
Performance of the nitrogen reduction reaction on metal bound g-C6N6: a combined approach of machine learning and DFT†
Moumita
Mukherjee
a,
Sayan
Dutta
a,
Madhusudan
Ghosh
b,
Partha
Basuchowdhuri
*b and
Ayan
Datta
 *a
*a
aSchool of Chemical Sciences, Indian Association for the Cultivation of Science, Kolkata, West Bengal, India. E-mail: spad@iacs.res.in
bSchool of Mathematical and Computational Science, Indian Association for the Cultivation of Science, Kolkata, West Bengal, India. E-mail: partha.basuchowdhuri@iacs.res.in
First published on 27th June 2022
Abstract
Developing a cost-effective and environmentally benign substitute for the energy-intensive Haber–Bosch process for the production of ammonia is a global challenge. The electrocatalytic nitrogen reduction reaction (NRR) under ambient conditions through the six proton–electron process has attracted significant interest. Herein, a series of transition-metal (TM) based single atom catalysts (SAC) embedded on carbon nitride (C6N6) have been chosen to explore the NRR activity. The promising metals have been primarily screened through density functional theory (DFT) by calculating their adsorption energies on C6N6 – energies for dinitrogen binding and the barriers at the rate determining step. Based on these criteria, amongst the 18 metal centers, Ta based C6N6 emerges as a good candidate for the reduction of nitrogen to NH3. On the other hand, for the Machine Learning (ML) regression models, the covalent radius and the d-band center of the TM have been identified as the most correlated descriptors for predicting the adsorption energy of nitrogen on the active metal center. Besides, probabilistic modeling using the soft voting technique in the classification model allows us to predict the most efficient single atom catalyst. Despite the realistic bottleneck of having only a limited number of TMs to choose from, this technique effectively predicts the best catalyst from a modest dataset. With the highest probabilistic score, Ta based C6N6 dominates over the other catalysts in a good agreement with DFT findings. This letter manifests the effectiveness of the soft voting technique in an ensemble-based classification model.
1. Introduction
Ammonia, the most important starting material for the food-security of the world is produced in huge quantities through the well-optimized century old Haber–Bosch-process utilizing nitrogen and hydrogen as the precursors.1,2 Shifting the equilibrium towards the product with cleavage of the triple bond in di-nitrogen demands high temperature (350–550 °C) and pressure (150–350 atm). This consumes a significant amount of global energy (∼1–2% of the total annual energy production).3 Additionally, a large volume of pure hydrogen is required which results in the emission of CO2 as the by-product, a greenhouse gas. Therefore, in recent years, a cost friendly approach for N2 reduction towards NH3 production suitable at ambient pressure and temperature has been actively pursued. Previous reports show that the nitrogenase enzymes as well as the molecular catalysts containing Fe, Co, Mo are favorable active sites for N2 reduction under mild conditions.4,5 Recently, electrocatalytic and photocatalytic approaches for the reduction have been widely explored because of their bio-friendly nature. These are generally accomplished using renewable sources of electricity under ambient conditions, making these approaches energy-efficient and thus, alleviating the harsh conditions of the traditional methods. In view of this, the electro/photo catalytic reduction of nitrogen has drawn huge attention and several experimental and theoretical works have been reported.6,7The electrocatalytic nitrogen reduction reaction (NRR) proceeds via activation of dinitrogen followed by breaking of N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N towards the production of NH3 under normal atmospheric pressure and temperature. From a thermodynamic viewpoint two competing reactions, viz., the NRR and the HER (hydrogen evolution reaction) might occur at the same potential. Hence, a suitable catalyst must be such that it helps in lowering the binding free energy for N2-adsorption in comparison to H-adsorption and thereby, enhancing the NH3 selectivity. Since the discovery of transition metal (TM) – N2 complex in 1965, many studies have been reported on metal doped two-dimensional (2D) materials as a suitable catalyst for the NRR where the metal center acts as an active catalyst.8–11 Qiao et al. have explored the advantages of single atom catalysts (SACs).12 SACs serve the purpose by maximizing the metal usage efficiency, especially for noble metals. Moreover, the low-coordinated metal active center with high surface free energy dispersed on a large surface support inhibits the possibility of diffusion and agglomeration and, thus, makes it a suitable candidate for the NRR. On the other hand, backdonation of the d-electrons from the active metal center to the antibonding π* orbital of the NN bond facilitates the dissociation of the N2 bond. In order to search for a suitable catalyst towards the NRR, density functional theory (DFT) has been used for studying systematically a series of TMs supported on different suitable substrates for strong interactions. Wang et al. have reported that amongst a series of single metal embedded on graphitic carbon nitride (g-C3N4), tungsten (W) showed the highest catalytic activity towards the NRR.13 In another study by Zhao et al., molybdenum (Mo) and chromium (Cr) were found to be the most suitable metals supported on nitrogen doped graphene towards the production of ammonia.14 Liu and his group studied conversion of N2 to ammonia via several TMs anchored on boron sheets and reported that ruthenium (Ru) showed superior activity.15
N towards the production of NH3 under normal atmospheric pressure and temperature. From a thermodynamic viewpoint two competing reactions, viz., the NRR and the HER (hydrogen evolution reaction) might occur at the same potential. Hence, a suitable catalyst must be such that it helps in lowering the binding free energy for N2-adsorption in comparison to H-adsorption and thereby, enhancing the NH3 selectivity. Since the discovery of transition metal (TM) – N2 complex in 1965, many studies have been reported on metal doped two-dimensional (2D) materials as a suitable catalyst for the NRR where the metal center acts as an active catalyst.8–11 Qiao et al. have explored the advantages of single atom catalysts (SACs).12 SACs serve the purpose by maximizing the metal usage efficiency, especially for noble metals. Moreover, the low-coordinated metal active center with high surface free energy dispersed on a large surface support inhibits the possibility of diffusion and agglomeration and, thus, makes it a suitable candidate for the NRR. On the other hand, backdonation of the d-electrons from the active metal center to the antibonding π* orbital of the NN bond facilitates the dissociation of the N2 bond. In order to search for a suitable catalyst towards the NRR, density functional theory (DFT) has been used for studying systematically a series of TMs supported on different suitable substrates for strong interactions. Wang et al. have reported that amongst a series of single metal embedded on graphitic carbon nitride (g-C3N4), tungsten (W) showed the highest catalytic activity towards the NRR.13 In another study by Zhao et al., molybdenum (Mo) and chromium (Cr) were found to be the most suitable metals supported on nitrogen doped graphene towards the production of ammonia.14 Liu and his group studied conversion of N2 to ammonia via several TMs anchored on boron sheets and reported that ruthenium (Ru) showed superior activity.15
In addition to DFT calculations, Machine Learning (ML) has emerged as a potent tool in the search for new catalysts for important processes namely, CO2 reduction,16 N2 reduction,17,18 and O2 reduction reactions.19 Typically, ML algorithms can be modeled in two ways – either that demonstrating a ML model where the input set is experimental data or that available from electronic calculations or utilizing previously established ML models for specific surfaces or metals for training the required catalytic application. The output is a collection of a few descriptors based on which ML can predict the target class at a significantly reduced computational cost. The descriptors may be the intrinsic parameters of the constituent atoms and/or that obtained from electronic or structural calculations. Instead of computing the rigorous, time consuming, expensive computational calculations, one can predict the target properties by screening through a complete dataset using the ML algorithm. Bagherzadeh et al. have compared the decision tree algorithm viz. Random Forest (RF) and Gradient Boosting Machine (GBM), using the neural network algorithm (ANN) to predict the amount of nitrogen present in waste water and showed the accuracy of the algorithm for this prediction.20 In a combined study of ML predictions and DFT verifications, Kim and his group found the most stable catalyst for NRR from a large database (∼3040 surface) based on the elemental properties of the adsorbates.21 For the N2 reduction, Zafari et al. have suggested a deep neural network (DNN) to predict efficient catalysts in B-doped graphene SAC. By employing the most important features as the input in the light gradient boosting machine (LGBM) model they have successfully predicted the adsorption free energies of the adsorbates.22 Ensemble learning models are being used nowadays for its efficient predictions based on the results generated from different prediction models that have been already trained by the available datasets.23
Herein, a series of transition metals (3d, 4d, and 5d) embedded carbon nitride (C6N6) have been used as catalysts for the NRR mechanism. Based on the DFT computations, Mo, Nb, W, Re, Hf and Ta supported on C6N6 are found to be suitable candidates. In the machine learning algorithm, the regression model has been adopted for calculating the adsorption energy of nitrogen, with the aid of different possible descriptors including the structural, electronic and properties associated with the host metal centers. Using statistically significant and simple features, an ensemble-based classification model has been established. It has shown a high correlation with the DFT computations for a realistically modest dataset of 18 TMs. Ta@C6N6 has been found to be an excellent electrocatalyst for the NRR reaction.
2. Computational methods
Spin polarized DFT computations have been carried out using the Vienna Ab Initio Simulation package (VASP).24,25 We have considered the projector augmented wave (PAW) pseudopotential for representing core–electron interaction and Perdew–Burke–Ernzerhof (PBE) within the generalized gradient approximation (GGA) as the exchange correlation functional to describe the electronic interactions. A 500 eV kinetic energy cut-off has been set for plane wave expansion. The convergence criteria of energy and force for all calculations have been set as 10−5 eV and 0.02 eV Å−1. The van der Waals interaction (vdW) has been taken into account using the correction in Grimme's DFT-D2 scheme.26 For sampling the 2D Brillouin zone, a Monkhorst–Pack k-point grid of 5 × 5 × 1 has been used for geometry optimization. To construct a single atom catalyst, each metal atom has to be deposited into the core of a 2 × 2 × 1 supercell of C6N6. The adsorption energy (Eads) of the metal atom and the intermediates formed in the nitrogen reduction pathway has been obtained using the equation| Eads = Etotal − Esubstrate − Eadsorbate | (1) |
The Gibbs free energy change for each reaction step considering the computational hydrogen electrode model (CHE), has been calculated using the following expression:
| ΔG = ΔE + ΔZPE − TΔS + ΔGpH + eU | (2) |
In this work, the scikit-learn package has been employed for all the ML based calculations.28,29 Here, the machine learning algorithms have been categorized into two models – regression and classification. We have mainly applied ensemble-based techniques, such as gradient boost regression (GBR), and random forest regression (RFR) for the regression problem.29,30 These two methods are discussed in detail in the ESI.† The performance of the regression model has been quantified by the R2 value and root-mean squared error (RMSE) value using the following equations:
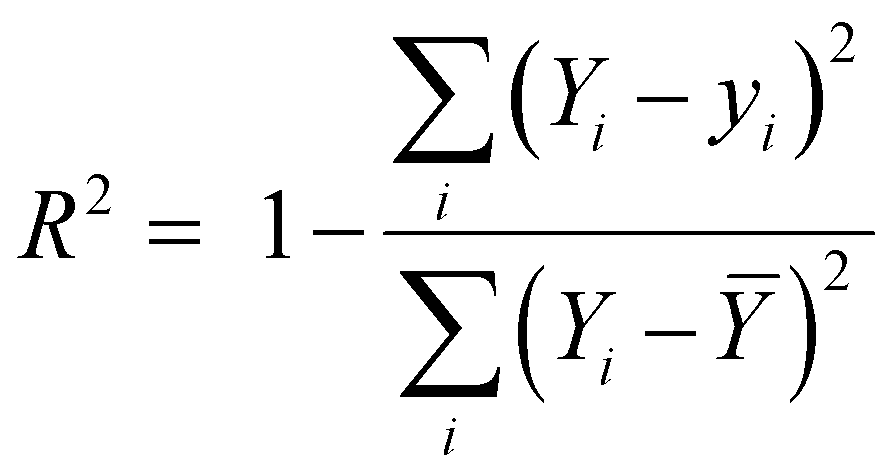 | (3) |
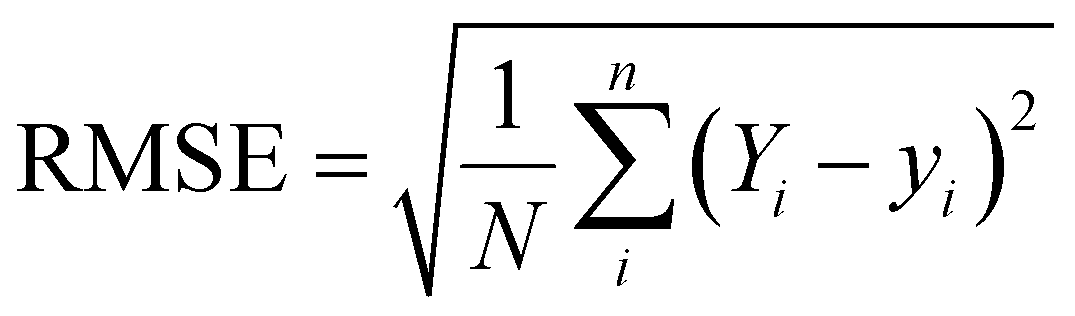 | (4) |
The classification model, on the other hand, calculated a probabilistic score for each data-point present in the full dataset. We have applied the majority voting principle to ensemble the algorithms such as logistic regression, support vector machine and random forest classifier used for the classification problem.30–32 The hyperparameters used here for the regression and classification model are listed in Table S1 (ESI†).
3. Results and discussion
The foremost factor that controls the catalytic activity of single atom catalysts is the adsorption energy of a metal atom with the substrate: the stronger the adsorption energy, the lower the possibility of aggregation. In this work, a 2 × 2 × 1 monolayer of C6N6 has been chosen as the substrate and a series of metal atoms from 3d, 4d and 5d transition rows have been taken into account for computing the adsorption energy by considering various possible binding sites on the monolayer. The adsorption energy of different metal atoms placed at 4 sites are shown in Table S2 (ESI†). Among all the configurations, position 2 (Fig. 1(a)), where, the single metal atom is placed nearer to the nitrogen atoms inside the C6N6 ring, is found to be the most stable one. The adsorption energies of the 18 different metal atoms are computed and summarized in Fig. 1(b). The negative Eads, range from −9.15 to −2.5 eV, indicate that the adsorption of metal atoms on C6N6 is thermodynamically favorable. Additionally, Bader charge analyses as shown in Fig. 1(c) shows that ∼0.51 e− to 1.67 e− is transferred from the TM to the C6N6 substrate making the TM positively charged for the coordination by N2 during the reduction.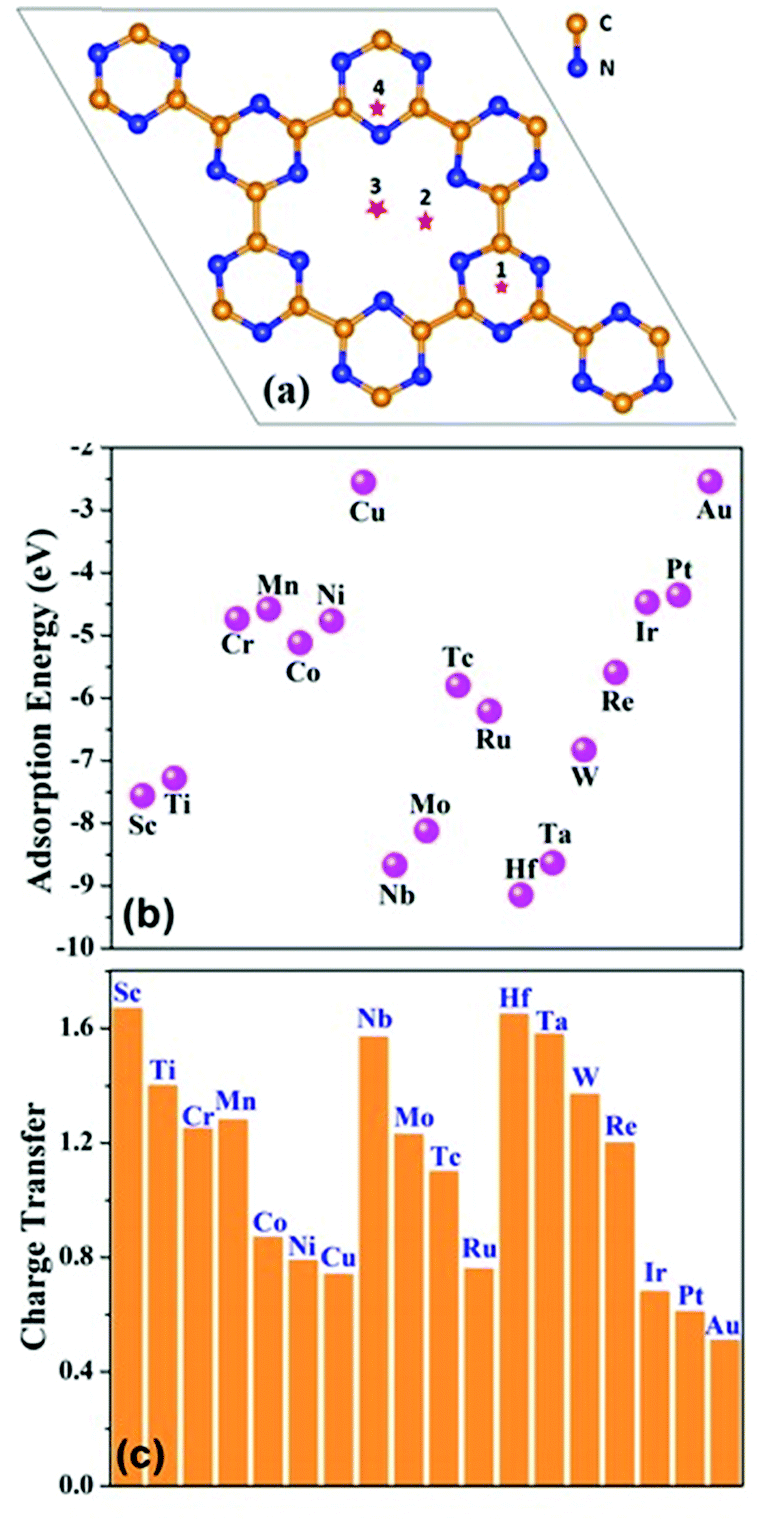 | ||
| Fig. 1 (a) Possible binding sites of metals on C6N6. (b) Adsorption energy of different metals on C6N6 at position 2. (c) Charge transfer from the active metal-center to C6N6. | ||
The adsorption of N2 on the TM doped catalyst surface is the initial step of electrocatalytic reduction of nitrogen, which plays an important role in complete pathway. N2 can be adsorbed in two different modes viz, end-on and side-on. The end-on mode is further classified into two pathways, namely, distal and alternating while side-on mode follows the enzymatic pathway.9 The end-on mode starts with the binding of one nitrogen with the TM and forms NN before going through the further steps towards reduction. In the distal mechanism, the hydrogenation occurs consecutively on the distal-N leading to the desorption of first NH3 from the surface followed by re-hydrogenation of the other N attached to the TM. On the other hand, in the alternating pathway, hydrogenation occurs alternatingly over the two N atoms. The side-on mode or enzymatic pathway proceeds through the binding of both the nitrogen with the TM and eventually hydrogenating them alternatively which liberates two NH3 consecutively. We have calculated adsorption energies of N2 in both the modes for 18 different SACs which indicates that the end-on mode is preferable over the side-on mode for binding of N2, as shown in Table S3 (ESI†). The potential determining step for most metal surfaces, as shown by Nørskov et al., would be either the first reduction step (formation of *NNH), or the final protonation step (*NH2 to *NH3).33 Also, Ling et al. proposed three key steps that include Gibbs free energy of N2 adsorption 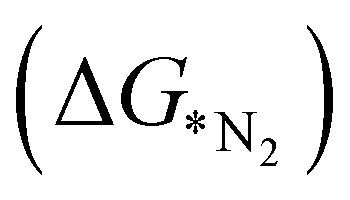 , first
, first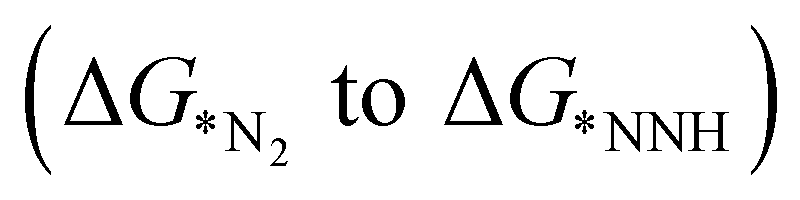 and final hydrogenation
and final hydrogenation 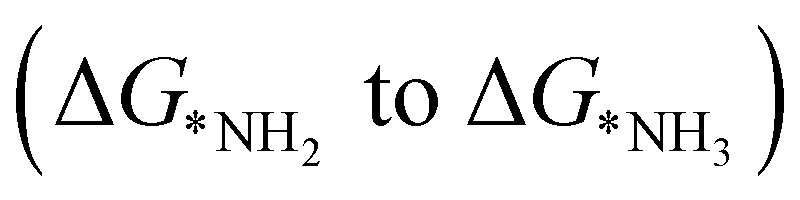 for finding a suitable electrocatalyst for the NRR.34 A screening through these essential steps can help to find a suitable electrocatalyst for the NRR. The transition metal atoms including Sc, Ti, Cr, Mn, Co, Ni, Cu, Nb, Mo, Tc, Ru, Hf, Ta, W, Re, Ir, Pt and Au doped on C6N6 have been considered in this study and the screening has been carried out through these three above mentioned steps. Fig. 2(a) shows Gibbs free energy changes (ΔG) for N2 adsorption which are found to be negative for Sc, Ti, Co, Ni, Cu, Nb, Mo, Tc, Ru, Hf, Ta, W, Re, Ir, and Pt doped substrates indicating that these TM doped catalyst surfaces are suitable for N2 activation, i.e., N2 to *N2 where * denotes the substrate. In the subsequent step for first hydrogenation, except Nb, Mo, Hf, Ta, W, Re doped C6N6, all other elements are ruled out because of the large endoergocity in the reduction of *N2 to *NNH, as shown in Fig. 2(b). In order to get more insight, the d-band center of each metal on TM@C6N6 has been computed and a relationship constructed with the barrier potential of the *N2 to *NNH step, as shown in Fig. 2(c). The catalytic activity shows a volcano type dependence which suggests that the six metals namely, Mo, Nb, W, Hf, Re, and Ta based SACs locate at the top of the diagram possessing lowest barrier potential and thereby demonstrating good NRR activity. Previous studies also showed a similar trend towards N2 reduction for the metals present in the middle of each period.35,36 Here, the catalysts having a positive d-band center (∼0.00–0.80 eV) around the Fermi level exhibits superiority. This can also be manifested in Fig. S1 (ESI†), where a linear relationship is observed between the d-band center and the binding energy of N2. Though the R2 value is not good, it roughly estimates that the d-band center plays an important role in evaluating the catalytic activity. According to the d-band model as predicted by Norskov et al., with the shifting of the d-band center above the Fermi level, the antibonding states begin to appear. These antibonding states are empty and as the number increases, the bond between the adsorbate and the metal becomes stronger. It can be quantitatively demonstrated from the d-band center values, as shown in Table S4 (ESI†) that Ta has a moderately positive d-band center value indicating moderate binding with N2.37 Therefore the d-band center correlates with the adsorption energy of the intermediate, that connects with the catalytic activity of SACs. Furthermore, NRR selectivity may be hindered by the competing hydrogen evolution reaction (HER) under acidic conditions. To find out the appropriate catalyst for nitrogen reduction, the adsorption energies of *H and *N2 are compared, as shown in Fig. 2(d). The catalyst is considered to be the best catalyst for HER in which
for finding a suitable electrocatalyst for the NRR.34 A screening through these essential steps can help to find a suitable electrocatalyst for the NRR. The transition metal atoms including Sc, Ti, Cr, Mn, Co, Ni, Cu, Nb, Mo, Tc, Ru, Hf, Ta, W, Re, Ir, Pt and Au doped on C6N6 have been considered in this study and the screening has been carried out through these three above mentioned steps. Fig. 2(a) shows Gibbs free energy changes (ΔG) for N2 adsorption which are found to be negative for Sc, Ti, Co, Ni, Cu, Nb, Mo, Tc, Ru, Hf, Ta, W, Re, Ir, and Pt doped substrates indicating that these TM doped catalyst surfaces are suitable for N2 activation, i.e., N2 to *N2 where * denotes the substrate. In the subsequent step for first hydrogenation, except Nb, Mo, Hf, Ta, W, Re doped C6N6, all other elements are ruled out because of the large endoergocity in the reduction of *N2 to *NNH, as shown in Fig. 2(b). In order to get more insight, the d-band center of each metal on TM@C6N6 has been computed and a relationship constructed with the barrier potential of the *N2 to *NNH step, as shown in Fig. 2(c). The catalytic activity shows a volcano type dependence which suggests that the six metals namely, Mo, Nb, W, Hf, Re, and Ta based SACs locate at the top of the diagram possessing lowest barrier potential and thereby demonstrating good NRR activity. Previous studies also showed a similar trend towards N2 reduction for the metals present in the middle of each period.35,36 Here, the catalysts having a positive d-band center (∼0.00–0.80 eV) around the Fermi level exhibits superiority. This can also be manifested in Fig. S1 (ESI†), where a linear relationship is observed between the d-band center and the binding energy of N2. Though the R2 value is not good, it roughly estimates that the d-band center plays an important role in evaluating the catalytic activity. According to the d-band model as predicted by Norskov et al., with the shifting of the d-band center above the Fermi level, the antibonding states begin to appear. These antibonding states are empty and as the number increases, the bond between the adsorbate and the metal becomes stronger. It can be quantitatively demonstrated from the d-band center values, as shown in Table S4 (ESI†) that Ta has a moderately positive d-band center value indicating moderate binding with N2.37 Therefore the d-band center correlates with the adsorption energy of the intermediate, that connects with the catalytic activity of SACs. Furthermore, NRR selectivity may be hindered by the competing hydrogen evolution reaction (HER) under acidic conditions. To find out the appropriate catalyst for nitrogen reduction, the adsorption energies of *H and *N2 are compared, as shown in Fig. 2(d). The catalyst is considered to be the best catalyst for HER in which 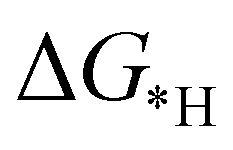 tends to its ideal value
tends to its ideal value 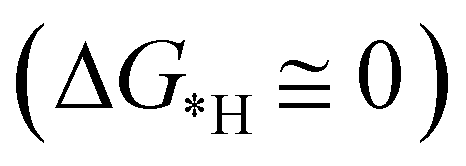 leading to the desorption of hydrogen easily from the catalyst surface either through the Tafel or the Heyrovsky step, which in turn, leaves the catalyst surface prepared for the successive adsorption of hydrogen for further reaction to proceed.38 Here, the values of
leading to the desorption of hydrogen easily from the catalyst surface either through the Tafel or the Heyrovsky step, which in turn, leaves the catalyst surface prepared for the successive adsorption of hydrogen for further reaction to proceed.38 Here, the values of 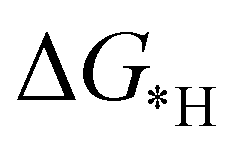 , as shown in the Fig. 2(d), vary from −0.2 eV to −0.79 eV which indicates that the H binds strongly with the catalyst surface that hinders the HER to proceed further. On the other hand, nitrogen in end-on configuration binds stronger than hydrogen that makes surface concentration of the *N2 higher, facilitating NRR to carry out further. Eventually, the positive value of
, as shown in the Fig. 2(d), vary from −0.2 eV to −0.79 eV which indicates that the H binds strongly with the catalyst surface that hinders the HER to proceed further. On the other hand, nitrogen in end-on configuration binds stronger than hydrogen that makes surface concentration of the *N2 higher, facilitating NRR to carry out further. Eventually, the positive value of 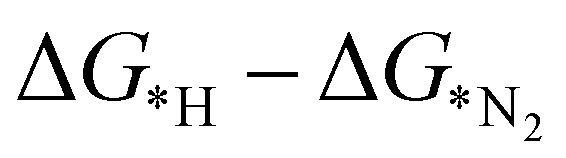 for each of the six catalysts suggest their selectivity of the NRR. Moreover, among these suitable six catalysts, the Ta based C6N6 is found to be the best having a small potential barrier ∼0.39 eV for the *N2 to the *NNH step. The same for Mo, W, Nb, Hf and Re based SACs are remarkably higher for this first coupled proton and electron transfer step. The corresponding values are 0.62, 0.60, 0.54, 0.76, and 0.61 eV respectively. Therefore, only Ta based C6N6 is studied further for the final hydrogenation step, i.e., *NH2 to *NH3, which has again a reasonable potential barrier of ∼0.30 eV. The alternating pathway has also been evaluated and the potential barrier for the rate determining step becomes 0.91 eV, and therefore neglected in this study. Having the lowest potential barrier, among the 18 different metal centers, Ta@C6N6 is found to be suitable for NRR. In order to determine the thermal stability of the Ta based catalyst, ab initio molecular dynamics (AIMD) simulations compiled using the VASP simulation package (VASP) are carried out at 300 K for a time period of 15 ps with a time step of 0.5 fs. The root-mean-square (RMSD) plot is presented in Fig. 3(a) exhibits that the fluctuations equilibrate within a few picoseconds which confirms the stability of Ta@C6N6. The free energy diagram for NRR on Ta@C6N6 in the end-on configuration is shown in Fig. 3(b). After the first hydrogenation step, the next two consecutive (H+ + e−) transfers occur on the same distal N atom with an energy release of 0.28 eV and 0.42 eV respectively, shown as downhill in the energy profile. Consequently, *NNH becomes *NNH2 and *N with the release of the first NH3. This remaining *N gets hydrogenated consecutively thrice before the release of the second NH3 molecule. The corresponding ΔG values for these steps are −0.46 eV, +0.15 eV, and +0.30 eV respectively. The optimized structures of different intermediates adsorbed on the metal center of Ta@C6N6 is presented in Fig. S2 (ESI†). The free energy changes of each step in the N2 reduction reaction have been assessed considering the solvent effect which is plotted in Fig. 3(b). It is evident that ΔG of each step changes under this condition and more negative values of ΔG imply more stabilization under aqueous conditions. The potential barrier of *N2 to *NNH is found to be ∼0.38 eV. This small change in the limiting potential has no effect on the NRR pathway and, therefore, the solvation effect is not considered further for this reduction, which is in accordance with the previous report.39
for each of the six catalysts suggest their selectivity of the NRR. Moreover, among these suitable six catalysts, the Ta based C6N6 is found to be the best having a small potential barrier ∼0.39 eV for the *N2 to the *NNH step. The same for Mo, W, Nb, Hf and Re based SACs are remarkably higher for this first coupled proton and electron transfer step. The corresponding values are 0.62, 0.60, 0.54, 0.76, and 0.61 eV respectively. Therefore, only Ta based C6N6 is studied further for the final hydrogenation step, i.e., *NH2 to *NH3, which has again a reasonable potential barrier of ∼0.30 eV. The alternating pathway has also been evaluated and the potential barrier for the rate determining step becomes 0.91 eV, and therefore neglected in this study. Having the lowest potential barrier, among the 18 different metal centers, Ta@C6N6 is found to be suitable for NRR. In order to determine the thermal stability of the Ta based catalyst, ab initio molecular dynamics (AIMD) simulations compiled using the VASP simulation package (VASP) are carried out at 300 K for a time period of 15 ps with a time step of 0.5 fs. The root-mean-square (RMSD) plot is presented in Fig. 3(a) exhibits that the fluctuations equilibrate within a few picoseconds which confirms the stability of Ta@C6N6. The free energy diagram for NRR on Ta@C6N6 in the end-on configuration is shown in Fig. 3(b). After the first hydrogenation step, the next two consecutive (H+ + e−) transfers occur on the same distal N atom with an energy release of 0.28 eV and 0.42 eV respectively, shown as downhill in the energy profile. Consequently, *NNH becomes *NNH2 and *N with the release of the first NH3. This remaining *N gets hydrogenated consecutively thrice before the release of the second NH3 molecule. The corresponding ΔG values for these steps are −0.46 eV, +0.15 eV, and +0.30 eV respectively. The optimized structures of different intermediates adsorbed on the metal center of Ta@C6N6 is presented in Fig. S2 (ESI†). The free energy changes of each step in the N2 reduction reaction have been assessed considering the solvent effect which is plotted in Fig. 3(b). It is evident that ΔG of each step changes under this condition and more negative values of ΔG imply more stabilization under aqueous conditions. The potential barrier of *N2 to *NNH is found to be ∼0.38 eV. This small change in the limiting potential has no effect on the NRR pathway and, therefore, the solvation effect is not considered further for this reduction, which is in accordance with the previous report.39
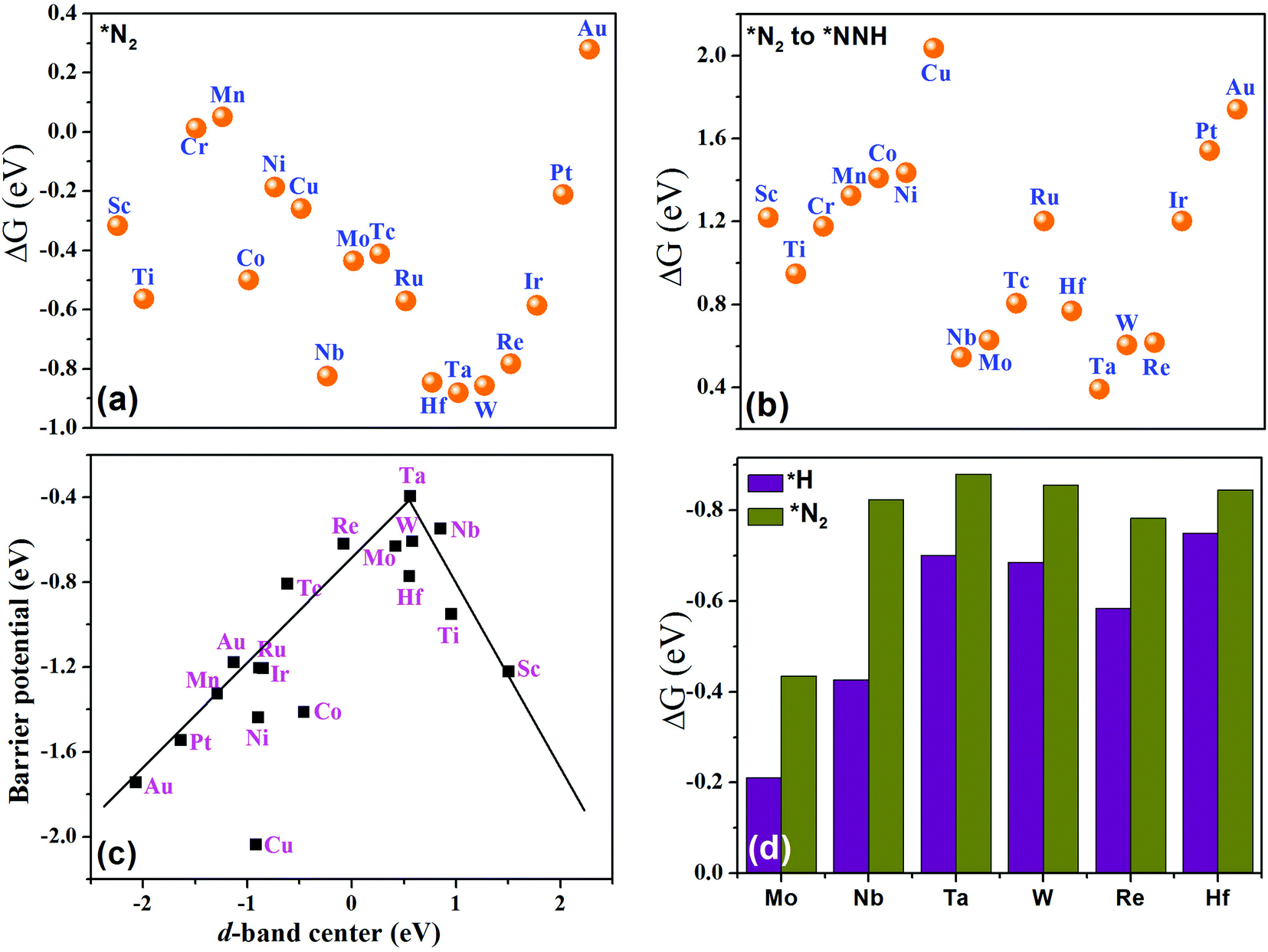 | ||
| Fig. 2 Free energy change of (a) N2 adsorption, and (b) first hydrogenation. (c) Relationship between the d-band center and the barrier potential. (d) Free energy change of the HER and NRR. | ||
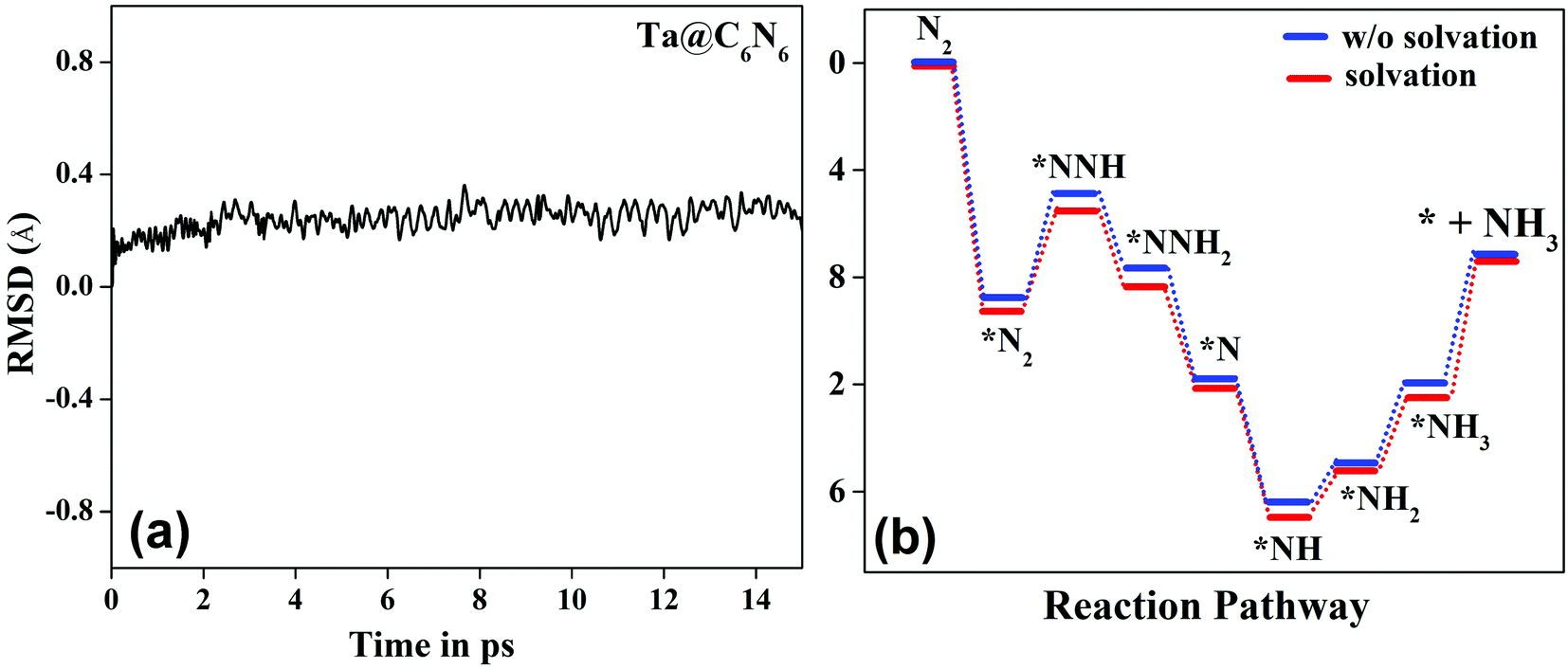 | ||
| Fig. 3 (a) AIMD plot of Ta@C6N6. (b) Free energy diagram for Ta@C6N6 at U = 0 V. | ||
In order to elucidate a correlation between the intrinsic properties of active metal center with its catalytic activity, ML has been adopted in assistance with DFT calculations. A set of features/descriptors based on the intrinsic properties of metal atoms, viz., the ionization energy (IE), electron affinity (EA), electronegativity (EN), covalent radius of the metal atom (rcov), Zunger radius (dz),40 and d-electron count (θd) have been considered as features for building the ML model to predict 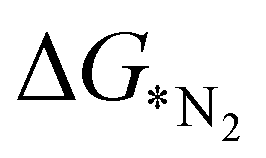 . A few additional descriptors have chosen based on the DFT calculations. As the chemical environment is important for the catalytic activity of the active site, we have considered the distance between the active metal center and nearest nitrogen (dM–N) as a salient parameter for demonstrating the ML model.41 Additionally, since the positive charge of the metal center plays a crucial role towards the selectivity of the NRR over the other competing reaction, HER, it is reasonable to consider the amount of charge transfer (q) as obtained through Bader charge analyses (in Fig. 1(c)) as an important descriptor. Moreover, Hammer et al. have suggested that the metal d-band centre (εd) acts as the key parameter to gauge the strength of the bonding of the adsorbate and therefore, we consider the d-band centre of the metal atoms to be a prime descriptor.42 These nine features for the 18 different metal atoms based C6N6 have been taken into account for training and testing in ML to predict
. A few additional descriptors have chosen based on the DFT calculations. As the chemical environment is important for the catalytic activity of the active site, we have considered the distance between the active metal center and nearest nitrogen (dM–N) as a salient parameter for demonstrating the ML model.41 Additionally, since the positive charge of the metal center plays a crucial role towards the selectivity of the NRR over the other competing reaction, HER, it is reasonable to consider the amount of charge transfer (q) as obtained through Bader charge analyses (in Fig. 1(c)) as an important descriptor. Moreover, Hammer et al. have suggested that the metal d-band centre (εd) acts as the key parameter to gauge the strength of the bonding of the adsorbate and therefore, we consider the d-band centre of the metal atoms to be a prime descriptor.42 These nine features for the 18 different metal atoms based C6N6 have been taken into account for training and testing in ML to predict 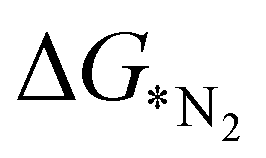 . The dataset of the features taken for this study is illustrated in Table S4 (ESI†). To analyze the strength of the correlation between each of the features and eliminate further the redundant ones, the Pearson correlation coefficient (p) has been calculated using the following relation:
. The dataset of the features taken for this study is illustrated in Table S4 (ESI†). To analyze the strength of the correlation between each of the features and eliminate further the redundant ones, the Pearson correlation coefficient (p) has been calculated using the following relation:
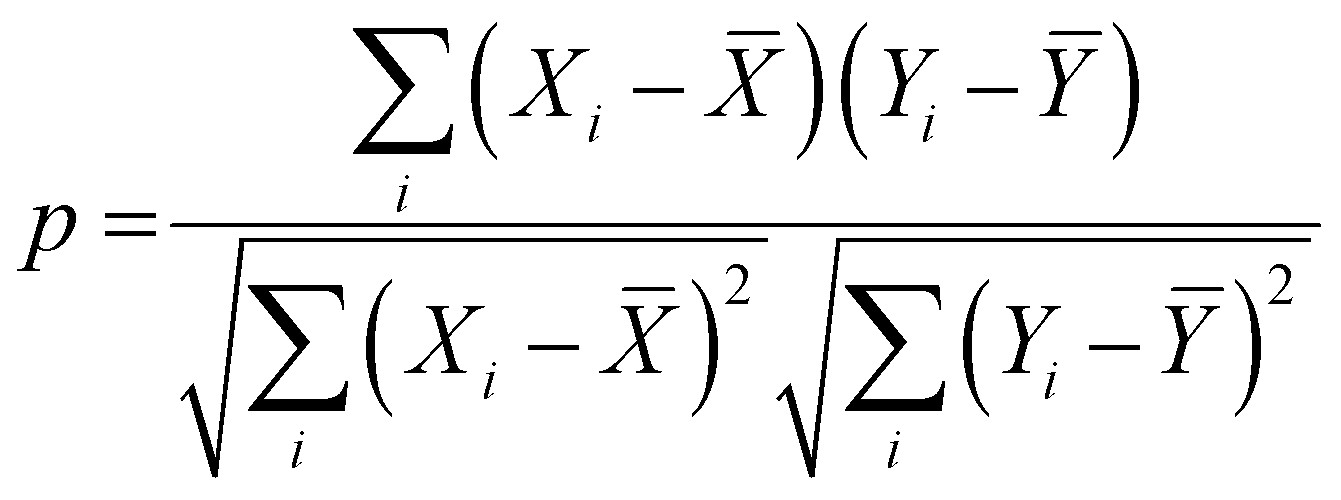 | (5) |
![[X with combining macron]](https://www.rsc.org/images/entities/i_char_0058_0304.gif) and Ȳ are their mean values respectively. The value of p may vary from −1.0 to +1.0, and a higher |p| represents a stronger correlation.
and Ȳ are their mean values respectively. The value of p may vary from −1.0 to +1.0, and a higher |p| represents a stronger correlation.
The heat map, as presented in Fig. 4, demonstrates a significant correlation between rcov and εd with θd. A previous study by Zhao et al. has shown that elimination of the redundant features can enhance the performance in ML.43 Hence, θd has been omitted here from the regression models. Initially, different regression models have been counted on for cross validation over 50 different distributions of training and test split, and their RMSE values are compared in Fig. S3 (ESI†). Having low bias and low variance, the ensemble learning algorithm namely, GBR and RFR are found to be good among all of them. Henceforth, these two models are put forward in estimating the target. These 18 metal doped C6N6 are further divided into 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 as training and testing dataset. The
:4 as training and testing dataset. The 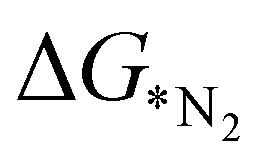 values predicted via the ML algorithm and obtained from DFT have been compared. The GBR model performs comparatively well with high R-square (R2) value (0.87) and low RMSE (0.005) value respectively, as shown in Fig. 5(a). The contribution of the features towards the prediction of
values predicted via the ML algorithm and obtained from DFT have been compared. The GBR model performs comparatively well with high R-square (R2) value (0.87) and low RMSE (0.005) value respectively, as shown in Fig. 5(a). The contribution of the features towards the prediction of 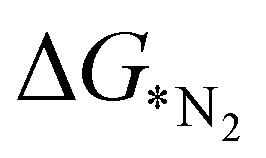 is shown in Fig. 5(b). It shows that the d-band center (εd) has the maximum feature importance ∼56.8%. Also, the covalent radius of the atom has a significant importance (17.7%). The contribution of other features is relatively low towards the target properties. By using the mutual information method as shown in Fig. 5(c), we have found that the d-band center become one of the most determining factor.
is shown in Fig. 5(b). It shows that the d-band center (εd) has the maximum feature importance ∼56.8%. Also, the covalent radius of the atom has a significant importance (17.7%). The contribution of other features is relatively low towards the target properties. By using the mutual information method as shown in Fig. 5(c), we have found that the d-band center become one of the most determining factor.
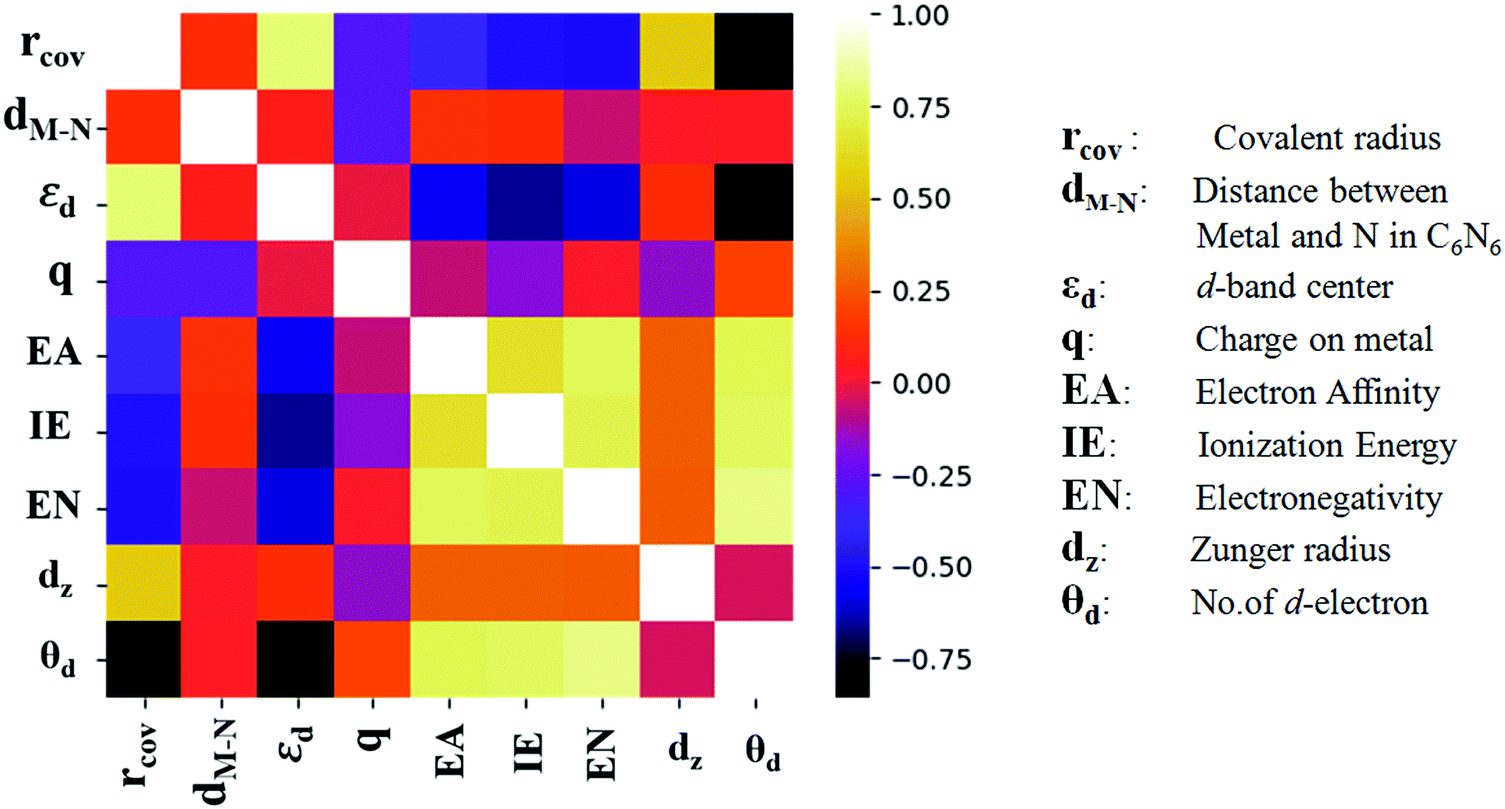 | ||
| Fig. 4 Heat map of Pearson correlation coefficients. | ||
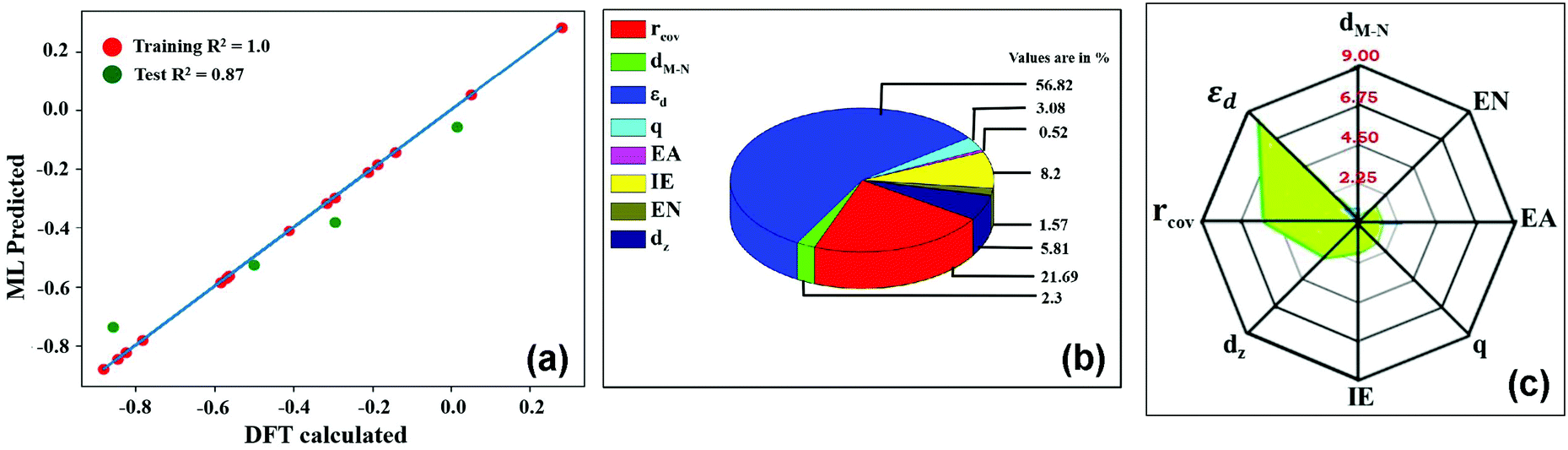 | ||
Fig. 5 (a) Comparison of 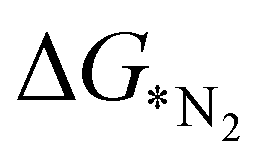 values (in eV) as predicted from ML and calculated from DFT. (b) Relative importance of the features to predict values (in eV) as predicted from ML and calculated from DFT. (b) Relative importance of the features to predict 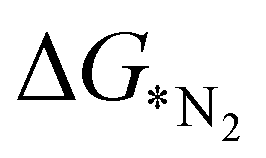 using the GBR model. (c) important features predicted by mutual information. using the GBR model. (c) important features predicted by mutual information. | ||
In addition to 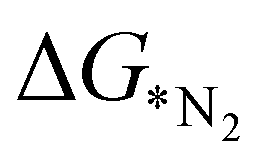 , the first hydrogenation step,
, the first hydrogenation step, 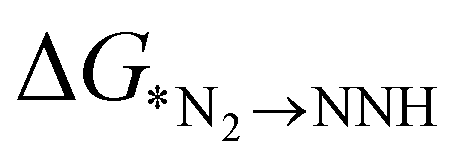 plays a significant role towards prediction of efficient electrocatalysts. The ensemble-based classification model using the soft voting technique has been considered to predict the best catalyst with such a small dataset. The main goal of our classification model is to produce a probabilistic score in search of the efficient catalysts based on important key steps. Additionally, we have compared the results predicted via ML with that via DFT. Based on the approach proposed by Ling. et al., we adopted the two key step method, i.e, Gibbs free energy of nitrogen binding and the first protonation step to find out the active electrocatalyst.32 The cut-off barrier for these steps have been set as
plays a significant role towards prediction of efficient electrocatalysts. The ensemble-based classification model using the soft voting technique has been considered to predict the best catalyst with such a small dataset. The main goal of our classification model is to produce a probabilistic score in search of the efficient catalysts based on important key steps. Additionally, we have compared the results predicted via ML with that via DFT. Based on the approach proposed by Ling. et al., we adopted the two key step method, i.e, Gibbs free energy of nitrogen binding and the first protonation step to find out the active electrocatalyst.32 The cut-off barrier for these steps have been set as
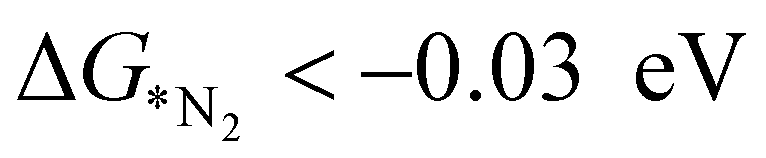
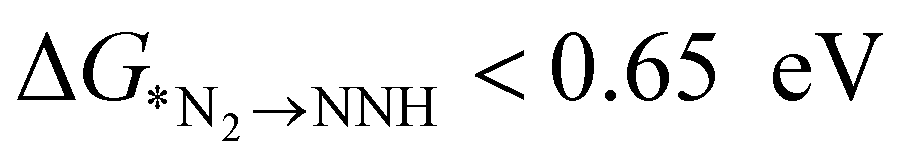
The catalysts fulfilling the above criteria have been considered as an efficient catalyst and labeled as class 1; otherwise, it is considered as an inefficient catalyst and designated as class 0. From the DFT computation, out of 18 SACs, 6 catalysts (Mo, Nb, W, Re, Hf, and Ta) are labeled as class 1 and rest of them belonged to the class 0 category. Thus, we have framed our problem statement as a simple binary classification problem thereby enabling us to apply ML based classification methods. Here, we proposed ensemble models using logistic regression, support vector machine and random forest classifier and also apply the cross validation strategy with a splitting ratio of 14:4 on the total dataset. The best outcome has been obtained by bagging three classification models using the soft voting principle. The schematic flowchart of the ML process in the classification model is shown in Fig. S4 (ESI†). The atomic descriptors such as ionization energy, electronegativity, electron affinity, covalent radius of the metal atom, and Zunger radius are used to get a high probability score for the best catalyst. In the soft voting scenario, every individual classifier provides the probability score for the catalysts which are then taken as input and voting is calculated based on this probability to ensure the class in which the specific catalyst belongs. We have applied grid search cross validation strategy on each individual classification model to tune the required hyper-parameters. In order to train our classification model using cross-validation, 10 different pairs of training and test sets using random sampling without replacement have been taken from the complete dataset (shown in Table S4, ESI†). Each time, we compare the probability score (calculated using eqn (6)) of the predicted class based on our model with that of the result obtained from DFT.
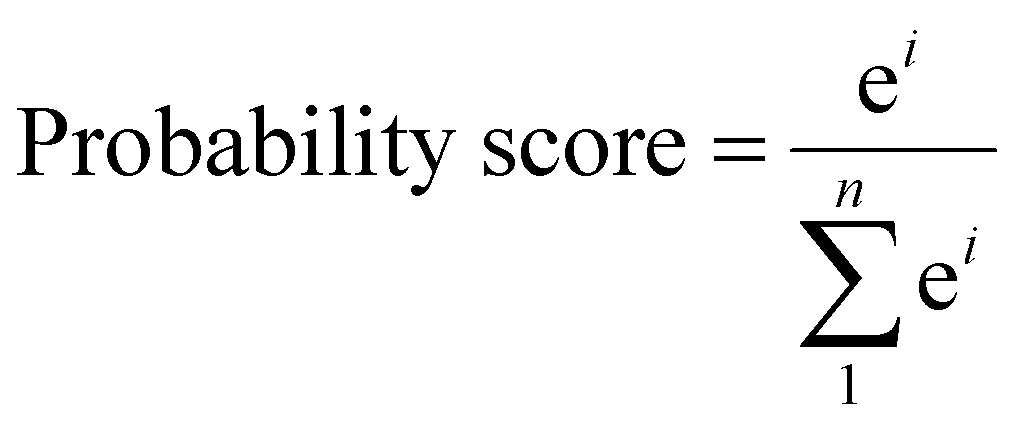 | (6) |
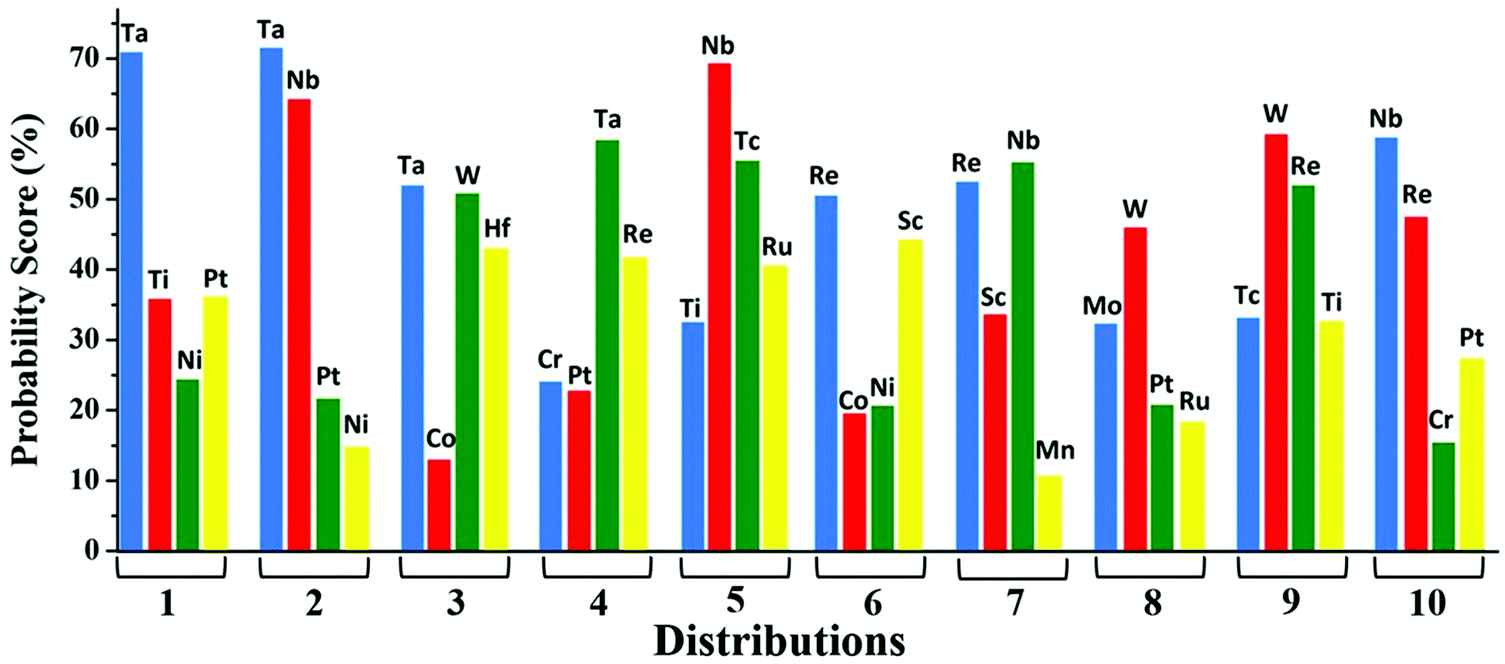 | ||
| Fig. 6 Probabilistic distributions for SAC at C6N6 based on the soft-voting technique. | ||
4. Conclusions
In conclusion, a series of single atom doped C6N6 substrates have been chosen with the intention of finding out the best catalyst for N2 reduction towards the production of NH3. A systematic investigation performed using DFT based on the first hydrogenation step reveals that the Ta based C6N6 is a superior catalyst amongst the 18 SACs. Complementing this approach, ensemble-based machine learning algorithms aided by a regression model and a soft voting technique in classification model have been utilized as well. The descriptors for the regression model have been chosen from structural and electronic properties as well as the properties of metal centers that have been obtained directly from the periodic table. Also, based on the DFT generated dataset for the d-band center and model training, the GBR model shows the best prediction accuracy with R2 = 0.87. On the other hand, considering the intrinsic properties of metals like the electronegativity, ionization energy, covalent radius, Zunger radius and d-electron count, the probabilistic score is found in the order Ta > W > Nb > Mo which is in harmony with the DFT results. The classification model successfully predicts the best catalyst. The initialization of the key intermediate step can be predicted using the above intrinsic descriptors that greatly reduces the computational cost. We believe that a combined approach of DFT and various ML models can unravel highly effective catalysts for other important reactions.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
M. M. acknowledges financial support from the Council of Scientific and Industrial Research (CSIR), India, vide order no.: 09/080(1152)/2020-EMR-I. A. D. thanks TRC-DST and SERB grant no. DIA/2018/000013 and CRG/2020/000301 for partial funding.References
- D. E. Canfield, A. N. Glazer and P. G. Falkowski, The evolution and future of Earth's nitrogen cycle, Science, 2010, 330, 192 CrossRef CAS PubMed.
- R. Schlögl, Catalytic synthesis of ammonia-a “never-ending story”?, Angew. Chem., Int. Ed., 2003, 42, 2004 CrossRef PubMed.
- H. P. Jia and E. A. Quadrelli, Mechanistic aspects of dinitrogen cleavage and hydrogenation to produce ammonia in catalysis and organometallic chemistry: Relevance of metal hydride bonds and dihydrogen, Chem. Soc. Rev., 2014, 43, 547 RSC.
- B. K. Burgess and D. J. Lowe, Mechanism of molybdenum nitrogenase, Chem. Rev., 1996, 96, 2983 CrossRef CAS PubMed.
- K. Arashiba, E. Kinoshita, S. Kuriyama, A. Eizawa, K. Nakajima, H. Tanaka, K. Yoshizawa and Y. Nishibayashi, Catalytic reduction of dinitrogen to ammonia by use of molybdenum–nitride complexes bearing a tridentate triphosphine as catalysts, J. Am. Chem. Soc., 2015, 137, 5666 CrossRef CAS PubMed.
- X. Cui, C. Tang and Q. Zhang, A Review of electrocatalytic reduction of dinitrogen to ammonia under ambient conditions, Adv. Energy Mater., 2018, 8, 1800369 CrossRef.
- V. Kyriakou, I. Garagounis, E. Vasileiou, A. Vourros and M. Stoukides, Progress in the electrochemical synthesis of ammonia, Catal. Today, 2017, 286, 2 CrossRef CAS.
- A. D. Allen and C. V. Senoff, Nitrogenopentammineruthenium(II) complexes, Chem. Commun., 1965, 621 RSC.
- K. Bhattacharyya and A. Datta, Visible light driven efficient metal free single atom catalyst supported on nanoporous carbon nitride for nitrogen fixation, Phys. Chem. Chem. Phys., 2019, 21, 12346 RSC.
- X. Ma, J. Hu, M. Zheng, D. Li, H. Lv, H. He and C. Huang, N2 reduction using single transition-metal atom supported on defective WS2 monolayer as promising catalysts: A DFT study, Appl. Surf. Sci., 2019, 489, 684 CrossRef CAS.
- S. H. Talib, Z. Lu, B. Bashir, S. Hussain, K. Ahmad, S. Khan, S. Haider, Z. Yang, K. Hermansson and J. Li, CO oxidation on MXene (Mo2CS2) supported single-atom catalyst: A termolecular Eley-Rideal mechanism, Chin. Chem. Lett., 2022 DOI:10.1016/j.cclet.2022.04.010.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Single-atom catalysis of CO oxidation using Pt1/FeOx, Nat. Chem., 2011, 3, 634 CrossRef CAS PubMed.
- S. Wang, W. Wei, X. Lv, B. Huang and Y. Dai, W supported on g-CN manifests high activity andselectivity for N2 electroreduction to NH3, J. Mater. Chem. A, 2011, 3, 634 CrossRef PubMed.
- M. D. Zott, P. G. Barros and J. Peters, Electrocatalytic ammonia oxidation mediated by a polypyridyl iron catalyst, ACS Catal., 2019, 9, 3419 CrossRef.
- C. Liu, Q. Li, J. Zhang, Y. Jin and D. R. MacFarlane, Conversion of dinitrogen to ammonia on Ru atoms supported on boron sheets: A DFT study, J. Mater. Chem. A, 2019, 7, 4771 RSC.
- A. Chen, X. Zhang, L. Chen, S. Yao and Z. Zhou, A machine learning model on simple features for CO2 reduction electrocatalysts, J. Phys. Chem. C, 2020, 124, 22471 CrossRef CAS.
- B. Hoar, S. Lu and C. Liu, Machine-learning-enabled exploration of morphology influence on wire-array electrodes for electrochemical nitrogen fixation, J. Phys. Chem. Lett., 2020, 11, 4625 CrossRef CAS PubMed.
- L. Ge, W. Xu, C. Chen, C. Tang, L. Xu and Z. Chen, Rational prediction of single metal atom supported on two-dimensional metal diborides for electrocatalytic N2 reduction reaction with integrated descriptor, J. Phys. Chem. Lett., 2020, 11, 5241 CrossRef CAS PubMed.
- C. Deng, Y. Su, F. Li, W. Shen, Z. Chen and Q. Tang, J. Mater. Chem. A, 2020, 8, 24563 RSC.
- F. Bagherzadeh, M. J. Mehrani, M. Basirifard and J. Roostaei, Comparative study on total nitrogen prediction in wastewater treatment plant and effect of various feature selection methods on machine learning algorithms performance, J. Water Process. Eng., 2021, 41, 102033 CrossRef.
- M. Kim, B. C. Yeo, Y. Park, H. M. Lee, S. S. Han and D. Kim, Artificial intelligence to accelerate the discovery of N2 electroreduction catalysts, Chem. Mater., 2020, 32, 709 CrossRef CAS.
- M. Zafari, D. Kumar, M. Umer and K. S. Kim, Machine learning-based high throughput screening for nitrogen fixation on boron-doped single atom catalysts, J. Mater. Chem. A, 2020, 8, 5209 RSC.
- M. H. Alobaidi, M. A. Meguid and F. Chebana, Predicting seismic-induced liquefaction through ensemble learning frameworks, Sci. Rep., 2019, 9, 11786 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed.
- C. Chowdhury and A. Datta, Silicon-doped nitrogen-coordinated graphene as electrocatalyst for oxygen reduction reaction, J. Phys. Chem. C, 2018, 122, 27233 CrossRef CAS.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss and V. Dubourg, Scikit-learn: Machine learning in python, J. Mach. Learn. Res., 2011, 12, 2825 Search PubMed.
- J. H. Friedman, Greedy function approximation: A gradient boosting machine, Ann. Stat., 2001, 29, 1189 Search PubMed.
- T. K. Ho, Random decision forests, Proceedings of 3rd international conference on document analysis and recognition, 1995, 1, 278.
- D. R. Cox, The regression analysis of binary sequences, J. R. Stat. Soc. Series B Stat. Methodol., 1958, 20, 215 Search PubMed.
- C. Cortes and V. Vapnik, Support-vector networks, Mach. Learn., 1995, 20, 273 Search PubMed.
- J. H. Montoya, C. Tsai, A. Vojvodic and J. K. Nørskov, The challenge of electrochemical ammonia synthesis: A new perspective on the role of nitrogen scaling relations, ChemSusChem, 2015, 8, 2180 CrossRef CAS PubMed.
- C. Ling, Y. Ouyang, Q. Li, X. Bai, X. Mao, A. Du and J. A. Wang, General two-step strategy–based high-throughput screening of single atom catalysts for nitrogen fixation, Small Methods, 2019, 3, 1800376 CrossRef.
- Z. Chen, J. Zhao, C. R. Cabrera and Z. Chen, Computational screening of efficient single-atom catalysts based on graphitic carbon nitride (g-C3N4) for nitrogen electroreduction, Small Methods, 2019, 3, 1800368 CrossRef.
- X. Liu, Y. Jiao, Y. Zheng, M. Jaroniec and S.-Z. Qiao, Building up a picture of the electrocatalytic nitrogen reduction activity of transition metal single atom catalysts, J. Am. Chem. Soc., 2019, 141, 9664 CrossRef CAS PubMed.
- B. Hammer and J. K. Nørskov, Electronic factors determining the reactivity of metal surfaces, Surf. Sci., 1995, 343, 211 CrossRef CAS.
- S. H. Talib, X. Yu, Z. Lu, K. Ahmad, T. Yang, H. Xiao and J. Li, A polyoxometalate cluster-based single-atom catalyst for NH3 synthesis via an enzymatic mechanism, J. Mater. Chem. A, 2022, 10, 6165 RSC.
- X. Guo, J. Gu, S. Lin, S. Zhang, Z. Chen and S. Huang, Tackling the activity and selectivity challenges of electrocatalysts toward the nitrogen reduction reaction via atomically dispersed biatom catalysts, J. Am. Chem. Soc., 2020, 142, 5709 CrossRef CAS PubMed.
- A. Zunger, Systematization of the stable crystal structure of all AB-type binary compounds: A pseudopotential orbital-radii approach, Phys. Rev. B: Condens. Matter Mater. Phys., 1980, 22, 5839 CrossRef CAS.
- T. A. A. Batchelor, J. K. Pedersen, S. H. Winther, I. E. Castelli, K. W. Jacobsen and J. Rossmeisl, High-entropy alloys as a discovery platform for electrocatalysis, Joule, 2019, 3, 834 CrossRef CAS.
- B. Hammer, Y. Morikawa and J. K. Nørskov, Co chemisorption at metal surfaces and overlayers, Phys. Rev. Lett., 1996, 76, 2141 CrossRef CAS PubMed.
- X. Zhao, L. Wang and Y. Pei, Single metal atom catalyst supported on g-C3N4 for formic acid dehydrogenation: A combining density functional theory and machine learning study, J. Phys. Chem. C, 2021, 125, 22513 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: List of hyperparameters of the regression model, adsorption energy of nitrogen in the end-on mode and side-on mode, feature statistics of the dataset considered for machine learning method, schematic diagram of work-flow for the classification model, cross validation result for different classification models. See DOI: https://doi.org/10.1039/d2cp01901a |
| This journal is © the Owner Societies 2022 |