
Compartmentalisation of molecular catalysts for nonorthogonal tandem catalysis
Peiyuan
Qu
 a,
Jacob W.
Cleveland
b,
Eman
Ahmed
a,
Fangbei
Liu
a,
Sage
Dubrawski
a,
Christopher W.
Jones
*b and
Marcus
Weck
*a
a,
Jacob W.
Cleveland
b,
Eman
Ahmed
a,
Fangbei
Liu
a,
Sage
Dubrawski
a,
Christopher W.
Jones
*b and
Marcus
Weck
*a
aMolecular Design Institute and Department of Chemistry, New York University, 100 Washington Square East New York, NY 10003, USA. E-mail: marcus.weck@nyu.edu
bSchool of Chemical & Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA 30332-0100, USA. E-mail: christopher.jones@chbe.gatech.edu
First published on 9th December 2021
Abstract
The development of nonorthogonal tandem catalysis enables the use of a combination of arbitrary catalysts to rapidly synthesize complex products in a substainable, efficient, and timely manner. The key is to compartmentalise the molecular catalysts, thereby overcoming inherent incompatibilities between individual catalysts or reaction conditions. This tutorial review analyses the development of the past two decades in the field of nonorthogonal tandem catalysis with an emphasis on compartmentalisation strategies. We highlight design principles of functional materials for compartmentalisation and suggest future directions in the field of nonorthogonal tandem catalysis.
 Peiyuan Qu | Peiyuan Qu received his BS at Peking University working with Prof. Jianbo Wang on the development of transition metal-catalysed carbene transformations. He obtained his PhD in 2021 under the direction of Prof. Marcus Weck at New York University where he developed functional polymeric nanoreactors for non-orthogonal tandem catalysis. He joined the group of Prof. Joanna Aizenberg at Harvard University as a postdoctoral fellow, where he is currently working on the synthesis of adaptive microstructures based on liquid crystal elastomers as well as developing inversed-opal structures for catalytic applications. |
 Jacob W. Cleveland | Jacob W. Cleveland is a doctoral candidate under the supervision of Prof. Christopher W. Jones in the School of Chemical & Biomolecular Engineering at the Georgia Institute of Technology. He received his BS in Chemical Engineering at Tennessee Tech University in 2017. While at TTU, he worked with Prof. Jesse Carrick on the amination of 6-bromo-pyridinyl-1,2,4-triazine scaffolds. His current work focuses on using controlled radical polymers and porous materials to engineer multicatalytic systems for non-orthogonal tandem catalysis. |
 Eman Ahmed | Eman Ahmed is a doctoral candidate under Prof. Marcus Weck at the Molecular Design Institute, New York University. She received her BA in chemistry from Macalester College in 2018. At Macalester, her research focused on the self-assembly of carbon cages using covalent interactions. Her current work focus is on synthesizing functional amphiphilic polymers that assemble into multicompartment micelles with unique morphologies for non-orthogonal cascade catalysis. |
 Fangbei Liu | Fangbei Liu is a doctoral candidate under Prof. Marcus Weck at the Molecular Design Institute, New York University. She earned her BS in Material Science & Technology at Zhengzhou University in China in 2017 and her MS in Polymer Science at University of Akron in 2019. Her current research in the Weck group focuses on photoresponsive shell cross-linked micelles for catalytic applications. |
 Sage Dubrawski | Sage Dubrawski is a doctoral candidate under Prof. Marcus Weck at the Molecular Design Institute, New York University. She earned her BS in Chemistry from the University of Rhode Island in 2020. At the University of Rhode Island, her research focused on the synthesis of various TADF dyes for terahertz spectroscopy under Dr Dugan Hayes. She also worked under Dr Hyvl at the University of Hawaii synthesizing asymmetric Bismuth compounds to be used as chiral Lewis acid catalysts. Her current work focuses on multiresponsive core–shell micelles for nonorthogonal tandem catalysis. |
 Christopher W. Jones | Christopher W. Jones earned a PhD in 1999 from Caltech working with Mark E. Davis. After postdoctoral work with Davis and John E. Bercaw, he joined the faculty at Georgia Institute of Technology, where he currently serves as the John H. Brock School Chair & Professor of Chemical & Biomolecular Engineering. His research focuses on synthetic materials chemistry with emphases in catalysis, reaction engineering and adsorptive separations. |
 Marcus Weck | Marcus Weck obtained his PhD degree in 1998 from Caltech with Robert H. Grubbs. After a postdoctoral stay at Harvard University with George M. Whitesides, he joined the faculty at Georgia Tech. In 2007, he moved to New York University, where he is a professor in the chemistry department. His research interests are in organic and polymer chemistry as well as materials science. The main foci of his group are supported catalysis, the introduction of complexity through the use of orthogonal functionalisation methods, and to synthesize polymers, organized assemblies, biomaterials, and nanostructures. |
Key learning points1. Current strategies for compartmentalisation of molecular catalysts.2. Principles of designing support structures for nonorthogonal tandem catalysis. 3. Methods to immobilize incompatible molecular catalysts. 4. The state-of-the-art nonorthogonal tandem catalytic transformations. 5. Perspective on the future direction of tandem catalysis and multistep synthesis. |
1. Introduction
In a global effort to strive towards greater economic and environmental efficiency, it is imperative to re-evaluate how chemical synthesis is conducted in academia and industry. In a conventional multistep synthesis, each reaction occurs in a separate vessel and iterative physical separation of products from the reaction mixture is required at each stage of the synthetic route. Sequential set-up and work-up of a multistep synthesis consumes time, energy, and resources, which can include toxic and environmentally harmful organic solvents. For example, the average kilogram of pharmaceutical product via a multistep synthesis generates substantial chemical waste (25–100 kg).1 To address these challenges, chemists have designed multicatalytic systems for efficient multistep synthesis with significant financial and environmental benefits.One strategy is to transition to a one-pot setup with a combination of catalysts for sustainable tandem reactions. Tandem catalysis is the stepwise chemical process comprised of at least two consecutive reactions that occur via distinct catalytic mechanisms, where each subsequent reaction occurs only in virtue of the chemical products formed in the previous step.2 A one-pot system encompasses all the catalytic species from the onset of the desired multistep transformation. An ideal organic synthesis should be performed by the synergistic use of a combination of catalysts. Complex molecules can be synthesized from simple starting materials rapidly and efficiently in one pot fashion under sustainable reaction conditions (e.g. water as only solvent), providing maximum use of resources and simplifying the overall reaction workup.
Despite its elegance and potential, one-pot tandem catalysis has limited practical use to date. The primary reason is that in most tandem reactions, individual catalytic sequences are nonorthogonal to one another. The term orthogonal, originally meaning perpendicular, describes events that do not affect one another in terms of outcome. Nonorthogonal tandem catalysis then is the combination of two or more independent catalytic processes that interfere with each other in a detrimental way, resulting in lower or no yields and/or selectivities. In a tandem sequence, each catalytic transformation has its own optimal reaction conditions such as pH, temperature, additives, and solvent(s). An active catalyst may react with other species (e.g. other catalysts, substrates, intermediates, solvents, and/or additives) in a deleterious manner (e.g. redox chemistry, ligand exchange, and acid–base neutralization), rendering them inactive. Current synthetic procedures for pharmaceutical compounds include multiple nonorthogonal catalytic transformations. For example, the synthesis of Pregabalin, a commercialised therapeutic agent, entails the use of multiple incompatible catalytic species (e.g. acid and base, oxidation and reduction, organocatalysts and enzymes, and transition metal catalysts) in discrete reaction vessels.1 Furthermore, while using different catalysts in a one-pot system, selectivity becomes an issue since multiple catalytic mechanisms can occur concurrently and lead to competitive reaction pathways. Such competitive reaction pathways can result in undesired intermediates and side-products that complicate or impede the purification process, leading to lower yields of the desired products. Hence, the successful execution of one-pot tandem catalysis relies on addressing two issues of nonorthogonality: the incompatibility between individual catalysts and/or reaction conditions and the competition between catalytic pathways. Striving towards an ideal multistep synthesis, it is critical to develop a general strategy to solve the issue of nonorthogonality between catalysts. Compartmentalisation is key to realizing this goal.
The concept of compartmentalisation is adapted from eukaryotic cells where active species, such as enzymes, are positioned in sub-cellular compartments.3 Chemical reactions mediated by enzymes are confined to such cellular compartments, with each compartment potentially having different environments such as pH, salt content, substrate concentration, etc. The compartments are separated from each other by semipermeable membranes, yet placed in a close proximity for streamlined metabolic pathways.3 It has been a long-standing goal to implement compartmentalisation within artificial structures.
Supported catalysis is a strategy that immobilizes catalysts on synthetic structures to tune the reactivity, selectivity, and specificity of a catalyst. The use of an appropriate support allows for multiple catalysts to be positioned in close proximity to optimize substrate diffusion and active species migration and facilitates tandem catalysis yet isolating each catalytic species to prevent mutual deactivation. Immobilizing molecular catalysts on supports can also enhance their stability, recyclability, and reactivity.4 Further environmental benefits of designing a support for tandem catalysis is the potential to carry out all transformations in an aqueous environment. Over the years, material science has enabled the realization of three-dimensional support structures with different chemical environments and topographies across all size scales, ranging from the nanoscale to the mesoscale. Support structures can be divided into insoluble and soluble systems, micro-sized and nano-sized systems, or organic and inorganic supports. Catalysts can be immobilized onto a support by covalent linkages, van der Waals interaction, ionic interactions, or encapsulation.4
To successfully execute nonorthogonal tandem catalysis, compartmentalised support structures, analogous to membrane-bound compartments in a cell, are preferred for efficient site-isolation of incompatible active species. The principle of designing a successful support for catalyst compartmentalisation is to introduce orthogonal functionalities within the support structure for catalyst immobilisation. Herein, we characterize the compartmentalisation strategies for non-orthogonal tandem catalysis into three approaches (Table 1): Approach-1, compartmentalisation of a catalytic entity on a discrete support while the remaining active species are in bulk solution; Approach-2, compartmentalisation of two or more catalytic entities on multiple discrete supports; and Approach-3, compartmentalisation of two or more catalytic entities within different domains of a single support.
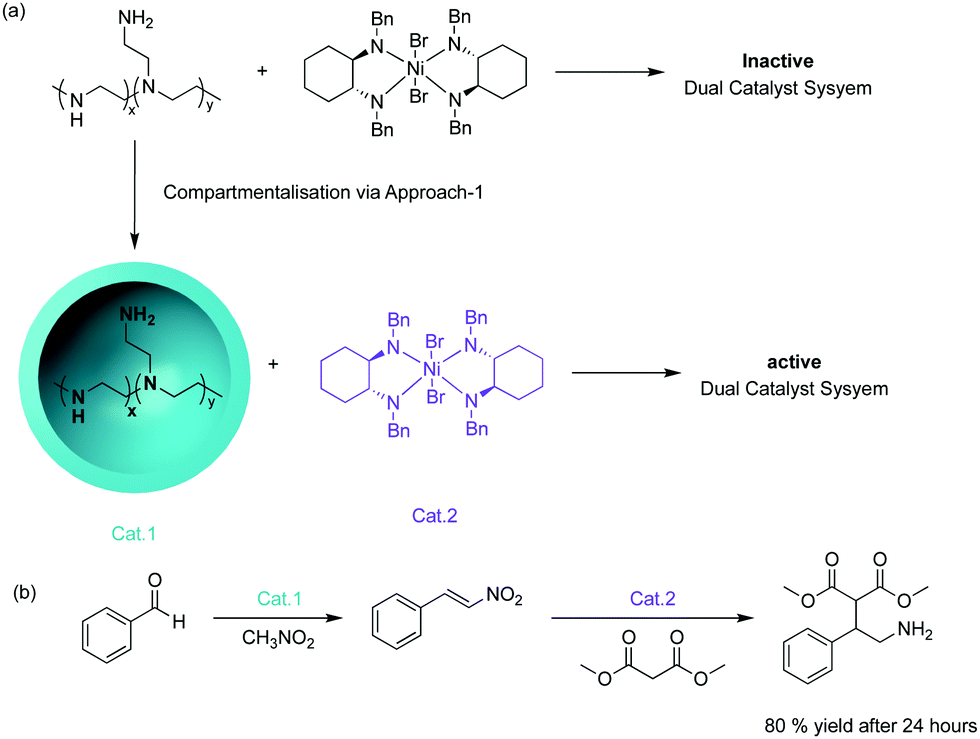 | ||
| Fig. 1 Microcapsule compartmentalised multicatalytic system. (a) The compartmentalisation of two incompatible catalysts: microencapsulated PEI and a nickel-based Michael addition catalyst. (b) One-pot tandem catalysis involving amine-catalysed nitroaldol reaction followed by nickel catalysed Michael addition. | ||
In this tutorial review, using selected examples from the literature, we analyse the developments of the past two decades in the field of nonorthogonal tandem catalysis by categorizing the multicompartmentalisation strategies using the three strategies for isolation described in Table 1 with an emphasis on catalyst orthogonality. Each compartmentalisation strategy will be further grouped into inorganic and organic supports. Enzymes as catalysts are covered elsewhere5 and are not reviewed here. Metal–organic frameworks6 and polymeric aromatic frameworks7 are not discussed in this review either since there are no explicit catalyst compartmentalisations described to date. We elucidate and describe guidelines to design multifunctional catalytic materials that promote sustainable and efficient chemical synthesis, allowing the design of new strategies towards tandem catalysis.
| Approach | Compartmentalisation strategy | Pros | Cons | Graphical illustration |
|---|---|---|---|---|
| 1 | Compartmentalisation of a catalytic entity on a discrete support while the remaining active species are in bulk solution | Easy synthesis of supported catalysts, simple reaction set-up, and facile recovery of catalysts | Catalysts in solution are unstable |

|
| 2 | Compartmentalisation of two or more catalytic entities on multiple discrete supports | Efficiently limit mutual deactivation between antagonistic catalytic species | Effective reaction rates can be hindered by diffusion rates |

|
| 3 | Compartmentalisation of two or more catalytic entities within different domains of a single support | Afford higher catalytic efficiency by cooperative behavior, regulation of substrate diffusion, and substrate channeling | Relatively complex synthetic procedures |

|
2. Compartmentalisation of molecular catalysts on monofunctional supports
2.1 Compartmentalisation of a catalyst on a discrete support
The concept of supporting chemical reagents for sequential incompatible transformations was first demonstrated in the 1970s by the “wolf and lamb” reaction.8 The objective was to site-isolate stoichiometric or even excess amounts of two incompatible reagents into separate synthetic supports to avoid quenching followed by the use of a mixture of those solid-phase reagents to perform multistep syntheses. This strategy of supported systems also enables facile post-synthetic work-up techniques, such as filtration, that can be employed to remove excess supported reagents, simplifying the synthetic process.One strategy to compartmentalise incompatible catalysts is to encapsulate one catalytic entity with the remaining active species in bulk solution. McQuade and co-workers successfully demonstrated this strategy by performing a nonorthogonal amine – Lewis acid tandem catalysis.9 They encapsulated amine catalysts within a microcapsule of poly(ethyleneimine) (PEI) in a solution where an incompatible nickel-based catalyst was dispersed (Fig. 1). The microencapsulation provided effective site-isolation to prevent mutual interaction between the catalysts. The desired Michael addition product was formed in 80% yield while the non-supported tandem catalysis yielded trace product.
While this strategy is viable and has several advantages (Table 1) including easier synthesis of supported catalysts, simple reaction set-up, and facile recovery of catalysts, there are obvious limitations. The catalysts that are dispersed in solution can leach into the encapsulated core interacting or interfering with the supported catalyst, eventually resulting in catalyst deactivation. Additionally, unsupported catalysts, such as transition metal catalysts, are less stable and easier to deactivate over time, compared to their supported counterparts.
2.2 Compartmentalisation of multi-catalysts on discrete monofunctional supports
Compartmentalisation of incompatible catalysts on discrete monofunctional supports (Approach-2) can efficiently limit physical interaction between antagonistic catalytic species and impede mutual deactivation (Table 1). A multitude of synthetic supports have been reported (Fig. 2), including inorganic supports such as porous and nonporous oxides, silicates, and Pickering emulsions. Organic supports such as poly(styrene) (PS) resins, star polymers, poly(dimethylsiloxane) (PDMS) membranes, and hydrogels have been used. | ||
| Fig. 2 Timeline of the development of monofunctional supports for nonorthogonal tandem catalysis. | ||
The sol–gel method is a process where liquid precursor(s) from solution are formed into a gel and subsequently form a dry inorganic structure.11 Co-condensation of functional organosilanes during this process generates reactive sites in the pores, accessible via diffusion. This method can protect mutually destructive catalysts from one another since incompatible catalysts can be entrapped in different SiO2 sol–gel matrices. For example, Avnir and co-workers used two separated silica sol–gel matrices to immobilize two incompatible catalysts, the base H2N(CH2)2NH(CH2)3Si(OMe)3 and the rhodium catalyst RhCl[P(C6H5)3]3 (Wilkinson catalyst).11 Wilkinson's catalyst was physically entrapped in the support and the base covalently incorporated by co-condensation of the aminosilane structure with tetramethylorthosilicate. A mixture of antagonistic Rh and base-catalysed dehydroiodination and alkene hydrogenation were performed in one-pot.
Silicates are materials commonly found in nature and can be synthesized easily in distinct forms. Incorporation of cations within their frameworks generates active catalytic sites. Kaneda and co-workers fabricated a layer of Ti4+-exchanged montmorillonite phyllosilicate materials (Ti4+-mont) as a solid Brønsted acid catalyst and a second layer of hydrotalcites (HTs) as a solid base catalyst.12 These layered materials were used as catalysts for a variety of nonorthogonal acid–base tandem reactions in one-pot with a simple workup procedure and sufficient reusability (at least five times with retention of high reactivity and selectivity).
Droplets in an oil–water emulsion can serve as catalytic microcompartments for nonorthogonal tandem catalysis.13 Emulsions can be stabilized using solid emulsifier particles, such as SiO2, to form Pickering emulsions. In a water-in-oil Pickering emulsion, a reagent, e.g., acid catalyst, can be dissolved in the water droplets while another incompatible reagent, e.g., base catalyst, is dissolved in another layer of water droplets. In this fashion, the layered architecture positions the incompatible emulsion droplets in different regions to avoid mutual deactivation of the catalysts while the continuous phase allows other reagents to diffuse freely. In 2015, Yang and co-workers used Pickering emulsions for a series of nonorthogonal tandem catalysis, including deacetalisation-reduction, deacetalisation-Knoevenagel, deacetalisation-Henry, and diazotization–iodization tandem reactions.13 More recently, the authors improved the stability of the Pickering emulsion by cross-linking the emulsifier particles, forming an inorganic shell around the droplets to avoid coalescence.14 The reinforced Pickering emulsion enabled a nonorthogonal deacetalisation-Knoevenagel reaction sequence using ethylenediamine as base catalyst, which was not viable for the uncross-linked counterparts under the same reaction conditions. This study demonstrates the effectiveness of site-isolation of acid and base droplets in the reinforced Pickering emulsion. Additionally, the comparison between cross-linked and uncross-linked catalytic systems indicates that the stability of the support structures can lead to more efficacious compartmentalisation.
Mesoporous silica SBA-15 is a widely investigated material for supported catalysis because of its well-defined, ordered framework, high surface area and pore volume, and tuneable pore size.10 Using functionalised mesoporous silica as the stationary phase in flow reactors enables a flow-through process that benefits from compartmentalisation of incompatible catalysts and the tenets of green chemistry. To fulfil this goal, Pericas and co-workers packed the bed of a flow reactor with the acidic SBA-SO3H and basic SBA-NH2 materials separately, for a flow-version of nonorthogonal acid–base tandem catalysis. Acid catalysed acetal hydrolysis, followed by base catalysed Henry reaction and Knoevenagel condensation were investigated.15 The system demonstrated good substrate scope (10 examples) with high yields (>90%), long-term stability (>3 days operation), and recyclability through simple regeneration.
Early efforts developing organic supports for tandem catalysis focused on using insoluble structures, with insoluble PS-based resins been the most frequently used polymeric supports in heterogeneous catalysis.16 Styrene-based monomers with different functionalities can fabricate polymeric catalysts with different catalytic reactivity, such as Amberlyst 15 (A15) containing sulfonic acids as a strong acidic catalyst and PS-BEMP (BEMP = 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2 diazaphosphorine) as a base catalyst. Dixon and co-workers exploited a combination of A15 and PS-BEMP for a one-pot, base-catalysed Michael addition, followed by acid-mediated N-acyl iminium ion cyclization cascades with pro-nucleophiles to afford synthetically useful heterocyclic products.17 In another example, to achieve better control over a multistep reaction as well as maximum catalyst recovery, Jones and co-workers demonstrated a combination of insoluble organic and inorganic supported catalysts in one-pot.18 A superparamagnetic spinel ferrite nanoparticle with amine functionality was prepared as the base catalyst and an acid PS resin as the acid catalyst. Platinum on alumina was used as a solid hydrogenation catalyst. The reaction sequences of nonorthogonal tandem catalysis can be controlled by the combination of different solid catalysts. Moreover, catalysts can be recovered in pure form via orthogonal methods, such as magnetic, gravimetric, and membrane separation methods and can be reused in other transformations for the formation of different products (Fig. 3).
 | ||
| Fig. 3 Expanding the utility of one-pot nonorthogonal acid–base tandem catalysis via compartmentalisation. | ||
Aiming at investigating supported catalysts with highly tuneable solubility and reactivity, Hawker, Fréchet and co-workers pioneered a series of core-functionalised star polymers. They reported a soluble, highly branched, multi-arm star polymer as catalyst support (Fig. 4).19 First, they introduced a hydrophilic macromolecular “arm” to increase solubility. This “arm” was then used as a macroinitiator to co-polymerize functional monomers to afford either core-confined p-toluenesulfonic acid (PTSA) or 4-(dimethylamino)pyridine (DMAP) containing star polymers. Control experiments against molecular DMAP or PTSA demonstrated sterically constrained environments of the star polymers that efficiently impeded mutual interference between the two incompatible catalysts. This methodology allowed for acetal hydrolysis followed by a Baylis–Hillman reaction sequence. Later, Fréchet and co-workers extended this concept of soluble yet non-interpenetrating star polymer catalysts for an one-pot combination of iminium, enamine, and hydrogen-bond catalysis.20 This was the first example of nonorthogonal tandem catalysis that involved multiple asymmetric reactions and generated cascade products with multiple chiral centres. In 2010, Fréchet and co-workers modified the previous star polymer structure with a “clickable” core that provided viability and tunability of the core microenvironments.21 Water-soluble poly(ethylene glycol) (chiral proline catalyst), molecular PTSA, and pyrrolidine were functionalised at the core of the water dispersible star polymers to form a chiral polymeric catalyst, an acid polymeric catalyst, and a base polymeric catalyst, respectively. In this way, not only was nonorthogonal acid–base tandem catalysis achieved, but also the diffusion of hydrophobic compounds and asymmetric catalysis was successfully demonstrated.
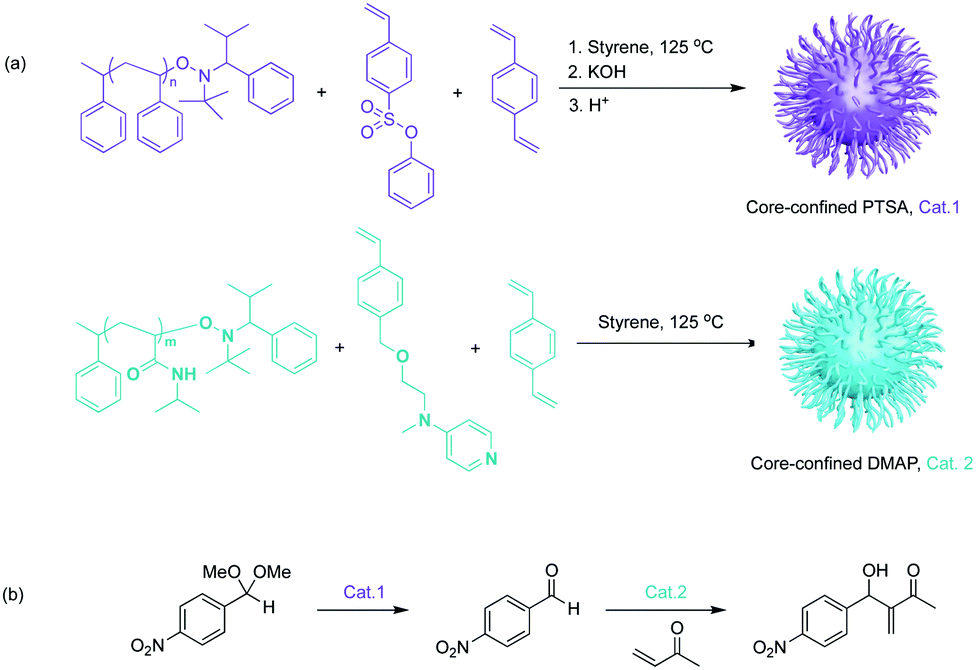 | ||
| Fig. 4 Soluble star polymers with core-confined catalysts for nonorthogonal tandem catalysis. (a) Synthesis of star polymers that contain core-confined PTSA or DMAP. (b) One-pot tandem catalysis involving acid-catalysed acetal hydrolysis followed by the base-catalysed Baylis–Hillman reaction. | ||
Besides focusing on nanosized supports with different solubility, material scientists also synthesized mesoscale organic materials for nonorthogonal tandem catalysis (Fig. 2), such as thimbles,22 self-sorted hydrogels,23 microporous hyper-cross-linked polymers,24 and electrospun and two-dimensional (2D) printing structures.25 The key is to introduce orthogonality for compartmentalisation of molecular catalysts at a mesoscale level. Bowden and co-workers introduced PDMS thimbles (Fig. 5).22 PDMS is hydrophobic and selectively permeable for neutral compounds with low molecular weight. Bowden and co-workers added polymer-bound or molecular PTSA and substrates to the interior of the PDMS thimble, while adding polymer-bound DMAP to the exterior of the thimble. The polymer-bound catalysts or charged molecules have very low flux through the PDMS membrane, while neutral organic molecules have high flux. This results in the compartmentalisation of multiple incompatible catalysts while substrates can react with catalysts on either side of the membrane. The acid–base catalysed acetalization and Baylis–Hillman tandem reactions succeeded with up to 93% conversion.
 | ||
| Fig. 5 PDMS thimble enabled compartmentalisation for nonorthogonal tandem catalysis. (a) The experimental set-up with a PDMS thimble contained in a glass vial is shown. (b) One-pot tandem catalysis involving acid-catalysed acetal hydrolysis (interior of the thimble) followed by the base-catalysed Baylis–Hillman reaction (exterior of the thimble). | ||
3. Compartmentalisation of molecular catalysts on multifunctional supports
Over the past two decades, the focus has transitioned from using a combination of discrete monofunctional supports to single multifunctional supports that are integrated with several active sites that are nonorthogonal but can operate in tandem. One of the potential benefits of co-localizing multiple incompatible catalysts on one support, compared to discrete monofunctional supports, is cooperative behaviour between active sites, which often affords higher reactivities.26 Additionally, orchestrated arrangements of the positions of catalysts within a single structure can improve catalytic efficiency by regulating substrate diffusion and substrate channelling, both of which are nearly impossible to achieve by using physical mixtures of monofunctional supports (Table 1).In this section, we describe the development of both inorganic and organic multi-functional supports using both a hierarchical as well as a chronological order (Fig. 6 and 7). Initial efforts focused on the co-existence of incompatible catalytic sites without compartmentalisation. With the innovation of more advanced synthetic techniques, compartmentalisation was introduced into the support structure through the isolation of opposing molecular catalysts into physically separated domains. Synthetic techniques are generally unique to either inorganic or organic structures yet contain commonality in terms of design principles: generation of heterogeneity in the material followed by orthogonal reactions to immobilize catalysts. These design principles enable precise attachment and spatial confinement of catalysts throughout the three-dimensional structure, such as the internal pore/exterior surface, the core–shell confinements, or via hierarchical porosities. Furthermore, besides compartmentalisation, recent designs of multifunctional supports mimic the responsive regulation, substrate screening, and substrate channelling in Nature to tackle grander challenges in multistep synthesis.
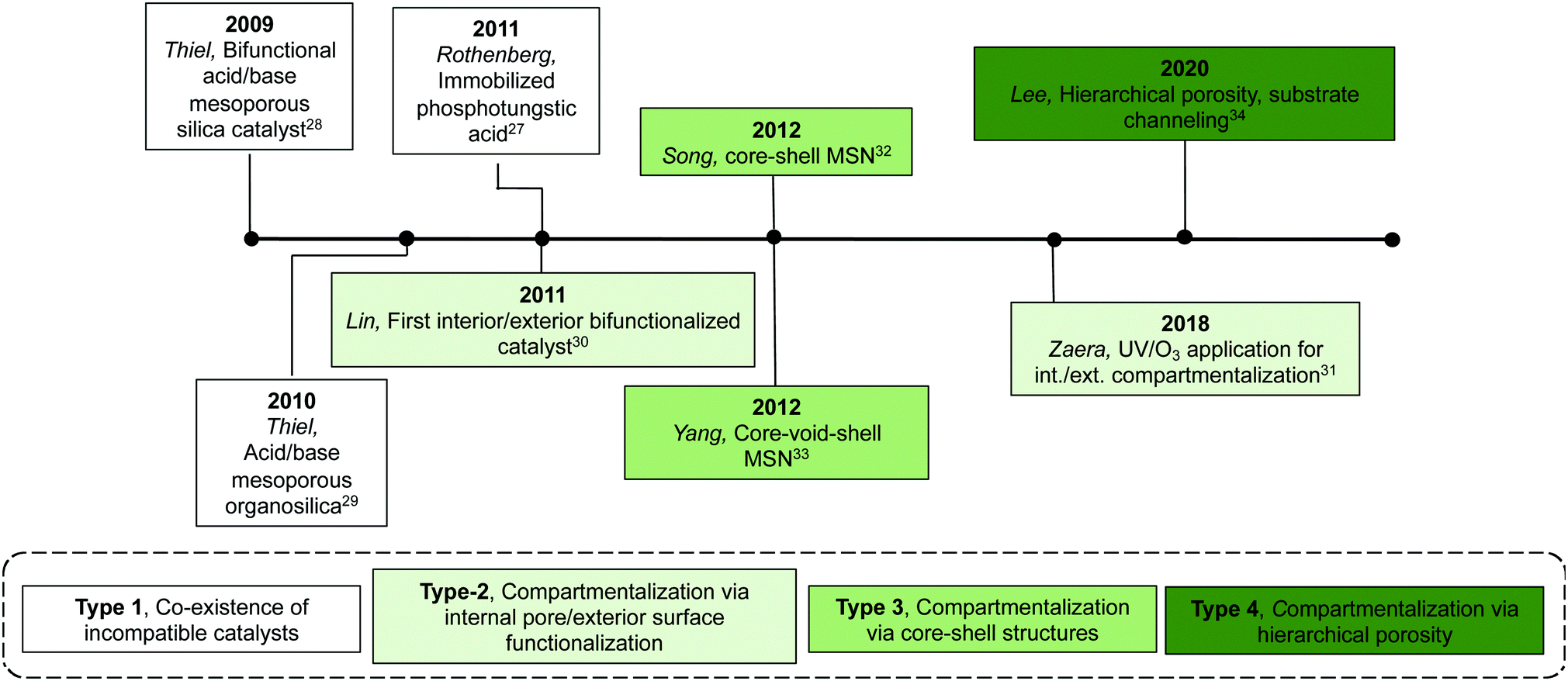 | ||
| Fig. 6 Timeline of the development of multifunctional inorganic supports for nonorthogonal catalysis. | ||
 | ||
| Fig. 7 Timeline of the development of multifunctional organic supports for nonorthogonal catalysis. | ||
3.1 Compartmentalisation using inorganic multifunctional supports
Table 2 shows current inorganic multifunctional supports that are classified into four types based on the topological distribution of two incompatible reactive sites in a single support: Type 1, random co-existence of incompatible catalysts; Type 2, compartmentalisation via internal pore/exterior surface functionalisation; Type 3, compartmentalisation via core–shell structures; and Type 4, compartmentalisation via hierarchical porosity.| Type | Compartmentalization approach | Pros | Cons | Graphical illustration |
|---|---|---|---|---|
| 1 | No direct approach used, sparse, undirected functionalization | Simple synthesis, effective catalysts | Ineffective control over compartmentalization of active sites, requires low catalyst loadings, clustered organosilanes result in quick deactivation |

|
| 2 | Interior pore structures plus exterior surfaces of particles | More effective method of physical isolation, amenable to any type of organosilanes | Requires particles with high external surface areas (small particles) |

|
| 3 | Core–shell compartments using primary and secondary growth stages | Efficient method to create physically isolated domains for catalysts, amenable to many more layers than two, intra-structure diffusion facilitate tandem catalysis | Depending on pore sizes and silica layer thicknesses, effective reaction rates could be hindered by diffusion rates, more complex synthesis |

|
| 4 | Hierarchical porosity and selective functionalization of meso and macropores | Excellent method to physically isolate catalysts in compartments, substrate channeling-pore hierarchy dictates the reaction sequence | Relatively complex synthetic procedures |
|
Even though Type-1 materials are feasible for nonorthogonal tandem catalysis, it is inevitable that a significant portion of the supported acids and bases are mutually quenched by one another, resulting in significant catalyst deactivation, thus failing to realize the full potential of the materials. This drawback was also reflected in using Type-1 materials in a flow reactor, as discussed in Section 2.2.1, where the authors found that using physical mixtures of discrete supported catalysts was more efficient in catalysing nonorthogonal tandem catalysis.15 The observed activity in these materials is likely the result of sufficient spacing between some of each incompatible species due to low grafting density or statistical variations of the surface distribution of different functional groups.
 | ||
| Fig. 8 Bifunctionalised mesoporous materials for compartmentalisation of acids and bases for hydrolysis–Henry reaction. (a) Synthetic strategy for dual acid–base catalysts. (b) One-pot tandem catalysis involving acid-catalysed acetal hydrolysis followed by the base-catalysed nitroaldol reaction. | ||
Type-2 materials represent the initial attempt to physically isolate a pair of incompatible catalysts inside a multifunctional inorganic support. Synthetic approaches are effective in achieving compartmentalisation and have generality for other types of nonorthogonal tandem catalysis due to facile synthesis and commercial availability of functional organosilanes. This strategy, however, relies on using materials with high external surface areas to achieve desired catalyst loadings, which limits the particle size of materials appropriate for this method.
The Type-3 material can be described as a core–shell mesoporous silica particle that is synthesised via the initial formation of the core, a secondary shell growth, and post-functionalisation modifications to install active sites. Two incompatible catalytic species are compartmentalised into the core and the shell domain of the structure. This core–shell layered arrangement of catalysts can simplify the diffusion pathway between catalytic centres to yield faster reaction rates compared with other multicatalytic systems. This system can be easily applied to other types of tandem sequences by using different functional organosilanes during co-condensation procedures. Additionally, there is potential to increase the number of catalytic sites by increasing the number of shells.
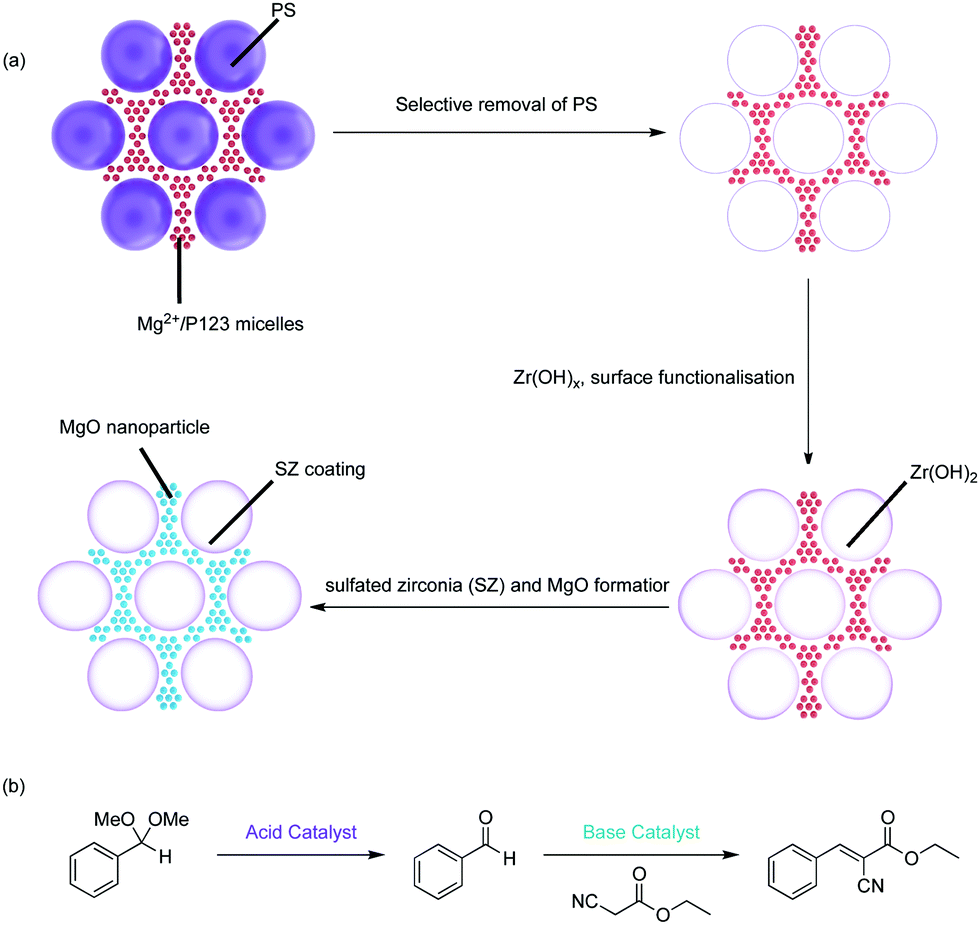 | ||
| Fig. 9 Spatially orthogonal hierarchically porous structure for compartmentalisation of acid–base catalyst for nonorthogonal tandem catalysis. (a) Synthetic route to a spatially orthogonal, acid–base hierarchically porous framework. (b) One-pot tandem catalysis involving acid-catalysed acetal hydrolysis followed by the base-catalysed Knoevenagel condensation. | ||
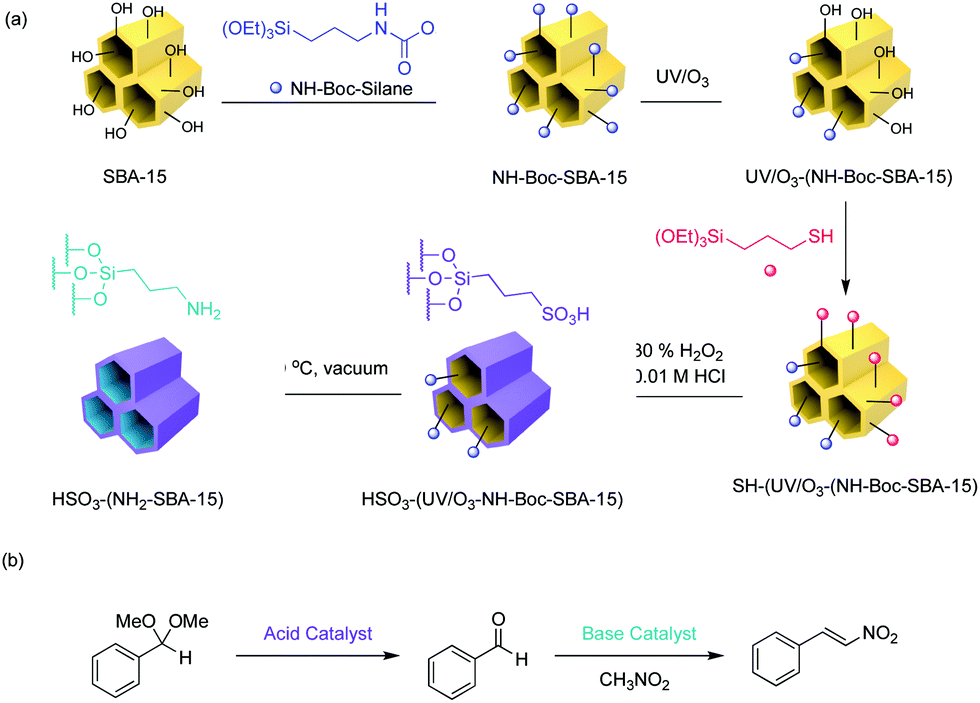
The Type-4 material uses pore hierarchy to implement the concept of substrate channelling, similar to biological systems. The substrate channelling efficiently directed the diffusion path and minimized wasted encounters between substrates and catalyst sites, which allows nonorthogonal tandem reactions as well as other catalytic processes involving antagonistic species. Additionally, using inorganic catalytic species such as Zr(OH)x and MgO is potentially beneficial in the cases of extreme reaction conditions, such as high temperatures.
3.2 Compartmentalisation using organic multifunctional supports
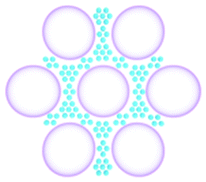
One of the goals in chemistry is to design artificial catalysts that rival biological systems in terms of structural complexity, intricate functionality, and catalytic efficiency. Organic materials with multiple functionalities have shown tremendous potential in the recreation of complex aspects of living systems.35 In an attempt to emulate a cell-like microenvironment using organic materials, techniques that are widely used in Nature, such as self-assembly and templating, are used to create structures with discrete compartments such as multicompartmental micelles, double-shelled hollow nanospheres, and core–shell colloids. Recently, compartmentalised organic materials have been designed with features such as substrate specificity and responsiveness that are difficult to achieve using conventional inorganic materials (Fig. 7).Early efforts in multifunctional organic supports pivoted from using a mixture of monofunctional structures to the simultaneous coexistence of incompatible active sites in one single support particle, without site-specific isolation. As mentioned in Section 2.2.2, in 2017, Gu, Tan, and co-workers reported the synthesis of microporous acid (sulfonic acid) and base (benzylamine) catalysts on a monofunctional PS-based hyper-crosslinked polymer (HCP), and used the mixture of catalysts for acid-based tandem catalysis.24 Later that year, the same group synthesised a hollow spherical nanostructure with a harmonious conjugation of sulfonic acid and amine sites, using similar HCPs.36 SiO2 particles were used as templates and then sacrificed to create the hollow core, followed by partial sulfonation on the HCP nanosphere. The authors subtly used the orthogonal reactivity between different functional aromatic rings to place the acid and base catalysts in their respective locations. The electron withdrawing nature of the sulfonate groups prevented amination on the same aromatic rings, thereby selectively functionalising the PS. In this way, two antagonistic species were covalently attached to the different aromatic rings of the HCP nanospheres, minimizing their interaction, and preventing mutual deactivation. The bi-functionalised HCP displayed excellent reactivity and chemical stability in series of nonorthogonal tandem catalysis (hydrolysis/Henry and hydrolysis/Knoevenagel) toward water and many organic solvents.
Benefiting from the advances in synthetic chemistry, well-defined polymeric materials can be used to compartmentalise multiple incompatible catalysts within one particle (Fig. 7). Block copolymers that self-assemble into nano-aggregates in selective solvents have long been investigated to mimic key attributes of biological systems such as hydrophobic pockets and compartmentalisation.37 Block copolymers can be synthesised via living polymerisation, offering precise control over key parameters, such as molecular weight, molecular weight distribution, composition, and functionality. These attributes lay the foundation to access well-defined nanostructured polymeric materials as well as to introduce orthogonality into the polymeric materials.38 Core–shell micelles are one of the most accessible and well-studied polymeric nanostructures, possessing two orthogonal nanodomains: a hydrophobic core and hydrophilic corona.38 To achieve microenvironments for catalysis, Weck and co-workers developed a series of shell cross-linked micelles (SCMs) for aqueous nonorthogonal tandem catalysis (Fig. 10). The micellar structures were composed of functional poly(2-oxazoline) that were synthesized via cationic ring-opening polymerisation (CROP), incorporating orthogonal functional groups along the polymer backbone. Upon micelle formation, these orthogonal functional groups can be placed in different nano-environments throughout the three-dimensional structure, which was stabilized via covalent cross-linking. The functional handles were further used for compartmentalisation of multiple incompatible molecular catalysts with great spatial resolution via orthogonal reactions. In 2015, SCMs were employed as the first example that enabled compartmentalisation of two incompatible transition metal catalysts in one nanoparticle for an enantioselective nonorthogonal tandem catalysis in one-pot.39 Co-porphyrin and Rh-N-tosylated 1,2-diphenyl-1,2-ethylenediamine (Rh-TsDPEN) were immobilized in the core and the shell, respectively, for a tandem alkyne hydration and asymmetric transfer hydrogenation (ATH). This bifunctional SCM converted the alkynes to enantioenriched secondary alcohols with excellent yields and enantiomeric excess (ee) (up to 99% yield, 97% ee). The SCM-enabled compartmentalisation strategy was further extended to three other one-pot nonorthogonal tandem reactions: an acid–base deacetalisation-nitroaldol reaction,40 a redox-driven one-pot deracemizations of secondary alcohols,41 and an enantioselective three-step tandem catalysis (Fig. 10).42 Besides compartmentalisation of incompatible species, SCMs as supports advance all nonorthogonal tandem catalysis through the use of water as the only solvent, enhanced reactivity of molecular catalysts, and structure recyclability.
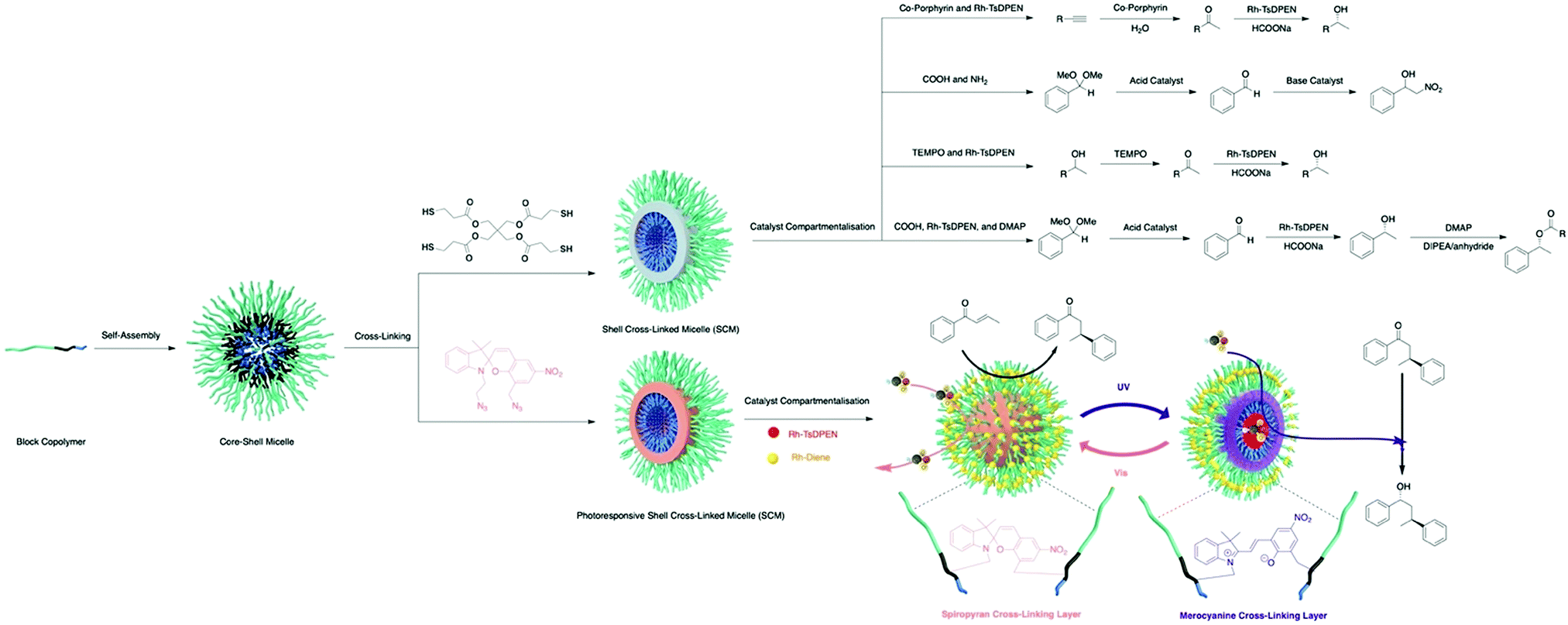 | ||
| Fig. 10 Synthesis of SCMs and SCM-enabled compartmentalisation and photoregulation for aqueous nonorthogonal tandem catalysis. | ||
In addition to SCMs, various types of multifunctional organic supports have been utilized for efficient compartmentalisation of incompatible molecular catalysts. Hollow mesoporous double-shelled polymeric nanospheres (HMOPNs) have a double layer structure and can be synthesized via emulsion polymerisation of functional styrene derivatives to site-isolate incompatible active sites into the layers. Ma and co-workers used HMOPNs to compartmentalise carboxylic acid and chiral proline species on the outer shell and chiral amine organocatalysts on the inner shell for the Michael addition and α-amination, respectively.43 The control experiments performed against homogeneous molecular catalysts indicated that it was critical to compartmentalise multiple catalysts for higher stereoselectivity of nonorthogonal enantioselective Michael addition/α-amination tandem catalysis in one-pot. Additionally, colloidal particles can be synthesized on a large scale and are promising candidates for scale-up processes. Gröschel and co-workers reported a scalable synthesis (up to 25 g) of core–shell micro-sized colloidal particles with an acidic core and a basic shell, using a two-step, surfactant-free, emulsion polymerisation technique.44 The resulting bifunctional catalysts can be used in the one-pot acid–base catalysed deacetalisation-Knoevenagel tandem reaction on a 10 gram scale. This work is a step towards addressing scalability limitations for industrial applications of compartmentalised structures for nonorthogonal tandem catalysis.
Aiming at further improving the efficacy of a multistep synthesis, researchers have incorporated strategies for molecular discrimination such as responsive regulation and molecular imprinting into the design of advanced compartmentalised multicatalytic systems. These efforts aid in overcoming challenges such as competitive reaction pathways and substrate selectivity. Weck and co-workers developed a photoresponsive SCM for compartmentalisation and photoregulating pathways of two incompatible and competing enantioselective catalysis in aqueous media (Fig. 10).45 By fabricating orthogonal yet dynamic domains, two features are incorporated: compartmentalisation of incompatible catalysts into two domains and activation of desired synthetic pathways through external triggers. In detail, a photoresponsive spiropyran cross-linking layer was introduced within a micelle. Rh-Diene-catalysed asymmetric 1,4-addition of phenylboronic acids was confined in the shell, while Rh-TsDPEN-catalysed ATH was confined in the core. The spiropyran cross-linking can respond to different light irradiation, resulting in switching the physicochemical properties of the SCMs for spatiotemporal modulation of mass transport for tandem catalysis. As a result, the SCM catalytic system can direct the reaction pathway toward a multi-chiral product with high conversions and enantioselectivities. Other regulation, such as thermoregulation, can also be achieved using thermoresponsive polymers for selective Suzuki coupling and ATH, for example.46,47
In addition to responsive regulation, molecular imprinting is another strategy to achieve substrate selectivity. Molecular imprinting creates template-shaped cavities in cross-linked matrices with predetermined selectivity and high affinity and is widely used for substrate recognition.48 Zhao and co-workers created molecularly imprinted nanoparticles (MINPs) for a substrate selective nonorthogonal tandem catalysis.48 The MINPs were based on cross-linked surfactant micelles with orthogonal functional sites. A basic amine catalyst was installed covalently on the surface of the micelles while a sulfonic acid catalyst was immobilized within the imprinted binding site in the hydrophobic core. The resulting bifunctional MINPs overrode the intrinsic reactivity of substrate to catalyse a highly selective acid–base deacetalisation-aldol tandem reaction. Besides molecular imprinting, surface functionalisation on the support can also enable the feature of substrate selectivity. Li and co-workers synthesised multicompartmental photonic spheres for substrate selective nonorthogonal tandem catalysis.49 The photonic spheres were synthesised via a sacrificial SiO2 template followed by successive etching and grafting techniques to isolate organic acid and amine catalysts in different polymeric layers within the photonic sphere. Additionally, various charged polymers were grafted on the surface of the pores by surface-initiated atom transfer radical polymerisation. Due to electrostatic repulsion, the charged polymer brushes only allowed neutral substrates to penetrate for hydrolysis and prevented charged substrate from reacting. As a comparison, without the polymer brush layer, there was no discrimination between a neutral substrate and a positively charged one. In addition to substrate selectivity, reaction progress monitoring can also be achieved by using the photonic properties of the particles. As the reagents diffuse within the layers of the photonic multicompartmental system, swelling of the pore structure is observed that shifted the photonic band gap and facilitated tandem reaction monitoring.
More recently, Jones and co-workers have demonstrated an approach that combines silica and polymer supported catalysts to create multi-domain hybrid systems for nonorthogonal tandem catalysis (Fig. 11).50 Mesoporous silica supports contain ordered channels that can be functionalised with active sites and are used as a catalytic host. Polymers not only provide additional sites for catalyst compartmentalisation but also offer a dynamic domain that can be tuned by external stimuli and regulate mass transfer within the silica structure. The authors functionalised the pores of SBA-15 or MCM-41 with polymers via a grafting-to process to yield a silica grafted Brønsted acid and polymer-supported Lewis base hybrid structure. Molecular dynamic simulations supported the experimental observation that the molecular weight of polymers was crucial for the fabrication of effective structures in terms of solubility, pore blockage, and mass transfer. The resulting bifunctional mesoporous silicate–polymer composite materials were then used for nonorthogonal acid–base catalysed deacetalisation–Knoevenagel condensation.
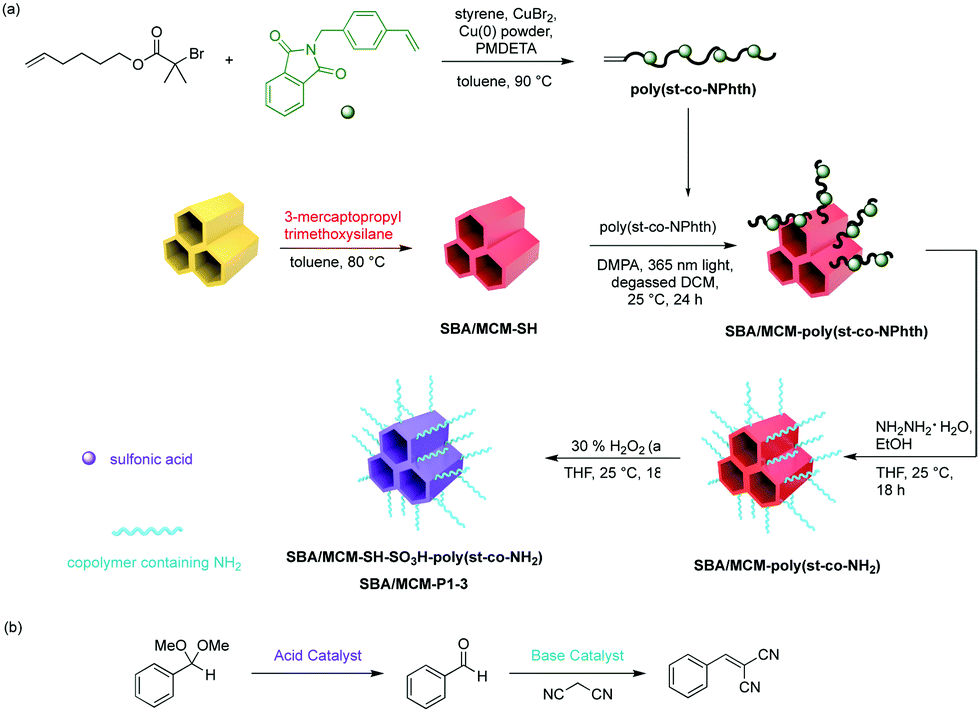 | ||
| Fig. 11 Creation of discrete active site domains via mesoporous silica–PS composite materials for nonorthogonal tandem catalysis. (a) Synthetic route for acid–base silica polymer composite catalysts. (b) One-pot tandem catalysis involving acid-catalysed acetal hydrolysis followed by the base-catalysed Knoevenagel condensation. | ||
4. Conclusions
Exploring and refining efficient multi-step catalytic processes is paramount for evolving the field of chemistry towards a more sustainable and environmentally friendly state. The development of nonorthogonal tandem catalysis provides an opportunity to use multiple incompatible catalysts in one-pot for a multistep synthesis and to eliminate tedious purifications of intermediates, thus improving financial and environmental costs. To achieve successful nonorthogonal tandem catalysis, innovation of materials that enable compartmentalisation of multiple catalysts is vital and a key to designing a compartmentalised material is to introduce orthogonality into the system. In this tutorial review, we outlined an expansive array of synthetic structures from the literature for nonorthogonal tandem catalysis using molecular catalysts. Earlier work began with the site isolation of one catalyst on a scaffold with the other catalyst in bulk solution, followed by the isolation of two or more catalysts on multiple discrete supports, and finally the compartmentalisation of two or more catalysts within a singular support particle. Researchers have mimicked compartmentalisation witnessed in Nature as well as substrate selectivity, substrate channelling, and reaction regulation through the exploration of both organic and inorganic supports for nonorthogonal tandem catalysis.Significant progress has been made toward synthesizing various compartmental supports with tailor-made microenvironments for nonorthogonal tandem catalysis. Design of future catalytic materials should consider the following: What catalysis challenge is the material designed to overcome? Is the goal to support as many catalysts as possible, regardless of compatibility, to perform one-pot procedures to achieve a high-throughput screening of valuable compounds? If so, then the focus should be on increasing the number of orthogonal functionalities for catalyst attachment and the microenvironments for more efficient compartmentalisation. For example, multicompartmental supports with more than two distinct nanodomains, such as multicompartmental micelles, hybrid materials that are comprised of polymers and inorganic domains, such as silica, have potential to support three or more antagonistic catalysts. Additionally, scale-up syntheses of the supported catalysts, such as the use of emulsion polymerisation as well as the use of continuous flow reactors or meso-scale materials have the potential to enable safe, large-scale, and efficient high-throughput screening. If the goal, however, is to develop tandem catalysis that targets the synthesis of complex compounds, such as natural products, then designing multiple orthogonal dynamic microenvironments in the catalytic system is vital to control the reaction sequence to yield products with high conversion and selectivity. It has been demonstrated that introducing responsive regulation can generate spatial-temporal controls over tandem catalysis and direct reaction pathways. Fabrication of multiresponsive catalytic materials with dynamic domains that respond to multiple, yet orthogonal stimuli have the potential to generate different products based on the manipulation of reaction sequences via the order/types of stimuli. Artificial intelligence (AI) and machine learning have emerged as powerful tools for retro-synthetic analysis and for designing efficient synthetic pathways in both academia and industry. Progress in the field of nonorthogonal tandem catalysis can enrich the library and combination of catalysts for AI, which can potentially lead to more efficient and sustainable AI-driven synthesis.
Synthesis of the next generation of supported catalysts calls for a better fundamental understanding of materials. Consider selectivity in supported tandem catalysis, this is not only governed by the intrinsic reactivity of catalysts but rather driven by kinetic selectivity disguised as mass-transfer and other factors. From macroscopic convection to molecular diffusion, every component in the diffusion process can have a substantial impact on the encounter between substrates/reagents and the catalysts, resulting in different reaction rates, and therefore different selectivity of tandem catalysis to form different products. Unfortunately, catalyst parameters such as catalyst spatial distribution and catalyst loading are not easily accessible and the understanding of compound diffusion in a confined nanoenvironment is limited. More research efforts need to focus on precise characterisation of these typically disordered, non-crystalline catalytic structures as well as study the relationship between material properties (e.g. pore diameters, cross-linking density, surface polymer chain length, and morphology of the support) and catalytic performance. A comprehensive comparison across all types of support structures is lacking and will provide insights into the advantages and limitations of different materials in supported catalysis. Additionally, using simulations side-by-side with experimental work can also deepen the understanding of the properties of the materials as well as provide guidelines for designing future catalytic supports.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding provided by the U.S. Department of Energy, Office of Basic Energy Sciences, through Catalysis Science Contract DE- FG02-03ER15459, is gratefully acknowledged.Notes and references
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC
.
- T. L. Lohr and T. J. Marks, Nat. Chem., 2015, 7, 477–482 CrossRef CAS PubMed
- C. M. Agapakis, P. M. Boyle and P. A. Silver, Nat. Chem. Biol., 2012, 8, 527–535 CrossRef CAS PubMed
- M. Heitbaum, F. Glorius and I. Escher, Angew. Chem., Int. Ed., 2006, 45, 4732–4762 CrossRef CAS PubMed
- J. Shi, Y. Wu, S. Zhang, Y. Tian, D. Yang and Z. Jiang, Chem. Soc. Rev., 2018, 47, 4295–4313 RSC
- Y.-B. Huang, J. Liang, X.-S. Wang and R. Cao, Chem. Soc. Rev., 2017, 46, 126–157 RSC
- Y. Tian and G. Zhu, Chem. Rev., 2020, 120, 8934–8986 CrossRef CAS PubMed
- B. J. Cohen, M. A. Kraus and A. Patchornik, J. Am. Chem. Soc., 1977, 99, 4165–4167 CrossRef CAS
- S. L. Poe, M. Kobašlija and D. T. McQuade, J. Am. Chem. Soc., 2006, 128, 15586–15587 CrossRef CAS PubMed
- P. Verma, Y. Kuwahara, K. Mori, R. Raja and H. Yamashita, Nanoscale, 2020, 12, 11333–11363 RSC
- F. Gelman, J. Blum and D. Avnir, J. Am. Chem. Soc., 2000, 122, 11999–12000 CrossRef CAS
- K. Motokura, N. Fujita, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2005, 127, 9674–9675 CrossRef CAS PubMed
- H. Yang, L. Fu, L. Wei, J. Liang and B. P. Binks, J. Am. Chem. Soc., 2015, 137, 1362–1371 CrossRef CAS PubMed
- N. Xue, G. Zhang, X. Zhang and H. Yang, Chem. Commun., 2018, 54, 13014–13017 RSC
- P. Borah, M. Fianchini and M. A. Pericàs, ACS Catal., 2021, 11, 6234–6242 CrossRef CAS
- J. Lu and P. H. Toy, Chem. Rev., 2009, 109, 815–838 CrossRef CAS PubMed
- A. W. Pilling, J. Boehmer and D. J. Dixon, Angew. Chem., Int. Ed., 2007, 46, 5428–5430 CrossRef CAS PubMed
- N. T. S. Phan, C. S. Gill, J. V. Nguyen, Z. J. Zhang and C. W. Jones, Angew. Chem., Int. Ed., 2006, 45, 2209–2212 CrossRef CAS PubMed
- B. Helms, S. J. Guillaudeu, Y. Xie, M. McMurdo, C. J. Hawker and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2005, 44, 6384–6387 CrossRef CAS PubMed
- Y. Chi, S. T. Scroggins and J. M. J. Fréchet, J. Am. Chem. Soc., 2008, 130, 6322–6323 CrossRef CAS PubMed
- V. Rodionov, H. Gao, S. Scroggins, D. A. Unruh, A.-J. Avestro and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 2570–2572 CrossRef CAS PubMed
- A. L. Miller II and N. B. Bowden, Adv. Mater., 2008, 20, 4195–4199 Search PubMed
- N. Singh, K. Zhang, C. A. Angulo-Pachón, E. Mendes, J. H. van Esch and B. Escuder, Chem. Sci., 2016, 7, 5568–5572 RSC
- K. Wang, Z. Jia, X. Yang, L. Wang, Y. Gu and B. Tan, J. Catal., 2017, 348, 168–176 CrossRef CAS
- M. O. Pretscher, G. Sitaru, M. Dietel, H. Schmalz, S. Gekle and S. Agarwal, ACS Appl. Polym. Mater., 2021, 3, 1349–1357 CrossRef CAS
- V. E. Collier, N. C. Ellebracht, G. I. Lindy, E. G. Moschetta and C. W. Jones, ACS Catal., 2016, 6, 460–468 CrossRef CAS
- N. R. Shiju, A. H. Alberts, S. Khalid, D. R. Brown and G. Rothenberg, Angew. Chem., Int. Ed., 2011, 50, 9615–9619 CrossRef CAS PubMed
- S. Shylesh, A. Wagner, A. Seifert, S. Ernst and W. R. Thiel, Chem. – Eur. J., 2009, 15, 7052–7062 CrossRef CAS PubMed
- S. Shylesh, A. Wagener, A. Seifert, S. Ernst and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 184–187 CrossRef CAS PubMed
- Y. Huang, S. Xu and V. S.-Y. Lin, Angew. Chem., Int. Ed., 2011, 50, 661–664 CrossRef CAS PubMed
- Z. Weng, T. Yu and F. Zaera, ACS Catal., 2018, 8, 2870–2879 CrossRef CAS
- P. Li, C.-Y. Cao, Z. Chen, H. Liu, Y. Yu and W.-G. Song, Chem. Commun., 2012, 48, 10541–10543 RSC
- Y. Yang, X. Liu, X. Li, J. Zhao, S. Bai, J. Liu and Q. Yang, Angew. Chem., Int. Ed., 2012, 51, 9164–9168 CrossRef CAS PubMed
- M. A. Isaacs, C. M. A. Parlett, N. Robinson, L. J. Durndell, J. C. Manayil, S. K. Beaumont, S. Jiang, N. S. Hondow, A. C. Lamb, D. Jampaiah, M. L. Johns, K. Wilson and A. F. Lee, Nat. Catal., 2020, 3, 921–931 CrossRef CAS
- M. Marguet, C. Bonduelle and S. Lecommandoux, Chem. Soc. Rev., 2013, 42, 512–529 RSC
- Z. Jia, K. Wang, B. Tan and Y. Gu, ACS Catal., 2017, 7, 3693–3702 CrossRef CAS
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC
- P. Qu, M. Kuepfert, E. Ahmed, F. Liu and M. Weck, Eur. J. Inorg. Chem., 2021, 1420–1427 CrossRef CAS
- J. Lu, J. Dimroth and M. Weck, J. Am. Chem. Soc., 2015, 137, 12984–12989 CrossRef CAS PubMed
- L.-C. Lee, J. Lu, M. Weck and C. W. Jones, ACS Catal., 2016, 6, 784–787 CrossRef CAS
- P. Qu, M. Kuepfert, S. Jockusch and M. Weck, ACS Catal., 2019, 9, 2701–2706 CrossRef CAS
- M. Kuepfert, A. E. Cohen, O. Cullen and M. Weck, Chem. – Eur. J., 2018, 24, 18648–18652 CrossRef CAS PubMed
- G. Xie, J. Zhang and X. Ma, ACS Catal., 2019, 9, 9081–9086 CrossRef CAS
- C. Chen, N. Janoszka, C. K. Wong, C. Gramse, R. Weberskirch and A. H. Gröschel, Angew. Chem., Int. Ed., 2021, 60, 237–241 CrossRef CAS PubMed
- P. Qu, M. Kuepfert, M. Hashmi and M. Weck, J. Am. Chem. Soc., 2021, 143, 4705–4713 CrossRef CAS PubMed
- X. Li, Y. Sun, S. Wang and X. Jia, ACS Appl. Mater. Interfaces, 2020, 12, 44094–44102 CrossRef CAS PubMed
- J. Meng, F. Chang, Y. Su, R. Liu, T. Cheng and G. Liu, ACS Catal., 2019, 9, 8693–8701 CrossRef CAS
- I. Bose and Y. Zhao, ACS Appl. Polym. Mater., 2021, 3, 2776–2784 CrossRef CAS PubMed
- K. Zhou, T. Tian, C. Wang, H. Zhao, N. Gao, H. Yin, P. Wang, B. J. Ravoo and G. Li, J. Am. Chem. Soc., 2020, 142, 20605–20615 CrossRef CAS PubMed
- J. W. Cleveland, D. R. Kumar, J. Cho, S. S. Jang and C. W. Jones, Catal. Sci. Technol., 2021, 11, 1311–1322 RSC
| This journal is © The Royal Society of Chemistry 2022 |