
Tuning fast excited-state decay by ligand attachment in isolated chlorophyll a†
Elisabeth
Gruber
 ,
Ricky
Teiwes
,
Christina
Kjær
,
Steen
Brøndsted Nielsen
and
Lars H.
Andersen
*
,
Ricky
Teiwes
,
Christina
Kjær
,
Steen
Brøndsted Nielsen
and
Lars H.
Andersen
*
Department of Physics and Astronomy, Aarhus University, Aarhus 8000C, Denmark. E-mail: LHA@phys.au.dk
First published on 8th December 2021
Abstract
Excited-state dynamics plays a key role for light harvesting and energy transport in photosynthetic proteins but it is nontrivial to separate the intrinsic photophysics of the light-absorbers (chlorophylls) from interactions with the protein matrix. Here we study chlorophyll a (4-coordinate complex) and axially ligated chlorophyll a (5-coordinate complex) isolated in vacuo applying mass spectrometry to shed light on the intrinsic dynamics in the absence of nearby chlorophylls, carotenoids, amino acids, and water molecules. The 4-coordinate complexes are tagged by quaternary ammonium ions while the charge is provided by a formate ligand in the case of 5-coordinate complexes. Regardless of excitation to the Soret band or the Q band, a fast ps decay is observed, which is ascribed to the decay of the lowest excited singlet state either by intersystem crossing (ISC) to nearby triplet states or by excited-state relaxation on the excited-state potential-energy surface. The lifetime of the first excited state is 15 ps with Mg2+ at the chlorophyll center, but only 1.7 ps when formate is attached to Mg2+. When the Soret band is excited, an initial sup-ps relaxation is observed which is ascribed to fast internal conversion to the first excited state. With respect to ISC, two factors seem to play a role for the reduced lifetime of the formate-chlorophyll complex: (i) The Mg ion is pulled out of the porphyrin plane thus reducing the symmetry of the chromophore, and (ii) the first excited state (Q band) and T3 are tuned almost into resonance by the ligand, which increases the singlet–triplet mixing.
1 Introduction
The photosynthetic apparatus is designed to efficiently harvest sunlight and transfer the excitation energy to a reaction center where charge separation occurs.1,2 In nature, the key pigments for the light-harvesting process are chlorophylls and bacteriochlorophylls.3 It is well known that the spectroscopic features as well as the dynamics of chlorophylls are substantially affected by the surrounding molecular environment.4–6 In light-harvesting photosystem complexes PSI and PSII, the chromophores are embedded in protein pockets and experience electrostatic interactions with nearby amino acids, neighboring pigment molecules, and nearby water molecules. Moreover, 80% of the chlorophylls are five-coordinated, i.e. a fifth bond is formed between the magnesium at the center of the porphyrin ring and an axial ligand like water or an amino acid residue.7 Interactions between the chlorophylls and the environment influence the energy transfer; the exact mechanism is, however, still under debate.8Apart from the importance of efficient energy transfer for energy harvesting, interactions between pigments play a key role for photoprotection.9 Regulation mechanisms that rapidly dissipate excessively absorbed light are necessary because of high fluctuations in incident sunlight. Thus, as chlorophyll is excited by light, energy may be transferred to carotenoids in their first excited state, which may decay to the ground state either after excitation-energy transfer (EET) or through charge-transfer (CT) quenching, both on the tens of picosecond-time scale.10 This is faster and hence more efficient than the intrinsic radiative stabilization of chlorophyll which takes place on the ns-time scale. Alternatively, intersystem crossing (ISC) in the photoexcited chlorophyll may lead to triplet–triplet energy transfer from chlorophylls to neighbor carotenoid pigments. This also stabilizes the excess energy and at the same time avoids generation of reactive singlet oxygen causing photodamage.11
The large variety of pigments and their derivatives in photoactive organisms12,13 and the complexity of the surrounding proteins make it difficult to distinguish aspects of the photophysics related to intrinsic properties of the chlorophylls from those of environmental interactions. In the gas phase, the concentration is sufficiently low that quenching of excitation by ground-state chlorophyll molecules and molecular oxygen is negligible, but for an extensive period of time, studies of the structure, spectroscopy and excited-state dynamics of chlorophylls were limited to natural systems or solvated molecules. In 2011, the first absorption measurements of chlorophyll isolated in the gas phase were conducted.14 Some years later, the blue-shift of isolated chlorophylls was confirmed in other experimental setups. Photodissociation-action spectra of gaseous chla/b in a single-pass experiment15–18 as well as in an electrostatic ion-storage ring have been obtained.19,20 As chlorophyll molecules are neutral, quaternary ammonium cations were tagged onto the chlorophylls to perform action spectroscopy with isolated neutral chlorophylls. It was shown experimentally and by DFT calculations that the charge tag has little influence on the electronic system of the porphyrin ring.15,16 The spectroscopic properties of chlorophyll dimers as well as complexes between a chlorophyll pigment and an axial ligand have been studied,17,18,20 and fluorescence studies21 have demonstrated that photo-physical properties of chlorophylls are sensitive to small changes directly on the chlorophyll chromophore and in the local environment.
In the present work we focus on the intrinsic chlorophyll a (chla) excited-state decay which happens due to internal conversion in the singlet manifold or by intersystem-crossing from singlet to triplet states. We investigate to what degree a ligand bound to the Mg2+ center of chla may change the dynamics. In particular we discuss factors that may influence the triplet branch of energy dissipation. It was found earlier that the attachment of a water ligand to the magnesium center22 and metal substitution23 affect the triplet-state lifetimes, as the triplet intersystem crossing rates are controlled by the spin–orbit coupling involving the singlet σπ* states, which are associated with the pyrrole nitrogen-metal center complex.22 So far, singlet and triplet excited-states dynamics of photosynthetic pigments have been investigated in various solvents,24,25 where fluorescence lifetimes cover several ns. The reported triplet excited-state lifetime varies from single μs to hundreds of μs, depending on the aerobic environment. Previous gas-phase studies with porphyrin ions have reported ms-long lifetimes of the triplet-excited states,26 and long-lived triplet states were also seen in chla- and chlb-TMA complexes.19 Chlorophyll chromophores and their absorption spectra in gas phase are shown in Fig. 1.
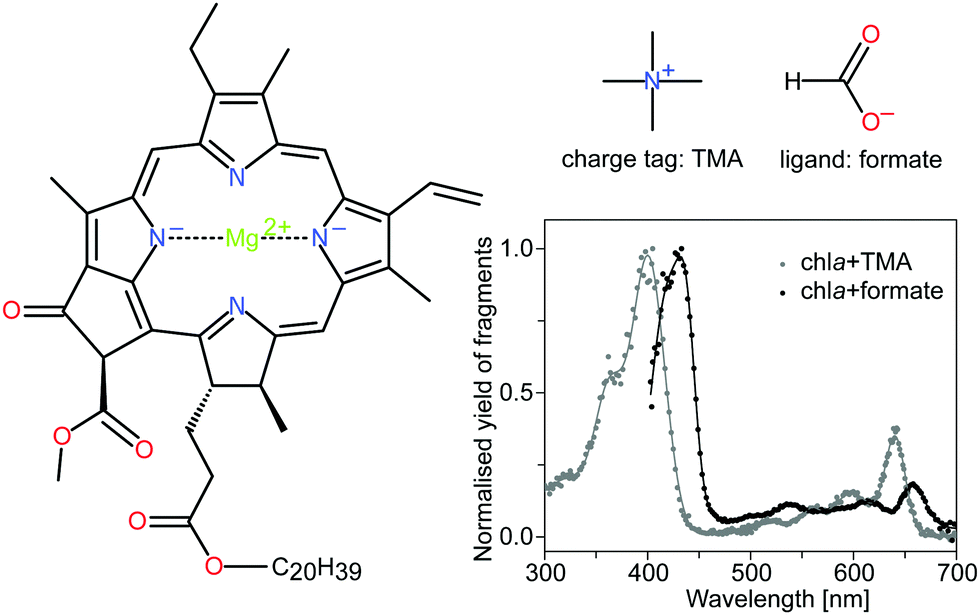 | ||
| Fig. 1 Structure of chla, the charge tag tetramethylammonium cation (TMA) and the formate ligand HCOO−. Chla is the most abundant light-harvesting pigment in oxygenic photosynthetic organisms. It consists of a tetrapyrrole ring with a four-coordinated Mg2+ ion in the center. The spectroscopic properties are largely associated with a π → π* electronic transition, polarized along the x and y-axes of the macrocycle. The two major absorption bands, the Soret and Q bands, appear at around 400 nm and 650 nm, respectively. DFT calculations have shown that the charge tag is attached to the oxygen atoms near the phytyl tail (C20H39) of the chlorophyll.15 The formate HCOO−, on the other hand, is bound to the magnesium ion at the center of the porphyrin ring and causes perturbations of the electronic system. The gas-phase action absorption spectra of the chlorophyll complexes with TMA and formate are shown for completeness (data taken from Kjær et al.20). | ||
It is challenging to measure photo-physical properties of gas-phase chlorophyll ions due to low target densities. Hence, in action-absorption spectroscopy, a unique detectable signal (action) is identified that can be related to the absorption of one or more photons.27 By using ion-storage devices, a time-resolved action signal may be detected as a function of time after initial photoexcitation and used to differentiate between various channels.28 To determine excited-state lifetimes, we identify a detectable signal that may be related to action from combined absorption of photons from a pump and a delayed probe light pulse. The absorption of a probe photon upon initial excitation by a pump photon corresponds to transient absorption, where the probe photon only is absorbed as long as the system is in a particular excited state. The challenge is to identify a signal that reports on such a transient-absorption event. In the present work we use fast two-photon induced dissociation fragments of stored chlorophyll ions as such a uniquely identifiable action signal. As shown in the ESI,† two-photon absorption is distinguishable from one-photon absorption as it leads to prompt dissociation on the μs-time scale instead of slow statistical dissociation on the ms-time scale. This is used in the SAPHIRA ion-storage ring,29–32 which is illustrated in Fig. 2. Here our data reveal that a dominant fraction of the gas-phase molecules remains excited for nanoseconds, whereas a fraction decays on the picosecond time scale, depending on the micro-environment. We assign the long component to population in the triplet manifold. The short ps component corresponds to transient population of the first excited state, which decays by intersystem crossing (ISC) or internal conversion (IC) in the singlet manifold. Axial ligation is found to change the fast decay rate by an order of magnitude. Without detailed knowledge about the first excited-state potential-energy surface, it is difficult to predict the dynamics in the singlet state. We focus here on the role of triplet states and intersystem crossing induced by spin–orbit coupling.
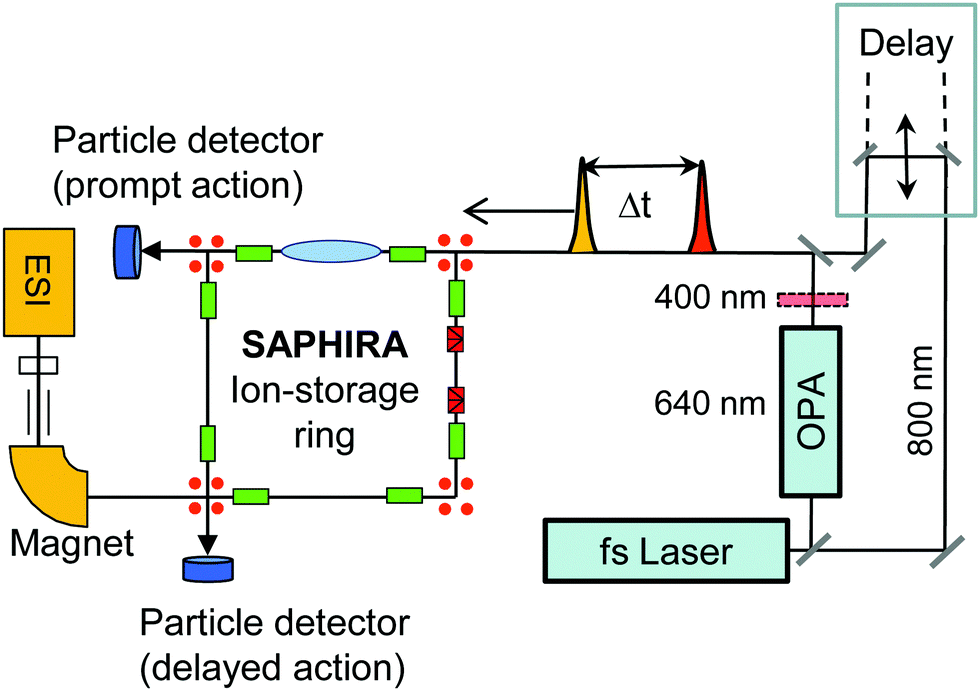 | ||
| Fig. 2 Schematic of the experimental setup for probing excited-state dynamics of stored mass selected isolated gas-phase chromophore ions in the SAPHIRA ion-storage ring. The chromophores are produced in an electrospray ionization (ESI) source. A delay stage is used to delay a 800 nm probe pulse with respect to a pump pulse of either 400 nm or 640 nm produced by a BBO crystal and in an optical-parametric amplifier, respectively. After photoabsorption, the chromophore ions decay in the ring by dissociation, hence producing neutral fragments, detectable by single-particle detectors. Transient absorption by the 800 nm probe pulse is registered by the appearance of prompt two-photon dissociation fragments detected right after the laser-ion interaction region (prompt action). | ||
To investigate the influence of a perturbation on the central Mg ion in chla, we apply the charge-tagging method33–36 using the tetramethylammonium (TMA) cation, (see Fig. 1) rendering a planar structure at the Mg center. First-principle calculations of TMA tagged chla complexes have shown that the energetically favored location of the charge tag is near the oxgygen atoms of the phytyl tail. When the tag was initially placed above the magnesium atom in the center of the porphyrin ring, the tag was observed to move to this site during geometry optimization.15 We use a formate anion (HCOO−) to study the effect of a ligand attached to the Mg ion. Formate mimics the binding of glutamate and aspartate with side-chains containing carboxylate, and importantly it displaces the Mg ion and breaks the planar symmetry (see ESI†).
2 Results and discussion
The absorption of two photons specifically leads to prompt fragmentation for both complexes (see ESI†), which makes the fs-pump–probe time-resolved action-spectroscopy setup at SAPHIRA an ideal instrument to study the dynamics of these gas-phase complexes in the femtosecond to nanosecond-time domain.For simplicity, we shall use the term Q for the Qx as well as the Qy state and similarly, B is used for the Bx as well as the nearby By state. This simplification is justified as it is known from condensed-phase measurements that Qx will internally convert to the lower Qy state in about 100 fs,37 and Bx and By will convert very fast to the Qy state by internal conversion.38 Similar fast processes can be expected in the gas phase. Moreover, the used pump–probe schematics in our measurements (see discussion below) leads only to population and probing of the Qy or By state and higher vibrational states of those, but no excitation into Qx or Bx is reached with the applied pump pulses.
The lifetime of the first excited state is obtained by populating the Q band through absorption of a 640 nm pump photon, and then monitor the decay out of this state by absorption of an 800 nm probe photon which may be absorbed while the chromophore is still in the singlet-excited state (see Fig. 3). With a given branching ratio, the first excited state may undergo intersystem crossing and populate triplet states (Tn) from where the 800 nm probe pulse likewise may cause excitation in the triplet manifold. When both photons (pump and probe) are absorbed, the system will eventually end up in a hot electronic ground state after IC, ISC, and intermolecular vibrational redistribution (IVR) and provide a prompt μs-fragmentation signal which is measured as a function of the pump–probe laser-pulse delay to reveal the excited-state dynamics. In a similar way, the Soret band may be excited by a 400 nm pump photon and probed by an 800 nm photon (see Fig. 4). Note that the probe pulse may excite the chromophore complexes both in the singlet and in the triplet manifold, and it is the different absorption-cross sections that make their respective populations visible in the pump–probe measurement.
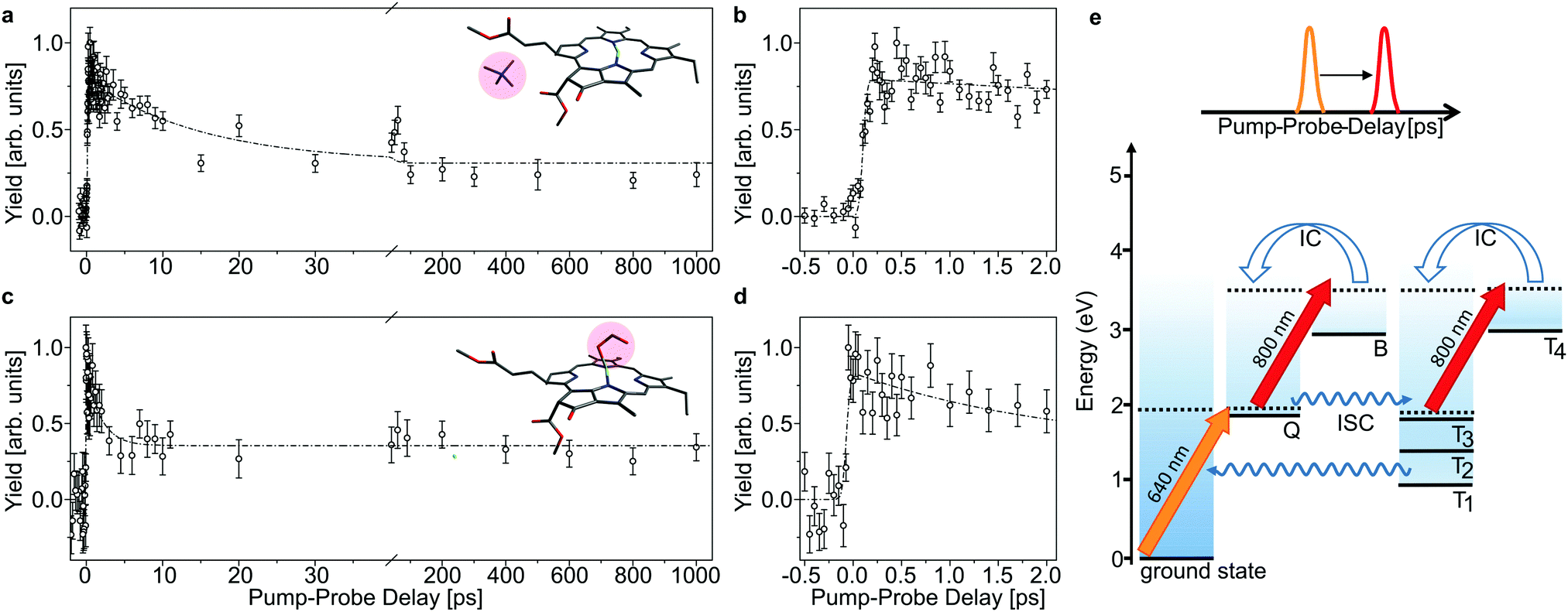 | ||
| Fig. 3 Time-resolved action data as a function of laser-pulse delay of chla-TMA (a and b) and chla-HCOO− (c and d) complexes after excitation with a 640 nm laser pulse into the Q band. A delayed 800 nm pulse is used to probe the excited-state dynamics. The error bars represent the standard deviation of the mean value of the obtained yield. The dashed curves represent bi-exponential fits. (b and d) Zoom-in on the first 2 ps of the pump–probe delay, which shows instant (within the laser-pulse duration) excitation of Q. (e) Schematic energy levels and the applied pump–probe scheme. A 640 nm pulse excites Q. The 800 nm delayed pulse probes the population in Q and Tn = 1–3, where Tn = 1–3 becomes populated by intersystem crossing (ISC) from Q. The 800 nm absorption in Q and Tn differs, which makes the population in these states discernible in the data (a and c). | ||
 | ||
| Fig. 4 As Fig. 3, here with initial 400 nm-pump excitation into the Soret (B) band. Note that there is an initial delay of about 250 fs which is ascribed to internal conversion of B into Q (b and d). The time constants obtained by fitting the data in the Q band are used for the fits shown as dashed curves in panels (a and c) for comparison. | ||
In Fig. 3 and 4, the pump–probe action spectra are shown for the two chla complexes for initial excitation into the Q band (640 nm pump) and the Soret band (400 nm pump), both being probed by 800 nm photons. The yield is here given by the number of prompt neutral fragments obtained when both a pump and a (delayed) probe pulse are applied, after subtraction of single-pulse events (obtained with only pump and only probe laser pulses) and events from background collisions (obtained without any laser pulses). The overall experiment was running at a repetition rate of 100 Hz (divided in 4 cycles) over several hours. At first glance, the decay curves look quite similar, featuring a short (ps) and a long (more than 1 ns) component, regardless of initial excitation to the Q or B-band. The reason is that regardless of the excitation wavelength, the system will end up in the Q band by internal conversion38 and in both cases it is the population and decay of the first excited state together with T1 which is probed by the 800 nm photon. Naturally, the dynamics takes place with hotter ions due to initial internal conversion (B → Q) when the high-energy Soret band is initially excited. Note that the 800 nm pump photons cannot excite the electronic ground state.
In the case, where the chla complexes are excited by 400 nm laser pulses, we measure an initial delay of about 250 fs, ascribed to internal conversion of B to Q. Such a delay is absent when Q is directly excited by the pump pulse (see Fig. 3). We therefore consider the decay of Q with progression out of the Franck–Condon region to regions on the first excited-state potential-energy surface where the 800 nm probe pulse no longer can excite the chromophore further, and the decay by ISC from Q to nearby Tn states. The 640 nm pump – 800 nm probe data give an exponential decay with a lifetime of 15.0 ± 3.7 ps for the chla-TMA complex and 1.7 ± 0.6 ps for the chla-formate complex (see Fig. 3). The uncertainties of the lifetimes are given by the standard deviation of the fit. Strikingly, the decay is almost ten times faster for the chla-formate complex than for the chla-TMA complex. The decay involves both nuclear and electronic degrees of freedom, and we first notice that there may be a reasonable nuclear overlap between vibrational levels in Q and Tn enabling high coupling rates between these states relative to those for the Q → ground-state and T1 → ground-state radiationless transitions.39 This is consistent with a high triplet quantum yield in the gas phase of 0.7–0.8.19 It should be noticed that for the chla-TMA as well as for the chla-formate complex we used the same pump–probe scheme with 640 nm + 800 nm, although the absorption bands of the chla-formate are redshifted in comparison to that of the chla-TMA complex, and the absorption maximum of the Q band lies around 660 nm for the chla-formate complex and around 640 nm for the chla-TMA complex (see Fig. 1). In case of the chla-formate, the pump pulse at 640 nm prepares an excited state Q with additional internal energy (population of higher vibrational states of the Q band), which might increase the coupling to a triplet state. As we repeated the measurements by using a pump–probe scheme with 400 nm + 800 nm, and the decay curves look quite similar to that obtained by the 640 nm + 800 nm pump–probe scheme, we suggest that the excitation into higher vibrational states of the Q-band in the case of the chla-formate complex has no crucial influence on the dynamics and cannot account alone for the much faster decay (almost ten times) out of the excited state.
The importance of the spin–orbit coupling was recognized already in 1959 where it was suggested that a shoulder on the red side of the main ππ* absorption band was due to a nπ* singlet transition with a corresponding nπ* spin-forbidden triplet emission.40 The n, π and π* molecular orbitals are based on atomic states of the nitrogen and carbon atoms (see ESI† for a representation of the HOMO and LUMO of chla and chla-formate).39 The electronic configurations of the excited singlet and triplet states are mixtures of nπ* and ππ* configurations with antiparallel and parallel spin of the open shell electrons, respectively. The singlet–triplet mixing is provided directly by the spin–orbit coupling and by vibrations that may lift the orthogonal properties of the orbitals.
To discuss the ISC rate for the chla-formate complex, we consider here specifically the spin–orbit matrix elements:
| VSOCn,m = < Sn|HSO|Tm > | (1) |
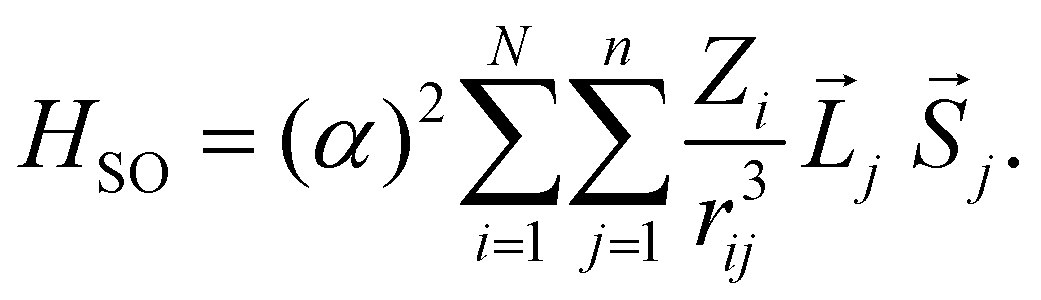 | (2) |
The summation (i) is over N nuclei and (j) over n electrons of the molecule. ![[L with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_004c_20d1.gif) j and
j and ![[S with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0053_20d1.gif) j are the orbital and spin momenta of the electrons, and α the fine-structure constant. rij is the distance between nucleus i and electron j. The ISC-rate constant is proportional to (VSOCn,m)2 times the density of states and the Franck–Condon overlap of the involved vibrational levels. We calculate the spin–orbit coupling matrix elements for the bare chla and the chla-formate complex to understand the increased ISC-decay rate for the chla-formate complex.
j are the orbital and spin momenta of the electrons, and α the fine-structure constant. rij is the distance between nucleus i and electron j. The ISC-rate constant is proportional to (VSOCn,m)2 times the density of states and the Franck–Condon overlap of the involved vibrational levels. We calculate the spin–orbit coupling matrix elements for the bare chla and the chla-formate complex to understand the increased ISC-decay rate for the chla-formate complex.
The vertical energies are obtained through a TDDFT calculation by the Gaussian16 software package41 using the ωB97XD functional and the TZVP basis.42 The structures of chla and the chla-formate complexes are optimized at the same level of theory, and a summary of the calculations is given in the ESI† and in Table 1. In terms of spectroscopy, we find for both systems that there are three triplet states (Tn = 1−3) below the first singlet state. The significant red shift of the ground state → B excitation may be ascribed to the effect of the negatively charged formate and the charge-transfer character of this transition, where electron density is transferred from the center region to the periphery of the chromophore.20
| Molecule | S n E n (eV) | T m E m (eV) | V SOC1,m (cm−1) | V SOC2,m (cm−1) |
|---|---|---|---|---|
| chla | S1 2.14 | T1 1.02 | 0.29 | 1.35 |
| S2 2.58 | T2 1.61 | 1.28 | 0.12 | |
| S3 3.48 | T3 2.08 | 0.82 | 0.17 | |
| chla-formate | S1 2.16 | T1 0.98 | 0.43 | 1.54 |
| S2 2.47 | T2 1.51 | 1.34 | 0.20 | |
| S3 3.41 | T3 2.13 | 1.24 | 0.18 |
The output from the DFT calculations is used as input for a direct calculation of the spin–orbit coupling matrix elements using the PySOC code.42 The highest coupling is found between Q and T2. We specifically note that the spin–orbit coupling systematically gets stronger when the formate anion is attached to the chlorophyll which results in an increased ISC rate. The ligand interaction causes the chlorophyll to be nonplanar with the Mg ion pulled out towards the ligand (see ESI†). This breaks the central planar symmetry of chla and reduces its C2v symmetry.43 For the matrix elements of the spin–orbit coupling (eqn (1)) to vanish, the direct product of the irreducible representations of the singlet-state Sn (Q or B) and the triplet-state Tm must contain the irreducible representation of one of the components of the orbital-angular momentum operator Lx,Ly or Lz.44 Under the reduced symmetry of the chla-ligand complex, these selection rules are relaxed, hence potentially causing higher ISC rates. This is fully consistent with the finding that highly symmetric planar conjugated chromophores in general have low ISC rates.44 On the other hand, nπ* and ππ* mixing and ultrasmall energy gaps can provide high intersystem-crossing rates of the order 1010 to 1011 s−1,45 In the case of chla, Q and T3 are almost degenerate (see Table 1), the energy difference being 60 meV for chla but only 30 meV for the chla-formate complex. The small energy difference makes the Q → T3 decay particular important despite the fact that other transitions may have a somewhat higher spin–orbit coupling matrix element.
We note that the intrinsic Q excited-state lifetime of the chla-formate complex stays in the several picosecond time regime, long enough to establish conditions for excitation-energy transfer in a picosecond and subpicosecond time domain in photosystems.46 For the long component, ascribed to trapping in T1 after ISC and triplet internal conversion, we cannot observe any decay of the excited-state population within the nanosecond time-delay considered here, fully consistent with previous (ms) single-pulse measurements in gas phase.19,26
3 Experimental
Complexes between chla and the TMA charge tag or the formate are produced in the gas phase by electrospray ionization of a methanol solution containing chlorophyll (from spinach and provided by Sigma-Aldrich) and the salt of tetramethylammonium chloride or sodium formate, respectively. A few grains of chlorophyll were dissolved in 2–3 droplets of N,N-dimethylformamide and 1 mL methanol, and a few grains of tetramethylammonium chloride or sodium formate were added.The pump–probe experiments are performed at the SAPHIRA29,47 electrostatic ion-storage ring at Aarhus University. Molecular ions are accumulated in a multipole radio frequency ion trap with a He buffer gas at room temperature. Following extraction from the trap, the ions are accelerated to a kinetic energy of 4 keV, mass-to-charge selected by a magnet and injected into the storage ring. The ion bunch is stored for several ms before photoexcitation. The absorption of photons is registered by detecting neutral photofragments leaving the storage ring and hitting a detector (action spectroscopy). Prompt as well as delayed action is measured by using two separate detectors positioned on opposite sites of the storage ring (see Fig. 2).
At SAPHIRA, fs-laser pulses are generated by a Ti:Sapphire based Libra HE-UFS oscillator and regenerative amplifier system from Coherent, delivering 3.8 mJ, 50 fs pulses with up to 1 kHz repetition rate. The pulses are split 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to separately provide pump and probe pulses. A TOPAS-C optical-parametric amplifier, generates 640 nm pump pulses (a BBO crystal is used for 400 nm pump pulses). The 800 nm probe-laser beam is delayed by a motorized delay stage – see Fig. 2. For details see ref. 32.
:1 to separately provide pump and probe pulses. A TOPAS-C optical-parametric amplifier, generates 640 nm pump pulses (a BBO crystal is used for 400 nm pump pulses). The 800 nm probe-laser beam is delayed by a motorized delay stage – see Fig. 2. For details see ref. 32.
4 Conclusions
In summary, by applying a fs pump–probe scheme at an ion-storage ring, we used time-resolved action spectroscopy to study excited-state dynamics for two chla complexes to unravel the effect of a ligand-interaction with the central Mg ion of isolated gas phase chlorophyll chromophores. When excited to B, we observe a few hundred fs delayed action, ascribed to the B → Q internal conversion time. Apart from that, ps and >ns components are registered. Previous studies have shown very long (ms) lifetimes and high triplet-quantum yields of gas-phase chloroplylls which support our interpretation of the long-lived >ns decay component as being due to trapping in the triplet manifold. The ps component is ascribed to excited-state dynamics in the Q state, where the system progresses out of a region accessible to 800 nm probe absorption. The depletion of Q is by intersystem crossing to the triplet manifold as well as internal conversion and possibly fluorescence. The ligand significantly reduces the excited-state lifetime which in part may be ascribed to loss of planar symmetry in chla and a corresponding increase in transition rate Q → Tn = 1–3 by the spin–orbit coupling. At the applied level of theory, one state, T3, may be particular important as it is tuned almost in resonance with Q in the chla-ligand complex. After initial Q → Tn intersystem crossing, the excited-state population remains for >ns, corresponding to trapping in the triplet manifold. After internal conversion the population eventually will occupy the lowest T1 state. The study shows that a ligand not only fine tunes the spectroscopy of chlorophyll, it may also influence the amount of triplet trapping, which in photosystems is part of energy destabilization mechanisms involving couplings with carotenoids.Author contributions
Experiments were designed by E. G. and L. H. A. and executed by E. G., R. T., and C. K. Data were analyzed by E. G. and R. T. All authors have discussed the results and commented on the paper, written primarily by E. G. and L. H. A.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by research grant (17512) from VILLUM FONDEN. E. G. received support from the Austrian Science Fund, FWF (Project number T1181) during the writing process.References
- R. E. Blankenship, Molecular mechanisms of photosynthesis, John Wiley & Sons, 2014 Search PubMed.
- R. Croce and H. Van Amerongen, Nat. Chem. Biol., 2014, 10, 492–501 CrossRef CAS PubMed.
- H. Scheer, An Overview of Chlorophylls and Bacteriochlorophylls: Biochemistry, Biophysics, Functions and Applications. In: B. Grimm, R. J. Porra, W. Rüdiger, H. Scheer (eds) Chlorophylls and Bacteriochlorophylls. Advances in Photosynthesis and Respiration, 2006, vol 25, Springer, Dordrecht Search PubMed.
- R. Croce and H. van Amerongen, Photosynth. Res., 2013, 116, 153–166 CrossRef CAS PubMed.
- Z. Liu, H. Yan, K. Wang, T. Kuang, J. Zhang, L. Gui, X. An and W. Chang, Nature, 2004, 428, 287–292 CrossRef CAS PubMed.
- T. Morosinotto, J. Breton, R. Bassi and R. Croce, J. Biol. Chem., 2003, 278, 49223–49229 CrossRef CAS PubMed.
- J. Heimdal, K. P. Jensen, A. Devarajan and U. Ryde, J. Biol. Inorg. Chem., 2007, 12, 49–61 CrossRef CAS PubMed.
- Y. C. Cheng and G. R. Fleming, Annu. Rev. Phys. Chem., 2009, 60, 241–262 CrossRef CAS PubMed.
- B. Demmig-Adams and W. W. Adams, Science, 2002, 298, 2149–2153 CrossRef CAS PubMed.
- S. Park, C. J. Steen, D. Lyska, A. L. Fischer, B. Endelman, M. Iwai, K. K. Niyogi and G. R. Fleming, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 3385–3390 CrossRef CAS PubMed.
- H. Hashimoto, C. Uragami and R. J. Cogdell, Subcell. Biochem., 2016, 79, 111–139 CAS.
- J. K. Hoober, L. L. Eggink and M. Chen, Photosynth. Res., 2007, 94, 387–400 CrossRef CAS PubMed.
- M. Senge, A. Ryan, K. Letchford, S. MacGowan and T. Mielke, Symmetry, 2014, 6, 781–843 CrossRef.
- N. Shafizadeh, M. H. Ha-Thi, B. Soep, M. A. Gaveau, F. Piuzzi and C. Pothier, J. Chem. Phys., 2011, 135, 114303 CrossRef CAS PubMed.
- B. F. Milne, Y. Toker, A. Rubio and S. Brøndsted Nielsen, Angew. Chem., Int. Ed., 2015, 54, 2170–2173 CrossRef CAS PubMed.
- M. H. Stockett, L. Musbat, C. Kjær, J. Houmøller, Y. Toker, A. Rubio, B. F. Milne and S. Brøndsted Nielsen, Phys. Chem. Chem. Phys., 2015, 17, 25793–25798 RSC.
- B. F. Milne, C. Kjær, J. Houmøller, M. H. Stockett, Y. Toker, A. Rubio and S. Brøndsted Nielsen, Angew. Chem., Int. Ed., 2016, 55, 6248–6251 CrossRef CAS PubMed.
- C. Kjær, M. H. Stockett, B. M. Pedersen and S. Brøndsted Nielsen, J. Phys. Chem. B, 2016, 120, 12105–12110 CrossRef PubMed.
- E. Gruber, C. Kjær, S. Brøndsted Nielsen and L. H. Andersen, Chem. – Eur. J., 2019, 25, 9153–9158 CrossRef CAS PubMed.
- C. Kjær, E. Gruber, S. Brøndsted Nielsen and L. H. Andersen, Phys. Chem. Chem. Phys., 2020, 22, 20331–20336 RSC.
- S. M. J. Wellman and R. A. Jockusch, Chem. – Eur. J., 2017, 23, 7728–7736 CrossRef CAS PubMed.
- R. Clarke, S. Hotchandani, S. Jagannathan and R. M. Leblanc, Chem. Phys. Lett., 1982, 89, 37–40 CrossRef CAS.
- R. H. Clarke and H. A. Frank, Chem. Phys. Lett., 1977, 51, 13–17 CrossRef CAS.
- D. M. Niedzwiedzki and R. E. Blankenship, Photosynth. Res., 2010, 106, 227–238 CrossRef CAS PubMed.
- D. Kosumi, T. Nishiguchi, Y. Amao, R. Cogdell and H. Hashimoto, J. Photochem. Photobiol., A, 2018, 358, 374–378 CrossRef CAS.
- M. R. Calvo, J. U. Andersen, P. Hvelplund, S. B. Nielsen, U. V. Pedersen, J. Rangama, S. Tomita and J. S. Förster, J. Chem. Phys., 2004, 120, 5067–5072 CrossRef CAS PubMed.
- A. M. Rijs and J. Oomens, Gas-phase IR spectroscopy and structure of biological molecules, Springer, 2015, vol. 364 Search PubMed.
- A. V. Bochenkova and L. H. Andersen, Photo-initiated dynamics and spectroscopy of the deprotonated Green Fluorescent Protein chromophore, Springer, 2013, pp. 67–103 Search PubMed.
- H. B. Pedersen, A. Svendsen, L. S. Harbo, H. Kiefer, H. Kjeldsen, L. Lammich, Y. Toker and L. H. Andersen, Rev. Sci. Instrum., 2015, 86, 063107 CrossRef CAS PubMed.
- H. V. Kiefer, H. B. Pedersen, A. V. Bochenkova and L. H. Andersen, Phys. Rev. Lett., 2016, 117, 243004 CrossRef PubMed.
- A. Svendsen, H. V. Kiefer, H. B. Pedersen, A. V. Bochenkova and L. H. Andersen, J. Am. Chem. Soc., 2017, 139, 8766–8771 CrossRef CAS PubMed.
- H. V. Kiefer, E. Gruber, J. Langeland, P. A. Kusochek, A. V. Bochenkova and L. H. Andersen, Nat. Commun., 2019, 10, 1–9 CrossRef CAS PubMed.
- C. S. Jensen, A. I. S. Holm, H. Zettergren, J. B. Overgaard, P. Hvelplund and S. Brøndsted Nielsen, J. Am. Soc. Mass Spectrom., 2009, 20, 1881–1889 CrossRef CAS PubMed.
- J. Rajput, D. B. Rahbek, L. H. Andersen, T. Rocha-Rinza, O. Christiansen, K. B. Bravaya, A. V. Erokhin, A. V. Bochenkova, K. M. Solntsev, J. Dong, J. Kowalik, L. M. Tolbert, M. Aaxman Petersen and M. Brøndsted Nielsen, Phys. Chem. Chem. Phys., 2009, 11, 9996–10002 RSC.
- L. Lammich, M. Å. Petersen, M. Brøndsted Nielsen and L. H. Andersen, Biophys. J., 2007, 92, 201–207 CrossRef CAS PubMed.
- I. B. Nielsen, M. Å. Petersen, L. Lammich, M. Brøndsted Nielsen and L. H. Andersen, J. Phys. Chem. A, 2006, 110, 12592–12596 CrossRef CAS PubMed.
- M. F. Khyasudeen, P. J. Nowakowski and H.-S. Tan, J. Phys. Chem. B, 2019, 123, 1359–1364 CrossRef CAS PubMed.
- W. P. Bricker, P. M. Shenai, A. Ghosh, Z. Liu, M. G. M. Enriquez, P. H. Lambrev, H.-S. Tan, C. S. Lo, S. Tretiak, S. Fernandez-Alberti and Y. Zhao, Sci. Rep., 2015, 5, 13625 CrossRef PubMed.
- G. M. Maggiora and L. L. Ingraham, Chlorophyll triplet states. In: C. K. Jørgensen, J. B. Neilands, R. S. Nyholm, D. Reinen, R. J. P. Williams (eds) Structure and Bonding. Structure and Bonding, 1967, vol 2, Springer, Berlin, Heidelberg Search PubMed.
- J. Fernandez and R. S. Becker, J. Chem. Phys., 1959, 31, 467–472 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian16 Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- X. Gao, S. Bai, D. Fazzi, T. Niehaus, M. Barbatti and W. Thiel, J. Chem. Theory Comput., 2017, 13, 515–524 CrossRef CAS PubMed.
- M. Senge, A. Ryan, K. A. Letchford, S. A. MacGowan and T. Mielke, Symmetry, 2014, 6, 781–843 CrossRef.
- D. Beljonne, Z. Shuai, G. Pourtois and J. L. Bredas, J. Phys. Chem. A, 2001, 105, 3899–3907 CrossRef CAS.
- R. W. Anderson, R. M. Hochstrasser, H. Lutz and G. W. Scott, J. Chem. Phys., 1974, 61, 2500–2506 CrossRef CAS.
- T. Bittner, K.-D. Irrgang, G. Renger and M. R. Wasielewski, J. Phys. Chem., 1994, 98, 11821–11826 CrossRef CAS.
- A. Svendsen, R. Teiwes, H. V. Kiefer, L. H. Andersen and H. B. Pedersen, Rev. Sci. Instrum., 2016, 87, 013111 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp04356k |
| This journal is © the Owner Societies 2022 |