
Nature of excited-state dependent hydrogen bonds and their critical role in determining the photophysical properties of aromatic thioketones†
Ye-Guang
Fang
ab and
Wei-Hai
Fang
 *a
*a
aKey Laboratory of Theoretical and Computational Photochemistry, Ministry of Education, College of Chemistry, Beijing Normal University, Beijing 100875, P. R. China. E-mail: fangwh@bnu.edu.cn
bLaboratory of Theoretical and Computational Nanoscience, CAS Key Laboratory of Nanosystem and Hierarchical Fabrication, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing 100190, P. R. China
First published on 24th June 2022
Abstract
In this work, how the excited-state dependent hydrogen bond (H-bond) interactions control photophysical processes have been uncovered by accurate electronic structure calculations for the five lowest-lying states (S0, S1, S2, T1, and T2) of three aromatic thioketones and their isomers. The difference in the H-bond nature between S2 and S1 gives rise to ultrafast S2 → S1 internal conversion via the two-state conical intersection. Strong S2 fluorescence observed usually in thiocarbonyl compounds is absent in aromatic thioketones with intramolecular H-bonds. Meanwhile, the relatively weak H-bond interactions in S1 and T1 states make the S1, T2, and T1 states degenerate or quasi-degenerate. As a result, the T2 state acts as a relay and enables both forward S1 → T1 and reverse T1 → S1 processes to occur efficiently, which provides new insights into the mechanism of thermally activated delayed fluorescence (TADF), and could be used to improve the design principle of purely organic TADF materials.
1. Introduction
Since molecular photochemistry emerged as a discipline, carbonyl compounds have always been used as model molecules to explore the fundamental processes and general rules in the discipline. Consequently, the carbonyl compounds have their photophysics and photochemistry studied probably in more detail than any other functional groups.1–9 Thiocarbonyl compounds (also known as thioketones or thioaldehydes), where the carbonyl oxygen is replaced by sulfur, have been the subject of extensive experimental studies.10–20 The results obtained from these experimental studies have revealed that thiocarbonyl compounds possess unique photophysical properties that are different from the properties of their parent compounds and other classes of organic compounds. Like their oxygen analogues, thiocarbonyl compounds can serve as suitable model molecules for exploring the basic principles of molecular photophysics and photochemistry.Upon irradiation of a thioketone in the near-ultraviolet (UV) region, the subsequent photophysical processes involve a minimum of the lowest-lying five electronic states (S0, S1, S2, T1, and T2). The excited states of S2, S1, T2, and T1 originate from the π → π* and n → π* excitations.17,20 As the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond is weaker than the CO bond, the n → π* excited states (S1 and T1) of a thioketone have relatively low excitation energies, as compared with those for the parent ketone. As a result, the energy gap between S2(1ππ*) and S1(1nπ*) is relatively large, which is usually comparable to that between S1 and S0. Consequently, the S2 → S1 internal conversion (IC) is relatively slow for thioketones in the gas phase or in inert media.12–14,18,20 The large energy gap between S2 and S1 is the crucial factor responsible for significant S2 fluorescence and chemical reactions starting from the S2 state, in violation of Kasha's rule.
S bond is weaker than the CO bond, the n → π* excited states (S1 and T1) of a thioketone have relatively low excitation energies, as compared with those for the parent ketone. As a result, the energy gap between S2(1ππ*) and S1(1nπ*) is relatively large, which is usually comparable to that between S1 and S0. Consequently, the S2 → S1 internal conversion (IC) is relatively slow for thioketones in the gas phase or in inert media.12–14,18,20 The large energy gap between S2 and S1 is the crucial factor responsible for significant S2 fluorescence and chemical reactions starting from the S2 state, in violation of Kasha's rule.
Irrespective of the S1, S2, or higher excited state populated by photoexcitation, the T1(3nπ*) phosphorescence has been always observed in most of the thioketones even at room temperature.10,11,16,17,19–25 Such a spectroscopic feature has been generally ascribed to the heavy-atom (sulfur) effect that significantly increases the rate constant of forward S1 → T1 intersystem crossing (F-ISC). On the other hand, thermally activated delayed fluorescence (TADF) from the S1 state has been observed for pyranthione (PT), benzopyranthione (BPT), xanthione (XT), and 2,2,3,3-tetramethylindanethione (TMIT) in different solvents.15,20 Reverse T1 → S1 intersystem crossing (R-ISC) is the pre-step of the TADF emission. Meanwhile, thermally activated phosphorescence has also been observed in the alicyclic thioketones of adamantanethione, thiofenchone, and thiocamphor.20,26 The phenomenon of thermally activated emission should be one of the striking features observed in thioketones. Because the radiative and nonradiative processes originating from the S1 state are extremely sensitive to intermolecular or intramolecular hydrogen bond (H-bond) interactions, experimentally, it is difficult to explore the underlying mechanism, which has never been investigated from the viewpoint of theory.
There is a large difference in the electronegativities of the O and S atoms. Hence, it could be hypothesized that the H-bond interaction between the carbonyl oxygen and H–X (X = O, N,…) is much stronger than that between the thiocarbonyl sulfur and H–X. However, the results from the experimental studies conclusively proved that the CS⋯H–O H-bond interactions have an important influence on the absorption and emission spectra of aromatic thioketones in methanol or water.27–30 The maxima of the S2 and S1 fluorescence bands were observed to be significantly red- and blue-shifted, respectively, due to the formation of H-bond complexes between BPT and H2O, which were supported by the ab initio calculated relative energies of the S2 and S1 states.27–29 Recently, various spectroscopic techniques, together with density functional theory (DFT) and the time-dependent DFT (TDDFT) method, have been used to investigate photophysical processes of 7-hydroxy-2,2-dimethyl-3,3-dihydro-1H-indene-1-thione (DM-7HIT).30 The unique excited-state behaviour of intramolecular H-bonds (CS⋯H–O) was determined to be responsible for dual room-temperature phosphorescence of DM-7HIT. It has been experimentally proven that the intermolecular and intramolecular H-bond interactions affect the S2 deactivation and the T1 phosphorescence processes of aromatic thioketones. As far as we know, the essential differences of the H-bond interactions between the ground and different excited states have not been studied up to date.
As pointed out by Maciejewski and Steer, “A detailed understanding of the photochemistry and photophysics of any system must be based on spectroscopic measurements and theoretical calculations”.20 Numerous experimental studies have been conducted on the photophysical processes of aromatic thioketones. To the best of our knowledge, there is a single report on the theoretical study of the H-bond complex between an aromatic thioketone and methanol. The structures and energies of the H-bonds and the CS and H–O stretching frequencies were determined by the DFT and TDDFT calculations.27 As a complement to experimental studies, the DFT and TDDFT methods have been used to characterize electronic structures and relative energies of excited states.30 On the basis of the DFT or TDDFT optimized structures, the single-point energies were calculated by means of the CISD and CCSD electronic correlation methods.28,29 It should be pointed out that almost all the density functionals used up to now are developed initially for the ground state rather than an excited state. The applicability of the TDDFT method to describe the excited-state properties of a molecule depends on the functionals used and the system under study. Even though the CISD, CCSD, and other single-reference-based methods are reliable for a description of a molecular system in the ground state, they usually failed to describe the excited-state properties.
In this work, aromatic thioketones of 2HTP (2-hydroxy-pivalo-thiophenone), 7HIT (7-hydroxy-2,2,3,3-tetrahydro-1H-indene-1-thione), DM-7HIT and their non-H-bonded isomers (DM-7HIT-iso, 7HIT-iso, and 2HTP-iso), as shown in Fig. 1, were taken as representatives. Their excited-state structures and properties have been explored using the multistate complete active space second-order perturbation theory (MS-CASPT2) and the extended MS-CASPT2 (XMS-CASPT2) method. It has been found that relatively weak H-bond interactions in S1 and T1 states make the S1, T2, and T1 states quasi-degenerate. The three states were discovered to intersect with each other, which could be a common feature of a wide variety of aromatic thioketones. Furthermore, the T2(3ππ*) state functions as a bridge and enables the F-ISC and R-ISC processes to occur effectively, which could play a decisive role in the generation of thermally activated delayed fluorescence (TADF) from the S1(1nπ*) state. In addition, this is the first report that describes the conical intersection between S2 and S1 of aromatic thioketones where intramolecular H-bonds are present, which could lead to ultrafast S2 → S1 internal conversion. Finally, we would like to emphasize that the essential differences in the nature of the intramolecular H-bonds between the ground and various excited states, and their critical role in determining the photophysical properties of aromatic thioketones were exclusively explored in the present work, which are relevant to aromatic thioketones in the inert media.
 | ||
| Fig. 1 Molecular structures of the aromatic thioketones studied in this work. | ||
2. Results and discussion
2.1 Minimum-energy structures and their H-bond dependent properties
Excited-state structures and their relative energies are basic but very important factors that help understand the photophysical properties of a molecule. However, there is a general lack of information on the excited-state structures of aromatic thioketones. Therefore, the minimum-energy structures of 2HTP, 7HIT, DM-7HIT, 2HTP-iso, 7HIT-iso, and DM-7HIT-iso in the lowest-lying five electronic states were determined herein, which were referred to as S0-min, S1-min, S2-min, T1-min, and T2-min hereafter. A lot of test MS-CASPT2 calculations have been done for 7HIT in the five states through the use of cc-pVDZ, aug-cc-pVDZ, and cc-pVTZ basis sets. Meanwhile, the active space has been reasonably selected, which includes twelve electrons distributed in ten orbitals (12,10) for the MS-CASPT2 optimization of the ground- and excited-state structures. The MS-CASPT2(12,10) calculated results have proven that structures and energies of 7HIT in the five states exhibit a small dependence on the basis sets used. Thus, the MS-CASPT2(12,10) optimizations have been performed with the cc-pVDZ basis set for the aromatic thioketones under study, referred to as MS-PT2(12,10)-DZ hereafter. The computational details and the active orbitals have been presented in the ESI.†As shown in Fig. 2, the S1 (T1) state of 7HIT originates from the n → π* one-electron excitation, which is mainly localized in the thiocarbonyl group. As a result, the C–S π bond is nearly broken in the S1-min or T1-min structure, and the C–S bond is considerably elongated from S0-min to S1-min or T1-min, which can be seen from the bond parameters listed in Table 1. It is generally accepted that the intramolecular H-bond is strong, when the H⋯S distance is short and the O–H⋯S arrangement is quasi-linear for 2HTP, 7HIT, and DM-7HIT in different states. Additionally, the O–H bond elongation and the C–O bond shrinkage are favourable for the formation of strong H-bonds. As listed in Table 1, the H⋯S distance is significantly increased, while the O–H bond length and the O–H⋯S angle are considerably decreased in the S1-min and T1-min structures of 7HIT, as compared with those in the S0-min structure. These changes in the structure show evidence that the intramolecular H-bond in S1-min or T1-min is much weaker than that in S0-min.
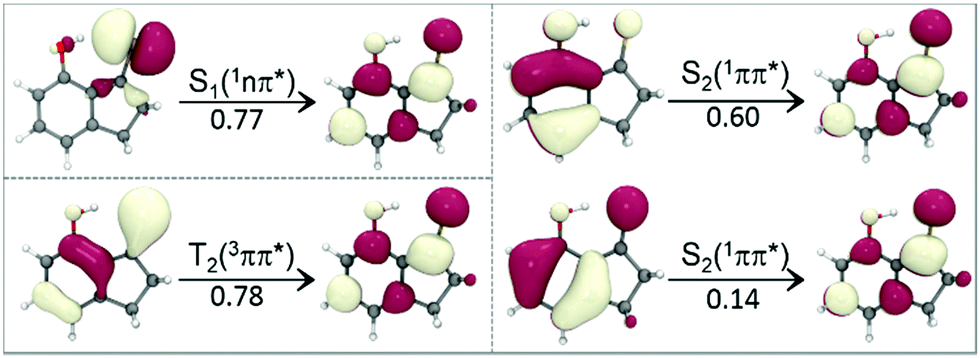 | ||
| Fig. 2 The dominant single- or double-excitation electronic configurations in the wavefunctions of the S1-min, T2-min and S2-min structures of 7HIT. | ||
C) in the MS-PT2(12,10)-DZ optimized S0-min, S1-min, S2-min, T1-min, T2-min, MECI-S2/S1, and MEI-S1/T2/T1 structures of 7HIT and 7HIT-iso
| S0-min | S1-min | S2-min | T1-min | T2-min | MECI-S2/S1 | MEI-S1/T2/T1 | ||
|---|---|---|---|---|---|---|---|---|

|
C–O | 1.335 | 1.367 | 1.308 | 1.369 | 1.347 | 1.295 | 1.354 |
| O–H | 0.996 | 0.969 | 1.122 | 0.968 | 0.982 | 1.084 | 0.981 | |
| H⋯S | 2.126 | 2.420 | 1.800 | 2.441 | 2.243 | 1.822 | 2.292 | |
| CS |
1.672 | 1.741 | 1.719 | 1.725 | 1.769 | 1.717 | 1.775 | |
| O–H⋯S | 155.0 | 149.0 | 162.0 | 148.0 | 154.0 | 161.0 | 151.0 | |

|
C–O | 1.353 | 1.369 | 1.332 | 1.371 | 1.359 | 1.342 | 1.359 |
| O–H | 0.970 | 0.969 | 0.972 | 0.969 | 0.970 | 0.971 | 0.973 | |
| CS |
1.658 | 1.741 | 1.686 | 1.723 | 1.784 | 1.662 | 1.762 | |
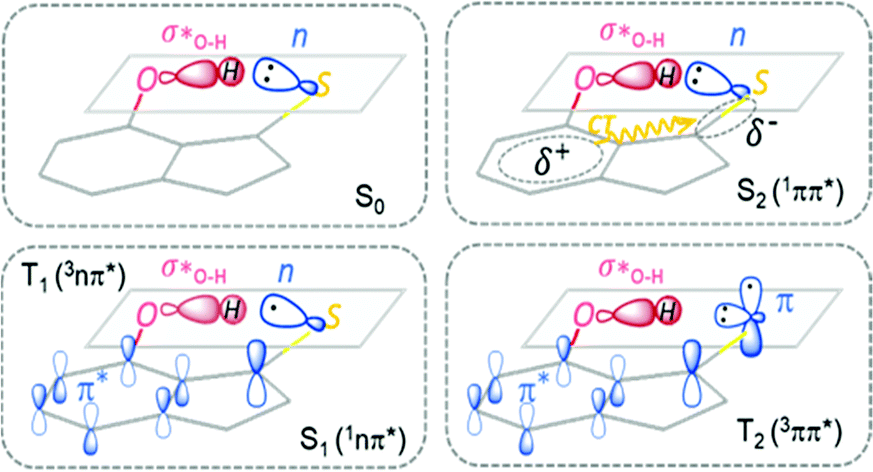
After the n → π* excitation, one unpaired electron remains at the hybrid sp2 orbital of the S atom, while another unpaired electron in the π* orbital is distributed in the aromatic ring with a large probability on the thiocarbonyl C atom. The distribution of the two unpaired electrons in the S1 or T1 state of 7HIT is schematically shown in Fig. 3, which is helpful for qualitatively understanding the origin of the excited-state dependent H-bond interaction. From the perspective of valence bond theory, the H-bond (CS⋯H–O) interaction in the S0 state originates mainly from the donation of the lone-pair electrons in the hybrid sp2 orbital of the S atom to the O–H σ* anti-bonding orbital. In the S1 and T1 states of 7HIT, however, only one electron is populated in the sp2 orbital. The decrease of electron number in the sp2 orbital is the main reason why the H-bond interaction is weaker in the S1-min and T1-min structures, as compared with that in the S0-min geometry.
 | ||
| Fig. 3 Schematic description of the electronic distributions on the n, π, and π* orbitals for the S0, S1(T1), T2, and S2 states of 7HIT, along with the CT characteristics of the S2 state. | ||
The T2(3ππ*) state of a thiocarbonyl compound is optically dark. Photophysical data obtained experimentally for this state are scarce. The T2(3ππ*) state of aromatic thioketones has never been investigated theoretically. Like the S1 or T1 state, the T2(3ππ*) state is of biradical character. As shown in Fig. 2 and 3, one unpaired π electron in the optimized T2-min structure of 7HIT is localized at the pz orbital of the S atom (the x–y plane is defined as the symmetric plane), which is different from the T1 or S1 state, where this unpaired electron is distributed in the hybrid sp2 orbital. Another unpaired π electron is delocalized into the aromatic ring, and the probability of this unpaired electron distributed in the thiocarbonyl C atom was estimated to be ∼0.6 from the calculated spin-density distribution in the T2-min structure (see the ESI,† Fig. S14). It should be pointed out that the distribution of electrons on the S atom is almost unchanged in going from S0-min to T2-min. Accordingly, the nature of the intramolecular H-bond in the T2 state is basically the same as that in the S0 state, although redistribution of π electrons from S0 to T2 could have a slight influence on the H-bond interaction of the T2 state.
From the perspective of valence bond theory, the intramolecular H-bond interaction in the S2 state is almost the same as that in the S0 state, because the lone-pair electrons are still distributed in the hybrid sp2 orbital. However, the Coulomb-repulsion interaction of the two unpaired electrons in the spatially overlapping π and π* orbitals makes them migrate partially to the thiocarbonyl group from the aromatic ring. The MS-PT2(12,10)-DZ calculations show that the double-excitation electronic configurations play a dominant role in the wavefunction of the S2 state, as shown in Fig. 2, which evidences that the S2 state is of the charge-transfer (CT) character. As a result, the electrostatic interaction has important contributions to the intramolecular H-bond (CS⋯H–O) of the S2 state. Thus, the H-bond of the S2 state, which contains both classical electrostatic and covalent chemical interactions, is much stronger than that of the S0 state for 2HTP, 7HIT, and DM-7HIT. The unusual H-bond nature of the S2 state makes the bond parameters of S2-min significantly different from those of T2-min, T1-min, and S1-min, as listed in Table 1.
The molecular orbital approximation has been adopted to understand qualitatively the differences in the intramolecular H-bond interactions between the ground and various excited states. Further evidence for the differences in the H-bond nature comes from the MS-PT2(12,10)-DZ calculated excited-state relative energies and their H-bond dependence, which will be discussed below.
2.2 The excited-state relative energies and their H-bond dependence
The MS-PT2(12,10)-DZ calculated adiabatic excitation energies from S0 to S1, S2, T1, and T2 are listed in Table 2 for the optimized structures of three aromatic thioketones and their non-bonded isomers. First, the energy differences between S1-min and T1-min have been found to be relatively small, which are in the range of 0.12–0.17 eV for the aromatic thioketones with and without the intramolecular H-bond. As pointed out before, both S1(1nπ*) and T1(3nπ*) states originate from the n → π* one-electron excitation. From a simple two-electron model, the energy difference (ΔE) between S1 and T1 is estimated to be equal to the exchange energy of the two unpaired electrons (Kn,π*),31i.e., ΔE ≈ Kn,π*. Because the n and π* orbitals are well separated in space, the electron-exchange energy is very small for the two unpaired electrons distributed in the n and π* orbitals. This is the main reason why the energy difference between S1 and T1 is small for the aromatic thioketones with and without the intramolecular H-bond. Actually, the S1(1nπ*) and T1(3nπ*) states were determined to be close to each other in energy for carbonyl compounds.2,3,5 However, it should be noted that the relative energies of S1-min and T1-min for 2HTP, 7HIT, and DM-7HIT are obviously higher than those (0.52–0.75 eV) for their non-H-bonded isomers (see Table 2). This is due to the fact that the intramolecular H-bond interactions destabilize significantly S1-min and T1-min structures of 2HTP, 7HIT, and DM-7HIT, which supports strongly that the H-bond interactions in the T1 and S1 states are much weaker than that in the S0 state.| S0-min | T1-min | S1-min | T2-min | S2-min | MECI-S2/S1 | MEI-S1/T2/T1 | |
|---|---|---|---|---|---|---|---|
| 7HIT | 0.0 | 2.20 | 2.33 | 2.32 | 2.91 | 2.94 | 2.34 |
| DM-7HIT | 0.0 | 2.19 | 2.36 | 2.31 | 2.92 | 2.93 | 2.37 |
| 2HTP | 0.0 | 2.18 | 2.30 | 2.27 | 2.75 | 2.75 | 2.37 |
| 7HIT-iso | 0.0 | 1. 64 | 1.77 | 2.17 | 3.54 | 4.24 | 2.58 |
| DM-7HIT-iso | 0.0 | 1.67 | 1.82 | 2.20 | 3.53 | 4.26 | 2.57 |
| 2HTP-iso | 0.0 | 1.43 | 1.58 | 2.16 | 3.49 | 3.88 | 2.37 |
Because the two unpaired electrons are distributed in the spatially overlapping π and π* orbitals, their Coulomb-repulsion interactions lead to the energy of the S2(1ππ*) state to be increased significantly. However, the T2(3ππ*) state is of biradical character where the two unpaired π electrons are mainly localized on the pz orbital of the S atom and the aromatic ring, respectively (see Fig. 3). The Coulomb-repulsion interaction in the T2(3ππ*) state is relatively weak. In addition, the T2(3ππ*) state is decreased in energy, due to the exchange energy (Kπ,π*) of the two unpaired electrons. The difference in the electronic distribution of the two states results in a large energy gap between S2(1ππ*) and T2(3ππ*), which reasonably explains remarkable differences in the MS-PT2(12,10)-DZ calculated relative energies between S2-min and T2-min (see Table 2).
Experimentally, it has been well established that the large energy gap between S2 and S1 is one of the most striking features of thiocarbonyl compounds in the gas phase or inert media. This feature is well reproduced by the MS-PT2(12,10)-DZ calculations for 2HTP-iso, 7HIT-iso, and DM-7HIT-iso. However, the situation is quite different for 2HTP, 7HIT and DM-7HIT, where the intramolecular H-bonds are present. As can be seen from Table 2, the energy gap between S2-min and S1-min is 0.45/0.58/0.56 eV for 2HTP/7HIT/DM-7HIT, which is much smaller than 1.91/1.77/1.71 eV for 2HTP-iso/7HIT-iso/DM-7HIT-iso. As discussed previously, the S2(1ππ*) and S1(1nπ*) states are significantly stabilized and destabilized by the intramolecular H-bond interactions, respectively, which are responsible for a significant decrease in the energy gap between S2 and S1 from the non-H-bonded to H-bonded aromatic thioketones.
As plotted in Fig. 3, there is one unpaired electron in the n orbital of the T1 state, while this unpaired electron is redistributed into the pz orbital of the T2 state, which is the main difference between the T1 and T2 electronic structures. The energy difference between T2 and T1 (S1) is estimated to be small from their difference in the electronic structure. As listed in Table 2, T2-min is 0.40/0.38/0.58 eV higher than S1-min in energy for 7HIT-iso/DM-7HIT-iso/2HTP-iso. However, it should be emphasized that T2-min is slightly lower in energy than S1-min for 7HIT, DM-7HIT, and 2HTP. Actually, S1-min, T2-min and T1-min are energetically close to each other. The main reason for these is the intramolecular H-bond interaction that destabilizes T1 and S1 states significantly, but has a little influence on the T2 state.
The S0 → S1 absorption and the T2 → S0 phosphorescence were explored for BPT in H2O and CH3CN.32,33 The absorption peak was observed to be blue-shifted by more than 2000 cm−1 for BPT in H2O, as compared with that for BPT in CH3CN, while the peak positions of the phosphorescence bands for BPT in the two solvents are almost identical. The intermolecular interaction between BPT and CH3CN can be expected to be weak, which has little influence on the absorption and emission bands of BPT. However, the H-bond interaction between BPT and H2O is relatively strong, the appreciable blue shift of the S0 → S1 absorption band for BPT in H2O could be ascribed to significant weakening of the H-bond (BPT⋯H2O) interaction from S0 to S1. It can be expected from the previous discussion that the H-bond (BPT⋯H2O) interaction in the T2(3ππ*) state is almost the same as that in the S0 state, which is the reason why the peak positions of the phosphorescence bonds for BPT in H2O and CH3CN are almost identical. The absorption and emission spectral properties of BPT in the two different solvents reported experimentally provide strong support for the excited-state dependent H-bond interactions predicted theoretically for 2HTP, 7HIT, and DM-7HIT.
2.3 Minimum-energy conical intersection between S2 and S1 states
The intramolecular H-bond interactions present in 2HTP, 7HIT, and DM-7HIT result in a significant decrease of the S2–S1 energy gap. This gives us a hint that the S2 and S1 states could intersect with each other. The minimum-energy conical intersection between the S2 and S1 states (MECI-S2/S1) was optimized at the MS-PT2(12,10)-DZ level of theory. The key bond parameters of the optimized MECI-S2/S1 structures for 7HIT and 7HIT-iso are listed in Table 1. The XMS-CASPT2 method has been demonstrated to be well suited for the description of conical intersections of potential energy surfaces. For comparison, the MECI-S2/S1 structure of 7HIT was further optimized at the XMS-PT2(12,10)-DZ level. It was found that the XMS-PT2(12,10)-DZ optimized MECI-S2/S1 structure is well consistent with the structure optimized at the MS-PT2(12,10)-DZ level, except for a slight fluctuation (<0.01 Å) in the XMS-PT2(12,10)-DZ optimized bond lengths. Thus, the MECI-S2/S1 structures were determined at the MS-PT2(12,10)-DZ level for the aromatic thioketones under study. The optimized MECI-S2/S1 structure of 7HIT is shown in Fig. S13 of the ESI.† The detailed structural parameters for all aromatic thioketones investigated in this work can be found in the ESI.†The MS-PT2(12,10)-DZ optimized MECI-S2/S1 and S2-min structures are very similar for 2HTP, 7HIT and DM-7HIT, but the MECI-S2/S1 structure exhibits some S1-min-like features. Thus, the H-bond interaction in the optimized MECI-S2/S1 structure is a little weaker than that in the S2-min structure. Meanwhile, the π electrons in the MECI-S2/S1 structure are redistributed in the aromatic ring and the thiocarbonyl group, as compared with the π electron distribution in the S2-min structure. As a result, there is a small difference in energy between S2-min and MECI-S2/S1, which is predicted to be smaller than 0.03 eV for 2HTP, 7HIT and DM-7HIT by the MS-PT2(12,10)-DZ calculations. It is evident that MECI-S2/S1 and S2-min are energetically degenerate or quasi-degenerate. Extensive theoretical endeavors regarding dynamics at the conical intersection of two states have shown that internal conversion (IC) occurs in the vicinity of the MECI structure very efficiently in most organic molecules, with a typical timescale of a picosecond.34,35 Considering that the differences in structure and energy between MECI-S2/S1 and S2-min are very small, the S2 → S1 internal conversion could be expected to be an ultrafast process within 2HTP, 7HIT and DM-7HIT systems.
Apparently, the conical intersection between S2 and S1 in the vicinity of the S2 minimum originates from the dependence of the intramolecular H-bond interaction on the excited state. The existence of such a conical intersection leads to the short-lived S2 state, which reasonably explains why there is no measurable fluorescence from the S2 state for DM-7HIT in cyclohexane.30 Once the OH group of DM-7HIT is methylated, DM-7MIT is formed. As the intramolecular H-bond is absent in DM-7MIT, the S2 → S0 fluorescence band was observed for DM-7MIT in cyclohexane with the emission maximum at ∼380 nm.30 The absorption and emission spectroscopies measured for DM-7HIT and DM-7MIT in cyclohexane provide clear evidence that the conical intersection between S2 and S1 is the crucial factor responsible for the mechanism of the S2 deactivation.
The photophysical processes of BPT in water have been investigated via fs-fluorescence upconversion and transient absorption spectroscopies.33 The S2 decay kinetics was fitted to a single exponential function with time constants of 1.0 ± 0.1 and 2.1 ± 0.15 ps for the H-bonded complex of BPT in water and deuterated water, respectively. In comparison with the S2 time constants of 14.9 and 180.0 ps measured for BPT in acetonitrile and perfluoro-n-hexane, respectively,32,36 the extremely short S2 lifetime for BPT in H2O indicates that the S2 deactivation could occur within the H-bonded BPT⋯H2O complex. The observed isotope effect on the S2 lifetime has shown that O–H or O–D stretching modes play an important role in the non-radiative deactivation of the BPT⋯H2O complex in the S2 state. The O–H elongation interrupted on the S2 surface at the conical intersection and the subsequent O–H shortening along the S1 pathway have been proposed to be responsible for the fast deactivation of the BPT⋯H2O complex in the S2 state.32,33 The above fs-fluorescence up-conversion and transient absorption studies provide evidence that the intermolecular H-bond interaction can result in the conical intersection between S2 and S1 in the S2 Franck–Condon region, and the conical intersection could be a funnel for the fast radiationless decay of the S2 state, which supports the S2 deactivation mechanism predicted by the MS-PT2(12,10)-DZ calculations for 2HTP, 7HIT, and DM-7HIT, where the intramolecular H-bond is present.
For comparison, the MECI-S2/S1 structures of 2HTP-iso, 7HIT-iso, and DM-7HIT-iso are also optimized at the MS-PT2(12,10)-DZ level of theory. The optimized MECI-S2/S1 structures are significantly different from their S2-min structures. Meanwhile, MECI-S2/S1 is higher than S2-min in energy (see Table 2), which is calculated to be 0.39, 0.70, and 0.73 eV for 2HTP-iso, 7HIT-iso, and DM-7HIT-iso, respectively. It is evident that the S2 → S1 internal conversion occurs with relatively low efficiency in these non-H-bonded aromatic thioketones, which enables the S2 state to fluoresce with a high quantum yield. In fact, significant fluorescence emission has been observed experimentally for a lot of aromatic thioketones that have no intramolecular or intermolecular H-bonds.20
2.4 Minimum-energy intersection of the S1, T2, and T1 states and its unique role in the TADF mechanism
The T2(3ππ*) state is optically dark and has been inferred to be close to the S1 state in energy for thiocarbonyl compounds in the gas phase and different solvents.20 Such a feature has been well reproduced by the MS-PT2(12,10)-DZ calculations. In particular, the T2 state lies energetically between the S1 and T1 states for the aromatic thioketones with intramolecular H-bonds, as discussed before. The minimum-energy intersection structure of the S1, T2, and T1 states, referred to as MEI-S1/T2/T1 hereafter, was determined for 2HTP, 7HIT, or DM-7HIT by means of MS-PT2(12,10)-DZ optimization. As listed in Table 2, the MEI-S1/T2/T1 structure is only 0.07, 0.01, and 0.01 eV higher than the S1-min structure for 2HTP, 7HIT, and DM-7HIT, respectively. According to the energy-gap law for radiationless transition in a molecular system, both the S1 → T1 and S1→T2 ISC processes could take place efficiently in the vicinity of the S1-min or MEI-S1/T2/T1 structure. Because of their spin-forbidden nature, however, the rate constants of the two ISC processes are largely controlled by the spin-orbit coupling (SOC) interaction. The SOC constants between the singlet (S1) and triplet (T1 and T2) states were calculated at the MS-PT2(12,10)-DZ level of theory, and the calculated results are listed in Fig. 4. In the MEI-S1/T2/T1 structure, the SOC constant between S1 and T2 is 158.1 cm−1, which is much larger than that between S1 and T1 (12.1 cm−1). Thus, the S1 → T1 nonadiabatic transition cannot be in competition with the S1 → T2 ISC process. Once the T2 state is populated via the S1 → T2 transition, the subsequent T2 → T1 internal conversion (IC) could proceed on a timescale of picosecond or shorter at the MEI-S1/T2/T1 structure,34,35 because this three-state intersection is bound to be a conical intersection between T2 and T1. Because of the existence of the three-state intersection, the T2 state functions as a bridge and enables both forward S1 → T1 and reverse T1 → S1 processes (F-ISC and R-ISC) to proceed very efficiently for the aromatic thioketones with the intramolecular H-bond. The linear interpolation internal coordinate (LIIC) pathways among S1-min, T2-min, and T1-min of 7HIT have been determined by the stepwise MS-PT2(12,10)-DZ calculations. From the MS-PT2(12,10)-DZ calculated SOC constants and energy differences for various structures on the LIIC pathways (see Fig. 4), we can come to the conclusion that the T2 state acts as relay and enables both F-ISC and R-ISC processes to proceed very efficiently in a wide structural region. | ||
| Fig. 4 The calculated SOC constants (cm−1) between S1 and T2 (T1) for various structures on the LIIC pathways of 7HIT and for MEI-S1/T2/T1. | ||
For the non-H-bonded aromatic thioketones (2HTP-iso, 7HIT-iso, and DM-7HIT-iso), the relative energies of the MEI-S1/T2/T1 structures were calculated to be 0.21–0.41 (0.75–0.81) eV higher than the relative energies of the T2-min (S1-min) structures (see Table 2). In this case, the T2 state exerts little influence on the S1 → T1 ISC process. The time constant of the S1 → T2 transition has been experimentally found to be 3.4 ps for DM-7HIT in cyclohexane, while the S1 → T1 ISC process was found to be slow for DM-7HIT-iso in cyclohexane,30 which are consistent with the calculated SOC constants between the singlet (S1) and triplet (T1 and T2) states.
As discussed before, both the S1(1nπ*) and T1(3nπ*) states of the aromatic thioketones originate from n → π* electronic transition, while the T2 state is of 3ππ* character. The change of orbital angular momentum from S1(1nπ*) to T2 (3ππ*) could help flip the spin of one electron, which favors the first-order SOC interaction between the S1(1nπ*) and T2 (3ππ*) states.6 However, the ISC process from S1(1nπ*) to T1(3nπ*) involves an exclusively spin flip of one electron and does not generate any change in orbital angular momentum. According to the EI-Sayed rule,6 there is no first-order SOC interaction between the S1(1nπ*) and T1(3nπ*) states. The above discussion explains reasonably why the SOC constants between the S1 and T2 states are considerably larger than those between the S1 and T1 states.
A class of purely organic molecular materials was reported by Adachi and co-workers in 2012.37 These materials exhibit prompt fluorescence and efficient TADF emission by harvesting both singlet and triplet excitons. Meanwhile, they are free of heavy metals and are still able to achieve high quantum efficiencies, which led to a major revolution in the field of organic electroluminescent materials. The reverse intersystem crossing (R-ISC) from T1 to S1 is the pre-step for TADF emission. The energy gap (ΔEST) and the SOC constant between S1 and T1 have been recognized as the two key factors that are responsible for efficient TADF emission. Diverse strategies and tips have been reported for designing purely organic TADF materials.38–43 Although the energy gap between S1 and T1 can be controlled to be smaller than 0.1 eV by modifying the molecular structure, adjusting polarization effects of the solid-state environment and regulating the localized and delocalized properties of one-electron transition, the SOC constants between the excited singlet and triplet states of the designed organic TADF materials are still much smaller than those for iridium-trisphenylpyridine complexes used as emitters of the second-generation organic light-emitting diode (OLED).41 As shown in Fig. 4, the calculated SOC constants between S1 and T2 on various structures of the LIIC pathways are larger than 156.0 cm−1, which is close to 150.0 cm−1 evaluated for iridium-trisphenylpyridine complexes. Because of small energy gaps between S1 and T1 and relatively large SOC constants for a wide variation of nuclear configurations, both F-ISC and R-ISC processes can occur very effectively. The above comparison and discussion give us a reason to expect that the aromatic thioketone systems could be used for the development of the desired organic TADF molecular materials. More importantly, the new TADF mechanism reported herein, which involves a three-state intersection or quasi-degenerate, could be used to improve the design principle of purely organic TADF materials as OLED emitters.
2.5 Crucial role of the excited-state dependent H-bond interaction in determining the photophysical properties of aromatic thioketones
The MS-PT2(12,10)-DZ calculated results, combined with a detailed analysis of the dependence of the excited-state structures and their relative energies on the H-bond interactions, proved that the essential differences in the nature of intramolecular H-bonds between the ground state and different excited states result in a dramatic change in the photophysical properties of 7HIT, DM-7HIT and 2HTP. As representatives of aromatic thioketones with and without the intramolecular H-bond, the photophysical processes of 7HIT and 7HIT-ISO are summarized in Fig. 5. For the H-bonded aromatic thioketones (such as 7HIT), the ultrafast S2 → S1 internal conversion occurs in the vicinity of the minimum-energy conical intersection (MECI-S2/S1), once the S2 state is populated by near-UV irradiation. However, the fluorescence emission primarily contributed to the deactivation of the S2 state for the non-H-bonded aromatic thioketones (such as 7HIT-iso), while the S2 → S1 internal conversion is a slow process. Subsequently, the T1 → S0 phosphorescence could be observed for 7HIT-iso via the S1 → T1 intersystem crossing. The essential differences in the intramolecular H-bond interactions between the S1 (T1) and S0 states of 7HIT have been found to be the key factors that lead to the degeneracy or near-degeneracy of the S1, T2, and T1 states. Once the S1 state is populated through the S2 → S1 internal conversion, the T2-mediated S1 → T1 and T1 → S1 ISC processes could take place very efficiently. Because of the small energy difference and ultrafast interconversion between S1 and T1, the S1 population could be comparable to that of the T1 state. The TADF emission from the S1 state and phosphorescence from the T1 state could be observed with a comparable quantum yield. Because the S2 emission is the dominant deactivation pathway, only weak T1 phosphorescence can be observed for the aromatic thioketones without the intramolecular H-bond.30 Evidently, the dramatic changes in the photophysical properties of aromatic thioketones can be attributed to the differences in the nature of the intramolecular H-bond interactions between the ground state and various excited states. | ||
| Fig. 5 Schematic diagram of photophysical processes for the non-H-bonded (7HIT-iso) and H-bonded (7HIT) aromatic thioketones. | ||
3. Conclusions
In this work, we have reported high-accuracy MS-CASPT2 calculations for the five lowest-lying electronic states of three aromatic thioketones (2HTP, 7HIT, and DM-7HIT) and their non-H-bonded isomers (2HTP-iso, 7HIT-iso, and DM-7HIT-iso). The calculated results have revealed that the dramatic changes in the photophysical properties of the aromatic thioketones originate from the dependence of the intramolecular H-bond interactions on the excited electronic states. The H-bond interaction in the S2 state of 2HTP, 7HIT, or DM-7HIT is significantly strengthened, while this interaction in the S1 or T1 state is considerably weakened, as compared with that in the S0 state. As a result, the energy gap between S2 and S1 is remarkably reduced, and the minimum-energy conical intersection of the S2 and S1 states was determined for 2HTP, 7HIT, and DM-7HIT. Because of the existence of the conical intersection near the S2 minimum, the S2 → S1 internal conversion becomes an ultrafast process, and there is no measurable fluorescence from the S2 state, which is supported by the experimental findings. The T2 state is a little stabilized by the intramolecular H-bond interaction, which is different from the S1 and T1 states that are destabilized by the H-bond interaction. Accordingly, the S1, T2, and T1 states are quasi-degenerate in a wide structural region. The minimum-energy intersection structures of the three states have been discovered for the aromatic thioketones with the intramolecular H-bond. Meanwhile, this three-state intersection is bound to be a conical intersection between the T1 and T2 states. In this case, the T2 state functions as a bridge and enables both forward S1 → T1 and reverse T1 → S1 ISC processes to occur efficiently in the three-state intersection and quasi-degenerate regions.The S2 → S1 internal conversion is the dominant pathway for the S2 deactivation of 2HTP, 7HIT and DM-7HIT, and the subsequent T2-state mediated interconversion between the S1 and T1 states is responsible for TADF from the S1 state and the phosphorescence from the T1 state. For the non-H-bonded aromatic thioketones (2HTP-iso, 7HIT-iso, and DM-7HIT-iso), however, the S2 → S0 fluorescence primarily contributed to the deactivation of the S2 state, while the S2 → S1 internal conversion is a slow process, which is followed by the S1 → T1 intersystem and the T1 → S0 phosphorescence. Finally, it should be pointed out that the new TADF mechanism reported herein could help improve the design principle of purely organic TADF molecular materials.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We are grateful for the financial support from the NSFC (Grant No.: 22288201).References
- E. W. G. Diau, J. L. Herek, Z. H. Kim and A. H. Zewail, Science, 1998, 279, 847–851 CrossRef CAS PubMed.
- W. H. Fang, J. Am. Chem. Soc., 1999, 121, 8376–8384 CrossRef CAS.
- X. B. Chen and W. H. Fang, J. Am. Chem. Soc., 2004, 126, 8976–8980 CrossRef CAS PubMed.
- R. Srinivasan, J. S. Feenstra, S. T. Park, S. J. Xu and A. H. Zewail, Science, 2005, 307, 558–563 CrossRef CAS PubMed.
- W.-H. Fang, Acc. Chem. Res., 2008, 41, 452–457 CrossRef CAS PubMed.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, Viva Books University Science Books, Sausalito, 2017 Search PubMed.
- R. K. Venkatraman and A. J. Orr-Ewing, J. Am. Chem. Soc., 2019, 141, 15222–15229 CrossRef CAS PubMed.
- D. Townsend, S. A. Lahankar, S. K. Lee, S. D. Chambreau, A. G. Suits, X. Zhang, J. Rheinecker, L. B. Harding and J. M. Bowman, Science, 2004, 306, 1158–1161 CrossRef CAS PubMed.
- C.-H. Yang, S. Bhattacharyya, L. Liu, W.-H. Fang and K. Liu, Chem. Sci., 2020, 11, 6423–6430 RSC.
- A. H. Maki, P. Svejda and J. R. Huber, Chem. Phys., 1978, 32, 369–380 CrossRef CAS.
- D. M. Burland, J. Chem. Phys., 1981, 75, 2635–2642 CrossRef CAS.
- A. Maciejewski and R. P. Steer, J. Am. Chem. Soc., 1983, 105, 6738–6740 CrossRef CAS.
- L. W. Molenkamp, D. P. Weitekamp and D. A. Wiersma, Chem. Phys. Lett., 1983, 99, 382–387 CrossRef CAS.
- A. Maciejewski, D. R. Demmer, D. R. James, A. Safarzadehamiri, R. E. Verrall and R. P. Steer, J. Am. Chem. Soc., 1985, 107, 2831–2837 CrossRef CAS.
- A. Maciejewski, M. Szymanski and R. P. Steer, J. Phys. Chem., 1986, 90, 6314–6318 CrossRef CAS.
- R. P. Steer and V. Ramamurthy, Acc. Chem. Res., 1988, 21, 380–386 CrossRef CAS.
- M. Szymanski, A. Maciejewski and R. P. Steer, J. Phys. Chem., 1988, 92, 2485–2489 CrossRef CAS.
- C. J. Ho, A. L. Motyka and M. R. Topp, Chem. Phys. Lett., 1989, 158, 51–59 CrossRef CAS.
- M. Szymanski, A. Maciejewski and R. P. Steer, J. Photochem. Photobiol., 1991, 57, 405–418 CrossRef CAS.
- A. Maciejewski and R. P. Steer, Chem. Rev., 1993, 93, 67–98 CrossRef CAS.
- A. A. Ruth, F. J. Okeeffe, R. P. Brint and M. W. D. Mansfield, Chem. Phys., 1997, 217, 83–98 CrossRef CAS.
- A. A. Ruth, F. J. Okeeffe, M. W. D. Mansfield and R. P. Brint, J. Phys. Chem. A, 1997, 101, 7735–7741 CrossRef CAS.
- A. A. Ruth, F. J. Okeeffe, M. W. D. Mansfield and R. P. Brint, Chem. Phys. Lett., 1997, 264, 605–613 CrossRef CAS.
- A. A. Ruth, T. Fernholz, R. P. Brint and M. W. D. Mansfield, Chem. Phys. Lett., 1998, 287, 403–411 CrossRef CAS.
- R. P. Brint, W. G. Doherty and A. A. Ruth, Mol. Spectrosc., 2002, 216, 151–158 CrossRef CAS.
- K. J. Falk and R. P. Steer, J. Phys. Chem., 1990, 94, 5767–5772 CrossRef CAS.
- G.-J. Zhao and K.-L. Han, ChemPhysChem, 2008, 9, 1842–1846 CrossRef CAS PubMed.
- E. Krystkowiak, J. Koput and A. Maciejewski, Phys. Chem. Chem. Phys., 2012, 14, 8842–8851 RSC.
- E. Krystkowiak, R. A. Bachorz and A. Maciejewski, Phys. Chem. Chem. Phys., 2016, 18, 492–502 RSC.
- Z.-Y. Liu, J.-W. Hu, C.-H. Huang, T.-H. Huang, D.-G. Chen, S.-Y. Ho, K.-Y. Chen, E. Y. Li and P.-T. Chou, J. Am. Chem. Soc., 2019, 141, 9885–9894 CrossRef CAS PubMed.
- A. O. Szabo and N. S. Ostlund, Modern Quantum Chemistry: Introduction to Ad-vanced Electronic Structure Theory, 1989 Search PubMed.
- G. Burdzinski, A. Maciejewski, G. Buntinx, O. Poizat and C. Lefumeux, Chem. Phys. Lett., 2004, 384, 332–338 CrossRef CAS.
- G. Burdzinski, A. Maciejewski, G. Buntinx, O. Poizat, P. Toele, H. Zhang and M. Glasbeek, Chem. Phys. Lett., 2004, 393, 102–106 CrossRef CAS.
- W. Domcke and D. R. Yarkony, Annu. Rev. Phys. Chem., 2012, 63, 325–352 CrossRef CAS PubMed.
- M. S. Schuurman and A. Stolow, in Annual Review of Physical Chemistry, ed. M. A. Johnson and T. J. Martinez, 2018, vol. 69, pp. 427–450 Search PubMed.
- G. Burdzinski, A. Maciejewski, G. Buntinx, O. Poizat and C. Lefumeux, Chem. Phys. Lett., 2003, 368, 745–753 CrossRef CAS.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- C. M. Marian, J. Phys. Chem. C, 2016, 120, 3715–3721 CrossRef CAS.
- P. K. Samanta, D. Kim, V. Coropceanu and J.-L. Bredas, J. Am. Chem. Soc., 2017, 139, 4042–4051 CrossRef CAS PubMed.
- X.-K. Chen, D. Kim and J.-L. Bredas, Acc. Chem. Res., 2018, 51, 2215–2224 CrossRef CAS PubMed.
- D.-H. Kim, A. D'Aleo, X.-K. Chen, A. D. S. Sandanayaka, D. Yao, L. Zhao, T. Komino, E. Zaborova, G. Canard, Y. Tsuchiya, E. Choi, J. W. Wu, F. Fages, J.-L. Bredas, J.-C. Ribierre and C. Adachi, Nat. Photonics, 2018, 12, 98–104 CrossRef CAS.
- D. S. M. Ravinson and M. E. Thompson, Mater. Horiz., 2020, 7, 1210–1217 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The computational details, Cartesian coordinates, absolute energies, tables and figures that show the results of the test calculations. See DOI: https://doi.org/10.1039/d2cp02016e |
| This journal is © the Owner Societies 2022 |