
Optical/electrochemical methods for detecting mitochondrial energy metabolism
Wenhui
Ji†
a,
Xiao
Tang†
a,
Wei
Du†
 a,
Yao
Lu
a,
Nanxiang
Wang
a,
Qiong
Wu
*a,
Wei
Wei
b,
Jie
Liu
a,
Haidong
Yu
c,
Bo
Ma
d,
Lin
Li
*ace and
Wei
Huang
*ace
a,
Yao
Lu
a,
Nanxiang
Wang
a,
Qiong
Wu
*a,
Wei
Wei
b,
Jie
Liu
a,
Haidong
Yu
c,
Bo
Ma
d,
Lin
Li
*ace and
Wei
Huang
*ace
aKey Laboratory of Flexible Electronics (KLOFE) & Institute of Advanced Materials (IAM), Nanjing Tech University (NanjingTech), Nanjing 211816, China. E-mail: iamqwu@njtech.edu.cn; iamlli@njtech.edu.cn; iamwhuang@njtech.edu.cn
bDepartment of General Surgery, Jiangsu Cancer Hospital & Jiangsu Institute of Cancer Research & The Affiliated Cancer Hospital of Nanjing Medical University, Nanjing 210009, China
cFrontiers Science Center for Flexible Electronics, Xi’an Institute of Flexible Electronics (IFE) and Xi’an Institute of Biomedical Materials & Engineering, Northwestern Polytechnical University, 127 West Youyi Road, Xi'an 710072, China
dSchool of Pharmaceutical Sciences, Nanjing Tech University (NanjingTech), 30 South Puzhu Road, Nanjing 211800, China
eThe Institute of Flexible Electronics (IFE, Future Technologies), Xiamen University, Xiamen 361005, China
First published on 18th November 2021
Abstract
This review highlights the biological importance of mitochondrial energy metabolism and the applications of multiple optical/electrochemical approaches to determine energy metabolites. Mitochondria, the main sites of oxidative phosphorylation and adenosine triphosphate (ATP) biosynthesis, provide the majority of energy required by aerobic cells for maintaining their physiological activity. They also participate in cell growth, differentiation, information transmission, and apoptosis. Multiple mitochondrial diseases, caused by internal or external factors, including oxidative stress, intense fluctuations of the ionic concentration, abnormal oxidative phosphorylation, changes in electron transport chain complex enzymes and mutations in mitochondrial DNA, can occur during mitochondrial energy metabolism. Therefore, developing accurate, sensitive, and specific methods for the in vivo and in vitro detection of mitochondrial energy metabolites is of great importance. In this review, we summarise the mitochondrial structure, functions, and crucial energy metabolic signalling pathways. The mechanism and applications of different optical/electrochemical methods are thoroughly reviewed. Finally, future research directions and challenges are proposed.
 Wenhui Ji | Wenhui Ji received his BSc degree in Pharmacy from South-Central Minzu University in 2018. He is now a PhD candidate under the supervision of Prof. Lin Li in the Institute of Advanced Materials (IAM), Nanjing Tech University (NanjingTech). His current research interests are the flexible electronic devices for biological applications. |
 Xiao Tang | Xiao Tang received his BSc degree in Applied Chemistry from Nanjing University (Jinling College) in 2018. He received his MSc degree in Organic Chemistry in Prof. Lin Li's group in the Institute of Advanced Materials (IAM), Nanjing Tech University (NanjingTech). His current research interests are synthetic NIR organic small molecule fluorescent probes. |
 Wei Du | Dr Wei Du received his BSc and PhD in Chemistry from Fuyang Normal University (2013) and Anhui University (2018), respectively. He carried out postdoctoral studies with Academician Wei Huang in Northwestern Polytechnical University (NPU) from 2018 to 2020. He obtained his current postdoctoral position under the supervision of Prof. Lin Li in Nanjing Tech University (NanjingTech) from 2020. His current research interests are design of fluorescent sensors for the detection of biomolecules and assembly of nanocarriers for protein drug delivery. |
 Qiong Wu | Prof. Qiong Wu received her BSc in biotechnology from Nanjing Tech University in 2006. She completed her PhD in Chemistry from Ewha Womans University in 2012 and received postdoctoral training at the above two universities under the supervision of Prof. Jinheung Kim and academician Wei Huang, respectively. Then she obtained her current position in 2018. Her research interests includes flexible electronic devices for biological applications and rapid pre-examination of cardiovascular risk and mitochondrial diseases. |
 Lin Li | Prof. Lin Li received his BSc degrees (Chemistry) and PhD degrees (Chemistry) from Anhui University (AHU, China) in July 2004 and July 2009, respectively. He carried out postdoctoral training (Chemical Biology, 2009–2015) with Prof. Shao Q. Yao at National University of Singapore (NUS, Singapore). After that, he obtained his current full professor position in Nanjing Tech University (NanjingTech, China) in April 2015 after joining Academician Huang’s group. He is the young committee member of BioMedical Photonics Committee of Chinese Society of Biomedical Engineering (BMPC of CSBME). His current research interest is “Organic Biomedical Photonics”, which mainly focuses on the synthetic small-molecule indicators/drugs for biomarkers identification/detection/regulation in physiological and pathological environments, especially in the mitochondrion. |
 Wei Huang | Prof. Wei Huang received his PhD from Peking University in January 1992. Then he began his postdoctoral research with the National University of Singapore (NUS). In April 1995, he joined the Institute of Materials Research and Engineering (IMRE), which was newly founded by Prof. SHIH Choon Fong, to establish a laboratory of Organic Electronics and Information Displays. Afterwards, he moved to Fudan University where he founded the Institute of Advanced Materials (IAM). In June 2006, he was appointed as the Deputy President of the Nanjing University of Posts and Telecommunications. Then he assumed his duty as the President of Nanjing Tech University (NanjingTech) in July 2012. Then he was appointed as the Deputy President and Provost of the Northwestern Polytechnical University in April 2017. His research interests include organic/plastic/printable/bio-/flexible electronics, nano-materials and nanotechnology. |
1. Introduction
A dynamic change in metabolism involves ions, metabolic intermediates, products of biochemical reactions, proteins, and nucleic acids, and usually reflects the metabolic status of the cells, organs and human body; these changes can be used as discovery tools or as biomarkers of diseases.1,2 Mitochondria, derived from an α-proteobacteria-related ancestor, are essential for metabolism in eukaryotic cells, as they provide the majority of energy that aerobic cells need to maintain their physiological activity. Additionally, they are crucial to metabolic pathways such as fatty acid oxidation, pyruvate oxidation, and the tricarboxylic acid (TCA) cycle that are also related to numerous diseases, including Alzheimer's disease (AD),3 Parkinson's disease (PD),4 Huntington's disease, diabetes,5 and cancer.6 Therefore, mitochondrial energy metabolism is gradually becoming a research hotspot in medicine, biology, and chemistry.During energy generation, most of the biological oxidation occurs in the mitochondria, except for the glycolysis step, which occurs in the cytoplasm. Mitochondrial energy metabolism is regulated by various factors. Therefore, out-of-control mitochondrial energy metabolism results in mitochondrial dysfunction and eventually, mitochondrial diseases. In general, abnormal mitochondrial energy metabolism is divided into two groups. The first involves a decrease in enzymatic or protein activities or a fluctuation in the concentration of biochemical substances during adenosine triphosphate (ATP) generation, including oxidative phosphorylation, fatty acid oxidation, pyruvate metabolism, and amino acid metabolism, and the production of excess reactive oxygen species (ROS). The second involves the alteration of several factors involved in regulating ATP energy metabolism, including mitochondrial DNA (mtDNA) mutations, mitophagy, fusion, and division. In short, many of the mitochondrial metabolic substances are often directly or indirectly associated with human health or diseases and can be targeted as therapeutics. Furthermore, the decrease in mitochondrial abundance and activity is also a marker of aging in different organisms.7 In addition, targeting mitochondrial energy metabolism is considered to be a promising strategy for killing cancer cells by inhibiting the production of ATP, generating excessive ROS, and affecting the functions of mitochondrial antiapoptotic proteins.8
Mitochondria feature interconnected metabolic pathways, which link to other metabolic and regulatory pathways in the cells. Therefore, real-time analysis methods with high sensitivity, high selectivity, and subcellular organelle specificity are urgently needed to understand specific functions and variations in physiological metabolite concentration, and to systematically map the mitochondrial energy metabolism networks. This could potentially reveal novel ways to investigate the mechanisms in mitochondrial diseases associated with abnormal mitochondrial energy metabolism. Over the years, numerous efforts have been made to diagnose and monitor changes in mitochondrial energy metabolites in different biological samples (e.g., cells, cell lysates, serum, and tissues), as well as to develop diverse assays, including enzyme-linked immunosorbent assays (ELISA),9 immunofluorescence assays,10 chromatography, mass spectrometry,11 and gel electrophoresis.12 However, these conventional methods have some drawbacks and are thus not conducive to dynamic, specific, and highly sensitive monitoring of mitochondrial metabolite changes in complex samples. For example, ELISA has low accuracy, chromatography or mass spectrometry requires destruction of the sample and is costly, and gel electrophoresis demands specialised personnel and is time-consuming. To solve these problems, optical/electrochemical-based detection methods have been developed, which have shown remarkable merits regarding the detection performance, accessibility, and practicality for continuous or frequent early detection of disease models.13–17 However, several disadvantages of these optical/electrochemical methods may limit their application, for example, irrelevant molecules interfere with the target recognition process when applying electrochemical methods.18 Therefore, optical/electrochemical methods may enable researchers to develop novel detection methods for the diagnosis of mitochondrial diseases.
In this review (Scheme 1), we summarise the crucial molecular pathways of mitochondrial energy metabolism, as well as various optical/electrochemical approaches and their bioapplications in the determination of mitochondrial energy metabolites. Section 1 provides a brief introduction to the background, Section 2 introduces the mitochondrial structure and five metabolic signalling pathways, Section 3 summarises the mechanism of diversified optical/electrochemical methods, and Section 4 reviews the applications of these methods for detecting mitochondrial energy metabolism. Finally, the potential limitations and prospects of the analytical methods for the detection of mitochondrial energy metabolism are evaluated.
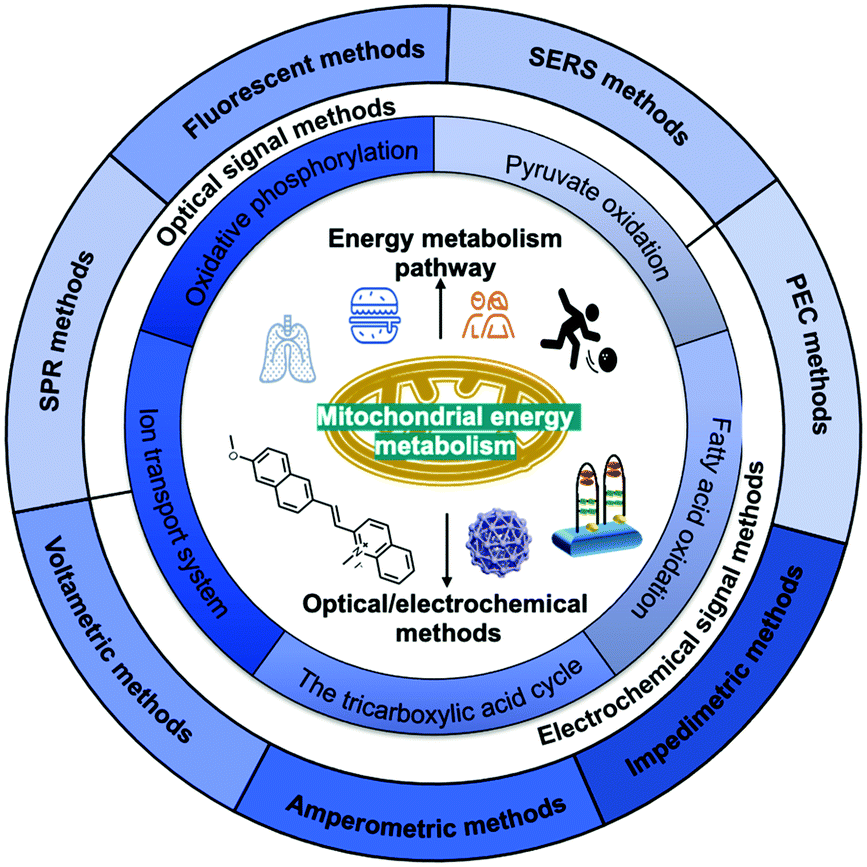 | ||
| Scheme 1 Scheme showing the main structure and themes of this article. In this review, we comprehensively summarized the biological functions of mitochondrial energy metabolism, the five major energy metabolic pathways and the application of multiple optical/electrochemical approaches to determine energy metabolites. | ||
2. Mitochondrial structure, function, and energy metabolism pathways
2.1 Mitochondrial structure and function
Mitochondria are unusual, small, dynamic subcellular organelles, consisting of two major membranes and their own genome, called mtDNA. In terms of structure, the outer membrane contains a channel-forming protein known as the voltage-dependent anion channel (VDAC) that is permeable to ions, sugars, amino acids, and other hydrophilic molecules. The inner membrane is folded into impermeable cristae and provides the site of oxidative phosphorylation (OXPHOS), which is carried out by complexes I–IV and ATP synthase, and complexes I–IV form the electron transport chain (ETC). Between the two membranes is the intermembrane space, containing various biochemical substrates such as soluble enzymes, cofactors, and the mitochondrial matrix, where the citric acid cycle produces reduced substrates.19 Mitochondria have multiple functions, including producing the largest amount of cellular ATP in the majority of aerobic eukaryotic cells via several crucial metabolic pathways, including pyruvate oxidation, fatty acid oxidation, TCA, and transport system (Fig. 1).20 Important metabolic pathways and their metabolites are summarised in Table 1.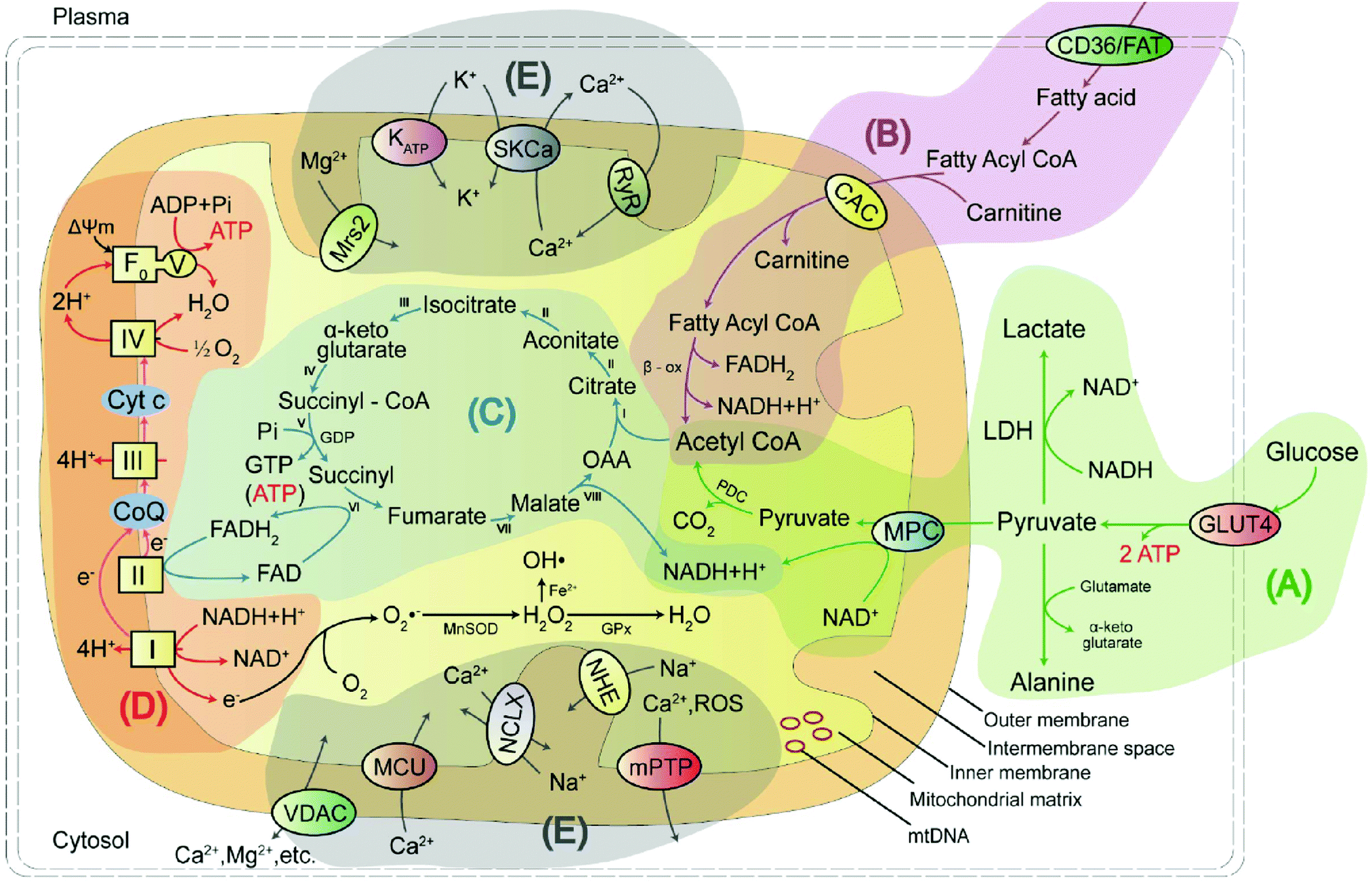 | ||
| Fig. 1 The mitochondrial structure consists of an outer membrane, intermembrane space, inner membrane, matrix, mitochondrial DNA (mtDNA), and molecular energy pathways, which include (A) the pyruvate oxidation, (B) fatty acid oxidation, (C) tricarboxylic acid cycle (TCA), (D) oxidative phosphorylation (OXPHOS) and (E) ion transport systems. GLUT4: glucose transporter type 4; LDH: lactate dehydrogenase; FAT: fatty acid translocase; CAC: carnitine/acylcarnitine carrier; TCA: (I) citrate synthase; (II) aconitase; (III) isocitrate dehydrogenase; (IV) α-ketoglutarate dehydrogenase complex; (V) succinyl-CoA synthetase; (VI) succinate dehydrogenase; (VII) fumarase; and (VIII) malate dehydrogenase. | ||
| Energy metabolic pathway | Metabolites/biochemicals |
|---|---|
| Pyruvate oxidation pathway | Pyruvate, alanine, lactate, acetyl-CoA, mitochondrial pyruvate carrier, α-lipoic acid, etc. |
| Fatty acid oxidation pathway | Fatty acid, fatty acyl-CoA, carnitine, acetyl-CoA, fatty acid oxidation, p53, etc. |
| Tricarboxylic acid cycle (TCA) pathway | Oxaloacetic acid, citrate, aconitate, isocitrate, α-ketoglutarate, succinyl-CoA, succinyl, fumarate, malate, NADH/NAD+, NADPH/NADP+, FAD/FADH2, malate dehydrogenase, etc. |
| Oxidative phosphorylation (OXPHOS) | Coenzyme Q, cytochrome c, reactive oxygen species, reactive nitrogen species, reactive sulphur species, adenosine triphosphate, caspase-3, γ-glutamyl transpeptidase, nitroreductase, thioredoxin reductase 2, methionine sulphoxide reductases, etc. |
| Transport system | K+, H+, Ca2+, Zn2+, Cu+/2+, Fe2+, Na+etc. |
Pyruvate oxidation is one of the main sources of mitochondrial energy and is the metabolic hub of three major nutrients. It has two pathways to achieve antioxidant function in cells. Defects in this pathway, or in the related markers, may play an essential function in disease progression. Fatty acid oxidation is another main source of mitochondrial energy, and a deficiency in enzyme activity may lead to disorder in energy metabolism. The TCA cycle feeds into other pathways and provides a mutual transformation of various metabolites. Disruption of the key rate-limiting enzymes and important substrates involved in energy metabolism will result in the TCA cycle being blocked. OXPHOS is the primary source of mitochondrial ATP and the main site of ROS production. Excess ROS levels cause oxidative stress, which affects the ETC process and hinders ATP synthesis. The ions in mitochondria not only maintain the basic structure of proteins, nucleic acids, enzymes, and other biomolecules but also play a regulatory role, thus affecting the normal metabolism of the body.
2.2 Mitochondrial energy metabolism pathways
As shown in Fig. 1A, pyruvate produced by glycolysis enters the mitochondria via a mitochondrial pyruvate carrier (MPC) and is catalysed by the pyruvate dehydrogenase complex (PDC) to produce acetyl coenzyme A (acetyl-CoA). Acetyl-CoA then combines with oxaloacetic acid (OAA) and participates in the TCA cycle. This mechanism may be involved in increasing autophagy by weakening the activity of mammalian target of rapamycin (mTOR) in neurons and inhibiting neuroinflammation that directly obstructs MPC.23 It has also been suggested that Ca2+ uptake via the mitochondrial calcium uniporter can stimulate intramitochondrial pyruvate dehydrogenase phosphatase (PDP), which activates pyruvate dehydrogenase (PDH) and increases ATP production via dephosphorylation of serine residues in the E1α subunit of PDH.24 More importantly, pyruvate exhibits neuroprotective effects in neuronal cells and has antioxidant functions. The antioxidant effect of pyruvate occurs mainly through two pathways: (1) the inhibition of hydrogen peroxide through a non-enzymatic decarbonylation reaction; and (2) the enhancement of the TCA cycle with supplemented pyruvate, which subsequently leads to more citric acid production. This inhibits phosphofructokinase and produces nicotinamide adenine nucleotide phosphate (NADPH), ultimately increasing the capacity of the glutathione (GSH) antioxidant system.
Intracellular metabolic status is commonly used to assess disease models and mitochondrial status. For example, the lactate, NAD+/NADH ratio is most commonly used as an indicator for the assessment of cancer or aging in animal models.30,31 This stems from two main aspects; on the one hand, recently, recent numerous studies indicated that NAD+ levels are reduced in aging mouse models and that supplementation with exogenous NAD+ delays aging in mice.32,33 As the NAD+ molecules are large and do not easily cross the cell membrane, it is difficult to supplement NAD+ directly in the human body. On the other hand, most cancer cells use the Warburg effect, in which the cells exhibit increased glycolysis and lactic acid production even under aerobic conditions. Therefore, the above indicators are commonly used to evaluate the corresponding models.
p53 activates glutaminase 2 (GLS2), thereby accelerating the conversion of glutamine to glutamic acid and increasing the production of α-ketoglutarate (α-KA), a key product in the TCA cycle, to promote OXPHOS.34 p53 can also polymerise cytochrome c (cyt c) oxidase, reduce oxygen consumption during aerobic respiration, and produce ATP, and plays a vital role in the energy metabolism.35
The mitochondrial OXPHOS provides the majority of the energy required by aerobic cells for survival. It includes NADH–ubiquinone (complex I), succinic–ubiquinone reductase (complex II), ubiquinone–cytochrome c reductase (complex III), cytochrome c oxidase (complex IV), and ATP synthase (complex V), whose functions are to carry out electron transport, H+ transport, oxygen utilisation, and H2O and ATP production. Briefly, NADH and FADH2 stemming from glycolysis, fatty acid oxidation, and the citric acid cycle, are used as electron donors. Electrons move towards enzyme complexes and release energy, part of which is used to generate ATP, while the rest is lost as heat energy. Protons are pumped from the matrix to the intermembrane space via proton pumps (complexes I, III, and IV) which generate an electrochemical gradient consisting of proton concentration gradient and membrane potential. When the protons re-enter the mitochondrial matrix through complex V, ADP and Pi are driven by ATP synthase to generate ATP using the stored energy from the mitochondrial membrane potential (MMP, ΔΨm).36 Approximately 30–40% of mitochondrial diseases are caused by deficiencies in these mitochondrial OXPHOS enzyme complexes. The mitochondrial ETC has several redox sites, including complexes I, II, and III. If the electrons leak to O2 during their transfer from these complexes, O2 is reduced to form superoxide, which is then converted to hydrogen peroxide (H2O2) by MnSOD in the matrix or superoxide dismutase (Cu/Zn-SOD) in the intermembrane space, which is the major source of ROS in mitochondria. When the levels of these oxides increase in the cells, the oxidant and antioxidant systems become dysregulated and the cells enter an oxidative stress state. Excessive ROS can uncouple the mitochondrial ETC, downregulate mitochondrial energy metabolism, upregulate the expression of the pro-apoptotic protein Bax, and finally rupture the outer mitochondrial membrane and cause apoptosis.37
Targeting OXPHOS can be used to develop not only different disease models but also disease therapeutics. On the one hand, some oxidative phosphorylation inhibitors, such as ETC inhibitors, phosphorylation inhibitors, and uncoupling agents, are usually utilised to develop specific disease models. For example, rotenone is a highly lipid-soluble inhibitor of mitochondrial ETC complex I. It can block electron transport and ATP synthesis and is often used in PD models, owing to its ability to selectively induce nigrostriatal dopamine degeneration.38 On the other hand, tumour cells are capable of bioenergetics and substance synthesis via OXPHOS, which can be used to treat diseases. IACS-010759, a small molecule inhibitor of mitochondrial ETC complex I, has shown high selectivity in phase I clinical settings. Compound screening and structural optimisation have revealed its anti-tumour mechanism in acute myelogenous leukaemia (AML), malignant glioma, and neuroblastoma.39
Mitochondria produce ATP through OXPHOS, while changes in the concentration of Ca2+ in mitochondria will dynamically regulate the rate of ATP production, the number of Ca2+-related proteins and enzymes, and the involvement in the TCA cycle, fatty acid oxidation and apoptosis. The influx of Ca2+ is mainly regulated by the VDAC, mitochondrial calcium uniporter (MCU) and mitochondrial ryanodine receptor (mRYR) transporter.42,43 Of these, VDAC allows Ca2+ and some small molecules to enter and exit the mitochondria, and MCU is a mitochondrial calcium selective channel that regulates the balance of Ca2+ concentration between the cytoplasm and the mitochondrial matrix, thus preventing excessive or insufficient calcium uptake into the mitochondria. Mitochondrial Ca2+ efflux is primarily the responsibility of mitochondrial Ca2+/H+ transport proteins (e.g., Letm1), Na+/Ca2+ exchange proteins (e.g., NCLX) and the mitochondrial permeability transition pore (mPTP).44,45
K+ in mitochondria has an important role in maintaining volume homeostasis, with its ability to prevent excessive mitochondrial swelling or shrinkage. Transport channels that regulate mitochondrial K+ influx, including mitochondrial ATP-sensitive K+ (mitoKATP) channels, Ca2+-activated K+ (KCa) channels, voltage-gated K+ (Kv) channels, and TWIK-related acid-sensitive K+ channel 3 (mitoTASK-3) have been identified.46,47 K+ efflux occurs through the mitochondrial K+/H+ exchanger (KHE), which is activated as the mitochondrial volume expands, in order to prevent excessive matrix swelling.48
Na+ not only acts as a regulator of membrane potential, but also modulates OXPHOS function and ROS production by regulating the fluidity of the inner mitochondrial membrane.49 Mitochondrial Na+ efflux is governed by the mitochondrial Na+/H+ exchanger (NHE).50 In contrast, Na+ influx into the mitochondria is primarily mediated by the mitochondrial Na+/Ca2+ exchanger (NCLX).51 Specifically, elevated Na+ concentrations promote mitochondrial Ca2+ efflux via the NCLX, which is an important mechanism for maintaining the Na+–Ca2+ homeostasis within the mitochondria.
Many enzymatic reactions in mitochondrial ATP production are dependent on Mg2+, which serves not only as an important enzyme cofactor, but also as an antagonist of Ca2+. The influx of Mg2+ is regulated by the mitochondrial RNA splicing protein 2 (MRS2) transporter located in the inner mitochondrial membrane, which selectively transports Mg2+ and is involved in the apoptotic process.52 The efflux of Mg2+ is regulated by SLC41A3 and Mme1 proteins, which together with the Mg2+ internal transporters regulate mitochondrial Mg2+ homeostasis.53
Mitochondria contain 20–50% of the total cellular iron. Important mitochondrial iron-containing proteins are all involved in the ETC function, including the subunits of succinate dehydrogenase, Rieske iron–sulphur protein, and Fe–S. Thus, changes in mitochondrial iron concentration can lead to alterations in iron–sulphur (Fe–S) homeostasis, impaired Fe–S biogenesis, impaired haem synthesis, mitochondrial dysfunction, and increased oxidative stress. Currently, some of the reported transporters of iron ions include MFRN (SLC25A37) and transferrin/transferrin receptor 2 (Tf/TfR2), all of which may be involved in the uptake of mitochondrial iron ions.54,55
3. Detection mechanisms of optical/electrochemical methods
Numerous efforts have been made to develop advanced techniques for qualitatively or quantitatively detecting or profiling key mitochondrial energy metabolites in different biological samples. These include cells, tissues, cell lysates, and human serum or plasma. Generally, the signal generated by a recognition element (e.g., antibody and adaptor) interacting with the target analytes can be divided into optical signals (e.g., absorption and emission) and electrical signals (e.g., current, impedance, and potential) as shown in Fig. 2. Methods based on optical strategies exhibit high signal-to-noise ratios and are simple to construct. Comparatively, methods based on electrical strategies are highly sensitive and specific and have a fast response. However, nonspecific signals in biological samples need to be minimised. In Section 3, we will focus on the mechanisms and methodologies of optical approaches, including fluorescence, surface-enhanced Raman scattering (SERS), and surface plasmon resonance (SPR). In addition, we will discuss the electrochemical approaches involving electrochemical methods such as voltammetry, amperometry, electrochemical impedance spectroscopy (EIS), and photoelectrochemical (PEC) analysis.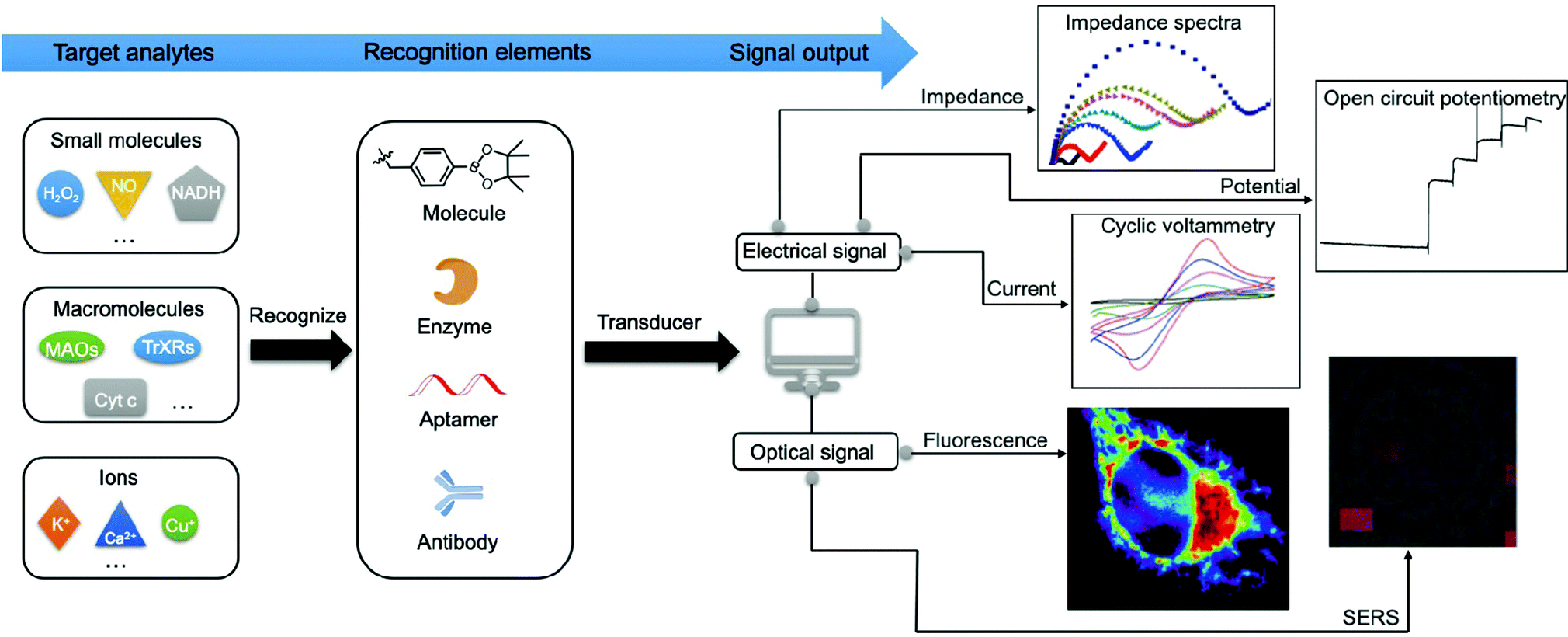 | ||
| Fig. 2 Schematic showing the detection methods based on optical and electrical signals. The signal generated by recognition elements (e.g., antibody and adaptor) interacting with specific target analytes can be divided into optical signals (e.g., fluorescence and SERS) and electrical signals (e.g., current, impedance, and potential). | ||
3.1. Optical signal methods
(1) Organic small molecular fluorescent probes. Organic small molecular fluorescent probes are widely applied in biological detection and imaging because of their excellent biocompatibility, easy modification, and fast response times.56 They are mainly composed of fluorophores, recognition groups, linkers, and sometimes targeting groups. A fluorophore is quenched when the recognition group is not bound to the target, and the fluorescence is restored through the effects of fluorescence resonance energy transfer (FRET), photo-induced electron transfer (PET), intramolecular charge transfer (ICT), and excited-state intramolecular proton transfer (ESIPT) while binding to the target, thereby converting biological signals into optical signals (Fig. 3A).57
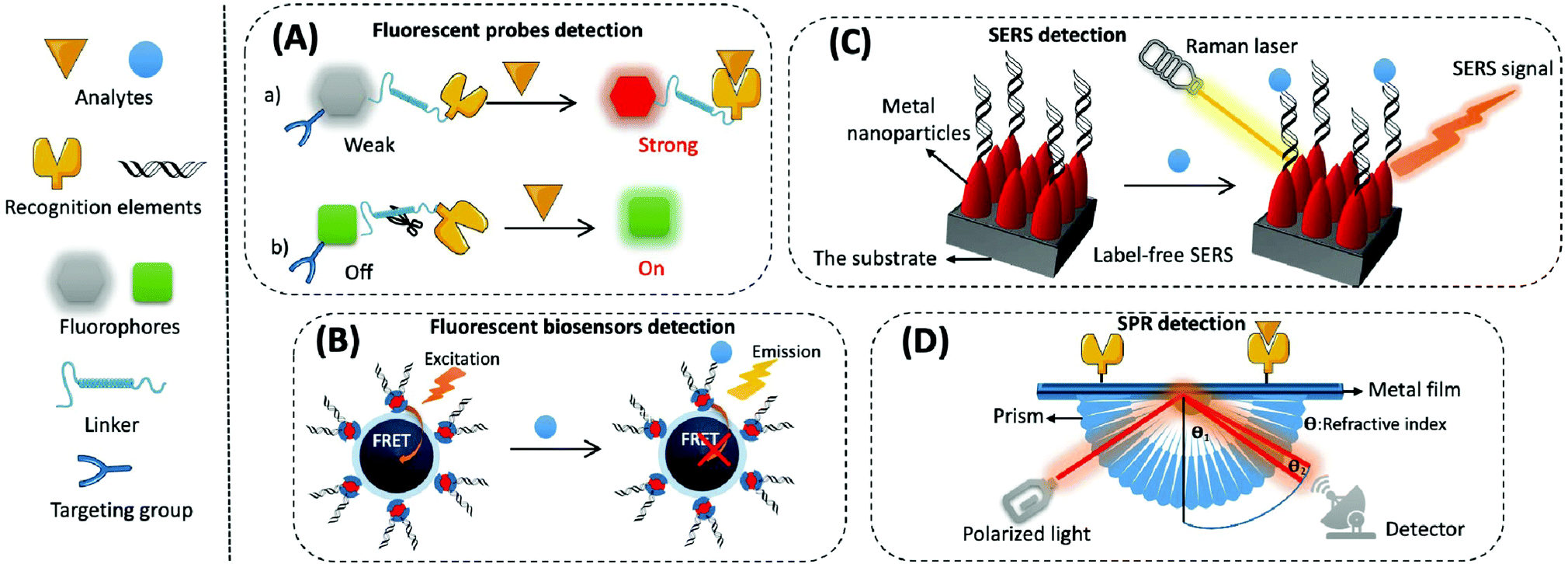 | ||
| Fig. 3 Schematic diagram of the mechanism of the optical-based detection method. (A) Fluorescent probe method. (a) Based on the response of the recognition group to the probe, and its pre- and post-reaction fluorescence intensity from weak to strong; (b) based on the specific catalytic reaction between the target and the probe to achieve the pre-/post-recognition fluorescence signal from off to on. (B) Fluorescent biosensor method. A fluorescent sensor based on FRET response mechanism, where the FRET effect is blocked after the reaction of the target with the recognition group. (C) SERS method. The label-free SERS sensor achieves the detection of the target by amplifying the local electric field around the metal particle under the excitation of laser. (D) SPR method. The target analyte binds to the recognition element on the sensing layer, causing a change in the refractive index on the surface of the metal film, which shifts the position of the SPR resonance peak of the reflection spectrum. | ||
Most existing fluorescent probes targeting mitochondria rely on a cationic group as the binding unit. When the ETC on the inner mitochondrial membrane carries out electron transfer, it pumps protons from the matrix to the membrane gap, creating a difference in proton concentration on both sides of the inner membrane. This, in turn, makes the MMP negative (about −180 mV), and lipophilic cations can be enriched at the mitochondria by electrostatic force. Several cations, including triphenylphosphine cation (TPP+), quaternary ammonium ion, pyridinium ion and rhodamine B skeleton, are available. In addition, there are also mitochondria-targeting peptides such as Szeto–Schiller (SS) peptides.58,59 TPP+, which has a simple structure and is easily attached to the target molecule, is now the most widely used mitochondria-targeting group. However, the electrical potential in tumour and normal cells is very similar; therefore, probes based on lipophilic cations may also be enriched in normal cells. To address this issue, a more precise targeting of mitochondria in tumour cells, using the ROS-responsive N-alkylaminoferrocene moiety, was developed.60
The recognition part of the fluorescent probe can detect the corresponding small molecules by binding or by chemical reaction, thereby resulting in a change in fluorescence. Two types of probes are used for targeting mitochondrial enzymes: (a) fluorescent probes based on the recognition of enzyme inhibitor groups in the molecule, resulting in changes in fluorescence signal and (b) fluorescent probes based on the specific catalytic reaction of the enzyme and the substrate/probe to achieve activation of the fluorescence signal before or after recognition.
There is a wide variety of organic molecular fluorophores, most of which have easily adjustable structures and a wide wavelength range. Commonly used fluorophores include anthocyanin dyes, rhodamine derivatives, coumarins and their derivatives, naphthalimides, fluoroborodipyrroles, and aggregation-induced emission (AIE) type fluorophores such as tetraphenylene.61,62 The excitation and emission wavelengths of fluorophores are the main considerations in probe design. Different fluorophores have specific excitation and emission wavelengths. The excitation and emission wavelengths available for classical fluorescence imaging mostly range in the visible region (400–650 nm), resulting in weak tissue penetration (<3 mm) because of the absorption and scattering effects of biological tissues and the strong autofluorescence of specific biomolecules.63 Hence, scientists have gradually developed multi-photon, first-window near-infrared I (NIR-I), and NIR-II probes in order to improve the detection depth, resolution, and sensitivity. These longer wavelengths are also beneficial for improving the signal-to-noise ratios.
The linker is the bridge that connects the recognition element to the fluorophore. The linker controls the overall size and structural rigidity of the probe and, thus, affects molecular solubility, fluorescence, structural stability and probe sensitivity.
(2) Fluorescent biosensors. Fluorescent biosensors can detect fluorescence signals, including recognition elements and signal transduction elements. First, the recognition element identifies the target analyte and reacts with it to form different modes of interactions, which involves protein–protein interactions in most immunoassays, nucleotide hybridisation or polymerase-based gene amplification for the determination of nucleic acids, and chemical reactions of small molecules. Then, biological signals are converted into readable fluorescence signals through the transduction element for the quantitative assay of an analyte. For example, when constructing fluorescent biosensors for assaying mitochondrial cyt c, a cyanine 5 (cy5)-labelled DNA aptamer is usually applied as the specific recognition element, which produces significant changes in fluorescence intensity before or after the reaction. Nanomaterials/polymerisation with signal amplification is combined with biorecognition elements to enhance the sensitivity and selectivity of biosensors.64 The strategy based on FRET is often used to construct fluorescent sensors with high sensitivity and a signal-to-noise ratio. This is composed of a pair of donor and acceptor chromophores in close proximity to one other, and the designated acceptor (for example DNA or an antibody) is used to recognise the target (Fig. 3B).65 There are two main categories of FRET: one that shows an increase in signal when an analyte is present, and the other that exhibits a decrease or disappearance in signal when the analyte is present. Most commercial fluorophores with adjustable emission, wide absorption, and long fluorescence life can be utilised as FRET donors or acceptors.66,67 This includes the rhodamine derivatives, near-infrared dyes, fluorescent proteins (e.g., green fluorescent protein), and some fluorescent nanoparticles (e.g., QDs).
SERS-based biosensors typically consist of two key components, namely the substrate and the SERS probe.70 The substrate is used to modify the SERS probe (e.g., antibody, aptamer, etc.) to capture specific analytes in the sample. The SERS probe is used to specifically identify, capture, and provide a SERS signal for the quantitative detection of the analyte. SERS biosensors contain four main elements: metal nanoparticles with amplified signals, Raman reporters with intrinsically high scattering cross-sections, protective layers with reduced Raman reporter loss coupled to target molecules, and target molecules with specific recognition sites (Fig. 3C). The SERS-based sensors can either be labelled or label-free. Labelled SERS sensors are cumbersome but suitable for most analytes; they possess higher sensitivity and require a Raman reporting labelled probe to detect the analytes.71 Label-free SERS sensors, which significantly simplify the procedures but need efficient SERS substrates, directly measure the intrinsic Raman spectrum of the target.
The process for common SPR detection is to first immobilise the recognition element on a metal film and allow the sample to contact this film. The target analyte then combines with the recognition element on the sensing layer, causing the refractive index of the metal surface to change. This shift in the position of the SPR resonance peak on the reflection spectrum reveals the concentration of the target. The reading is obtained from the relationship between the concentration of the solution and its refractive index (Fig. 3D).73 However, it is difficult to directly detect target analytes when their concentration is less than 1 pM or if their molecular weight is less than 8 kDa.74,75 Thus, like several other biosensor amplification modules, SPR-based sensing performance can be enhanced by modifying various nanomaterials (e.g., Au or Ag nanoarrays, nanorods, and graphene oxide) on the sensing substrate or by using nanomaterials (e.g., magnetic nanoparticles of Fe3O4 and carbon nanotubes (CNTs)) as amplification tags in combination with the recognition elements.
3.2. Electrochemical signal methods
Generally, electrochemical methods consist of three elements: recognition, transduction, and sensing.77 The recognition element, which usually includes enzymes, antibodies, aptamers, nano-enzymes, and substances with electrocatalytic activity that can accurately and efficiently capture and fix the target subject through chemical or biological reactions and generate electrons, should be efficient, simple, and selective. For example, with the help of O2, glucose oxidase, which can catalyse the conversion of glucose to gluconolactone and H2O2, is usually immobilised on the sensing element. H2O2 can then be converted to H2O and electrons.78 The transduction element is mainly used for fixing various conductive materials with high efficiency and good biocompatibility. A typical sensing element of an electrochemical biosensor is comprised of a three-electrode system: a working electrode (WE), a counter electrode (CE), and a reference electrode (RE). The RE has a known potential and is non-polarised (Fig. 4A). The CE and WE form a circuit to ensure that the current flows through, and the reactions occur at the WE. The WE is the essential element of an electrochemical biosensor because it plays the role of electron donor or acceptor, thus simulating the electron transfer and metabolic processes during the biochemical reactions, which consequently enables the measurements of relevant thermodynamic and kinetic variables. The WE is available in different materials, such as screen-printed carbon electrodes (SPCEs), glassy carbon electrodes (GCEs), indium tin oxide (ITO), nanowire electrodes, flexible electrodes, and noble metal (e.g., platinum and gold) electrodes, that convert electrons into electrical signal outputs (Fig. 4B). Electrochemical biosensors usually possess three types of detection mechanisms. The first one is the direct reaction of the target with the recognition group, the second one is the addition of an artificial electronic mediator after recognition, and the third one is a sandwich structure based on labelled secondary antibodies. In summary, the electrochemical biosensors are powerful tools for the determination of mitochondrial metabolites by combining the strengths of specific biological reactions and the high sensitivity of electrochemical approaches (Fig. 4C).
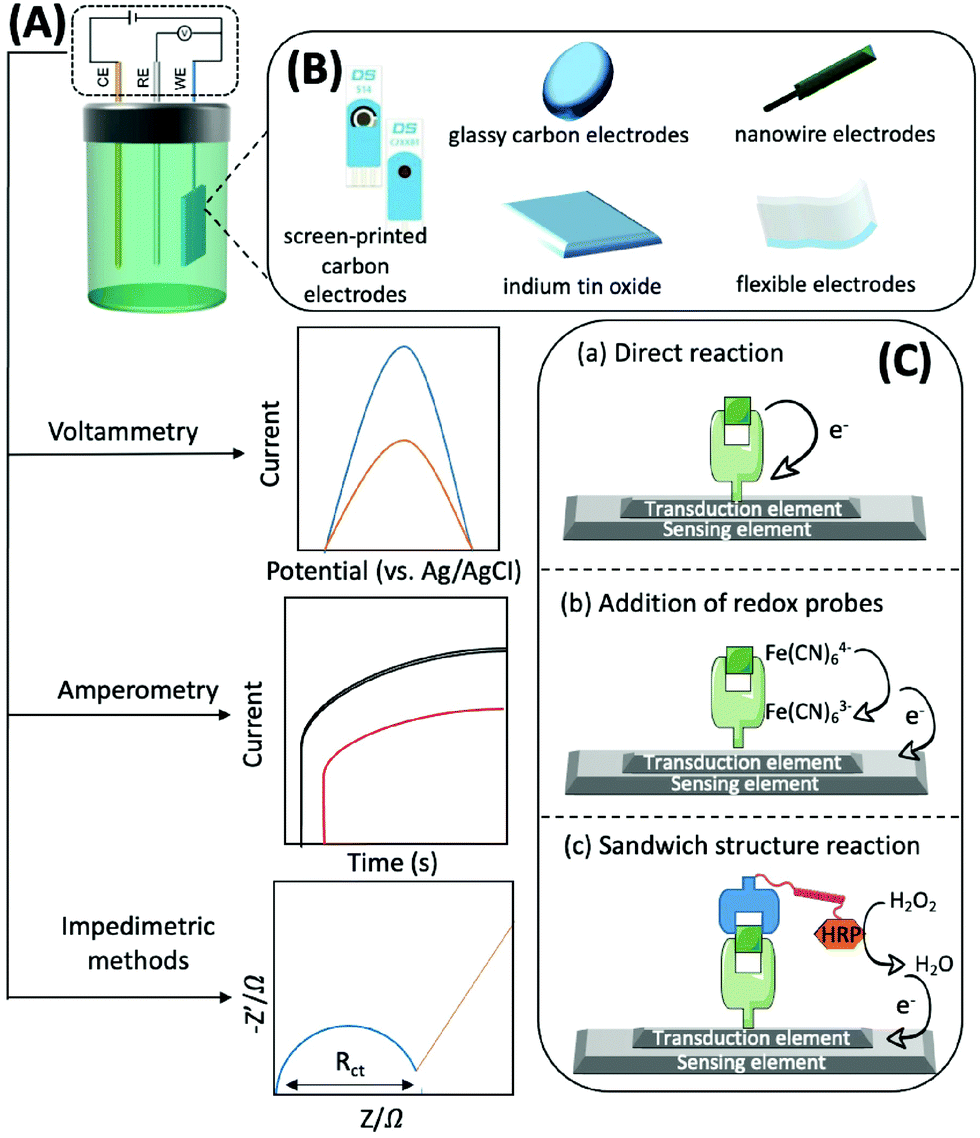 | ||
| Fig. 4 Schematic diagram of the mechanism based on the electrochemical method. (A) Three commonly employed detection methods of electrochemical methods, including voltammetry, amperometry and impedance; (B) frequently available working electrodes of different materials; (C) electrochemical biosensors based on different detection mechanisms. (a) The direct redox reaction between the target and the recognition group at a specific bias potential generates current, which is usually used to detect small molecules. (b) The target reacts with the recognition group and then a redox probe is added to achieve the detection of the target, which is often used for the detection of large molecules such as proteins based on the EIS method; (c) the reaction of sandwich structures is often applied to construct electrochemical immunosensors, and the HRP-labeled secondary antibody generates a current by decomposing hydrogen peroxide and thus indirectly detects the target. | ||
Voltammetry, amperometry, and impedimetric methods are generally adopted for electrochemical characterisation in the identification process, as the current, voltage or impedance will shift as the concentration of the analyte increases (Fig. 4A).79 Herein, we briefly introduce the principles of the commonly used electrochemical analysis methods and their advantages and disadvantages. Voltammetry is a technique that uses voltage to scan between the working and reference electrodes, and the generated current is used to calculate the target concentration. It has the advantages of good selectivity, simultaneous analysis of multiple substances, as well as low detection limits; thus, it can be used for detecting traces of analytes. However, this technique may trigger a background reaction that interferes with the desired signal. In addition, the signal analysis method is complex.80 Amperometry involves the utilization of a bias voltage at the working electrode and uses redox reactions to produce an electric current for detecting the target. The advantage of this detection method, and its signal processing, is that it is simple and has low power consumption owing to the use of media to detect the necessary potential. Its disadvantage is that it is not suitable for the detection of trace substances, particularly those with concentrations below the micromolar level.81 By applying a sinusoidal voltage to the surface of the sensor, the electrochemical impedance of target binding can be determined, thus indicating the concentration. Measuring impedance is advantageous, as it does not require coupling of electrochemically inactive substances on the electrode prior to detection. However, this method has the disadvantage of a longer analysis time, complex signal processing, and lower sensitivity.82
In voltammetry and amperometry, binding of the target to the recognition element is detected by a current generated at a fixed voltage, generally through differential pulse voltammetry (DPV), cyclic voltammetry (CV), chronoamperometry, or square wave voltammetry (SWV). The CV response curve can be used to identify the reversibility of the reaction at the electrode, the availability of intermediate or new phase formation, and the nature of the coupled chemical reaction within the voltage scan range. In new electrochemical systems, CV is the preferred method for studying the nature, mechanism, and kinetic parameters of the electrode reactions. DPV technology can determine the reversibility of the electrode processes, the adsorption phenomena on the electrode surfaces, and the quantitative detection of trace substances.
EIS is a rapidly developing impedimetric electrochemical technique used to detect biological reactions at the electrode interface of modified biomolecules, which is useful for interface characterisation. It is a non-destructive analytical tool that records the changes in the electrical properties on the surface of conductors or semiconductors to measure the biochemical reaction kinetics and the analytes on the modified electrode. To analyse impedance, several models have been proposed, Randles equivalent circuit being the most popular. In this model, Rs is the solution resistance independent of the presence or identification of a biological target, Rct is the charge transfer resistance, Zw is the Warburg impedance, and CDL refers to the double layer capacitance. The Rct of a biosensor increases (in most cases) or decreases with target recognition.83 In faradaic EIS, the commonly used redox probes include ferricyanide [Fe(CN)6]4−/3− and ruthenium hexamine. Typically, these probes are used at a much higher concentration (mM) than that present in the target analyte and interact with receptor-modified electrodes biased at or near their half-wave potential. The resulting perturbation in the current is generated by reversible electron transfer.84
Most electrochemical biosensors typically employ three ways to detect mitochondrial energy metabolites. (1) Mitochondria are extracted from cells and prepared as a suspension, or they are isolated and purified, and then incubated on the electrode surface while drugs are added to induce the production of different metabolites.85 (2) Determination of apoptotic factors (e.g., cyt c) released from the mitochondria of cells into the culture medium.86 (3) Using bright-field microscopy, microelectrodes are inserted into cells and positioned in areas where mitochondria are abundant.87,88
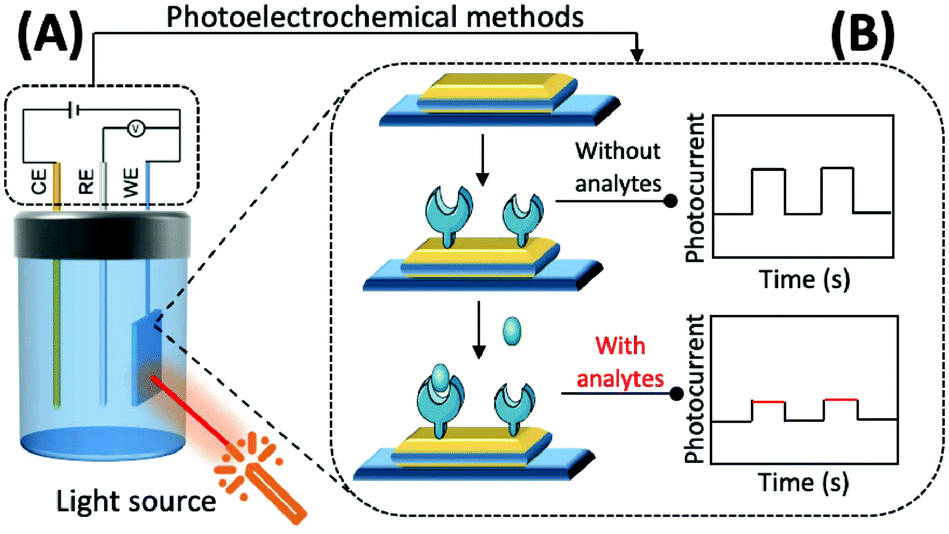 | ||
| Fig. 5 Schematic diagram of the mechanism based on photoelectrochemistry. (A) Photochemical reaction of the photoelectric active materials on the surface of the working electrode under the excitation of external light source to generate photocurrent; (B) construction process of a photoelectrochemical biosensor. | ||
The preparation of photoactive materials with different properties and the construction of signal amplification strategies have key roles in improving the analytical capability of PEC sensors. Photoactive materials are the core components of these sensors and their performance, and photoelectric conversion efficiency is directly reflected in their response to photocurrent intensity and the stability of the materials.
In addition to this, the intrinsic properties of photoactive materials, such as morphology, solubility, energy level, and forbidden bandwidth, should also be considered in scientific research and practical applications. Currently, the available photochemical materials can be broadly divided into three categories. (1) Inorganic photoelectrochemical materials: this mainly refers to inorganic semiconductors and some elemental monomers such as silicon and germanium. Inorganic semiconductors, such as TiO2,90 CdTe,91 and CdS,92 are suitable for the construction of most biosensors because of their rich morphology, simple preparation method, easy association, and good biocompatibility. (2) Organic photoelectrochemical materials: these are mainly organic small molecules and polymers that are rich in carbon atoms and have a conjugated π system.93,94 Compared with inorganic photoactive materials, the bandwidth of organic photoactive materials is narrow, the electron–hole pairs are easy to compound, and the preparation usually requires more stringent conditions. (3) Composite photoelectrochemical materials: by combining different kinds of photoelectrochemical materials researchers can select the respective advantages of each material to enhance light absorption, promote the separation of electron–hole pairs within the material and the directional transfer of free electrons in the system, and enhance the strength and stability of the material in response to photocurrents, thereby improving the analytical and detection performance of PEC sensors based on composite material.95–97
4. Detection of mitochondrial energy metabolites
Low concentration levels, short half-life, easy conversion, and external metabolite interference are the main challenges that hinder the real-time, accurate detection of mitochondrial energy metabolites both in vivo and in vitro. Hence, it is crucial to monitor the dynamic variations in mitochondrial metabolites using various optical/electrochemical methods that are accurate, sensitive, and specific. In Section 4, we will detail the application of various optical/electrochemical methods for the detection of mitochondrial energy metabolites, including small molecules, macromolecules, and ions as listed in Table 2. Specific detection mechanisms will also be summarised.| Detection method | Analytes | Transducer/fluorophore | Receptor | LODa | Models | Ref. | |
|---|---|---|---|---|---|---|---|
| a The lowest LOD of the analyte using the same detection method. | |||||||
| Optical detection | Fluorescent probes | ATP | Rhodamine | Diethylenetriamine | 0.1 mM | HeLa cells | 116 |
| Acetyl-CoA/CoA | Rhodamine | Amino | — | HeLa cells | 131 | ||
| α-KA | NBD | Hydrazino group | 0.9 μM | Yeast cells, human serum | 136 | ||
| NADH | Dicyanoisophorone | Quinolinium | 12 nM | HepG2 cells, HL-7702 cells | 155 | ||
| O2 | Rhodamine B isothiocyanate | Platinum(II) octaethylporphyrin | — | HeLa cells | 166 | ||
| O2˙− | 5-Carboxy-2,4,5,7-tetrafluorofluorescein | Trifluoromethanesulphonate group | 23 nM | HCT116 cells, BV-2 cells, RAW 264.7 cells and zebrafish embryos | 181 | ||
| H2O2 | Hemicyanine | Boronic acid | 0.348 μM | HeLa cells, RAW 264.7 cells | 198 | ||
| HOCl | Nile blue | Amido | 10.8 pM | HeLa cells | 202 | ||
| ˙OH | Rhodamine | N2O-Type Bobpy | 0.68 μM | HeLa cells and zebrafish | 211 | ||
| 1O2 | Silicon-containing | Anthracene | — | HeLa cells | 214 | ||
| NO | BODIPY | N-Benzyl-4-hydroxyaniline | 4.8 nM | HeLa cells, RAW 264.7 cells | 228 | ||
| ONOO− | Coumarin | TP-NIR | 11.3 nM | HeLa cells, living hepatic tissue and inflammation model in mice | 235 | ||
| HNO | Coumarin | Diphenyl(o-benzoyl)phosphine | 18 nM | HeLa cells | 239 | ||
| H2S | Dimethylaniline and pyridine | Azide | 0.17 μM | HeLa cells | 242 | ||
| SO2 | HMII | Aldehyde | 13.1 nM | HeLa cells and zebrafish | 251 | ||
| GSH | IR-780 | Naphthalimide | 15.7 μM | HeLa cells | 258 | ||
| FAO | Coumarin | Nonanoic acid (C9) chain | — | Hepatocytes in the NASH disease model | 260 | ||
| MAO-A | Resorufi | Substituted phenol of clorgyline and propylamine | 2.7 ng mL−1 | HepG2 cells, and SH-SY5Y cells | 281 | ||
| MAO-B | Acedan (2-methylamino-6-acetylnaphthalene) | Alkylated propylamine | — | HepG2 cells, SH-SY5Y cells, and PD mouse model | 284 | ||
| m-MDH | Cy5 | — | — | HCT116 cells | 291 | ||
| GGT | Amide and indocyanine | Amide | 0.16 U L−1 | Cancerous colons of mice | 293 | ||
| NTR | 2,3-Dihydro-1H-xanthene-6-ol | 4-Nitrobenzene group | 3.5 μg mL−1 | HeLa cells | 297 | ||
| TrxR2 | Naphthalimide | Disulphide | 50 nM | HeLa cells and cell extracts | 301 | ||
| Msrs | Methyl phenyl sulphoxide and methylpyridinium | Sulphoxide | 0.01 μg mL−1 | HeLa cells and PC12 cells | 304 | ||
| Ca2+ | Fura-2 | BAPTA | 10 mM | HeLa cells | 305 | ||
| K+ | BODIPY | ACLE | 10 mM | HeLa cells and L02 cells | 309 | ||
| Zn2+ | Fluorescein diacetate | Acetyl | 0.6 nM | Human cancerous cells | 306 | ||
| Cu+/2+ | BODIPY | Azatetrathia | 7.2 pM | SCO1 and SCO2 patient fibroblasts | 310 | ||
| Fe2+ | Fluorescein | N-Oxidation | — | HepG2 cells and HEK293 cells | 312 | ||
| Mitochondrial membrane potential (MMP) | FixD and LA | Cationic groups | — | A549 cells | 325 | ||
| Fluorescent biosensor | ATP | MNQI | Cationic quinolinium | — | HeLa cells | 326 | |
| Pyruvate | Rhodamine-based dye | Cationic and lipophilic group | — | HEK cells | 327 | ||
| Acetyl-CoA/CoA | Cy3 | DNA aptamer for ATP | 3.7 μM | HeLa ells | 110 | ||
| α-KA | mTFP and Venus | LutR | — | HEK293 cells | 123 | ||
| NTR | Melatonin | Cu2+ | 12 nM | Human serum samples | 132 | ||
| Zn2+ | Compound 1′ | Hydrazino | 0.61 μM | — | 138 | ||
| SERS biosensor | ATP | BODIPY | p-Nitrobenzyl thioether | 0.0168 μg mL−1 | A549 cells | 298 | |
| Pyruvate | cpGFP | ZF1 and ZF2 | — | HeLa cells | 307 | ||
| ROS | Fe3O4@AgNPs | — | 0.1 pM | — | 122 | ||
| Cyt c | Fe3O4@AgNPs | — | 1 pM | SGC cells, HepG2 cells and MCF-7 cells | 122 | ||
| Au@AgNPs | 4-Mercaptobenzonitrile | — | MCF-7 cells | 171 | |||
| Gold nanotriangles | Cyt c aptamer | 0.02 μM | HepG2 cells | 270 | |||
| MAO-B | Cysteamine | Phenethylamine | 1.7 μM | — | 286 | ||
| Electrical detection | Electrochemical biosensor | ATP | GCE/AuNPs | DNA1/S1 | 0.45 pM | Human serum sample | 102 |
| Pyruvate | Gold electrode | Pox | 0.67 μM | Sera sample | 121 | ||
| Acetyl-CoA/CoA | GCE/AgNCs | CoA–Ag(I) CP | 0.067 nM | Human serum sample | 128 | ||
| α-Lipoic acid (α-LA) | Graphene electrode | MnO2 | 0.42 μM | — | 139 | ||
| NADH | GCE | MIP | 20 pM | Mouse blood sample | 153 | ||
| O2 | AuNPs electrode | Nafion | — | C2C12 cells | 170 | ||
| ROS | Nanopipette | G4-DNAzyme | — | CHO cells | 173 | ||
| O2˙− | SPCE/Pt–Pd | SOD | 0.13 μM | HeLa cells | 175 | ||
| H2O2 | GCE graphene | PtPb/graphene | 2 nM | RAW 264.7 cells | 189 | ||
| ˙OH | Tungsten nanoneedle | HAT | 0.33 nM | RAW 264.7 cells | 212 | ||
| NO | Carbon fibre | Methyltrimethoxysilane/17-FTMS | 12.1 nM | Brain of rats | 225 | ||
| GSH | Carbon paste electrode/MgO/SWCNT | 2-CDHPMA | 10 nM | Haemolysed erythrocyte sample | 256 | ||
| p53 | GCE/AuNPs/GO | Antibody p53 | 30 fM | Human serum sample | 262 | ||
| Cyt c | GCE/RuSiO2 NPs | Fc–aptamer | 0.48 pM | Human serum sample | 269 | ||
| Caspase-3 | GCE/AuNPs | Biotinylated peptide | 5 fM | Hematopoietic stem cells | 276 | ||
| CS | GCE/graphene | CoA–Ag(I) | 0.00165 U μL−1 | — | 288 | ||
| SPR biosensor | NADH | Aluminium | — | — | MC3T3-E1 cells | 157 | |
| Cyt c | SPR gold surface | 76-mer DNA aptamer | 50 pM | HepG2 cells | 272 | ||
| Photoelectrochemical biosensor | p53 | AuNPs/TiO2 NTs | ALP-a-p53 | 0.05 ng mL−1 | Patient serum sample | 264 | |
| Cyt c | TiO2 NTA | Apt Cyt c | 5 pM | Human serum sample | 273 | ||
4.1 Detection of small molecules
The use of ATP aptamers to construct sensitive electrochemical aptasensors is a widely used strategy because they can form a G-quadruplex structure while binding with and recognising ATP, which further results in considerable changes in the detection signals. Concomitantly, different signal amplification strategies have been developed to improve the sensitivity of ATP aptamer sensors. Kashefi-Kheyrabadi et al. used a gold electrode as the sensing element, silver nanoparticles (AgNPs) as the transduction element, and an ATP-conjugated aptamer as the recognition unit in an electrochemical aptasensor sandwich assay. Importantly, this was the first time AgNPs were used as a redox tag in an electrochemical aptasensor. Gold electrode surface immobilization of aptamer 1 was performed by covalent binding between 3-mercaptopropionic acid and amino-labeled aptamer 1, followed by the addition of AgNP-modified aptamer 2 together with ATP, and finally the oxidation current of AgNPs was taken as the signal for detecting ATP.100 In contrast to this sandwich assay, Bao et al. designed an electrochemical aptasensor for ATP determination by amplifying the signal of exonuclease III (Exo III)-catalysed target recycling. This aptasensor consisted of hairpin DNA that encoded an ATP aptamer and a methylene blue (MB) label at the 5′-terminus. The 3′ end of the hairpin was single-stranded so that Exo III could not cleave the hairpin DNA. A G-quadruple was produced by hybridizing five prominent single nucleotides of the 5′ end with the trapped DNA and MB-labelled hairpin DNA. Exo III selectively cleaved the 3′ end of the double-stranded DNA by the mechanism of target-induced structural transition, which enabled the disruption of the G-quadruplex structure and the release of ATP for recycling, thus providing signal amplification.101 Unfortunately, the authors did not apply this sensor to mitochondrial or patient serum ATP assays. This Exo III cyclic catalytic amplification strategy can also be extended to the detection of other small molecules. Another signal amplification strategy is enzyme-free DNA recycling amplification. However, this method typically utilises only a single DNA amplification cycle to produce output DNA, somewhat limiting the target conversion rate. To address this issue, Xie et al. designed an ultrasensitive electrochemical dual amplification biosensor for ATP determination based on targeting induction and Mg2+-dependent DNAase recycling (Fig. 3A).102 Briefly, the binding of two fractured ATP aptamers launched the first cycle of catalytic hairpin assembly (CHA) and enlargement. This converted the input ATP to an enzyme (called HP1–HP2) that subsequently hybridised with the substrate hairpin HP3 to produce Mg2+-dependent DNA nuclease. The availability of Mg2+ resulted in a second cycle of HP3 scission to produce DNA, thereby achieving a transformation rate of one ATP to a large amount of DNA. The released DNA then hybridised with DNA1 and MB-labelled DNA2 through a ternary “Y” linkage structure, producing a significant electrochemical signal. The detection limit of this ATP sensor (0.45 pM) was substantially higher than that of the Exo III-based ATP sensor (34 pM), and the detection range was wider (Fig. 6A). This suggested that these “Y” structures are expected to improve the detection of lower levels of ATP with high sensitivity, while the closer the distance between the redox probe (e.g., MB) and the electrode surface, the higher the electrochemical signal that is obtained, thus improving sensitivity.
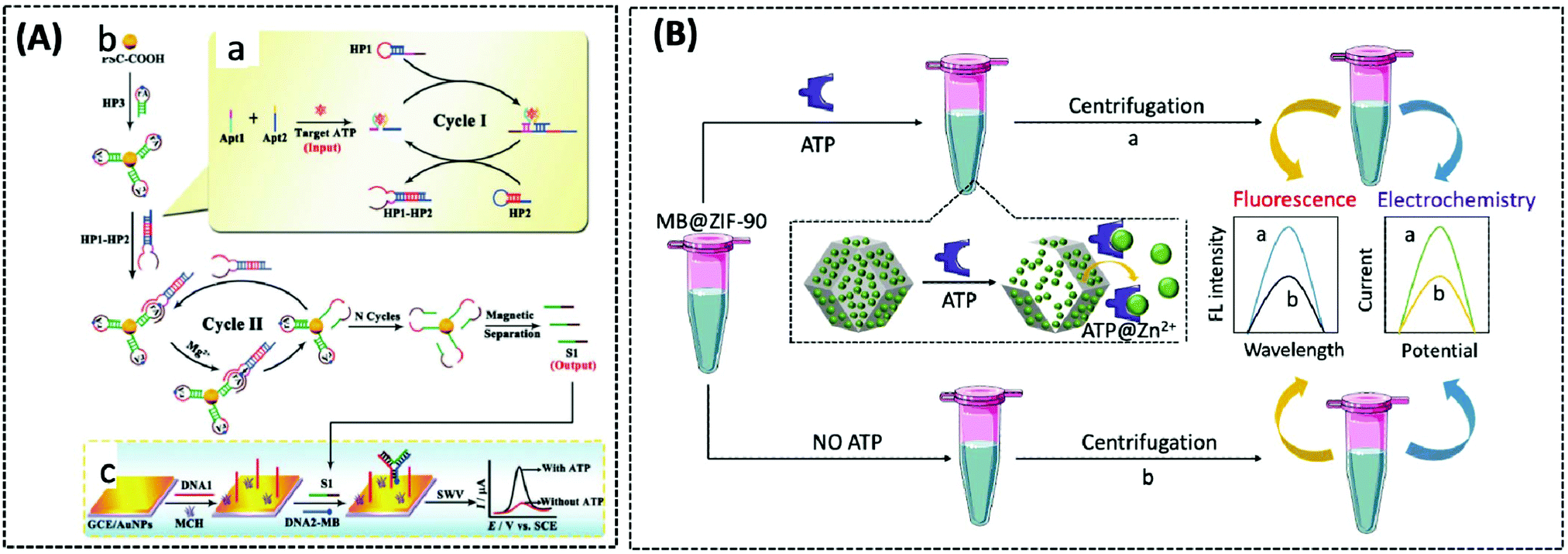 | ||
| Fig. 6 (A) Schematic illustration of the electrochemical biosensor for ATP detection on the basis of a highly effective molecule converting strategy (Chem. Commun., 2017, 53, 8368–8371), adapted with permission from ref. 102, copyright © 2017 Royal Society of Chemistry. (B) Using MB@ZIF-90 to construct electrochemical and fluorescent sensors for detecting ATP (Anal. Chem., 2020, 92, 8959–8964). | ||
The three above-described aptamer-based ATP biosensors are not cost-effective owing to their stringent DNA probe design, complex and expensive labelling process, and the need for bio-enzymes to improve detection sensitivity. Furthermore, it is difficult to achieve fast responses. Due to these factors, the development of label- and enzyme-free ATP biosensors is of particular interest. In recent years, metal–organic frameworks (MOFs) have been broadly implemented in drug delivery and biosensing, owing to their functionality, large porosity and surface area, and adjustable pore size.103–105 Zeolite imidazole frameworks (ZIF) are a class of MOF with a wide range of biomedical applications. Deng et al. reported a nanoscale ZIF-90 framework for mitochondrial ATP sensing.106 The authors synthesized ZIF-90 using self-assembling Zn2+ and imidazole-2-carboxylic acid (2-ICA). Rhodamine B (RhB) was encapsulated in ZIF-90 to achieve inhibition of RhB emission due to a “self-quenching” effect. The phosphate and adenosine groups in ATP showed a stronger coordination force with Zn2+ than the imidazole group in 2-ICA, leading to the disruption of ZIF-90 and the release of RhB to detect ATP. In vitro experiments demonstrated that the fluorescence intensity of RhB increased as the concentration of ATP increased. Finally, RhB/ZIF-90 was applied to detect the variations in ATP in mitochondria induced by different drugs. Under the induction of pentachlorophenol (PCP) and sodium azide (a drug that inhibits ATPase), the fluorescence intensity of ATP was remarkably diminished in comparison with that of the control cells. However, the mitochondrial ATP fluorescence intensity was significantly increased under the induction of Ca2+, which increases the concentration of NADH and thus the production of ATP.
In 2020, Chang et al. similarly designed a label-, enzyme-, and reagent-free fluorescent and electrochemical dual-signal ATP biosensor.107 MB, as affinity material and signal source, was encapsulated in ZIF-90 using a one-step reaction. Following the addition of ATP, MB was released from ZIF-90 and exhibited strong fluorescence signals at 683 nm (λex = 610 nm) and electrochemical signals with an oxidation voltage of −0.23 V. The detection limits of the fluorescence and electrochemical methods were 0.38 nM and 0.027 nM, respectively. Meanwhile, the proposed sensor detected 5.63 mM and 5.75 mM of ATP in human serum and MCF-7 cell lysates, respectively, which were in agreement with previously reported assays, and also showed significant selectivity and stability (Fig. 6B).
Photoregulation is an attractive mode of stimulation because it can be operated remotely and can minimise contamination of the response system.108 Zhang et al. designed an electrochemical aptasensor that combined three elements (recognition, signal-transformation, and regeneration elements) in one DNA oligonucleotide chain.109 The recognition, signal conversion and regeneration elements of the biosensor are DNA aptamer, ferrocene (Fc) and azobenzene group, respectively. Under different wavelengths of ultraviolet light, azobenzene changes between the trans and cis forms, thereby altering the conformation of the aptamer (Fig. 7A). Then, by dissociating the four base pairs, the hairpin structure of the aptamer could be disrupted, and the electrode could easily be regenerated for the next ATP. Importantly, the transition of the hairpin configuration, upon recognition of ATP, allows the distance between the redox group Fc and the electrode to be reduced, thus speeding up electron transfer. The electrochemical photoregulation methods have demonstrated the feasibility of temporal control and in vitro detection based on aptamer ATP sensors. However, considering the different subcellular distribution and dynamics of the metabolites such as ATP, the in vivo spatiotemporal control and targeted detection of ATP are even more important. Hence, Hong et al. developed a photoregulation aptamer biosensor based on photocleavable (PC) linkers and liposomes targeting mitochondria for intracellular ATP detection (Fig. 7B).110 Additionally, to avoid the problem that many fluorescent aptamers are “always on”, the fluorescence of the ATP DNA aptamer was prevented by hybridization with a portion of the complementary DNA strand consisting of nitrobenzene of the PC linker. The hybridisation between PC-Blocker-Q and the ATP aptamer was so strong before it reached the mitochondria that even in the presence of ATP in other subcellular organelles, the aptamer conformation could not be formed and ATP could not bind. Under a wavelength of 365 nm, the PC linker undergoes photolysis splitting the PC-Blocker-Q into two DNA segments. The ATP binding region was exposed when the shorter DNA segment was dehybridised with the ATP aptamer. The PC-Blocker-Q fragment was cleaved from the aptamer chain due to the binding of the aptamer to ATP, enabling the recovery of Cy3 fluorescence. In the in vitro experiments, the proposed sensor was incubated with ATP in a cell lysate buffer without any change in fluorescence intensity; however, after irradiation for 20 min with 365 nm light, the fluorescence intensity was enhanced more than 10-fold and remained stable for 12 h. The authors combined the negatively charged backbone of the aptamers with the positively charged mitochondria-targeted liposomes, the DQAsomes, through electrostatic interaction to achieve a spatiotemporal control of ATP levels in live-cell mitochondria.
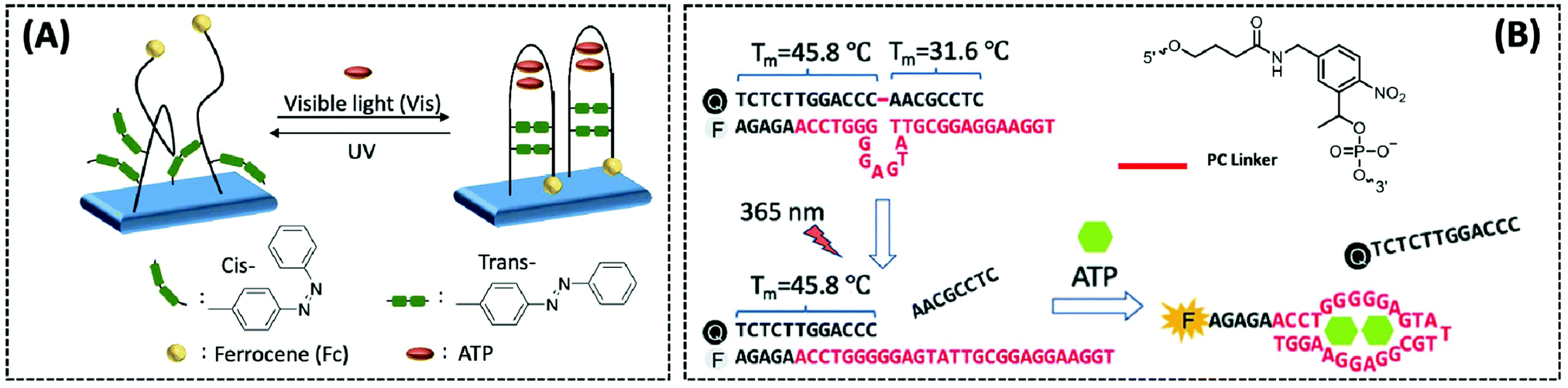 | ||
| Fig. 7 (A) Schematic diagram of a photo-renewable electrochemical sensor using an azobenzene system and ferrocene (Fc)-labeled aptamer as probes for ATP detection (Anal. Chem., 2018, 90, 4968–4971). (B) Design of a photo-regulated aptamer probe to detect mitochondrial ATP in living cells, and the chemical structure of the PC linker (Chem. Sci., 2020, 11, 713–720), adapted with permission from ref. 110, copyright © 2020 Royal Society of Chemistry. | ||
The main mechanism of organic small molecule fluorescent probes for ATP determination is the electrostatic interaction between the positively charged recognition group of the probe and the negatively charged group of the ATP phosphate. Nevertheless, interference can be caused by polynucleotides or negatively charged biomolecules present in the organism, thus reducing the selectivity, stability, and capability of the probe. Although several fluorescent probes have been already developed for ATP assay, they either were not capable of targeting mitochondria111,112 or could target mitochondria but suffered from poor selectivity113 and a narrow linear range.114
Therefore, many researchers have proposed a multisite combination strategy to improve the selectivity and linear range of mitochondrial ATP detection. Wang et al. reported probe 1 (λex/λem = 510/570 nm) for detecting ATP by taking advantage of the fact that phenylboronic acid has a reversible reaction with diols and introduces it into RhB (Fig. 8A).115 There is no fluorescence before the reaction owing of the ring-closed structure of the probe. When the probe reacts with ATP, the covalent bonding between boric acid and ribose, the π–π interaction between xanthan and adenine, and the electrostatic interaction between the amino and phosphate groups jointly contribute to the open-ring reaction of the probe, producing a strong fluorescence. An 18.1-fold increase in fluorescence intensity of the probe when reacted with 10 μM ATP that was the strongest fluorescence response among the three probes. This may be attributed to the larger steric hindrance of ortho-phenylboronic, which facilitates the open-ring reaction of the probe. Finally, using probe 1, the authors observed not only the reduction of mitochondrial ATP induced by KCN and starvation, but also the increase of mitochondrial ATP during initial apoptosis.
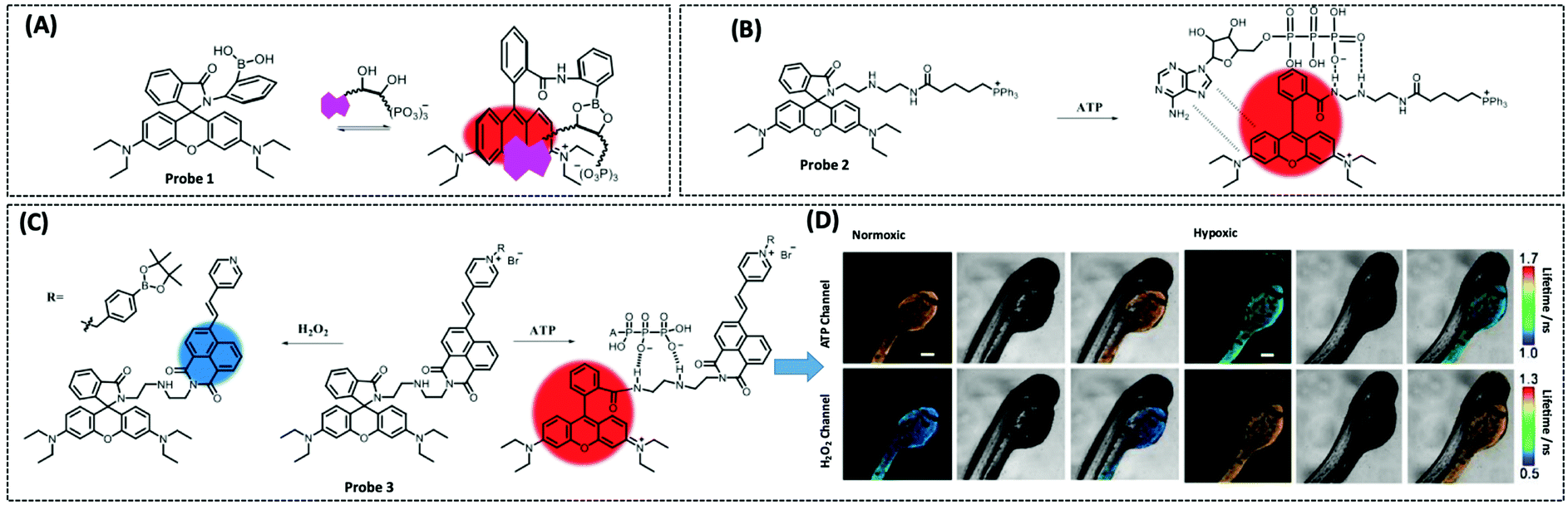 | ||
| Fig. 8 Structures and the response mechanism of mitochondrial (A) fluorescent probe 1 (Angew. Chem., Int. Ed., 2016, 55, 1773–1776) and (B) probe 2 (Anal. Chem., 2017, 89, 1749–1756) to detect ATP. (C) Structure and the response mechanism of the two-photon mitochondrial fluorescent probe 3 (J. Am. Chem. Soc., 2020, 142, 7532–7541) to detect ATP and H2O2, and (D) the fluorescence lifetime images of zebrafish dispersed under the normoxic and hypoxic conditions after 2 h with probe 3. Scale bars: 200 μm. Adapted with permission from ref. 117, copyright © 2020 American Chemical Society. The colors of the probes represent fluorophores. | ||
Probe 1 was used to target mitochondria using the positive charge of the post-reaction product. However, the disadvantage of this targeting strategy is that the probe may react with the same biomarker in other organelles before binding to the mitochondrial biomarker, providing a false positive result. Therefore, based on the same multisite combination strategy, Tan et al. reported a dual-responsive ATP probe 2 (λex/λem = 515/583 nm). This probe also used rhodamine as the fluorophore and modified diethylenetriamine as the response group for ATP on the lactam of rhodamine. TPP was applied as the mitochondria-targeting motif (Fig. 8B).116 When the probe was hydrogen-bonded to ATP, the ATP adenine group and the xanthene portion of the probe showed π–π stacking interactions and the spirolactam opened to distinguish ATP from ADP. The probe exhibited high sensitivity with an 81-fold increase in fluorescence intensity upon reaction with ATP, while the detection range (0.1–10 mM) overlapped the mitochondrial ATP concentration range (1–5 mM). In particular, this probe could be specifically localised in the mitochondria and demonstrated its effectiveness in determining ATP fluctuation in the mitochondria of HeLa cells under carbonyl cyanide m-chlorophenyl hydrazine (CCCP) and apyrase induction.
On the one hand, there is an intricate causal relationship between various signalling molecules in mitochondrial energy metabolism and single marker assays are no longer sufficient for the current exploration of the relationships between mitochondrial metabolites. On the other hand, in mitochondria, H2O2 and ATP have close interactions and both influence each other. However, the different detection ranges and intracellular positions of H2O2 and ATP render it hard to monitor their changes simultaneously within the mitochondria. To solve this problem, Wu et al. designed a single-molecule two-photon fluorescent probe 3 (λex = 710 nm, λem = 470 nm for H2O2, and λem = 590 nm for ATP), which can simultaneously monitor mitochondrial H2O2 and ATP concentrations at different emission wavelengths (Fig. 8C).117 The probe featured a naphthalimide derivative with high selectivity for H2O2 cleavage, a rhodamine derivative with multiple amino groups for easy ATP capture, and an intermediate pyridinium cation for targeting mitochondria. This probe showed great selectivity and response in the detection range of 0.4–10 μM for H2O2 and 0.5–15 mM for ATP. Finally, the probe measured mitochondrial neuronal H2O2 and ATP fluctuations after O2˙− stimulation, which resulted in excess H2O2 production and inhibition of ATP synthesis. The longer the stimulation time, the longer the recovery time and the lower the recovery of H2O2 and ATP levels. The probe was also used to simultaneously assay H2O2 and ATP levels in zebrafish under normoxia and hypoxia conditions, with the former increasing and the latter decreasing.
The electrochemical biosensors used for pyruvate detection are usually based on pyruvate oxidase (POx). POx is combined with two substances (phosphate ions and thiamine pyrophosphate) in the working solution to make POx-based electrochemical biosensor complexes.119 The reaction is catalysed by POx to generate acetyl phosphate and hydrogen peroxide, which further continues to be catalysed at high voltage to generate electrons. Therefore, the challenges associated with detecting pyruvate include maintaining the activity and stability of POx, immobilising POx on the electrode, and minimising the influence of interferents. Gajovic et al. used a dialysis membrane made of acetate and nitrocellulose to protect against ascorbate interference, and immobilised recombinant POx by cross-linking with poly(4-vinyl pyridine) to construct a POx-based “wired” electrochemical biosensor. The probe showed higher sensitivity than those used earlier; moreover, the results were not affected by undiluted calf serum components or the change in pH.120
However, the direct immobilisation of POx on the electrodes still has certain drawbacks, including enzyme denaturation and low stability. Enzyme nanoparticles can overcome these drawbacks due to their strengthened electrical, mechanical, and optical properties. Malik et al. designed POx nanoparticles, named POxNPs, using a desolvation method. A clean gold electrode was placed into a suspension of POxNPs in order to make the cysteamine functionalised. POxNPs were immobilised on the Au electrode through a covalent bond between the –SH group of POxNPs and the –OH group of the gold electrode.121 Using CV to verify the catalytic process, the authors found that the bare gold electrode did not show any obvious redox peaks, while the POxNP-modified gold electrode exhibited obvious redox peaks. This indicated that POxNPs were successfully immobilised on the gold electrode and served as a good catalyst. The Rct of the POxNP-modified gold electrode (370 Ω) was larger than that of the bare gold electrode (250 Ω) in potassium ferricyanide solution. This is because the biological enzyme is a non-conducting substance that hinders electron transfer. The detection range of the proposed sensor was 0.01 μM to 5000 μM, while the detection limit was 0.67 μM. Recently, Kucherenko et al. reviewed electrochemical biosensors for lactate and pyruvate detection, which will help researchers understand the construction strategies for lactate and pyruvate biosensors.119
In recent years, single-cell metabolomics has provided a more visual and dynamic picture of cellular response to external environmental stresses. Droplet-based microfluidic devices allow the encapsulation of individual cells in microliter droplets to eliminate the disturbance of the external environment, while permitting the incorporation of multiple sensors. Thus, Sun et al. implemented the simultaneous unlabelled monitoring of pyruvate, ATP, and lactic acid within a single cell through the SERS and droplet-based microfluidic platform because each metabolite possesses a characteristic SERS fingerprint. (Fig. 9).122 The platform is a multifunctional magnetic SERS base made up of AgNPs decorated on Fe3O4 to form Fe3O4@AgNPs. This could enhance SERS by ensuring high detection sensitivity of metabolites. This magnetic-metal composite matrix facilitates efficient adsorption of single cell metabolites, rapid separation from complex matrices, and high SERS sensitivity. Moreover, due to its relatively large size, it can prohibit endocytosis during prolonged off-chip cell incubation. Plasma coupling between closely arranged AgNPs on a single Fe3O4 microsphere greatly enhances SERS analysis. The improved SERS activity using the Fe3O4@AgNP nanocomposites is a contribution from two factors: one is that AgNPs are spontaneously and densely arranged due to their strong inherent magnetic field, and the other is that tightly packed AgNPs lead to proton coupling between neighbouring AgNPs on each magnetic microsphere. A single cell was used to embed Fe3O4@AgNPs in water-in-oil droplets. The metabolites of single cells were directly adsorbed onto the Fe3O4@AgNPs. Due to the strong Fe3O4 magnetic field generated, they can be spontaneously collected. The SERS spectra of each metabolite in the aggregated state were obtained when the absorbance of metabolites was balanced. Finally, the authors used this platform to explore how cell–cell interactions affect metabolite production and found that these interactions affect the metabolic activity of the cells and shows different trends in different cell types.
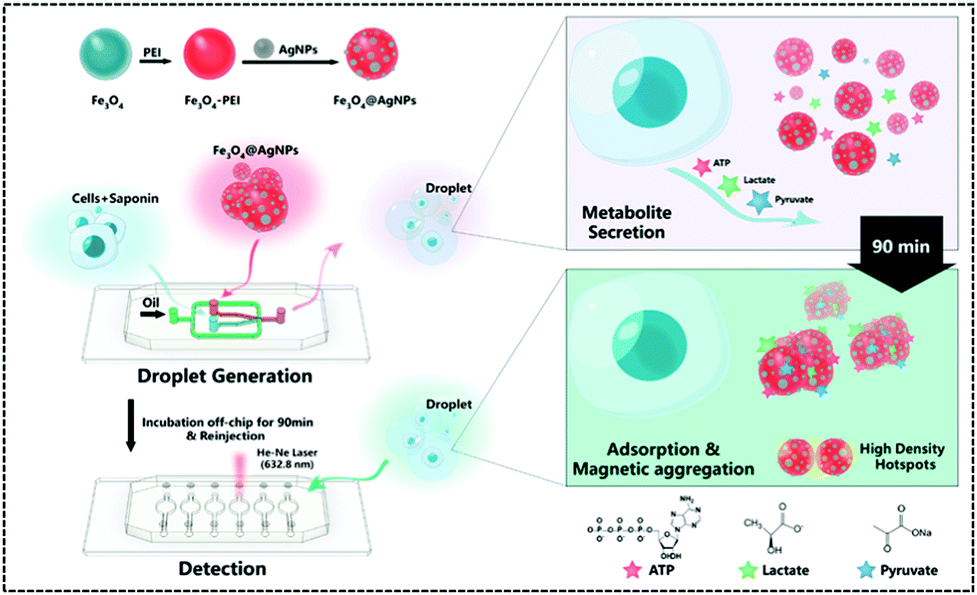 | ||
| Fig. 9 Workflow of the SERS-microfluidic droplet platform for single-cell encapsulation and simultaneous detection of pyruvate, ATP, and lactic acid produced by a single cell (Anal. Chem., 2019, 91, 15484–15490), adapted with permission from ref. 122, copyright © 2019 American Chemical Society. | ||
The lactate/pyruvate ratio is one of the indicators of the redox status between glycolysis and mitochondrial oxidative metabolism and is therefore commonly used to monitor the state of intracellular energy metabolism. Galaz et al. reported a genetically encoded FRET-based sensor named Lapronic, which used LutR that can bind to lactate/pyruvate as an internal biosensor.123 LutR is flanked by mTFP–Venus fluorophore pairs connected by short linkers. The fluorescence intensity of the mTFP–Venus ratio reflected the lactate/pyruvate ratio (mTFP: λex/λem = 440/492 nm, Venus: λex/λem = 440/526 nm). After modulating the orientation of the FRET acceptor, the dipole–dipole coupling of the FRET acceptor and donor was enhanced, thus improving the binding between the sensor and pyruvate/lactate and facilitating the detection of slight fluorescence changes. The in vitro experiments demonstrated that pyruvate binding resulted in a decrease in the mTFP signal and an increase in the Venus signal, while the opposite was true for lactate. This is due to the competitive behaviour between pyruvate and lactate at the binding domain of LutR. Simultaneous interference experiments demonstrated that Lapronic is insensitive to changes in pH and temperature. Finally, the experiments in HEK293 cells demonstrated that Lapronic has a homogeneous cytoplasmic localisation. Using the COX IV mitochondrial signal peptide to target Lapronic to mitochondria in COS7 cells, a uniform mitochondrial localisation was observed. This novel FRET fluorescent biosensor exhibits great potential for applications in quantitative evaluations of glycolysis and oxidative metabolism in living organisms and biological fluids.
In recent years, coordination polymers (CPs) of metals with thiol compounds have been attracting increasing interest owing to their distinctive physical characteristics and widespread applications.127 Thiol–Ag(I) CPs are a typical representative and CoA is also a thiol compound. Hu et al. designed a nucleic acid simulating CoA–Ag(I) CP, which is found to be remarkably akin to polyadenylated single-stranded nucleic acid (ssNA) (Fig. 10A).128 Therefore, as there is a strong interaction between ssNA and two-dimensional materials, the CoA–Ag(I) CP and the graphene oxide (GO) surface are able to respond to each other through π–π stacking. Using electrochemical reduction, the authors modified the GCE with GO and CoA–Ag(I) CP to prepare a CP/GO/GCE electrochemical biosensor. This sensor exhibited effective reduction of H2O2, which was caused by CoA–Ag(0) CP or the little silver nanoclusters (AgNC), during the electrochemical scan. For this reason, the authors utilised this sensor for electrochemical detection of different CoA concentrations by fixing the concentration of Ag(I) at 100 μM. The proposed biosensor was capable of measuring CoA in the range of 0.1–100 μM, with a detection limit of 0.05 μM and a high selectivity for CoA. The recovery of CoA in human serum ranged from 96.7–108.5%. Recently, studies employing a similar CoA detection mechanism, but combining different electrochemical detection methods, have been reported.127,129
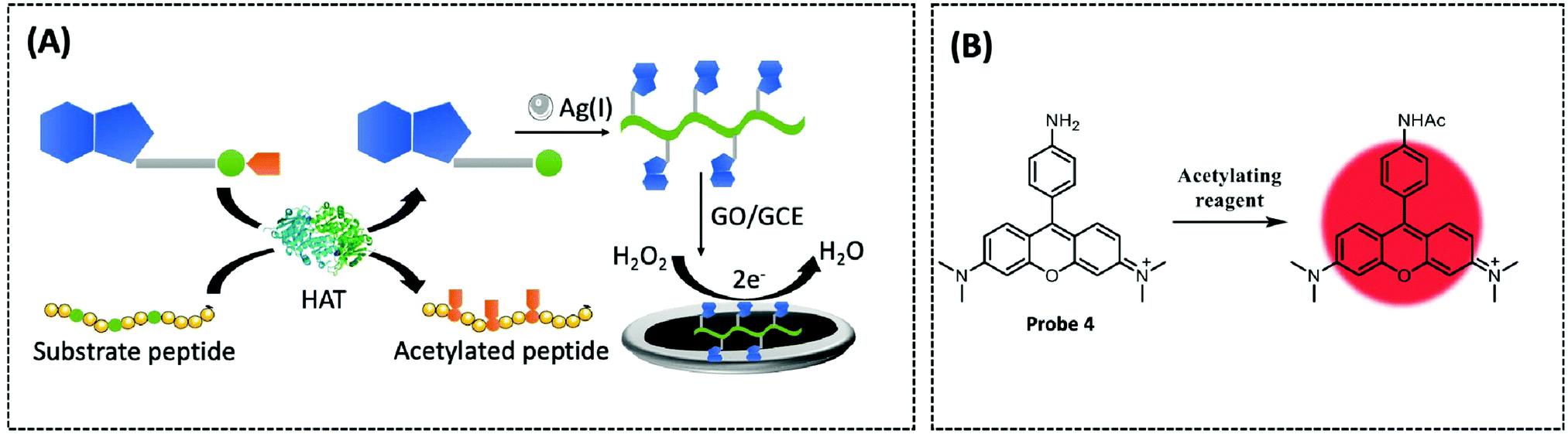 | ||
| Fig. 10 (A) Schematic of the electrochemical biosensor for probing HAT activity based on GO and CoA–Ag(I) CP (Chem. Commun., 2015, 51, 17611–17614). (B) Fluorescent probe 4 (Chem. Commun., 2013, 49, 2876–2878) is based on N,N,N′,N′-tetramethylaminorhodamine and an acyl transfer promoter (PBu3) to detect acetyl-CoA. | ||
Tributylphosphine (PBu3), an artificial reaction promoter that has been shown to promote acetylation reactions in vitro, also facilitates the interaction between acetyl-CoA and the exogenously introduced fluorescent probes within the mitochondria.130 Komatsu et al. designed a fluorescent probe 4 (λex/λem = 546/570 nm) based on N,N,N′,N′-tetramethylaminorhodamine and an acyl transfer promoter (PBu3) to detect acetyl-CoA (Fig. 10B).131 Upon measuring the changes in fluorescence intensity with or without the existence of various acyl transfer promoters, the results revealed that PBu3 accelerated the fluorescence intensity in a time-dependent manner in comparison to the promoter-free control. PBu3 was the most active acetylation promoter of the probe, resulting in a 5-fold enhancement in fluorescence intensity within five minutes, while this increase could only be induced by the acyl transfer promoter. Finally, the structure and positive charge of the rhodamine probe enabled it to be well targeted to mitochondria, which is highly advantageous for studying acetyl-CoA in mitochondria. Therefore, the authors co-incubated the probe and PBu3 with HeLa cells and found that the mitochondria had a significant enhancement in fluorescence intensity as PBu3 promoted the acetylation of the probe via physiological acetyl-CoA. However, the concentration of the artificial promoter in this study was comparatively high; therefore, the probe structure or reaction conditions need to be optimised for future use.
Although several biosensors incorporating different nanomaterials have been applied to detect CoA, some of them have complicated preparation processes. Therefore, in 2021, Xu et al. fabricated nitrogen-doped carbon dots (N-CDs) (λex/λem = 360/480 nm) using carbon dots (CDs) with high resistance to photobleaching, good water solubility and chemical stability and a high biocompatibility for in vitro detection of CoA by a simple and fast pyrolysis method.132 The fluorescence produced by the prepared N-CDs after excitation at 340–380 nm was always in the range of 470–480 nm. This indicated excitation-dependent stages, which is probably attributable to the size heterogeneity of N-CDs. The blue fluorescence of N-CDs was quenched by Cu2+via electron or energy transfer to constitute N-CDs/Cu2+ complex. A relatively robust CoA/Cu2+ compound emerged by adding CoA, and the fluorescence of N-CDs was restored. Finally, the authors were able to apply the presented sensor for the determination of CoA in human serum.
Jin et al. designed and synthesized an α-KA probe 5 (λex/λem = 430/552 nm) and applied it to a microfluidic chip to detect α-KA in human serum.135 The probe used naphthalimide derivatives as the fluorophore and the hydrazine group as the response element (Fig. 11A). The hydrazine group generates a Schiff base with the carbonyl group of α-KA, which causes the PET effect to turn off and restores fluorescence. Three different alkylamino groups were incorporated into the naphthalimide derivatives and three different probes were designed. The first probe was poorly water soluble; thus, diglycolamine (DGA) was introduced to develop a second probe. This probe was not suitable for in vivo applications owing to its reduced reaction rate and the need for additional organic solvents. Finally, a long carbon chain probe was designed. The positive charge on the surface of the micelles can aid the gathering of α-KA. Therefore, the authors introduced the surfactant cetyltrimethylammonium bromide (CTAB) into the probe to form micelles. This enabled a 3.2-fold enhancement in fluorescence signal in 10 min in a nonpolar solvent-like microenvironment. The microfluidic system included two syringe pumps (pushing chemical sensors and pre-treated serum), sensor channels, laser emitters, and detectors (Fig. 11B). The sensor channel was divided equally into two branches. One served as a reaction channel for serum and micelles, while the other was designated as the reference. When the two channels were full, the pump stopped and the sensor detected the signals of the two channels in sequence.
 | ||
| Fig. 11 (A) Structure and the response mechanism (naphthalimide derivatives as the fluorophore and the hydrazine group as the response element) of mitochondrial fluorescent probe 5 (Chem. Sci., 2014, 5, 4012–4016) to detect α-KA, and (B) rational design of fluorescent chemosensors for α-KA detection, and diagram of the microfluidic system. Adapted with permission from ref. 135, copyright © 2014 Royal Society of Chemistry. Structures and the response mechanism of mitochondrial (C) fluorescent probe 6 (Acta Pharmacol. Sin., 2017, 38, 1683–1690) and (D) probe 7 (Sens. Actuators, B, 2017, 241, 1035–1042) to detect α-KA. | ||
Additionally, by exploiting the Schiff base reaction between the carboxyl and amino groups of α-KA, Gan et al. designed a two-component system probe 6 (λex/λem = 439/495 nm) to detect α-KA based on the calcium ion indicator benzothiazole-coumarin (BTC) (Fig. 11C).136 Firstly, the probe was incubated with α-KA and Ca2+ in ethanol. The intermediate structure produced by the reaction between the probe and α-KA is similar to BTC, and it generated a strong fluorescence under Ca2+ to measure α-KA concentration in serum. However, the sensitivity of the probe was significantly reduced when detecting α-KA in the serum, which the authors considered could be due to two reasons. One is that some components in the serum may interfere with the response of the probe to α-KA, and the second is the poor solubility of the probe in the aqueous system, which limits the ability to improve sensitivity by increasing the concentration of the probe.
The disadvantages of the two previous fluorescent probes in α-KA detection are the complex treatment with surfactants and low sensitivity, which limits the accurate and sensitive quantification of α-KA in the biological samples. In 2017, Fu et al. developed a unique mechanism that combined PET with a large absorption red-shift to develop a versatile probe 7 (λex/λem = 475/540 nm), with a low detection limit (0.9 μM).137 This is a surfactant-free system for the simultaneous absorbance-colorimetric, fluorescence, and colorimetric detection of α-KA in the biological samples (Fig. 11D). The probe consisted of 4-nitrobenzo-2-oxa-1,3-diazole (NBD) as a fluorophore to synthesise probe 4-hydrazinyl-7-itrobenzo[c][1,2,5]oxadiazole. When the hydrazine group at position-7 of NBD combines with the carboxyl group of α-KA to form a Schiff base, it inhibits PET, leading to a significantly enhanced fluorescence (60-fold). To demonstrate that the reaction mechanism is PET rather than ICT, the authors inhibited the process by neutralising the amino group by the addition of H+. Upon protonation of the diazinyl group, the amino nitrogen produces a blue shift in fluorescence, suggesting an ICT process. The probe exhibited excellent selectivity for α-KA even in the presence of 20 interferents. Finally, the orange-red colour (under daylight) and green fluorescence (under UV) were observed in the serum samples upon the addition of α-KA and the probe allowed monitoring of α-KA (2.0–900 μM) changes with the naked eye. Furthermore, the large redshift (100 nm) facilitated the observation of significant fluorescence changes in yeast cells and tissues.
All three previously described organic small molecule fluorescent probes require 10–60 min incubation time for practical in vitro detection of α-KA in serum, which limits the rapid clinical detection of the metabolite. In 2020, Das et al. designed and synthesized a hydrazinyl functionalised Al(III)-based DUT-5-N2H3 MOF fluorescent sensor (λex/λem = 330/470 nm).138 The reaction between the hydrazinyl groups of MOF and α-KA was extremely fast (60 s), with a detection limit of 0.61 μM. The morphology of the material remained stable before and after the reaction in methanol/water with α-KA, thus ruling out the mechanism of structural collapse. Then, the existence of the FRET effect between the electron-rich functionalised MOF and electron-deficient α-KA was examined. Experimental results revealed that the overlap between the emission spectra of KA and MOF was quite small, indicating that the resonance energy transfer from MOF to α-KA was too small to satisfy the sensing mechanism of FRET. Finally, the authors investigated whether there was a PET effect between MOF and α-KA, with the help of time-dependent density functional theory (TD-DFT). The results demonstrated that the PET effect between MOF and α-KA is caused by the hydrogen bond formed between the –NHNH2 linker and α-KA.
The general principle of α-LA detection employing an electrochemical approach is that it can undergo redox reactions on the surface of the electrode at a specific bias potential. However, conventional electrochemical detection methods for α-LA often have problems such as the need for higher voltage, lower sensitivity, and easy contamination of the electrode surface. Therefore, researchers have developed a variety of nanomaterials with excellent electrical conductivity and catalytic properties and modified them on the electrode surface, thus achieving highly sensitive determination of α-LA at low potentials. Marin et al. developed the manganese(IV) oxide-modified screen-printed graphene sensors (MnO2/SPGEs) to detect coenzyme Q10 (CoQ10) and α-LA simultaneously. The addition of MnO2 greatly improves the oxidation performance of the electrode for CoQ and α-LA. This is mainly due to the ability of graphene and MnO2 to accelerate the electron transfer rate, in addition to increasing the area of electrocatalytic activity. The detection range of the constructed sensor for α-LA was 0.3–25 μg mL−1, and the detection limit was 0.088 μg mL−1.140
NAD+ is used more as an oxidant for catabolism and NADPH is used more as a reducing agent for anabolism. The redox status of the cells, especially the level of NAD+/NADH or NADP+/NADPH, directly influences major life processes such as cell rhythm, aging, cancer, and death.141,142 In addition, the multi-enzyme redox system, involving NAD+/NADH and NADP+/NADPH, is the dominant oxidation system in the ETC. Most of the oxidation reactions involved in the decomposition of the three major metabolites, carbohydrates, lipids and proteins, are also accomplished through this system.143,144 Therefore, researchers have designed and developed many classical methods for the detection of NAD+ or NADH, and NAD+/NADH, such as the enzymatic cycling assay,145 high performance liquid chromatography,146 capillary electrophoresis,147 and spectrophotometric assays.148 As their intracellular and extracellular concentrations may vary between 10 nM and 1 mM, methods to quantify these metabolites need to be fast, reproducible, robust and span a wide concentration range.149
NADH exhibits slow electron transfer kinetics on untreated or modified electrodes, necessitating the application of high overpotentials for its detection (for instance, +1.1 V on the carbon electrode). In addition, the substances accumulated during NADH oxidation can contaminate and hinder the reaction at the electrode surface. Therefore, to overcome these problems, the approach commonly implemented is the modification of the electrode surface with redox mediators or various nanomaterials with excellent conductive sensing properties.
For example, in 2017, Han et al. prepared a graphene (Gr)–pyrroloquinoline quinone (PQQ)/chitosan (CS) composite by ultrasonically dispersing Gr–PQQ in CS using a facile and convenient self-assembly method. The composite was dried naturally at room temperature by dropwise addition on GCE. This facile self-assembly method not only ensured enzymatic activity and stability for electron transfer but also had a synergistic effect with Gr to promote the electrocatalytic oxidation of NADH. The Gr–PQQ/CS composite with a larger active electrode area was able to adsorb more PQQ, while the composite conductive materials exert the catalytic effect synergistically. The biosensor was able to measure NADH spiked in human serum samples within 2 s and exhibited a wide linear range of 0.32 to 220 μM, with a low detection limit of 0.16 μM. More importantly, it featured excellent selectivity and eliminated some common electroactive interferents.150 However, an enzyme (PQQ) was employed in the study, and the non-enzymatic NADH sensor was superior in terms of stability and cost compared to the enzymatic sensor. In 2019, Wang et al. prepared aluminium hydroxide/iron, hydroxide/multi-walled carbon nanotube (AH/IH/MWCNTs) composites using a facile synthetic method and modified them on GCE to assay NADH at +0.15 V which is a lower potential. AH/IH not only expedites the electron transfer but also boosts the capability of NADH determination through the electrostatic interaction between iron hydroxide and the phosphate group on NADH to increase the sensitivity of the proposed biosensor. Meanwhile, MWCNTs provide the built-in conductor as a bonding agent, thereby facilitating electron transfer. The proposed sensor had an exceptional electrocatalytic performance in detecting NADH, with a linear concentration range of 0.5–220 μM and a low detection limit of 0.30 μM.151
In another study, Thiruppathi et al. fabricated an enzyme-free electrochemical biosensor based on an electrochemical probe using the small molecule 4-aminophenol (AP) to achieve simultaneous detection of NADH and H2O2. The SPCE was first pre-anodised before adding H2O2 to the electrode surface, followed by the addition of 1 mM 4-aminophenylboronic acid (APBA).152 The electrode was then baked to generate AP. This is due to the ability of H2O2 to selectively convert APBA to AP by boronic acid deprotection. The generated AP was adsorbed onto the surface of the pre-anodised SPCE through hydrophobic reaction to form the SPCE*/AP sensor. The pre-anodised SPCE has edge/defective sites with different oxygen functions that contribute to the adsorption of AP. The electrocatalytic performance of the sensor was verified with CV, and the results showed that the oxidation of NADH occurred at +0.15 V, while the reduction of H2O2 occurred at −0.3 V. When targets were added, the detection of both targets could be achieved simultaneously without significant interference. Obvious redox peaks were observed due to the interconversion of AP and APBA at different voltages. Finally, the authors used the constructed electrochemical sensor to monitor normal and cancer cells and found that the concentrations of NADH and H2O2 in cancer cells were significantly higher than those in normal cells, which was consistent with previous reports.
Molecularly imprinted polymers (MIPs) have been extensively employed in electrochemical sensing, because of their acid and base resistance, good stability, ease of preparation, and high selectivity. MIPs serve as sensitive recognition units to modify WE and significantly improve the selectivity of these sensors to facilitate the detection and analysis of complex samples. Therefore, based on this technique, Liu et al. developed multiple MIP sensors for the detection of endogenous redox pairs, including glutathione disulphide (GSSG), GSH, cysteine (Cys), cystine (CySS), NADP+, and NADPH, in blood samples from lung cancer patients.153 This simultaneous detection of multiple redox pairs by multiple biosensors provides a better insight into the overall redox homeostasis levels in cancer cells. The MIP/GCE sensor was prepared by electro-polymerisation in the molecular monomer O-phenylenediamine (OPD) and template molecule (six targets) solution. The embedded template molecules were then extracted in NaOH by CV scanning at −0.8 to 0.8 V. Using these sensors, the authors assayed blood samples from lung cancer patients. They found that the levels of GSH and NADPH in cancer patients were significantly lower than those in healthy subjects, while the level of GSSG was significantly higher than that in healthy individuals. This suggests that GSH/GSSG and NADPH/NADP+ can be used as markers for the early diagnosis of lung cancer.
Real-time monitoring of NADH under different environmental stress levels is more useful for interpreting the function of NADH in mitochondrial energy metabolism. It is also useful for evaluating or probing the mechanism of action of anti-cancer drugs. Jiang et al. developed an electrochemical sensor that can detect the in situ real-time changes in mitochondrial NADH at the single-cell level. Based on a previous work related to SiC@C nanowire electrodes (NWE) (Fig. 12A),154 the SiC@C-NWE electrode was modified with conductive polymers [poly(3,4-ethylendioxythiophene), PEDOT] and CNTs. PEDOT is positively charged, allowing more negatively charged NADH to be enriched around the electrode by electrostatic interaction. CNTs have good electrical conductivity and promote electron transfer, thus enabling the catalytic oxidation of NADH at lower potentials while avoiding contamination by deposition of reaction products. The authors inserted the sensor in MCF-7 human breast cancer cells to monitor the glucose- and paclitaxel-induced NADH levels. They found that glucose-induced NADH levels increased significantly, as glucose can increase NADH levels through intracellular metabolism, while paclitaxel-induced NADH levels did not change noticeably after glucose stimulation. This is due to the ability of paclitaxel to hinder the above-mentioned metabolic process. The sensor was inserted in MCF-7 cells and localised around mitochondria. Cyclosporine A (CsA, an inhibitor of mPTP opening)-treated cells showed no detectable changes in current signals prior to stimulation with RSV, suggesting that RSV may open mPTP and further increase the intracellular NADH level.
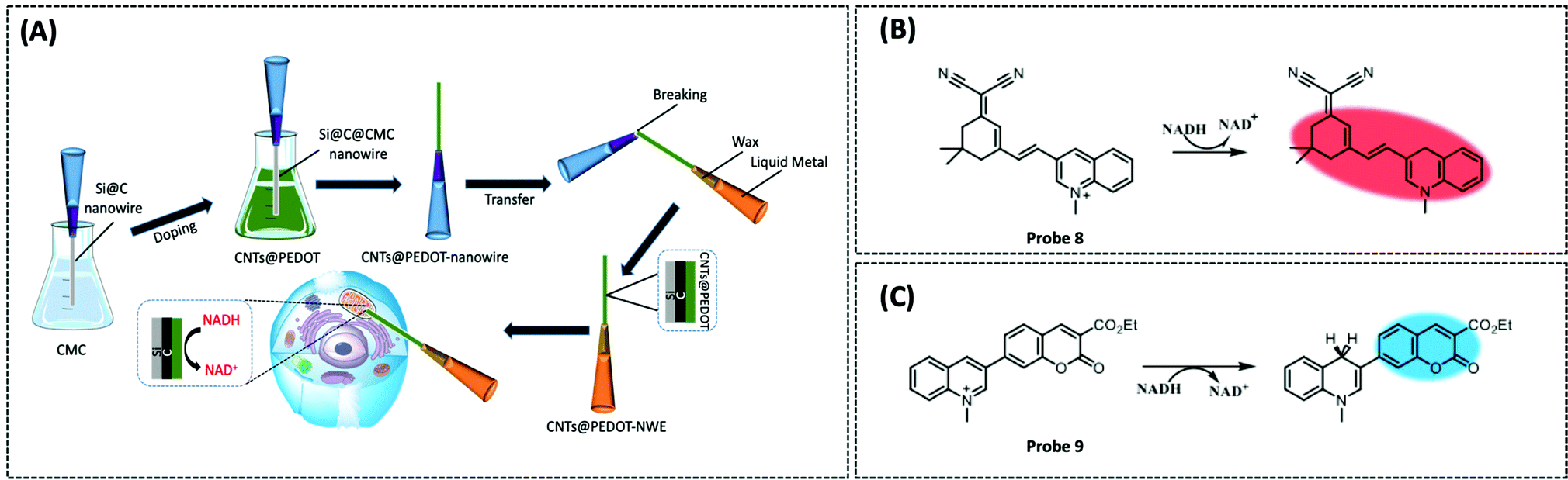 | ||
| Fig. 12 (A) Schematic diagram showing the main fabrication process of the electrochemical biosensor (SiC@C-NWE electrode modified with PEDOT and CNT) and amperometric monitoring of NADH from intracellular mitochondria (Chem. Sci., 2020, 11, 8771–8778). Structures and the response mechanism of mitochondrial NIR (B) fluorescent probe 8 (Anal. Chem., 2019, 91, 1368–1374) and (C) two-photon fluorescent probe 9 (Sens. Actuators, B, 2020, 324. 128637) to detect NADH. | ||
Zhao et al. reported a NIR mitochondrial-targeting probe 8 (λex/λem = 568/660 nm) to detect NADH. The probe consisted of a dicyanoisophorone electron-withdrawing group as the NIR fluorescent chromophore and 1-methylquinolinium cation as the NAD(P)H response group (Fig. 12B). The recognition group 1-methylquinolinium is reduced to 1,4-dihydroquinoline by NADH, leading to a red-shift, to 660 nm. This is mainly due to the ICT effect of the recognition group transforming the probe into a typical donor–π–acceptor (D–π–A) structure, resulting in a clear NIR fluorescence signal.155 In live cell experiments, the probe was able to differentiate the changes in NADH content in normal and cancer cells. Specifically, the fluorescence intensity of NADH in cancer cells (HepG2 cells) was approximately more than double that in normal cells (HL-7702 cells). The level of NADH in cancer cells is largely dependent on glycolysis, which refers to the consumption of glucose to produce NADH. Based on this, the probe was applied to track changes in the cellular NADH levels. When glucose was added to cancer cells and then allowed to react with the probe for 15 minutes, the fluorescence intensity of NADH was dramatically higher than that of the control subjects. Finally, owing to the near-infrared nature, low toxicity, and good cell membrane penetration of the probe, the authors used the probe to detect endogenous NADH content in a mouse model of cancer (H22-tumor-bearing mice).
In a separate work, Podder et al. synthesized a two-photon fluorescent probe 9 (λex/λem = 860/460 nm), consisting of a two-photon fluorophore coumarin and the recognition group quinoline, by the Suzuki reaction.156 An intra-molecular ICT effect occurs when the probe reacts with NADH, causing the probe to switch from “turn off” to “turn on”, resulting in a distinct blue fluorescence (Fig. 12C). The authors then used this probe to investigate NADH imaging in different cell lines (colon cancer HT-29, melanoma cancer B16F10 and murine leukaemia RAW 264.7) and showed that NADH expression varies with cell line. During glycolysis, the fluorescence intensity of NADH gradually increases with increasing concentration of glucose, decreases with an increasing concentration of pyruvate, and conversely, gradually increases with increasing lactate concentration, demonstrating that the level of NAD+/NADH is influenced by the substrate or metabolite in the glycolysis pathway. Finally, the probe was applied to track NADH changes in the oxidative phosphorylation pathway and in GSH crosstalk. These results confirmed that rotenone increases NADH levels by inhibiting complex I of the ETC. In contrast, an uncoupling agent (CCCP) induced NADH depletion. Dynamic expression of NAD(P)H increased upon GSH inhibition of H2O2 to balance the mitochondrial redox potential.
In recent years, autofluorescence-excited deep-ultraviolet (DUV) light with high photon energy has been used to excite the structures of numerous kinds of biochemical substances in cells. Based on this, Kikawada et al. used DUV-SPR to achieve enhanced autofluorescence and highly sensitive imaging of organelles in different cells.157 First, DUV-SPR was applied to the MC3T3-E1 cells and yeast cells cultured on aluminium. After two days of incubation, the autofluorescence spectrum of the MC3T3-E1 cells was found to exhibit two emission peaks following DUV laser excitation: the peak at 330 nm originated from nucleic acids and the peak at 500 nm belonged to mitochondria. Owing to the enhanced autofluorescence of cellular structures by DUV-SPR, both the nucleic acid and mitochondria in the MC3T3-E1 cells can be visualized with high sensitivity. The authors then compared conventional fluorescence microscopy and DUV-SPR for the identification of mitochondria in the MC3T3-E1 cells. DUV-SPR enhanced the autofluorescence of live yeast cells and was used to observe tryptophan and NADH in their mitochondria, which showed autofluorescence at 335 and 440 nm, respectively. Although DUV probably damages proteins, this technique requires less energy, is label-free, and can reduce the observation time. Therefore, DUV-SPR allows for highly sensitive, label-free observation of the structures of various biochemicals that are not excitable by visible light, while allowing for real-time in situ analysis of these biochemicals.
Mitochondria are the primary O2-consuming organelles in the cell and the central site of cellular metabolism, providing sufficient energy and substances for the cell. Therefore, the concentration of O2 should be accurately maintained at an appropriate level. Excessive ROS are produced from excess O2, which is detrimental to the mitochondria. The insufficient supply of O2 affects mitochondrial metabolism, which is closely related to cancer.163,164 Therefore, accurate detection of the mitochondrial oxygen consumption rate (OCR) and concentration of dissolved oxygen is important.
The methods available for the measurement of O2 include electrochemical and optical methods. Classic optical commercial instruments include the XF series from Agilent Inc.165 In addition to optical commercial instruments, scientists have designed and developed optical nanosensors for the detection of O2 concentration in mitochondria. Lian et al. constructed a fluorescent nanosensor for real-time in situ detection of proton concentration and O2 concentration in mitochondria using polystyrene nanomaterials.166 They obtained polystyrene nanoparticles with active amino groups on their surface by copolymerising styrene and 2-aminoethyl methacrylate hydrochloride. The oxygen-sensitive probe platinum(II) octaethylporphyrin (PtOEP) and the reference probe rhodamine B isothiocyanate (RBITC) were encapsulated inside the polystyrene nanoparticles during the polymerisation process, thus reducing the interference of the surrounding solution environment with O2 detection. The pH-sensitive probe fluorescein isothiocyanate (FITC) and TPP were covalently attached to the surface of the particle by reaction with the surface amino groups of the nanoparticles, thus increasing the accuracy of proton concentration detection. The fluorescent sensor was able to measure HeLa cells at 0–40.4 mg L−1 O2 concentration with a response time of <10 s, indicating that the method is reliable for detecting intracellular O2 concentration. The sensor's pH measurements range from 5.0–9.0, which allowed it to be used to measure the pH of mitochondria and other intracellular organelles. At the same time, it showed very low biotoxicity and high mitochondrial targeting activity, which enabled it to be applied to measure mitochondrial pH and O2 concentration in HeLa cells (Fig. 13A). This work provides insights into the design of multi-parameter and structurally simple nanosensors. Wang et al. reported a detailed review of the optical methods for sensing and imaging O2, which provided the necessary guidance for detecting O2.167
 | ||
| Fig. 13 (A) pH fluorescence images and oxygen lifetime images of HeLa cells costained with fluorescent nanosensor incubated with CCCP (20 μM) for 0, 10, 20, and 30 min (Anal. Chem., 2021, 93, 8291–8299), adapted with permission from ref. 166, copyright © 2021 American Chemical Society. (B) Cellular oxygen consumption rate (OCR) comparison after injecting different drugs (Sens. Actuators, B, 2020, 306, 127465), adapted with permission from ref. 170, copyright © 2020 Elsevier Ltd. | ||
The most widespread electrode used in electrochemical biosensors is the Clark-type electrode.168,169 Clark-type electrochemical sensors typically use a liquid electrolyte with some problems such as electrolyte leakage and drying, as well as degradation of analytical performance during long-term storage. Therefore, Han et al. designed a 24-well electrochemical dissolved oxygen sensor array with Nafion as the solid polymer electrolyte, which not only solved the above problems but also acted as a protective layer.170 The Nafion membrane was used to cover the surface of the working electrode; it absorbs water and oxygen dissolved in water, thus allowing the reaction to occur at the reduction potential of O2 (−0.7 V vs. Ag/AgCI), achieving the detection of dissolved oxygen. The prepared sensor was able to detect the changes in the cellular respiration rates in 5000, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 30000 C2C12 cells under the stimulation of carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) and rotenone (Fig. 13B). The experimental results showed that the average ratio of uncoupled OCR to coupled OCR obtained from the sensor array was 2.18, which was higher than the average value of 1.70 obtained from XF-24. The detection speed of the sensor was also twice as fast as that of XF-24. Notably, the variation in O2 concentration is several hundred pmol μL−1 due to cellular respiration, which implies the high sensitivity of the prepared sensor.
000 and 30000 C2C12 cells under the stimulation of carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) and rotenone (Fig. 13B). The experimental results showed that the average ratio of uncoupled OCR to coupled OCR obtained from the sensor array was 2.18, which was higher than the average value of 1.70 obtained from XF-24. The detection speed of the sensor was also twice as fast as that of XF-24. Notably, the variation in O2 concentration is several hundred pmol μL−1 due to cellular respiration, which implies the high sensitivity of the prepared sensor.
Shen et al. designed and synthesized a core–shell SERS nanosensor in three steps (Fig. 14A).171 4-Mercaptobenzonitrile (MBN), whose Raman band is located in the Raman-silent region (1800–2800 cm−1) and can thus avoid interference of other molecules in the cell, was selected as the non-interfering Raman reporter molecule and incorporated into the Au (13 nm) core–Ag (5 nm) shell nanoparticles (Au@Ag NPs). Meanwhile, the Ag shell protects the MBN reporter molecule from ROS attack and is etched by ROS, resulting in a decreased SERS intensity of MBN; this process, as such, can serve as one of the sensing mechanisms to detect ROS. The peak of MBN at 2226 cm−1 gradually decreases with the gradual increase in hydrogen peroxide concentration. This is due to the reduction in thickness and the localized surface plasmon resonance (LSPR) effect of the Ag shell, which leads to weak coupling between the Au core and the Ag shell layer. Subsequently, the mitochondrial-targeting peptide MLS was decorated on the surface of the Au@MBN@Ag nanoparticles to achieve mitochondrial targeting. All experimental data further showed that the MBN peak at 2226 cm−1 was much lower in the treated cells than in the untreated cells. Finally, Au@MAN@Ag was successfully used to dynamically monitor ROS production during photothermal therapy (PTT), which is crucial for understanding ROS in this process.
 | ||
| Fig. 14 (A) Schematic illustration of the preparation of the sandwich SERS nanoprobe and the response mechanism for the detection of ROS (Sens. Actuators, B, 2019, 285, 84–91). (B) Schematic illustration of the G4-DNAzyme modified nanopipette for the detection of ROS using ABTS as the substrate and chemical formulae of ABTS before and after oxidation (Angew. Chem., Int. Ed., 2018, 57, 13226–13230), adapted with permission from ref. 173, copyright © 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
ROS can be electrochemically reduced or oxidised by using appropriate electrode potentials, resulting in the production of electron transfer that can be directly recorded. Electrochemical methods have several advantages over traditional methods, including no indicator compounds, instant response to ROS, and real-time detection, and have proven to be strong tools for detecting ROS in vivo.172 Currently, nanoelectrodes are applied to monitor the variations of ROS concentration in cells owing to their high temporal and spatial resolution. Jiang et al. developed a platinised SiC@C nanowire electrode that could be applicable for in-situ detection of mitochondrial ROS under different environmental stresses. In addition, ROS production induced by an antineoplastic agent (paclitaxel) was also successfully recorded, providing an insight into the variations in the subcellular ROS levels during chemotherapy.87
Nanopore-based, electrochemical, bioanalytical techniques based on ion current rectification (ICR) have proven to be powerful molecular sensing tools. Song et al. reported a 30 nm ICR-nanopipette with nanopores decorated with 3-aminopropyltriethoxysilane (APTES) and glutaraldehyde and then cross-linked with the G-quadruplex (G4) DNAase (Fig. 14B).173 G4 DNAase can catalyse H2O2-mediated oxidation of ABTS and is very stable compared to HRP. ABTS can be oxidised by ROS to generate metastable radicals, which have a high affinity for DNA. ABTS reacts with ROS to generate large amounts of ABTS˙−, resulting in an enhanced charge-density on the inner wall of the electrode, which can be quantified using the current–potential (I–V) curve for local ROS. The rectification ratio (I−1.0V/I+1.0V) is most noticeable for a similar concentration of H2O2 in a neutral environment compared to that in an alkaline environment, indicating that it is useful for monitoring the cellular environment. Finally, the authors used the ICR-nanopipette to monitor the oxidative stress status of different cell lines under capsaicin induction. It was found that the current in normal HEK293 cells without TRPV1 was almost unchanged, indicating that capsaicin does not affect normal cells. In contrast, MCF-7 cells with upregulated TRPV1 expression exhibited a much higher current than ROS from CHO cells that were stably transfected with TRPV1, demonstrating a relationship between TRPV1 expression and cell sensitivity.
O2˙− plays a critical role in several diseases, including atherosclerosis, diabetes, inflammation, and neurodegenerative disease, and is regarded as a second messenger in various signalling pathways. Its normal concentration ranges between 10 and 100 nM. In particular, O2˙− may cause irreversible damage to tissue when its accumulative concentration exceeds 0.1 mM.174
Superoxide dismutase (SOD) is responsible for catalyzing the decomposition of O2˙− to O2 and H2O2. Zhu et al. developed a rapid, selective biosensor based on SPCE modified Pt–Pd–SOD and Nafion to monitor O2˙− in cells.175 The porous Pt–Pd nanomaterial not only has a large electrochemical surface area to accelerate electron transfer but can also better support the load of SOD, so that the redox centre of SOD can be in full contact with O2˙− to achieve better catalysis. The SOD/Pt–Pd SPCE O2˙− detection mechanism is mainly described as a direct electron transfer (DET) between copper (Cu) or zinc-superoxide dismutase (Zn-SOD) and the modified electrode. The oxidation current and the reduction current gradually increased when various concentrations of KO2 were applied, indicating that the sensor had a good catalytic ability for O2˙−. Subsequently, the continuous dropwise addition of O2˙− was applied at −0.1 V using the amperometric method. The Michaelis–Menten constant Kappm is commonly used to assess the affinity of an enzyme to the substrate, and the lower the value, the higher the affinity. This is one of the indicators to be evaluated for enzyme-based sensors. The Kappm of the prepared sensor was 1.37 μM, indicating a great catalytic ability and affinity for O2˙−.
In addition to enzyme-based electrochemical sensors, researchers have likewise developed numerous nanomaterial-based enzyme-free electrochemical sensors for the determination of O2˙−. Herein, we present three materials: silica-based materials, cobalt nanomaterials, and MOF materials. Researchers have prepared novel enzyme-free electrochemical biosensors for the detection of O2˙− in cells using silicon–manganese phosphate (SiO2–Mn3(PO4)2) nanoparticles that mimic Mn-superoxide dismutase (MnSOD) activity in combination with MWCNTs.176 Zhang et al. also prepared cobalt nanoparticle-doped porous cubic carbon (Co-PCC) using zeolitic imidazole framework-67 (ZIF-67), which was modified on SPCE to prepare an enzyme-free electrochemical sensor for the detection of O2˙−.177 Qiu et al., for the first time, employed the shear exfoliation method to construct exfoliated 2D MOF nanosheets (ELM-12 nanosheets) (Fig. 15A). They fabricated a flexible porous graphene electrode on a polyimide substrate using the laser-induced method, and then added the ELM-12 nanosheet solution to the working electrode. The result of CV demonstrated that ELM-12 nanosheets exhibited better electrocatalytic ability towards O2˙− in comparison to their bulky counterparts. The current response trends were recorded by successive dropwise addition of various O2˙− concentrations at −0.1 V using i–t curves. The EML-12 sensor showed a wide detection range (10–1200 μM) and a low detection limit (3.01 μM).178
 | ||
| Fig. 15 (A) Schematic illustration of the ELM-12 nanosheet based flexible electrochemical sensor for the detection of O2˙− (ACS Appl. Mater. Interfaces, 2020, 12, 5429–5436), adapted with permission from ref. 178, copyright © 2020 American Chemical Society. Structure and response mechanism of mitochondrial (B) fluorescent probe 10 (J. Am. Chem. Soc., 2015, 137, 6837–6843), and (C) probe 11 (Biomaterials, 2018, 156, 134–146) to detect O2˙−. | ||
Although the aforementioned studies on the electrochemical detection of O2˙− fail to spotlight mitochondria, they provide us with inspiration for the detection of O2˙− in mitochondria. (1) Choice of material: enzyme-based electrochemical sensors can use SOD to catalyse O2˙− while combining materials with excellent electrical conductivity to catalyse the decomposition of H2O2; enzyme-free electrochemical sensors can be constructed with low toxicity materials and exhibit excellent catalytic properties. (2) Drug models: O2˙− production is usually stimulated with Zymosan A, but this model is not a cellular model that specifically detects O2˙− production by mitochondria. It is well-known that mitochondrial complexes I, II, and III are redox centres for O2˙− production; therefore, inhibitors of all four mitochondrial complexes (e.g., rotenone, thenoyltrifluoroacetone, antimycin A, KCN) can be used to induce O2˙− production by mitochondria.
The groups that respond to O2˙− in the probe typically include the following: pyridinium-containing molecules,179 catechol,180 and trifluoromethanesulphonamide groups.181 Hu et al. synthesized a mitochondrial O2˙− targeting probe 10 (λex/λem = 509/534 nm) based on the fluorescein structure.182 The authors used 5-carboxy-2′,4′,5′,7′-tetrafluorofluorescein as the fluorophore, trifluoromethanesulphonate as the identification group, and TPP+ as the mitochondrial-targeting group. The introduction of trifluoroester groups at the 3′ and 6′ positions of xanthene inhibits fluorescence because of the formation of a lactone ring, and the free xanthene is released upon reaction with O2˙−, leading to the “turn-on” of fluorescence (Fig. 15B). First, the authors evaluated the reactivity and selectivity of probe 10 (R1). Probe 10 (R1) could reach equilibrium with O2˙− within 10 min and the fluorescence intensity of the reaction product displayed no variation over 60 min, indicating that the probe responds rapidly and is chemically highly stable. The results showed that the fluorescence intensity of the solution increased linearly with the concentration of O2˙− (0–13 μM), while the detection limit was as low as 23 nM. The probe exhibited a 66–100-fold selectivity for O2˙− compared to other interferents. Subsequently, the authors conducted a range of experiments within the cells by using probe 10 (R2). Four mitochondrial complex inhibitors were used to induce mitochondrial O2˙− production in three different cell lines (HCT116, human colon cancer cells; BV-2, mouse microglia; and RAW 264.7, mouse macrophages). Two-photon confocal imaging revealed that the efficiency of O2˙− production by the four inhibitors was as follows: antimycin A > FCCP > rotenone > malonic acid, from high to low. Probe 10 (R3) enabled the rapid time-delayed visualisation of the mitochondrial O2˙− level induced by antimycin A in THP-1 cells, using low laser power output, due to the introduction of the mitochondrial targeting motif TPP+. Finally, the authors used probe 10 (R2) to achieve dynamic monitoring of protein kinase C (PKC)-regulated NADPH oxidase (NOX)-derived O2˙− production, and two-photon imaging of O2˙− induced by PMA or antimycin A in zebrafish.
In another study, Han et al. synthesized a ratiometric NIR mitochondria-targeting probe 11 (F742/F790), for dynamic monitoring of mitochondrial O2˙− during ischemia/reperfusion (I/R).181 The probe was composed of the NIR fluorophore heptamethine cyanine, the O2˙− recognition group trifluoromethanesulphonamide, and TPP+ (Fig. 15C). When the probe reacted with O2˙− a significant blue-shift occurred. The absorption spectrum shifted from 762 to 606 nm, while the emission spectrum shifted from 790 nm to 742 nm. Importantly, the probe reacted with O2˙− within three minutes to reach the maximum fluorescence intensity, which is satisfactory for the in vivo imaging of O2˙− with a fast metabolism rate. The authors constructed four I/R cell models using glucose, serum, oxygen, and glucose-serum deprivation/reperfusion, as well as a cell model cultured under normal conditions. The concentration of O2˙− in these five I/R cell models was separately monitored by using probes, which indicated that different levels of reperfusion induce different levels of O2˙− float. Significantly, the authors used probes to explore the molecular changes in oxidative stress caused by excess O2˙− in mitochondria. They found that the O2˙−-induced oxidative stress causes changes in Ca2+ concentration and pH level. This further disrupts Na+–H+ and Na+–Ca2+ exchange, resulting in Ca2+ influx and collapse of the MMP ultimately causing an increase in the level of apoptosis proteins. These results indicated that I/R is the main factor that causes apoptosis. Finally, the probe was applied to evaluate the association between O2˙− concentration and the level of tissue injury during I/R in vivo, as well as the protective effect of ischemic preconditioning and post-treatment.
H2O2 can be considered as a potential marker for disease diagnosis. When the concentration of H2O2 is at subtoxic levels, it can act as a second messenger to maintain normal cellular function;183 however, when excess H2O2 is produced in mitochondria, it causes oxidative stress and inflammation, which are related to various disorders such as cancer, PD, and AD.184 In this section, we will highlight several classical materials for electrocatalytic hydrogen peroxide decomposition as well as their mechanisms and reasons. Electrochemical biosensors usually employ horseradish peroxidase (HRP), immobilised on the electrode, to catalyse H2O2 because of its great selectivity and sensitivity and rapid reaction.185 Some conductive materials, including AuNPs, CNTs and graphene, are coated on the biosensor to facilitate electron transfer, and improve conductivity and catalytic performance. Bai et al. constructed an electrochemical biosensor by immobilising HRP on dendritic mesoporous silica nanoparticles to detect H2O2 in PC12 cells. In particular, the highly accessible internal surface areas in the biosensor increased the electronic transfer, while the large pore volume of dendritic mesoporous silica nanoparticles enhanced catalytic activity and showed high enzymatic loading.186
HRP-based electrochemical sensors suffer from the disadvantages of easy denaturation and complex and strict immobilisation processes, even to the extent that the rate of electron transfer between the enzyme and the electrode is limited by the deep burial of the enzyme reactive sites. Hence, developing a nonenzymatic, rapid, and stable biosensor for monitoring H2O2in vivo and in vitro is urgently required.187 Since 2007, a number of nanomaterials that can mimic the catalytic properties of natural HRP, including noble metals (e.g., AuNPs), metal oxides (e.g., Fe3O4), and carbon-based nanomaterials (e.g., single-walled carbon nanotubes), which can be used to fabricate nanozyme biosensors at a low-cost, have good stability, and are easy to modify, have been developed.188
The intermetallic PtPb nanoplates were assembled on graphene through self-assembly to form PtPb/G nanocomposites. This biosensor showed a wide linear detection range of 2–2.5 mM and a low detection limit of 2 nM. The outstanding electrocatalytic activity was due to the following reasons: (1) PtPb nanoplates and graphene have unique high-density electrocatalytic active areas for the reaction of H2O2; and (2) PtPb and graphene facilitate the electron transfer to electrodes and exhibit a synergistic effect for electrocatalysis of hydrogen peroxide.189 Zhang et al. prepared NS-GQD/G nanosheets using graphene quantum dots (GQDs), GO and thiourea by hydrothermal self-assembly and thermal annealing method, which exhibited high electrocatalytic capacity for the reduction of H2O2. The superior electrocatalytic capacity of NS-GQD/G to H2O2 was attributed to the extremely strong electron transport capability of N and S coplanar, the large number of oxygen-containing functional groups of GQD, and the bent surface of NS-GQD/G. The high electrical conductivity of graphene sheets provides a conductive substrate for the interconnection of GQDs, resulting in higher efficiency and speed of electron transfer.190 Lu et al. first prepared Fe3O4@ZIF-8 composite nanomaterials modified by molybdenum disulfide (MoS2) nanosheets, and then decorated the composite nanomaterials with gold nanoflowers (AuNFs) using electrodeposition, and finally coated the nanomaterials on the electrodes to prepare an enzyme-free H2O2 electrochemical biosensor. Fe3O4@ZIF-8 nanomaterial was selected because of the ability of magnetic Fe3O4 nanoparticles to mimic HRP activity, and the large pore size and surface area of ZIF-8. Nonetheless, the Fe3O4@ZIF-8 nanomaterial is poorly connected with the electrode surface and is easily soluble in the electrolyte solution. Therefore, the authors introduced MoS2 nanosheets and one-step electrodeposited gold nanostructures, which not only significantly enhanced the stability, conductivity and electrocatalytic ability of the catalyst but also maintained the surface morphology and structure of the Fe3O4@ZIF-8 nanomaterial. The constructed sensor possessed an ultra-wide detection range of 5 μM to 120 mM and a low detection limit of 0.9 μM.191 This demonstrated that AuNFs/Fe3O4@ZIF-8-MoS2 has great potential as a novel HRP mimetic enzyme to replace conventional enzymes, and the wide detection range and low detection limits provide an important tool for monitoring cellular variations in H2O2 when treated with different drugs.
The design of the H2O2 probe recognition unit includes boric acid or boronic acid pinacol ester,192,193 α-ketoamide,194 and phenol.195 For example, when H2O2 reacts with the probe, boric acid or boric acid pinacol ester is shed, resulting in changes in the molecular structure and the fluorescence signal of the probe. The use of H2O2 for seleno-Mislow–Evans rearrangement with allyl selenides followed by acetal hydrolysis to generate green fluorescent molecules within 8 s, has recently been reported in the literature.196
Yang et al. reported probe 12, which not only targets mitochondria but also proteins, nucleic acids, and H2O2. The signals of each do not interfere with one another. When the probe enters the mitochondria, H2O2 cleaves the borate group, causing the probe to emit an orange fluorescence (λex/λem = 405/580 nm) (Fig. 16A).197 The hydrophobic part of the probe was combined with the exposed hydrophobic pocket of the protein, causing it to produce a red fluorescence (λex/λem = 530/632 nm). When the probe interacted with negatively charged nucleic acids (DNA and RNA) through electrostatic interaction and π-conjugation, it condensed and aggregated on the nucleic acid, emitting a deep red fluorescence (λex/λem = 580/688 nm). Although the product generated from the binding of the probe to H2O2 was also able to bind to protein, the emitted light was blue-shifted, and additionally the affinity of the protein to the probe was greater than that of the reaction product; hence, protein detection was not affected. The authors used PMA to induce the production of endogenous H2O2, and included a negative control with the addition of H2O2 scavenger. A commercial mitochondrial dye (mitochondrial tracker) was also used to detect the intracellular fluorescence signal of the three substances, and the results showed a high co-localisation coefficient as well as overlapping spectra. These results demonstrated that the probes could target mitochondria and detect hydrogen peroxide, proteins and nucleic acids in mitochondria. The authors monitored the dynamic changes in proteins and nucleic acids in mitochondria and observed the behaviour of proteins and nucleic acids under H2O2 stimulation by using super-resolution fluorescence microscopy technology-stimulated emission depletion (STED) optical microscopy. The finding that protein dissociation from nucleic acids increased with increasing H2O2 concentration suggests that H2O2 is a key upstream signal transducer that regulates the separation of mitochondrial nucleoproteins.
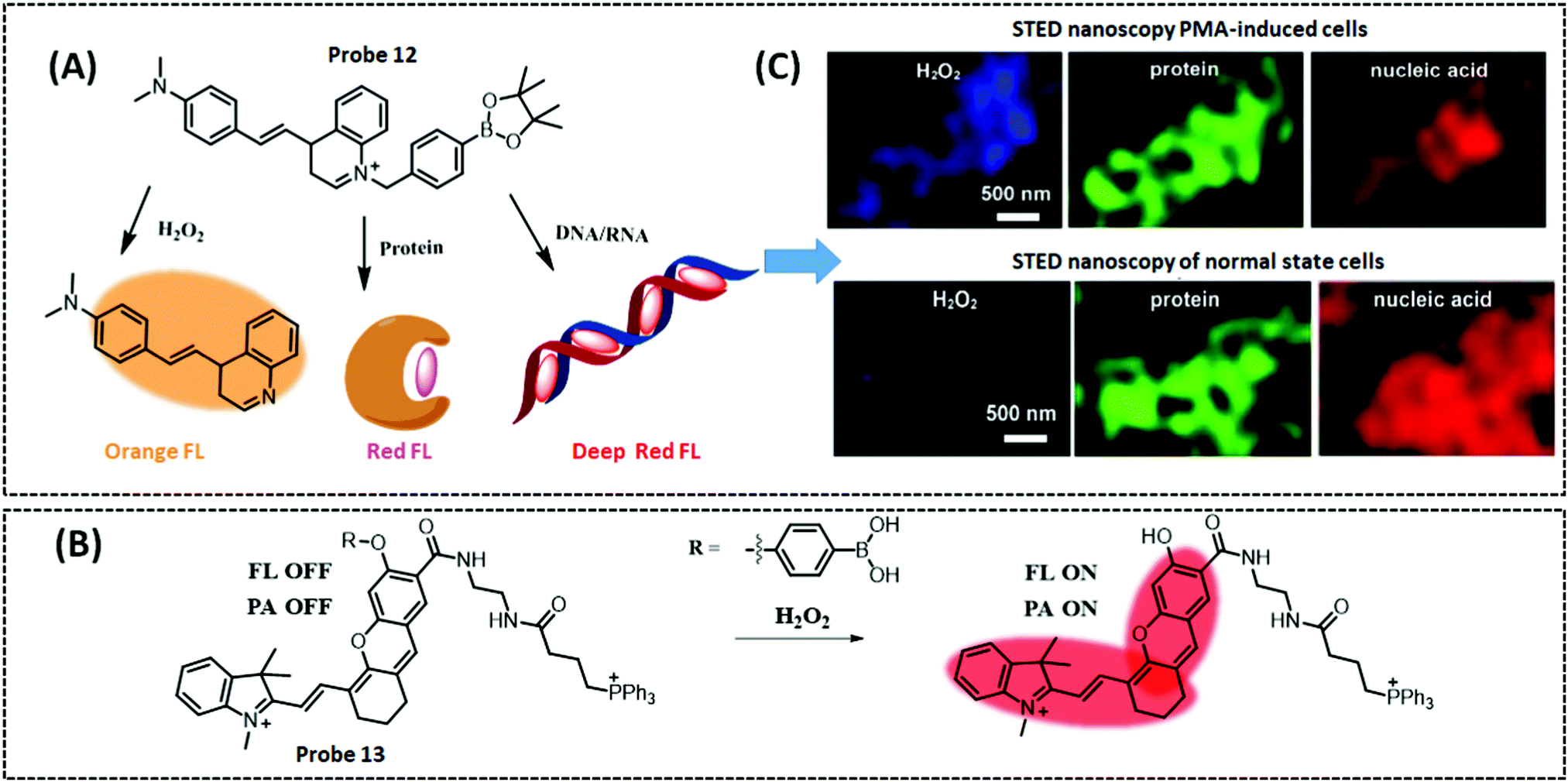 | ||
| Fig. 16 Structures of mitochondrial (A) fluorescent probe 12 (Angew. Chem., Int. Ed., 2020, 59, 16154–16160), and (B) probe 13 (Anal. Chem., 2020, 92, 14244–14250) to detect H2O2. (C) STED super resolution nanoscopy image of H2O2, proteins and nucleic acids in mitochondria with probe 12, adapted with permission from ref. 197 copyright © 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Chen et al. designed a NIR probe 13 (λex/λem = 600/706 nm) that targets mitochondria, and made it available for fluorescence/photoacoustic (FL/PA) dual-modality imaging of excess H2O2 in an inflammatory mouse model (Fig. 16B).198 The probe comprised three components: a boronic acid (BOH) moiety as the H2O2 response moiety, the near-infrared dye hemicyanine (HCy) to provide signals for FL and PA, and TPP+ as the mitochondrial targeting moiety. The ICT effect is inhibited prior to the reaction because the hydroxyl group of HCy is blocked. Upon reaction with H2O2, BOH is rapidly released, liberating free HCy and restoring the ICT effect to provide FL and PA signals. The in vitro experiments demonstrated that the probe had a low detection limit (0.348 μM) and a high kinetic rate constant for H2O2 (kobs = 4.72 × 10−3 s−1), which were superior to the most recently reported data for H2O2 probes. Cell experiments demonstrated a 2.4-fold increase in the FL signal and a 4.7-fold increase in the PA signal in HeLa cells, and a 2.1-fold increase in the FL signal and a 3.3-fold increase in the PA signal in RAW 264.7 cells. In an LPS-induced inflammation mouse model, the probe increased FL sensitivity by 1.6-fold in the abdomen, and PA sensitivity by 2-fold in the liver.
Hypochlorous acid (HOCl) is produced by H2O2 and Cl− under the catalysis of myeloperoxidase (MPO). HOCl is an unstable, weak acid that decomposes into hypochlorite (OCl−) in a physiological environment. The detection mechanisms of the fluorescent probes for HOCl can be divided into the following reaction types: oxidative desulphurisation,199 oxidative unsaturated and double bond reaction,200 oxidation of phenol or aniline,201,202 oxidation of thioether or thiolactone,203 and the oxidation of dibenzoyl hydrazine.204
Yuang et al. firstly synthesized three probes with different responsive HOCI groups, namely oxathiolane, mercaptal, and oxime groups, and found that the probe with oxathiolane possessed the best responsiveness. This was due to the single sulphur atom in 1,3-oxathiolane that required less HOCl for complete oxidation of the probe. Hence, they designed a mitochondrial-targeting two-photon fluorescent “turn-on” probe 14 (λex/λem = 375/500 nm) to monitor HOCl levels in live cells and in a mouse model for inflammation (Fig. 17A).205 The probe was comprised of three units: the two-photon fluorophore acedan, the HOCI response group oxathiolane, and the mitochondrial-targeting group, TPP. This probe responded to HOCl within a few seconds and had exceptional sensitivity and selectivity. The authors used a typical “push–pull” (amine-ketone) structure for the two-photon fluorophore acedan and used 2-mercaptoethanol to protect the ketone structure of acedan, which quenched the fluorophore. When the probe reacted with HOCl, the oxathio deprotection of cyclopentane exposed the ketone and restored fluorescence. This study successfully used probes to detect HOCl in the mitochondria of RAW 246.7 cells. Finally, the authors probed HOCI in vivo changes in a mouse model of inflammation using this two-photon probe.
 | ||
| Fig. 17 Structures and response mechanism of mitochondrial (A) fluorescent probe 14 (J. Am. Chem. Soc., 2015, 137, 5930–5938), and (B) probe 15 (Biosens. Bioelectron., 2018, 107, 218–223) to detect HOCl. (C) Structures and the response mechanism of mitochondrial fluorescent probe 16 (Anal. Chem., 2020, 92, 11103–11110) to simultaneously detect HOCl (red), cysteine/homocysteine (green), and GHS (blue). | ||
Zhu et al. synthesized a mitochondrial-targeting NIR fluorescent probe 15 (λex/λem = 600/672 nm).202 It measured ClO− with a sensitivity in the range of 0–200 pM and a detection limit of 10.8 pM. To our knowledge, this is the first fluorescent probe that is capable of determining ClO− at picomolar levels. In addition, the probe had a fast response to ClO− (<5 s) (Fig. 17B). The probe consisted of a Nile Blue dye with lipophilic cations and a ClO− responsive amido group. The colour of the probe turned from blue to pink upon reaction with ClO−, allowing colour changes to be visualized with the naked eye. The fluorescence spectral overlap coefficient between this probe and the commercial mitochondrial dye, Mito-Tracker Green, was 0.93, indicating that the probe possessed good mitochondrial targeting ability. Finally, the authors used the probe to track endogenous and exogenous ClO− in HeLa cells, and demonstrated that the probe can successfully and dynamically track the changes in endogenous and exogenous ClO− concentrations in mitochondria. However, this probe has a detection limit of picomolar, picomolar concentrations of ClO− are usually not related to their physiological activity, but to residual concentrations. Therefore, the design and application of the probes should not only aim for ultra-low detection limit or detection range but also take into consideration the actual physiological concentration of the target. Each metabolite has its normal physiological range of concentrations (nM to μM) within the cell, at which it exerts biological function; hence, any assay should take full account of the biologically relevant concentration of the target to render the assay more significant.
Multi-biomarker imaging using a single probe not only eliminates complex procedures, but also ensures that the probes are distributed spatiotemporally and metabolically identical. Zhu et al. constructed a single-molecule fluorescent probe 16 that can simultaneously monitor the key indicators cysteine/homocysteine (Cys/Hcy) (λex/λem = 405/417–477 nm) and HOCI (λex/λem = 488/499–529 nm), as well as the MMP (λex/λem = 543/553–618 nm), and the opening of the mPTP in cancer cells (Fig. 17C).206 In short, the structure of the probe consisted of NBD–coumarin–imidazolium salt–rhodamine. NBD, coumarin, and rhodamine are the groups that respond to Cys/Hcy, the mitochondrial state and HOCl, respectively. The cationic linker of imidazolium salts facilitates the probe to target mitochondria. The probe was converted to its derivative upon reaction with GSH or Cys/Hcy. Reaction with GSH generated a blue fluorescence (F405) from the release of 7-hydroxycoumarin. Unlike GSH, the Cys/Hcy reaction produced green fluorescence (F488). When the probe reacted with HOCI, it resulted in red fluorescence (F543) from rhodamine. The F405 revealed that that the probe could not only differentiate MCF-7 cells from HBL-100 cells but could also measure MMP and mPTP opening. Finally, the authors exploited probes to investigate Cys/Hcy, HOCI and the mitochondrial status concurrently, and the results demonstrated that HOCl can distinctively induce the opening of mPTP and reduction of MMP in different cells.
Hydroxyl radicals (˙OH) have a very strong ability to attract electrons, which is known as oxidising ability. They are the second most reactive oxidants in nature after fluorine. Hydroxyl radicals have high reactivity and can easily attack high electron cloud density points. The current hydroxyl radical response groups include, but are not limited to, the following: terephthalic acid,207 merocyanine dyes,208 phenothiazine,209 2,2,6,6-tetramethyl-1-piperidinoxyl (TEMPO),210 and N2O type benzopyrrole boron complex (Bobpy).211
Recently, Deng et al. proposed a new ˙OH-responsive group, a N2O-type Bobpy structure, which is susceptible to ˙OH attack and thus leads to fluorescence “turn-off”.211 Based on this, the authors designed a FRET ratiometric fluorescent probe 17 (F590/F630), which was composed of rhodamine B as the donor and N2O-type Bobpy as the acceptor (Fig. 18A). The authors utilised MitoTracker Green to co-incubate with the probe and revealed a co-localisation factor of 0.88, indicating that the probe possessed excellent mitochondrial targeting. After generating ˙OH using the Fenton reaction of H2O2 and EDTA–Fe2+, cells were first cultured with the probe and then added to the above-mentioned system. Cell imaging showed diminished red fluorescence and increased green fluorescence, indicating that variations in the fluorescence ratio of the probe can be employed to track ˙OH variations. The probe was used to evaluate the effect of ˙OH production by dihydroartemisinin (DHA) and its derivative synthetic triphenyl phosphine (DHA–TPP), and demonstrated that DHA–TPP produced more ˙OH than DHA. Finally, the authors examined the ability of the probe to track ˙OH in an animal model. Using zebrafish, which survive under normal conditions with ˙OH present mainly in the gastrointestinal tract, it was found that the addition of the antioxidant NAC attenuated ˙OH production.
 | ||
| Fig. 18 (A) Structure and response mechanism of mitochondrial fluorescent probe 17 (Chem. Commun, 2020, 56, 4432–4435) to detect ˙OH; (B) schematic illustration of the HAT-modified tungsten nanoelectrode for single-cell analysis of ˙OH (Anal. Chem., 2020, 92, 2543–2549), adapted with permission from ref. 212, copyright © 2020 American Chemical Society. | ||
In the section describing the electrochemical detection of ROS, we introduced nanoelectrodes for ROS detection. The oxidation potential of ˙OH is approximately 2.8 eV (vs. standard hydrogen electrode, SHE) which is sufficient to oxidise the majority of ROS/RNS or small molecules such as uric acid, which will interfere with the detection. Ding et al. reported a novel tungsten nanoelectrode to assay ˙OH, by fixing AuNPs and the self-assembled monolayer (SAM) of 1-hexyl mercaptan (HAT) on the external side of the conical tungsten substrate (Fig. 18B).212 The HAT on the surface of the nanoelectrode is available to be selectively disrupted by ˙OH, causing the electrode interface to convert from hydrophobic to hydrophilic. Thus, the level of electrochemical signal recovery of the redox probe is proportional to the degradation of the SAM of HAT. DPV was employed to quantify ˙OH, and the current peak increased progressively with increasing concentration of ˙OH. The nanoelectrodes were inserted into different locations under a microscope to record LPS-induced ˙OH production. The authors found that when inserted into the mitochondria, a large amount of ˙OH was generated, which led to an increase in current, while when inserted into the nucleus, the current was significantly reduced. Interestingly, the nanoelectrodes not only measured changes in the Aβ-stimulated ˙OH levels in single cell, but also demonstrated that Aβ can lead to increased intracellular ˙OH in macrophages, revealing the relationship between ROS and Aβ in the pathogenesis of AD.
Singlet oxygen (1O2) is the lowest excited state of the oxygen molecule and has high reactivity. It can oxidise various biological substrates, including proteins, certain amino acids, and nucleic acids. 1O2 has also become a powerful tool for photodynamic therapy (PDT) in tumour treatment.213 Current probes for detecting singlet oxygen include anthracene,214,215 diphenylisobenzofuran (DPBF),216 HOCD cycloaddition,217 1,4-cycloaddition of the imidazole ring of histidine,218 and the europium(III) complex.219 DPBF is now commonly used as a singlet oxygen trapping agent in PDT experiments and can be used to detect the in vitro quantity of singlet oxygen produced by the photosensitiser. Anthracene can quench fluorescence through electron transfer with certain fluorophores as a selective intramolecular switch to 1O2 through the [2+4] cycloaddition reaction, resulting in internal peroxide and enhanced fluorescence intensity.
Kim et al. used the 1O2-responsive group 9,10-dimethylanthracene (DMA)-modified silicon-containing rhodamine to construct a two-photon probe 18 (λex/λem = 532/640 nm) (Fig. 19A).214 The PET effect between the silodamine fluorophore and DMA led to the quenching of silodamine fluorescence. When DMA was oxidised by 1O2 into intramolecular peroxide, the PET effect disappeared and fluorescence was recovered. The fluorescence intensity of the probe enhanced 17-fold upon reaction with 1O2 due to the formation of endoperoxide on the anthracene group. The probe also possessed excellent mitochondrial localisation and was suitable for monitoring 1O2 production during PDT, as mitochondria are the main target of PDT. The authors evaluated the effect of three different localised photosensitisers in 1O2 production during PDT and found that the probe could only respond to 1O2 produced by 5-aminolevulinic acid (ALA)-derived PpIX photosensitisers localised in the mitochondria. In addition, the authors assessed three different types of photosensitisers using the probe and found that mitochondria-targeted KillerRed and lysosomal porphyrins failed to elicit changes in the fluorescence of the probe. In summary, this probe is dependent on the localisation and type of photosensitiser, mainly due to the limited 1O2 diffusion range and the insignificant 1O2 production by type I photosensitisers. Liu et al. synthesized a mitochondrial-targeted 1O2 two-photon probe using the detection mechanism. In this work, the authors exploited the excellent tissue penetration of two-photon to probe the floating of 1O2 in liver tissue under Ce6 induction.215
 | ||
| Fig. 19 Structures and the response mechanism of mitochondrial (A) fluorescent probe 18 (J. Am. Chem. Soc., 2014, 136, 11707–11715) and (B) probe 19 (Chem. Commun., 2018, 54, 13997–14000) to detect 1O2, and pseudocolour fluorescence images of HeLa cells with probe 18, adapted with permission from ref. 214, copyright © 2014 American Chemical Society. | ||
Flores-Cruz et al. synthesized a dual-emission fluorescent probe 19 (λex/λem = 430/500 nm, λex/λem = 430/670 nm) to dynamically track the production and localisation of 1O2-ROS between the mitochondria and the nucleolus (Fig. 19B).220 The dual fluorescence response of the probe is not only due to the binding of chromium and coumarin 334 but also the result of electron transfer between spirolactone and propionic acid structures. Once the probe is oxidised, a coumarin moiety is released to generate fluorescence. The coumarin exits the mitochondria to obtain a distinct nucleolus distribution. Variations in 1O2 were monitored by differential organelle (mitochondria–nucleolus) distribution following redox inputs, and the probe displayed excellent resistance to highly relevant interferents. In localisation experiments, the probe was found to exhibit good mitochondrial localisation at 5–10 μM and endoplasmic reticulum and Golgi localisation at higher levels (20 μM), having the same properties as commercial Mito-Trackers. With the spectrally resolved confocal imaging, the authors observed localised probe oxidation and changes in structural information that could separate the fluorescence signal of the target from the interferents. Importantly, the probe was used to monitor subtle changes in 1O2 during PDT by altering the MMP using oligomycin and antimycin A inhibitors.
NO is a free radical molecule produced by nitric oxide synthase (NOS) in biological systems.222 Based on its concentration, it mediates various physiological and pathological processes that may be beneficial or harmful. However, the precise concentrations produced by these biological activities remain unclear. In this section, we focus on the various materials used for the electrochemical detection of NO and their detection principles. Pashai et al. immobilised AuNPs, CS, cyt c, and Nafion on a gold electrode surface to produce a Nafion/cyt c/CS–3-mercaptopropionic acid (MPA)–AuNPs/cysteamine–MPA–Au electrode (Fig. 20A).223 Among them, cyt c serves as a NO recognition element. The constructed sensor exhibited significant electrocatalytic ability for the reduction of NO, with a detection range of 0.1–2.15 μM and a detection limit of 45 nM. This demonstrated that the CS–MPA–AuNP nanocomposite not only exerts to augment the electron transfer rate, but also fully maintains the catalytic activity of cyt c. However, the authors did not apply this biosensor to cells or serum samples; thus, considering the sensitivity of the prepared sensor and the biocompatibility of the material, the next stage is to prepare it as a nanoelectrode for the in situ real-time monitoring of mitochondrial NO.
 | ||
| Fig. 20 (A) Schematic representation of the fabrication of the Nafion/cyt c/CS–MPA–AuNPs/SAMs-Au electrode to detect NO (Int. J. Biol. Macromol., 2018, 108, 250–258). (B) Schematics for the overall detection process of NO with the CAS/SOD/MP/MWCNT-PTTCA/AuNPs biosensor (Biosens. Bioelectron., 2010, 26, 1080–1086). | ||
A limitation of the Nafion-coated electrode approach is the reduction in sensitivity. In addition, the CS modified electrode may also affect the conductivity of the sensor. Hence, Abdelwahab et al. used microperoxidase (MP), a small redox oligopeptide obtained from a haem group for the electrochemical reduction of NO, as a sensing element (Fig. 20B). Catalase and SOD subsequently were introduced rather than Nafion to protect the NO detection from the interference of H2O2 and O2−.224 Recently, Meiller et al. synthesized fluorinated xerogels to produce a screening layer in the NO biosensor, which also protected against endogenous redox molecules in vivo. The fluorinated xerogels were composed of methyltrimethoxysilane and (heptadecafluoro-1,1,2,2-tetrahydrodecyl)trimethoxysilane (17-FTMS). Carbon fibre microelectrodes combined with nickel porphyrin and fluorinated xerogels are able to very effectively and specifically electrocatalyse NO. The in vivo and in vitro results showed that the selectivity of this microelectrode is preferable to that of the Nafion-modified microelectrodes. Additionally, this microelectrode was highly specific for the detection of NO evoked by N-methyl-D-aspartate (NMDA) in the brain.225
The groups of the probe that respond to NO include, but are not limited to, the following: O-phenylenediamine,226 2-amino-3′-dimethylaminobiphenyl,227 secondary aromatic amines,228 and Schiff base.229
Sun et al. synthesized a “turn-on” fluorescent probe 20 (λex/λem = 570/616 nm), which introduced o-phenylenediamine dihydrochloride (OPD) into the pyronine fluorophore as a receptor for NO.226 This probe regulates fluorescence switching through the PET effect. The probe possessed a positive charge and was easily targeted to mitochondria to detect NO in this important organelle. It is worth noting that with the reducing species Cys and GSH, the fluorescent probe oxidation products could further react to produce bright green fluorescence (λmax = 536 nm) for Cys and red fluorescence (λmax = 618 nm) for GSH (Fig. 21A). Thus, the probe could detect intracellular NO from both green and red channels with the assistance of Cys and GSH in the cells. To confirm that the probe is a mitochondrial-targeting probe for NO, B16 cells were incubated with the probe and MitoTracker Red FM, respectively, and the cells were handled with DEA·NONOate for 30 min. The results definitively confirmed that the probe is capable of targeting mitochondrial NO. Simultaneous nuclear staining with DIPA demonstrated that the cells remained viable during the experiment.
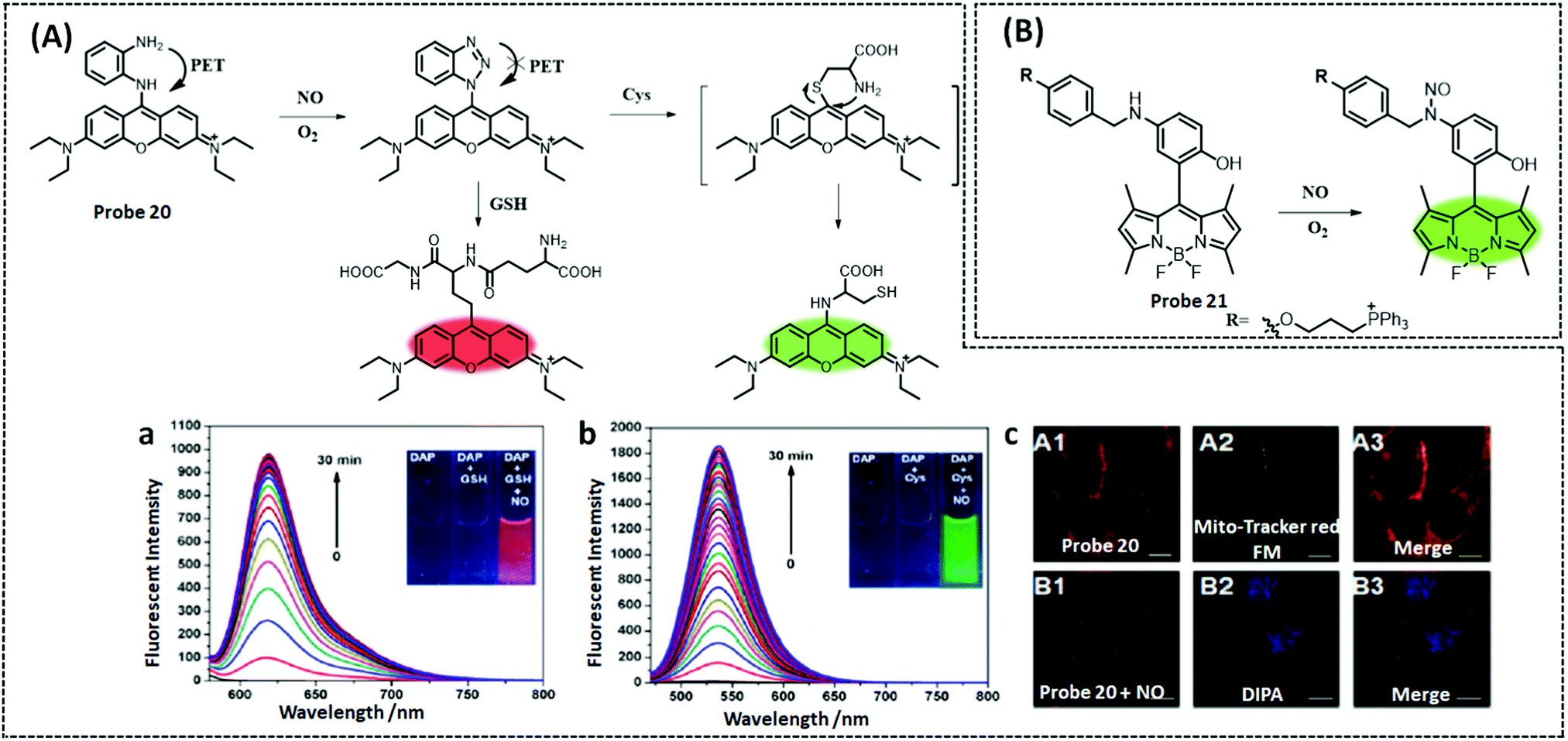 | ||
| Fig. 21 Structures and response mechanism of mitochondrial (A) fluorescent probe 20 (J. Am. Chem. Soc., 2014, 136, 12520–12523) and (B) probe 21 (Biomaterials, 2016, 78, 11–19) to detect NO and time-dependent fluorescence spectra of probe 20 upon the addition of DEA·NONOate in the presence of GSH (a) and Cys (b), respectively. (c) Fluorescence images of B16 cells with probe 20. Scale bar: 10 μm, adapted with permission from ref. 226, copyright © 2014 American Chemical Society. | ||
Miao et al. reported a mitochondrial-targeting NO fluorescent probe 21 (λex/λem = 490/518 nm) based on the BODIPY structure.228 The probe used boron dipyrromethene (BODIPY) as the fluorophore, secondary aromatic amine N-benzyl-4-hydroxyaniline as the NO response unit, and TPP (Fig. 21B). The authors fabricated NO fluorescent probes by the N-nitrosylation reaction of aromatic secondary amines and NO under oxygen enrichment. When the probe reacts with NO, the fluorescence at 518 nm is significantly enhanced (92-fold), indicating that the probe has excellent recognition and response. Subsequently, the authors evaluated the capability of the probe in RAW 264.7 macrophages. Upon incubation with the cells, the probe emitted weak fluorescence; however, stimulation with a drug significantly enhanced the fluorescence.
ONOO− is a strongly oxidising and nucleophilic RNS, which is created by the bond formation between NO and superoxide anion radicals in a diffusion-controlled manner. ONOO− also converts to secondary free radicals with high reactivity, including ˙OH, nitrogen dioxide (˙NO2), and carbonate radicals (CO3˙−), which damage proteins, nucleic acids, lipids, and many other biologically active substances.230 To date, the strategies for detecting ONOO− include oxidation of unsaturated bonds,231 oxidation of phenylhydrazine,232 and decomposition of α-ketoamide.233
Xie et al. reported a near-infrared two-photon mitochondrial ONOO−probe 22 (λex/λem = 570/630 nm).234 The authors designed the probe based on the response of α-ketoamide to ONOO−. When the α-ketoamide bridge is cleaved by mitochondrial ONOO−, the PET effect disappears and the fluorophore is released (Fig. 22A). Following pre-treatment of the H9C2 cells with 3-morpholinone imine hydrochloride (SIN-1), an ONOO− generator, the probe was co-incubated with the cells and a distinct red fluorescence was observed. Additionally, pre-treating H9C2 cells with SIN-1 and uric acid (an ONOO− scavenger), followed by co-incubation with the probe, significantly suppressed the fluorescence. A Pearson correlation coefficient of 0.93 for the co-localisation assay demonstrated the good mitochondrial targeting ability of the probe. Next, the authors employed the probe to monitor the floating of ONOO− in a H9C2 cardiomyocyte model during anthracycline-induced cardiotoxicity. When doxorubicin (Dox) and epirubicin (Epi) were added, the mitochondrial ONOO− levels significantly increased and the increase was dose-dependent. Finally, the authors successfully monitored changes in ONOO− using the probe in a mouse model of cardiotoxicity.
 | ||
| Fig. 22 Structures and the response mechanism of mitochondrial (A) fluorescent probe 22 (Anal. Chem., 2018, 90, 11629–11635), (B) probe 23 (J. Am. Chem. Soc., 2017, 139, 285–292) and (C) probe 24 (Anal. Chem., 2020, 92, 10792–10799) to detect ONOO−. (D) Structure and response mechanism of mitochondrial fluorescent probe 25 (Chem. Commun., 2017, 53, 1723–1726) to detect HNO. | ||
Cheng et al. designed and synthesized a mitochondrial-targeted ONOO−probe 23 (F473/F651) exploiting the FRET effect.235 Due to its unique design, this probe had excellent selectivity for ONOO− and could respond specifically to ONOO− under other ROS and RSS (Fig. 22B). The probe used coumarin as the donor and the two-photon near-infrared fluorescent dye TP-NIR (4-(2-carboxyphenyl)-7-(diethylamino)-2-phenylchromen-ylium) as the acceptor. When reacting with ONOO−, TP-NIR undergoes nucleophilic addition, oxidation, elimination, and hydrolysis reactions, finally resulting in the removal of the enoic acid product. In the absence of ONOO−, the probe exhibited a robust emission region at 651 nm and a weak coumarin featured emission region at 473 nm. When ONOO− was added, the emission region positioned at 651 nm almost vanished, while the coumarin emission was remarkably strengthened, and the colour of the probe solution simultaneously shifted from red to blue-green. The final probe could ratiometrically monitor ONOO− in HepG2 and RAW 264.7 cells by single- or two-photon fluorescence confocal microscopy. In addition, the probe was able to monitor ONOO− production in 110 μm thick tissue slices from an LPS-induced inflammatory mouse model.
Liu et al. proposed a novel phenolate-based D–π–A fluorophore (A–DOH) that has brilliant NIR fluorescence in living cells by taking advantage of the distinctive halogen effect.236 First, three A–DOH fluorophores based on dicyanomethylene-4h-chromium (DCM), which have different phenolic groups as electron donors, were designed. Meanwhile, a clickable azide group was configured for the DCM to form an azide-DCM, which is more functional through a click reaction. A fluorescent probe 24 (λex/λem = 520/670 nm) was synthesized by introducing a mitochondrial targeting group and a trigger group for the ONOO− reaction, on the basic structural backbone (Fig. 22C). When ONOO− was added, the fluorescence was increased 308-fold due to the intramolecular ICT effect. The detection range of the probe was 0–20 μM and the detection limit was 15 nM. The authors demonstrated that the unique halogen effect significantly increases quantum yields, resulting in a significantly brighter fluorescence of the probe than that of the conventional halogen-free analogues. Notably, due to the flexibility of click chemistry and phenol caging chemistry, the probe will be structurally modified for live cell imaging of a wider variety of biomarkers.
HNO is the electron reduced form of NO and is engaged in critical physiological processes such as neuromodulation. There are probes designed to detect HNO by the reaction of reducing Cu2+ to Cu+ or nitroxide to hydroxylamine,237,238 but other reducing substances can interfere. HNO probes based on the Staudinger reaction overcome this selectivity problem and other reducing substances do not interfere with the reaction. Sunwoo et al. reported a mitochondrial-targeted two-photon HNO probe 25 (λex/λem = 405/452 nm) based on the Staudinger reaction, using 2-(diphenylphosphino)benzyl as the HNO recognition group, coumarin as the fluorophore, and TPP+ (Fig. 22D).239 The probe was incubated with HNO and various active substances together separately for 20 min. The fluorescence intensity of other active substances was very weak; however, the fluorescence intensity of HNO was significantly high. When the concentration of GSH exceeded that of HNO, the probe still had an excellent selectivity for HNO. The authors then used the probe to monitor HNO changes in HeLa cells. The cells were first preconditioned with DEA·NONOate in the presence of sodium ascorbate, as it converts NO to HNO. After incubation with the probe, it was found that the probe was mainly localised in the mitochondria and was able to detect endogenous HNO in the mitochondria.
The biological origin of H2S is L-cysteine or homocysteine, which is catalysed by the H2S-generating enzyme cystathionine-β-synthase and other enzymes. The probes for H2S detection include a reduction reaction of the azide or nitro groups,242–244 thiolysis,245,246 Cu2+ precipitation,247,248 or a nucleophilic attack reaction of H2S on the probe.249,250
Chen et al. designed and synthesized a ratiometric mitochondrial-targeting H2S probe 26 (F510/F652).249 The authors used the nucleophilic addition properties of H2S to synthesise this probe by combining coumarin and cyanine dyes (Fig. 23A). Under neutral conditions, H2S rapidly undergoes nucleophilic addition to the cyanine component, destroying the cyanine structure and changing the probe spectrum. The probe and H2S reacted completely within 30 seconds, even when the H2S concentration was quite low (10 μM). When the concentration of H2S was in the range of 100–200 μM, complete reaction was achieved within 2 s. Interference experiments demonstrated the ability of the probe to specifically select H2S without reacting with other interferents. To verify the mitochondrial targeting ability of the probe, the authors incubated the probe and Mito-Tracker Deep Red separately in MCF-7 cells and found that the spectra of the probe and the commercial dye overlapped (overlap coefficient of 0.95), indicating that the probe was mainly localised in the mitochondria.
 | ||
| Fig. 23 Structures and response mechanism of mitochondrial (A) fluorescent probe 26 (Angew. Chem., Int. Ed., 2013, 52, 1688–1691) and (B) probe 27 (Anal. Chem., 2018, 90, 9418–9425) to detect H2S, and (C) fluorescence images of HeLa cells with probe 27. Scale bar: 10 μm, adapted with permission from ref. 242, copyright © 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
It is reported that variability of viscosity and H2S levels in mitochondria are relevant to the overexpression of amylin (Aβ), which has a vital function in the development of AD. Therefore, Li et al. reported a dual-responsive fluorescent probe 27 (λex/λem = 405/525 nm) to monitor the viscosity and H2S levels in mitochondria (Fig. 23B).242 The internal rotation of the probe rendered it non-fluorescent, but when the viscosity increased the probe failed to rotate, releasing a vigorous red fluorescence signal. The authors used these probes to explore the crossover effects of viscosity and H2S in the mitochondria of HeLa cells. When HeLa cells were incubated with the probe alone there was a distinct red fluorescence and a distinct green fluorescence. When the cells were incubated with nystatin, the addition of the probe caused a dramatic increase in red fluorescence and a significant decrease in green fluorescence, implying that the increased viscosity probably accounts for the reduction of mitochondrial H2S. Conversely, a significant improvement in green fluorescence and a considerable decrease in red fluorescence were noticed when stimulating the cells with Cys, suggesting that an increase in H2S may lead to a decrease in mitochondrial viscosity. These experimental results indicated that the concentration of H2S exhibits a negative correlation with viscosity in mitochondria.
SO2 plays a vital role, as an antioxidant, in regulating the redox state; however, excessive SO2 can induce oxidative stress. Nucleophilic addition of an unsaturated double bond is now the primary detection mechanism of the probes.
Chen et al. designed a mitochondrial-targeting SO2probe 28 (λex/λem = 460/555 nm). The authors added a weak electron-withdrawing carbonyl group to the fluorophore and used the ICT effect to quench the fluorophore (Fig. 24A).251 With the nucleophilic addition of SO2 and ketone groups, the carbonyl electron-withdrawing function disappears, the ICT effect turns on, and a strong signal change occurs. The probe was successfully applied to study the role of mitochondrial SO2 in apoptosis, and the results showed that it regulates early apoptosis by decreasing the MMP. In addition, the probe is capable of tracking endogenous SO2 in zebrafish and, therefore, may serve as an excellent optical instrument to quantify SO2 in various biological samples.
 | ||
| Fig. 24 Structures and response mechanisms of mitochondrial (A) fluorescent probe 28 (Anal. Chem., 2018, 90, 12442–12448), (B) probe 29 (Biomaterials, 2020, 241, 119910) and (C) probe 30 (Chem. Commun., 2020, 56, 7710–7713) to detect SO2/HSO3−. | ||
Through-bond energy transfer (TBET) is a promising approach to prepare ratiometric fluorescent probes with improved energy transfer efficiency, which is without the requirement of spectrum overlap between the energy donor and acceptor. A TBET-based ratiometric probe 29 (F493/F611) was developed by Hu et al.252 The probe consists of the following three parts: a coumarin derivative as the energy donor owing to efficient two-photon absorption, a hemi-anthocyanin derivative as the energy acceptor and the SO2 response group. The energy donor and acceptor were connected with a twistable alkyne bond (Fig. 24B). Furthermore, the probe can be localised to the mitochondria due to its positive charge. Upon reaction with SO2, the probe performed a ratiometric signal enhancement of about 30-fold with a low detection limit of 0.09 μM. Changes in the red/green fluorescence ratio clearly indicated the enhanced level of apoptosis with respect to the increase in SO2 concentration and the prolongation of incubation time. Finally, the probe was applied to detect the variations of SO2 concentration in mouse brain tissue; the experimental results further demonstrated the protective effect of NS-398 (an anti-inflammatory agent) against SO2-induced neuroinflammation.
Niu et al. reported a three-channel mitochondrial-targeted two-photon probe 30 (λex/λem = 544/585 nm) with a dual-response to SO2 and HOCl.253 Based on the FRET principle, the probe was composed of three fluorophores: pyrazoline, coumarin, and hemicyanine (Fig. 24C). The coumarin–pyrazoline is the FRET energy donor, while the hemicyanine is the energy acceptor. When the probe reacted with HOCl, the coumarin was partially removed and the probe showed hemicyanine and pyrazoline fluorescence. When the probe was combined with an SO2 regenerative nucleophile, the cyanine structure was destroyed, the FRET effect was extinguished, and the probe showed coumarin fluorescence. The authors proved with the probe that intracellular HSO3− and ClO− are always in dynamic homeostasis. When the concentration of one species is excessive, which results in a stress response, then the cell self-generates the other species to maintain the equilibrium of the cell. Specifically, during excessive ClO− or LPS-induced oxidative stress in mitochondria, SO2 operates effectively as an antioxidant to decelerate the progression of oxidative damage. When SO2 is in excess in mitochondria, it induces a stress response, resulting in ClO− generation in the cell.
The determination and identification of the reduced and oxidised forms of GSH is currently a difficult task. Tahernejad-Javazmi et al. introduced a novel mediator, 2-chloro-N′-[1-(2,5-dihydroxyphenyl)methylidene]aniline (2-CDHPMA), in the fabrication of a carbon paste electrode modified with MgO/SWCNTs. The electrocatalytic interaction between 2-CDHPMA and GSH was used as a factor in the detection of GSH in real samples. GSH was detected with the proposed biosensor with a limit of detection of 10.0 ± 0.3 nM.256 Several electrochemical methods for the detection of GSH and GSSG are usually used in conjunction with separation methods such as HPLC, or absence of evoked oxidative stress in human plasma. To solve this issue, Moya et al. fabricated a electrochemical biosensor to determinate simultaneously the GSH/GSSG ratios based on GCE modified cobalt phthalocyanine (CoPc) and MWCNT functionalised with carboxyl groups.257 This biosensor eliminates the requirement to resort to isolation approaches in the presence of oxidative stress. CoPc has electrocatalytic ability for the electroreduction of GSSG, and its electrocatalytic effect can be improved by incorporating CNTs, which have a greater electroactive area to promote charge transfer. In an interference study, they found that the signals of the reduced GSSG, the redox process of CoPc, and the oxidised GSH remained visualized independently with or without interferences. Different concentrations of GSH and GSSG mixed in a synthetic solution were investigated using DPV. The results showed that the peak at −0.1 V corresponded to the oxidised GSH and the peak at −0.72 V corresponded to the reduced GSSG. The peak connected with the Co2+/Co1+ redox process appeared at −0.5 V and shifted in a positive direction as a result of GSH. They used the proposed biosensor to simultaneously determine GSH and GSSG concentrations and the GSH/GSSG ratios in Wistar rat urine and plasma samples, pre- and post-lead intoxication, for estimating the oxidative stress level.
In a previous study, Yoon et al. found that naphthalimide derivatives, containing sulphonamide, were highly selective to determinate GSH. Following this, Xu et al. introduced anthocyanin derivatives to develop a dual-channel open fluorescent probe 31 (visible channel: λex/λem = 370/495 nm; near-infrared channel: λex/λem = 700/795 nm) that is highly selective for the detection of GSH and is unaffected by cysteine and homocysteine (Fig. 25A).258 The probe was composed of naphthalimide as the GSH response group and visible fluorophore, an anabolic dye IR-780 as the NIR fluorophore, and a sulphonamide-like thiol group as the linker. GSH combined with the sulphonamide group in the probe to form an intermediate product, Then, the sulphur–nitrogen bond of this intermediate product was cleaved, resulting in a stable near-infrared luminescent product and a reactive intermediate which released SO2 to generate a stable visible luminescent product. Thus, the reaction of the probe with GSH produced fluorescence in two channels: a green fluorescence in the visible channel (442 nm) and a red fluorescence in the NIR channel (810 nm) emitted by the IR-780. The fluorescence of both the channels increased significantly with the increase in GSH concentration. Additionally, since the probe was positively charged, it exhibited a remarkable ability to target mitochondria and the dual channels tracked intracellular GSH in a spatiotemporal and synchronous manner.
 | ||
| Fig. 25 Structures and response mechanisms of mitochondrial (A) fluorescent probe 31 (Chem, 2018, 4, 1609–1628) and (B) probe 32 (J. Am. Chem. Soc., 2020, 142, 17069–17078) to detect GSH. | ||
Zhang et al. further designed and prepared a class of “carbon-dipyrrole methylene dyes” (Cardipys) with positive ionic properties by single-atom modification of the conventional boron dipyrrole methylene dyes (Bodipys). The new dyes not only retain the excellent photophysical properties of classical Bodipys but also exhibit better water solubility and light stability. Interestingly, in contrast to the widely used positive ionic rhodamine dyes, Cardipys are not subject to the TICT fluorescence quenching mechanism. Based on this, the authors selected one of the dyes to prepare probe 32 (λex/λem = 570/593 nm) (Fig. 25B).259 Owing to its positive ionic properties, the probe could penetrate the cell membrane very easily and specifically target the mitochondria. Upon entry into the mitochondria, the probe was able to undergo a Michael addition reaction with GSH catalysed by glutathione-S-transferase (GST) to produce a GSH adduct. This product subsequently migrated to the lysosome via the cellular transport mechanism and underwent a trans-Michael addition reaction, ultimately releasing Cardipys and GSH. The authors used this probe to demonstrate that the “mitochondrial-to-lysosomal” transport rate of Cardipys was not affected by an increasing intracellular GSH concentration, because it was closely related to the intracellular GST expression level.
4.2 Detection of macromolecules
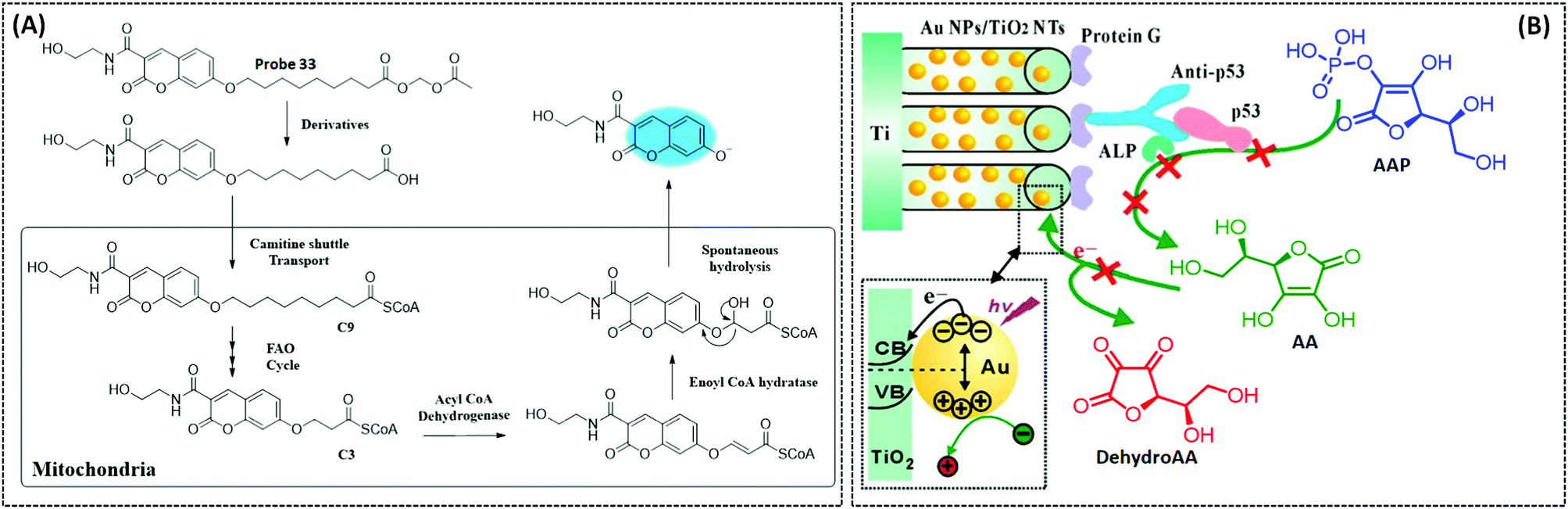 | ||
| Fig. 26 (A) Structure and response mechanism of the fluorescence detection of FAO activity in living cells by probe 33 (Chem. Commun., 2020, 56, 3023–3026). (B) Schematic diagram of the Au NPs–TiO2 NTS PEC biosensor for the detection of p53 (Anal. Chem., 2016, 88, 5626–5630), adapted with permission from ref. 264, copyright © 2016 American Chemical Society. | ||
Afsharan et al. developed a sandwich immunosensor to ultra-sensitively assay p53. Briefly, biotinylated capture anti-p53 was immobilised on an electrode modified with thiolated GO/streptavidin–AuNPs.262 The HRP-labelled secondary antibody was attached to the p53 protein by antibody–antigen interaction, and finally thionine was employed as the electronic mediator and H2O2 as the signal amplifying molecule. HRP catalyses the decomposition of H2O2 so as to produce electrons, which are transmitted through the electron mediator thionine, and thus H2O2 amplifies the reduction current of thionine. This kind of biosensor is a classic sandwich immunosensor with remarkable LOD (30 fM) and LOQ (115 fM). The reason for this ultra-high sensitivity is GO and streptavidin-modified AuNPs, which not only boost the electron transfer rate but also augment the captured antibody p53. Finally, the authors employed the constructed immunosensor to assay the p53 protein in human serum samples as well as normal and cancerous human skin fibroblasts. Although the detection limits of the prepared sensors reach the fM level, their sandwich structure design renders the sensor preparation and detection processes slightly complicated. In the future, it will be necessary to achieve high sensitivity and detection limits while also simplifying the preparation and handling steps.
The possibility of recognising different mutant p53 conformations and understanding their biological functions can contribute to develop new personalised therapeutic approaches and to improve diagnosis. Based on the current knowledge, native p53 has a small current peak, while an open isoform exhibits an improvement in the electrochemical current. Therefore, Tonello et al. developed two, label-free and label-based, electrochemical biosensors to investigate p53 conformation and redox chemicals. The electrochemical peaks are associated with diverse protein conformations and are well-supported by the specific amino acid electrocatalysis. The label-free electrochemical biosensor demonstrated that wild-type, oxidised, nitrated and denatured p53 possess specific potentials, respectively. Also, the labelled electrochemical biosensor evidenced the above results.263
Zhu et al. prepared a label-free amplified PEC immunosensor to assay p53 with high sensitivity and specificity.264 Firstly, by depositing AuNPs on hierarchically ordered TiO2 nanotubes (TiO2 NTs) using a facile polydiallyldimethylammonium chloride (polyDDA)-mediated adsorption technique, a photoresponsive transduction module was constructed. Then, a protein G molecular membrane was applied to immobilise alkaline phosphatase (ALP)-labelled anti-p53 (ALP-a-p53), as it not only specifically binds antibodies but also adsorbs efficiently on the Au NPs–TiO2 NT electrode (Fig. 26B). Meanwhile, ALP-a-p53 can maintain the ability of hydrolase to catalyse the in situ generation of ascorbic acid from ascorbic acid 2-phosphate, while also maintaining its specific binding to p53. The plasma charge separation from the conduction band (CB) of AuNPs to TiO2 NTs was significantly affected when p53 formed an immune complex with the antibody, thereby reducing the plasma photocurrent response. The magnitude of signal inhibition exhibited a positive dependence on p53 concentration. The photoexcitation of AuNPs led to the collective oscillation of hot electrons, allowing for the plasma charge separation on the surface of AuNPs. These electrons could be pumped spatially from AuNPs into the CB of TiO2 NTs when in a tight encounter with TiO2 NTs, and the remaining oxidised AuNPs were reduced by AA as an electron donor. The authors further investigated the PEC properties using the transient photocurrent response. The chronoamperometric I–t curves calculated under visible light irradiation, at wavelengths greater than 410 nm, revealed that the Au NPs–TiO2 NT electrode produced significant photocurrent enhancement with the “turn on” and “turn off” of light, further demonstrating the excellent photoactivity of the hybridised Au NPs–TiO2 NTs.
Poturnayova et al. used thickness shear mode (TSM) acoustic methods to fabricate an electrochemical biosensor for detecting cyt c by DNA aptamer. The authors analysed the binding response of three different aptamers specific to cyt c: apt 40, apt 61, and apt 76 immobilised by biotin–neutravidin on a gold electrode using the TSM technique. They found that apt 76 showed the highest affinity for cyt c. Furthermore, they showed that apt 76 is composed of a multiple-loop configuration that demonstrates binding specifically to cyt c. They then examined an apt 76-based sensor in spiked samples of human plasma by electrochemical methods. EIS was applied to detect cyt c using [Fe(CN)6]4−/3− as a redox probe, and showed that the LOD was 5.0 ± 0.2 nM.266
The electrochemiluminescence (ECL) method combines the advantages of both electrochemical and chemiluminescence methods including no external light source, easy control of excitation potential, ease of handling and high sensitivity, and it is therefore also considered to be a powerful tool for the detection of biomarkers.267 Ru(bpy)32+ and its derivatives have the advantages of good water solubility, stable luminescence performance, high luminescence efficiency, electrochemical reversibility, repeatable excitation, and easy labelling and fixation with different functional groups (such as amino and carboxyl groups). They are usually conjugated with co-reactants and numerous nanomaterials, thus enhancing the ECL signal.268 Sha et al. constructed a sandwiched ECL aptasensor based on Ru(bpy)32+-doped silica nanoparticles (RuSiO2 NPs) with a ferrocene carboxylic acid–aptamer (Fc–aptamer) to detect cyt c (Fig. 27A).269 The detection mechanism of the ECL biosensors involves the oxidation of Fc–COOH to form stable Fc–COOH+ that can react with Ru(bpy)32+* to prevent Fc–COOH+ + Ru(bpy)32+* → Fc–COOH + Ru(bpy)33+, quenching the ECL signals. In brief, RuSiO2 NPs were dropped on the electrode and the cyt c aptamer was immobilised on the GCE by a crosslinking reaction between the amino groups. Cyt c could then be recognised via an immune recognition reaction. Subsequently, Fc–aptamer composites were linked to ECL sensors via an immune recognition reaction. EIS and three-dimensional ECL spectroscopy were used to explore the ECL signal of the stepwise assembly procedure, and the results showed that the layer-by-layer assembly process was successful.
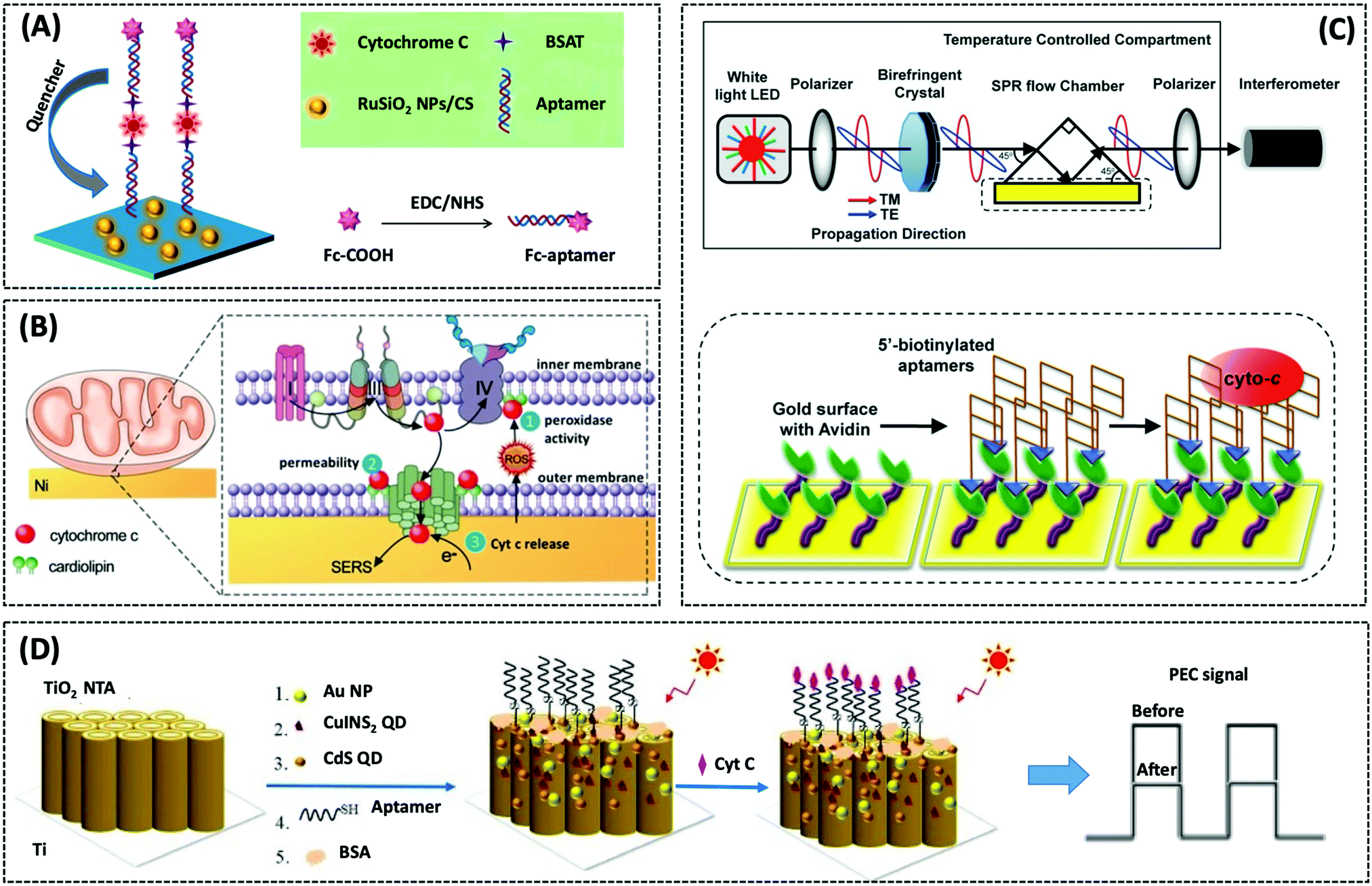 | ||
| Fig. 27 (A) The fabrication procedures of this proposed sandwich-type electrochemiluminescence aptasensor for the detection of cyt c, adapted with permission from ref. 269 (Biosens. Bioelectron., 2019, 132, 203–209), copyright © 2019 Elsevier Ltd. (B) Schematic of SERS-based investigation of cyt c–CL interactions and their correlation with cyt c release, adapted with permission from ref. 271 (Angew. Chem., Int. Ed., 2019, 58, 16499–16503), copyright © 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (C) An aptasensor using common-path SPR spectral interferometry for cyt c detection, adapted with permission from ref. 272 (Sens. Actuators, B, 2014, 198, 416–423), copyright © 2014 Elsevier Ltd. (D) Schematic illustration of the preparation and mechanism of the QD-based PEC aptasensor, adapted with permission from ref. 273 (Sens. Actuators, B, 2019, 281, 1088–1096), copyright © 2019 Elsevier Ltd. | ||
Zhang et al. developed a SERS-fluorescence and FRET “on–off” dual-mode nanosensor. Dual-signal detection was implemented by assembling AuNTs that were constructed from cyt c aptamers and some of their complementary sequences, which were modified with the Raman, and fluorescent reporter, Cy5. AuNTs provide enhanced SERS signals and fluorescence quenching effects. To verify that the aptamer–AuNTs can specifically recognise cyt c, the authors added (10 μM) cyt c with eight separate interferents (1 mM) to the sensor. They found that the cyt c SERS intensity at 1366 cm−1 was lower than those of other interferents, while the fluorescence intensity at 670 nm was higher than those of other interferents. This is because when exposed to external stimuli, cyt c can specifically bind to its aptamer, separating the cy5-modified complementary strand, which results in a weakened SERS signal and a restored fluorescence signal. The authors then used this sensor to quantify the changes in cyt c production in HeLa cells induced by aflatoxin B1 (AFB1 is a drug that induces apoptosis in mitochondria resulting in cyt c production) by using SERS and confocal imaging. As the incubation time with AFB1 increased, the complementary chain was shed due to specific binding of cyt c to the ligands on the surface of AuNTs, resulting in enhanced fluorescence signal and decreased SERS signal. This study observed cyt c transport events in apoptotic cells through two signals, which proved that they could use the sensor to quantitatively measure the release of cyt c by apoptotic cells.270
The interaction of cyt c with cardiolipin (CL) is known to have a crucial function in the initial stages of apoptosis. Therefore, Zhu et al. explored the relationship between SERS on nickel substrates and cyt c release through the interaction between CL and cyt c in the redox state (Fig. 27B).271 The nickel nanoparticles not only have the ability to induce apoptosis by the production of ROS, but the nickel nanostructures also show enhanced Raman signals. Based on the experimental results showing that the interaction between Fe2+cyt c and CL results in greater protein conformational changes than Fe3+cyt c alone, the authors further demonstrated that binding of Fe2+cyt c to CL induces higher peroxidase activity. The SERS signal of cyt c during mitochondrial apoptosis was monitored by placing the isolated mitochondria on the Ni surface. Meanwhile, mitochondria were pre-treated with an oxidising (H2O2) or reducing (sodium hyposulphite, SD) agent and then added to the Ni substrate. The results showed a cyt c SERS band at 748 cm−1 in SD-treated mitochondria only, which indicated that the reduced state of cyt c was essential for triggering apoptosis.
Loo et al. designed an aptamer sensor for drug screening by combining it with the common-path phase surface plasmon resonance (CPSPR) sensing technology (Fig. 27C).272 The authors chose a 76-mer DNA ligand as the recognition element to specifically capture cyt c. The aptamer is anchored to the SPR glass surface by biotin affinity interaction. CPSPR is a novel technique for SPR excitation in the visible to near-infrared spectrum to measure the phase change of the SPR in a label-free manner at optimized coupling wavelengths. The authors determined that the optimal wavelength for SPR is 653.8 nm, and subsequently used this sensor to detect cancer cells by isolating mitochondrial cyt c. This method is commonly used to screen for antitumor drugs. Polyphyllin D is known to cause an increase in cyt c release from the cells. Lastly, the authors isolated mitochondria and induced cyt c production with PD, and found that mitochondria from drug-resistant R-HepG2 cells released more cyt c than those from drug-sensitive HepG2 cells.
A label-free cyt c photoelectric aptamer sensor was prepared by Wang et al. Firstly, CdS/CuInS2/AuNPs/TiO2 nanotube arrays (NTAs) were prepared by deposition and impregnation techniques, continuous ion layer adsorption and reaction methods.273 The interfacial electron transfer rate of CdS and CuInS2 were enhanced by AuNPs. This stimulated the formation of the ideal step-band edge structures of TiO2, thus allowing efficient transfer of excited electrons (Fig. 27D). Cyt c aptamers were then attached to the NTAs via Au–S, Cd–S, and Cu–S bonds, and non-specifically closed using 6-mercapto-1-hexanol (MCH). The structure of CdS/CuInS2/AuNPs/TiO2 facilitates electron/hole transfer when cyt c binds to the aptamers through the formation of complex that spatially impedes the production of photogenerated electrons/holes and complexes at the photoelectrode interface, resulting in a lower photocurrent. The detection range of the PEC sensor with excellent selectivity and stability is 5 pM–100 nM and the detection limit is 5 pM. Finally, the authors used the constructed sensor to detect the level of cyt c in serum. Although the prepared sensor has a wide detection range as well as picomolar sensitivity, the authors applied it only to detect cyt c recovery in serum samples not to detect cells; therefore, future studies should validate the ability of the platform to monitor the cellular level. Biocompatibility of new photoelectric materials in cellular experiments deserves considerable attention.
Khalilzadeh et al. developed an HRP-based biosensor to detect caspase-3 activity.276 First, a new AuNP functionalised mobile crystalline material (MCM-41) (AuNPs-MCM-41) was modified on GCE by electrodeposition, and then activated biotinylated peptide (BP) solution was dropped on the electrode. Finally, streptavidin covered magnetic beads (MBs) were incubated with biotinylated HRP (B-HRP) to produce the HRP-MB-BP-AuNPs-MCM-41-GCE biosensor. The MBS on the BP surface resulted in a dense HRP load on the biosensor, which significantly amplified the electrochemical signal. This biosensor had a low limit of quantification (5 fM), which was employed to determine the caspase-3 activity in the stem cell differentiation. Notably, caspase-3 is present in cells as an inactive precursor, which is cleaved to produce the active caspase-3 enzyme following induction of apoptosis. This form of the enzyme may not show activity, and therefore those methods that rely solely on detecting the concentration of the active form may not indicate the true activity of caspase-3.277
Deng et al. fabricated a sign-on electrochemical biosensor to assay caspase-3 and evaluate the apoptosis via the production of amino terminal Cu(II) and Ni(II)-binding (ATCUN) motif ATCUN–Cu(II) complexes.278 These complexes were produced by the interaction of the ATCUN peptide fragment and Cu(II), as the electrocatalysts for water oxidation on the surface of the GO electrode. The peptide sequence Ac-GDEVDSK(FFFF)H was employed as the caspase-3 substrate. The free C-terminal carboxyl group of the peptide promoted the turnover of ATCUN–Cu(II) and enhanced the catalytic oxidation of H2O. Caspase-3 was able to dislodge Ac-GDEVD from the electrode surface to expose the ATCUN group (SGH), and the remaining peptide fragment was capable of binding to Cu(II) to form the ATCUN–Cu(II) complex, which exhibited favorable electrocatalytic ability for water oxidation. This amplification strategy was simple and more environmentally-friendly than enzyme or nanomaterial labeled biosensors. Finally, a biosensor with a linear range of 0.5–2000pg mL−1 was used to evaluate caspase-3 activity and cell apoptosis induced by different drugs. Khalilzadeh et al. wrote a detailed review on the electrochemical biosensors, based on nanomaterials, used for the detection of caspase-3 in 2017.279
Wu et al. designed a probe 34 (λex/λem = 550/586 nm) for the specific monitoring of MAO-A (Fig. 28A).281 The recognition part of the probe is the specific inhibitor substituted phenol of clorgyline from MAO-A, while binding to propylamine further enhances the specific recognition ability. The reaction mechanism of the probe with MAO-A is followed by oxidative deamination, then β-elimination, and 1,6-alignment. The experiments demonstrated that the probe could clearly image MAO-A activity in different types of cells, including SH-SY5Y with high MAO-A expression and HepG2 with high MAO-B expression. The probe exhibited a substantial fluorescence signal in SH-SY5Y, which was significantly reduced with the addition of clorgyline, an inhibitor of MAO-A. There was almost no fluorescence signal in HepG2 cells. Cell transfection experiments confirmed that the probe possesses better performance in identifying MAO-A and MAO-B. This study used the inhibitory structure of proteases to develop new recognition units, which provided a new general strategy for designing highly selective recognition units for other proteases. Based on the same inhibitor structure-guided synthesis strategy, Yang et al. designed and synthesized a NIR fluorescent probe 35 (λex/λem = 530/675 nm) that specifically recognises MAO-A. The probes could detect endogenous MAO-A levels with high selectivity, high sensitivity and rapid response. Importantly, the probes allowed for fluorescence imaging of the changes in the endogenous MAO-A levels of tumors and zebrafishes (Fig. 28B).282
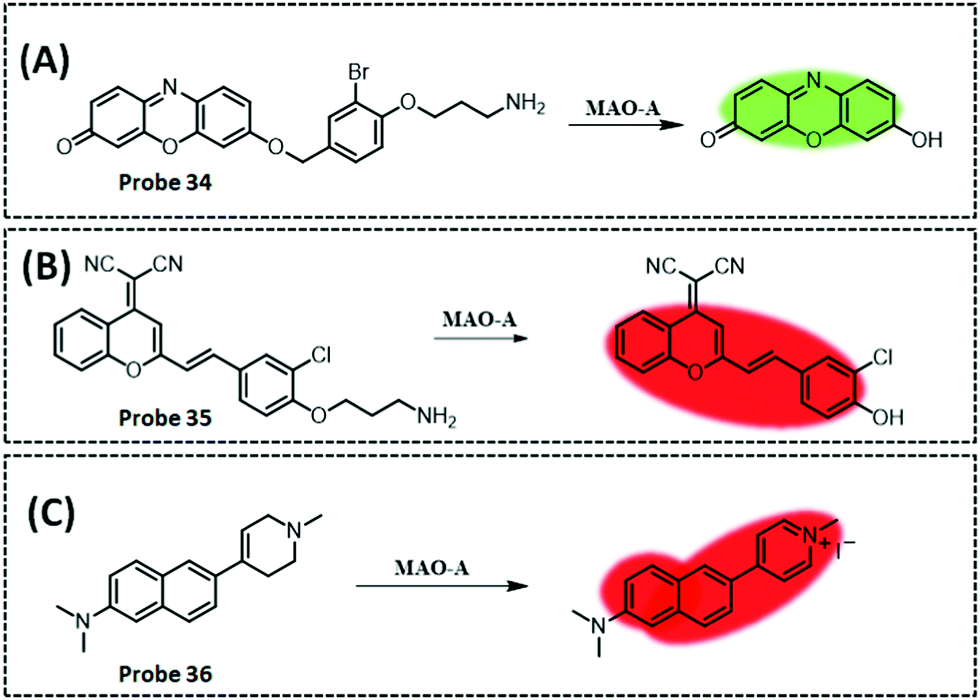 | ||
| Fig. 28 Structures and the response mechanisms of mitochondrial (A) fluorescent probe 34 (Angew. Chem., Int. Ed., 2017, 56, 15319–15323), (B) probe 35 (Chem. Commun., 2019, 55, 2477–2480) and (C) probe 36 (Angew. Chem., Int. Ed., 2020, 59, 7536–7541) to detect MAO-A. | ||
Based on the concept of “spatial configuration switching”, Fang et al. designed and synthesized a new MAO-A specific two-photon fluorescent probe 36 (λex/λem = 430/620 nm) (Fig. 28C).283 This probe selected the tetrahydropyridine of MPTP, a known MAO substrate, as the detection group, and the change in fluorescence signal was generated through a “transient conjugated structure construction” mechanism. The purpose was to detect the activity of MAO-A through the enhancement of the fluorescence signal. According to docking experiments, the nitrogen atom on the tetrahydropyridine only points to the coenzyme FAD when probe is bound to MAO-A, thus completing enzymatic oxidation and generating a fluorescence signal. We successfully confirmed the selectivity of the probe for MAO-A using different types of mammalian cell lines that regulate MAO expression via CRISPR/Cas9. We further explored the application of the probe on tissues and imaged fresh mouse brain tissue, SH-SY5Y tumour tissue, and human glioma tissue (paraneoplastic tissue sections as reference), respectively, and showed that the imaging depth could reach 220 μm. We found that the addition of a MAO-A specific inhibitor (clogyline) could largely inhibit the enhancement of the fluorescence signal. The activity of MAO-A in paraneoplastic tissues was much lower than in other tissue samples, which is consistent with previously reported results.
Li et al. designed and synthesized the first two-photon fluorescent probe 37 (λex/λem = 304/449 nm), for specific detection and imaging of MAO-B in a PD model (Fig. 29A).284 The probe employed the typical two-photon fluorophore acedan (2-methylamino-6-acetylnaphthalene) as the reporter, propylamine as the MAO-B responsive group, and the urethane structural unit as a linker between the reporter and response group. The probe was highly selective for MAO-B and allowed monitoring of MAO-B activity in mammalian lysates and recombinant proteins as well as endogenous MAO-B activity in deep tissues such as mouse brain. Finally, the probe was successfully applied to examine the level of MAO-B in PD patients, and it was found that only isolated B-lymphocytes from PD patients possessed a high level of MAO-B, indicating that the level of MAO-B in peripheral blood can serve as a biomarker for the rapid detection of PD.
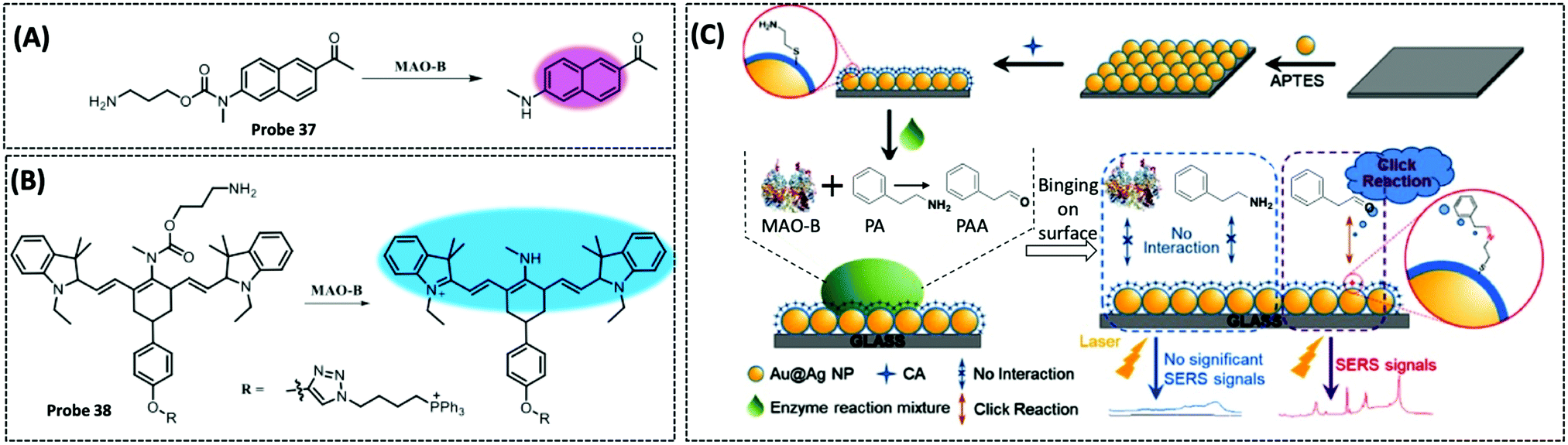 | ||
| Fig. 29 Structures and the response mechanisms of mitochondrial (A) fluorescent probe 37 (Nat. Commun., 2014, 5, 3276) and (B) probe 38 (Anal. Chem., 2018, 90, 4054–4061) to detect MAO-B. (C) Schematic illustration of the fabrication of the SERS sensor and its mechanism for specific detection of MAO-B activity (Anal. Chem., 2020, 92, 15050–15058), adapted with permission from ref. 286, copyright © 2020 American Chemical Society. | ||
Wang et al. designed and synthesized a novel ratiometric NIR probe 38 (F750/F803) to monitor MAO-B and its effects on oxidative stress in cells and mice.285 The probe was composed of three parts: the NIR fluorophore heptamethonium cyanide, the responsive group propionamide, and TPP+. The probe underwent an amine oxidation and β-elimination reaction with MAO-B, resulting in a significant increase in fluorescence and a colour change from green to blue through the ICT effect. The probe was applied to detect changes in the MAO-B levels in aging cells and mouse models, which showed that the level of MAO-B increases with age. Finally, the probe evaluated the therapeutic effects of pagiline and selegiline in a mouse model (Fig. 29B).
Wu et al. designed and fabricated a novel SERS sensor to specifically determine MAO-B activity by the SERS signal of enzyme-catalysed reaction products using an amine-aldol click reaction (Fig. 29C).286 The specific preparation and detection processes are as follows: first, core–shell gold–silver nanoparticles (Au@Ag NPs) were coated on APTES-modified glass, and then a cysteamine (CA) monolayer was modified on the nanoparticle with Ag–S bond as the linker. Using phenylethylamine (PA) as the specific substrate of MAO-B, the enzyme product phenylethylaldehyde (PAA) reacts with CA amine aldehyde under 532 nm laser irradiation to produce significant Raman signal. Other molecules such as MAO-B or PA are unable to form tight interactions with the nanoparticles due to the presence of the CA layer and therefore no signal output is observed. The authors used this sensor to specifically detect MAO-B activity in clinical blood samples, as well as to evaluate MAO-B inhibitors. This strategy offers great potential for the simultaneous detection of multiple enzymes for enzyme-related disease diagnosis and drug screening.
Wang et al. designed a label-free and highly sensitive electrochemical method to detect CS activity.288 The basic principle of this method is that the nucleic acid-mimetic CP formed by the repetition of CoA–Ag(I) units, exhibits a significant electrocatalytic effect on H2O2. First, CS catalyses the condensation reaction of oxaloacetate (OAA) and acetyl-CoA to form citrate and CoA. Then, CoA–Ag(I) CP is formed in the presence of Ag(I), due to the strong complexation ability of the CoA thiol group. The generated CoA–Ag(I) CP exhibited nuclease activity and possessed multiple adenine bases as the side groups, which allowed self-assembly on the GO-modified GCE surface by π–π stacking interactions, thus exhibiting a high electrocatalytic ability towards H2O2 (Fig. 30A). The activity of CS could therefore be indirectly quantified by monitoring the production of CoA during the enzymatic reaction. CP/GO/GCE catalyses the decomposition of H2O2 at −0.7 V to generate a current, which gradually increased with an increasing concentration of CS. The detection range was 0.0033–0.264 U μL−1, and the detection limit was 0.00165 U μL−1. Finally, the authors used this sensor to screen for inhibitors of CS. Taking ATP as an example, ATP competed with Ac-CoA thus leading to a decrease in the electrical current and the affinity of CS for Ac-CoA.
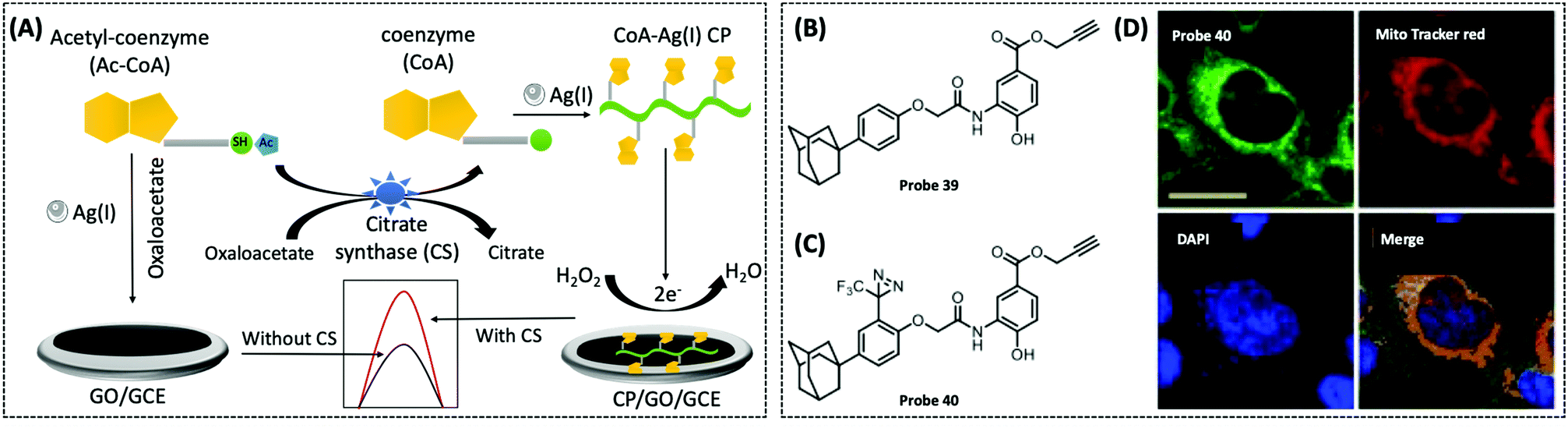 | ||
| Fig. 30 (A) Schematic illustration of activity analysis of CS based on CoA–Ag(I) CP (Talanta, 2016, 161, 583–591). (B) and (C) Structures of mitochondrial fluorescent probe 39 (Angew. Chem., Int. Ed., 2013, 52, 10286–10289) to detect m-MDH and its photoactivatable probe 40. (D) Localization of probe 39 was detected through a click reaction using azide-linked Alexa Fluor 488 in HCT116 cells. Scale bars: 20 μm, adapted with permission from ref. 291, copyright © 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Malate dehydrogenase (m-MDH), which mainly catalyses malate dehydrogenation in mitochondria, is an important enzyme in the tricarboxylic acid cycle, playing a crucial role in the complete oxidation or mutual transformation of nutrients in the body.289 The magnitude of m-MDH elevation correlates with disease severity, and is also associated with malignant liver metastases, traumatic shock, renal disease, rheumatoid arthritis and haematological disorders, among others.290
Lee et al. designed and synthesized a chemical probe 39 (λex/λem = 488/519 nm), which uses click chemistry by introducing an alkyne bond.291 Based on the authors’ previous studies showing that a similar structure could inhibit the accumulation of hypoxia-inducible factor-α (HIF-α), the authors found that probe 39 could localise in cells and inhibit the accumulation of HIF-α by click chemistry (Fig. 30B–D). The authors then incubated the probe or MitoTracker with cells and found a high co-localisation coefficient for probe 39, indicating that it localised in the mitochondria. Subsequently, the authors introduced a photoaffinity group, trifluoromethyl diazirine, into probe 39 to synthesise probe 40. The MDH2 protein can bind to Cy5 of probe 40; however, the binding ability decreases when probe 39 is present, indicating that probe 40 is directly bound to MDH2 and thus identifying MDH2 as the target protein. The experimental results suggest that probe 39 and MDH2 inhibitor suppress hypoxia-induced HIF-α accumulation by inhibiting mitochondrial respiration.
Park et al. designed a mitochondria-targeting ratiometric fluorescent probe 41 (F565/F520) by replacing the acid group of the previously reported probe, AP-Glu, with an ester group.293 The probe was targeted to mitochondria due to its positive charge, and bound the amide group directly to the indigo fluorophore (Fig. 31A). The probe initially produced a relatively weak fluorescence due to its amide function; however, when the probe reacted with γGGT, an intense fluorescence was emitted through an enzyme-mediated amide bond cleavage reaction. Co-localisation experiments using the probe and MitoTracker Deep Red demonstrated excellent mitochondrial targeting. To image tumours, the authors used two carcinogens, dextran sodium sulphate (DSS) and azoxymethane (AOM), in a mouse model of colon cancer. No fluorescence was observed in normal mice, while intense fluorescence was seen in cancerous mice.
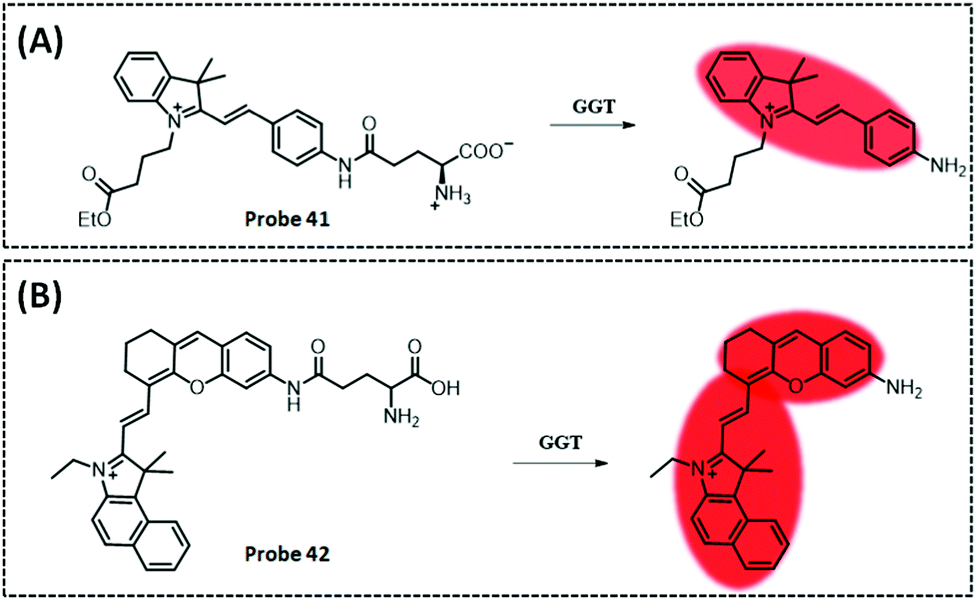 | ||
| Fig. 31 Structures and the response mechanisms of mitochondrial (A) fluorescent probe 41 (Chem. Commun., 2016, 52, 10400–10402) and (B) probe 42 (Analyst, 2018, 143, 5530–5535) to detect GGT. Both emitted a red fluorescence on reaction with GGT. | ||
Liu et al. synthesized a new mitochondrial-targeted near-infrared fluorescent probe 42 (λex/λem = 680/727 nm) for the detection of γGGT.294 The probe consisted of a hemi-anthocyanin fluorophore with benzindole as the absorbing electron unit, a positively charged mitochondrial targeting fraction, and a glutamate-masked amido-dihydroxyanthracene as the γGGT response group (Fig. 31B). The probe itself was not fluorescent because the presence of the amide bond greatly diminished the electron-donating capacity of flavonamide and destroyed the push–pull structure of the probe. However, when γGGT cleaves the γ-glutamyl bond to release the flavonamide fraction, a fluorescence signal is generated. The authors examined the ability of the probe to image γGGT in HeLa cells; however, the probe could detect endogenous γGGT. When 6-diazo-5-oxo-L-norleucine (DON, an inhibitor of γGGT) was added, the fluorescence was significantly suppressed.
In 2016, Chevalier et al. synthesized NTR-activated probe 43 (λex/λem = 572/703 nm) using ortho-, meta-, and para-substituted 4-nitrobenzyloxy modified on quinone-cyanine 7 (QCy7) dye (Fig. 32A).296 The positive nitrogen ion of QCy7 provides good mitochondrial targeting ability. When the probe was transported to the mitochondria, the nitro group was reduced by NTR and the alkylation product was removed by a rearrangement reaction, thus converting the non-fluorescent precursor into QCy7 dye and increasing fluorescence. The experiments showed that the meta-substitution product 43b could not be activated by NTR, and that the activation of para-substituted 43c was much faster than the ortho-substituted 43a. The probes were used to image NTR activity in the mitochondria of A549 cells (human lung adenocarcinoma cells). The authors detected a type I NTR in the mitochondria of A549 cells. Inspired by this, the authors developed a prodrug for the mitochondrial poison antimycin A using para-substituted 4-nitrobenzyloxy as the protecting group. They showed that the prodrug had better mitochondrial transport efficiency and cell death rate.
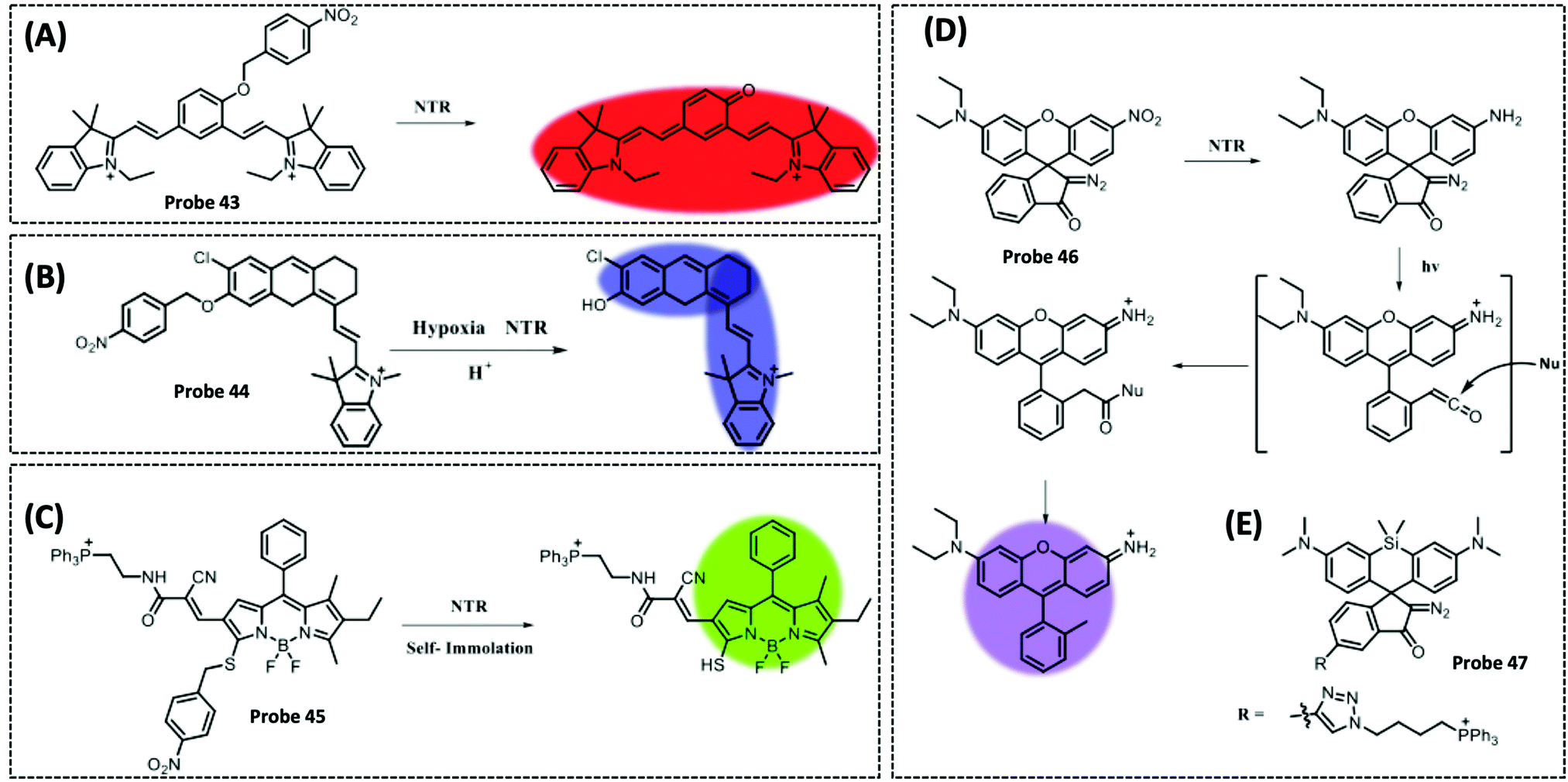 | ||
| Fig. 32 Structures and the response mechanisms of mitochondrial (A) fluorescent probe 43 (J. Am. Chem. Soc., 2016, 138, 12009–12012), (B) probe 44 (Chem. Sci., 2018, 9, 5347–5353) and (C) probe 45 (Chem. Commun., 2020, 56, 7761–7764) to detect NTR. (D) and (E) Structures and the response mechanisms of mitochondrial fluorescent probe 46 and probe 47 (Angew. Chem., Int. Ed., 2019, 58, 11474–11478) to detect NTR. | ||
An NIR fluorescent probe 44 (λex/λem = 670/710 nm) was designed and synthesized and had dual recognition sites: NTR and pH (Fig. 32B).297NTR is overexpressed when cells are under hypoxic conditions. Here, NTR reduced the p-nitrophenyl group of the probe to p-aminobenzene. Following aminobenzene removal by a 1,6-rearrangement elimination reaction, the deprotonated hydroxyl group was exposed and could exhibit ratiometric pH changes. This probe could simultaneously visualise different degrees of hypoxic microenvironments and distinguish corresponding mitochondria in living cells, due to the unique pH changes observed in mitochondria in anoxic microenvironments.
Zhu et al. designed another NTR activated fluorescent probe 45 composed of a BODIPY reporter group, a p-nitronitrophenyl sulphide moiety as the NTR response site, and TPP (Fig. 32C).298 NTR reduces p-nitronitrophenyl sulphide to p-aminobenzyl sulphide, and then 1,6-elimination reaction is performed to release the fluorophore, which results in a red shift of 155 nm (λex/λem = 495/560 nm; λex/λem = 650/713 nm), thus showing the near infrared emission and dual-channel ratiometric fluorescence. This probe was used for high-precision imaging of real-time monitoring of nitroreductase activity in cancer cell mitochondria.
Thiel et al. designed and synthesized a photoactivatable probe 46 (λex/λem = 405/561 nm) using a diazoindanone-modified rhodamine analogue (Fig. 32D and E).295 The nitro group on the probe generated a non-fluorescent product upon photoirradiation. The reduction of the nitro group to an amino group by NTR upon photoirradiation and nucleophile attack, a Wolff rearrangement reaction, produced a strong fluorescent product. Furthermore, the probe covalently binds to the nucleophilic residues of the protein, thus immobilizing the fluorescence signal for a long time. The authors used the light-crosslinked fluorescent probe to show that nitroreductase activity occurs in the mitochondrial microdomains. These authors also prepared probe 47 (λex/λem = 405/640 nm), a far-red emitting probe targeting mitochondria. The authors used probe 47 and probe 46 and to carry out two-colour, three-dimensional STORM imaging. After minimising the diffusion of probe 46 following enzymatic conversion, and obtaining superimposable signals of the two imaging channels, super-resolved images of both the mitochondria and nitroreductase activity were obtained.
Mitochondrial thioredoxin reductase (TrxRs) is present in all living cells and is a member of the pyridine nucleotide-disulphide bond oxidoreductase family. There are three forms of TrxRs: TrxR1 mainly exists in the cytoplasm and nucleus, TrxR2 is mainly located in the mitochondria, and TrxR3 is expressed primarily in the testis. Mitochondrial TrxRs are essential for removing the oxidatively damaged mitochondrial proteins, regulating the transition of mitochondrial permeability, and inhibiting apoptosis.299 Abnormal mitochondrial TrxRs levels are directly related to mitochondrial dysfunction, leading to increased ROS and apoptosis rates.300
Lee et al. reported a mitochondria-targeting Trx probe 48 (λex/λem = 428/540 nm). The probe used naphthalene diformamide as the fluorophore, TPP+, and a disulphide bond as the Trx reaction site (Fig. 33A).301 Following disruption of the disulphide bond, naphthalene diimide fluorescence was restored. In vitro kinetics of disulphide bond cleavage indicated that the secondary rate constant was (4.04 ± 0.26) × 103 (M s)−1, which was about 5000-fold higher than GSH. To verify that this probe could assay Trx in cells, the authors co-incubated the cells with different concentrations of PX-12, a Trx inhibitor. They found that the higher the PX-12 concentration, the lower the fluorescence intensity emitted by the cells, indicating that Trx was able to break the probe-disulphide bond. Co-localisation experiments using the probe and MitoTracker Red showed that the probe was localised in the mitochondria. Liu et al. used a similar probe to observe the severe inhibition of TrxR2 activity in PD, revealing the potential link between TrxR2 dysfunction and PD aetiology.302
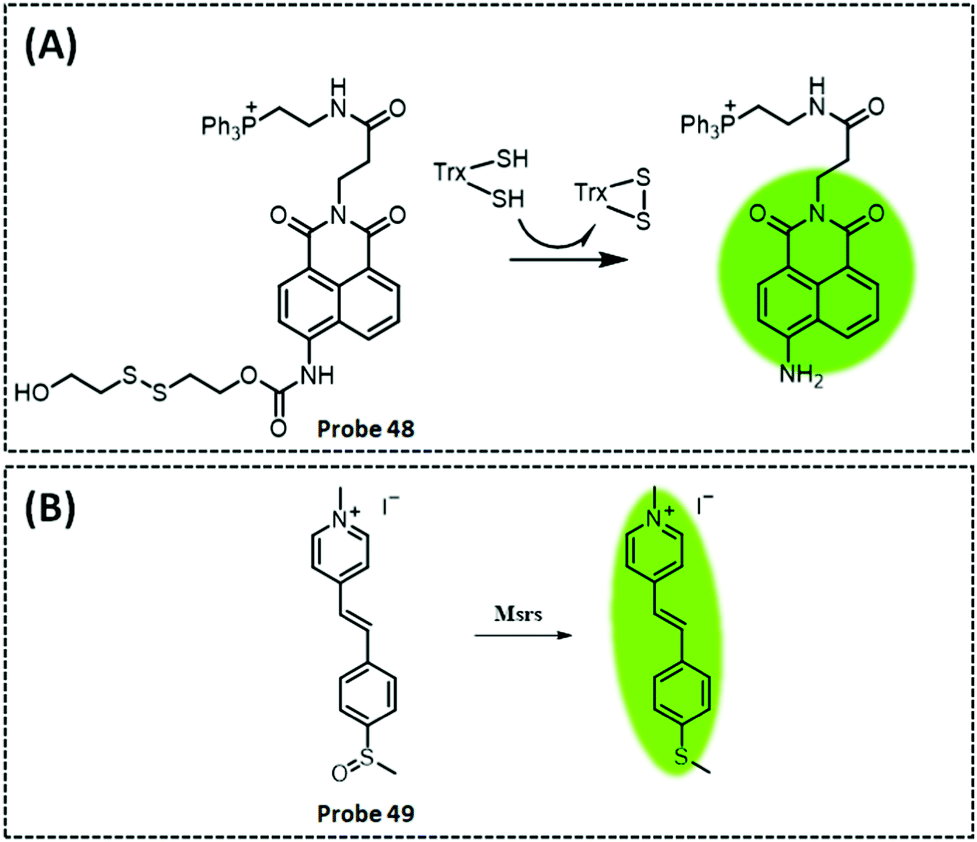 | ||
| Fig. 33 (A) Structure and the response mechanism of mitochondrial fluorescent probe 48 (J. Am. Chem. Soc., 2012, 134, 17314–17319) to detect TRxR2. (B) Structure and response mechanism of mitochondrial fluorescent probe 49 (Anal. Chem., 2019, 91, 5489–5493) to detect Msrs. Both probes emitted a green fluorescence upon reaction with their target analyte. | ||
Methionine sulphoxide reductases (Msrs), as an important antioxidant defence system, play a crucial role in regulating mitochondrial energy metabolism and function. Msrs consist of methionine sulphoxide reductase A (MsrA) and B (MsrB), which can reduce methionine thiols to the corresponding isomers assisted by electron donors.303 Abnormal concentration of Msrs is related with aging and neurodegenerative diseases.
The catalytic site of Msrs is mainly located on relatively small and restricted protein surfaces. Inspired by a well-known substrate of Msrs and methyl phenyl sulphoxide (MS), it is hypothesised that small-sized fluorescent probes are more suitable for the catalytic site. Xiang et al. designed Msr fluorescent probe 49 (λex/λem = 385/547 nm) for detecting mitochondrial Msrs (Fig. 33B).304 The probe was non-emissive for the acceptor–acceptor electronic structure. After Msrs reduced the electron-accepting sulphoxide to the electron-donating sulphide, a donor–acceptor fluorophore structure with large Stokes shift was generated and the fluorescence signal turned on based on the principle of ICT. The probe exhibited high specificity, high sensitivity and rapid response for Msrs in vitro. Furthermore, hydrophobic methylpyridinium confers excellent mitochondrial targeting ability to the probe. In addition, the probe could detect and image mitochondrial Msrs in living cells; moreover, reduced Msrs activity was successfully detected by the probe in a PD cell model.
4.3 Detection of mitochondrial ions and membrane potential
Metal ions are widely distributed in cells, tissues, and body fluids and are very important for human physiology and pathology. The ions in mitochondria not only maintain the basic structure of biomolecules such as proteins, nucleic acids, and enzymes but also collaborate to perform catalytic or regulatory functions, thereby influencing the normal metabolism of the organism. Most of the probes employed to detect ions are based on three reaction mechanisms: complexation, chemical bond breaking, and chemical bond formation.The design of probes based on complexation involves two strategies: direct complexation and competitive substitution complexation. Some metals or anions can selectively bind to certain chemical sites due to their unique electrophilic, sulphophilic, or nucleophilic properties and undergo hydrolysis or electron rearrangement reactions, leading to the cleavage of certain chemical bonds. In addition, some ions can also facilitate the formation of chemical bonds. In this section, we focus on the biological function and detection of mitochondrial calcium (Ca2+), potassium (K+), zinc (Zn2+), copper (Cu+/2+), and iron (Fe2+) ions.
At present, most probes cannot distinguish between Ca2+ and magnesium ions. Direct detection of mitochondrial Ca2+ content is crucial. Pendin et al. reported a mitochondrial Ca2+probe 50 (λex/λem = 345/520 nm) (Fig. 34A) based on furan derivatives.305 To determine the best mitochondrial localisation strategy for providing appropriate localisation and in situ functionality, they synthesized and screened several furan-2 derivatives containing one, two, or three TPP groups, and ligated them to the probe at different positions. They found that the best-performing probe contained two TPP lipophilic cations, one of which was attached to ethylenedioxy via n-butyl intervals and the other to the carboxylate component of the fluorine group via n-propyl intervals. The probe showed a specific response to Ca2+. When the probe was chelated to Ca2+, there was a 20 nm red shift. The authors successfully detected Ca2+ levels in mouse cardiomyocytes and observed an increase of mitochondrial Ca2+ stimulated by potassium chloride. Furthermore, the increase of mitochondrial Ca2+ in astrocytes stimulated by ATP was also observed.
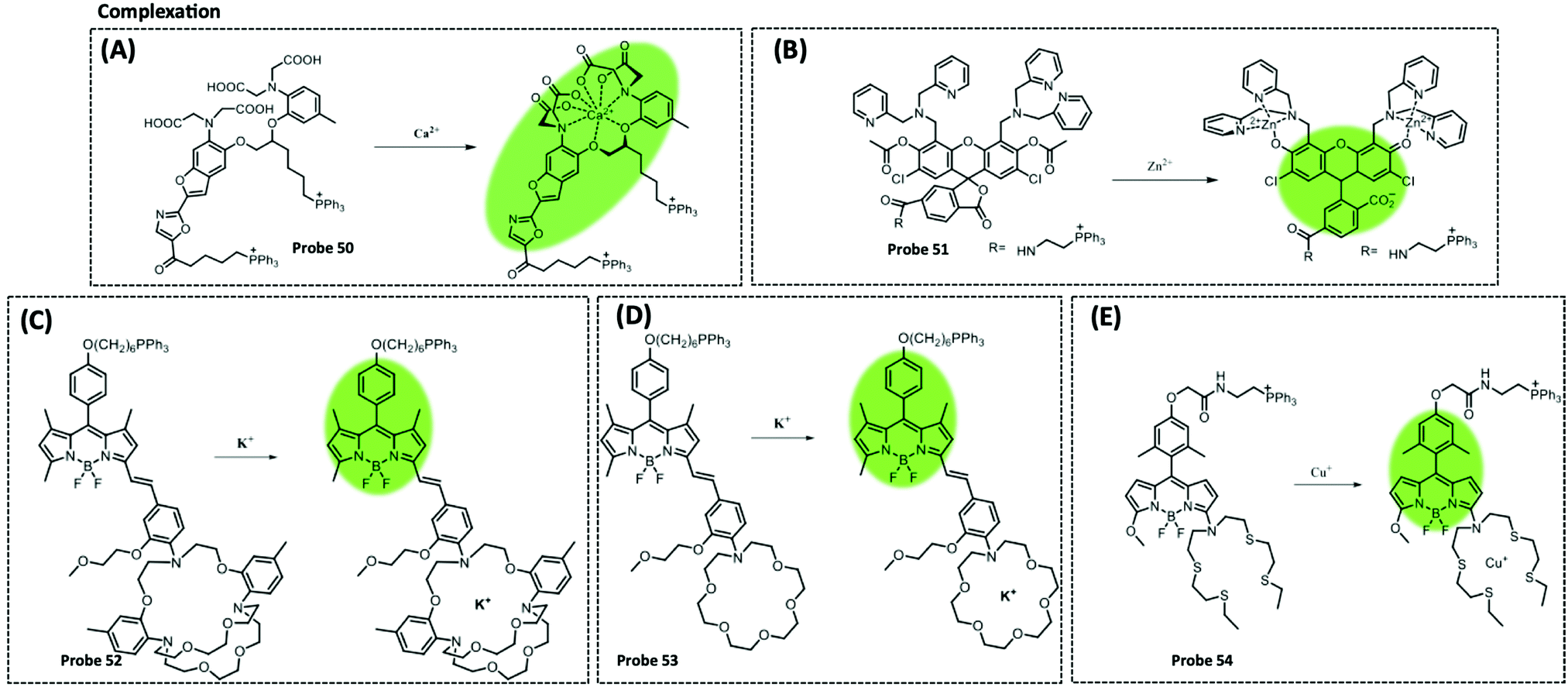 | ||
| Fig. 34 Detection of ions based on the principle of complexation. Structure and the response mechanism of mitochondrial fluorescent (A) probe 50 (Angew. Chem., Int. Ed., 2019, 58, 9917–9922) to detect Ca2+, (B) probe 51 (Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 143–148) to detect Zn2+, (C) probe 52 (Angew. Chem., Int. Ed., 2015, 54, 12053–12057), (D) probe 53 (Sens. Actuators, B, 2020, 307, 127659.) to detect K+, and (E) probe 54 (J. Am. Chem. Soc., 2011, 133, 8606–8616) to detect Cu+. Each probe emitted green fluorescence upon complexation with its corresponding ion. | ||
In addition to binding to the mitochondrial ETC complex and affecting enzyme activity and ion transferases, Zn2+ ions are involved in mitochondrial protection in cardiomyocytes. Chyan et al. synthesized a reversible off-label probe 51 (λex/λem = 488/530 nm).306 The fluorescence of the PET effect quenched the probe and the complex fluorescence was formed by chelating to the Zn2+ ions. To create a derivative that is optimised for mitochondrial localisation, they designed a reaction-based probe based on fluorescein diacetate (Fig. 34B). Modification of fluorescein with ether or esters derivatives is a common strategy to improve the cell sensitivity, retention and permeability of fluorescein probes. Their approach used Zn2+ to promote acetyl cleavage to weaken the quenching of PET on the DPA arm and restore fluorescence. The probe could detect Zn2+ in HeLa cells through fluorescence imaging. Cell studies showed that, unlike healthy prostate cells, carcinogenic cells cannot accumulate free divalent Zn2+ in the mitochondria.
Different from fluorescent probes, Fudge et al. designed a series of GZnP2 derived biosensors for the determination of the unstable Zn2+ concentration in the cytosol, mitochondrial intermembrane space and mitochondrial matrix in different cell lines. The unstable Zn2+ concentration was ∼100 pM in the IMS and cytosol, while being less than 1 pM in matrix, in depolarised INS-1 cells.307 This indicated that the IMS and cytosol were overloaded with Zn2+ when exposed to high Zn2+ concentrations, but the mitochondrial matrix was incapable of preserving extra destabilized Zn2+. Their work emphasized the importance of detecting unstable Zn2+ concentrations in the mitochondrial IMS and matrix.
The biological functions of K+ ions are outlined in Section 2. The traditional methods for studying potassium channels (KCh) are mostly indirect experimental methods with many uncertainties. Kong et al. designed a fluorescent probe 52 (λex/λem = 540/572 nm) for sensing of K+ ions in mitochondria at the cellular level (Fig. 34C).308 The probe used BODIPY as the fluorophore and a triphenylphosphine cation as the targeting group, with the reported highly selective K+ ligand (TAC) as the recognition group. When the probe was first chelated with K+ ions, there was a PET effect between BODIPY and the lone pair electrons of the nitrogen atom in the ligand. Following the reaction of the probe with K+ ions, the PET effect was turned off and fluorescence was restored. Probe 52 had good selectivity for K+, with a 130-fold increase in fluorescence in response to the ion. The authors used this probe to observe K+ influx and efflux under various stimuli; however, the complexity of TAC synthesis limits the widespread use of this probe.
Ning et al. optimised the recognition group of probe 52 to generate probe 53 (λex/λem = 540/572 nm), which uses phenylaza-crown-6-lariat-ether with 2-methoxyethoxy (ACLE) as the recognition group. Probe 53 had a higher selectivity compared to probe 52, with a 160-fold increase in fluorescence upon reaction, 0.2 s response time, and wide detection range of 10–500 mM (Fig. 34D).309
Copper-induced toxicity leads to cell apoptosis, which is caused by ion imbalance affecting mitochondrial ETC, ROS formation, mitochondrial membrane depolarization, lipid peroxidation, and cyt c release. Dodani et al. designed a new type of mitochondrial fluorescent probe 54 (λex/λem = 540/558 nm) for detecting Cu+ (Fig. 34E).310 The probe has a thioether recognition group and a high selectivity for Cu+ in an aqueous solution. The fluorescence intensity of the probe was increased 10-fold after adding Cu+. This probe was used in the model cell line HEK 293T, as well as for real-time detection of mitochondrial Cu+ in human fibroblasts. The researchers have shown that the cells can be stabilised by modulating copper content in mitochondria, even in the face of severe copper deficiency. Wang et al. previously designed a novel mitochondria-specific fluorescent probe 55 (λex/λem = 450/525 nm) to accurately visualise Cu2+ in the mitochondria of living cells (Fig. 35A).311 The alkyl-linked fluorescein hydrazide was a fluorescently responsive, photocage version of Cu2+ with 2-nitrobenzyl units that were efficiently photodissociated under UV light. This not only increased the cell permeability of the probe, but also provided additional spatial and temporal control for mitochondrial Cu2+ imaging. In this study, a simple photodissociated 2-nitrobenzyl group was selected as the embedding molecule, which might be easily replaced by a two-photon embedding molecule. TPP internalised the probe into the mitochondria. Subsequent UV irradiation then forced the photocage group away to release the uncaged probe, which was then hydrolysed by mitochondrial Cu2+, producing specific localisation and intense fluorescence signal. Imaging showed that the probe could be used to visualise mitochondrial Cu2+ in living mammalian cells in a photo-controlled manner that successfully avoided the effects of cytoplasmic Cu2+ in mitochondria-specific assays.
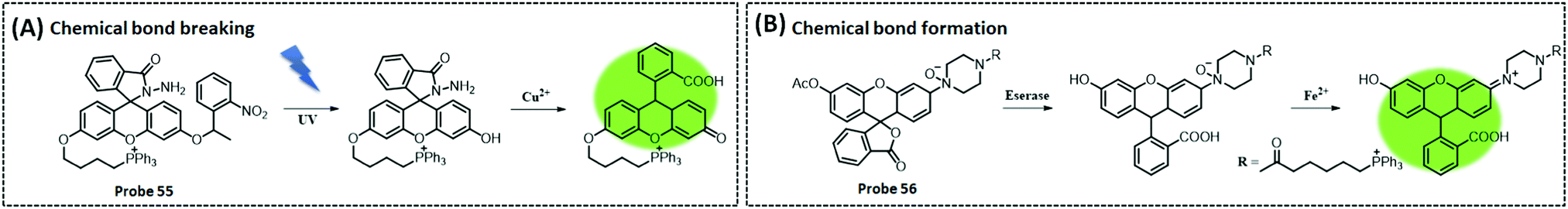 | ||
| Fig. 35 Detection of ions based on the principle of chemical bond breakage or formation. (A) Structure and the response mechanism of mitochondrial fluorescent probe 55 (Chin. Chem. Lett., 2017, 28, 1965–1968) to detect Cu2+. (B) Structure and response mechanism of mitochondrial fluorescent probe 56 (Metallomics, 2018, 10, 794–801) to detect Fe2+. Both probes fluoresced green upon reaction with the ions. | ||
The biological functions of Fe2+ are outlined in Section 2. For detection of Fe2+, Hirayama et al. reported a new mitochondria-targeting fluorescent probe 56 (λex/λem = 510/535 nm) that acted as an Fe2+-selective fluorogenic switch.312 Piperazinylrhodol N-oxide was used as an Fe2+ sensing unit because the acyl piperazine could accelerate Fe2+-triggered deoxygenation (Fig. 35B). The Fe2+ response unit was linked to a TPP group. The phenolic hydroxyl group was protected by an acetyl group to promote cellular uptake efficiency. The acetyl group of probe 56 was cleaved by esterases in the cell and enriched in the mitochondria by TPP action. Furthermore, the probe was able to detect variations in endogenously accumulated Fe2+ caused by the inhibition of heme synthesis.
Mitochondria provide energy to aerobic cells through aerobic respiration, and in this process, the proton and other ion concentrations on both sides of the inner membrane are asymmetrically distributed to form MMP, which is an indicator of the health of the cell.313 Normal MMP usually fluctuates between 150 and 180 mV and is important for cellular functions. However, any abnormalities in the MMP levels can lead to mitochondrial dysfunction; therefore, dynamic monitoring of cellular MMP variations is of great significance for biochemical studies and the diagnosis of relevant diseases.
Electrochemical sensors measure MMP usually by potentiometric analysis, which is commonly used with cation-modified ion-selective electrodes in a suspension of mitochondria, such as TPP+ ion-selective electrodes.314 This is due to the ability of TPP+ to diffuse into the inner mitochondrial membrane, thus creating a potential difference that enables the measurement of MMP according to the Nernst equation. For example, Lim et al. combined microfluidics with electrochemical sensors to construct a TPP+ ion-selective sensor, which enabled the monitoring of MMP in isolated mitochondria in the presence of different ETC inhibitors.315 However, microelectrode-based measurements of MMP often require the isolation of mitochondria from the cell and provide measured parameters that are not real cellular parameters. Therefore, the organic fluorescent probe-based fluorescence imaging provides a powerful tool for the in situ, real-time, dynamic and non-invasive monitoring of MMP.316
The general fluorescent probes for MMP can be divided into two main categories according to their commercial names: rhodamine and rhodamine derivatives (including TMRM, TMRE and Rh123)317 and carbocyanines (including JC-1 and DiOC6).318,319 However, these commercially available fluorescent probes have certain disadvantages. For example, Rh123 non-specifically targets mitochondria. It is difficult for JC-1 to form aggregated states at low concentrations and JC-1 is poorly water soluble at higher concentrations, thus making it difficult to be eluted. A detailed review by Perry et al. discusses the best usage and experimental notes on commonly available commercial MMP dyes, and provides an in-depth introduction to MMP determination.320 Simultaneously, a variety of novel fluorescent probes have been designed and synthesized to address the monitoring problems of commercial MMP fluorescent probes, thus better enabling the in situ and real-time monitoring of MMP changes in living cells.321–324
Feng et al. designed and synthesized a FRET probe 57 (FixD and LA) for MMP ratiometric fluorescence monitoring, which consisted of a FRET donor molecule FixD and a FRET acceptor molecule LA (Fig. 36A).325 FixD (λex/λem = 405/560 nm) was constructed using a benzyl chloride moiety and a carbazole-pyridium with a green fluorophore. FixD could be attached to the sulfhydryl group of mitochondrial proteins and anchored in the mitochondria independently of MMP floating. They also designed LA (λex/λem = 560/675 nm) with green absorption and deep red fluorescence emission, and LA was an MMP-dependent mitochondrial probe with phenylmethyl used as the side chain to increase cell membrane permeability. As the MMP decreases, the LA was gradually shed from the mitochondria, and the FixD still remained in the mitochondria. The distance between the molecules gradually blocked the FRET between the FixD and LA molecules, and thus the deep red fluorescence was gradually reduced and the green fluorescence emission was gradually increased. Based on the FRET mechanism, the dynamics of MMP could be monitored by the green and deep red fluorescence changes. However, one limitation of the FixD/LA detection system was the non-specific mitochondrial targeting of LA, which might remain in depolarised mitochondria and interfere with the detection of MMP.
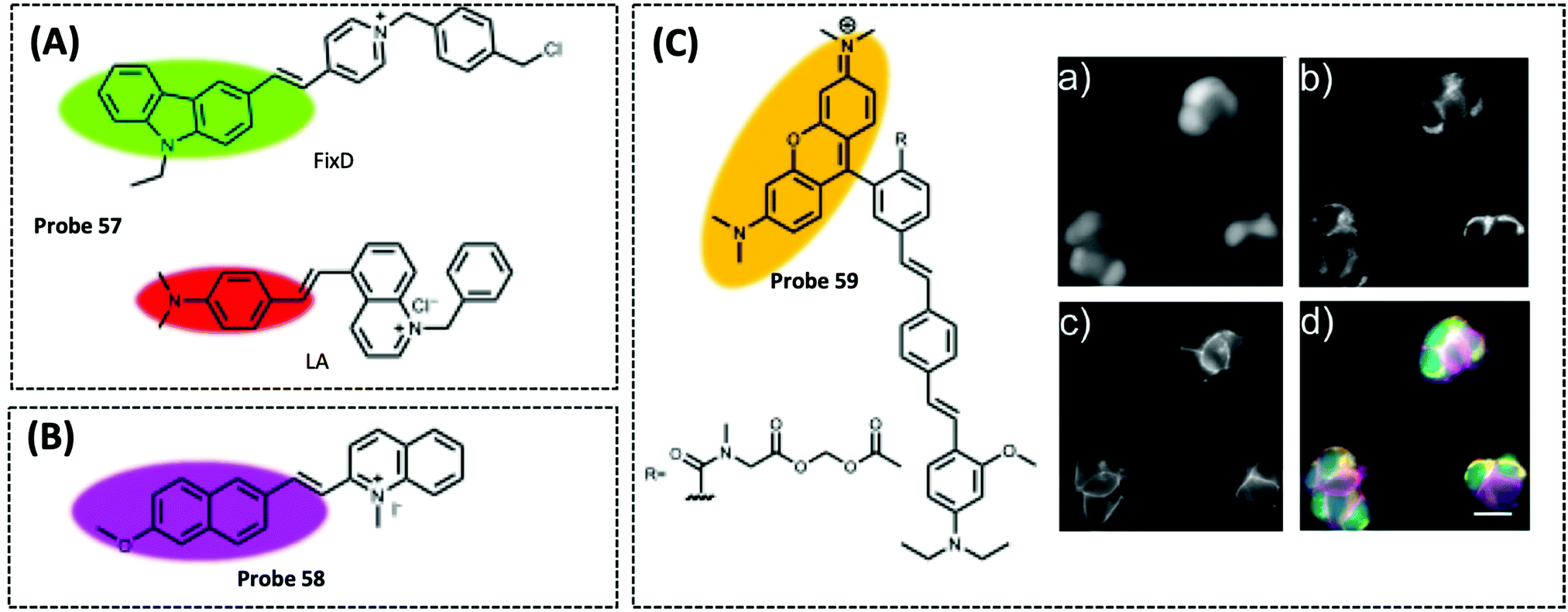 | ||
| Fig. 36 The probes of mitochondrial membrane potential (MMP). (A) Structure of mitochondrial fluorescent probe 57 (Anal. Chem., 2019, 91, 3704–3709) consisted of FixD and LA. (B) Structure of mitochondrial fluorescent probe 58 (Angew. Chem., Int. Ed., 2018, 57, 16506–16510). (C) Structure of mitochondrial fluorescent probe 59 (J. Am. Chem. Soc., 2021, 143, 4095–4099). Simultaneous, multi-color imaging of MMP, cytosolic Ca2+, and plasma membrane potential in mammalian cells. Wide-field epifluorescence images of HEK cells stained with (a) OGB, (b) probe 59, and (c) BeRST 1, (d) an overlay of the images showing cytosolic localization of OGB (green), mitochondrial localization of probe 59 (yellow), and plasma membrane localization of BeRST (magenta). Scale bar is 20 μm, adapted with permission from ref. 327, copyright © 2021 American Chemical Society. | ||
Tian et al. designed and synthesized a novel dual-emission fluorescent probe 58 based on a dual-targeting strategy (Fig. 36B).326 The probe was composed of a weak electron donor and a quinoline cationic moiety. The hydrophobic and positive charged probe could target MMP in healthy cells. As a result, the probe targets mitochondria in healthy cells and emits deep red fluorescence (λex/λem = 488/663 nm). When the MMP levels decreased due to the addition of CCCP, the probe was released from the mitochondria and migrated to the nucleus to produce green fluorescence (λex/λem = 488/550 nm). When the CCCP was removed and the MMP levels were restored, the green signal in the nucleus gradually disappeared and migrated back to the mitochondria. Finally, the authors successfully used the probe to monitor the process of rotenone induced apoptosis. Remarkably, this is the first probe capable of reversibly detecting cell health status by positional migration and fluorescence color change, which provides an important guide for the development of such fluorescent probes.
Klier et al. reported the first fluorescent probe 59 (λex/λem = 525/500–700 nm) with a transient response to MMP changes that was not dependent on MMP accumulation (Fig. 36C).327 The membrane potential response mechanism of this probe was achieved by PET mechanism. The probe employed esterified tetramethylrhodamine as the fluorophore, diethylamine as the electron-donating group and a lipophilic molecular chain in the middle to drive membrane localization. In order to solve the problem of rhodamine signal being affected by MPP, the authors masked the carboxylic acid in tetramethylrhodamine using an acetoxymethyl ester, which was unstable and easily hydrolysed by esterases into negative carboxylate ions upon entering the mitochondrial matrix. The probe entering the matrix were not affected by MPP change. Then, the authors used the probe to dynamically and reversibly monitor the MMP depolarization and hyperpolarization processes induced by FCCP. Finally, the authors combined the probe with Oregon Green BAPTA (OGB, to detect intracellular Ca2+), and BeRST (to monitor changes in cell membrane potential) to achieve a multi-parameter monitoring of FCCP-induced cells. Notably, this is the first probe with a voltage-sensitive fluorophore to study MMP changes by the PET mechanism.
5. Conclusions and outlooks
In this review, we have summarised and highlighted recent advances in the study of biological functions of mitochondrial energy metabolism, and the various methods used to detect mitochondrial metabolites and intermediates. The advantages and disadvantages of these detection methods were highlighted, compared, and objectively discussed. We believe that this review sheds substantial light on how to design a novel approach or strategy to monitor mitochondrial energy metabolism in complex biological matrices, which is of great significance for preventing, diagnosing, and elucidating the mechanisms of various metabolic diseases.Development of new technologies, instrumentation, and data analysis methods will distinctly improve and expand the detection methods required to understand the dynamic information contained within the entire mitochondrial energy metabolism network, or to quantify and reproduce specific mitochondrial metabolic pathways under different conditions. With the emergence of new diseases (e.g., COVID-19) and the increase in ageing of the population, rapid, accurate, highly sensitive, and specific mitochondrial energy metabolism detection methods will play an important role in the improvement of medicine and health. However, there remain several limitations and challenges that urgently need to be addressed in order to accelerate the development and future clinical applications of analytical methods for mitochondrial energy metabolism.
(1) Improving the response sensitivity: metabolites or intermediates are usually present at low concentrations for short durations and are easily converted in the biological environment; therefore, analytical methods should be ultra-sensitive in order to detect small changes in the biochemical substances before the onset of the disease, to enable effective intervention and treatment. Constructing and using multiple, effective biological analysis and signal amplification strategies in parallel can improve detection sensitivity. These strategies include nucleic acid amplification, co-reaction reagent amplification, nanomaterial and biomolecule binding, and dual signal ratio amplification. For electrochemical methods, conductive materials are modified on the electrodes, which is the most common approach. For example, conductive nanomaterials are modified on the electrode by electrodeposition or spin coating, which can increase the efficiency of electron transport, reduce the distance between the active centre of the biomaterials and the electrode, and improve the reaction speed and sensitivity. In addition, increasing the detection method or the parameters of the detection instrument also improves sensitivity. Synthesis of near-infrared, two-photon, or even multi-photon fluorescent probes that increase penetration and reduce auto-fluorescence interference will allow for the imaging of tissues and living organisms in situ and will reduce background interference. At present, many fluorescent probes targeting mitochondria reported in the literature require the addition of many exogenous analytes or cells stimulated by the corresponding exogenous stimuli when detecting endogenous ultra-low target concentrations. Currently, there are only a few probes that can selectively detect and image at ultra-low endogenous concentrations, which hinders the understanding of these biochemical substances in mitochondrial energy metabolism.
(2) Improving biocompatibility: biocompatibility refers to the effect of a biological material on a specific biological tissue environment once it has entered the organism. It depends primarily on the toxicity, solubility and stability of the material in the biological environment. The principle of biocompatible design is to stimulate the biological material to perform a specific function while maintaining a remarkably low level of toxicity. Excellent biocompatibility is an important feature to ensure that the optical and electrochemical sensors can accurately detect metabolites without affecting the normal function of living cells/tissues. Poorly biocompatible materials can be toxic, irritating, and teratogenic and cause local inflammation when interacting with living cells/tissues, which can seriously interfere with the detection results. Currently, scientists are increasingly focusing on improving the biocompatibility of sensors. Flexible biodegradable biosensors are currently a key trend. Flexible sensors can be used not only for real-time in situ assays at the live cell/tissue level, but can also be implanted in the body for continuous measurements, which can provide an important basis for drug guidance, cell metabolism, etc. For example, Yin et al. used a flexible transient electrochemical biosensor designed and implanted in mammals to enable continuous long-term monitoring of NO concentrations in the bone and joint cavities.328 In addition, more and more materials with excellent conductivity are being used in electrochemical sensors, but the biocompatibility of these materials should also be considered when designing and preparing electrochemical sensors, for example by examining their biocompatibility using approaches that test for cytotoxicity (e.g., the MTT method). For optical biosensors, especially organic small molecule fluorescent probes, the issue of phototoxicity also deserves attention. The phototoxicity of fluorescent probes generally arises from the first excited triplet state (T1), which is generated during excitation by the first excited singlet state (S1) after interlinear scramble. The T1 state interacts with oxygen to generate ROS such as singlet oxygen, which can damage biomolecules or cell structures. Detailed characterisation of properties such as probe phototoxicity has often been neglected in previous fluorescent probe designs in favour of photobleaching. For example, Chen et al. found that groups capable of reducing photobleaching, such as nitrobenzene, did not necessarily reduce phototoxicity.329 Therefore, both phototoxicity and photobleaching need to be fully considered in the design of fluorescent probes. In addition, there are a number of natural biomaterials that can be considered when designing fluorescent nanosensors, including peptides, exosomes, and cell membranes. For instance, cell membrane coating technology is the combination of natural cell membrane properties with artificial materials, thus greatly enhancing biocompatibility while enabling long-lasting circulation and targeted delivery in vivo.330
(3) Enhancing specific targeting: the surrounding environment and non-target objects in the sample will affect the recognition process when these analytical methods are applied in practice, thereby affecting the sensitivity and accuracy of detection. Thus, avoiding the influence of interference and improving specific recognition and detection are the key steps needed for the development of mitochondrial energy metabolism detection. Using a variety of markers to form logical gates to identify specific biochemical substances, or cells, is a new way of improving specificity. For biosensors, these specific elements are usually biomaterials such as antibodies, enzymes, or peptides. These are favourable in terms of high specificity and good responsiveness, but unfavourable for recovery and improvement over the working lifetime. Taking antibodies as an example, it is necessary to have the same spatial orientation to keep the antigen binding site as far away from the solid surface as possible. Therefore, using genetic engineering technology to alter the specific antibody sites could be the future of antibody fixation technology. In addition, the recognition and sensing mechanisms between the target and recognition groups need further research. New biomaterials have been developed and rapidly applied to construct various detection models. Most of the research has focused on the synthesis and characterisation of materials, while the specific recognition and sensing mechanisms of new biomaterials and targets are still unclear. A number of other experimental techniques or methods including X-ray, electron paramagnetic resonance or electron microscopy can help researchers understand these mechanisms.
(4) Combining with new technology: existing detection methods need to be combined with new technological approaches to help drive interdisciplinary and creative detection methods and to reveal and track dynamic changes in mitochondrial energy metabolism and the interactive networks of various substances. For instance, combining artificial intelligence, big data, organic chemistry, and biochemistry to design fluorescent probes for high-throughput screening and simulation calculations will be a promising approach. Scientists have developed an artificial intelligence robotic chemist capable of completing hundreds of experiments and synthesising new chemical catalysts in a short time.331 CRISPR (clustered regularly interspaced short palindromic repeats) can be coupled with other approaches to monitor or screen the different states of the metabolites in mitochondria. For instance, Mootha et al. used genome-wide CRISPR screens in the presence of different mitochondrial inhibitors to reveal a chemical-genetic map in the mitochondria.332 In addition, flexible electronics have also been developed in recent years. Flexible devices can be combined with a variety of detection methods, such as flexible electrochemical or fluorescent sensors, which are not only used in cells, tissues, and other samples for real-time monitoring but can also be used to monitor key metabolites of mitochondrial diseases.
(5) Multimodal diagnosis and combination treatment: diagnosis and treatment of diseases is a complex process. In many cases, it is difficult to achieve ideal diagnosis and therapeutic effects using a single method. Multimodal diagnosis can greatly improve accuracy while reducing misdiagnosis, and may provide more useful information for treatment. For example, Yang et al. used triple-modal fluorescence/magnetic resonance (MR)/photoacoustic tomography (PAT) for in vivo tumour imaging to reveal high passive tumour accumulation of their nanoprobe using an enhanced permeability and retention (EPR) effect. The nanoparticles, as an MRI probe, were used to guide photothermal therapy (PTT).333 Combined treatment provides additional avenues of exploration for the diagnosis and treatment of complex diseases; in addition, it also provides great benefits for improving therapeutic efficacy. For example, Wang et al. employed MnO2-laden black phosphorus nanosheets (BPNs) for nuclear MRI guided synergistic PDT, PTT, and chemotherapy. The MnO2-laden BPNs provided a more reliable tumour microenvironment-activated MRI, and simultaneously realised more efficient combined phototherapies than singlet organic or inorganic photosensitisers.334 The detection of mitochondrial energy metabolism using multimodal diagnosis can help to accurately identify the pathogenic mechanism of mitochondria-related diseases. Multimodal diagnosis and combination therapy will be the main means of clinical diagnosis and disease treatment (including mitochondrial diseases) in the future.
Mitochondrial energy metabolism will always play a crucial role in metabolism, particularly in mitochondrial diseases. Thus, designing and developing rapid, highly sensitive, dynamic, and specific analytical tools or methods for determining mitochondrial metabolites are urgently needed, not only to lay the foundation for the development, screening, and evaluation of drug-related mitochondrial diseases but also to deepen the understanding of the pathogenesis of mitochondrial diseases.
Author contributions
W. Ji, X. Tang, Q. Wu, and L. Li prepared the outline of this review. W. Ji wrote the Abstract, Introduction, Conclusions and outlooks, “Detection mechanisms of optical/electrochemical methods” section, and “Detection of mitochondrial energy metabolites” section about the application of electrochemical methods. X. Tang, and Dr W. Du wrote the “Detection of mitochondrial energy metabolites” section about the application of optical methods. Y. Lu, and N. Wang wrote the “Mitochondrial structure, function, and energy metabolism pathways” section. W. Ji, X. Tang, Dr W. Du, Y. Lu, and N. Wang prepared the graphic design and tables. Q. Wu, W. Wei, J. Liu, H. D. Yu, B. Ma, and L. Li revised and edited the draft. Q. Wu, L. Li, and W. Huang conceived and supervised this project. All authors participated in the discussion of the draft and carefully revised the draft before the final submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key R&D Program of China (2020YFA0709900), the National Natural Science Foundation of China (22077101, 21675085 and 21402219), the Joint Research Funds of Department of Science & Technology of Shaanxi Province and Northwestern Polytechnical University (2020GXLH-Z-008, 2020GXLH-Z-021, 2020GXLH-Z-023), Key Research and Development Program of Shaanxi (2020ZDLGY13-04), Open Research Fund of Anhui Key Laboratory of Tobacco Chemistry (20181140), China-Sweden Joint Mobility Project (51811530018) and Fundamental Research Funds for the Central Universities.Notes and references
- M.-E. Harper and M.-E. Patti, Nat. Metab., 2020, 2, 476–477 CrossRef PubMed.
- A. Cambiaghi, M. Ferrario and M. Masseroli, Briefings Bioinf., 2016, 18, 498–510 Search PubMed.
- E. F. Fang, Y. Hou, K. Palikaras, B. A. Adriaanse, J. S. Kerr, B. Yang, S. Lautrup, M. M. Hasan-Olive, D. Caponio, X. Dan, P. Rocktäschel, D. L. Croteau, M. Akbari, N. H. Greig, T. Fladby, H. Nilsen, M. Z. Cader, M. P. Mattson, N. Tavernarakis and V. A. Bohr, Nat. Neurosci., 2019, 22, 401–412 CrossRef CAS PubMed.
- L. Puspita, S. Y. Chung and J.-W. Shim, Mol. Brain, 2017, 10, 53 CrossRef PubMed.
- X. Guo, X. Sun, D. Hu, Y.-J. Wang, H. Fujioka, R. Vyas, S. Chakrapani, A. U. Joshi, Y. Luo, D. Mochly-Rosen and X. Qi, Nat. Commun., 2016, 7, 12646 CrossRef CAS PubMed.
- J. Lee, A. E. Yesilkanal, J. P. Wynne, C. Frankenberger, J. Liu, J. Yan, M. Elbaz, D. C. Rabe, F. D. Rustandy, P. Tiwari, E. A. Grossman, P. C. Hart, C. Kang, S. M. Sanderson, J. Andrade, D. K. Nomura, M. G. Bonini, J. W. Locasale and M. R. Rosner, Nature, 2019, 568, 254–258 CrossRef CAS PubMed.
- D. R. Green, L. Galluzzi and G. Kroemer, Science, 2011, 333, 1109–1112 CrossRef CAS PubMed.
- S. E. Weinberg and N. S. Chandel, Nat. Chem. Biol., 2015, 11, 9–15 CrossRef CAS PubMed.
- O. Teijido, Y. T. Ganesan, R. Llanos, A. Peton, J.-B. Urtecho, A. Soprani, A. Villamayor, B. Antonsson, S. Manon and L. Dejean, Anal. Biochem., 2016, 497, 90–94 CrossRef CAS PubMed.
- J. Wang, P. Zhu, R. Li, J. Ren, Y. Zhang and H. Zhou, Theranostics, 2020, 10, 384–397 CrossRef CAS PubMed.
- H. W. Rhee, P. Zou, N. D. Udeshi, J. D. Martell, V. K. Mootha, S. A. Carr and A. Y. Ting, Science, 2013, 339, 1328–1331 CrossRef CAS PubMed.
- M. G. Dibley, L. E. Formosa, B. Lyu, B. Reljic, D. McGann, L. Muellner-Wong, F. Kraus, A. J. Sharpe, D. A. Stroud and M. T. Ryan, Mol. Cell. Proteomics, 2020, 19, 65–77 CrossRef CAS.
- W.-T. Wu, H. Jiang, Y.-T. Qi, W.-T. Fan, J. Yan, Y.-L. Liu and W.-H. Huang, Angew. Chem., Int. Ed., 2021, 60, 19337–19343 CrossRef CAS PubMed.
- Y.-L. Ying, Y.-X. Hu, R. Gao, R.-J. Yu, Z. Gu, L. P. Lee and Y.-T. Long, J. Am. Chem. Soc., 2018, 140, 5385–5392 CrossRef CAS PubMed.
- Q. Zeng, Q. Guo, Y. Yuan, Y. Yang, B. Zhang, L. Ren, X. Zhang, Q. Luo, M. Liu, L.-S. Bouchard and X. Zhou, Anal. Chem., 2017, 89, 2288–2295 CrossRef CAS.
- O. S. Ahmad, T. S. Bedwell, C. Esen, A. Garcia-Cruz and S. A. Piletsky, Trends Biotechnol., 2019, 37, 294–309 CrossRef CAS PubMed.
- Z. Li, J. R. Askim and K. S. Suslick, Chem. Rev., 2019, 119, 231–292 CrossRef CAS PubMed.
- Y. Dai and C. C. Liu, Angew. Chem., Int. Ed., 2019, 58, 12355–12368 CrossRef CAS PubMed.
- S. S. Liew, X. Qin, J. Zhou, L. Li, W. Huang and S. Q. Yao, Angew. Chem., Int. Ed., 2021, 60, 2232–2256 CrossRef CAS.
- A. Mottis, S. Herzig and J. Auwerx, Science, 2019, 366, 827–832 CrossRef CAS PubMed.
- T. Bender, G. Pena and J. C. Martinou, EMBO J., 2015, 34, 911–924 CrossRef CAS PubMed.
- K. S. McCommis, A. Kovacs, C. J. Weinheimer, T. M. Shew, T. R. Koves, O. R. Ilkayeva, D. R. Kamm, K. D. Pyles, M. T. King, R. L. Veech, B. J. DeBosch, D. M. Muoio, R. W. Gross and B. N. Finck, Nat. Metab., 2020, 2, 1232–1247 CrossRef CAS PubMed.
- E. Quansah, W. Peelaerts, J. W. Langston, D. K. Simon, J. Colca and P. Brundin, Mol. Neurodegener., 2018, 13, 28 CrossRef PubMed.
- N. Lander, M. A. Chiurillo, M. S. Bertolini, M. Storey, A. E. Vercesi and R. Docampo, J. Biol. Chem., 2018, 293, 17402–17417 CrossRef CAS PubMed.
- S. M. Houten, S. Violante, F. V. Ventura and R. J. Wanders, Annu. Rev. Physiol., 2016, 78, 23–44 CrossRef CAS PubMed.
- M. Wajner and A. U. Amaral, Biosci. Rep., 2016, 36, 13 CrossRef PubMed.
- S. M. Nowinski, J. G. Van Vranken, K. K. Dove and J. Rutter, Curr. Biol., 2018, 28, R1212–R1219 CrossRef CAS PubMed.
- M. P. Murphy and R. C. Hartley, Nat. Rev. Drug Discovery, 2018, 17, 865–886 CrossRef CAS PubMed.
- C. T. Walsh, B. P. Tu and Y. Tang, Chem. Rev., 2018, 118, 1460–1494 CrossRef CAS.
- A. Garrido and N. Djouder, Trends Cancer, 2017, 3, 593–610 CrossRef CAS.
- M. V. Liberti and J. W. Locasale, Trends Biochem. Sci., 2016, 41, 211–218 CrossRef CAS PubMed.
- K. Yaku, K. Okabe and T. Nakagawa, Ageing Res. Rev., 2018, 47, 1–17 CrossRef CAS PubMed.
- E. Katsyuba, M. Romani, D. Hofer and J. Auwerx, Nat. Metab., 2020, 2, 9–31 CrossRef CAS PubMed.
- W. W. Hu, C. Zhang, R. Wu, Y. Sun, A. Levine and Z. H. Feng, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7455–7460 CrossRef CAS PubMed.
- V. J. N. Bykov, S. E. Eriksson, J. Bianchi and K. G. Wiman, Nat. Rev. Cancer, 2018, 18, 89–102 CrossRef CAS PubMed.
- J. E. Walker, Biochem. Soc. Trans., 2013, 41, 1–16 CrossRef CAS PubMed.
- S. S. Sabharwal and P. T. Schumacker, Nat. Rev. Cancer, 2014, 14, 709–721 CrossRef CAS PubMed.
- L. Li, C.-W. Zhang, J. Ge, L. Qian, B.-H. Chai, Q. Zhu, J.-S. Lee, K.-L. Lim and S. Q. Yao, Angew. Chem., Int. Ed., 2015, 54, 10821–10825 CrossRef CAS PubMed.
- J. R. Molina, Y. Sun, M. Protopopova, S. Gera, M. Bandi, C. Bristow, T. McAfoos, P. Morlacchi, J. Ackroyd, A. A. Agip, G. Al-Atrash, J. Asara, J. Bardenhagen, C. C. Carrillo, C. Carroll, E. Chang, S. Ciurea, J. B. Cross, B. Czako, A. Deem, N. Daver, J. F. de Groot, J. W. Dong, N. Feng, G. Gao, J. Gay, M. G. Do, J. Greer, V. Giuliani, J. Han, L. Han, V. K. Henry, J. Hirst, S. Huang, Y. Jiang, Z. Kang, T. Khor, S. Konoplev, Y. H. Lin, G. Liu, A. Lodi, T. Lofton, H. Ma, M. Mahendra, P. Matre, R. Mullinax, M. Peoples, A. Petrocchi, J. Rodriguez-Canale, R. Serreli, T. Shi, M. Smith, Y. Tabe, J. Theroff, S. Tiziani, Q. Xu, Q. Zhang, F. Muller, R. A. DePinho, C. Toniatti, G. F. Draetta, T. P. Heffernan, M. Konopleva, P. Jones, M. E. Di Francesco and J. R. Marszalek, Nat. Med., 2018, 24, 1036–1046 CrossRef CAS PubMed.
- X. Wang, P. An, Z. Gu, Y. Luo and J. Luo, Int. J. Mol. Sci., 2021, 22, 7525 CrossRef CAS PubMed.
- I. Szabo, M. Zoratti and L. Biasutto, Redox Biol., 2021, 42, 101846 CrossRef CAS PubMed.
- B. O’Rourke, S. Cortassa and M. A. Aon, Physiology, 2005, 20, 303–315 CrossRef.
- K. N. Belosludtsev, M. V. Dubinin, N. V. Belosludtseva and G. D. Mironova, Biochemistry, 2019, 84, 593–607 CAS.
- P. Bernardi and A. Rasola, Subcell. Biochem., 2007, 45, 481–506 CAS.
- S. Austin and K. Nowikovsky, Trends Biochem. Sci., 2019, 44, 648–658 CrossRef CAS PubMed.
- A. R. Cardoso, B. B. Queliconi and A. J. Kowaltowski, Biochim. Biophys. Acta, Bioenerg., 2010, 1797, 832–838 CrossRef CAS PubMed.
- I. Inoue, H. Nagase, K. Kishi and T. Higuti, Nature, 1991, 352, 244–247 CrossRef CAS PubMed.
- K. D. Garlid and P. Paucek, Biochim. Biophys. Acta, Bioenerg., 2003, 1606, 23–41 CrossRef CAS.
- P. Hernansanz-Agustín, C. Choya-Foces, S. Carregal-Romero, E. Ramos, T. Oliva, T. Villa-Piña, L. Moreno, A. Izquierdo-Álvarez, J. D. Cabrera-García, A. Cortés, A. V. Lechuga-Vieco, P. Jadiya, E. Navarro, E. Parada, A. Palomino-Antolín, D. Tello, R. Acín-Pérez, J. C. Rodríguez-Aguilera, P. Navas, Á. Cogolludo, I. López-Montero, Á. Martínez-Del-Pozo, J. Egea, M. G. López, J. W. Elrod, J. Ruíz-Cabello, A. Bogdanova, J. A. Enríquez and A. Martínez-Ruiz, Nature, 2020, 586, 287–291 CrossRef PubMed.
- S. F. Pedersen and L. Counillon, Physiol. Rev., 2019, 99, 2015–2113 CrossRef CAS PubMed.
- L. Boyman, G. S. Williams, D. Khananshvili, I. Sekler and W. J. Lederer, J. Mol. Cell. Cardiol., 2013, 59, 205–213 CrossRef CAS.
- L. Merolle, G. Sponder, A. Sargenti, L. Mastrototaro, C. Cappadone, G. Farruggia, A. Procopio, E. Malucelli, P. Parisse, A. Gianoncelli, J. R. Aschenbach, M. Kolisek and S. Iotti, Metallomics, 2018, 10, 917–928 CrossRef CAS.
- L. Mastrototaro, A. Smorodchenko, J. R. Aschenbach, M. Kolisek and G. Sponder, Sci. Rep., 2016, 6, 27999 CrossRef.
- G. C. Shaw, J. J. Cope, L. Li, K. Corson, C. Hersey, G. E. Ackermann, B. Gwynn, A. J. Lambert, R. A. Wingert, D. Traver, N. S. Trede, B. A. Barut, Y. Zhou, E. Minet, A. Donovan, A. Brownlie, R. Balzan, M. J. Weiss, L. L. Peters, J. Kaplan, L. I. Zon and B. H. Paw, Nature, 2006, 440, 96–100 CrossRef CAS.
- H. Kawabata, Free Radical Biol. Med., 2019, 133, 46–54 CrossRef CAS PubMed.
- L. Li, J. Ge, H. Wu, Q. H. Xu and S. Q. Yao, J. Am. Chem. Soc., 2012, 134, 12157–12167 CrossRef CAS.
- D. Chen, W. J. Qin, H. X. Fang, L. Wang, B. Peng, L. Li and W. Huang, Chin. Chem. Lett., 2019, 30, 1738–1744 CrossRef CAS.
- J. Zielonka, J. Joseph, A. Sikora, M. Hardy, O. Ouari, J. Vasquez-Vivar, G. Cheng, M. Lopez and B. Kalyanaraman, Chem. Rev., 2017, 117, 10043–10120 CrossRef CAS PubMed.
- L. Brulikova, S. Krupkova, M. Labora, K. Motyka, L. Hradilova, M. Mistrik, J. Bartek and J. Hlavac, RSC Adv., 2016, 6, 23242–23251 RSC.
- V. Reshetnikov, S. Daum, C. Janko, W. Karawacka, R. Tietze, C. Alexiou, S. Paryzhak, T. Dumych, R. Bilyy, P. Tripal, B. Schmid, R. Palmisano and A. Mokhir, Angew. Chem., Int. Ed., 2018, 57, 11943–11946 CrossRef CAS PubMed.
- R. R. Zhang, A. B. Schroeder, J. J. Grudzinski, E. L. Rosenthal, J. M. Warram, A. N. Pinchuk, K. W. Eliceiri, J. S. Kuo and J. P. Weichert, Nat. Rev. Clin. Oncol., 2017, 14, 347–364 CrossRef CAS PubMed.
- Y. Wang, Y. Zhang, J. Wang and X. J. Liang, Adv. Drug Delivery Rev., 2019, 143, 161–176 CrossRef CAS PubMed.
- C. Li, G. Chen, Y. Zhang, F. Wu and Q. Wang, J. Am. Chem. Soc., 2020, 142, 14789–14804 CrossRef CAS PubMed.
- Ł. Rodzik-Czałka, J. Lewandowska-Łańcucka, V. Gatta, I. Venditti, I. Fratoddi, M. Szuwarzyński, M. Romek and M. Nowakowska, J. Colloid Interface Sci., 2018, 514, 479–490 CrossRef PubMed.
- Y. Wang, L. Bao, Z. Liu and D. W. Pang, Anal. Chem., 2011, 83, 8130–8137 CrossRef CAS PubMed.
- W. R. Algar, N. Hildebrandt, S. S. Vogel and I. L. Medintz, Nat. Methods, 2019, 16, 815–829 CrossRef CAS PubMed.
- X. Zhang, Y. Hu, X. Yang, Y. Tang, S. Han, A. Kang, H. Deng, Y. Chi, D. Zhu and Y. Lu, Biosens. Bioelectron., 2019, 138, 111314 CrossRef CAS PubMed.
- W. Kim, S. H. Lee, Y. J. Ahn, S. H. Lee, J. Ryu, S. K. Choi and S. Choi, Biosens. Bioelectron., 2018, 111, 59–65 CrossRef CAS PubMed.
- H. Sun, S. Cong, Z. Zheng, Z. Wang, Z. Chen and Z. Zhao, J. Am. Chem. Soc., 2019, 141, 870–878 CrossRef CAS.
- L. A. Lane, X. Qian and S. Nie, Chem. Rev., 2015, 115, 10489–10529 CrossRef CAS PubMed.
- Z. Wang, S. Zong, L. Wu, D. Zhu and Y. Cui, Chem. Rev., 2017, 117, 7910–7963 CrossRef CAS PubMed.
- S. Zeng, D. Baillargeat, H. P. Ho and K. T. Yong, Chem. Soc. Rev., 2014, 43, 3426–3452 RSC.
- J. F. Masson, ACS Sens., 2017, 2, 16–30 CrossRef CAS PubMed.
- P. Wang, M. E. Nasir, A. V. Krasavin, W. Dickson, Y. Jiang and A. V. Zayats, Acc. Chem. Res., 2019, 52, 3018–3028 CrossRef CAS PubMed.
- A. V. Kabashin, P. Evans, S. Pastkovsky, W. Hendren, G. A. Wurtz, R. Atkinson, R. Pollard, V. A. Podolskiy and A. V. Zayats, Nat. Mater., 2009, 8, 867–871 CrossRef CAS PubMed.
- F. Vajhadin, S. Ahadian, J. Travas-Sejdic, J. Lee, M. Mazloum-Ardakani, J. Salvador, G. E. Aninwene, P. Bandaru, W. Sun and A. Khademhossieni, Biosens. Bioelectron., 2020, 151, 111984 CrossRef CAS PubMed.
- Y. Dai and C. C. Liu, Angew. Chem., Int. Ed., 2019, 58, 12355–12368 CrossRef CAS PubMed.
- J. Wang, Chem. Rev., 2008, 108, 814–825 CrossRef CAS PubMed.
- M. Bariya, H. Y. Y. Nyein and A. Javey, Nat. Electron., 2018, 1, 160–171 CrossRef.
- W. Gao, H. Y. Y. Nyein, Z. Shahpar, H. M. Fahad, K. Chen, S. Emaminejad, Y. Gao, L.-C. Tai, H. Ota, E. Wu, J. Bullock, Y. Zeng, D.-H. Lien and A. Javey, ACS Sens., 2016, 1, 866–874 CrossRef CAS.
- S. Emaminejad, W. Gao, E. Wu, Z. A. Davies, H. Yin Yin Nyein, S. Challa, S. P. Ryan, H. M. Fahad, K. Chen, Z. Shahpar, S. Talebi, C. Milla, A. Javey and R. W. Davis, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 4625–4630 CrossRef CAS PubMed.
- X. Liu, P. A. Duckworth and D. K. Wong, Biosens. Bioelectron., 2010, 25, 1467–1473 CrossRef CAS.
- Y. Xu, X. Xie, Y. Duan, L. Wang, Z. Cheng and J. Cheng, Biosens. Bioelectron., 2016, 77, 824–836 CrossRef CAS PubMed.
- X. Luo and J. J. Davis, Chem. Soc. Rev., 2013, 42, 5944–5962 RSC.
- M. Magro, D. Baratella, N. Pianca, A. Toninello, S. Grancara, R. Zboril and F. Vianello, Sens. Actuators, B, 2013, 176, 315–322 CrossRef CAS.
- M. R. Karimi Pur, M. Hosseini, F. Faridbod, M. R. Ganjali and S. Hosseinkhani, Sens. Actuators, B, 2018, 257, 87–95 CrossRef CAS.
- H. Jiang, X. W. Zhang, Q. L. Liao, W. T. Wu, Y. L. Liu and W. H. Huang, Small, 2019, 15, 1901787 CrossRef CAS.
- S. Ben-Amor, E. Vanhove, F. S. Belaidi, S. Charlot, D. Colin, M. Rigoulet, A. Devin, N. Sojic, J. Launay, P. Temple-Boyer and S. Arbault, Electrochim. Acta, 2014, 126, 171–178 CrossRef CAS.
- J. Shu and D. Tang, Anal. Chem., 2020, 92, 363–377 CrossRef CAS PubMed.
- R. Y. Wang, L. Wang, Y. Zhou and Z. G. Zou, Appl. Catal., B, 2019, 255, 117738 CrossRef CAS.
- M. J. Li, Y. N. Zheng, W. B. Liang, Y. L. Yuan, Y. Q. Chai and R. Yuan, Chem. Commun., 2016, 52, 8138–8141 RSC.
- Y. X. Chu, A. P. Deng, W. J. Wang and J. J. Zhu, Anal. Chem., 2019, 91, 3619–3627 CrossRef CAS PubMed.
- H. Wang, M. Li, Y. Zheng, T. Hu, Y. Chai and R. Yuan, Biosens. Bioelectron., 2018, 120, 71–76 CrossRef CAS PubMed.
- L. Y. Xia, Y. N. Zheng, W. B. Liang, M. J. Li, T. Hu, R. Yuan and Y. Q. Chai, Chem. – Eur. J., 2019, 25, 4087–4092 CrossRef CAS PubMed.
- L. Meng, M. Liu, K. Xiao, X. Zhang, C. Du and J. Chen, Chem. Commun., 2020, 56, 8261–8264 RSC.
- R. Atchudan, N. Muthuchamy, T. Edison, S. Perumal, R. Vinodh, K. H. Park and Y. R. Lee, Biosens. Bioelectron., 2019, 126, 160–169 CrossRef CAS PubMed.
- S. Z. Lv, K. Y. Zhang, L. Zhu and D. P. Tang, Anal. Chem., 2020, 92, 1470–1476 CrossRef CAS PubMed.
- P. Gao, W. Pan, N. Li and B. Tang, Chem. Sci., 2019, 10, 6035–6071 RSC.
- M. H. R. Ludtmann, P. R. Angelova, M. H. Horrocks, M. L. Choi, M. Rodrigues, A. Y. Baev, A. V. Berezhnov, Z. Yao, D. Little, B. Banushi, A. S. Al-Menhali, R. T. Ranasinghe, D. R. Whiten, R. Yapom, K. S. Dolt, M. J. Devine, P. Gissen, T. Kunath, M. Jaganjac, E. V. Pavlov, D. Klenerman, A. Y. Abramov and S. Gandhi, Nat. Commun., 2018, 9, 16 CrossRef PubMed.
- L. Kashefi-Kheyrabadi and M. A. Mehrgardi, Biosens. Bioelectron., 2012, 37, 94–98 CrossRef CAS PubMed.
- T. Bao, H. Shu, W. Wen, X. Zhang and S. Wang, Anal. Chim. Acta, 2015, 862, 64–69 CrossRef CAS PubMed.
- H. Xie, Y. Q. Chai, Y. L. Yuan and R. Yuan, Chem. Commun., 2017, 53, 8368–8371 RSC.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Ferey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS.
- T. Simon-Yarza, A. Mielcarek, P. Couvreur and C. Serre, Adv. Mater., 2018, 30, 1707365 CrossRef.
- M. T. Zhao, Y. Huang, Y. W. Peng, Z. Q. Huang, Q. L. Ma and H. Zhang, Chem. Soc. Rev., 2018, 47, 6267–6295 RSC.
- J. Deng, K. Wang, M. Wang, P. Yu and L. Mao, J. Am. Chem. Soc., 2017, 139, 5877–5882 CrossRef CAS PubMed.
- J. Chang, W. Lv, Q. Li, H. Li and F. Li, Anal. Chem., 2020, 92, 8959–8964 CrossRef CAS PubMed.
- J. Zhao, J. Gao, W. Xue, Z. Di, H. Xing, Y. Lu and L. Li, J. Am. Chem. Soc., 2018, 140, 578–581 CrossRef CAS PubMed.
- X. Zhang, C. Song, K. Yang, W. Hong, Y. Lu, P. Yu and L. Mao, Anal. Chem., 2018, 90, 4968–4971 CrossRef CAS PubMed.
- S. Hong, X. Zhang, R. J. Lake, G. T. Pawel, Z. Guo, R. Pei and Y. Lu, Chem. Sci., 2020, 11, 713–720 RSC.
- Z. Xu, N. J. Singh, J. Lim, J. Pan, H. N. Kim, S. Park, K. S. Kim and J. Yoon, J. Am. Chem. Soc., 2009, 131, 15528–15533 CrossRef CAS PubMed.
- X. Li, X. Guo, L. Cao, Z. Xun, S. Wang, S. Li, Y. Li and G. Yang, Angew. Chem., Int. Ed., 2014, 53, 7809–7813 CrossRef CAS PubMed.
- Y. Kurishita, T. Kohira, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2012, 134, 18779–18789 CrossRef CAS PubMed.
- P. Srivastava, S. S. Razi, R. Ali, S. Srivastav, S. Patnaik, S. Srikrishna and A. Misra, Biosens. Bioelectron., 2015, 69, 179–185 CrossRef CAS PubMed.
- L. Wang, L. Yuan, X. Zeng, J. J. Peng, Y. Ni, J. C. Er, W. Xu, B. K. Agrawalla, D. D. Su, B. Kim and Y. T. Chang, Angew. Chem., Int. Ed., 2016, 55, 1773–1776 CrossRef CAS PubMed.
- K.-Y. Tan, C.-Y. Li, Y.-F. Li, J. Fei, B. Yang, Y.-J. Fu and F. Li, Anal. Chem., 2017, 89, 1749–1756 CrossRef CAS PubMed.
- Z. Wu, M. Liu, Z. Liu and Y. Tian, J. Am. Chem. Soc., 2020, 142, 7532–7541 CrossRef CAS PubMed.
- C. S. Pundir, V. Narwal and B. Batra, Biosens. Bioelectron., 2016, 86, 777–790 CrossRef CAS PubMed.
- I. S. Kucherenko, Y. V. Topolnikova and O. O. Soldatkin, Trac, Trends Anal. Chem., 2019, 110, 160–172 CrossRef CAS.
- N. Gajovic, G. Binyamin, A. Warsinke, F. W. Scheller and A. Heller, Anal. Chem., 2000, 72, 2963–2968 CrossRef CAS PubMed.
- M. Malik, R. Chaudhary and C. S. Pundir, Enzyme Microb. Technol., 2019, 123, 30–38 CrossRef CAS PubMed.
- D. Sun, F. Cao, Y. Tian, A. Li, W. Xu, Q. Chen, W. Shi and S. Xu, Anal. Chem., 2019, 91, 15484–15490 CrossRef CAS PubMed.
- A. Galaz, F. Cortes-Molina, R. Arce-Molina, I. Romero-Gomez, G. Antonio Mardones, L. F. Barros and A. San Martin, Anal. Chem., 2020, 92, 10643–10650 CrossRef CAS PubMed.
- P. K. Mishra and D. G. Drueckhammer, Chem. Rev., 2000, 100, 3283–3310 CrossRef CAS PubMed.
- M. Can, F. A. Armstrong and S. W. Ragsdale, Chem. Rev., 2014, 114, 4149–4174 CrossRef CAS PubMed.
- F. Pietrocola, L. Galluzzi, J. M. Bravo-San Pedro, F. Madeo and G. Kroemer, Cell Metab., 2015, 21, 805–821 CrossRef CAS PubMed.
- D. Hu, T. Zhan, Z. Guo, S. Wang and Y. Hu, Sens. Actuators, B, 2021, 327, 128896 CrossRef CAS.
- Y. Hu, S. Chen, Y. Han, H. Chen, Q. Wang, Z. Nie, Y. Huang and S. Yao, Chem. Commun., 2015, 51, 17611–17614 RSC.
- H. Chen, X. Liu, W. Li, Y. Peng and Z. Nie, Biosens. Bioelectron., 2019, 126, 535–542 CrossRef CAS PubMed.
- Y. Shindo, H. Komatsu, K. Hotta, K. Ariga and K. Oka, Sci. Rep., 2016, 6, 29224 CrossRef CAS PubMed.
- H. Komatsu, Y. Shindo, S. A. Kawashima, K. Yamatsugu, K. Oka and M. Kanai, Chem. Commun., 2013, 49, 2876–2878 RSC.
- F. Xu, H. Tang, J. Yu and J. Ge, Talanta, 2021, 224, 121838 CrossRef CAS PubMed.
- W. Xu, H. Yang, Y. Liu, Y. Yang, P. Wang, S. H. Kim, S. Ito, C. Yang, P. Wang, M. T. Xiao, L. X. Liu, W. Q. Jiang, J. Liu, J. Y. Zhang, B. Wang, S. Frye, Y. Zhang, Y. H. Xu, Q. Y. Lei, K. L. Guan, S. M. Zhao and Y. Xiong, Cancer Cell, 2011, 19, 17–30 CrossRef CAS PubMed.
- A. Makahleh, G. M. Ben-Hander and B. Saad, Bioanalysis, 2015, 7, 713–723 CrossRef CAS PubMed.
- P. Jin, C. Jiao, Z. Guo, Y. He, S. Zhu, H. Tian and W. Zhu, Chem. Sci., 2014, 5, 4012–4016 RSC.
- L.-L. Gan, L.-H. Chen and F.-J. Nan, Acta Pharmacol. Sin., 2017, 38, 1683–1690 CrossRef CAS PubMed.
- Q. Fu, Z. Chen, X. Ma, G. Chen, Y. Liu, Z. Cao, F. Qu, D. Yang, X. Zhao, Z. Sun, G. Li, S. Zhang, F. Qu, R. Kong, H. Wang and J. You, Sens. Actuators, B, 2017, 241, 1035–1042 CrossRef CAS.
- A. Das, M. Alam, C. Gogoi, R. Dalapati and S. Biswas, Dalton Trans., 2020, 49, 16928–16934 RSC.
- A. Maczurek, K. Hager, M. Kenklies, M. Sharman, R. Martins, J. Engel, D. A. Carlson and G. Münch, Adv. Drug Delivery Rev., 2008, 60, 1463–1470 CrossRef CAS.
- K. Charoenkitamorn, S. Chaiyo, O. Chailapakul and W. Siangproh, Anal. Chim. Acta, 2018, 1004, 22–31 CrossRef CAS PubMed.
- A. R. Cameron, L. Logie, K. Patel, S. Erhardt, S. Bacon, P. Middleton, J. Harthill, C. Forteath, J. T. Coats, C. Kerr, H. Curry, D. Stewart, K. Sakamoto, P. Repiscak, M. J. Paterson, I. Hassinen, G. McDougall and G. Rena, Redox Biol., 2018, 14, 187–197 CrossRef CAS PubMed.
- A. J. Covarrubias, R. Perrone, A. Grozio and E. Verdin, Nat. Rev. Mol. Cell Biol., 2021, 22, 119–141 CrossRef CAS PubMed.
- Y. G. Shen, D. Kapfhamer, A. M. Minnella, J. E. Kim, S. J. Won, Y. T. Chen, Y. Huang, L. H. Low, S. M. Massa and R. A. Swanson, Nat. Commun., 2017, 8, 13 CrossRef.
- D. V. Titov, V. Cracan, R. P. Goodman, J. Peng, Z. Grabarek and V. K. Mootha, Science, 2016, 352, 231–235 CrossRef CAS PubMed.
- P. Brunnbauer, A. Leder, C. Kamali, K. Kamali, E. Keshi, K. Splith, S. Wabitsch, P. Haber, G. Atanasov, L. Feldbrügge, I. M. Sauer, J. Pratschke, M. Schmelzle and F. Krenzien, Sci. Rep., 2018, 8, 16110 CrossRef PubMed.
- V. B. Ritov, E. V. Menshikova and D. E. Kelley, Anal. Biochem., 2004, 333, 27–38 CrossRef CAS PubMed.
- W. Xie, A. Xu and E. S. Yeung, Anal. Chem., 2009, 81, 1280–1284 CrossRef CAS PubMed.
- A. S. Veskoukis, N. V. Margaritelis, A. Kyparos, V. Paschalis and M. G. Nikolaidis, Redox Rep., 2018, 23, 47–56 CrossRef CAS PubMed.
- Y. Liu, J. Clement, R. Grant, P. Sachdev and N. Braidy, Biochim. Biophys. Acta, Gen. Subj., 2018, 1862, 2527–2532 CrossRef CAS.
- S. Han, T. Du, H. Jiang and X. Wang, Biosens. Bioelectron., 2017, 89, 422–429 CrossRef CAS.
- Y. Wang, L. Yin, X. Li, R. Shang, X. Yang, X. Zhou and Y. Chen, Micro Nano Lett., 2020, 15, 997–1002 CrossRef CAS.
- M. Thiruppathi, P. Y. Lin, Y. T. Chou, H. Y. Ho, L. C. Wu and J. A. Ho, Talanta, 2019, 200, 450–457 CrossRef CAS PubMed.
- J. Liu, Y. Wang, X. Liu, Q. Yuan, Y. Zhang and Y. Li, Talanta, 2019, 199, 573–580 CrossRef CAS PubMed.
- H. Jiang, Y.-T. Qi, W.-T. Wu, M.-Y. Wen, Y.-L. Liu and W.-H. Huang, Chem. Sci., 2020, 11, 8771–8778 RSC.
- Y. Zhao, K. Wei, F. Kong, X. Gao, K. Xu and B. Tang, Anal. Chem., 2019, 91, 1368–1374 CrossRef CAS PubMed.
- A. Podder, N. Thirumalaivasan, Y. K. Chao, P. Kukutla, S.-P. Wu and S. Bhuniya, Sens. Actuators, B, 2020, 324, 128637 CrossRef CAS.
- M. Kikawada, A. Ono, W. Inami and Y. Kawata, Anal. Chem., 2016, 88, 1407–1411 CrossRef CAS.
- D. Nolfi-Donegan, A. Braganza and S. Shiva, Redox Biol., 2020, 37, 101674 CrossRef CAS.
- C. Blajszczak and M. G. Bonini, Toxicology, 2017, 391, 84–89 CrossRef CAS PubMed.
- D. B. Zorov, M. Juhaszova and S. J. Sollott, Physiol. Rev., 2014, 94, 909–950 CrossRef CAS PubMed.
- P. R. Angelova and A. Y. Abramov, FEBS Lett., 2018, 592, 692–702 CrossRef CAS PubMed.
- P. H. Willems, R. Rossignol, C. E. Dieteren, M. P. Murphy and W. J. Koopman, Cell Metab., 2015, 22, 207–218 CrossRef CAS PubMed.
- A. L. Harris, Nat. Rev. Cancer, 2002, 2, 38–47 CrossRef CAS PubMed.
- P. Lee, N. S. Chandel and M. C. Simon, Nat. Rev. Mol. Cell Biol., 2020, 21, 268–283 CrossRef CAS PubMed.
- A. A. Gerencser, A. Neilson, S. W. Choi, U. Edman, N. Yadava, R. J. Oh, D. A. Ferrick, D. G. Nicholls and M. D. Brand, Anal. Chem., 2009, 81, 6868–6878 CrossRef CAS PubMed.
- Y. Lian, Z. Lin, Z. Zhang and X.-D. Wang, Anal. Chem., 2021, 93, 8291–8299 CrossRef CAS PubMed.
- X.-d. Wang and O. S. Wolfbeis, Chem. Soc. Rev., 2014, 43, 3666–3761 RSC.
- A.-J. Hsueh, S. Park, T. Satoh, T. Shimizu, K. Koiwai, M. Nakashima, Y. Morimoto, M. Kinoshita and H. Suzuki, Anal. Chem., 2021, 93, 5577–5585 CrossRef CAS PubMed.
- F. Liebisch, A. Weltin, J. Marzioch, G. A. Urban and J. Kieninger, Sens. Actuators, B, 2020, 322, 128652 CrossRef CAS.
- J.-H. Han, S. Kim, J. Choi, S. Kang, Y. K. Pak and J. J. Pak, Sens. Actuators, B, 2020, 306, 127465 CrossRef CAS.
- Y. Shen, L. Liang, J. Zhang, Z. Li, J. Yue, J. Wang, W. Xu, W. Shi and S. Xu, Sens. Actuators, B, 2019, 285, 84–91 CrossRef CAS.
- C. Calas-Blanchard, G. Catanante and T. Noguer, Electroanalysis, 2014, 26, 1277–1286 CrossRef CAS.
- J. Song, C. H. Xu, S. Z. Huang, W. Lei, Y. F. Ruan, H. J. Lu, W. Zhao, J. J. Xu and H. Y. Chen, Angew. Chem., Int. Ed., 2018, 57, 13226–13230 CrossRef CAS PubMed.
- N. Bézière, Y. Frapart, A. Rockenbauer, J. L. Boucher, D. Mansuy and F. Peyrot, Free Radical Biol. Med., 2010, 49, 437–446 CrossRef PubMed.
- X. Zhu, T. Liu, H. Zhao, L. Shi, X. Li and M. Lan, Biosens. Bioelectron., 2016, 79, 449–456 CrossRef CAS PubMed.
- X. Shen, Q. Wang, Y. Liu, W. Xue, L. Ma, S. Feng, M. Wan, F. Wang and C. Mao, Sci. Rep., 2016, 6, 28989 CrossRef CAS PubMed.
- H. Zhang, X. Cai, H. Zhao, W. Sun, Z. Wang and M. Lan, J. Electroanal. Chem., 2019, 855, 113653 CrossRef CAS.
- Q. Qiu, H. Chen, Z. You, Y. Feng, X. Wang, Y. Wang and Y. Ying, ACS Appl. Mater. Interfaces, 2020, 12, 5429–5436 CrossRef CAS PubMed.
- X. Lu, Z. Chen, X. Dong and W. Zhao, ACS Sens., 2018, 3, 59–64 CrossRef CAS PubMed.
- W. Zhang, P. Li, F. Yang, X. Hu, C. Sun, W. Zhang, D. Chen and B. Tang, J. Am. Chem. Soc., 2013, 135, 14956–14959 CrossRef CAS PubMed.
- X. Han, R. Wang, X. Song, F. Yu, C. Lv and L. Chen, Biomaterials, 2018, 156, 134–146 CrossRef CAS PubMed.
- J. J. Hu, N. K. Wong, S. Ye, X. Chen, M. Y. Lu, A. Q. Zhao, Y. Guo, A. C. Ma, A. Y. Leung, J. Shen and D. Yang, J. Am. Chem. Soc., 2015, 137, 6837–6843 CrossRef CAS PubMed.
- S. G. Rhee, S. W. Kang, W. Jeong, T.-S. Chang, K.-S. Yang and H. A. Woo, Curr. Opin. Cell Biol., 2005, 17, 183–189 CrossRef CAS PubMed.
- T. Zhang, Y. Xing, Y. Song, Y. Gu, X. Yan, N. Lu, H. Liu, Z. Xu, H. Xu, Z. Zhang and M. Yang, Anal. Chem., 2019, 91, 10589–10595 CrossRef CAS PubMed.
- E. Suraniti, S. Ben-Amor, P. Landry, M. Rigoulet, E. Fontaine, S. Bottari, A. Devin, N. Sojic, N. Mano and S. Arbault, Angew. Chem., Int. Ed., 2014, 53, 6655–6658 CrossRef CAS PubMed.
- G. Bai, X. Xu, Q. Dai, Q. Zheng, Y. Yao, S. Liu and C. Yao, Analyst, 2019, 144, 481–487 RSC.
- T. Deepalakshmi, D. T. Tran, N. H. Kim, K. T. Chong and J. H. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 35847–35858 CrossRef CAS PubMed.
- J. Wu, X. Wang, Q. Wang, Z. Lou, S. Li, Y. Zhu, L. Qin and H. Wei, Chem. Soc. Rev., 2019, 48, 1004–1076 RSC.
- Y. Sun, M. Luo, X. Meng, J. Xiang, L. Wang, Q. Ren and S. Guo, Anal. Chem., 2017, 89, 3761–3767 CrossRef CAS PubMed.
- T. Zhang, Y. Gu, C. Li, X. Yan, N. Lu, H. Liu, Z. Zhang and H. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 37991–37999 CrossRef CAS PubMed.
- J. Lu, Y. Hu, P. Wang, P. Liu, Z. Chen and D. Sun, Sens. Actuators, B, 2020, 311, 127909 CrossRef CAS.
- B. C. Dickinson and C. J. Chang, J. Am. Chem. Soc., 2008, 130, 9638–9639 CrossRef CAS.
- Y. Liu, L. Bai, Y. Li, Y. Ni, C. Xin, C. Zhang, J. Liu, Z. Liu, L. Li and W. Huang, Sens. Actuators, B, 2019, 279, 38–43 CrossRef CAS.
- X. Xie, X. Yang, T. Wu, Y. Li, M. Li, Q. Tan, X. Wang and B. Tang, Anal. Chem., 2016, 88, 8019–8025 CrossRef CAS.
- S. I. Reja, M. Gupta, N. Gupta, V. Bhalla, P. Ohri, G. Kaur and M. Kumar, Chem. Commun., 2017, 53, 3701–3704 RSC.
- D. Pham, U. Basu, I. Pohorilets, C. M. St Croix, S. C. Watkins and K. Koide, Angew. Chem., Int. Ed., 2020, 59, 17435–17441 CrossRef CAS PubMed.
- G. Yang, Z. Liu, R. Zhang, X. Tian, J. Chen, G. Han, B. Liu, X. Han, Y. Fu, Z. Hu and Z. Zhang, Angew. Chem., Int. Ed., 2020, 59, 16154–16160 CrossRef CAS PubMed.
- X. Chen, X. Ren, L. Zhang, Z. Liu and Z. Hai, Anal. Chem., 2020, 92, 14244–14250 CrossRef CAS PubMed.
- Q. Xu, C. H. Heo, J. A. Kim, H. S. Lee, Y. Hu, D. Kim, K. M. Swamy, G. Kim, S. J. Nam, H. M. Kim and J. Yoon, Anal. Chem., 2016, 88, 6615–6620 CrossRef CAS PubMed.
- G. Cheng, J. Fan, W. Sun, K. Sui, X. Jin, J. Wang and X. Peng, Analyst, 2013, 138, 6091–6096 RSC.
- R. Zhang, J. Zhao, G. Han, Z. Liu, C. Liu, C. Zhang, B. Liu, C. Jiang, R. Liu, T. Zhao, M. Y. Han and Z. Zhang, J. Am. Chem. Soc., 2016, 138, 3769–3778 CrossRef CAS PubMed.
- B. Zhu, L. Wu, M. Zhang, Y. Wang, C. Liu, Z. Wang, Q. Duan and P. Jia, Biosens. Bioelectron., 2018, 107, 218–223 CrossRef CAS PubMed.
- Y. Koide, Y. Urano, K. Hanaoka, T. Terai and T. Nagano, J. Am. Chem. Soc., 2011, 133, 5680–5682 CrossRef CAS PubMed.
- X. Chen, X. Wang, S. Wang, W. Shi, K. Wang and H. Ma, Chem. – Eur. J., 2008, 14, 4719–4724 CrossRef CAS PubMed.
- L. Yuan, L. Wang, B. K. Agrawalla, S. J. Park, H. Zhu, B. Sivaraman, J. Peng, Q. H. Xu and Y. T. Chang, J. Am. Chem. Soc., 2015, 137, 5930–5938 CrossRef CAS PubMed.
- N. Zhu, X. Guo, S. Pang, Y. Chang, X. Liu, Z. Shi and S. Feng, Anal. Chem., 2020, 92, 11103–11110 CrossRef CAS PubMed.
- B. Tang, L. Zhang and Y. Geng, Talanta, 2005, 65, 769–775 CrossRef CAS PubMed.
- L. Yuan, W. Lin and J. Song, Chem. Commun., 2010, 46, 7930–7932 RSC.
- F. Liu, J. Du, D. Song, M. Xu and G. Sun, Chem. Commun., 2016, 52, 4636–4639 RSC.
- P. Li, T. Xie, X. Duan, F. Yu, X. Wang and B. Tang, Chem. – Eur. J., 2010, 16, 1834–1840 CrossRef CAS PubMed.
- T. Deng, X. Wang, S. Wu, S. Hu, W. Liu, T. Chen, Z. Yu, Q. Xu and F. Liu, Chem. Commun., 2020, 56, 4432–4435 RSC.
- S. Ding, M. Li, H. Gong, Q. Zhu, G. Shi and A. Zhu, Anal. Chem., 2020, 92, 2543–2549 CrossRef CAS PubMed.
- N. M. Idris, M. K. Gnanasammandhan, J. Zhang, P. C. Ho, R. Mahendran and Y. Zhang, Nat. Med., 2012, 18, 1580–1585 CrossRef CAS PubMed.
- S. Kim, T. Tachikawa, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2014, 136, 11707–11715 CrossRef CAS PubMed.
- H. W. Liu, S. Xu, P. Wang, X. X. Hu, J. Zhang, L. Yuan, X. B. Zhang and W. Tan, Chem. Commun., 2016, 52, 12330–12333 RSC.
- D. Song, S. Cho, Y. Han, Y. You and W. Nam, Org. Lett., 2013, 15, 3582–3585 CrossRef CAS PubMed.
- K. Xu, L. Wang, M. Qiang, L. Wang, P. Li and B. Tang, Chem. Commun., 2011, 47, 7386–7388 RSC.
- K. Xu, L. Wang, M. Qiang, L. Wang, P. Li and B. Tang, Chem. Commun., 2011, 47, 7386–7388 RSC.
- B. Song, G. Wang, M. Tan and J. Yuan, J. Am. Chem. Soc., 2006, 128, 13442–13450 CrossRef CAS PubMed.
- R. Flores-Cruz and A. Jiménez-Sánchez, Chem. Commun., 2018, 54, 13997–14000 RSC.
- X. Chen, F. Wang, J. Y. Hyun, T. Wei, J. Qiang, X. Ren, I. Shin and J. Yoon, Chem. Soc. Rev., 2016, 45, 2976–3016 RSC.
- B. B. Mishra, V. A. Rathinam, G. W. Martens, A. J. Martinot, H. Kornfeld, K. A. Fitzgerald and C. M. Sassetti, Nat. Immunol., 2013, 14, 52–60 CrossRef CAS PubMed.
- E. Pashai, G. Najafpour Darzi, M. Jahanshahi, F. Yazdian and M. Rahimnejad, Int. J. Biol. Macromol., 2018, 108, 250–258 CrossRef CAS PubMed.
- A. A. Abdelwahab, W. C. Koh, H. B. Noh and Y. B. Shim, Biosens. Bioelectron., 2010, 26, 1080–1086 CrossRef CAS PubMed.
- A. Meiller, E. Sequeira and S. Marinesco, Anal. Chem., 2020, 92, 1804–1810 CrossRef CAS PubMed.
- Y. Q. Sun, J. Liu, H. Zhang, Y. Huo, X. Lv, Y. Shi and W. Guo, J. Am. Chem. Soc., 2014, 136, 12520–12523 CrossRef CAS PubMed.
- X. Lv, Y. Wang, S. Zhang, Y. Liu, J. Zhang and W. Guo, Chem. Commun., 2014, 50, 7499–7502 RSC.
- J. Miao, Y. Huo, X. Lv, Z. Li, H. Cao, H. Shi, Y. Shi and W. Guo, Biomaterials, 2016, 78, 11–19 CrossRef CAS PubMed.
- C. Xu, C. Xin, C. Yu, M. Wu, J. Xu, W. Qin, Y. Ding, X. Wang, L. Li and W. Huang, Chem. Commun., 2018, 54, 13491–13494 RSC.
- G. Ferrer-Sueta and R. Radi, ACS Chem. Biol., 2009, 4, 161–177 CrossRef CAS PubMed.
- X. Jia, Q. Chen, Y. Yang, Y. Tang, R. Wang, Y. Xu, W. Zhu and X. Qian, J. Am. Chem. Soc., 2016, 138, 10778–10781 CrossRef CAS PubMed.
- H. Li, X. Li, X. Wu, W. Shi and H. Ma, Anal. Chem., 2017, 89, 5519–5525 CrossRef CAS PubMed.
- D. Cheng, W. Xu, L. Yuan and X. Zhang, Anal. Chem., 2017, 89, 7693–7700 CrossRef CAS PubMed.
- X. Xie, F. Tang, G. Liu, Y. Li, X. Su, X. Jiao, X. Wang and B. Tang, Anal. Chem., 2018, 90, 11629–11635 CrossRef CAS PubMed.
- D. Cheng, Y. Pan, L. Wang, Z. Zeng, L. Yuan, X. Zhang and Y. T. Chang, J. Am. Chem. Soc., 2017, 139, 285–292 CrossRef CAS PubMed.
- M. Liu, W. Zhai, H. Chen, H. Zhang and C. Li, Anal. Chem., 2020, 92, 10792–10799 CrossRef CAS PubMed.
- J. Rosenthal and S. J. Lippard, J. Am. Chem. Soc., 2010, 132, 5536–5537 CrossRef CAS PubMed.
- A. T. Wrobel, T. C. Johnstone, A. Deliz Liang, S. J. Lippard and P. Rivera-Fuentes, J. Am. Chem. Soc., 2014, 136, 4697–4705 CrossRef CAS PubMed.
- K. Sunwoo, K. N. Bobba, J. Y. Lim, T. Park, A. Podder, J. S. Heo, S. G. Lee, S. Bhuniya and J. S. Kim, Chem. Commun., 2017, 53, 1723–1726 RSC.
- N. Lau and M. D. Pluth, Curr. Opin. Chem. Biol., 2019, 49, 1–8 CrossRef CAS PubMed.
- K. R. Olson, Y. Gao, F. Arif, K. Arora, S. Patel, E. R. DeLeon, T. R. Sutton, M. Feelisch, M. M. Cortese-Krott and K. D. Straub, Redox Biol., 2018, 15, 74–85 CrossRef CAS PubMed.
- S. J. Li, Y. F. Li, H. W. Liu, D. Y. Zhou, W. L. Jiang, J. Ou-Yang and C. Y. Li, Anal. Chem., 2018, 90, 9418–9425 CrossRef CAS PubMed.
- S. K. Bae, C. H. Heo, D. J. Choi, D. Sen, E. H. Joe, B. R. Cho and H. M. Kim, J. Am. Chem. Soc., 2013, 135, 9915–9923 CrossRef CAS PubMed.
- H. A. Henthorn and M. D. Pluth, J. Am. Chem. Soc., 2015, 137, 15330–15336 CrossRef CAS PubMed.
- Y. L. Pak, J. Li, K. C. Ko, G. Kim, J. Y. Lee and J. Yoon, Anal. Chem., 2016, 88, 5476–5481 CrossRef CAS PubMed.
- X. Cao, W. Lin, K. Zheng and L. He, Chem. Commun., 2012, 48, 10529–10531 RSC.
- K. Sasakura, K. Hanaoka, N. Shibuya, Y. Mikami, Y. Kimura, T. Komatsu, T. Ueno, T. Terai, H. Kimura and T. Nagano, J. Am. Chem. Soc., 2011, 133, 18003–18005 CrossRef CAS PubMed.
- J. Liu, X. Guo, R. Hu, X. Liu, S. Wang, S. Li, Y. Li and G. Yang, Anal. Chem., 2016, 88, 1052–1057 CrossRef CAS PubMed.
- Y. Chen, C. Zhu, Z. Yang, J. Chen, Y. He, Y. Jiao, W. He, L. Qiu, J. Cen and Z. Guo, Angew. Chem., Int. Ed., 2013, 52, 1688–1691 CrossRef CAS PubMed.
- Y. Qian, J. Karpus, O. Kabil, S. Y. Zhang, H. L. Zhu, R. Banerjee, J. Zhao and C. He, Nat. Commun., 2011, 2, 495 CrossRef PubMed.
- G. Chen, W. Zhou, C. Zhao, Y. Liu, T. Chen, Y. Li and B. Tang, Anal. Chem., 2018, 90, 12442–12448 CrossRef CAS PubMed.
- W. Hu, L. Zeng, S. Zhai, C. Li, W. Feng, Y. Feng and Z. Liu, Biomaterials, 2020, 241, 119910 CrossRef CAS PubMed.
- H. Niu, J. Tang, X. Zhu, Z. Li, Y. Zhang, Y. Ye and Y. Zhao, Chem. Commun., 2020, 56, 7710–7713 RSC.
- M. C. Areias, K. Shimizu and R. G. Compton, Analyst, 2016, 141, 2904–2910 RSC.
- M. Hanko, Ľ. Švorc, A. Planková and P. Mikuš, Anal. Chim. Acta, 2019, 1062, 1–27 CrossRef CAS PubMed.
- F. Tahernejad-Javazmi, M. Shabani-Nooshabadi and H. Karimi-Maleh, Talanta, 2018, 176, 208–213 CrossRef CAS PubMed.
- P. M. Olmos Moya, M. Martínez Alfaro, R. Kazemi, M. A. Alpuche-Avilés, S. Griveau, F. Bedioui and S. Gutiérrez Granados, Anal. Chem., 2017, 89, 10726–10733 CrossRef CAS PubMed.
- Z. Xu, X. Huang, X. Han, D. Wu, B. Zhang, Y. Tan, M. Cao, S. H. Liu, J. Yin and J. Yoon, Chem, 2018, 4, 1609–1628 CAS.
- H. Zhang, J. Liu, Y. Q. Sun, M. Liu and W. Guo, J. Am. Chem. Soc., 2020, 142, 17069–17078 CrossRef CAS PubMed.
- S. Uchinomiya, N. Matsunaga, K. Kamoda, R. Kawagoe, A. Tsuruta, S. Ohdo and A. Ojida, Chem. Commun., 2020, 56, 3023–3026 RSC.
- V. J. N. Bykov, S. E. Eriksson, J. Bianchi and K. G. Wiman, Nat. Rev. Cancer, 2018, 18, 89–102 CrossRef CAS PubMed.
- H. Afsharan, B. Khalilzadeh, H. Tajalli, M. Mollabashi, F. Navaeipour and M.-R. Rashidi, Electrochim. Acta, 2016, 188, 153–164 CrossRef CAS.
- S. Tonello, F. Stradolini, G. Abate, D. Uberti, M. Serpelloni, S. Carrara and E. Sardini, Sci. Rep., 2019, 9, 17347 CrossRef PubMed.
- Y. C. Zhu, N. Zhang, Y. F. Ruan, W. W. Zhao, J. J. Xu and H. Y. Chen, Anal. Chem., 2016, 88, 5626–5630 CrossRef CAS PubMed.
- E. Bossy-Wetzel, D. D. Newmeyer and D. R. Green, EMBO J., 1998, 17, 37–49 CrossRef CAS PubMed.
- A. Poturnayova, G. Castillo, V. Subjakova, M. Tatarko, M. Snejdarkova and T. Hianik, Sens. Actuators, B, 2017, 238, 817–827 CrossRef CAS.
- L. Hu and G. Xu, Chem. Soc. Rev., 2010, 39, 3275–3304 RSC.
- M. M. Chen, S. B. Cheng, K. Ji, J. Gao, Y. L. Liu, W. Wen, X. Zhang, S. Wang and W. H. Huang, Chem. Sci., 2019, 10, 6295–6303 RSC.
- H. Sha, Y. Zhang, Y. Wang, H. Ke, X. Xiong, H. Xue and N. Jia, Biosens. Bioelectron., 2019, 132, 203–209 CrossRef CAS PubMed.
- J. Zhang, X. Ma and Z. Wang, Anal. Chem., 2019, 91, 6600–6607 CrossRef CAS PubMed.
- J. Zhu, M. Jiang, H. Ma, H. Zhang, W. Cheng, J. Li, L. Cai, X. X. Han and B. Zhao, Angew. Chem., Int. Ed., 2019, 58, 16499–16503 CrossRef CAS PubMed.
- F.-C. Loo, S.-P. Ng, C.-M. L. Wu and S. K. Kong, Sens. Actuators, B, 2014, 198, 416–423 CrossRef CAS.
- L. Wang, W. Gu, P. Sheng, Z. Zhang, B. Zhang and Q. Cai, Sens. Actuators, B, 2019, 281, 1088–1096 CrossRef CAS.
- A. K. Frank, E. C. Pietsch, P. Dumont, J. Tao and M. E. Murphy, Cancer Biol. Ther., 2011, 11, 740–745 CrossRef CAS PubMed.
- S. Takano, S. Shiomoto, K. Y. Inoue, K. Ino, H. Shiku and T. Matsue, Anal. Chem., 2014, 86, 4723–4728 CrossRef CAS PubMed.
- B. Khalilzadeh, H. N. Charoudeh, N. Shadjou, R. Mohammad-Rezaei, Y. Omidi, K. Velaei, Z. Aliyari and M.-R. Rashidi, Sens. Actuators, B, 2016, 231, 561–575 CrossRef CAS.
- D. E. Prasuhn, A. Feltz, J. B. Blanco-Canosa, K. Susumu, M. H. Stewart, B. C. Mei, A. V. Yakovlev, C. Loukov, J. M. Mallet, M. Oheim, P. E. Dawson and I. L. Medintz, ACS Nano, 2010, 4, 5487–5497 CrossRef CAS PubMed.
- D. Deng, Y. Hao, S. Yang, Q. Han, L. Liu, Y. Xiang, F. Tu and N. Xia, Sens. Actuators, B, 2019, 286, 415–420 CrossRef CAS.
- B. Khalilzadeh, N. Shadjou, H. N. Charoudeh and M.-R. Rashidi, Microchim. Acta, 2017, 184, 3651–3662 CrossRef CAS.
- M. B. Youdim, D. Edmondson and K. F. Tipton, Nat. Rev. Neurosci., 2006, 7, 295–309 CrossRef CAS PubMed.
- X. Wu, W. Shi, X. Li and H. Ma, Angew. Chem., Int. Ed., 2017, 56, 15319–15323 CrossRef CAS PubMed.
- Z. Yang, W. Li, H. Chen, Q. Mo, J. Li, S. Zhao, C. Hou, J. Qin and G. Su, Chem. Commun., 2019, 55, 2477–2480 RSC.
- H. Fang, H. Zhang, L. Li, Y. Ni, R. Shi, Z. Li, X. Yang, B. Ma, C. Zhang, Q. Wu, C. Yu, N. Yang, S. Q. Yao and W. Huang, Angew. Chem., Int. Ed., 2020, 59, 7536–7541 CrossRef CAS PubMed.
- L. Li, C. W. Zhang, G. Y. Chen, B. Zhu, C. Chai, Q. H. Xu, E. K. Tan, Q. Zhu, K. L. Lim and S. Q. Yao, Nat. Commun., 2014, 5, 3276 CrossRef PubMed.
- R. Wang, X. Han, J. You, F. Yu and L. Chen, Anal. Chem., 2018, 90, 4054–4061 CrossRef CAS PubMed.
- X. Wu, Y. Li, J. Wang, H. Zhou, X. Tang, Y. Yang, Z. Wang, D. Chen, X. Zhou, J. Guo, H. Cai, J. Zheng and P. Sun, Anal. Chem., 2020, 92, 15050–15058 CrossRef CAS PubMed.
- T. Nunoura, Y. Chikaraishi, R. Izaki, T. Suwa, T. Sato, T. Harada, K. Mori, Y. Kato, M. Miyazaki, S. Shimamura, K. Yanagawa, A. Shuto, N. Ohkouchi, N. Fujita, Y. Takaki, H. Atomi and K. Takai, Science, 2018, 359, 559–563 CrossRef CAS PubMed.
- Q. Wang, H. Chen, Y. Li, H. Wang, Z. Nie, Y. Hu and S. Yao, Talanta, 2016, 161, 583–591 CrossRef CAS PubMed.
- C. R. Goward and D. J. Nicholls, Protein Sci., 1994, 3, 1883–1888 CrossRef CAS PubMed.
- P. Minárik, N. Tomásková, M. Kollárová and M. Antalík, Gen. Physiol. Biophys., 2002, 21, 257–265 Search PubMed.
- K. Lee, H. S. Ban, R. Naik, Y. S. Hong, S. Son, B. K. Kim, Y. Xia, K. B. Song, H. S. Lee and M. Won, Angew. Chem., Int. Ed., 2013, 52, 10286–10289 CrossRef CAS PubMed.
- P. Song, Y. Inagaki, Z. Wang, K. Hasegawa, Y. Sakamoto, J. Arita, W. Tang and N. Kokudo, Medicine, 2015, 94, e810 CrossRef CAS PubMed.
- S. Park, D. J. Bae, Y. M. Ryu, S. Y. Kim, S. J. Myung and H. J. Kim, Chem. Commun., 2016, 52, 10400–10402 RSC.
- H. Liu, F. Liu, F. Wang, R. Q. Yu and J. H. Jiang, Analyst, 2018, 143, 5530–5535 RSC.
- Z. Thiel and P. Rivera-Fuentes, Angew. Chem., Int. Ed., 2019, 58, 11474–11478 CrossRef CAS PubMed.
- A. Chevalier, Y. Zhang, O. M. Khdour, J. B. Kaye and S. M. Hecht, J. Am. Chem. Soc., 2016, 138, 12009–12012 CrossRef CAS PubMed.
- Y. Liu, L. Teng, L. Chen, H. Ma, H. W. Liu and X. B. Zhang, Chem. Sci., 2018, 9, 5347–5353 RSC.
- N. Zhu, G. Xu, R. Wang, T. Zhu, J. Tan, X. Gu and C. Zhao, Chem. Commun., 2020, 56, 7761–7764 RSC.
- V. Scalcon, A. Bindoli and M. P. Rigobello, Free Radical Biol. Med., 2018, 127, 62–79 CrossRef CAS PubMed.
- A. Patenaude, M. R. Ven Murthy and M. E. Mirault, J. Biol. Chem., 2004, 279, 27302–27314 CrossRef CAS PubMed.
- M. H. Lee, J. H. Han, J. H. Lee, H. G. Choi, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2012, 134, 17314–17319 CrossRef CAS PubMed.
- Y. Liu, H. Ma, L. Zhang, Y. Cui, X. Liu and J. Fang, Chem. Commun., 2016, 52, 2296–2299 RSC.
- L. Lai, J. Sun, S. Tarafdar, C. Liu, E. Murphy, G. Kim and R. L. Levine, Free Radical Biol. Med., 2019, 145, 374–384 CrossRef CAS PubMed.
- M. H. Xiang, H. Huang, X. J. Liu, Z. X. Tong, C. X. Zhang, F. Wang, R. Q. Yu and J. H. Jiang, Anal. Chem., 2019, 91, 5489–5493 CrossRef CAS PubMed.
- D. Pendin, R. Norante, A. De Nadai, G. Gherardi, N. Vajente, E. Basso, N. Kaludercic, C. Mammucari, C. Paradisi, T. Pozzan and A. Mattarei, Angew. Chem., Int. Ed., 2019, 58, 9917–9922 CrossRef CAS PubMed.
- W. Chyan, D. Y. Zhang, S. J. Lippard and R. J. Radford, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 143–148 CrossRef CAS PubMed.
- D. H. Fudge, R. Black, L. Son, K. LeJeune and Y. Qin, ACS Chem. Biol., 2018, 13, 1897–1905 CrossRef CAS PubMed.
- X. Kong, F. Su, L. Zhang, J. Yaron, F. Lee, Z. Shi, Y. Tian and D. R. Meldrum, Angew. Chem., Int. Ed., 2015, 54, 12053–12057 CrossRef CAS PubMed.
- J. Ning and Y. Tian, Sens. Actuators, B, 2020, 307, 127659 CrossRef.
- S. C. Dodani, S. C. Leary, P. A. Cobine, D. R. Winge and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 8606–8616 CrossRef CAS PubMed.
- L. Wang, B. Chen, P. Peng, W. Hu, Z. Liu, X. Pei, W. Zhao, C. Zhang, L. Li and W. Huang, Chin. Chem. Lett., 2017, 28, 1965–1968 CrossRef CAS.
- T. Hirayama, S. Kadota, M. Niwa and H. Nagasawa, Metallomics, 2018, 10, 794–801 CrossRef CAS PubMed.
- L. D. Zorova, V. A. Popkov, E. Y. Plotnikov, D. N. Silachev, I. B. Pevzner, S. S. Jankauskas, V. A. Babenko, S. D. Zorov, A. V. Balakireva, M. Juhaszova, S. J. Sollott and D. B. Zorov, Anal. Biochem., 2018, 552, 50–59 CrossRef CAS PubMed.
- A. Labajova, A. Vojtiskova, P. Krivakova, J. Kofranek, Z. Drahota and J. Houstek, Anal. Biochem., 2006, 353, 37–42 CrossRef CAS PubMed.
- T. S. Lim, A. Dávila, D. C. Wallace and P. Burke, Lab Chip, 2010, 10, 1683–1688 RSC.
- X. Li, Y. Zhao, J. Yin and W. Lin, Coord. Chem. Rev., 2020, 420, 213419 CrossRef CAS.
- R. C. Scaduto, Jr. and L. W. Grotyohann, Biophys. J., 1999, 76, 469–477 CrossRef.
- M. T. Binet, C. J. Doyle, J. E. Williamson and P. Schlegel, J. Exp. Mar. Biol. Ecol., 2014, 452, 91–100 CrossRef CAS.
- J. S. Teodoro, I. F. Machado, A. C. Castela, A. P. Rolo and C. M. Palmeira, Methods Mol. Biol., 2020, 2184, 197–213 CrossRef CAS PubMed.
- S. W. Perry, J. P. Norman, J. Barbieri, E. B. Brown and H. A. Gelbard, Biotechniques, 2011, 50, 98–115 CrossRef CAS PubMed.
- A. A. Gerencser, C. Chinopoulos, M. J. Birket, M. Jastroch, C. Vitelli, D. G. Nicholls and M. D. Brand, J. Physiol., 2012, 590, 2845–2871 CrossRef CAS PubMed.
- Z. Xue, H. Zhao, J. Liu, J. Han and S. Han, Chem. Sci., 2017, 8, 1915–1921 RSC.
- O. Murata, Y. Shindo, Y. Ikeda, N. Iwasawa, D. Citterio, K. Oka and Y. Hiruta, Anal. Chem., 2020, 92, 966–974 CrossRef CAS PubMed.
- N. Zhao, S. Chen, Y. Hong and B. Z. Tang, Chem. Commun., 2015, 51, 13599–13602 RSC.
- R. Feng, L. Guo, J. Fang, Y. Jia, X. Wang, Q. Wei and X. Yu, Anal. Chem., 2019, 91, 3704–3709 CrossRef CAS PubMed.
- M. Tian, J. Sun, B. Dong and W. Lin, Angew. Chem., Int. Ed., 2018, 57, 16506–16510 CrossRef CAS PubMed.
- P. E. Z. Klier, J. G. Martin and E. W. Miller, J. Am. Chem. Soc., 2021, 143, 4095–4099 CrossRef CAS PubMed.
- R. Li, H. Qi, Y. Ma, Y. Deng, S. Liu, Y. Jie, J. Jing, J. He, X. Zhang, L. Wheatley, C. Huang, X. Sheng, M. Zhang and L. Yin, Nat. Commun., 2020, 11, 3207 CrossRef CAS PubMed.
- Z. Yang, L. Li, J. Ling, T. Liu, X. Huang, Y. Ying, Y. Zhao, Y. Zhao, K. Lei, L. Chen and Z. Chen, Chem. Sci., 2020, 11, 8506–8516 RSC.
- Y. Liu, J. Luo, X. Chen, W. Liu and T. Chen, Nano-Micro Lett., 2019, 11, 100 CrossRef CAS PubMed.
- B. Burger, P. M. Maffettone, V. V. Gusev, C. M. Aitchison, Y. Bai, X. Wang, X. Li, B. M. Alston, B. Li, R. Clowes, N. Rankin, B. Harris, R. S. Sprick and A. I. Cooper, Nature, 2020, 583, 237–241 CrossRef CAS PubMed.
- T. L. To, A. M. Cuadros, H. Shah, W. H. W. Hung, Y. Li, S. H. Kim, D. H. F. Rubin, R. H. Boe, S. Rath, J. K. Eaton, F. Piccioni, A. Goodale, Z. Kalani, J. G. Doench, D. E. Root, S. L. Schreiber, S. B. Vafai and V. K. Mootha, Cell, 2019, 179, 1222–1238 CrossRef CAS PubMed.
- K. Yang, L. Hu, X. Ma, S. Ye, L. Cheng, X. Shi, C. Li, Y. Li and Z. Liu, Adv. Mater., 2012, 24, 1868–1872 CrossRef CAS PubMed.
- Q. Wu, G. Chen, K. Gong, J. Wang, X. Ge, X. Liu, S. Guo and F. Wang, Matter, 2019, 1, 496–512 CrossRef.
Footnote |
| † W. Ji, X. Tang and Dr W. Du contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |