
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular engineering of confined space in metal–organic cages
James E. M.
Lewis

School of Chemistry, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK. E-mail: j.e.m.lewis@bham.ac.uk
First published on 30th November 2022
Abstract
Metal–organic cages (MOCs) have become an intensely studied class of abiotic host molecules. This is due to the ability to generate a myriad of polyhedral architectures from relatively simple, and minimal numbers of, components in high yield and under thermodynamic control. The encapsulation of molecular guests within the nanoscale, confined cavities of these cages frequently draws comparisons with enzymatic binding sites. In this regard, the ostensible ease with which chemical modifications can be made to these internal cavities adds to their attractiveness, as they can be readily tailored with a high degree of precision. In this Feature Article, the ways in which the cavities of MOCs can be engineered at the molecular level will be looked at. The discussion will be divided across three key parameters: size, shape and functionality. Most concepts will be exemplified with a focus on the Pd2L4 class of assemblies due to their relative structural simplicity and the wealth of studies reported in the literature. The core principles discussed will, however, be generalisable to other classes of MOCs, and abiotic host systems as a whole. Gaining increasing mastery over the fine tuning of MOC cavity properties, whilst retaining facile, high-fidelity self-assembly processes, will lead to ever more precise engineering of the cavities of artificial host systems with complex and highly specific functionality.
 James E. M. Lewis | Jamie Lewis obtained his PhD (2014) from the University of Otago, New Zealand, under the supervision of Prof. James Crowley. He then spent three years in the group of Prof. Stephen Goldup at the University of Southampton, initially as a PDRA and subsequently a Marie Skłodowska-Curie Fellow. He began his independent career in 2017 as an Imperial College Research Fellow. In late 2022 Jamie joined the School of Chemistry at the University of Birmingham as a Royal Society University Research Fellow and Assistant Professor in Supramolecular Chemistry. Jamie's research interests in synthetic supramolecular chemistry are focussed on mechanically interlocked molecules and metallosupramolecular architectures. |
Introduction
Metal–organic cages (MOCs) are discrete, porous host architectures that self-assemble, generally under thermodynamic control, from metal ions/nodes and organic ligands. A number of conceptual approaches to the design and synthesis of MOCs have been developed in the last four decades or so. These include the symmetry interaction,1 directional bonding,2 molecular panelling,3 and weak-link4 approaches (the latter of which enables access to kinetically metastable species). Details of these different approaches have been given in recent review articles5 – towards which the reader is directed – and will not be repeated here.The prevailing interest in MOCs is due in large part to the accessible confined nanospaces, or cavities, described by the cage frameworks. These cavity environments can be used to bind a range of guest molecules and modulate their properties, which can be exploited for tasks such as promoting chemical transformations. This long-standing intrigue in using the confined cavities of MOCs for catalysis6 has prompted parallels to be drawn with the active site of enzymes. The drastically reduced structural sophistication of these supramolecular hosts in comparison to biological systems is counterbalanced by their ostensibly much more readily tuneable chemical features.
The outcomes of self-assembly processes are determined by the thermodynamic energy landscape of the assembling mixture, with balances of enthalpic and entropic factors readily disturbed by changing conditions. Consequently, in order to promote high-fidelity self-assembly and the homogenous formation of desired products, component numbers tend to be kept to a minimum, their symmetry high, and additional functionality eschewed. For MOCs the simplest assembly mixtures will consist of a single metal ion/node, and a single, high-symmetry, ligand. Using these “principles of simplicity”, an impressive range of MOCs of varying geometries, nuclearities and sizes have been realised. Investigations into their utility have led to reports on the stabilisation of reactive species,7 modulation of chemical reaction rates and product distributions,8 binding of pollutants9 and potential as drug delivery agents.10
Despite the successes of high-symmetry and minimalist MOCs, questions are now being raised by the community about what might be achieved if more structurally and functionally sophisticated assemblies could be accessed with the same ease.11 In this manner, the cavity spaces of MOCs could be tailored for more selective guest binding, and functionally tuned to promote more specific reactivities and chemical properties. Whilst tuning the chemical environment within MOCs is conceptually facile, in reality minor alterations to e.g. a ligand structure or metal ion identity, can have profound effects on self-assembly outcomes.
In this Feature Article, approaches to, and significant challenges posed by, modulating key properties of cavity spaces for the binding of guest molecules will be examined. These properties will be covered under the three broad headings of cavity size, shape and functionality. Palladium(II)-based architectures, popularised by seminal work from the Fujita group, are one of the most studied classes of MOCs.12 The popularity of Pd(II) stems from its preferred square-planar coordination geometry, diamagnetic nature, and favourable kinetic profile. Within this review most examples will be of Pd(II)-based cages and, in particular, a focus placed on the Pd2L4 – “paddle-wheel” or “lantern” – class of cages.13 The core concepts, however, are applicable to other MOCs, and supramolecular host systems in general.
Modulating cavity size
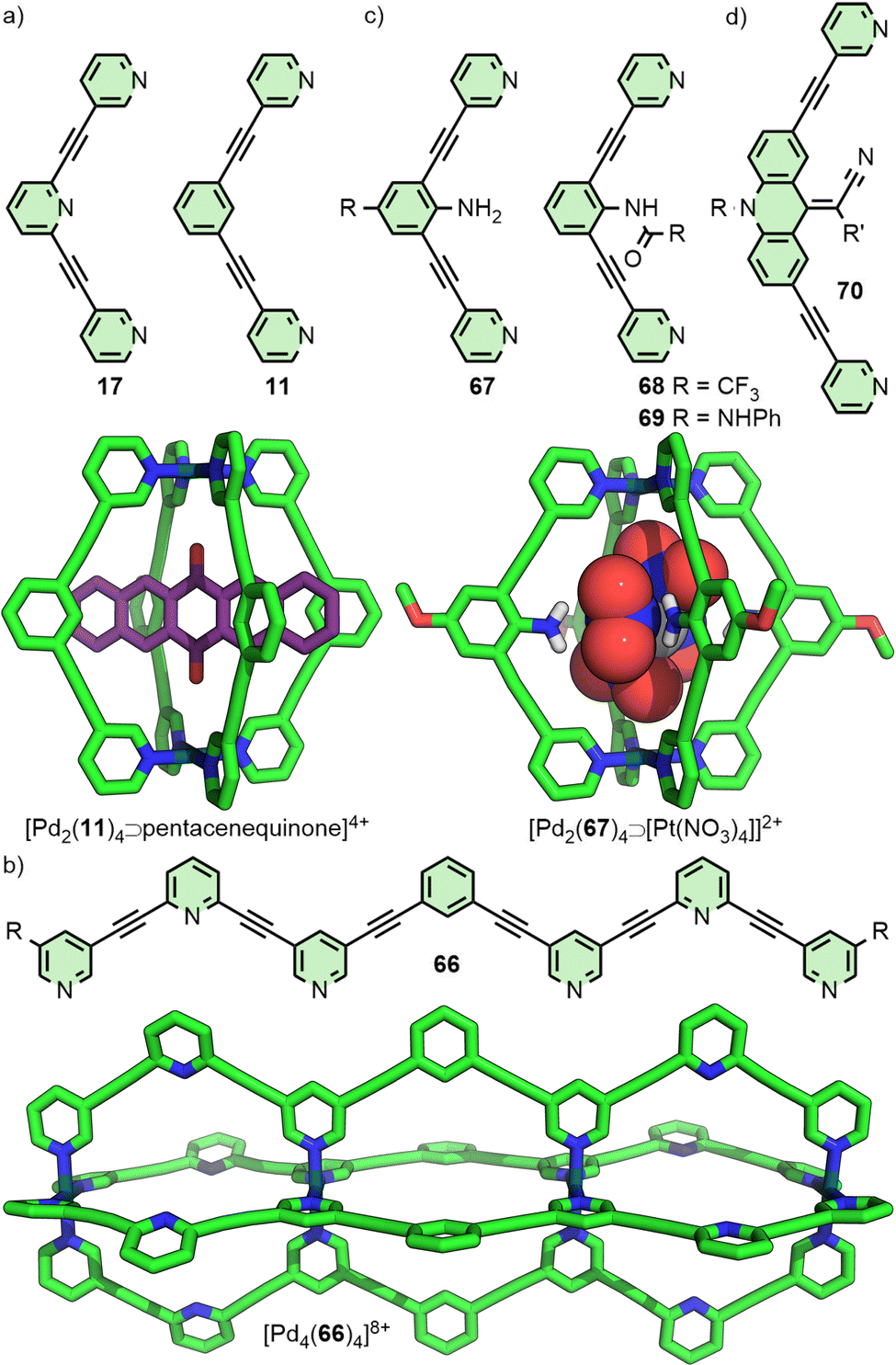
Tuning the size of the cavity space within MOCs is perhaps the most obvious way to control selectivity of guest binding. If a guest is larger than the cage cavity, encapsulation will be impossible (although partial clathration, or interactions with the external surface of the cage, may be possible).14 Conversely, if the cavity is significantly larger than the guest, interactions with the host will likely be weak and non-specific. Nitschke and co-workers, for example, have demonstrated selective sequestration by an MOC of coronene from a mixture of PAHs, likely due to a better size match of the cavity compared with smaller hydrocarbon species.15 MOCs have been reported capable of encapsulating guests ranging in size from single, monoatomic anions16 through to (via covalent tethering strategies) whole proteins.17 Two main approaches to tailor the size of MOCs are considered here: changing the size of the constituent ligand(s) whilst maintaining the same geometry, and changing the ligand design to target assembly of higher nuclearity structures.Tuning cage nuclearity
Fujita and co-workers have extensively investigated PdnL2n-type species assembled from rigid, dipyridyl ligands and square planar Pd(II) ions.12 It has been observed that the thermodynamically favoured value of n is highly correlated with the angle (Θ) between the coordination vectors of the pyridine units (Fig. 1). Ligand 1 (Θ = 127°), for example, assembles into a Pd12L24 cuboctahedron;18Θ = 131–134° was found to be a critical point,19 with ligands exceeding this value, such as 2a (Θ = 149°), forming Pd24L48 rhombicuboctahedral structures.20 The largest of these structures reported thus far, obtained from ligand 3 (Θ = 152°), was a Pd48L96 Goldberg polyhedron.21 Interestingly, [Pd24(2b)48]48+ was subsequently found to be a kinetically trapped species, with the thermodynamically favoured structure actually being the [Pd30(2b)60]60+ icosidodecahedron, a class of polyhedra that could be selectively targeted through further ligand design.22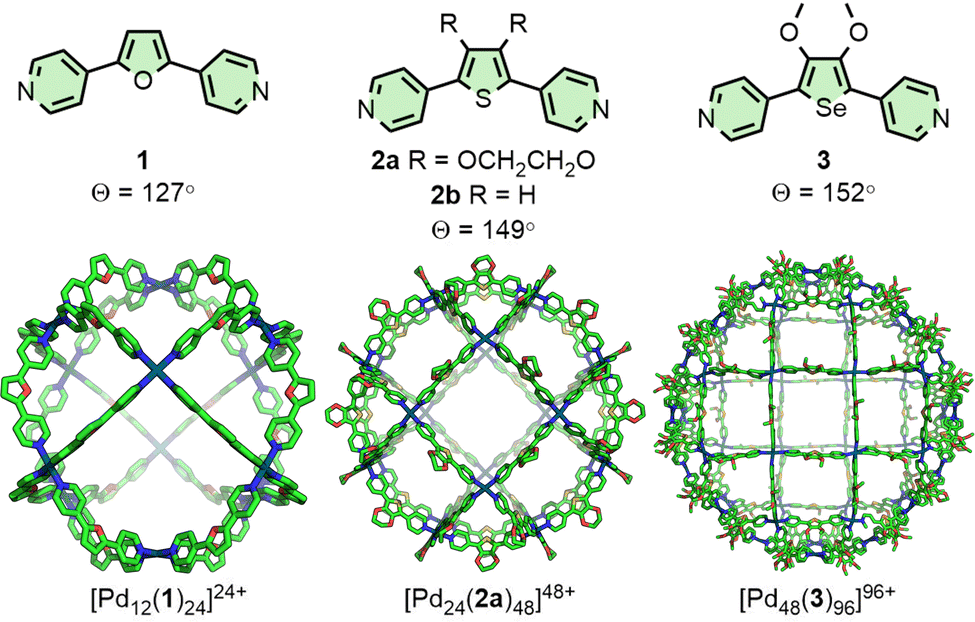 | ||
| Fig. 1 Dipyridyl ligands 1, 2, and 3 self-assemble with Pd(II) ions to form Pd12L24, Pd24L48 and Pd48L96 assemblies (SCXRD structures), respectively, with the angle between coordinating groups determining the nuclearity of the thermodynamically favoured product. | ||
Tailoring ligand size
For dipyridyl ligands where Θ approaches 120°, self-assembly with Pd(II) tends to result in the formation of Pd12L24 cuboctahedra.18 Each of the ligands 4, 5 and 6 (Fig. 2(a)) assemble into such architectures, with varying diameters estimated to be 3.6,18 4.6,23 and 6.3 nm,24 respectively, as they maintain the angle between coordinating pyridyl groups whilst extending the distance between them. This feature was even exploited in the synthesis of a cage-in-cage architecture assembled from ligand 7, designed to incorporate ditopic ligands 4 and 6 tethered to each other (Fig. 2(b)).25 Mukherjee and co-workers used this cage-in-cage design to stabilise an assembly that did not form otherwise. Ligand 8 assembles with Pd(II) to give a Pd24L24 species that can be visualised as a Pd12L24 cuboctahedron, assembled from bridging pyrimidine ligands from one arm of the ligand (Fig. 2(c), shown in pink), encapsulated within a larger Pd12L24 architecture formed through monodentate coordination to Pd(II) of the remaining two arms (Fig. 2(c), shown in green). Notably, when self-assembly of pyrimidine (9) itself with Pd(II) was attempted, only the mononuclear species [Pd(9)4]2+ formed, with monodentate coordination of 9.26 Thus, tethering to the larger ligand scaffold altered the coordination properties of the bis-monodentate pyrimidine unit. The larger Pd12L24 structure effectively templates assembly of the smaller cage through enforced proximity and arrangement of the internal pyrimidine units, enabling formation of an architecture that was independently inaccessible.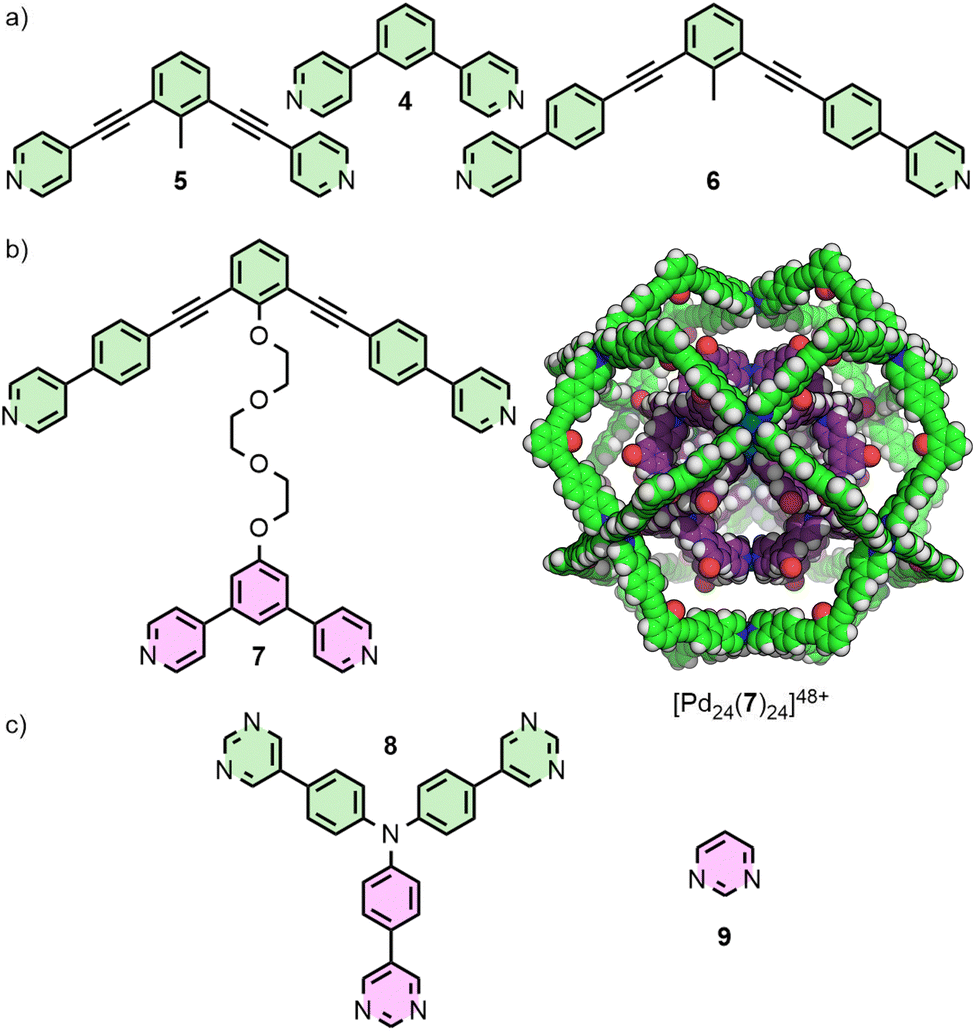 | ||
| Fig. 2 (a) Ligands 4, 5 and 6 all have Θ values of ∼120° and assemble to give Pd12L24 species. (b) Tethered tetratopic ligand 7 forms the cage-in-cage complex [Pd24(7)24]48+ (SCXRD structure). (c) Ligand 8 assembles into a similar Pd24L24 species, with monodentate coordination through two of the pyrimidine units (green), and bis-monodentate through the third (pink). Combining pyrimidine itself (9) with Pd(II) yielded only the mononuclear complex [Pd(9)4]2+. | ||
At the other end of the scale spectrum, the lowest nuclearity PdnL2n-type MOCs are the Pd2L4 cages, first reported by McMorran and Steel from ligand 10 (Fig. 3(a)).27 This helicate assembly was shown in the solid state to be able to alter its helical pitch, and thus the distance between Pd(II) ions, to best accommodate various anions (Pd⋯Pd 8.8 and 7.4 Å for PF6− and I−, respectively).28
 | ||
| Fig. 3 (a) Bis-3-pyridyl ligands 10–13 and their Pd2L4 assemblies (SCXRD structures). Larger organic ligands (b) 14 and 15 also assemble into Pd2L4 cages, whilst (c) 16, in the presence of suitably sized anions (A−), yields the quadruply-interlocked metallo-catenane [Pd4(16)8⊃(A)3]5+ (SCXRD structure – encapsulated anions not shown). | ||
Lusby and co-workers have investigated the effects of solvent and anion identity on the binding strength of quinone-type guests within [Pd2(11)4]4+ (Fig. 3(a)), originally reported by Hooley.29 Critically, anions such as OTf− had previously been observed to bind within the cavity of cage. Metathesis with the much larger and more weakly interacting anion BArF− (tetrakis[3,5-bis(trifluoromethyl)phenyl]borate) allowed access to the cage system without endohedral anions. This anion exchange, in combination with switching solvent from CD3CN to CD2Cl2, enhanced the binding constant of the cage with naphthoquinone by a remarkable three orders of magnitude, from 2.1 × 102 M−1 to 3.5 × 105 M−1.30
Within [Pd2(11)4]4+ the Pd(II) ions are far enough apart (∼12 Å) that a guest anion is not required to stabilise the structure. As the Pd⋯Pd distance in these Pd2L4 systems decreases, guest molecules may be necessary to negate the electrostatic repulsion between the palladium cations. Chand and co-workers, for example, found that ligand 12 (Fig. 3(a)) would assemble into Pd2L4 cages only in the presence of a suitably sized anion template, such as NO3− or Cl− (Pd⋯Pd distance in the SCXRD structure of the Cl− adduct was 6.6 Å), whilst BF4− was unable to template cage formation.31 Lusby and Hiraoka investigated the self-assembly of ligand 13 (Fig. 3(a)) with Pd(II). In this work, the Pd2L4 cage (Pd⋯Pd 9.8 Å) was found to not be the thermodynamically preferred species, but could nonetheless be obtained in high yield as a kinetically trapped, metastable product, provided a suitable anion template (BF4−) and a weakly coordinating solvent (CH3NO2) were used.32
Substantially larger Pd2L4 cages have been reported through the use of elongated organic ligands (Fig. 3(b)), such as an adamantyl-cored system reported by Preston and Kitchen ([Pd2(14)4]4+; Pd⋯Pd 19 Å),33 and a 28 Å “molecular peanut” from the Yoshizawa group ([Pd2(15)4]4+).34 A phenomenon, first reported for Pd2L4-type cages by Kuroda,35 can occur once cage ligands reach a certain size. In the presence of suitable templates, it is possible for some Pd2L4 cages to dimerise,36 giving quadruply-interlocked, Pd4L8 metallo-catenanes with three accessible cavity spaces. Ligand 16 self-assembles with Pd(II) as the NO3− salt, initially forming a transient kinetic product – the monomeric Pd2L4 cage – before converting to the quadruply-interlocked metallo[2]catenane (Fig. 3(c)). The identity of the anion was integral to this dimerisation process; PF6−, OTf− and 2-naphthalenesulfonate (ONs−) salts formed only as the Pd2L4 cage monomer, as these anions were too large to fit within the cavities of the dimer. Furthermore, partial conversion of the dimer [Pd4(16)8⊃(NO3)3]5+ to the monomeric [Pd2(16)4⊃(ONs)2]2+ cage was achieved upon addition of NaONs to the former.37 The Clever group have investigated Pd4L8-type metallo-catenanes extensively,38 and even reported a Pd6L8 system from the dimerisation of a double-cavity Pd3L4 cage.39
Thus, despite the need to consider the effects of non-covalent interactions between components that can inhibit access to target assemblies or promote formation of alternative species, a substantial range of cage sizes can be accessed through tailoring of the ligand structure. Importantly, however, even relatively minor changes can have substantial impacts on the chemistry of the cavities.
In 2012 the Crowley group reported cage [Pd2(17)4]4+ that was shown in solution and by single crystal X-ray diffraction (SCXRD) to bind two molecules of the anticancer drug cisplatin (Fig. 4(a)).40 Subsequently, the group prepared a structurally similar assembly, in which the alkyne units of ligand 17 were replaced with para-phenyl moieties (18).41 This resulted in an approximately 20% expansion of the cage cavity, with the Pd⋯Pd distance extending from 11.7 to 14.4 Å. Interestingly, cage [Pd2(18)4]4+ showed no binding affinity for cisplatin, assigned by the authors as due to steric hindrance from the linking aryl units. Thus, relatively small changes to the size of cavity spaces can be achieved through structural modifications, although these can dramatically affect the guest-binding properties of the cages.
In a similar vein, Yoshizawa and co-workers have used dipyridyl ligands 19 and 20 to assemble Pd2L4 cages with portals occluded by linking anthracene moieties (Fig. 4(b)), providing a large π surface for guests to interact with. Through changing the core unit from phenyl to naphthyl, anisotropic expansion of the cage cavity along the Pd⋯Pd axis could be achieved (13.6 and 16.1 Å for 19 and 20, respectively, based on the SCXRD structure of the “empty” cages). [Pd2(19)4]4+ was able to selectively bind C60 over C70,42 whilst [Pd2(20)4]4+ could efficiently encapsulate C70. Interestingly, mixing both cages in the presence of C60 led to the selective formation of the heteroleptic structure cis-[Pd2(19)2(20)2⊃C60]4+, templated by the guest molecule.43
As such, whilst it is possible to modulate size within MOCs to tailor the cavity for binding of guest species, in sufficiently labile systems guests may also induce transformations of the host(s). Whilst the potential desirability of this adaptable behaviour is clear, if unlooked for, such host rearrangements could lead to extinguishing of intended guest-binding properties.
 | ||
| Fig. 4 (a) Structurally similar ligands 17 and 18 assemble into analogous Pd2L4 cages (SCXRD structures); whilst [Pd2(17)4]4+ is able to encapsulate two molecules of cisplatin, [Pd2(18)4]4+ showed no recognition of the anticancer drug. (b) Cages [Pd2(19)4]4+ and [Pd2(20)4]4+ (SCXRD structure shown with –OCH2CH2OCH3 chains omitted for clarity) display different affinities for fullerene guests due to anisotropic expansion of the cavities along the Pd⋯Pd axis between the two. | ||
Anisotropic cavity space
Although MOCs of various geometries and metal:ligand stoichiometries have been described over the years, the majority are assembled from binary combinations of a single metal ion and single, symmetrical ligand. As a consequence, the cages themselves, and thus their cavities, are highly symmetrical. This is in contrast to enzymatic binding sites, which are defined by their high levels of anisotropy, aiding in high-fidelity discrimination and selectivity between substrates.44In this section, different approaches towards the realisation of lower symmetry architectures will be discussed. There are primarily two strategies towards this goal. The first is the incorporation of either more than one type of ligand, or more than one type of metal ion, into an assembly, yielding heteroleptic (mixed-ligand) and heteronuclear (mixed-metal) architectures, respectively. The second is the use of unsymmetrical ligands, as reducing the symmetry of the components should lead to lower symmetry assemblies. For both of these approaches, high-fidelity, integrative self-sorting of the components is essential to achieve high-yielding assembly of the desired product. This requires a careful consideration of entropic and enthalpic driving forces, in the absence of which either narcissistic self-sorting or statistical self-assembly (Fig. 5) may arise.
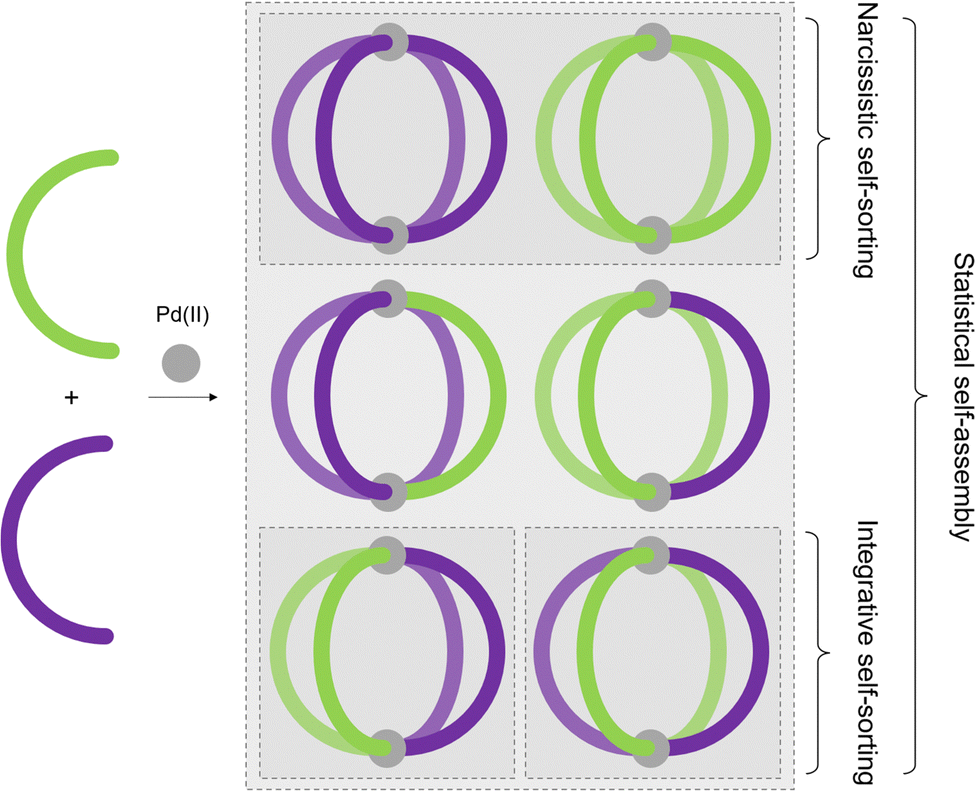 | ||
| Fig. 5 Self-assembly of ligand mixtures can result in (i) statistical mixtures of all possible homo- and heteroleptic species, (ii) narcissistic self-sorting to give exclusively the homoleptic species, or (iii) integrative, or social, self-sorting to yield defined heteroleptic species. | ||
Heteroleptic cages
Metal–organic assemblies incorporating more than one ligand type are of great interest not just due to the potential for generating more anisotropic MOCs, but also for introducing multiple sets of endo-/exohedral functionalities. In this article, strategies for generating heteroleptic structures will broadly be split into two approaches: ‘coordination sphere engineering’, and geometric complementarity.Although there has been a recent resurgence in interest in the area, it is noted that such strategies have been explored for some time. In 1993, Lehn and co-workers reported the integrative self-assembly of bis(bidentate) ligand 21 and tris(bidentate) ligand 22 with Cu(I) ions to form the heteroleptic cage [Cu6(21)3(22)2]6+ (Fig. 6(a)),45 (and subsequently extending this to multi-cavity systems).46 Importantly, the bulky phenyl substituents on 22 help inhibit metallo-polymer formation and promote integrative self-assembly,47 a concept that would become the key principle of Schmittel's HetPHEN approach to forming heteroleptic coordination environments.48 Of note, the principles of this strategy had previously been exploited by Sauvage in the development of passive metal template strategies for the synthesis of mechanically interlocked molecules.49 Although such approaches have been shown effective in the self-assembly of bidentate coordinating units, translation of these principles to monodentate donors has been more limited.
 | ||
| Fig. 6 Seminal examples of coordination sphere engineering in heteroleptic trigonal prisms: (a) Lehn's Cu(I) assembly (SCXRD structure), and (b) Fujita's side-chain directing approach with cis-protected Pd(II) ions (SCXRD structure). | ||
Fujita and co-workers used their side-chain directing approach50 to produce a series of heteroleptic trigonal prisms. The core principle of this strategy relies on the integrative self-assembly between a mixture of one ligand with unsubstituted pyridine donors, and another with 2,6-lutidine units, with cis-protected square planar metal ions ([Pd(en)]2+; en = ethylenediamine). The steric bulk of the lutidine methyl groups inhibits homoleptic coordination at the metal nodes, leading to heteroleptic structures such as the trigonal prismatic assembly [Pd6(en)6(23)3(24)2]12+ (Fig. 6(b)).51
Extending this principle to naked square planar metal ions, Clever and co-workers reported the self-assembly of ditopic ligands incorporating picolyl coordinating units with Pd(II) (Fig. 7(a)). Ligand 25 yielded a mixture of a Pd2L3S2 bowl, and a Pd2L2S4 metallocycle, as the steric bulk of the methyl groups prevented formation of the Pd2L4 cage (S = coordinating solvent molecule), whilst ligand 26 formed only a bowl assembly. Combining the two ligands in stoichiometric quantities with Pd(II), however, led to clean formation of a Pd2L2L′2 heteroleptic cage, concluded from DFT calculations to be the cis isomer.52
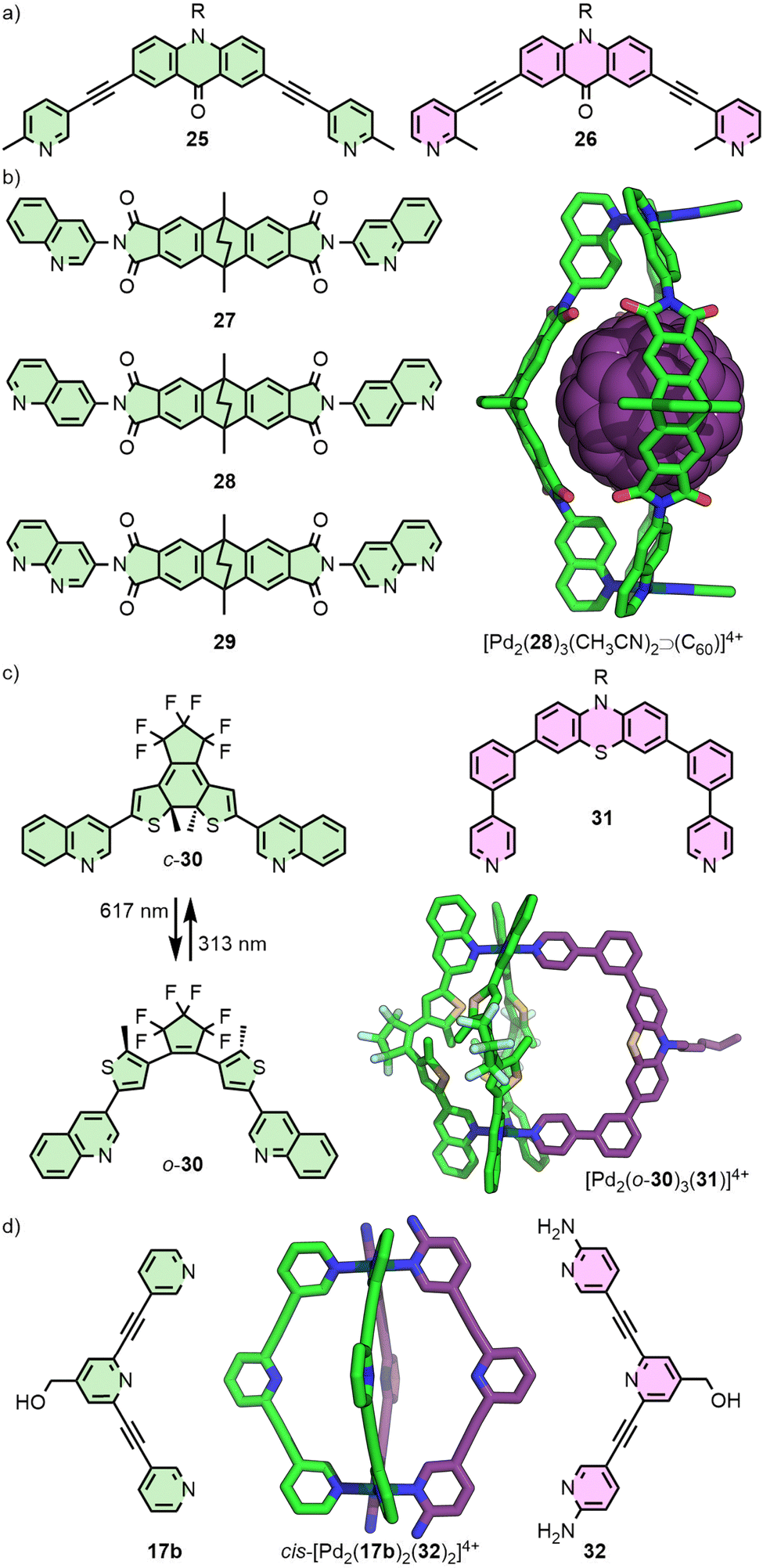 | ||
| Fig. 7 Coordination sphere engineering of heteroleptic cages using the steric bulk of (a) complementary picoline-based ligands (25 and 26), and quinoline donors (27, 28 and 30; SCXRD structure of [Pd2(28)3(CH3CN)2⊃C60]4+ shown) in combination with ligands with (b) appropriate hydrogen-bond acceptor units (29) or (c) less sterically demanding ligands (31; SCXRD structure of [Pd2(30)3(31)]4+ shown). (d) Heteroleptic cage cis-[Pd2(17b)2(32)2]4+ (DFT structure with CH2OH units omitted) was accessed through post-synthetic ligand displacement of homoleptic cage [Pd2(17b)4]4+. | ||
The steric bulk of quinoline donor groups has also proven effective in the engineering of heteroleptic systems (Fig. 7(b)). Ligand 28, with two 6-quinolyl donor units, was found to quantitatively form a [Pd2(28)3S2]4+ bowl capable of binding fullerenes C60 and C70.53 Interestingly, isomeric ligand 27 with 3-quinolyl units similarly formed a Pd2L3S2 bowl structure, but only as a kinetically trapped, metastable product – over time, quantitative rearrangement to give the Pd2L4 cage was observed.54
Post-assembly modification of [Pd2(28)3S2]4+ (S = MeCN) was achieved through exchanging the solvent ligands, S. Addition of two equivalents of Cl− led to the reduced charge [Pd2(28)3Cl2]2+ species, whilst terephthalate was able to act as a bridging ligand to form a dimerised cage.53 Addition of a naphthyridine ligand (29) to [Pd2(28)3(MeCN)2⊃C70]4+ led to an unusual Pd2L3L′ heteroleptic structure. Remarkably, self-assembly of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 mixture of Pd(II), 28 and 29 in the presence of C60 as a template yielded an alternative heteroleptic structure, trans-[Pd2(28)2(29)2⊃C60]4+. Both of these systems were stabilised by hydrogen-bonding between the C–H unit of 28 and the non-coordinated nitrogen of 29.55
:1:1 mixture of Pd(II), 28 and 29 in the presence of C60 as a template yielded an alternative heteroleptic structure, trans-[Pd2(28)2(29)2⊃C60]4+. Both of these systems were stabilised by hydrogen-bonding between the C–H unit of 28 and the non-coordinated nitrogen of 29.55
The generality of this approach was subsequently demonstrated with bowl complex [Pd2(30)3(MeCN)2]4+, assembled from photoswitchable ligand 30. Less sterically demanding ligands with pyridyl donor groups (e.g.31) were able to be incorporated into the structure, forming a series of Pd2L3L′ heteroleptic cages (Fig. 7(c)).56
An intriguing example of heteroleptic cis-Pd2L2L′2 cages from the Crowley group arose from ligand exchange of a preformed homoleptic cage (Fig. 7(d)). Addition of ligand 32 to [Pd2(17b)4]4+ resulted in displacement of only two of the 17b ligands (even upon addition of an excess of 32), giving the mixed-ligand system with hydrogen bonding interactions between the amino groups of 32 and exohedral ortho pyridyl protons of 17b stabilising the cage. Interestingly, DFT calculations suggested that this was a kinetically trapped metastable species, with full conversion to the homoleptic cage [Pd2(32)4]4+ the thermodynamically favoured outcome.57
:1:1 mixture of the two ligands and a Pd(II) source yielded the heteroleptic cage. The primary driving forces that make this assembly thermodynamically favourable are the lower energy conformation of ligand 33, compared to the homoleptic [Pd2(33)4] architecture, and the gain in entropy achieved through the incorporation of ligand 34 into the dinuclear Pd2L2L′2 cage, rather than the homoleptic [Pd4(34)8] species.58 This approach has subsequently been utilised in the assembly of other cis-Pd2L2L′2 cages incorporating fluorenone- and helicene-based ligands, exhibiting circularly-polarised luminescence (CPL),59 and systems in which endohedral carbazole N-substituents could be tuned to modulate binding of phosphate ester guests.60
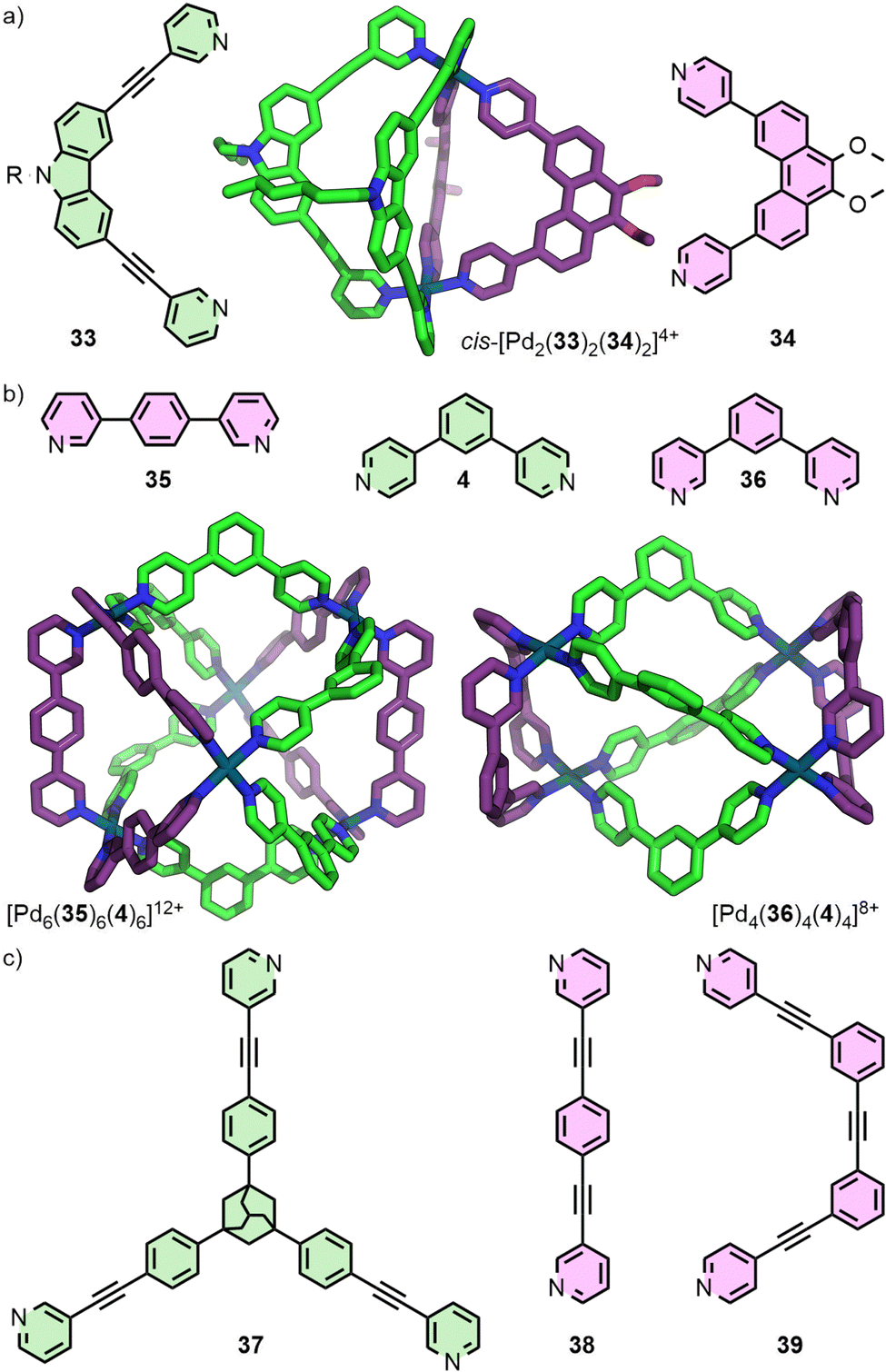 | ||
| Fig. 8 (a) Geometric complementarity between ditopic ligands 33 and 34 facilitates access to the heteroleptic cage cis-[Pd2(33)2(34)2]4+ (SCXRD structure). (b) Higher nuclearity heteroleptic structures have also been reported, such as [Pd6(35)6(4)6]12+ and [Pd4(36)4(4)4]8+ (SCXRD structures). (c) Through matching of ligand geometries, in combination with denticity mismatch, trigonal prismatic cages [Pd3(37)2(38/39)3]6+ have been realised. | ||
Geometric complementary between ligands has also been a successful strategy for generating higher nuclearity structures (Fig. 8(b)). Severin and co-workers reported the hexanuclear cage [Pd6(35)6(4)6], identified from the self-assembly products of a mixture of six ligands with Pd(II).61 Subsequent work from this group demonstrated selective formation of [Pdn(36)n(L′)n] assemblies where the value of n could be controlled through systematic variation of the angle (Θ) within dipyridyl L′ between coordinating units (e.g. L′ = 4, Θ = 120°, n = 4; L′ = 2b, Θ = 149°, n = 6; L′ = 1,4-bis(pyridin-4-yl)benzene, Θ = 180°, n = 8).62
Preston and co-workers reported an elegant example through a series of Pd3L2L′3 trigonal bipyramidal structures, obtained from the combined complementarity of both ligand geometry and denticity (Fig. 8(c)). Tritopic ligand 37 was shown to assemble with a series of complementary ditopic ligands (e.g.38, 39) to form homologous trinuclear heteroleptic cages, exploiting the differing denticities of the ligands to achieve heteroleptic cages with C3 symmetry.63
Unsymmetrical ligands
In the case of heteroleptic structures, lack of sufficient thermodynamic driving force to promote integrative self-assembly can lead to narcissistic self-sorting (selective formation of homoleptic species) or statistical self-assembly (Fig. 5). In contrast, self-assembly with unsymmetrical ligands, in addition to the potential to form species of varying nuclearity, can also result in possible formation of multiple diastereoisomers of MOCs with the same formulation. As discussed earlier, for example, Chand and co-workers have investigated the self-assembly of ditopic ligand 12 (Fig. 9(a)) to form a Pd2L4 structure. Due to the methylene ester backbone of the ligand, however, the cage can form as four diastereoisomers (Fig. 9(b)). With no energetic preference for any of the isomers, a mixture of all four formed in solution and crystallised as a statistical mixture.31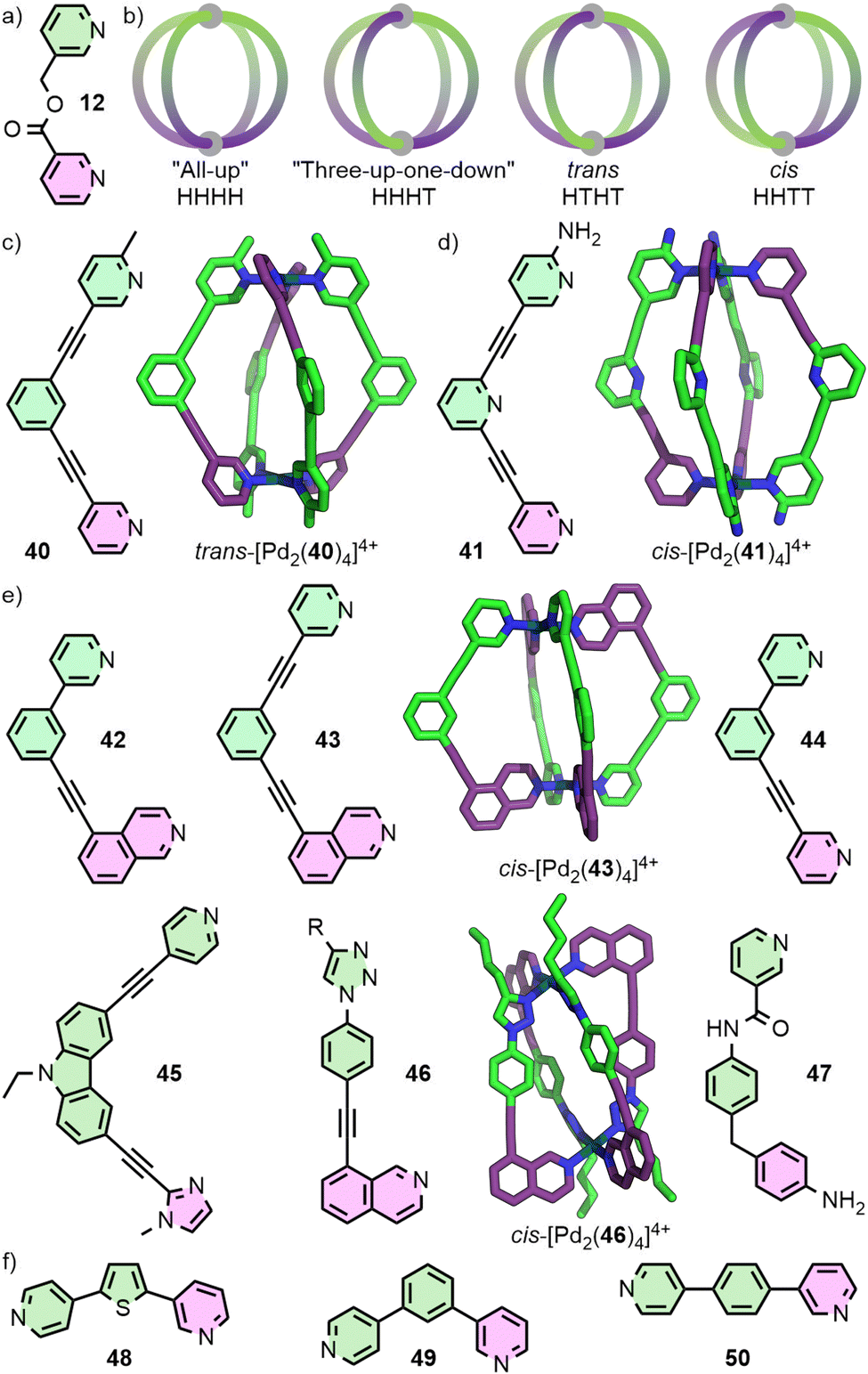 | ||
| Fig. 9 (a) Unsymmetrical ligand 12 self-assembles to give a statistical mixture of Pd2L4 cage isomers; (b) cartoon representations of the four possible configurational isomers. (c) Steric interactions between methyl groups in 40 led to formation of the trans (HTHT) cage (DFT structure), whilst (d) hydrogen bonding interactions between amino groups and pyridyl protons in 41 give the cis (HHTT) isomer (SCXRD structure). (e) Geometric principles were used in the design of ligands 42–47 to favour formation of cis cage isomers (SCXRD structures for 43 and 46 shown); for 44 the driving force was insufficient and a mixture of isomers was obtained. (f) Unsymmetrical ligands 48, 49 and 50 assemble into single isomers of Pd4L8, Pd6L12 and Pd12L24 species, respectively. | ||
Similar to engineering the integrative self-sorting of ligand mixtures to give defined heteroleptic assemblies (vide supra), design parameters to direct the self-assembly of unsymmetrical ligands can also be based around using non-covalent interactions at the site of coordination (coordination sphere engineering), or through geometric design of the ligand frameworks.64
For the controlled self-assembly of unsymmetrical ligands there are relatively few examples of successfully employing coordination sphere engineering approaches. Lewis and co-workers reported ditopic ligand 40 incorporating one pyridyl and one picolyl coordinating unit (Fig. 9(c)). Upon self-assembly with Pd(II) in CD3CN, the trans-Pd2L4 cage isomer (HTHT) was formed; assignment as the HTHT isomer was based on NMR data and DFT calculations. Interestingly, self-assembly in d6-DMSO solution yielded a mixture of the cis and trans isomers, which was suspected to be due to the differing polarities of the solvents.65 Crowley and co-workers were able to exploit interactions between unsubstituted pyridyl and ortho-aminopyridyl units, that had previously allowed them to access heteroleptic cage trans-[Pd2(17b)2(32)2]4+.57 Heating a mixture of 41 with Pd(II) gave the cis-Pd2L4 isomer (Fig. 9(d)). As with the heteroleptic system, DFT calculations suggested that the cis isomer (HHTT) was a kinetically metastable product, and that the “all-up” (HHHH) isomer should be thermodynamically preferred. Attempts to use solvophobic and dispersion forces to control isomer formation with ligands incorporating different substituents on the coordinating pyridyl groups were unsuccessful, yielding mixtures of isomeric Pd2L4 cages.66
Designing unsymmetrical ligands in which the coordinating units are geometrically matched to each other (Fig. 9(e)) has proved effective for the selective formation of specific MOC diastereoisomers. The Lewis group has developed principles for the design of unsymmetrical ditopic ligands that assemble with “naked” Pd(II) ions to selectively form cis-Pd2L4 cages of C2h symmetry. The underlying concept behind these design strategies relies on a cantilever effect, such that to best obtain a 180° angle between coordinating groups trans across the square planar Pd(II) ion, an anti-parallel arrangement of ligands is ideal.
Ligands 42 and 43, incorporating one pyridyl and one isoquinolyl coordinating unit, were designed such that the coordination vectors of these units were not coplanar with each other. Provided this deviation from coplanarity was large enough, it was anticipated that ligands trans from each other should favour the desired anti-parallel arrangement. For both ligands, self-assembly with Pd(II) exclusively gave the anticipated cis-Pd2L4 isomers as determined by NMR and, for 43, SCXRD. In contrast, ligand 44 formed a mixture of species as demonstrated by NMR, identified by MS and DOSY as being Pd2L4 species. Thus, a mixture of Pd2L4 isomers appeared to have formed, indicating the geometric parameters of the ligand were insufficient to prevent formation of significant quantities of alternative cage isomers. Indeed, DFT calculations suggested that whilst cis-[Pd2(43)4] was 4.4-26.8 kJ mol−1 lower in energy than the other three Pd2L4 isomers, for cis-[Pd2(44)4] this range reduced to 0.1–12.0 kJ mol−1, with the “three-up-one-down” isomer (HHHT) essentially isoenergetic.65
Based on this correlation between calculated isomer energy values and experimental observations, in collaboration with the Jelfs group, a high-throughput computational workflow was developed to search for ligand structures likely to exclusively form cis-Pd2L4 cages from unsymmetrical ditopic ligands. The relatively cheap, semi-empirical methods (GFN2-xTB)67 employed within this workflow proved effective at indicating exclusive formation of cis-Pd2L4 isomers, provided the calculated energy between this isomer and the next lowest energy isomer were sufficiently large (>6 kJ mol−1). From this, new cis-Pd2L4 cages were successfully synthesised. At lower energy differences (≤6 kJ mol−1), correlations between calculations and experimental results were less precise. The results from this work, however, point the way towards the use of relatively rapid computational studies to aid in the successful design and synthetic realisation of molecular systems.68
Unsymmetrical ligands with different coordination motifs have been reported (Fig. 9(e)). Ogata and Yuasa investigated ligand 45 with one pyridyl and one imidazolyl donor unit. Based on NMR data and DFT calculations, the authors demonstrated exclusive formation of the cis-Pd2L4 cage isomer upon self-assembly with Pd(II). Due to the stronger donor ability of the imidazole in comparison to the pyridine, a 1:4 ligand/metal ratio gave rise to the mononuclear [Pd(45)4] complex, with coordination exclusively through the imidazole.69 Lewis has shown a series of ligands with isostructural core frameworks consisting of both isoquinoline and 1,2,3-triazole donors (46) to form cis-Pd2L4 cages. The latter coordinating unit was installed using CuAAC ‘click’ chemistry, allowing a range of functional units to be appended to the exohedral face of the ligand. This enabled tuning of the donor strength of the triazole unit, and thus the self-assembly profile of the ligand.70 Chand and co-workers have investigate the self-assembly of ligand 47, incorporating pyridine and aniline moieties, with both “naked” Pd(II) ions and cis-protected Pd(en) nodes. For the former, formation of the cis-Pd2L4 isomer was observed, whilst the latter yielded a [Pd2(en)2L2] metallocycle with the ligands in an anti-parallel arrangement.71
Very recently, selective formation of specific isomers of larger PdnL2n cages from unsymmetrical ditopic ligands (Fig. 9(f)) have been reported. 48 formed a single Pd4L8 tetrahedron out of 35 possible isomers, 49 a Pd6L12 octahedron out of 112 isomers,72 and 50 a remarkable Pd12L24 cuboctahedron despite the potential 350 969 isomers of this assembly.73
An alternative approach to enforce a particular arrangement of ligands was recently reported by Lewis. Ligand 51 incorporates two copies of the unsymmetrical ditopic ligand 12 previously reported by Chand, but covalently held in a parallel relationship through a rigid tethering group. Self-assembly of this tetratopic ligand yielded the dinuclear cage [Pd2(51)2]4+ with the dipyridyl ligand fragments held in a trans (HTHT) arrangement. Thus, although the ligand employed for this system possess a C2 symmetry axis, the resultant cage can be considered as a pseudo-trans-Pd2L4 system (Fig. 10(a)).74
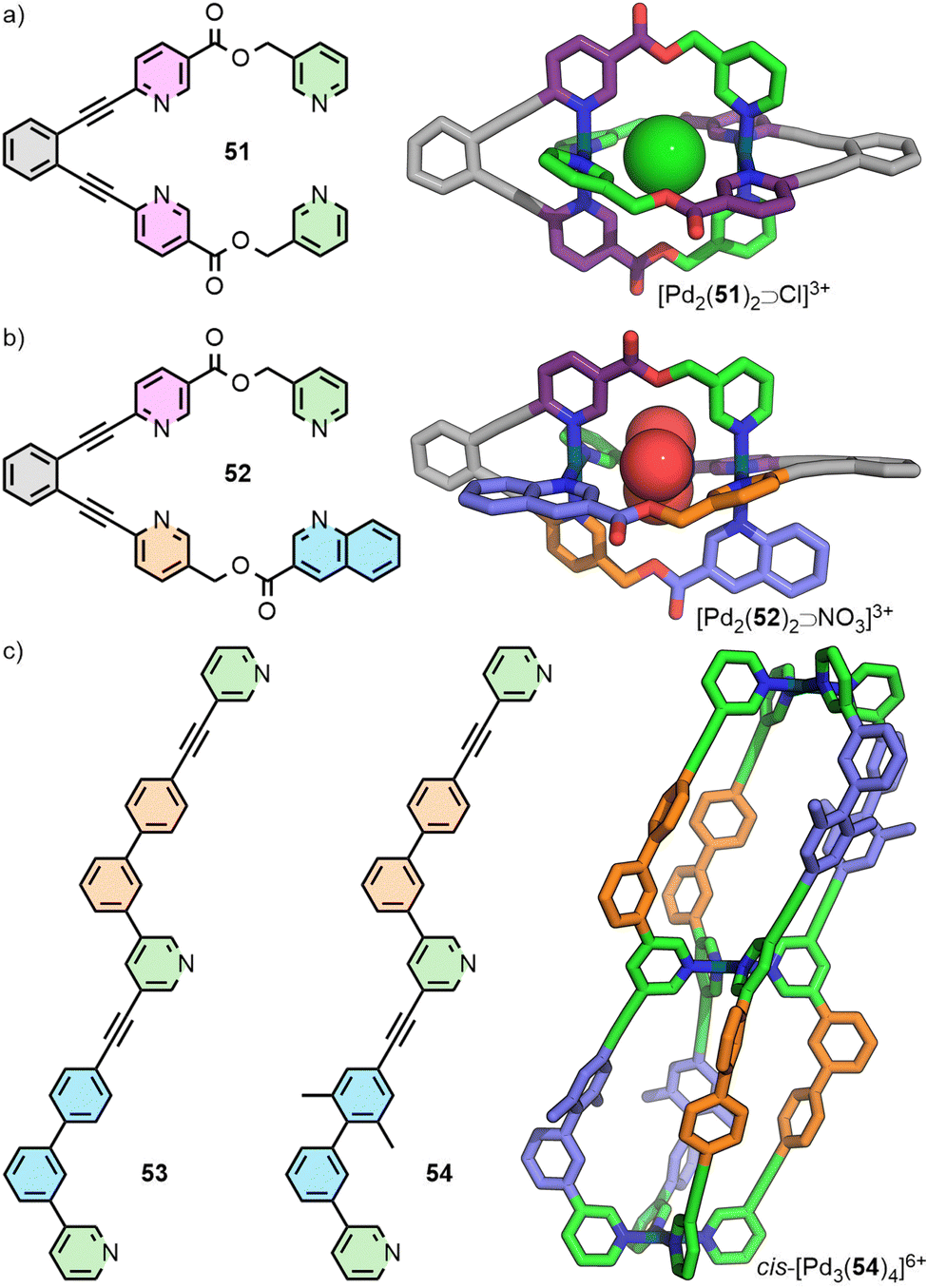 | ||
| Fig. 10 Covalent trans-tethering of (a) identical ditopic ligand fragments enables access to Pd2L2 cages with a pseudo-trans-Pd2L4 cavity (SCXRD structure), and (b) different ditopic units generates a pseudo-Pd2L2L′2 cavity of C2 symmetry (SCXRD structure). (c) Unsymmetrical tritopic ligands 53 and 54 assemble to give trinuclear cis-Pd3L4 cages (PM6 model) where each cavity can be described as a pseudo-Pd2L2L′2 environment. | ||
Pseudo-heterolepticity
The Lewis group has recently reported a new strategy towards the development of structurally sophisticated cages, whilst maintaining the simplicity of self-assembly process associated with homoleptic systems. The underlying principle of this approach relies on the realisation that it is the cavity environment itself, rather than the cage as a whole, which it is desirable to surround with multiple different functionalities.In an extension of the ligand-tethering strategy described above to enforce a particular arrangement of ligand fragments in a trans orientation, cage [Pd2(52)2]4+ was synthesised. This species is assembled from ligand 52 which has two different, unsymmetrical ditopic fragments (Fig. 10(b)). Consequently, the cavities are described by two different ligand environments, analogous to a Pd2L2L′2 cage with unsymmetrical ligands arranged in a defined configuration.74
Building on the principles of geometric complementarity with unsymmetrical ditopic ligands described above, tritopic ligands 53 and 54 were synthesised and shown upon self-assembly with Pd(II) ions to form cis-Pd3L4 double-cavity architectures (Fig. 10(c)). Due to the anti-parallel arrangement of ligands trans to each other across the Pd(II) ions, and the chemical inequivalence of the two “halves” of the ligands, each of the cavities is surrounded by two different, unsymmetrical ligand environments. This situation is analogous to the cavities prepared through integrative self-sorting of ligands in Pd2L2L′2 heteroleptic cages.74
In both of these strategies the cages are assembled from single ligands, thus are homoleptic. Each cavity, however, is surrounded by two different ligand environments, as occurs in heteroleptic Pd2L2L′2 cages. The term pseudo-heteroleptic was proposed to describe these systems. A significant advantage over true mixed-ligand systems is that integrative self-sorting of multiple components is no longer required, simplifying the self-assembly process, whilst enhancing the complexity of the confined spaces within the cages.
Heteronuclear structures
An alternative route to diversifying the structural composition of MOCs is to introduce different metal ions in the formation of heteronuclear assemblies. There have been several recent reviews that cover strategies to access systems from metallo-ligands;75 only select examples of Pd2L4-type cages will be discussed here.Dipyridyl metallo-ligands such as the Fe(II) clathrochelate 55 (Fig. 11(a)),76 and the bis-Ru(II) 564+ (Fig. 11(b)),77 assemble with Pd(II) to give Pd2L4-type architectures. Both of these approaches exploit different coordination environments and denticities of the organic components, and rely on the kinetic stability of the metallo-ligand in the presence of Pd(II). Although a useful strategy to incorporate potentially functional moieties into the ligand backbone, the resulting metallo-ligands are symmetrical. As such, the generation of these types of heteronuclear structures does not lead to an increase in anisotropy of the cavity space.
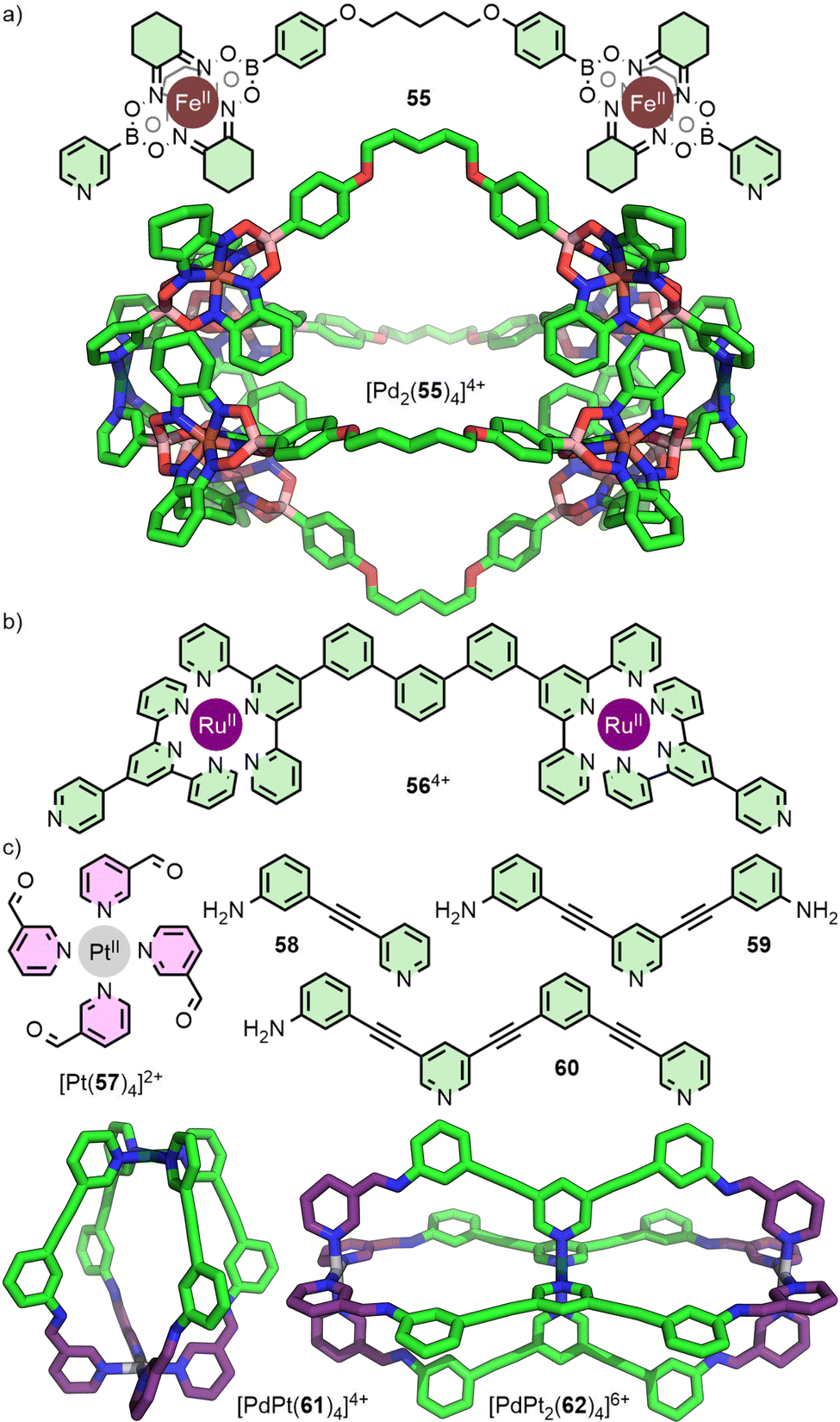 | ||
| Fig. 11 Kinetically stable metallo-ligands such as (a) Fe(II)-clathrochelate 55 (SCXRD structure), and (b) bis-Ru(II) 564+, each assemble with Pd(II) to give symmetrical Pd2L4 systems. (c) In contrast, the use of a subcomponent self-assembly strategy has allowed M2L4 and M3L4 cages (SCXRD structures) to be accessed in which Pd(II) and Pt(II) ions occupy distinct structural sites within these architectures, despite both residing in tetrapyridyl coordination environments. | ||
A variation on this strategy has been reported recently in which the metal ions occupy distinct structural sites within the MOC, rather than acting as components of a metallo-ligand. In this way, the differing sites of the metal ions aid in desymmetrisation of the cage structures. The Crowley group has investigated strategies to assemble MnL4 cages from bis- (n = 2) and tris-pyridyl (n = 3) ligand scaffolds, with different square planar metal ions coordinated to monodentate donors (Fig. 11(c)). To achieve this feat, they employed a subcomponent self-assembly strategy. The kinetically stable complex [Pt(57)4]2+ underwent imine condensation with anilines 58 and 59 which, in the presence of more labile Pd(II) ions, assembled to give heteronuclear cage [PdPt(61)4]4+ with C4v symmetry,78 and the double-cavity system [PdPt2(62)4]6+, respectively.79 Alternatively, self-assembly of [Pt(57)4]2+ with aniline 60 and Pd(II) ions gave an unsymmetrical Pd2PtL4 double-cavity system with two different cavities. By exploiting the difference in ligand exchange kinetics between Pt(II) and Pd(II) metal ions, this strategy does not rely on the use of ligands with different denticities, nor require self-sorting between different metal ions and donor groups, greatly simplifying the self-assembly process.
Two distinct approaches have been investigated to generate more anisotropic cavities within MOCs: the introduction of more than one ligand or metal ion, and the use of inherently lower symmetry building blocks. The combination of these two has recently been realised, in which covalent tethering of different, unsymmetrical ligand fragments has been used to generate formally homoleptic architectures with highly anisotropic cavities, termed pseudo-heterolepticity. Together, these strategies demonstrate how more anisotropic cavity environments may be engineered and may be exploited to generate shape-selective recognition sites within MOCs with precise control over functional group orientations.
Introducing functionality
For many MOCs, the ligands employed are kept as structurally simple as possible. “Functional groups” are only used as part of the structural backbone to effect formation of the desired self-assembled architecture. The inclusion of extraneous groups may impact the self-assembly process, leading to undesirable products.Towards the development of more functionally refined assemblies, however, the addition of particular chemical moieties may become attractive. Groups that protrude from the exterior surface of the cage or into the cavity space may be described as exohedral or endohedral, respectively.
The Crowley group have previously reported an azide-functionalised ditopic scaffold that could be diversified via CuAAC ‘click’ chemistry (Fig. 12(a)).80 Each of these series of ligands was shown to maintain the self-assembly profile of the parent ligand framework and form exohedrally functionalised Pd2L4 cages. In this manner, cages with emissive and electrochemical activity, water solubility, and decorated with biologically active units were realised. Suitably inert metallo-moieties with attractive photophysical properties could also be installed. Importantly, appending these exohedral groups did not have a significant impact on the guest-binding properties of the cavity; encapsulation of two molecules of cisplatin, as per the parent system [Pd2(17)4]4+, was demonstrated.
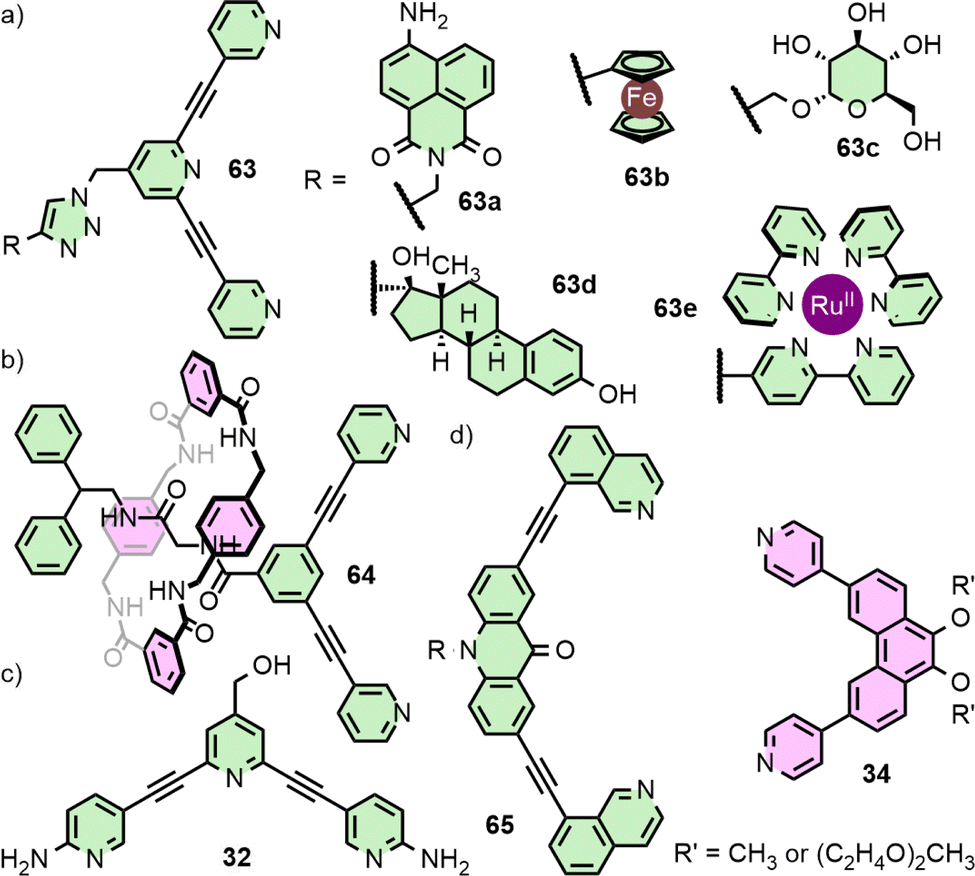 | ||
| Fig. 12 (a) Exo-functionalisation of a tripyridyl ligand fragment via CuAAC chemistry with a variety of functional groups allowed access to a series of Pd2L4 cages in which the host–guest chemistry of the core cage was unaffected. (b) [2]Rotaxane 64 assembled into a Pd2L4 cage decorated with four mechanically interlocked macrocycle units. (c) ortho-amino groups appended to the coordinating pyridine units in 32 induce a helical twist in the resultant Pd2L4 cage, altering the host–guest properties compared to the unfunctionalised [Pd2(17)4]4+ parent system. (d) Geometrically matched ligand 65 and 34 assemble to form heteroleptic cage cis-[Pd2(65)2(34)2]4+, but only if R′ = CH3. | ||
The Lewis group has been interested in the incorporation of mechanically interlocked components into MOCs that may be of use in gating the kinetics of guest exchange. As a proof of concept, [2]rotaxane ligand 64 (Fig. 12(b)) incorporated a dipyridyl unit as one of the stoppering groups, allowing it to assemble into a Pd2L4 cage decorated with four mechanically interlocked macrocycles.81
It might be assumed that functional moieties outside the cage would be innocent as far as the chemistry of the internal cavities is concerned. The impact that these groups have on the structure of the cage, however, may have knock-on effects for the host capabilities of the cavity space. Crowley and co-workers, for example, have reported cage [Pd2(32)4]4+ with ortho-amino groups appended to the pyridine rings (Fig. 12(c)). These served to enhance the ligand donor strength and increased the stability of the resultant Pd2L4 cage, prolonging its half-life in the presence of biologically relevant nucleophiles. Hydrogen-bonding interactions between the amino groups, however, induced a helical twist in the cage. This change in the shape and size of the cavity, combined with weakened hydrogen bond donor ability of the endohedral ortho-pyridyl protons, inhibited binding of cisplatin guests within the cage, in contrast to the unsubstituted, but structurally very similar, [Pd2(17)4]4+ cage.82
The mechanism through which ligand modifications impact the chemistry of the MOC, or even fundamentally the self-assembly profile of the ligands, may be more obscure. Clever and co-workers observed a dramatic effect on the self-assembly profile of ligands as a result of moieties on the exohedral face of ditopic ligands. Ligands 65 and 34 self-assembled in an integrative fashion to give a heteroleptic cis-Pd2L2L′2 cage, which itself assembled into vesicles. If the methoxy substituents of 34 were replaced with oligo-ethylene glycol chains, however, narcissistic self-sorting to give the homoleptic Pd2L4 cages was observed, despite no obvious reason for this effect.83
The remainder of this section will be split into two parts: (i) an examination of embedded functionality, in which functional units form structural components of the ligand framework, and (ii) appended functional moieties, where functional groups do not affect the core scaffold, but intrude on the cavity space. For both of these, a focus will be placed on the effects of the functional units on host–guest chemistry.
Embedded functionality
Although incorporating functionality into MOCs can entail the addition of obvious functional units, relatively small chemical alterations that do not result in significant structural changes can have important impacts on a system's utility. In their study40 on the binding of cisplatin within [Pd2(17)4]4+, Crowley and co-workers noted the failure of isostructural architecture [Pd2(11)4]4+29 to similarly demonstrate any appreciable binding of the anticancer drug, likely due to the loss of the hydrogen bond acceptor site provided by the uncoordinated pyridine of 17 (Fig. 13(a)). Exploiting this difference, triple-cavity cage 66 (Fig. 13(b)) was designed to enable segregated binding of cisplatin only within the outer, pyridine-functionalised cavities, and triflate anions within the central, benzene-functionalised space.84
 | ||
| Fig. 13 (a) Despite the structural similarity between ligands 17 and 11, the host–guest chemistries of their Pd2L4 cages are remarkably different (SCXRD structure of [Pd2(11)4⊃pentacenequinone]4+ shown). This difference was exploited in (b) triple-cavity cage [Pd4(66)4]8+ (MMFF model) in which the central cavity contains the framework of 11, whilst the two external cavities are akin to that of 17. (c) Endohedral functionalisation can enhance binding of particular guests, as with the amine hydrogen bond donors of 67 (SCXRD structure), but sufficiently bulky units can inhibit self-assembly as with ligands 68 and 69. The chemistry of endohedral units, such as the rotor in 70, can have their chemistry impacted by guest molecules. | ||
Lusby and co-workers have subsequently shown that this minor difference between the cages of 11 and 17 has a significant impact on their catalytic activity in various reactions. [Pd2(17)4]4+, for example, was able to catalyse Diels–Alder reactions with impressively high activity (kcat/kuncat > 1000) and turnover numbers (TON ∼1000). Cage [Pd2(11)4]4+, in contrast, was catalytically inactive, in spite of its stronger affinity for quinone guests (Fig. 13(a)). The difference between the two was rationalised as resulting from a combination of weaker substrate binding and better transition state stabilisation within [Pd2(17)4]4+.85 Conversely, [Pd2(11)4]4+ was better able to stabilise bound anionic species, leading to its use in the catalysis of radical-cation cycloadditions – stemming from the increased reduction potential of encapsulated quinones, facilitating electron transfer with substrates outside the cage cavity86 – and Michael additions.87
Thus, it can be seen from a comparison of these two cages the dramatic effect that a relatively simple change in chemical structure (in this instance pyridyl vs. phenyl units) can have on both guest recognition and modulation of chemical reactivity. In addition to inferences based on experimental data, computational studies have been useful in postulating about the origins of observed differences in activity between the cages assembled from ligands 11 and 17. Although the size and shape of the cages are highly similar, differences in flexibility and conformation dynamics arising from the minor difference in their chemical structures have been invoked to help explain their disparate behaviour and properties.88
Appended endohedral functionality
Endohedral functional groups can be introduced to modulate the size and chemical properties of a cavity. An important point of consideration is the impact that these groups may have on the self-assembly profile of the ligand scaffold. Following on from their initial report on a Pd2L4 cage assembled from ligand 11, Hooley and co-workers investigated the impact of endohedral groups on cage formation (Fig. 13(c)). An amine (67) was small enough to be incorporated into the cage structure, but larger triflimide (68) and phenyl urea (69) moieties inhibited formation of the homoleptic complexes. Combining mixtures of these ligands with Pd(II) ions, however, allowed access to a library of heteroleptic cage assemblies.89 Amouri and co-workers subsequently demonstrated the utility of the endohedral amine group of 67 as hydrogen bond donors capable of interacting with an encapsulated tetranitroplatinate(II) anion.90The importance of non-covalent interactions between host and guest was highlighted in recent work from the Clever group. Using a combined experimental and computational approach, it was possible to rationalise the impact of endohedral groups on the host–guest chemistry of heteroleptic cis-Pd2L4 cages. Exchanging the endohedral carbazole N–H of one ligand for a N–CH3 group led to a dramatic decrease in the binding constant of diphenyl phosphate anions (from 2 × 103 M−1 to 77 M−1) due to the loss of hydrogen bonding interactions between host and guest.60
Although encapsulation is generally considered from the point of view of the impact that confinement within the host has on the guest(s), guest-binding can also affect chemical groups of the host scaffold. Clever and co-workers examined cage [Pd2(70)4]4+, assembled from ditopic ligands with rotor units (Fig. 13(d)), in which binding of 2,7-naphthyl bissulfonate was shown to slow the spinning rate of the rotors.91
Adaptable cavities
MOCs are generally regarded as largely rigid species, in contrast to enzyme binding sites that are known to undergo a degree of induced fit – conformational change to adapt to binding of a particular guest. Investigations into conformational flexibility of MOCs, however, are increasing, and it is known that it is possible for cages to display a level of adaptability of cavity size and shape in response to stimuli. If this adaptive behaviour could be harnessed in a controlled manner then this may lead to MOCs possessing much more sophisticated properties. This topic has been covered in a previous review,92 but a limited number of recent examples will be highlighted here.An elegant example of the impact that guests can have on the size and shape of host cavities comes from Jung and co-workers, who extensively investigated the impact of changing anion (A−) identity within a double-cavity [Pd3L4⊃(A)n](6−n)+ system in which the ligands incorporated flexible alkyl linkers (Fig. 14(a)). The strikingly different ligand conformations accessible can be seen from the SCXRD structures of the PF6− adduct in comparison to NO3−. Between these two the Pd⋯Pd distance shrinks from 9.7 to ∼7.9 Å, and the angle described by the three Pd(II) ions diminishes from 180° to ∼140°.93
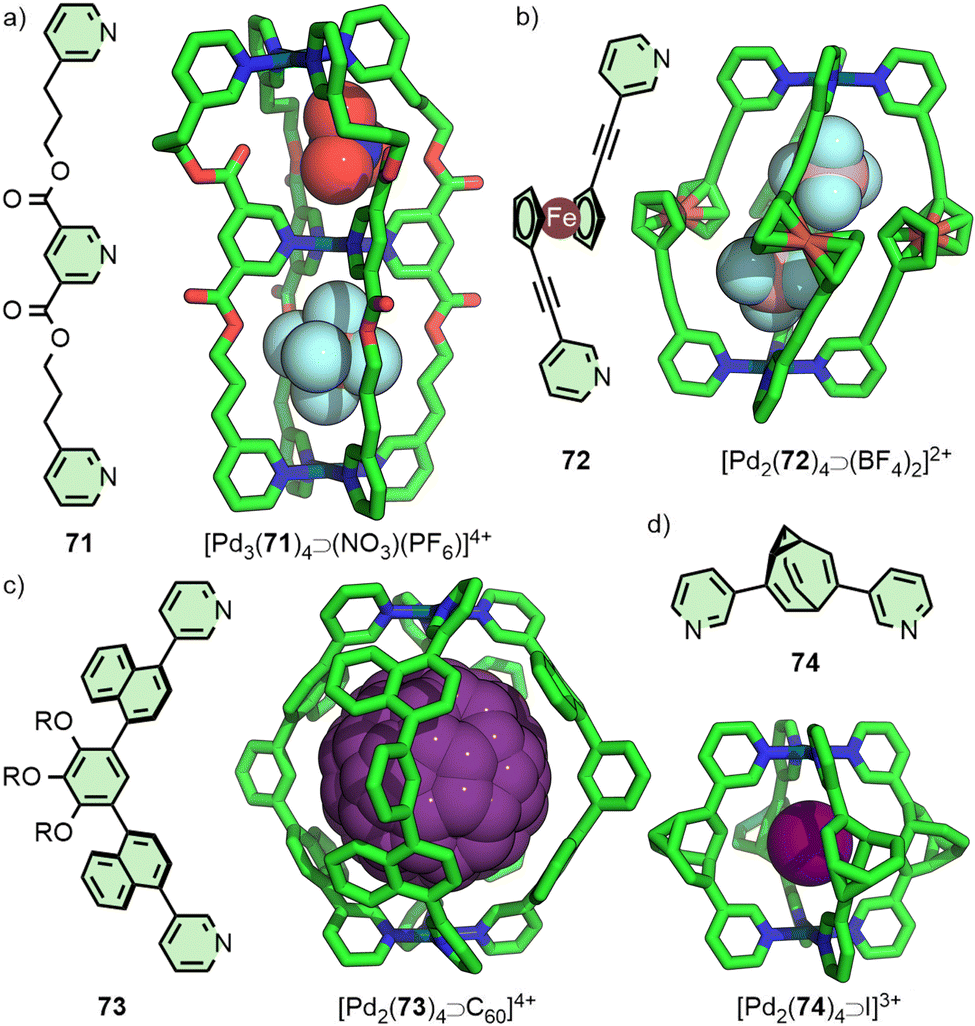 | ||
| Fig. 14 Conformationally adaptable cages have been accessed using ligands with (a) flexible alkyl linkers, (b) rotatable metallocene units, (c) atropisomeric components, and (d) configurationally fluxional groups (SCXRD structures). | ||
Crowley and co-workers incorporated a ferrocene unit into a dipyridyl ligand and exploited the ball-bearing attributes of the metallocene to generate a Pd2L4 cage in which the dihedral angle across the ferrocene impacted the distance between Pd(II) ions (Fig. 14(b)).94
The impact of guest-encapsulation on the atropisomerism of naphthalene-based ligand 73 within a Pd2L4 cage has been investigated by the Yoshizawa group (Fig. 14(c)). Rotation of the naphthalene groups meant there were, ostensibly, 3 possible atropisomers of 73, leading to 42 potential, interconverting isomers of the Pd2L4 structure. The binding of guest molecules within the cavity of the cage was shown to bias this equilibrium towards a particular isomer. Ultimately, using C60 as a guest led to quantitative conversion to a single cage isomer, the precise conformation of which was shown in the solid state by SCXRD.95
A related concept was recently examined by Bloch, Fallon and co-workers with a dipyridyl ligand featuring a bullvalene core (74, Fig. 14(d)). Theoretically the ligand could exist as an interconverting series of 15 isomers, capable of forming a library of ∼2 × 105 isomeric Pd2L4 cages, although VT NMR indicated that 74 actually exist as a dynamic equilibrium of 3 isomers. Indeed, the BF4− salt of the cage exhibited complex NMR spectra, indicative of a multitude of cage isomers. Addition of halide anions served to resolve the dynamic mixture into a single dominant species, suggesting the ligand existed predominantly as a single isomer (as a pair of enantiomers) within the cage assembly, confirmed by SCXRD structures of the Pd2L4 cage encapsulating different anions, all with the same ligand configuration.96
Conclusions
Abiotic host systems have fascinated chemists for decades due to their potential to act as readily tuneable artificial analogues of biological assemblies, such as enzymes. Metal–organic cages are particularly attractive due to their ostensibly facile and high-yielding synthesis via self-assembly under thermodynamic control.Within this Feature Article, three main structural properties amenable to tailoring for selective guest binding have been discussed: size, shape and functionality. Although the majority of examples used in this article to demonstrate key principles have been Pd2L4 cages, these general concepts can be applied to other MOCs, as well as alternative host systems such as covalent-organic cages.97
The development of design principles for increasingly sophisticated MOCs is an active area of study. Whilst MOCs are of significantly lower complexity than biological host systems, these efforts demonstrate the difficulty that structural and functional modification of MOCs present in reality. The continual development of synthetic strategies to molecularly engineer MOCs, and hence their cavities, in a well-controlled manner is enabling access to more readily tuneable host systems. Ultimately, complete mastery over these approaches would allow precision engineering of MOCs with unrivalled degrees of selectivity for guest binding and property modulation, leading to these systems finding more wide-ranging use as highly customisable molecular capsules.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Professors James Crowley and Steve Goldup are gratefully acknowledged for their continued mentorship, support and encouragement over the years. Past and present members of the Lewis Group are thanked for all their hard work, some of which has been discussed in this article. JEML is a Royal Society University Research Fellow.Notes and references
- D. L. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975–982 CrossRef CAS.
- S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972–983 CrossRef CAS PubMed.
- M. Fujita, K. Umemoto, M. Yoshizawa, N. Fujita, T. Kusukawa and K. Biradha, Chem. Commun., 2001, 509–518 RSC.
- N. C. Gianneschi, M. S. Masar and C. A. Mirkin, Acc. Chem. Res., 2005, 38, 825–837 CrossRef CAS PubMed.
- B. J. Holliday and C. A. Mirkin, Angew. Chem., Int. Ed., 2001, 40, 2022–2043 CrossRef CAS PubMed; T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef PubMed; M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC; B. S. Pilgrim and N. R. Champness, ChemPlusChem, 2020, 85, 1842–1856 CrossRef PubMed; A. J. McConnell, Chem. Soc. Rev., 2022, 51, 2957–2971 RSC.
- Y. Fang, J. A. Powell, E. Li, Q. Wang, Z. Perry, A. Kirchon, X. Yang, Z. Xiao, C. Zhu, L. Zhang, F. Huang and H.-C. Zhou, Chem. Soc. Rev., 2019, 48, 4707–4730 RSC.
- A. Galan and P. Ballester, Chem. Soc. Rev., 2016, 45, 1720–1737 RSC.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed.
- E. G. Percástegui, Chem. Commun., 2022, 58, 5055–5071 RSC.
- S. K. Samanta and L. Isaacs, Coord. Chem. Rev., 2020, 410, 213181 CrossRef CAS PubMed.
- S. Pullen, J. Tessarolo and G. H. Clever, Chem. Sci., 2021, 12, 7269–7293 RSC.
- K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703–6712 RSC.
- M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC; A. Schmidt, A. Casini and F. E. Kühn, Coord. Chem. Rev., 2014, 275, 19–36 CrossRef CAS; S. Saha, I. Regeni and G. H. Clever, Coord. Chem. Rev., 2018, 374, 1–14 CrossRef.
- M. D. Ludden and M. D. Ward, Dalton Trans., 2021, 50, 2782–2791 RSC.
- D. Zhang, T. K. Ronson, R. Lavendomme and J. R. Nitschke, J. Am. Chem. Soc., 2019, 141, 18949–18953 CrossRef CAS PubMed.
- N. L. S. Yue, D. J. Eisler, M. C. Jennings and R. J. Puddephatt, Inorg. Chem. Commun., 2005, 8, 31–33 CrossRef CAS.
- D. Fujita, K. Suzuki, S. Sato, M. Yagi-Utsumi, Y. Yamaguchi, N. Mizuno, T. Kumasaka, M. Takata, M. Noda, S. Uchiyama, K. Kato and M. Fujita, Nat. Commun., 2012, 3, 1093 CrossRef CAS PubMed; D. Fujita, R. Suzuki, Y. Fujii, M. Yamada, T. Nakama, A. Matsugami, F. Hayashi, J.-K. Weng, M. Yagi-Utsumi and M. Fujita, Chem, 2021, 7, 2672 Search PubMed.
- M. Tominaga, K. Suzuki, M. Kawano, T. Kusukawa, T. Ozeki, S. Sakamoto, K. Yamaguchi and M. Fujita, Angew. Chem., Int. Ed., 2004, 43, 5621–5625 CrossRef CAS PubMed.
- J. Bunzen, J. Iwasa, P. Bonkaderzadeh, E. Numata, K. Rissanen, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2012, 51, 3161–3163 CrossRef CAS PubMed.
- Q.-F. Sun, J. Iwasa, D. Ogawa, Y. Ishido, S. Sato, T. Ozeki, Y. Sei, K. Yamaguchi and M. Fujita, Science, 2010, 328, 1144–1147 CrossRef CAS PubMed.
- D. Fujita, Y. Ueda, S. Sato, N. Mizuno, T. Kumasaka and M. Fujita, Nature, 2016, 540, 563–566 CrossRef CAS PubMed.
- D. Fujita, Y. Ueda, S. Sato, N. Mizuno, T. Kumasaka and M. Fujita, Chem, 2016, 1, 91–101 CAS.
- M. Tominaga, K. Suzuki, T. Murase and M. Fujita, J. Am. Chem. Soc., 2005, 127, 11950–11951 CrossRef CAS PubMed.
- Q.-F. Sun, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2014, 43, 13510–13513 CrossRef PubMed.
- Q.-F. Sun, T. Murase, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2011, 50, 10318–10321 CrossRef CAS PubMed.
- I. A. Bhat, D. Samanta and P. S. Mukherjee, J. Am. Chem. Soc., 2015, 137, 9497–9502 CrossRef CAS PubMed.
- D. A. McMorran and P. J. Steel, Angew. Chem., Int. Ed., 1998, 37, 3295–3297 CrossRef CAS PubMed.
- P. J. Steel and D. A. McMorran, Chem. – Asian J., 2019, 14, 1098–1101 CrossRef CAS PubMed.
- P. Liao, B. W. Langloss, A. M. Johnson, E. R. Knudsen, F. S. Tham, R. R. Julian and R. J. Hooley, Chem. Commun., 2010, 46, 4932–4934 RSC.
- D. P. August, G. S. Nichol and P. J. Lusby, Angew. Chem., Int. Ed., 2016, 55, 15022–15026 CrossRef CAS PubMed.
- S. Samantray, S. Krishnaswamy and D. K. Chand, Nat. Commun., 2020, 11, 880 CrossRef CAS PubMed.
- T. Tateishi, S. Takahashi, A. Okazawa, V. Martí-Centelles, J. Wang, T. Kojima, P. J. Lusby, H. Sato and S. Hiraoka, J. Am. Chem. Soc., 2019, 141, 19669–19676 CrossRef CAS PubMed.
- D. Preston, K. M. Patil, A. T. O’Neil, R. A. S. Vasdev, J. A. Kitchen and P. E. Kruger, Inorg. Chem. Front., 2020, 7, 2990–3001 RSC.
- K. Matsumoto, S. Kusaba, Y. Tanaka, Y. Sei, M. Akita, K. Aritani, M.-A. Haga and M. Yoshizawa, Angew. Chem., Int. Ed., 2019, 58, 8463–8467 CrossRef CAS PubMed.
- M. Fukuda, R. Sekiya and R. Kuroda, Angew. Chem., Int. Ed., 2008, 47, 706–710 CrossRef CAS PubMed.
- M. Fujita, N. Fujita, K. Ogura and K. Yamaguchi, Nature, 1999, 400, 52–55 CrossRef CAS.
- R. Sekiya, M. Fukuda and R. Kuroda, J. Am. Chem. Soc., 2012, 134, 10987–10997 CrossRef CAS PubMed.
- M. Frank, M. D. Johnstone and G. H. Clever, Chem. – Eur. J., 2016, 22, 14104–14125 CrossRef CAS PubMed.
- R. Zhu, I. Regeni, J. J. Holstein, B. Dittrich, M. Simon, S. Prévost, M. Gradzielski and G. H. Clever, Angew. Chem., Int. Ed., 2018, 57, 13652–13656 CrossRef CAS PubMed.
- J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778–784 RSC.
- T. Y. Kim, N. T. Lucas and J. D. Crowley, Supramol. Chem., 2015, 27, 734–745 CrossRef.
- N. Kishi, Z. Li, K. Yoza, M. Akita and M. Yoshizawa, J. Am. Chem. Soc., 2011, 133, 11438–11441 CrossRef CAS PubMed.
- M. Yamashina, T. Yuki, Y. Sei, M. Akita and M. Yoshizawa, Chem. – Eur. J., 2015, 21, 4200–4204 CrossRef CAS PubMed.
- S. Kortagere, M. D. Krasowski and S. Ekins, Trends Pharmacol. Sci., 2009, 30, 138–147 CrossRef CAS PubMed.
- P. Baxter, J.-M. Lehn, A. Decian and J. Fischer, Angew. Chem., Int. Ed. Engl., 1993, 32, 69–72 CrossRef.
- P. N. W. Baxter, J.-M. Lehn, B. O. Kneisel, G. Baum and D. Fenske, Chem. – Eur. J., 1999, 5, 113–120 CrossRef CAS.
- P. N. W. Baxter, J.-M. Lehn, G. Baum and D. Fenske, Chem. – Eur. J., 1999, 5, 102–112 CrossRef CAS.
- S. De, K. Mahata and M. Schmittel, Chem. Soc. Rev., 2010, 39, 1555–1575 RSC; M. L. Saha, S. Neogi and M. Schmittel, Dalton Trans., 2014, 43, 3815–3834 RSC.
- C. O. Dietrich-Buchecker, J.-P. Sauvage and J. P. Kintzinger, Tetrahedron Lett., 1983, 24, 5095–5098 CrossRef CAS; C. O. Dietrich-Buchecker, J.-P. Sauvage and J. M. Kern, J. Am. Chem. Soc., 1984, 106, 3043–3045 CrossRef.
- M. Yoshizawa, M. Nagao, K. Kumazawa and M. Fujita, J. Organomet. Chem., 2005, 690, 5383–5388 CrossRef CAS.
- M. Yoshizawa, J. Nakagawa, K. Kumazawa, M. Nagao, M. Kawano, T. Ozeki and M. Fujita, Angew. Chem., Int. Ed., 2005, 44, 1810–1813 CrossRef CAS PubMed.
- R. Zhu, W. M. Bloch, J. J. Holstein, S. Mandal, L. V. Schäfer and G. H. Clever, Chem. – Eur. J., 2018, 24, 12976–12982 CrossRef CAS PubMed.
- B. Chen, J. J. Holstein, S. Horiuchi, W. G. Hiller and G. H. Clever, J. Am. Chem. Soc., 2019, 141, 8907–8913 CrossRef CAS PubMed.
- B. Chen, S. Horiuchi, J. J. Holstein, J. Tessarolo and G. H. Clever, Chem. – Eur. J., 2019, 25, 14921–14927 CrossRef CAS PubMed.
- B. Chen, J. J. Holstein, A. Platzek, L. Schneider, K. Wu and G. H. Clever, Chem. Sci., 2022, 13, 1829–1834 RSC.
- R.-J. Li, J. Tessarolo, H. Lee and G. H. Clever, J. Am. Chem. Soc., 2021, 143, 3865–3873 CrossRef CAS PubMed.
- D. Preston, J. E. Barnsley, K. C. Gordon and J. D. Crowley, J. Am. Chem. Soc., 2016, 138, 10578–10585 CrossRef CAS PubMed.
- W. M. Bloch, Y. Abe, J. J. Holstein, C. M. Wandtke, B. Dittrich and G. H. Clever, J. Am. Chem. Soc., 2016, 138, 13750–13755 CrossRef CAS PubMed; W. M. Bloch, J. J. Holstein, W. Hiller and G. H. Clever, Angew. Chem., Int. Ed., 2017, 56, 8285–8289 CrossRef PubMed.
- K. Wu, J. Tessarolo, A. Baksi and G. H. Clever, Angew. Chem., Int. Ed., 2022, 61, e202205725 Search PubMed.
- A. Platzek, S. Juber, C. Yurtseven, S. Hasegawa, L. Schneider, C. Drechsler, K. E. Ebbert, R. Rudolf, Q.-Q. Yan, J. J. Holstein, L. V. Schäfer and G. H. Clever, Angew. Chem., Int. Ed., 2022, 61, e202209305 CrossRef CAS PubMed.
- S. Sudan, R.-J. Li, S. M. Jansze, A. Platzek, R. Rudolf, G. H. Clever, F. Fadaei-Tirani, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2021, 143, 1773–1778 CrossRef CAS PubMed.
- R.-J. Li, F. Fadaei-Tirani, R. Scopelliti and K. Severin, Chem. – Eur. J., 2021, 27, 9439–9445 CrossRef CAS PubMed.
- J. A. Findlay, K. M. Patil, M. G. Gardiner, H. I. MacDermott-Opeskin, M. L. O’Mara, P. E. Kruger and D. Preston, Chem. – Asian J., 2022, 17, e202200093 CrossRef CAS PubMed.
- J. E. M. Lewis and J. D. Crowley, ChemPlusChem, 2020, 85, 815–827 CrossRef CAS PubMed.
- J. E. M. Lewis, A. Tarzia, A. White and K. E. Jelfs, Chem. Sci., 2020, 11, 677–683 RSC.
- R. A. S. Vasdev, D. Preston, C. A. Casey-Stevens, V. Martí-Centelles, P. J. Lusby, A. L. Garden and J. D. Crowley, Inorg. Chem., 2022 DOI:10.1021/acs.inorgchem.2c00937.
- M. Bursch, H. Neugebauer and S. Grimme, Angew. Chem., Int. Ed., 2019, 58, 11078–11087 CrossRef CAS PubMed.
- A. Tarzia, J. E. M. Lewis and K. E. Jelfs, Angew. Chem., Int. Ed., 2021, 60, 20879–20887 CrossRef CAS PubMed.
- D. Ogata and J. Yuasa, Angew. Chem., Int. Ed., 2019, 58, 18424–18428 CrossRef CAS PubMed.
- J. E. M. Lewis, Chem. – Eur. J., 2021, 27, 4454–4460 CrossRef CAS PubMed.
- S. S. Mishra, S. V. K. Kompella, S. Krishnaswamy, S. Balasubramanian and D. K. Chand, Inorg. Chem., 2020, 59, 12884–12894 CrossRef CAS PubMed.
- R.-J. Li, A. Marcus, F. Fadaei-Tirani and K. Severin, Chem. Commun., 2021, 57, 10023–10026 RSC.
- R.-J. Li, A. Tarzia, V. Posligua, K. E. Jelfs, N. Sanchez, A. Marcus, A. Baksi, G. H. Clever, F. Fadaei-Tirania and K. Severin, Chem. Sci., 2022, 13, 11912–11917 RSC.
- J. E. M. Lewis, Angew. Chem., Int. Ed., 2022, 61, e202212392 CrossRef CAS PubMed.
- H. Li, Z.-J. Yao, D. Liu and G.-X. Jin, Coord. Chem. Rev., 2015, 293–294, 139–157 CrossRef CAS; F. Li and L. F. Lindoy, Aust. J. Chem., 2019, 72, 731–741 CrossRef.
- S. M. Jansze, M. D. Wise, A. V. Vologzhanina, R. Scopelliti and K. Severin, Chem. Sci., 2017, 8, 1901–1908 RSC.
- C. Shen, A. D. W. Kennedy, W. A. Donald, A. M. Torres, W. S. Price and J. E. Beves, Inorg. Chim. Acta, 2017, 458, 122–128 CrossRef CAS.
- L. S. Lisboa, J. A. Findlay, L. J. Wright, C. G. Hartinger and J. D. Crowley, Angew. Chem., Int. Ed., 2020, 59, 11101–11107 CrossRef CAS PubMed.
- L. S. Lisboa, D. Preston, C. J. McAdam, L. J. Wright, C. G. Hartinger and J. D. Crowley, Angew. Chem., Int. Ed., 2022, 61, e202201700 CrossRef CAS PubMed.
- (a) J. E. M. Lewis, C. J. McAdam, M. G. G. Gardiner and J. D. Crowley, Chem. Commun., 2013, 49, 3398–3400 RSC; (b) J. E. M. Lewis, A. B. S. Elliott, C. J. McAdam, K. C. Gordon and J. D. Crowley, Chem. Sci., 2014, 5, 1833–1843 RSC; (c) A. B. S. Elliott, J. E. M. Lewis, H. van der Salm, C. J. McAdam, J. D. Crowley and K. C. Gordon, Inorg. Chem., 2016, 55, 3440–3447 CrossRef CAS PubMed.
- K. K. G. Wong, N. Hoyas Pérez, A. J. P. White and J. E. M. Lewis, Chem. Commun., 2020, 56, 10453–10456 RSC.
- D. Preston, S. M. McNeill, J. E. M. Lewis, G. I. Giles and J. D. Crowley, Dalton Trans., 2016, 45, 8050–8060 RSC.
- S. Saha, B. Holzapfel, Y.-T. Chen, K. Terlinden, P. Lill, C. Gatsogiannis, H. Rehage and G. H. Clever, J. Am. Chem. Soc., 2018, 140, 17384–17388 CrossRef CAS PubMed.
- D. Preston, J. E. M. Lewis and J. D. Crowley, J. Am. Chem. Soc., 2017, 139, 2379–2386 CrossRef CAS PubMed.
- V. Martí-Centelles, A. L. Lawrence and P. J. Lusby, J. Am. Chem. Soc., 2018, 140, 2862–2868 CrossRef PubMed.
- R. L. Spicer, A. D. Stergiou, T. A. Young, F. Duarte, M. D. Symes and P. J. Lusby, J. Am. Chem. Soc., 2020, 142, 2134–2139 CrossRef CAS PubMed.
- J. Wang, T. A. Young, F. Duarte and P. J. Lusby, J. Am. Chem. Soc., 2020, 142, 17743–17750 CrossRef CAS PubMed.
- T. A. Young, V. Martí-Centelles, J. Wang, P. J. Lusby and F. Duarte, J. Am. Chem. Soc., 2020, 142, 1300–1310 CrossRef PubMed.
- A. M. Johnson and R. J. Hooley, Inorg. Chem., 2011, 50, 4671–4673 CrossRef CAS PubMed; A. M. Johnson, O. Moshe, A. S. Gamboa, B. W. Langloss, J. F. K. Limtiaco, C. K. Larive and R. J. Hooley, Inorg. Chem., 2011, 50, 9430–9442 CrossRef PubMed.
- C. Desmarets, G. Gontard, A. L. Cooksy, M. N. Rager and H. Amouri, Inorg. Chem., 2014, 53, 4287–4294 CrossRef CAS PubMed.
- M. Krick, J. Holstein, C. Würtele and G. H. Clever, Chem. Commun., 2016, 52, 10411–10414 RSC.
- A. E. Martín Díaz and J. E. M. Lewis, Front. Chem., 2021, 9, 706462 CrossRef PubMed.
- G. Sarada, A. Kim, D. Kim and O.-S. Jung, Dalton Trans., 2020, 49, 6183–6190 RSC.
- R. A. Vasdev, J. A. Findlay, A. L. Garden and J. D. Crowley, Chem. Commun., 2019, 55, 7506–7509 RSC.
- T. Tsutsui, L. Catti, K. Yoza and M. Yoshizawa, Chem. Sci., 2020, 11, 8145–8150 RSC.
- A. P. Birvé, H. D. Patel, J. R. Price, W. M. Bloch and T. Fallon, Angew. Chem., Int. Ed., 2022, 61, e202115468 CrossRef PubMed.
- T. Hasell and A. I. Cooper, Nat. Rev. Mater., 2016, 1, 16053 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |