
Aqueous phase- and size-controlled synthesis, and secondary assemblies of CdS nanocrystals at room temperature†
Pengfei
Hu
 *a,
Dong
Zhou
a,
Shiqing
Xu
a,
Qianru
Ma
a,
Jiaqi
Yin
ab,
Yali
Cao
c and
Jing
Xu
*a
*a,
Dong
Zhou
a,
Shiqing
Xu
a,
Qianru
Ma
a,
Jiaqi
Yin
ab,
Yali
Cao
c and
Jing
Xu
*a
aLaboratory for Microstructures, Shanghai University, Shanghai 200444, P. R. China. E-mail: hpf-hqx@shu.edu.cn; xujingshu@shu.edu.cn; Fax: +86 21 66135030; Tel: +86 21 66135030
bNingbo Institute of Technology and Engineering, Chinese Academy of Sciences, Zhenjiang 315201, P. R. China
cKey Laboratory of Energy Materials Chemistry, Ministry of Education, Key Laboratory of Advanced Functional Materials, Xinjiang University, Urumqi, Xinjiang 830046, China
First published on 8th December 2021
Abstract
Room-temperature aqueous phase- and size-controlled synthesis of CdS nanocrystals was achieved by a simple strategy. The role of pH is to adjust the feeding of Cd2+ and S2− by modulating the 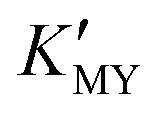 and the dissociation of TAA. Chelator ethylenediamine guides the secondary spherical and one-dimensional linear assembly of CdS nanocrystals.
and the dissociation of TAA. Chelator ethylenediamine guides the secondary spherical and one-dimensional linear assembly of CdS nanocrystals.
Driven by their unique size- and shape-dependent optical and electronic properties, the study of semiconductor nanocrystals (NCs) has been revolutionized by the development in recent decades of controllable synthesis strategies for tuning the size and morphology.1–4 Many works have studied the regulation of thermodynamic or kinetic parameters to explore their role in guiding the evolution of nanocrystals into specific shapes. For example, by adjusting the crystal planes around the single crystal seed, including the {100}, {111} and {110} crystal planes or other crystal planes, colloidal nanocrystals with desired shapes can be obtained, or manipulating the reaction kinetics by employing appropriate precursors can also be used to fabricate nanostructures with a specific morphology.5–7 One-dimensional (1D) nanostructures, which are quantum confined in two dimensions, have been in focus in the field of materials science.8,9 A lot of 1D NCs, such as CdSe,10 CdTe,11 PbSe,12,13 ZnO,14 ZnS,15 and so forth, have already been fabricated by the oriented attachment (OA) of primary chaining nanoparticles (NPs). Cadmium sulfide (CdS), which has two common crystalline phases of hexagonal wurtzite and cubic zinc blende, is an important direct band gap (2.42 eV) material with a number of striking properties, such as nonlinear optical behaviors and unusual fluorescence.16,17 It has a broad range of application prospects. Currently, based on solution-phase techniques (solution–liquid–solid and oriented attachment growth) that generally require costly organic reagents, numerous efforts are being devoted to the controlled preparation of CdS NCs with particular shapes and sizes.18–23 In spite of these reports, it is still a challenge to explore environment-friendly and cost-effective approaches to prepare a large quantity of 1D self-assembled CdS nanostructures.
In addition, apart from the size and shape, the crystal phase structure of CdS NCs also has an important influence on their optical and electrical properties.24,25 Control over the phase of CdS NCs has significance for fundamental science and applied studies. However, so far, there are only limited efforts on the fabrication of phase-controlled CdS NCs. It is therefore essential to develop an alternative viable approach for the synthesis of CdS NCs with control over the crystal phase.
In this communication, we address this issue and develop a room-temperature solution-based synthesis of CdS NCs that allows tuning of the crystal phase, size, and morphology. The synthesis procedure was carried out in an aqueous medium throughout. Different chelators are used to prepare precursor complexes that are responsive to the pH value. The supply rate of cadmium ions is regulated by adjusting the pH value to realize the kinetically-controlled synthesis of CdS nanocrystals. In this kinetically-controlled process, the supply rate of sulfur ions is also adjusted by changing the pH value. We also observed that monomeric NCs have a strong tendency to self-assemble to form pearl-chain-like structures, and even linear chains, under certain conditions. Here, the reaction factors for the anisotropic 1D assembly of CdS NCs were optimized. A surprising ability of this strategy is that the desired phases, sizes, and shapes of the products can be modulated by selecting different ligands as the chelating agent, adjusting the pH value of the solution, and employing flexible surfactants as modifiers. This method provides an economically feasible route for the controlled synthesis of CdS NCs, and may open up a new avenue to fabricate other semiconductor NCs.
Our synthetic approach (Scheme 1) follows the oriented attachment growth process, where thioacetamide (TAA) molecular sources are used as S reactants and a cadmium ion complex (CdLn) is used as a cadmium ion reactant (L: ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA), sulfosalicylic acid (SSA), and ethylenediamine (EN), respectively). Herein, a cadmium ion (Cd2+) and the ligand (L: EDTA, SSA, and EN) can form a CdLn complex to establish a dissociation equilibrium with a conditional stability constant (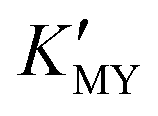 ), which can be adjusted by changing the pH of the solution. On the other hand, TAA also establishes a dissociation equilibrium that can be adjusted by changing the pH value of the solution, providing sulfur source reactants (S2−, HS−, and H2S) for CdS. In this way, the pH of the solution becomes a trigger for regulating the supply of cadmium source and sulfur source reactants to regulate the nucleation and growth of CdS NCs.
), which can be adjusted by changing the pH of the solution. On the other hand, TAA also establishes a dissociation equilibrium that can be adjusted by changing the pH value of the solution, providing sulfur source reactants (S2−, HS−, and H2S) for CdS. In this way, the pH of the solution becomes a trigger for regulating the supply of cadmium source and sulfur source reactants to regulate the nucleation and growth of CdS NCs.
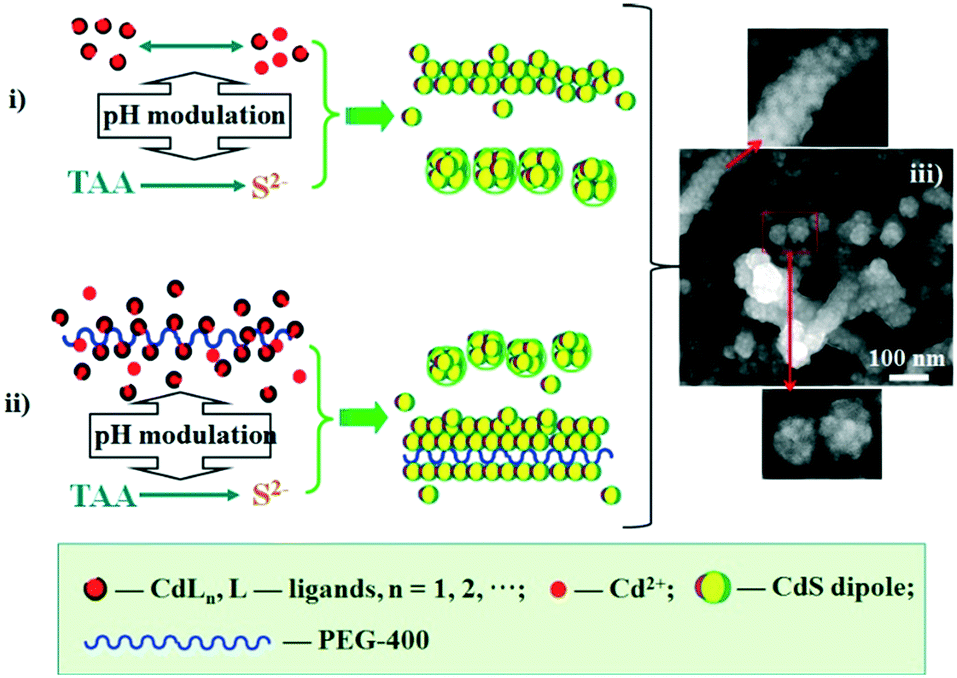 | ||
| Scheme 1 Proposed synthesis mechanism of CdS nanocrystals and their spherical and one-dimensional assembly processes. i) The surfactant-free path; ii) the path using PEG-400 as the surfactant; iii) scanning electron microscopy (SEM) photos of CdS nanocrystals synthesized without surfactants. | ||
In a typical experiment, under continuous magnetic stirring, an aqueous solution of EDTA was added dropwise into a cadmium acetate (Cd(OAc)2)/water solution with a certain volume and concentration to get a basic solution. The solution was adjusted to the desired pH value with sodium hydroxide (NaOH) or acetic acid (HOAc), followed by injection of a TAA/water solution under vigorous stirring. The mixture solution was maintained at room temperature for 6 h with stirring. The yellow precipitates were collected and washed with distilled water and alcohol, and then dried naturally. Extended synthesis was accomplished with the addition of appropriate amounts of the surfactant PEG 400 into the system before the influx of the TAA solution.
X-ray powder diffraction (XRD) was used to determine the evolution of the crystal phases. The XRD patterns of the CdS products synthesized at room temperature using the ligands EDTA, SSA, and EN, respectively, are depicted in Fig. 1. Panels a–c show the results at different pH values. It is widely known that the 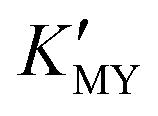 of complexes and the dissociation equilibrium of TAA can be modulated by changing the pH value of the solution.
of complexes and the dissociation equilibrium of TAA can be modulated by changing the pH value of the solution.
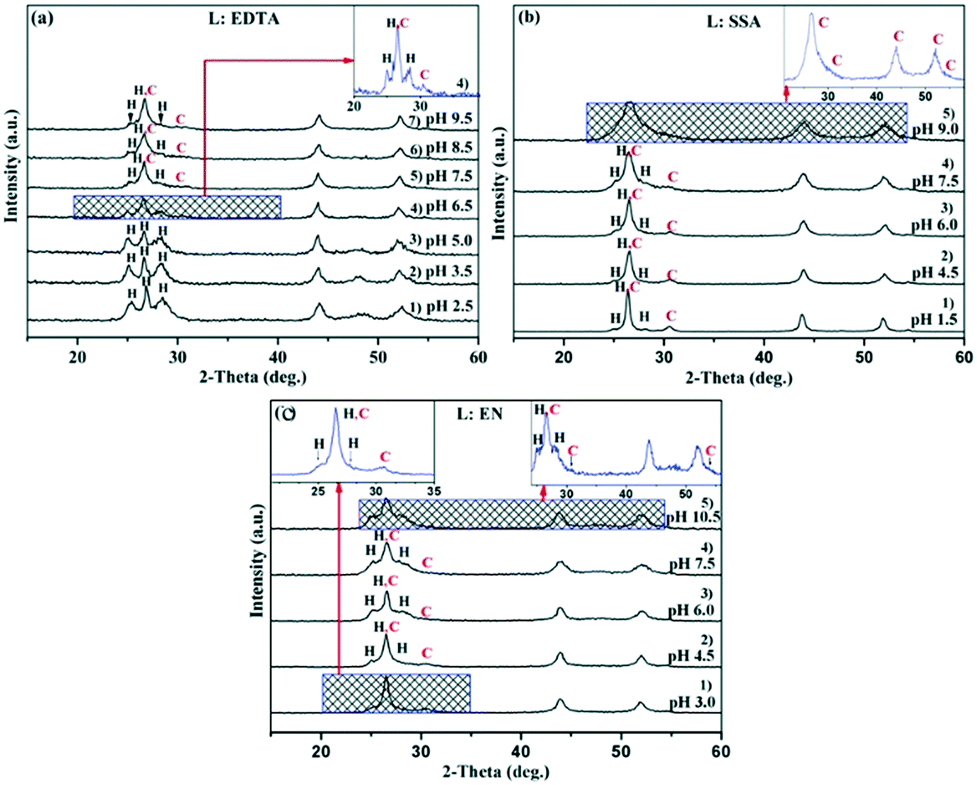 | ||
| Fig. 1 XRD patterns of CdS prepared at different pH with ligands (a) EDTA, among them, 1–7) represent XRD patterns at pH values of 2.5, 3.5, 5.0, 6.5, 7.5, 8.5 and 9.5 respectively; (b) SSA, among them, 1–5) represent XRD patterns at pH values of 1.5, 4.5, 6.0, 7.5, and 9.0 respectively; (c) EN, among them, 1–5) represent XRD patterns at pH values of 3.0, 4.5, 6.0, 7.5, and 10.5 respectively. The insets in (a–c) show the local amplification of the XRD patterns at pH 6.5 with EDTA, pH 9.0 with SSA, and pH 3.0 with EN, respectively. | ||
When EDTA was employed as a chelator, it is clear that a lower pH (lower 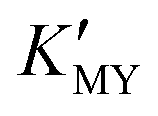 ) can give rise to hexagonal phase CdS NCs, whose diffraction patterns match well with those of hexagonal CdS (JCPDS Cards, No. 41-1049) (Fig. 1a-1)–3)). On increasing the pH value of the solution (increasing
) can give rise to hexagonal phase CdS NCs, whose diffraction patterns match well with those of hexagonal CdS (JCPDS Cards, No. 41-1049) (Fig. 1a-1)–3)). On increasing the pH value of the solution (increasing 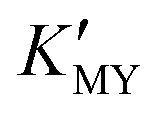 ), cubic phase NCs (JCPDS Cards, No. 65-2887) emerged (Fig. 1a-4)–7) and inset), but the hexagonal crystal phase was preponderant in the mixed phase structures.
), cubic phase NCs (JCPDS Cards, No. 65-2887) emerged (Fig. 1a-4)–7) and inset), but the hexagonal crystal phase was preponderant in the mixed phase structures.
In the present work, parallel studies on controlling the crystal phases were carried out using EN and SSA as chelating agents, respectively. From Fig. 1b and c, it is evident that both SSA and EN can also tune the crystal phases of the products on modulating the pH value. It is worth mentioning that for SSA, the evolutionary trend of the crystal phase with pH value is similar to that for EDTA. In short, when they are used as chelators, their corresponding 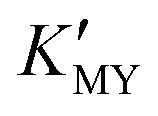 will increase with the increase of the pH value, the concentration of cadmium ions will decrease inversely (ESI† Tables S1 and S2), and the formation of cubic CdS will be promoted (Fig. 1a and b). Even absolute cubic phase NCs can be obtained at pH = 9 with SSA (Fig. 1b-5 and inset). On the other hand, the
will increase with the increase of the pH value, the concentration of cadmium ions will decrease inversely (ESI† Tables S1 and S2), and the formation of cubic CdS will be promoted (Fig. 1a and b). Even absolute cubic phase NCs can be obtained at pH = 9 with SSA (Fig. 1b-5 and inset). On the other hand, the 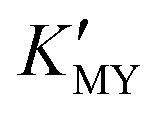 of the cadmium ion ethylenediamine complex increases with the decrease of the pH value, and the cubic CdS corresponds to solutions with a low pH value, i.e. high
of the cadmium ion ethylenediamine complex increases with the decrease of the pH value, and the cubic CdS corresponds to solutions with a low pH value, i.e. high 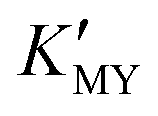 (Fig. 1c and inset). In conclusion, the experimental results of the crystalline phase response to pH show that the cubic phase of CdS corresponds to a high value of
(Fig. 1c and inset). In conclusion, the experimental results of the crystalline phase response to pH show that the cubic phase of CdS corresponds to a high value of 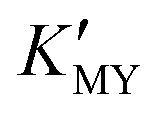 of these three complexes. In contrast, the nanocrystals of the hexagonal phase correspond to a low value of
of these three complexes. In contrast, the nanocrystals of the hexagonal phase correspond to a low value of 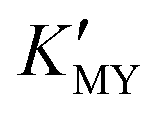 for them. From the dissociation equilibrium of the complex, the level of
for them. From the dissociation equilibrium of the complex, the level of 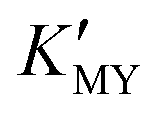 determines the supply concentration and speed of Cd2+. When
determines the supply concentration and speed of Cd2+. When 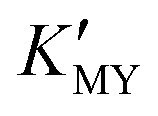 is high, the supply of Cd2+ is less, the grain growth of CdS is suppressed, and it is easy to form a small-size cubic phase. In this process, the supply rate of the sulfur source changes little with pH, and its regulation effect on the crystal phase can be ignored.
is high, the supply of Cd2+ is less, the grain growth of CdS is suppressed, and it is easy to form a small-size cubic phase. In this process, the supply rate of the sulfur source changes little with pH, and its regulation effect on the crystal phase can be ignored.
In addition, the XRD peaks of nanocrystalline products synthesized by various ligands all exhibit a red shift compared with the standard spectral peaks, which is due to the lattice shrinkage caused by the size effect of the nanocrystals. These small-size nanocrystals have a high surface energy, indicating that they will agglomerate or assemble to reduce the surface energy. The subsequent structural analysis section will verify the growth and assembly behaviors of the nanocrystals.
The two types of assembly mode, including spherical assembly and one-dimensional linear assembly of NCs, is another achievement in this work. Spherical assembly refers to the aggregation of the single crystalline CdS to form secondary spheres (SSs, shown in the bottom SEM photo of Scheme 1-iii). One-dimensional assembly behavior includes the linear assembly of single crystalline CdS particles to form rod shapes (shown in the upper SEM photo of Scheme 1-iii) and the linear assembly of SSs to form a pearl-chain-like configuration (shown in the middle SEM photo of Scheme 1-iii). Fig. 2a and FS1 present TEM and SEM images of spherical particles and one-dimensional nanowires prepared with EN at pH = 7.5. The diameter of the nanospheres is about 60–70 nm, while the diameter of the nanowires is about 60–70 nm and the length is hundreds of nanometers. High magnification SEM photos show that these nanospheres are multistage nanostructures assembled from NCs with sizes of several nanometers (bottom photo of Scheme 1-iii). HRTEM also clearly shows that the nanospheres have a high-order nanostructure, which is assembled from nanocrystals with sizes of several nanometers (Fig. 2d). Moreover, the straight chain, branched chain and flexural chains assembled by NCs or SSs are vividly displayed, reflecting the one-dimensional characteristics of the structure (middle photo of Scheme 1-iii and FS1-a).
 | ||
| Fig. 2 EM images of CdS NCs synthesized with EN as a chelating agent at pH = 7.5. a) TEM image of the sample prepared at room temperature; b) and c) TEM and SEM images of the sample synthesized with PEG400 as a modifier at room temperature; d) HRTEM of a nanosphere; and e) and f) HRTEM and structural analysis of individual joining regions of the CdS pearl-chain-like structure, i and ii are two docked nanoparticles. Scale bars of a–c, 100 nm. | ||
Additionally, we employed the surfactant polyethylene glycol 400 (PEG-400) to assist in the assembly of NCs in the original solution of CdLn (L = EN) at pH = 7.5. The results show that the products are still mainly two kinds of assembly objects of CdS NCs, that is, spherical particles and one-dimensional nanowires/nanopearls (TEM image in Fig. 2d and SEM image in Fig. 2e). Their particle size is close to that of the products synthesized in the absence of surfactant PEG. However, compared with the products without PEG, the yield of the linear assembly in the products synthesized with PEG was significantly improved, and the independent spherical assembly almost disappeared. The linear assembly is mainly in the shape of a long rod or a string of pearls. It can be inferred that the PEG-400 molecule is a right-hand man for the 1D self-assembly of CdS NCs. The PEG-400 molecule can guide the 1D combination of CdS NCs to form a complete long CdS wire- or pearl-chain-like structure. Fig. 2e shows a high-resolution TEM characterization of the interface between two nanoparticles in a pearl-chain-like structure. Local structural analysis indicates that the two docked nanocrystalline particles (i and ii) realize a link between them through the integration of the (110) crystal planes (Fig. 2f). There are obvious dislocations and defects in the eutectic region (shown by the orange dotted line), implying that the two nanocrystals show a common mismatch at the interface during the integration process. For linear assembly, the single crystal chain part was selected for HRTEM analysis (ESI† FS2). The results show that in some single crystal chains, the shape of the single crystals tends towards cuboid (Fig. S2-a†), and in some single crystal chains or quasi single crystal links, the boundaries of the nanocrystals are not clear and dislocations are serious (Fig. S2-b–d†).
Furthermore, we synthesized CdS NCs without surfactant at 80 °C to explore the impact of the temperature. The one-dimensional assembly trend is still obvious, but the particle size increases and the length of the 1D structures decreases (ESI† FS3). In addition, in the current work, the CdS NCs prepared with SSA and EDTA did not show the tendency of 1D combination (ESI† FS4-a and b; FS5-a and b). The reason why EN is different from the ligands EDTA and SSA in terms of the assembly mode of the nanocrystals may be related to the fact that the amino groups at both ends of the EN molecule will induce intermolecular links to form soft chains through hydrogen bonds, thus inducing the linear assembly of the CdS nanocrystals, but EDTA and SSA have no such possible chain induction.
According to studies on OA attachment of nanomonomers,26–31 it was speculated that the directional dipole moment of CdS NCs and van der Waals forces between NCs could direct their oriented attachment into linear chains or pearl-chain-like structures. Moreover, from the perspective of the solution molecules, the large number of hydrogen bonds in the ethylenediamine solution will play a positive guiding role in the self-assembly of nanocrystals. The proposed self-assembly process is illustrated in Scheme 1. In this system, the end-to-end directional force of the CdS NP array originates from the dipole moment of the CdS NCs and van der Waals forces (Scheme 1-i). It is a common view that PEG-400 is an effective “soft template”. As illustrated in Scheme 1, Cd2+ is firstly electrostatically adsorbed onto the PEG-400 chains. Subsequently, the CdS nanoparticles were formed after reacting with S2−. Finally, a large number of CdS nanoparticles form the 1D assembly structure of CdS NPs along the PEG molecular template (Scheme 1-ii). In contrast, the spherical assembly mode is attributed to the existence of multiple polarization points on the surface of the CdS NCs caused by them being irregular spheres. Generally, many nanocrystals synthesized in experiments are not perfect spheres, but exist as quasi-spheres. As shown in Scheme 2, many dipole points will exist on the surface of the particles. In this case, the one-dimensional self-assembly of these nanocrystals will be broken and they will progress towards spherical assembly with low surface free energy.
 | ||
| Scheme 2 Proposed assembly mechanism of the spherical assembly mode. | ||
Conclusions
To summarize, we have successfully synthesized CdS NCs in controllable phases and sizes at room temperature in an aqueous medium. The pH value has been used to control the sizes and phases of the products by regulating the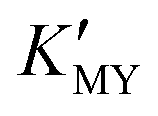 and the release rate of H2S or S2− from TAA. In the present work, cubic CdS NCs generally correspond to high
and the release rate of H2S or S2− from TAA. In the present work, cubic CdS NCs generally correspond to high 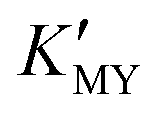 (EDTA and SSA at higher pH and EN at lower pH). CdS nanocrystals prepared with EN have a strong trend of secondary self-assembly, including spherical and linear modes for multistage nanospheres and nanowires/nanopearls. PEG-400 can effectively induce the linear assembly of the NPs. The scheme of preparing nanomaterials based on regulating the coordination equilibrium and ionization equilibrium can be extended to a variety of functional materials.
(EDTA and SSA at higher pH and EN at lower pH). CdS nanocrystals prepared with EN have a strong trend of secondary self-assembly, including spherical and linear modes for multistage nanospheres and nanowires/nanopearls. PEG-400 can effectively induce the linear assembly of the NPs. The scheme of preparing nanomaterials based on regulating the coordination equilibrium and ionization equilibrium can be extended to a variety of functional materials.
P. H. conceived and designed the project. D. Z., S. X., Q. M. and J. Y. conducted the experiments. P. H., J. X. and Y. C. wrote the manuscript and discussed the results with all the authors.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors greatly appreciate the financial support of this work from the National Nature Science Foundation of China (51471101 and 51472154).References
- B. D. Gates, Q. B. Xu, M. Stewart, D. Ryan, C. G. Willson and G. M. Whitesides, Chem. Rev., 2005, 105, 1171–1196 CrossRef CAS PubMed.
- C. Burda, X. B. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS PubMed.
- Y. W. Jun, J. S. Choi and J. Cheon, Angew. Chem., Int. Ed., 2006, 45, 3414–3439 CrossRef CAS PubMed.
- D. Zambo, A. Schlosser, P. Rusch, F. Luebkemann, J. Koch, H. Pfnuer and N. C. Bigall, Small, 2020, 16, 1906934 CrossRef CAS PubMed.
- Y. N. Xia, X. H. Xia and H. C. Peng, J. Am. Chem. Soc., 2015, 137, 7947–7966 CrossRef CAS PubMed.
- Y. Wang, W. S. Fu and X. Hu, CrystEngComm, 2015, 17, 6636–6640 RSC.
- J. Lim, W. K. Bae, K. U. Park, L. Z. Borg, R. Zentel, S. Lee and K. Char, Chem. Mater., 2013, 25, 1443–1449 CrossRef CAS.
- G. Sansone, L. Maschio and A. J. Karttunen, Chem. – Eur. J., 2017, 23, 15884–15888 CrossRef CAS PubMed.
- M. Telychko, J. Su, A. Gallardo, Y. W. Gu, J. L. Mendieta-Moreno, D. C. Qi, A. Tadich, S. T. Song, P. Lyu, Z. Z. Qiu, H. Y. Fang, M. J. Koh, J. S. Wu, P. Jelinek and J. Lu, Angew. Chem., Int. Ed., 2019, 58, 18591–18597 CrossRef CAS PubMed.
- N. Pradhan, H. Xu and X. Peng, Nano Lett., 2006, 6, 720 CrossRef CAS PubMed.
- Z. Tang, N. A. Kotov and M. Ciersig, Science, 2002, 297, 237 CrossRef CAS PubMed.
- K. S. Cho, D. V. Talapin, W. Gaschler and C. B. Murray, J. Am. Chem. Soc., 2005, 127, 7140 CrossRef CAS PubMed.
- Y. Yu, Y. T. Zhang, Z. Zhang, H. T. Zhang, X. X. Song, M. X. Cao, Y. L. Che, H. T. Dai, J. B. Yang, J. L. Wang, H. Zhang and J. Q. Yao, J. Phys. Chem. Lett., 2017, 8, 445–451 CrossRef CAS PubMed.
- D. Lee, M. Wolska-Pietkiewicz, S. Badoni, A. Grala, J. Lewinski and G. De Paepe, Angew. Chem., Int. Ed., 2019, 58, 17163–17168 CrossRef CAS PubMed.
- C. H. Xia, N. Winckelmans, P. T. Prins, S. Bals, H. C. Gerritsen and C. D. Donega, J. Am. Chem. Soc., 2018, 140, 5755–5763 CrossRef CAS PubMed.
- R. L. Brutchey, Acc. Chem. Res., 2015, 48, 2918–2926 CrossRef CAS PubMed.
- A. P. Alivisatos, Science, 1996, 271, 933 CrossRef CAS.
- M. Moniruddin, E. Oppong, D. Stewart, C. McCleese, A. Roy, J. Warzywoda and N. Nuraje, Inorg. Chem., 2019, 58, 12325–12333 CrossRef CAS PubMed.
- L. Piveteau, D. N. Dirin, C. P. Gordon, B. J. Walder, T. C. Ong, L. Emsley, C. Coperet and M. V. Kovalenko, Nano Lett., 2020, 20, 3003–3018 CrossRef CAS PubMed.
- H. Su, Y. M. Fang, F. Y. Chen and W. Wang, Chem. Sci., 2018, 9, 1448–1453 RSC.
- W. Lee, J. Oh, W. Kwon, S. H. Lee, D. Kim and S. Kim, Nano Lett., 2019, 19, 308–317 CrossRef CAS PubMed.
- S. Bisschop, P. Geiregat, T. Aubert and Z. Hens, ACS Nano, 2018, 12, 9011–9021 CrossRef CAS PubMed.
- L. J. Li, J. Zhang, M. Zhang, N. Rowell, C. C. Zhang, S. L. Wang, J. Lu, H. S. Fan, W. Huang, X. Q. Chen and K. Yu, Angew. Chem., Int. Ed., 2020, 59, 12013–12021 CrossRef CAS PubMed.
- K. Cheng, H. Wang, J. Bang, D. West, J. J. Zhao and S. B. Zhang, J. Phys. Chem. Lett., 2020, 11, 6544–6550 CrossRef CAS PubMed.
- D. X. Du, J. N. Shu, M. Q. Guo, M. A. Haghighatbin, D. Yang, Z. P. Bian and H. Cui, Anal. Chem., 2020, 92, 14113–14121 CrossRef CAS PubMed.
- M. L. Sushko, J. Mater. Res., 2019, 34, 2914–2927 CrossRef CAS.
- Y. Wang, S. I. I. Choi, X. Zhao, S. F. Xie, H. C. Peng, M. F. Chi, C. Z. Huang and Y. N. Xie, Adv. Funct. Mater., 2014, 24, 131–139 CrossRef CAS.
- P. Reiss and U. Koert, Acc. Chem. Res., 2013, 46, 2773–2780 CrossRef CAS PubMed.
- H. G. Liao, L. K. Cui, S. Whitelam and H. M. Zheng, Science, 2012, 336, 1011–1014 CrossRef CAS PubMed.
- Z. Y. Tang and N. A. Kotov, Adv. Mater., 2005, 17, 951–962 CrossRef CAS.
- R. L. Penn and J. F. Banfield, Science, 1998, 281, 969 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ce01276b |
| This journal is © The Royal Society of Chemistry 2022 |