997, 119, 12368 CrossRef CAS.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Álvarez, Dalton Trans., 2008, 2832 RSC.
- R. Taube and N. Stransky, Z. Chem., 1979, 19, 412 CrossRef CAS.
- M. Garçon, C. Bakewell, G. A. Sackman, A. J. P. White, R. I. Cooper, A. J. Edwards and M. R. Crimmin, Nature, 2019, 574, 390 CrossRef.
- R. J. Somerville, A. M. Borys, M. Perez-Jimenez, A. Nova, D. Balcells, L. A. Malaspina, S. Grabowsky, E. Carmona, E. Hevia and J. Campos, Chem. Sci., 2022, 13, 5268 RSC.
- P. P. Power, Nature, 2010, 463, 171 CrossRef CAS PubMed.
- (a) G. Bouhadir and D. Bourissou, Chem. Soc. Rev., 2016, 45, 1065 RSC; (b) D. You and F. P. Gabbaï, Trends Chem., 2019, 1, 485 CrossRef CAS.
- R. J. Somerville and J. Campos, Eur. J. Inorg. Chem., 2021, 3488 CrossRef CAS.
- S. Bajo, M. M. Alcaide, J. López-Serrano and J. Campos, Chem. – Eur. J., 2021, 27, 16422 CrossRef CAS PubMed.
- S. Bajo, C. A. Theulier and J. Campos, ChemCatChem, 2022, e202200157 CAS.
- (a) V. S. V. S. N. Swamy, S. Pal, S. Khan and S. S. Sen, Dalton Trans., 2015, 44, 12903 RSC; (b) T. A. Engesser, M. A. Lichtenthaler, M. Schleep and I. Krossing, Chem. Soc. Rev., 2016, 45, 789 RSC; (c) H. Fang, Z. Wang and X. Fu, Coord. Chem. Rev., 2017, 344, 214 CrossRef CAS.
- S. Bajo, M. M. Alcaide, J. López-Serrano and J. Campos, Chem. – Eur. J., 2020, 26, 15519 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |