
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymer nano-systems for the encapsulation and delivery of active biomacromolecular therapeutic agents
Marina
Machtakova
a,
Héloïse
Thérien-Aubin
 *ab and
Katharina
Landfester
*a
*ab and
Katharina
Landfester
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: landfester@mpip-mainz.mpg.de; therien@mpip-mainz.mpg.de
bDepartment of Chemistry, Memorial University of Newfoundland, St. John's, NL, Canada. E-mail: htherienaubin@mun.ca
First published on 11th November 2021
Abstract
Biomacromolecular therapeutic agents, particularly proteins, antigens, enzymes, and nucleic acids are emerging as powerful candidates for the treatment of various diseases and the development of the recent vaccine based on mRNA highlights the enormous potential of this class of drugs for future medical applications. However, biomacromolecular therapeutic agents present an enormous delivery challenge compared to traditional small molecules due to both a high molecular weight and a sensitive structure. Hence, the translation of their inherent pharmaceutical capacity into functional therapies is often hindered by the limited performance of conventional delivery vehicles. Polymer drug delivery systems are a modular solution able to address those issues. In this review, we discuss recent developments in the design of polymer delivery systems specifically tailored to the delivery challenges of biomacromolecular therapeutic agents. In the future, only in combination with a multifaceted and highly tunable delivery system, biomacromolecular therapeutic agents will realize their promising potential for the treatment of diseases and for the future of human health.
 Marina Machtakova | Marina Machtakova is currently a doctoral researcher at the Max Planck Institute for Polymer Research (MPIP). She obtained her BSc and MSc in chemistry from the Johannes-Gutenberg University in Germany. Her current research interest includes the synthesis of polymeric nano-systems for the delivery of active biomacromolecular therapeutic agents. |
 Héloïse Thérien-Aubin | Héloïse Thérien-Aubin is a professor in the Chemistry department at the Memorial University of Newfoundland, where her group study nanostructured polymer materials. She studied chemistry at the Université de Montréal. After her PhD she was a postdoctoral fellow at Cornell University, and then, at the University of Toronto. From 2016 to 2021, she was a group leader at the MPIP. |
 Katharina Landfester | Katharina Landfester received her doctoral degree in Physical Chemistry in 1995 after working with Prof. Spiess at the MPIP. After a postdoctoral stay at the Lehigh University, she worked at the MPI of Colloids and Interfaces in Golm leading the miniemulsion group. From 2003 to 2008, she was a professor at the University of Ulm. She joined the Max Planck Society in 2008 as one of the directors of the MPIP. Her research focuses on creating functional colloids for new material and biomaterial applications. |
1. Introduction
The use of biological macromolecules, such as peptides, proteins, and nucleic acids as therapeutic agents is emerging as a powerful option for treating diseases like cancer, immunological and infectious diseases, or metabolic disorders. One of the cornerstones of the efficacy of this new class of drugs relies on their high specificity leading to highly decreased off-target effects.1–3 New treatments based on those biological macromolecules have benefited from extensive advances in molecular biology, which enabled the large-scale production of such delicate biomolecules. This increased availability now offers an alternative to traditional small synthetic molecules, which often display both a low specificity and toxic side-effects in healthy tissues.4Given the unique advantages of biomacromolecular drugs over small molecule therapeutic agents, their implementation in new treatment strategies represents one of the most promising fields of development in biochemical and pharmaceutical science. Despite the remarkable potential of biomacromolecular therapeutic agents (BTAs), only a few examples of such treatments have successfully translated to clinical practice. The successful examples include insulin for the treatment of diabetes,5 somatropin used in growth hormone therapy,6 or monoclonal antibodies approved for the treatment of cancer and other diseases.7 In fact, among the more than 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 drugs currently approved by the FDA, less than 2% of them are BTAs.8 Although recent research developments have improved the efficiency of treatment based on the use of BTAs, successful clinical outcomes are rare. One of the crucial challenges currently encountered in many clinical trials is the inadequate and off-target delivery of these therapeutic agents.3,9,10 In particular, several studies attributed the failure of treatments based on the use of BTAs to their presumably poor delivery mechanism.11–13
000 drugs currently approved by the FDA, less than 2% of them are BTAs.8 Although recent research developments have improved the efficiency of treatment based on the use of BTAs, successful clinical outcomes are rare. One of the crucial challenges currently encountered in many clinical trials is the inadequate and off-target delivery of these therapeutic agents.3,9,10 In particular, several studies attributed the failure of treatments based on the use of BTAs to their presumably poor delivery mechanism.11–13
Amid today's global health crisis caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the need to design appropriate delivery systems for BTAs is more evident than ever. The new vaccines against COVID-19, like those developed by Pfizer/BioNTech and Moderna deliver the genetic sequence of the viral protein in the form of mRNA to the host cell.14 The mRNA induces the expression of the virus protein and leads to immunity against the original virus. However, using mRNA alone would lead to low efficacy, as mRNA is easily degraded by RNAses during circulation in the blood and cannot efficiently cross the cellular membrane due to its large size.15 If the mRNA vaccine formulation has been successful, it is in part due to the delivery vehicle, a lipid nanoparticle, a great achievement of nanotechnological engineering in and of itself, used as the delivery system.16 The encapsulation of the mRNA in the cationic lipid nanoparticles allows to protect the RNA from extracellular RNases and facilitates the uptake and endosomal release of the gene in the targeted cells. However, both the limited stability of the vaccine and the difficulties in scaling up the production are mainly associated with the ionizable lipid nanoparticles used as the delivery vehicle for the mRNA.17 It is clear that the scientific success of these vaccines providing a way out of the global crisis results from a strong collaboration between bio- and nanotechnology. This success also clearly displays the potential and benefits of biomacromolecular therapeutics but also highlights the urgent need for the design of efficient, stable, and versatile delivery systems for those molecules.18 Polymer-based drug delivery systems are being used in an increasing manner. In the last few decades, the first examples of those systems were successfully translated from the labs to the clinical applications, for both the delivery of small molecules (e.g., Atridox® an antibiotic, Abraxane® used in the treatment of breast and lung cancer, Sublocade® used to treat opioid addiction) and biomacromolecular therapeutic agent (e.g., Oncaspar® to treat leukemia).19–22 Some of the challenges faced in the design of new polymer DDSs for biomacromolecular therapeutic agents are shared with the DDSs design for small molecules. The continuing success in translating polymer DDSs toward clinical applications hinges both the design of more effective and efficient DDSs and the standardization of the production processes.23
Here, we highlight the challenges for the delivery of biomacromolecular therapeutics and the solutions polymer nanocarriers may provide for their delivery. Although the prominent success of the mRNA delivery in COVID-19 vaccines was accomplished with lipid-based nanocarrier, multiple smart polymer nanostructures are emerging as effective alternatives. Such drug delivery systems (DDSs) provide unmatched diversity and control in their design. In the review, we define nanocarriers as structures used to deliver the drug to desired tissues, organs, cells, and subcellular organs and potentially released the drug in a determined manner. The usual purpose of nanocarriers is to improve the pharmacological properties of the therapeutic drugs and to overcome challenges such as limited stability, low bioavailability, poor biodistribution, or the lack of selectivity. Excellent reviews on the general use of polymer nanostructures in drug delivery can be found elsewhere,24–27 here, the focus is on their use in the delivery of biomacromolecular therapeutics such as proteins, enzymes, and oligonucleotides.
2. Understanding the challenges in the delivery of active biomacromolecular therapeutic agents
The unique performance of treatments based on BTAs arises from the combination of their composition and structure. However, the structure of large biological molecules such as proteins and nucleic acids is complex. Due to natural evolution, the sequence of the individual building blocks is hierarchically defined and adapted to the inherent biological function. Moreover, the functionalities of those molecules depend not only on the primary structure, their chemical composition but also on the secondary, tertiary, and quaternary structures, i.e., how the molecules are folded and organized in complex 3D assemblies.28 Many non-covalent forces such as hydrogen bonds, van der Waals, electrostatic or hydrophobic interactions are present throughout the molecule and arrange the sequence of the building blocks in a specific three-dimensional folding.29 It is this unique structure that defines the activity and specificity of the BTAs.29,30One of the crucial challenges in the delivery of BTAs is to preserve this inherent sensitive structure of the BTAs during the delivery process.31 In general, biological macromolecules are vulnerable to the loss of their structure due to the effect of physicochemical triggers such as temperature, pH value, and the ionic strength of the surrounding environment. Those conditions can disrupt the specific intra- and inter-molecular interactions based on labile forces responsible for their complex structures. These physicochemical cues can cause aggregation, deamidation, isomerization, hydrolysis, oxidation, and denaturation, resulting in the irreversible loss of biological activity. Moreover, the primary structure also needs to be protected since it can be subjected to enzymatic degradation in vivo. Various proteases and nucleases found in the biological media can degrade the molecules into smaller fragments.32,33 This is a critical factor that leads to short in vivo half-life times, ranging from a few minutes to a few hours, exhibited by some BTAs.34 This is one of the reasons why the use of BTAs as medications administered systemically, like in the case of insulin to treat diabetes, often requires frequent and constant dosing to maintain an appropriate in vivo concentration.35
Another key challenge is the delivery of the large biomacromolecules, used as therapeutic agents, across the cellular membrane to the cytoplasm.36 After billions of years of evolution, the cell membrane has evolved to precisely regulate the transport of molecules in and out of the cytosol to protect the intracellular environment from extracellular interference.37 The high molecular weight and the intrinsic hydrophilicity of many BTAs are associated with a general reduction in their permeability across biological barriers such as cell membranes.38 Cellular membranes can selectively and actively allow small molecules and ions to enter, while the lipid bilayer is impermeable to high-molecular-weight substances.39 The intrinsic lipophilic nature of biological membranes is another major obstacle in the permeation of hydrophilic biomacromolecules to the targeted intracellular site. Furthermore, in the case of nucleic acid-based drugs, the active molecules bear the same surface charge as the negatively charged cellular surface, making their transport across the membrane nearly impossible without the aid of external forces.40 As a result, only a marginal fraction of BTAs can successfully reach their intracellular target, which leads to insufficient therapeutic efficacy.
3. Structures of polymer DDSs
For the past 50 years, lipid nanoformulations have dominated the field of nanosystems for drug delivery.41 The approval of Doxil® in 1995, a liposomal nanoformulation, is an important milestone in the development of nanomedicine. Originally developed as vehicles for the delivery of small molecules, liposomes and lipid formulations are now used to deliver a range of different payloads, including BTAs, as exemplified in the delivery of mRNA in COVID-19 vaccines.42,43 In this case, spherical lipid nanoparticles are made of ionizable lipids, which are able to complex the mRNA by electrostatic interactions. Due to the size and properties of the lipid nanoparticles, they are taken up by cells via endocytic pathways, and the cationic charge of the lipids at low pH values leads to the endosomal escape releasing the mRNA into the cell cytoplasm. The chemical composition of the lipid nanoparticles is very close to that of cell membranes, conferring to such DDSs excellent biocompatibility and biodegradability.Although lipid nanoparticles and liposomes have been used to deliver both small molecules and BTAs, some limitations remain to their widespread applications. One reason is that those formulations are based on self-assembling lipids and can suffer from destabilization or dissociation in biological media. Such dynamic dissociation might lead to both side effects due to off-target activation and the loss of the valuable therapeutic agent due to its degradation outside of the dissociated nanostructure. Hence, this inherent instability is one of the Achilles’ heels of lipid and liposomal delivery of BTAs.44,45 Another limitation in the design of liposomal delivery systems is the lack of structural diversity in the type of compatible lipid material used to generate the liposomes. The ideal drug delivery system might require the addition of smart functionalities such as stimuli-responsive structures for the precise and controlled release of the cargo or highly functional targeting moieties to overcome the hurdles toward efficient, specific, and targeted delivery.
Polymer nanostructures offer interesting alternatives. They provide a great versatility of building blocks and enable the incorporation of additional functionalities to the delivery systems. Furthermore, the organization of the polymer and the BTAs can adopt different structures leading to different DDSs architectures such as polymer-conjugates, polyplexes, layer-by-layer assemblies, nanogels, nanocapsules, and polymersomes (Fig. 1). The advances in the field have yielded a variety of structures with unique loading and functionalization, each displaying their own advantages for the successful delivery of BTAs (Table 1).
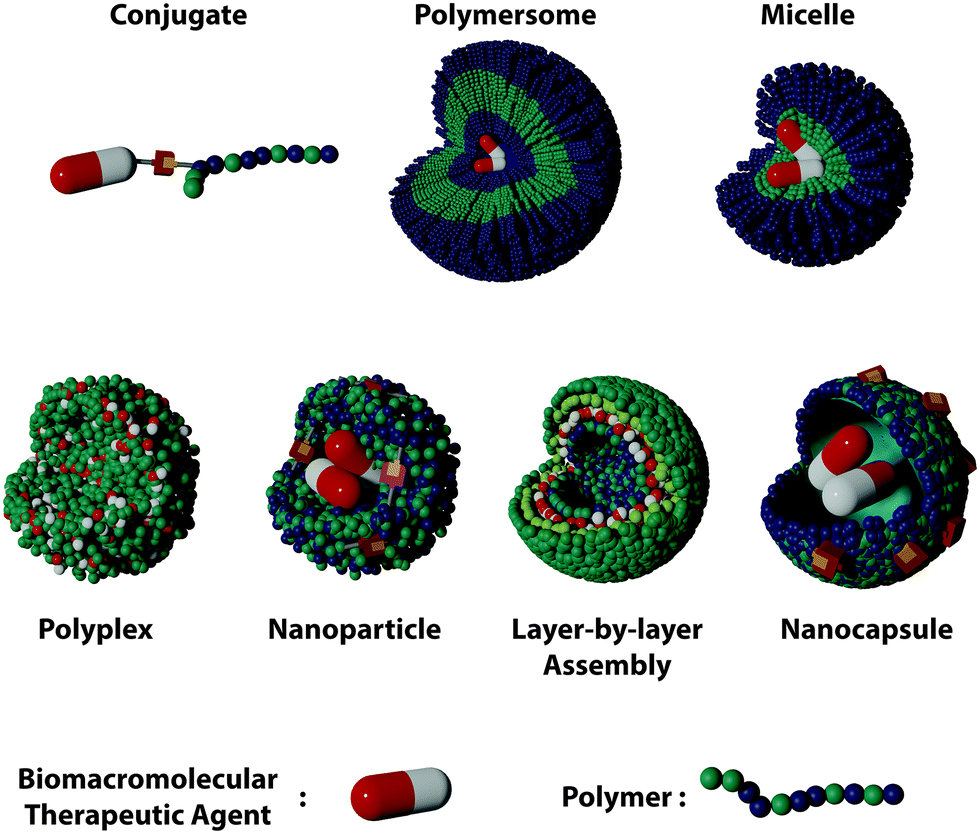 | ||
| Fig. 1 Types of polymer DDSs for the encapsulation and delivery of biomacromolecular therapeutic agents. | ||
| Polymer structure | Mode of formation | Example | Advantages | Limitations |
|---|---|---|---|---|
| Conjugate | Covalent attachment | PEGylation of proteins,54 | • No leakage of the drug | • Chemical modification of biomolecule |
| Stabilization of enzymes:55 | • High drug/polymer ratio | |||
| PEGylated asparaginase (FDA/EMA approved)56 | • Low degree of potentially harmful polymers | • High risk of denaturation | ||
| Peginterferon α-2b (FDA/EMA approved)57,58 | • Responsive conjugates enable controlled release | • Limited steric protection | ||
| Polyplex/coacervate | Electrostatic complexation of charged molecules | Delivery of mRNA59 | • Absence of chemical modification of the drug | • Limited to charged biomolecules |
| Delivery of siRNA60 | • Increased cellular uptake due to electrostatic interactions with the cells | • Electrostatic interactions influenced by external factors | ||
| Delivery of plasmidDNA61 | • High steric protection | • High risk of dissociation in blood | ||
| CYL-02: pDNA/PEI polyplex (phase II development for pancreatic cancer)62 | ||||
| CALAA-01 siRNA/cyclodextrin polyplex (phase I development for cancer therapy)63 | ||||
| LbL assembly | Electrostatic complexation of charged molecules | Co-delivery of antigens and adjuvants64 | • Absence of chemical modification of the drug | • Limited to charged biomolecules |
| Co-delivery of gene and anticancer drug65 | • Increased cellular uptake due to electrostatic interactions with the cells | • Electrostatic interactions influenced by external factors | ||
| • High risk of dissociation in blood | ||||
| Micelle and polymersome | Physical encapsulation | Controlled release of plasmidDNA66 | • Absence of chemical modification of the drug | • Limited encapsulation efficiencies |
| Targeted delivery of siRNA67 | • Variety of morphologies and architectures | • High risk of dissociation in blood | ||
| mRNA delivery (animal study)68 | • High protection | |||
| Polymersome delivery of the SARS-CoV-2 spike protein (preclinical trials)69 | ||||
| Nanoparticle/nanogel/nanocapsule | Physical encapsulation | Protein encapsulation70 | • Absence of chemical modification of the drug | • Risk of denaturation due to crosslinking reactions involved |
| Enzyme nanocapsules71 | • High protection | |||
| Delivery and controlled release of deoxyribonuclease I72 | • High stability | |||
| Vaccine development based on nanogel protein delivery (animal study)73 | • Easy incorporation of smart units | |||
| Iduronate 2-sulfatase delivery with PLGA nanoparticles (preclinical testing)74 | • Controlled release | |||
3.1 Polymer–drug conjugates
The conjugation of synthetic polymers with various BTAs is the simplest polymer DSS possible. Such polymer–drug conjugates are pharmacologically active constructs comprising one or more therapeutic agents covalently bound to a polymer chain.46 Since the first poly(ethylene glycol) (PEG)–protein conjugate, Adagen® (bovine pegademase), was approved by the FDA in 1990,47 functional polymer bioconjugates have been widely explored and are continuously evolving. A commonly used model of a polymer–drug conjugate consists of a biocompatible water-soluble polymer backbone as the main delivery vehicle tethered to the biomacromolecular agent (Fig. 2a). In this case, the polymer chain increases the aqueous solubility of the drug and protects the cargoes from a rapid exclusion from the body. Several types of BTAs can be employed to design polymer conjugates, irrespective of their size, charge, or chemical structure. Furthermore, one or several therapeutic molecules can be tethered to the same polymer backbone, and complementary moieties can also be attached to the polymer backbone, such as adjuvant drugs, fluorescent tags, PEG molecules, or targeting moieties.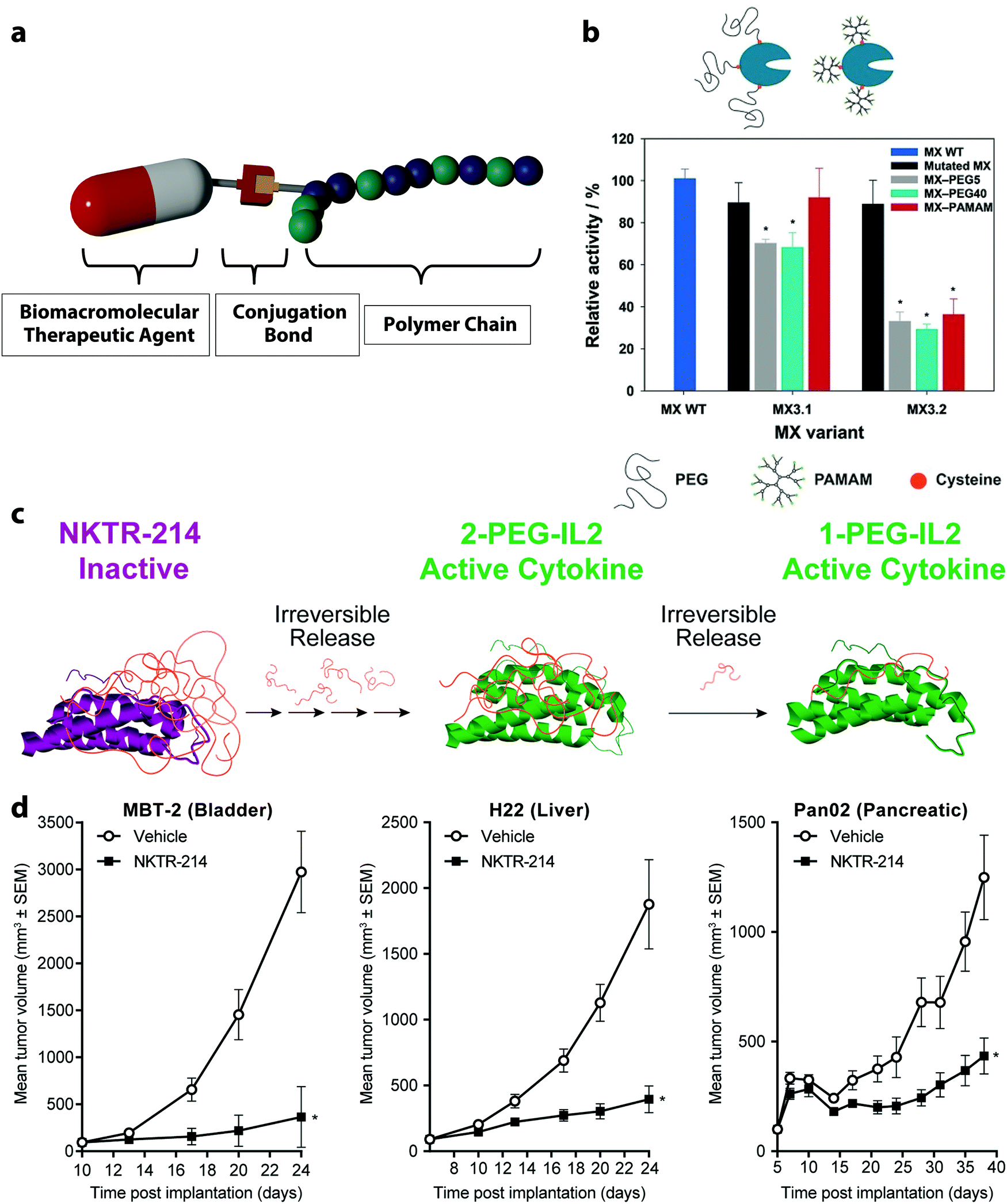 | ||
| Fig. 2 (a) Conjugation of a polymer with a biomacromolecular therapeutic agent, (b) enzymatic activity of a wild type of prolyl endopeptidase isolated from Myxococcus xanthus (MX WT) functionalized with polymer using thiol groups either on (MX 3.2) or outside the catalytic site (MX 3.1) before and after the conjugation of the polymers PEG and poly(amidoamine) (PAMAM, 6.5 kDa),51 (c) clinical stage prodrug NKTR-214 synthesized from the reversible PEGylation of interleukin-2 (IL-2), (d) NKTR-214 as a single-agent demonstrated significant tumor growth inhibition for bladder, liver and pancreatic carcinoma. (b) Reprinted with permission from ref. 51 Copyright 2016 Wiley; (c and d) reprinted with permission from ref. 83. Copyright 2017 PLOS. | ||
Alternatively, BTAs can be functionalized with non-endogenous amino acids or other appropriate external small molecules by using specific reactions. Those can be employed for the site-specific conjugation of polymers using bioorthogonal coupling chemistries. Many examples exploiting such a strategy exist. For instance, the unnatural amino acid p-azidophenylalanine was site-specifically incorporated into proteins.52 The azido end-group enabled a copper-medicated click-reaction with an alkyne functionalized PEG. In another example, a ketone-containing amino acid, p-acetylphenylalanine was attached to a human growth hormone, and the conjugation of an aminooxy-functionalized PEG optimized the pharmacological properties of this therapeutic.53 Many advantages are associated with the “grafting to” approach. Firstly, the synthesis of the polymer may occur in a non-aqueous solution before the conjugation step.
Secondly, this separate synthesis process enables the design of well-defined polymer building blocks for preparing advanced materials with controlled structures, often including several reaction steps. Thirdly, a wide variety of (bioorthogonal) reactions is available to efficiently preserve the sensitive structures of the BTAs during the conjugation process. However, this approach also suffers from shortcomings, such as a potential low yield of the reaction between the polymer and the biomacromolecule. Here, two large molecules must react together, and the reaction can be limited due to steric hindrance leading to insufficient functionalization yields.50 Moreover, the resulting product can be challenging to purify from unreacted starting material and by-products.75
As a consequence, the “grafting from” method has emerged for the synthesis of polymer–drug conjugates. The development of the “grafting from” methodology has benefited from advances in controlled and living polymerization techniques, including the reversible addition–fragmentation transfer (RAFT) and the atom transfer radical polymerization (ATRP).76 Typically, the “grafting from” approach involves the conjugation of the initiator to a BTA. This leads to a formation of a macroinitiator for the following polymerization and leads to a controlled growth of the polymer conjugate. The overall process results in high reaction yield since only small molecules, first the initiator and then the monomers one at a time, are coupled to the reactive site.77 Still, this method may hamper BTAs. For example, the conjugation method employed to prepare protein–polymer conjugates should not include reactive moieties undergoing side reactions with the amino acid functionalities of the protein. Additionally, the polymerization method should not lead to the denaturation of the BTAs, as induced by chemical modification or by inducing irreversible solubility changes.50
Controlled polymerizations, like ATRP and RAFT, have been extensively used to synthesize polymer with biomacromolecules since they combine a high degree of control over the resulting molecular weight and structure of the polymer chains and can be executed in aqueous media.50 Many BTAs have been functionalized in this way. For example, ATRP initiators were immobilized on proteins, which were used to initiate the growth of uniform poly(PEG–methacrylate) chains.78 In another example, the enzyme chymotrypsin was homogenously functionalized with nearly monodisperse polymer chains with a polydispersity index of 1.05. Furthermore, the protein–polymer conjugates synthesized by this method retained 50–86% of the original enzyme activity.79 Mild reaction conditions, using Cu(0)-mediated ATRP, was used to form Candida antarctica lipase B (CALB) conjugated to different polymers of acrylamides and acrylates.55 The conjugates CALB-poly(hydroxyethyl acrylamide) (HEAA), CALB-poly(N-isopropylacrylamide) (NIPAM) and CALB-poly(N-tert-butylacrylamide) (TBA) preserved the original biological activity of CALB, and the conjugates with poly(HEAA) and poly(NIPAM) even exhibited increased enzymatic activity by ∼1.5 and 2-fold. The lipolytic activity of CALB-conjugate was measured by the rate of hydrolysis of nitrophenyl palmitate in heptane. The activity enhancement was ascribed80,81 to the increased solubility in organic media and increased affinity of the enzyme to the substrate in heptane after the formation of the polymer-conjugate.55
Several examples of polymer-conjugates with BTAs have already been approved by the US Food and Drug Administration and the European Medicines Agency. For instance, the PEGylated enzyme aspargase (Oncaspar®) was approved to treat patients with acute lymphoblastic leukemia. The PEG-conjugation of the enzyme increased its circulation time, which allowed for less frequent administration of the treatment to obtain the same response as the unmodified enzyme.56 Another formulation of the recombinant interferon alfa-2b protein with PEG was approved to treat patients with melanoma.57 The conjugation of the interferon to the PEG polymer protects the protein from degradation by enzymes and increases its half-life. The treatment with the PEGinterferon led to a sustained and clinically meaningful longer relapse-free survival time after the surgical tumor removal.58
3.2 Coacervates and electrostatic polyplexes
The formation of supramolecular structures between two polymers based on specific interactions such as electrostatic, hydrogen bonding, and hydrophobic forces can produce coacervates.88 From the molecular point of view, coacervates can also be formed by the interaction between BTAs and other polymers.89 Polyelectrolyte complex coacervates, coacervates formed by oppositely charged polymers, are of particular interest in the development of polymer DDSs for BTAs since nucleic acids, and proteins often carry negative charges.90 The morphologies of the resulting complex display a large diversity depending on the balance of water, polymer, and salt ions within the complex.91 In general, they can range from loosely associated colloids with more liquid-like properties in the case of coacervates to denser precipitates in the case of polyplexes.92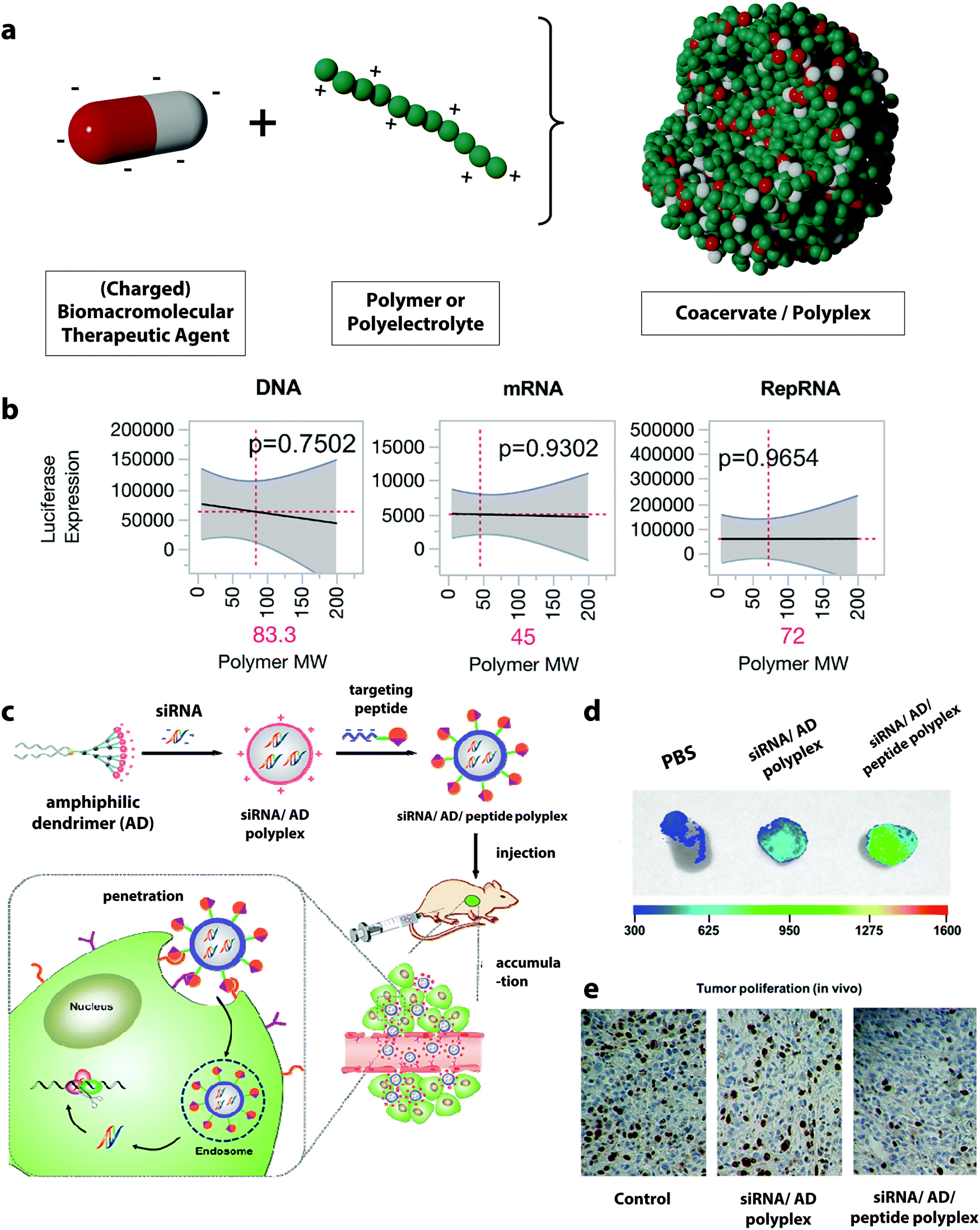 | ||
| Fig. 3 (a) Schematic representation of the formation of polyplexes, (b) prediction profiles for the optimal polymer molecular weight for different type of nucleic acid materials, with luciferase expression as the output. Gray shading indicates 95% confidence interval, and the p-value indicates statistical significance,100 (c) illustration of the formation of targeted complexes for siRNA delivery using an amphiphilic dendrimer (AD) and the targeting peptide E16G6RGDK,60 (d) targeted siRNA complexes (siRNA/AD/E16G6RGDK) accumulate more efficiently in tumor tissues from mice than non-targeted polyplexes (siRNA/AD),60 (e) targeted siRNA/AD/E16G6RGDK polyplexes are much more potent than the non-targeted siRNA/AD systems for anti-cancer activity in mice tumors: cancer cell proliferation in tumor tissue revealed by immunohistochemistry using Ki67 staining (brown-stained cells indicate Ki67 protein expression correlating with metastasis).60 (b) Reprinted with permission from ref. 100, Copyright 2018 American Chemical Society; (c–e) reprinted with permission from ref. 60. Copyright 2018 American Chemical Society. | ||
Many studies have investigated the influence of the polyanion/polycation ratio. For example, polyplexes prepared from DNA and poly-L-lysine showed that the highest DNA loading occurs at a DNA/poly-L-lysine mass concentration ratio between two to three.97 Other studies using PDMAEA or chitosan for the complexation of DNA showed similar results, suggesting that increasing the DNA/polymer ratio leads to the formation of durable complexes and provides a better coverage as well as a better release of DNA.98,99 Another key parameter in the formation of the polyplexes is the molecular mass of the polymer employed. Studies have investigated the influence of the molecular mass of polyethylenimine (PEI) on its complexation performance. PEI is one of the most widely employed polymers for the formation of polyplexes. It was shown that high molecular weight PEI (800 kDa) forms significantly more compact and more stable complexes with DNA and leads to an increased transfection efficiency compared to PEI with a lower molecular weight. However, the cell viability simultaneously decreased with the increasing molecular weight of the PEI, and a very tight complexation of the DNA by the PEI can also hinder the efficient intracellular release.59 Hence, a balance between high complexation efficiency, controlled dissociation at the targeted site, and low toxicity for PEI/DNA complex can be struck by tuning the molecular weight of the PEI. Furthermore, the ideal molecular weight for the complexation of one specific biomacromolecular therapeutic agent can differ from payload to payload. For example, the complexation and delivery of plasmid DNA, messenger RNA (mRNA), and replicon RNA (RepRNA) with a library of poly(2-ethyl-2-oxazoline)/poly(ethylene imine) copolymers with varying molar mass and charge densities showed that the optimal polymer design for each nucleic acid species was different and could significantly influence the transfection efficiency of the delivered nucleotide.100 Those BTAs differ significantly in their structural composition and charge density, and it was found that the polymer molecular weight forming the most efficient polyplexes decreased from 83, 72, and 45 kDa for DNA, RepRNA, and mRNA complexation (Fig. 3b).
Many studies have investigated the influence of the polyanion/polycation ratio. For example, polyplexes prepared from DNA and poly-L-lysine showed that the highest DNA loading occurs at a DNA/poly-L-lysine mass concentration ratio between two to three.97 Other studies using PDMAEA or chitosan for the complexation of DNA showed similar results, suggesting that increasing the DNA/polymer ratio leads to the formation of durable complexes and provides a better coverage as well as a better release of DNA.98,99 Another key parameter in the formation of the polyplexes is the molecular mass of the polymer employed. Studies have investigated the influence of the molecular mass of polyethylenimine (PEI) on its complexation performance. PEI is one of the most widely employed polymers for the formation of polyplexes. It was shown that high molecular weight PEI (800 kDa) forms significantly more compact and more stable complexes with DNA and leads to an increased transfection efficiency compared to PEI with a lower molecular weight. However, the cell viability simultaneously decreased with the increasing molecular weight of the PEI, and a very tight complexation of the DNA by the PEI can also hinder the efficient intracellular release.59 Hence, a balance between high complexation efficiency, controlled dissociation at the targeted site, and low toxicity for PEI/DNA complex can be struck by tuning the molecular weight of the PEI. Furthermore, the ideal molecular weight for the complexation of one specific biomacromolecular therapeutic agent can differ from payload to payload. For example, the complexation and delivery of plasmid DNA, messenger RNA (mRNA), and replicon RNA (RepRNA) with a library of poly(2-ethyl-2-oxazoline)/poly(ethylene imine) copolymers with varying molar mass and charge densities showed that the optimal polymer design for each nucleic acid species was different and could significantly influence the transfection efficiency of the delivered nucleotide.100 Those BTAs differ significantly in their structural composition and charge density, and it was found that the polymer molecular weight forming the most efficient polyplexes decreased from 83, 72, and 45 kDa for DNA, RepRNA, and mRNA complexation (Fig. 3b).
Polyplex gene delivery has lately also advanced to clinical trials. One of the first examples of a systemic administration of polyplex therapeutics applied to humans was a cationic cyclodextrin-based polyplex developed for the delivery of siRNA, which can reduce the expression of a ribonucleotide reductase and hence decreases tumor growth.63 In the phase I clinical trial 24 patients with different cancers were treated with the polyplex formulation and the clinical safety of the therapy was evaluated. The results showed that the formulation was well tolerated and the delivery system enabled a targeted delivery of the siRNA.
A currently ongoing phase II clinical trial (NCT02806687) is evaluating the ability of linear PEI complexed with plasmids encoding genes to achieve intratumoral gene transfer and to sensitize cancer cells to gemcitabine for pancreatic adenocarcinoma therapy.62 Phase 1 of the trial with 22 patients has demonstrated that the formulation can inhibit primary-tumor progression and dissemination when combined with additionally administered gemcitabine. Currently, the study measures the clinical benefits of the therapy, such as a progression-free survival and tumor response, the biological tumor markers, the biodistribution, and quality of life change in 100 patients with locally advanced tumors.102,103
However, this approach is also constrained by some factors. While gene materials are usually strongly charged and thus very suitable for the formation of polyplexes, the formation of polyplexes with certain proteins and polypeptides, which can be moderately charged or even neutral at physiological conditions depending on their isoelectric point, can be challenging.109 Additionally, the number of charged residues in a protein is limited and strongly varies from one protein to another; this can further hamper the formation of protein-based polyplexes.110 More generally, the systemic delivery of polyplexes can be hindered by their instability under physiological conditions. Indeed, various physiological salt and proteins found in blood can readily bind to the polyplexes and increase the risk for the premature release of the payload or even for the complete dissociation of the polyplex.111
3.3 Layer-by-layer assemblies
Another class of drug delivery platforms for BTAs based on electrostatic complexation is the layer-by-layer (LbL) assembly of polyelectrolytes. This method expands the simple electrostatic complexation by the design of multilayer architectures with layers of, usually, alternating charges with nanometer-scale precision. The technique is based on the alternating adsorption of complementary layers of building blocks, which can be synthetic or natural polyelectrolytes and BTAs (Fig. 4a).112,113 For applications in drug delivery, colloidal LbL particles and capsules are of particular interest. In these cases, the single layers can be assembled either on a core template, which can be a drug reservoir or a solid nanoparticle.114 To form an LbL particle system, a colloidal substrate bearing a defined charge or a polyelectrolyte in solution is mixed with complementary polyelectrolytes followed by washing, usually through centrifugation.115 These steps can be repeated several times, each time using an oppositely charged macromolecule to the charge of the previous step to achieve the desired loading of the BTA (Fig. 4b).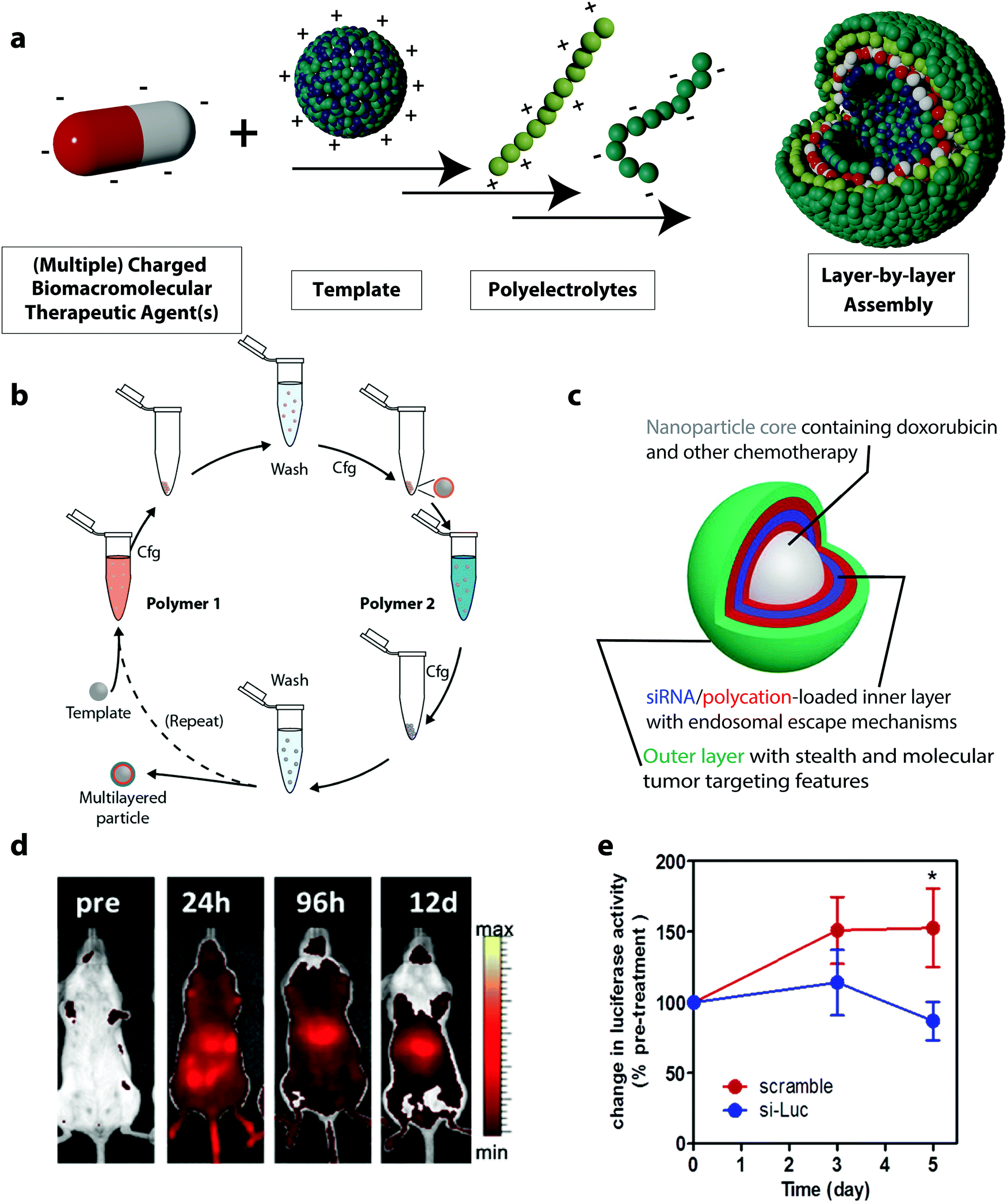 | ||
| Fig. 4 (a) Schematic representation of layer-by-layer (LbL) assemblies, (b) schematic illustration of immersive assembly on particulate substrates using centrifugation (cfg) in between washing steps,117 (c) schematic representation of a siRNA drug delivery platform based on LbL assembly,65 (d) the LbL nanoparticle cores in (c) were fluorescently labeled with AlexaFluor677 to assess the biodistribution using live animal fluorescent imaging (n = 4),65 (e) systemic delivery of siRNA using LbL nanoparticles in mice models: bioluminescence activities in subcutaneous mice tumors treated with a single dose tail vein injection of 1.4 mg kg−1 of luciferase-targeting siRNA-loaded LbL assemblies (si-Luc) and siRNA control (scramble).65 (b) Reprinted with permission from ref. 117, Copyright 2014 The American Chemical Society; (c–e) reprinted with permission from ref. 65. Copyright 2013 The American Chemical Society. | ||
LbL formulations used in the delivery of BTAs allow for controlled loading of the nanocarrier with the biomacromolecule. Since the LbL assembly of the BTA can be monitored, for instance, on a silicon oxide-coated quartz sensor as found in a quartz crystal microbalance with dissipation, the exact amount of adsorbed therapeutic agents can be calculated.116 In this regard, LbL assemblies can serve as a precise gene delivery platform. For example, this led to LbL systems being used to load a plasmid DNA encoding a green fluorescent protein (GFP) in defined quantities. The plasmid DNA was complexed on the surface of PLGA nanoparticles in combination with poly(L-lysine) (PLL) and poly(γ-glutamate) (PGA) via the LbL technique. A defined amount of 0.5 μg of the plasmid DNA in the LbL assemblies was loaded during the synthesis process. After the formulation of the plasmid vector in the LbL assembly, the gene delivery efficacy was tested by the expression of GFP in HEK293 cells. This DDS was compared to Lipofectamine, a commercial gene delivery system, loaded with 2 μg of GFP encoding plasmid DNA. No significant differences were observed for the transfection efficiency of the GFP expressing plasmid in HEK293 cells when using the LbL DDS or the Lipofectamine containing 4 times more plasmid. Hence, the LbL assemblies can deliver the plasmids at the same efficiency as Lipofectamine but require less plasmid material and also cause lower cytotoxic effects in HEK293 cells than the Lipofectamine.116
The LbL system can be built by integrating a variety of functions and molecules in the different layers. For instance, multiple BTAs and adjuvants can be easily incorporated in LbL systems. One therapeutic agent can be deposited in the core of the assembly and another one in the surrounding layers, thus creating an independently tunable device for the co-delivery of multiple therapeutic agents.65 This co-localization of drugs enables the construction of defined signal pathways and artificial architectures that can mimic biological functions found in natural organisms.118 For example, polystyrene (PS) carriers were used as the substrate for the assembly of glucose oxidase (GOX) and horseradish peroxidase (HRP) in conjunction with PEI and polystyrene sulfonate.119 Because both enzymes preserved their enzymatic activity during the formation of the LbL nanosystems, coupled enzymatic reaction between the GOX and the HRP could still be performed after the assembly, and the LbL nanoparticles were able to mimic the sequential cascade reactions observed inside cellular compartments.120
Moreover, to increase the efficiency of immunotherapeutic treatments, it can be helpful to deliver simultaneously multiple payloads.121 For example, in addition to the delivery of the biomacromolecular antigen, the co-delivery of adjuvant molecules is essential for the enhanced activation of immune cells. The flexibility of LbL structures has the potential to co-deliver a combination of antigen and adjuvant, even when they display diverse physicochemical properties in terms of molecular weight, hydrophilicity, and pharmacokinetics.122 LbL nanocarriers have been successfully used for such purpose due to their inherent ability to incorporate many active molecules in different layers within the same drug delivery system.65,123,124 In one of such systems, ovalbumin, a model protein antigen, and an immune-activating single-stranded synthetic DNA adjuvant were combined in one LbL assembly enabling the co-delivery of two components to induce a strong immune activation in mice.64
For this purpose, ovalbumin was coated with two bilayers of dextran sulfate and pLArg. Subsequently, the immunogenic anionic DNA was complexed to the outer pLArg layer and coated with additional pLArg. The LbL nanosystems loaded with ovalbumin and immune-activating DNA were readily internalized by dendritic cells. Furthermore, they promoted a strong upregulation of co-stimulatory markers combined with a vast secretion of pro-inflammatory and polarizing cytokines, whereas the OVA assemblies with dextran sulfate and pLArg lacking the DNA adjuvant totally failed to stimulate cytokine secretion.64
3.4 Polymer micelles and polymersomes
Polymer micelles and polymersomes are supramolecular nanostructures usually formed by the self-assembly of amphiphilic block copolymers.125 Classically, they are composed of block copolymers containing both hydrophilic and hydrophobic segments, and both polymer micelles and polymersome can be obtained from diblock, triblock or multiblock copolymers.126,127 In aqueous environments, the block copolymers assemble into nanostructures depending on the polymer composition. The different assemblies result from the relative volume occupied by the hydrophilic(s) and hydrophobic(s) blocks. This defines the geometric packing of block copolymers in the resulting copolymer assemblies in aqueous solution and controls the shape of the resulting assembly. For example, a diblock copolymer composed of a hydrophilic and a hydrophobic block can either assemble into spherical micelles, worm-like micelles, or vesicles (or polymersomes) as the length of the hydrophobic block increases.128In polymer micelles used for the delivery of BTAs, the assembly usually occurs in water, and the hydrophobic part of the block copolymer is excluded from the aqueous surrounding forming the micelle core. It is in this hydrophobic pocket at the center of the micelle that payloads, like BTAs, are typically encapsulated. The hydrophilic part of the block copolymer then builds the nanoparticle shell with a brush-like architecture (Fig. 5a).
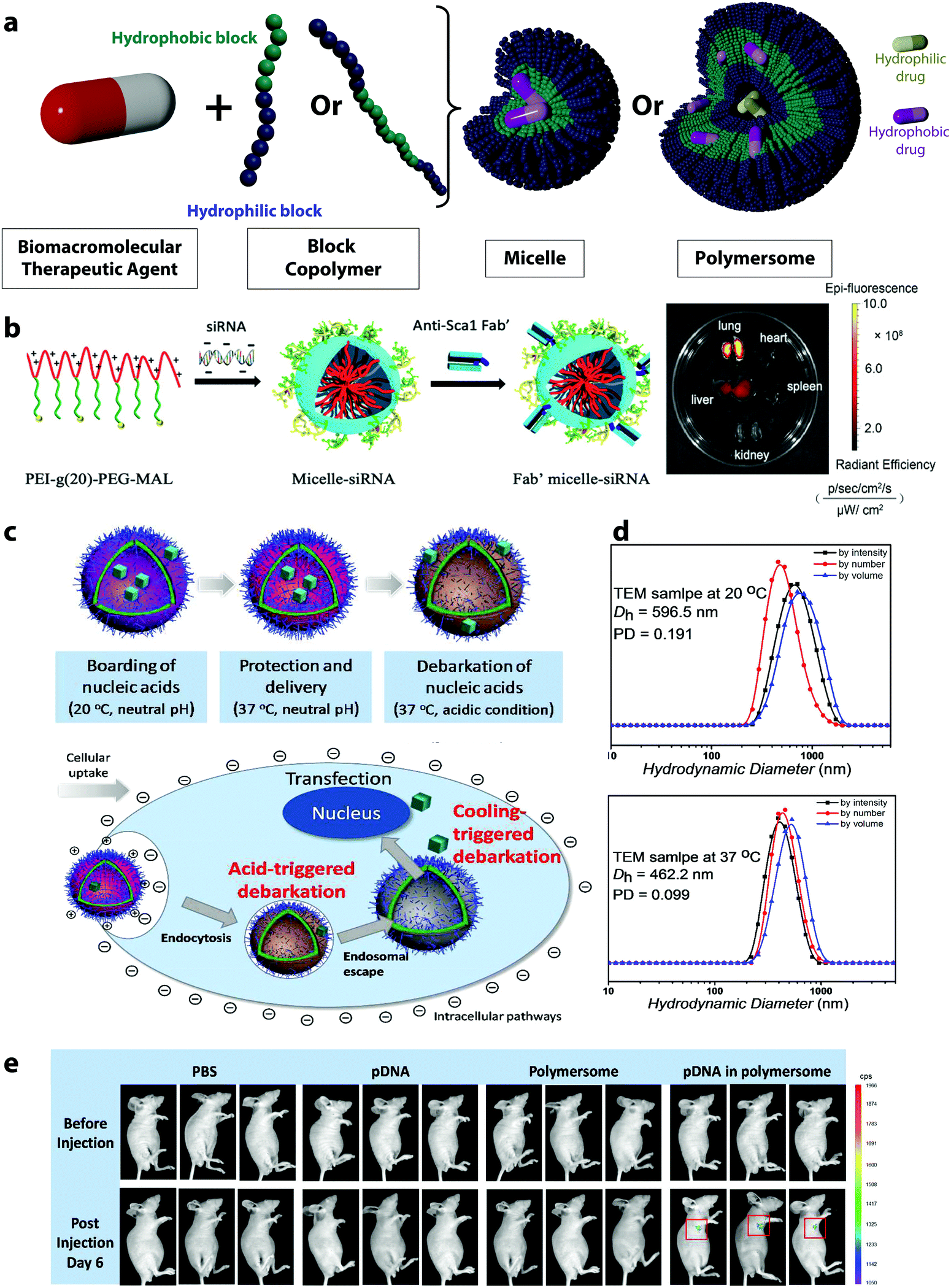 | ||
| Fig. 5 (a) Schematic representation of polymeric micelles and polymersomes, (b) scheme of the formation of siRNA-loaded micelles via the assembly of negatively charged siRNA and the positively charged block copolymers PEI-g(20)-PEG-MAL, followed by the conjugation of a stem cell antigen 1 antibody (Anti-sca 1 Fab′) for the targeting of lung mesenchymal stem cells and Alexa 647 fluorescence images of the organs of a mouse administrated with Fab′-conjugated Micelle,67 (c) self-assembly of polyNIPAM-polyDEA block copolymers into a polymersome with a “boarding gate” and a “release gate”,66 (d) DLS measurements of crosslinked polymersomes at different temperatures,66 (e) in vivo transfection and expression of plasmid DNA delivered by polymersomes and controls (experiments conducted with triplicates).66 (b) Reprinted with permission from ref. 67. Copyright 2021 Wiley, (c–e) reprinted with permission from ref. 66. Copyright 2018 The American Chemical Society. | ||
Polymersomes made from diblock copolymers self-assemble in a bilayer polymer membrane,129 reminiscent of the phospholipidic double layer of the liposome. The block copolymers used can be considered synthetic mimics of phospholipid to some extent.130 Typically, the polymersomes used in the delivery of BTAs have an aqueous core separated from the surrounding aqueous environment by a semi-permeable hydrophobic membrane (Fig. 5a). In the case of polymersomes, the payloads can be located either in the hydrophobic membrane or inside the hydrophilic core, providing more versatility to encapsulate a wide variety of payloads.129
However, the formation of the self-assembled structure, such as micelles and polymersomes, is not always straightforward. Different methods exist to convert a block copolymer solution into a suspension of self-assembled structures.126 Some of the most frequently used strategies involve a “solvent exchange”. The block copolymer is first dissolved in an organic solvent, which is chosen because it is a good solvent for all the blocks, and then the solution is transferred to the selective solvent, usually water when micelles and polymersome for the encapsulation of BTAs are prepared. The solution of block copolymer in the organic solvent can be added directly to water under vigorous stirring, slowly dialyzed against water, or water can be added in a step-wise manner to the organic solution. The resulting suspension is then extensively dialyzed against water to remove any traces of the organic solvent.132 Following the formation of the polymer aggregate, the extrusion of the resulting dispersion through nanoscale pores leads to uniformization of the size and may also be used to induce the transition from spherical to cylindrical geometries in the case of micelles.133 However, the formation of large polymersomes can be challenging, and more complex formation protocols need to be implemented.134
Most micelles and polymersomes are built through the delicate balance of hydrophilic/hydrophobic segments. Interestingly, the interaction of the block copolymer with the BTAs can itself be used as the driving force for the formation of micelles. The complexation of a charged BTA and an oppositely charged polymer can lead to the formation of a polyplex (Section 3.2). However, when the polymer used is a block copolymer composed of a poly(ionomer) segment and a neutral hydrophilic segment, the loss of hydration is associated with the formation of the ionic complex and the neutralization of the charged polymer block leads to the formation of polyion complex micelles. In such a case, the hydrophobic segment is the complex of the ionomer block of the copolymer and the BTA, which is then surrounded by the neutral hydrophilic block. The neutralization of the charge of the block copolymers by a charged payload induces the required amphiphilicity for the micelle assembly.135,136
The encapsulation of the payload often occurs through a passive loading mechanism for both polymer micelles and polymersomes.137 The hydrophilic BTAis embedded in the inner aqueous core of the polymersome or a hydrophobic agent can be encapsulated in the solid core of the micelles during the nanocarrier formation. The loading efficiency of this method is usually low, and although different studies successfully encapsulated oxygen transport proteins, like hemoglobin and myoglobin, into polymersomes while retaining their bioactivity, the encapsulation efficiencies did not exceed 30%.138–140 Inversely, the use of active loading procedures, where the loading usually takes advantage of diffusion properties following a gradient of concentration across the bilayers, results in a payload encapsulation with improved efficiency compared to passive methods. However, the efficiency of this method is impeded by the large hydrodynamic radius of BTAs, which results in low diffusivity of the molecules across the bilayer structures.
Polymersome DDSs for the delivery of BTAs have entered various preclinical testings. For instance, polymersomes prepared from a poly(ethylene glycol)-b-poly(dithiolane trimethylene carbonate-co-trimethylene carbonate)-b-polyethylenimine triblock copolymers were functionalized with apolipoprotein E derived peptides showing an excellent ability to cross the blood–brain barrier and aiming at the treatment of glioblastoma, a primary tumor inside the brain that still remains incurable.141 The polymersomes loaded with the therapeutic protein saporin displayed a highly specific and potent antitumor response for orthotopic human glioblastoma xenografts in animal studies. In another very recent study, polymersomes composed of polybutadiene-b-polyethylene glycol and 1,2-dioleoyl-3-trimethylammonium-propane block copolymers were developed to co-deliver the SARS-CoV-2 spike protein and a DNA nucleotide immune-stimulatory adjuvant as a vaccine against SARS-CoV-2.69 The animal study revealed that two doses of the polymersome formulation induced neutralizing antibody titers in mice for up to 40 days, while the co-administration of the adjuvant reduced the required dosage of the SARS-CoV-2 spike protein.
In polymersomes, the incorporation of functional polymer segments in the polymer chain allows for the design of smart membranes. The release of the encapsulated molecules can then be controlled by the functionalization of the membrane with molecules tuning the permeability of the membrane and increasing the mass transport of the active molecules. When dealing with macromolecular therapeutic agents, it is especially crucial to increase the permeability of the membrane.66 Several approaches have been developed to do so, such as the functionalization of the polymersome membrane with channel-forming transmembrane proteins,142 DNA nano-pores,143 or smart gates.66 For instance, inspired by biological cell membranes, where various proteins serve for ion transport, polymersomes with selective membrane permeability were designed by introducing membrane proteins, such as aquaporin ionomycin in the membrane shell.144,145 For this purpose, the aquaporin, a natural membrane protein forming pores due to their assembly as trimers, can be mixed with the block copolymers prior to the self-assembly. After the formation of the polymersome, the proteins incorporated in the membrane allow for a selective release of molecules.142 DNA nanopores provide another possibility for the controlled transport across polymersome membranes.146 They can be fabricated through the self-assembly of oligonucleotides, and they can build controlled holes in the polymer layer.143 They have been integrated into polymersomes by incubating the polymersomes with DNA fragments after their synthesis. The DNA pores insert into the membrane, first by tethering to the membrane, followed by complete insertion depending on the concentration of the oligonucleotides. The resulting nanopores act as gates controlling the selective release of payloads based on their size.143
However, the elaborate engineering of the polymersome membranes successfully leads to the controlled release of small molecules and drugs, but the release of biomacromolecules through polymersome pores is one of the current challenges. The design of smart membranes can provide one solution to this, as it can incorporate heterogeneous membrane sections with finely controllable and responsive behavior.66 By incorporating responsive polymers in the membrane, the permeability of the membrane can be controlled. The polymers used in such cases can adjust their properties in response to external stimuli which can lead to the swelling of the membrane and the controlled release of payload. One exemplary system forming smart gates in polymersome membranes exploits the responsive polymers polyNIPAM and poly(diethylamino)ethyl methacrylate (polyDEA).66 After the self-assembly in a mixture of water and tetrahydrofuran, the thermoresponsive polyNIPAM dehydrates and the pH-responsive polyDEA functionality is deprotonated. This results in polymersomes with closed gates in the membrane. The polyNIPAM-based segment enables the design of a “boarding gate” in the membrane, opening up below the LCST of PNIPAM (20–30 °C). Under these conditions, biomacromolecules can be loaded, and the gate can be closed at physiological temperature (37 °C) to lock the payload (Fig. 5c). The pH-sensitive polyDEA segments in the polymersome membrane were designed to serve as the “release gate”, allowing the release of the encapsulated plasmid DNA in acidic environment (such as in the endosomes) upon the protonation of the polyDEA. The protonation leads to a swelling of the polymersomes from around 450 nm at pH 6.8 (Fig. 5d) to 610 nm at pH 5.4. Such polymersomes have been used for the transfection of cells with GFP-encoding plasmid DNA and the successful release of the payload in vitro and in vivo (Fig. 5e).
3.5 Nanoparticles, nanogels, and nanocapsules
Over the past decade, covalently crosslinked polymer nanocarriers have attracted growing interest for their use in the delivery BTAs due to their flexibility and adaptability in terms of hydrophilicity, stability, size, charge.147,148 Crosslinked polymer nanocarriers are nanosized networks composed of polymer chains linked together through chemical or physical crosslinking points (Fig. 6a). In a polymer nanoparticle or polymer nanogel the polymer matrix is uniformly distributed in the entire nanocarriers, while in a nanocapsule, the polymer network is segregated at the carrier surface, creating a shell that can engulf either an aqueous or organic core. The main distinction between nanoparticles and nanogels is the swelling of the polymer network. In a nanoparticle, the defined three-dimensional polymer network is mostly desolvated, while in the case of nanogels, the network holds a large quantity of solvent.149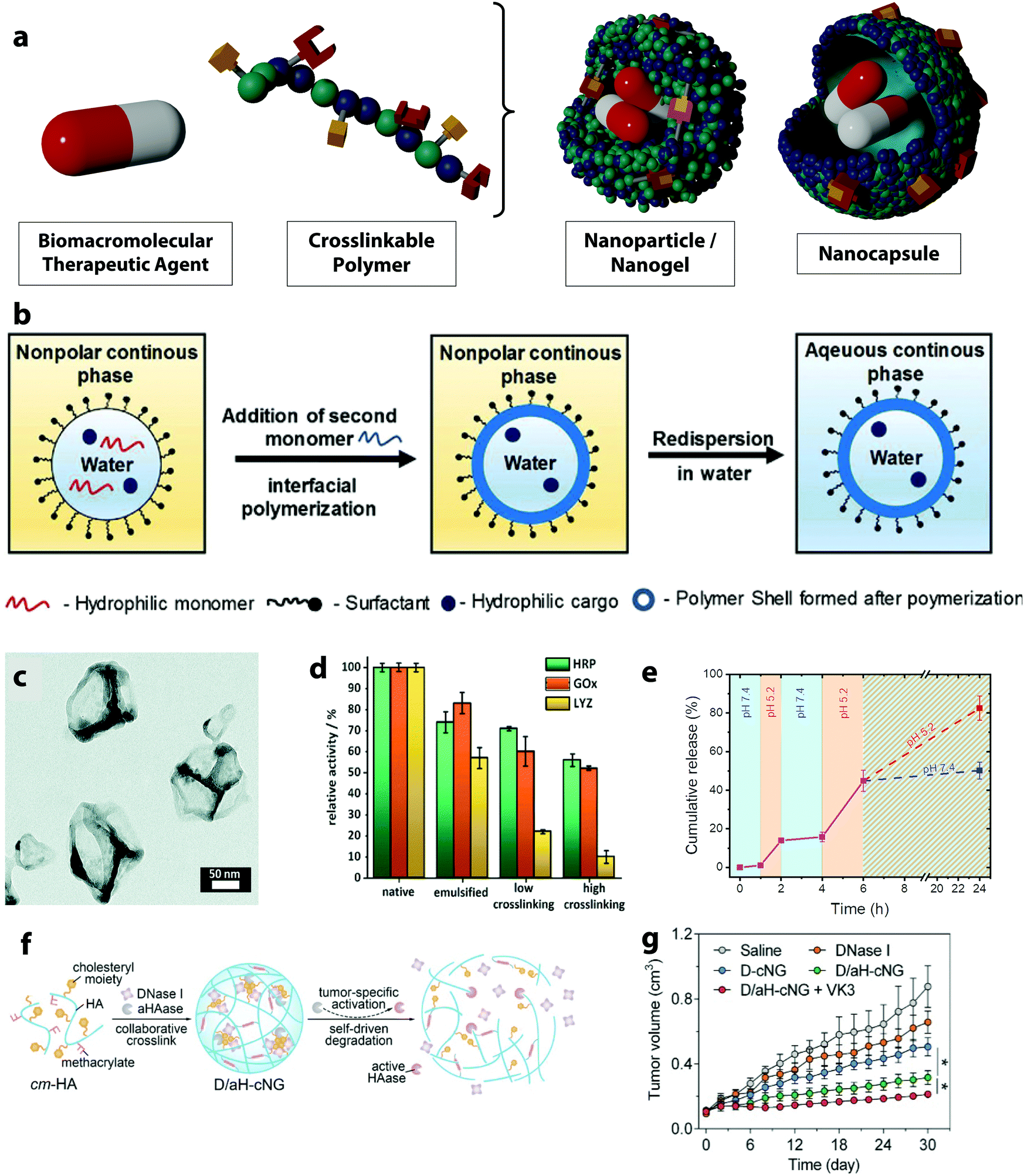 | ||
| Fig. 6 (a) Schematic representation of nanoparticles/nanogels and nanocapsules, (b) scheme of the nanocapsule formation through interfacial polymerization in miniemulsion droplets,25 (c) TEM image of horseradish peroxidase nanocapsules,71 (d) relative enzymatic activity of horseradish peroxidase, glucose oxidase and lysozyme after emulsification and crosslinking as nanocapsules,71 (e) stop-and-go release of FITC-albumin from dextran nanogels by the variation of the pH value of the environment between 5.2 and 7.4,70 (f) schematic of self-assembly and tumor-specific self-degradation of the collaboratively crosslinked, hyaluronidase (HAase)-degradable hyaluronic acid nanogels (D/aH-cNG), (g) tumor size variation of the tumor-bearing nude mice treated with different formulations: saline control, DNase I, DNase I loaded nanogels (D-cNG), nanogels loaded with DNase I and the inactive aHAase concurrently (D/aH-cNG) and D/aH-cNG nanogels with the addition of vitamin K3 (VK3).72 (b) Reprinted with permission from ref. 25. Copyright 2019 Wiley; (c and d) reprinted with permission from ref. 71. Copyright 2021 The Royal Chemical Society; (e) reprinted with permission from ref. 70. Copyright 2021 The American Chemical Society; (f and g) reprinted with permission from ref. 72. Copyright 2018 Wiley. | ||
Another technique for the preparation of nanoparticles is polymerization in heterogeneous media, like in a miniemulsion polymerization.156 When this method is applied to the encapsulation of BTAs, an aqueous solution of monomers, or already preformed polymers, is emulsified in a hydrophobic solvent, leading to an emulsion of water droplets in an oil phase stabilized by surfactants.157 The water droplets containing monomers or polymers are then polymerized or the preformed polymers are crosslinked to obtain solid polymer nanoparticles or nanogels. In contrast to the synthesis of nanoparticles and nanogels, nanocapsules can be obtained if the crosslinking agent is added to the continuous phase of the emulsion and that the reaction between the polymer and crosslinking agent occurs only at the interface of the two solvents (Fig. 6b). When the reaction kinetic is fast enough and the surface tension of the crosslinked polymer controlled, a solid polymer shell can be formed covering the precursor droplet.158 Many chemical reactions can occur at the droplet interface resulting in crosslinked core/shell structures.159 Various drugs can be loaded within the aqueous droplets, which usually results in very high encapsulation efficiencies.150,160 When mild reaction conditions are applied and the use of reagents and catalysts for the nanocapsule formation is controlled, sensitive BTAs can preserve their activity throughout this encapsulation process. For instance, such an approach allowed for the encapsulation of multiple enzymes in one nanocapsule with the aim of performing enzymatic cascade reactions.71 Similar strategies have been used to prepare both polymer and silica nanocapsules.71,161 All of the enzymes involved maintained their enzymatic activity after the encapsulation while they were successfully protected against proteases and heat. This strategy, of encapsulating biomacromolecular payload dissolved in the aqueous phase of an inverse miniemulsion by the formation of a shell at the droplet interface, has been used successfully to encapsulate enzyme and SiRNA in polymer and silica nanocapsules.162
Another technique yielding nanoparticles and nanogels without the aid of surfactants and organic solvents is precipitation polymerization. This process requires the use of a solvent, which can dissolve both the monomer and initiator or already preformed polymers and their crosslinker. Upon the initiation of the polymerization or crosslinking reaction, the polymer network becomes insoluble and precipitates as solid particles.71,157,163,164
Nanogels, usually made of hydrogel, are polymer matrices with the ability to absorb a high amount of water in their network. Such a dynamic network of swollen hydrophilic polymers can contain ions, salts, polysaccharides or other stabilizing molecules co-dissolved with the drug and provides an optimal environment during the delivery process. Conversely, nanoparticles are built with an unswollen network of rigid crosslinked polymers and usually display a very high stability during the delivery process. In all cases, the release of biological agents incorporated in those polymer systems can occur by diffusion and mass transport through the polymer network. However, the release process can also be triggered by either the degradation of the nanoparticle or by a change in the structure of the polymer nanosystems, which occurs as a consequence of environmental signals present at the intended release site.167 In either case, the crosslinking density of the polymer network decreases while the mesh size is increased. The mesh size controls steric interactions between the drugs and the polymer network. When the mesh is larger than the drug, the drug release process is dominated by the diffusion of the drug through the polymer network. By controlling the mesh size, molecules of different sizes can be released with great control. For example, the release of fluorescently labeled albumin from ketone- and aldehyde-functionalized dextran-based nanogels was controlled by a reversible crosslinking of the polymer network.70 The hydrazone network density resulting from the reaction with an adipic acid dihydrazide crosslinker was tuned by the pH-responsive equilibrium between hydrazide and hydrazone groups. Such dynamic covalent bonds undergo a reversible disassembly under an acidic pH value leading to the swelling of the nanogels by ca. 30%. As the effective crosslinking density decreases, the swelling is triggered. It is fully reversible and can be used to induce a stop-and-go release of proteins from the nanogels (Fig. 6e).
In another example, a tumor-specific self-degradable nanogel composed of a hyaluronic acid network entrapping an acid-activatable hyaluronidase enzyme for the systemic delivery of anticancer proteins was developed.168 The hyaluronidase was modified with citraconic anhydride, which shielded the enzymatic activity. It can be reversibly activated as induced by the hydrolysis of the citraconic amide under acidic pH value. The mild acidity of the tumor microenvironment hence activates the hyaluronidase enzyme resulting in the swelling of the nanogel and triggering the release of the enzyme. The released enzyme is capable of degrading the nanogel network and further induces the release of the encapsulated anticancer protein, deoxyribonuclease I intracellularly (Fig. 6f). Further, the deoxyribonuclease I digested the DNA in the cell leading to the tumor cell death and an enhanced antitumor efficacy (Fig. 6g).
Some examples of polymer nanoparticles and nanogels used for the delivery of BTAs have proceeded to preclinical trials. For instance, PLGA nanoparticles loaded with the enzyme iduronate 2-sulfatase were developed to treat the brain disease mucopolysaccharidosis type II by enzyme replacement therapy.74 This in vivo study, of a duration of six weeks, was performed on mice with mucopolysaccharidosis type II, showed a reduction of glycosaminoglycans typically accumulated in liver and brain tissues and an overall improvement of the brain pathology induced by the successful enzyme delivery after weekly administration of the nanoparticles. Another very recent study reported on the evaluation of cationic cholesteryl pullulan nanogels delivering the antigen protein pneumococcal surface protein A as a vaccine against Streptococcus pneumoniae bacteria, which can lead to respiratory infections.73 This study evaluated the immunogenicity and therapeutic efficacy of the nanogel vaccine in non-human primates (macaques). The results demonstrated that the vaccination of macaques with the nanogels induced high protection against pneumococcal infections. The macaques showed protection against pneumococcal intratracheal challenge by two mechanisms, the inhibition of possible lung inflammation and a decreased number of bacteria in the lungs.
4. Administration routes and toxicological evaluation of polymer drug delivery systems
Polymer-based drug delivery systems provide many advantages, including increased stability of the formulations and versatility in terms of biomacromolecular therapeutic agents or their combinations to be delivered, compared to alternative delivery systems. However, for their efficient transfer from the lab to the bedside, we need to evaluate their efficacy, using different potential administration routes and their resulting toxicity after administration. The efficacy and risks associated with polymer-based DDSs are influenced by both their chemical compositions and the method chosen for their administration. As discussed in the previous sections, the choice of the ideal polymer formulation is highly influenced by the nature of the biomacromolecular therapeutic agent used. The choice of the administration route is usually a function of both the therapeutic agent used and the targeted site of action.Administration routes
Potential administration routes for polymer DDSs include intravenous injections, oral administration, pulmonary inhalation, and transdermal delivery.172 Typically, the intravenous administration is accompanied by a rapid onset of action and high availability of the drug even with low doses.172 At a length scale of less than 200 nm, polymer DDSs with encapsulated BTAs can be injected intravenously without any concern about embolization and blockages of the blood vessels.173 After the injection, they are able to permeate and move through the different organs and tissues. Depending on their surface chemistry, they can bind to cell surface receptors and enter target cells for intracellular delivery of the cargos. However, there are some risks associated with intravenous injection administration because of the direct exposure of the DDSs in the systemic circulation. The challenge is to maintain the DDSs in circulation in the blood for a long enough duration to ensure that the DDSs reach the site of action.174 In general, immediately after the injection, the DDSs suffer from a passive accumulation in the liver and the kidney. Those organs have the natural function to remove foreign material, such as viruses, bacteria or the nanoparticles used as DDSs from the bloodstream.175 After their injections, many DDSs are subjected to a nonspecific uptake by the mononuclear phagocyte system, composed of different phagocytic cells, such as resident macrophages commonly found in the spleen, lymph nodes, and the liver.176 Both undesired effects depend on the size and surface functionalization of the DDSs and may be prevented by adjusting those properties.177Oral delivery is one of the administration routes associated with the largest patient compliance. Many biodegradable and non-biodegradable polymeric DDSs have been used for oral drug delivery. For instance, a study of PLGA nanoparticles encapsulating curcumin, a water-insoluble drug, analyzed the pharmacokinetics and bioavailability of the formulation in vivo and showed a 6-fold higher oral bioavailability of the nanoparticulate drug delivery system compared with the pure drug.178 In another example, polydopamine nanoparticles loaded with gambogenic acid, an anti-cancer drug, showed that after oral administration in mice, the polymer DDS increased the plasma drug concentration almost 3 times without apparent toxicity to the major organs.179 One of the major challenges associated with oral delivery is the transit through the gastrointestinal tract and the very acidic pH of this environment, which also contain a complex enzymatic system. Protecting the sensitive structure of the biomacromolecular therapeutic agent, such as genetic material, under such harsh conditions is difficult. Even ensuring the stability of the DDSs is a challenging task, even more so for systems like polyplexes and LBL assemblies relying on pH and ionic strength to prevent their dissociation. Consequently, they can be destabilized by the environmental cues encountered in the gastrointestinal tract. Different strategies have been implemented to improve the stability of the DDSs. For example, to increase the stability of a polyplex used for the treatment of diabetes by oral administration used the presence of an additive to stabilize the DDSs.180 In this system, a plasmid DNA was complexed with protamine, but the protamine used was functionalized with taurocholic acid. The resulting nonviral oral gene delivery polyplex protected the integrity of the plasmid in gastric juice and prevented its degradation by proteolytic enzymes in the environment. Furthermore, following administration, the results show significant gene expression in in vitro, in vivo, and ex vivo experiments. After administering a single oral dose of the formulation, normal blood glucose levels were maintained for up to 7 days in three diabetic mouse models and for 14 days in monkeys with diabetic conditions. Those impressive results were attributed to the incorporation of taurocholic acid into the delivery system, which led to an improvement in the stability of the DDS in a broad range of pH, preserving the integrity of the polyplex.
Pulmonary delivery also has some advantages since lungs have a high solute permeability, a large surface area for absorption, and that pulmonary delivery is non-invasive.181 For their use in pulmonary delivery, the DDSs should have the ability to be transferred into an aerosol, for example, by the nebulization of a colloidal suspension.182 Then, after the inhalation, the DDSs loaded with the BTA may provide a sustained release of the drug in the lung tissue, minimizing the required dosage frequency.181 For instance, insulin-loaded nanoparticles composed of poly(butyl cyanoacrylate) extended the blood glucose level reduction over 20 h compared to the non-encapsulated drug after pulmonary administration to rats.183 Furthermore, the local pulmonary administration of DDSs may reduce potential side effects on other organs. An interesting example showed the successful pulmonary administration of PEI-siRNA polyplexes using a liquid aerosol device. In tumor-bearing mice models, the local delivery of the siRNA polyplexes resulted in a significant accumulation of the siRNA in the lungs leading to an inhibition of the tumor growth of the metastatic lung cancer.182
Transdermal drug delivery is another possibility for a non-invasive systemic drug administration, but the inherent protective function of the skin often leads to low permeability toward external substances.184 In general, the molecular weight of the drug is a significant limitation for the dermal delivery of therapeutics, and drugs with a molecular weight of more than 500 Da cannot penetrate through the skin passively.185 However, the formulation of a drug or BTA with the appropriate polymer DDSs can increase the transdermal delivery. The penetration ability of the DDSs is affected mainly by their size.185 For instance, penetration experiments in porcine skin revealed that polystyrene nanoparticles of 20 nm size permeate more efficiently in the skin than bigger ones (200 nm).186 Furthermore, polymer DDSs are also able to penetrate skin follicles and hence allow for the opportunity of directed intrafollicular drug release.187 This release can be initiated by extrinsic or intrinsic triggers, such as low power density UVA radiation. For example, ultraviolet A-responsive polyurethane nanocapsules showed sufficient follicular penetration and intrafollicular drug release on an ex vivo porcine ear skin model while showing excellent biocompatibility before and after UVA-induced cleavage.187
Toxicology of polymer DDSs
The use of polymer-based DDSs for the delivery of biomacromolecular payloads suffers from the same limitations as DDSs used to deliver smaller therapeutic agents.188 In addition to the toxicity of the DDSs themselves, the toxicity of the polymers used and their degradation products need to be taken into account, as well as the toxicity potentially associated with an off-target delivery of the payload. The clinical application of polymer DDSs needs to be critically controlled, and the inherent toxicity of all components of the formulation has to be screened.189,190 However, there is a pervasive challenge in the study of polymer nanosystems used as DDSs; it is critical to understand and compare results obtained with different systems, there could be large variations in how such studies are performed and how the results are reported. This lack of standardization impede the community to draw clear conclusions on the interactions and process occurring at such bio-nano interface.191Several factors affect the toxicity of polymer DDSs. On the one hand, those include colloidal properties, such as the size and shape of the DDSs, the surface charge, and the surface functionalization. The size is one of the most critical parameters in influencing the toxicity of the DDS. Typically, the smaller the size of the nano-DDSs, the higher the associated toxicity will be. This phenomenon can be attributed to the increasing ability of smaller nanoparticles to cross biological barriers and reach different organs without being filtered by the liver or spleen.192 Inversely, if the DDSs are too large, they will be rapidly filtered out and cause local toxicity to the liver and spleen. Furthermore, the surface charge of the DDSs also significantly influences the interaction between the nanocarriers and the body. In general, nanocarriers with positive charges show larger toxicity compared to DDSs with negative surface charges.193
Other parameters affecting the toxicity of the DDSs are related to the properties of the polymer material. These include the origin of the polymer, the molecular weight, the hydrophilicity, the charge, and the biodegradability. Polymers from natural sources are usually more biocompatible and biodegradable compared to some synthetic polymers. Their degradation products are mostly non-toxic, even in high concentrations, since they are endogenous. Such systems made of, for example, hyaluronic acid, chitosan, starch or dextran, have been extensively used as non-toxic building blocks for the preparation of DDSs. For instance, the toxicity of chitosan DDSs upon repeated oral administrations was studied extensively. A large variety of chitosan nanosystems, such as nanoparticles composed of chitosan/alginate, chitosan/glutamic acid or oleoyl-carboxy methyl chitosan, as well as chitosan-coated PLGA DDSs, have been studied in different animal models, such rats, mice, and carps, all those studies revealed no or very limited in vivo toxicity.194–198
Many synthetic biodegradable polymer building blocks employed in the design of drug delivery systems, such as poly(lactic acid), poly(glycolic acid), and poly(lactide-co-glycolide), have already been approved by the US Food and Drug Administration and the European Medicines Agency due to their safety, biocompatibility, and successful biodegradation during in vitro and in vivo studies.190 Hence, the design of nanoparticulate formulations using such polymers shows a high potential for a satisfactory toxicological response. For instance, PLGA nanoparticles used in A549 human lung cancer cells at a large concentration range (300–5000 μg mL−1) showed no toxicity even at the highest concentration.199 Similarly, the administration of poly(n-butylcyano-acrylate) nanoparticles coated with polysorbate 80 in vivo in rats showed almost no cytotoxic or inflammatory effects at therapeutic concentrations lower than 500 μg mL−1.200 Additionally, the use of biological coating materials such as chitosan or serum proteins may even further increase the biocompatibility of polymer nanostructures. In this regard, an interesting study demonstrated a reduction of the cytotoxicity of PLGA nanoparticles stabilized with bovine serum protein when compared to synthetic coating materials in cultured lung cancer cells.201
At the opposite end of the spectra, charged polymers like those used to prepare delivery carriers based on electrostatic interaction, such as polyplexes or LbL assemblies, may exhibit critical toxicological properties. Cationic polymers are known to interact with the anionic molecules present at the surface of different types of cells, and the use of such polymers can damage cells and tissues.202 For instance, the toxicology of poly(ethyleneimine) was extensively studied and the results revealed two distinct stages of the cytotoxicity of PEI.203 The first stage of cytotoxicity is a rapid response; for example, PEI/pDNA polyplexes and free PEI polymers bound to cell membranes induced membrane damages in three clinically relevant human cell lines (Jurkat T cells, umbilical vein endothelial cells, and THLE3 hepatocyte-like cells) within 30min following the exposure.203 Then, the second stage of cytotoxicity occurs after longer exposition to the DDSs; for the same PEI polyplex, severe apoptotic changes of the cells were observed after 24h of exposure. Hence, to avoid the potential cytotoxicity, cationic polymers used in drug delivery systems could be designed with the inclusion of degradable functionalities. Some promising examples of this approach include the crosslinking of low-molecular-weight PEI with degradable linkers, such as ester bonds, disulfide bonds, and ketal functions.204,205 Anther possible approach is the coating of cationic polymer DDSs with neutral or anionic polymers in order to increase their biocompatibility. For instance, cationic polymer micelles composed of a diblock amphiphilic copolymer of PEI and poly(DL-lactic acid) were coated with polycarboxylic acid dextran after the binding of survivin-shRNA.206 The polymer micelles were used in anti-cancer therapy and resulted in significant inhibition of the tumor growth in C26 tumor-bearing mice without a sign of systemic toxicity as monitored by the bodyweight loss and survival rate of the mice during the treatment.
5. Conclusion
Biomacromolecular therapeutic agents are promising candidates for the treatment of many complex and severe diseases. However, due to their inherent low stability and poor delivery potential under in vivo conditions, the design of tailored and multi-functional DDSs is essential. The ideal drug delivery system for BTAs should overcome the following specific challenges: preserve and protect the vulnerable structure of the biomacromolecule, and enable the intracellular uptake of the therapeutic agent. Traditional DDSs, such as liposomal formulations, have shown great success in the delivery of BTAs. However, these DDSs are inherently unable to simultaneously solve all the challenges encountered in the delivery BTAs.Currently, because of advances in protein and oligonucleotide synthesis and isolation, the development and approval of new biomacromolecular therapeutic agents are increasing. Consequently, the design of appropriate delivery systems for such drugs remains a significant challenge to tackle. While polymer-conjugates of several biomacromolecular therapeutic agents have already been approved and are currently being used by patients, other types of polymer DDSs have primarily only been evaluated in in vitro and in vivo screenings, with a very small number of cases proceeding to preclinical and clinical trials. This situation can be mainly attributed to the complexity in their design, which can become a hurdle when it comes time to scale up the production while maintaining control over the chemistry, structure, and properties during this manufacturing step. The apparent dilemma in the field is that successful DDSs require a sophisticated structure and smart functionalities to simultaneously protect the biomacromolecular therapeutic agent, successfully deliver the payload to the intended site of action, and controlled the release, but such complex systems are challenging to scale up. Therefore, it is likely that DDSs combining the desired pharmacokinetic properties with some degree of design simplicity will more easily translate from the lab to applications. To allow for more complex DDSs to thrive and get approval by the authorities for human use, more attention has to be paid to the development of approaches with a high batch-to-batch reproducibility compatible with industrial scale-up production.207
The success rate of new treatments entering clinical trials is low no matter the type of drugs or delivery systems.208 However, this is particularly the case for nanodelivery systems where the translational efficiency and the clinical success rate are both (still) very low.22 In order to change this situation, a more systematic study of the nano-bio interaction is needed,191 and in addition to some degree of standardization in the field, a broader scope of models both in terms of diseases and animal models should be considered.209
Polymer drug delivery systems are unique among DDSs, since they can combine multiple functionalities in one nanosystem due to the flexibility in their synthesis. The resulting polymer DDSs can address all the challenges faced by the delivery of biomacromolecules. Their unique strength lies in the unlimited variety of potential starting materials resulting in precisely controlled and rationally designed systems. The different architectures of polymer DDSs provide versatile strategies for the successful delivery of the most challenging payloads.
Some key advantages of polymer DDSs are their high stability in complex environments even at low concentration, the ability to introduce active targeting ligands but also to tune passive targeting by controlling their size and shape, their high loading efficiency, their ability to co-deliver multiple therapeutics regardless of their hydrophilicity and molecular weight, and the selective and controlled release of cargo.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Else Kröner-Fresenius-Stiftung, the Max Planck Society, the MaxSynBio consortium, and the Deutsche Forschungsgemeinschaft (SFB1066) for their financial support. The help of Stefan Schuhmacher with graphical design is acknowledged. Open Access funding provided by the Max Planck Society.Notes and references
- S. Mitragotri, P. A. Burke and R. Langer, Nat. Rev. Drug Discovery, 2014, 13, 655–672 CrossRef CAS.
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS PubMed.
- J. A. Kulkarni, D. Witzigmann, S. B. Thomson, S. Chen, B. R. Leavitt, P. R. Cullis and R. van der Meel, Nat. Nanotechnol., 2021, 16, 630–643 CrossRef CAS PubMed.
- B. C. Evans, R. B. Fletcher, K. V. Kilchrist, E. A. Dailing, A. J. Mukalel, J. M. Colazo, M. Oliver, J. Cheung-Flynn, C. M. Brophy, J. W. Tierney, J. S. Isenberg, K. D. Hankenson, K. Ghimire, C. Lander, C. A. Gersbach and C. L. Duvall, Nat. Commun., 2019, 10, 1–19 CrossRef CAS PubMed.
- G. Walsh, Appl. Microbiol. Biotechnol., 2005, 67, 151–159 CrossRef CAS PubMed.
- C. Sheridan, Nat. Rev. Drug Discovery, 2006, 5, 445 CrossRef CAS PubMed.
- A. L. Nelson, E. Dhimolea and J. M. Reichert, Nat. Rev. Drug Discovery, 2010, 9, 767–774 CrossRef CAS PubMed.
- G. Walsh, Nat. Biotechnol., 2018, 36, 1136–1145 CrossRef CAS.
- A. Wolfram Julie and J. K. Donahue, J. Am. Heart Assoc., 2013, 2, 119 Search PubMed.
- I. Lostalé-Seijo and J. Montenegro, Nat. Rev. Chem., 2018, 2, 258–277 CrossRef.
- N. G. Rainov, Hum. Gene Ther., 2000, 11, 2389–2401 CrossRef CAS PubMed.
- W. J. Marks, J. L. Ostrem, L. Verhagen, P. A. Starr, P. S. Larson and R. A. Bakay, Lancet Neurol., 2008, 7, 400–408 CrossRef.
- R. T. Bartus, T. L. Baumann, J. Siffert, C. D. Herzog, R. Alterman, N. Boulis, D. A. Turner, M. Stacy, A. E. Lang, A. M. Lozano and C. W. Olanow, Neurology, 2013, 80, 1698 CrossRef CAS PubMed.
- F. P. Polack, S. J. Thomas, N. Kitchin, J. Absalon, A. Gurtman, S. Lockhart, J. L. Perez, G. Pérez Marc, E. D. Moreira, C. Zerbini, R. Bailey, K. A. Swanson, S. Roychoudhury, K. Koury, P. Li, W. V. Kalina, D. Cooper, R. W. Frenck, L. L. Hammitt, Ö. Türeci, H. Nell, A. Schaefer, S. Ünal, D. B. Tresnan, S. Mather, P. R. Dormitzer, U. Şahin, K. U. Jansen and W. C. Gruber, N. Engl. J. Med., 2020, 383, 2603–2615 CrossRef CAS PubMed.
- J. Houseley and D. Tollervey, Cell, 2009, 136, 763–776 CrossRef CAS.
- T. Kisby, A. Yilmazer and K. Kostarelos, Nat. Nanotechnol., 2021, 16, 843–850 CrossRef CAS PubMed.
- D. J. A. Crommelin, T. J. Anchordoquy, D. B. Volkin, W. Jiskoot and E. Mastrobattista, J. Pharm. Sci., 2021, 110, 997–1001 CrossRef CAS PubMed.
- F. Vahedifard and K. Chakravarthy, Emergent Mater., 2021, 4, 75–99 CrossRef CAS.
- A. C. Anselmo and S. Mitragotri, Bioeng. Trans. Med., 2019, 4, e10143 Search PubMed.
- O. S. Fenton, K. N. Olafson, P. S. Pillai, M. J. Mitchell and R. Langer, Adv. Mater., 2018, 30, 1705328 CrossRef.
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Pharm. Res., 2016, 33, 2373–2387 CrossRef CAS PubMed.
- H. Zhong, G. Chan, Y. Hu, H. Hu and D. Ouyang, Pharmaceutics, 2018, 10, 263 CrossRef CAS PubMed.
- K. R. Sims, J. P. Maceren, A. I. Strand, B. He, C. Overby and D. S. W. Benoit, RSC Adv., 2020, 10, 2513–2518 RSC.
- Z. Tang, C. He, H. Tian, J. Ding, B. S. Hsiao, B. Chu and X. Chen, Prog. Polym. Sci., 2016, 60, 86–128 CrossRef CAS.
- B. Iyisan and K. Landfester, Macromol. Rapid Commun., 2019, 40, 1800577 CrossRef PubMed.
- N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Chem. Rev., 2016, 116, 2602–2663 CrossRef CAS PubMed.
- J. Wang, Y. Li and G. Nie, Nat. Rev. Mater., 2021, 6, 766–783 CrossRef CAS.
- L. Zhao, T.-h. Ren and D. D. Wang, Acta Pharmacol. Sin., 2012, 33, 1339–1347 CrossRef CAS PubMed.
- K. A. Dill, Biochemistry, 1990, 29, 7133–7155 CrossRef CAS PubMed.
- S. Burge, G. N. Parkinson, P. Hazel, A. K. Todd and S. Neidle, Nucleic Acids Res., 2006, 34, 5402–5415 CrossRef CAS.
- S. Jung and I. Kwon, Polym. Chem., 2016, 7, 4584–4598 RSC.
- K. Samejima and W. C. Earnshaw, Nat. Rev. Mol. Cell Biol., 2005, 6, 677–688 CrossRef CAS.
- C. López-Otín and J. S. Bond, J. Biol. Chem., 2008, 283, 30433–30437 CrossRef PubMed.
- M. Werle and A. Bernkop-Schnürch, Amino Acids, 2006, 30, 351–367 CrossRef CAS PubMed.
- G. B. Bolli, D. Kerr, R. Thomas, E. Torlone, A. Sola-Gazagnes, E. Vitacolonna, J. L. Selam and P. D. Home, Diabetes Care, 2009, 32, 1170 CrossRef CAS.
- F. Scaletti, J. Hardie, Y.-W. Lee, D. C. Luther, M. Ray and V. M. Rotello, Chem. Soc. Rev., 2018, 47, 3421–3432 RSC.
- J. Lombard, Biol. Direct, 2014, 9, 32 CrossRef PubMed.
- P. Matsson and J. Kihlberg, J. Med. Chem., 2017, 60, 1662–1664 CrossRef CAS PubMed.
- N. J. Yang and M. J. Hinner, in Site-Specific Protein Labeling: Methods and Protocols, ed. A. Gautier and M. J. Hinner, Springer, New York, 2015 Search PubMed.
- F.-S. Du, Y. Wang, R. Zhang and Z.-C. Li, Soft Matter, 2010, 6, 835–848 RSC.
- G. Gregoriadis, N. Engl. J. Med., 1976, 295, 765–770 CrossRef CAS PubMed.
- Nat. Rev. Mater., 2021, 6, 99 Search PubMed.
- J. A. Zuris, D. B. Thompson, Y. Shu, J. P. Guilinger, J. L. Bessen, J. H. Hu, M. L. Maeder, J. K. Joung, Z.-Y. Chen and D. R. Liu, Nat. Biotechnol., 2015, 33, 73–80 CrossRef CAS PubMed.
- R. Münter, K. Kristensen, D. Pedersbæk, J. B. Larsen, J. B. Simonsen and T. L. Andresen, Nanoscale, 2018, 10, 22720–22724 RSC.
- D. J. McClements, Adv. Colloid Interface Sci., 2018, 253, 1–22 CrossRef CAS PubMed.
- S. H. Lee, Y. Y. Kang, H.-E. Jang and H. Mok, Adv. Drug Deliv. Rev., 2016, 104, 78–92 CrossRef CAS PubMed.
- I. Ekladious, Y. L. Colson and M. W. Grinstaff, Nat. Rev. Drug Discovery, 2019, 18, 273–294 CrossRef CAS PubMed.
- Y. Wu, D. Y. W. Ng, S. L. Kuan and T. Weil, Biomater. Sci., 2015, 3, 214–230 RSC.
- E. Steiert, J. Ewald, A. Wagner, U. A. Hellmich, H. Frey and P. R. Wich, Polym. Chem., 2020, 11, 551–559 RSC.
- I. Cobo, M. Li, B. S. Sumerlin and S. Perrier, Nat. Mater., 2015, 14, 143–159 CrossRef CAS PubMed.
- J. D. Schulz, M. Patt, S. Basler, H. Kries, D. Hilvert, M. A. Gauthier and J.-C. Leroux, Adv. Mater., 2016, 28, 1455–1460 CrossRef CAS PubMed.
- S. Averick, O. Karácsony, J. Mohin, X. Yong, N. M. Moellers, B. F. Woodman, W. Zhu, R. A. Mehl, A. C. Balazs, T. Kowalewski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2014, 53, 8050–8055 CrossRef CAS PubMed.
- H. Cho, T. Daniel, Y. J. Buechler, D. C. Litzinger, Z. Maio, A.-M. H. Putnam, V. S. Kraynov, B.-C. Sim, S. Bussell, T. Javahishvili, S. Kaphle, G. Viramontes, M. Ong, S. Chu, B. Gc, R. Lieu, N. Knudsen, P. Castiglioni, T. C. Norman, D. W. Axelrod, A. R. Hoffman, P. G. Schultz, R. D. DiMarchi and B. E. Kimmel, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9060 CrossRef CAS PubMed.
- A. C. Braun, M. Gutmann, T. D. Mueller, T. Lühmann and L. Meinel, J. Controlled Release, 2018, 279, 17–28 CrossRef CAS PubMed.
- C. Bao, J. Chen, D. Li, A. Zhang and Q. Zhang, Polym. Chem., 2020, 11, 1386–1392 RSC.
- Y.-A. Heo, Y. Y. Syed and S. J. Keam, Drugs, 2019, 79, 767–777 CrossRef CAS PubMed.
- T. M. Herndon, S. G. Demko, X. Jiang, K. He, J. E. Gootenberg, M. H. Cohen, P. Keegan and R. Pazdur, The Oncologist, 2012, 17, 1323–1328 CrossRef CAS.
- A. M. M. Eggermont, S. Suciu, M. Santinami, A. Testori, W. H. J. Kruit, J. Marsden, C. J. A. Punt, F. Salès, M. Gore, R. MacKie, Z. Kusic, R. Dummer, A. Hauschild, E. Musat, A. Spatz and U. Keilholz, The Lancet, 2008, 372, 117–126 CrossRef CAS.
- K. Kim, H. S. Hwang, M. S. Shim, Y.-Y. Cho, J. Y. Lee, H. S. Lee and H. C. Kang, Colloids Surf., B, 2019, 184, 110497 CrossRef CAS PubMed.
- Y. Dong, T. Yu, L. Ding, E. Laurini, Y. Huang, M. Zhang, Y. Weng, S. Lin, P. Chen, D. Marson, Y. Jiang, S. Giorgio, S. Pricl, X. Liu, P. Rocchi and L. Peng, J. Am. Chem. Soc., 2018, 140, 16264–16274 CrossRef CAS PubMed.
- O. K. Appelbe, B.-K. Kim, N. Rymut, J. Wang, S. J. Kron and Y. Yeo, Cancer Gene Ther., 2018, 25, 196–206 CrossRef CAS PubMed.
- L. Buscail, B. Bournet, F. Vernejoul, G. Cambois, H. Lulka, N. Hanoun, M. Dufresne, A. Meulle, A. Vignolle-Vidoni, L. Ligat, N. Saint-Laurent, F. Pont, S. Dejean, M. Gayral, F. Martins, J. Torrisani, O. Barbey, F. Gross, R. Guimbaud, P. Otal, F. Lopez, G. Tiraby and P. Cordelier, Mol. Ther., 2015, 23, 779–789 CrossRef CAS.
- J. E. Zuckerman, I. Gritli, A. Tolcher, J. D. Heidel, D. Lim, R. Morgan, B. Chmielowski, A. Ribas, M. E. Davis and Y. Yen, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11449 CrossRef CAS PubMed.
- B. G. De Geest, M. A. Willart, B. N. Lambrecht, C. Pollard, C. Vervaet, J. P. Remon, J. Grooten and S. De Koker, Angew. Chem., Int. Ed., 2012, 51, 3862–3866 CrossRef CAS PubMed.
- Z. J. Deng, S. W. Morton, E. Ben-Akiva, E. C. Dreaden, K. E. Shopsowitz and P. T. Hammond, ACS Nano, 2013, 7, 9571–9584 CrossRef CAS.
- F. Wang, J. Gao, J. Xiao and J. Du, Nano Lett., 2018, 18, 5562–5568 CrossRef CAS PubMed.
- Q. Ji, J. Hou, X. Yong, G. Gong, M. Muddassir, T. Tang, J. Xie, W. Fan and X. Chen, Adv. Mater., 2021, 33, 2007798 CrossRef CAS PubMed.
- C.-Y. Lin, F. Perche, M. Ikegami, S. Uchida, K. Kataoka and K. Itaka, J. Controlled Release, 2016, 235, 268–275 CrossRef CAS PubMed.
- J. H. Lam, A. K. Khan, T. A. Cornell, T. W. Chia, R. J. Dress, W. W. W. Yeow, N. K. Mohd-Ismail, S. Venkatraman, K. T. Ng, Y.-J. Tan, D. E. Anderson, F. Ginhoux and M. Nallani, ACS Nano, 2021, 15, 15754–15770 CrossRef CAS PubMed.
- M. S. Alkanawati, M. Machtakova, K. Landfester and H. Thérien-Aubin, Biomacromolecules, 2021, 22, 2976–2984 CrossRef CAS.
- M. Machtakova, S. Han, Y. Yangazoglu, I. Lieberwirth, H. Thérien-Aubin and K. Landfester, Nanoscale, 2021, 13, 4051–4059 RSC.
- Q. Zhu, X. Chen, X. Xu, Y. Zhang, C. Zhang and R. Mo, Adv. Funct. Mater., 2018, 28, 1707371 CrossRef.
- R. Nakahashi-Ouchida, Y. Uchida, Y. Yuki, Y. Katakai, T. Yamanoue, H. Ogawa, Y. Munesue, N. Nakano, K. Hanari, T. Miyazaki, Y. Saito, S. Umemoto, S.-I. Sawada, R. Mukerji, D. E. Briles, Y. Yasutomi, K. Akiyoshi and H. Kiyono, Vaccine, 2021, 39, 3353–3364 CrossRef CAS PubMed.
- L. Rigon, M. Salvalaio, F. Pederzoli, E. Legnini, J. T. Duskey, F. D’Avanzo, C. De Filippis, B. Ruozi, O. Marin, M. A. Vandelli, I. Ottonelli, M. Scarpa, G. Tosi and R. Tomanin, Int. J. Mol. Sci., 2019, 20, 214 CrossRef PubMed.
- H. Schlaad, Bio-synthetic Polymer Conjugates, Springer-Verlag, Berlin Heidelberg, 2013 Search PubMed.
- E. M. Pelegri-O’Day and H. D. Maynard, Acc. Chem. Res., 2016, 49, 1777–1785 CrossRef PubMed.
- W. Zhao, F. Liu, Y. Chen, J. Bai and W. Gao, Polymer, 2015, 66, 1–10 CrossRef.
- B. S. Lele, H. Murata, K. Matyjaszewski and A. J. Russell, Biomacromolecules, 2005, 6, 3380–3387 CrossRef CAS PubMed.
- K. M. Mansfield and H. D. Maynard, ACS Macro Lett., 2018, 7, 324–329 CrossRef CAS PubMed.
- A. Bastida, P. Sabuquillo, P. Armisen, R. Fernández-Lafuente, J. Huguet and J. M. Guisán, Biotechnol. Bioeng., 1998, 58, 486–493 CrossRef CAS PubMed.
- R. D. Schmid and R. Verger, Angew. Chem., Int. Ed., 1998, 37, 1608–1633 CrossRef.
- J. Khandare and T. Minko, Prog. Polym. Sci., 2006, 31, 359–397 CrossRef CAS.
- D. Charych, S. Khalili, V. Dixit, P. Kirk, T. Chang, J. Langowski, W. Rubas, S. K. Doberstein, M. Eldon, U. Hoch and J. Zalevsky, PLoS One, 2017, 12, e0179431 CrossRef PubMed.
- D. H. Charych, U. Hoch, J. L. Langowski, S. R. Lee, M. K. Addepalli, P. B. Kirk, D. Sheng, X. Liu, P. W. Sims, L. A. VanderVeen, C. F. Ali, T. K. Chang, M. Konakova, R. L. Pena, R. S. Kanhere, Y. M. Kirksey, C. Ji, Y. Wang, J. Huang, T. D. Sweeney, S. S. Kantak and S. K. Doberstein, Clin. Cancer Res., 2016, 22, 680 CrossRef CAS PubMed.
- H. Lv, S. Zhang, B. Wang, S. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS PubMed.
- M. Lucius, R. Falatach, C. McGlone, K. Makaroff, A. Danielson, C. Williams, J. C. Nix, D. Konkolewicz, R. C. Page and J. A. Berberich, Biomacromolecules, 2016, 17, 1123–1134 CrossRef CAS PubMed.
- X. Pang, Y. Jiang, Q. Xiao, A. W. Leung, H. Hua and C. Xu, J. Controlled Release, 2016, 222, 116–129 CrossRef CAS PubMed.
- S. L. Turgeon, C. Schmitt and C. Sanchez, Curr. Opin. Colloid Interface Sci., 2007, 12, 166–178 CrossRef CAS.
- C. G. de Kruif, F. Weinbreck and R. de Vries, Curr. Opin. Colloid Interface Sci., 2004, 9, 340–349 CrossRef CAS.
- E. Kizilay, A. B. Kayitmazer and P. L. Dubin, Adv. Colloid Interface Sci., 2011, 167, 24–37 CrossRef CAS PubMed.
- C. H. Porcel and J. B. Schlenoff, Biomacromolecules, 2009, 10, 2968–2975 CrossRef CAS.
- Q. Wang and J. B. Schlenoff, Macromolecules, 2014, 47, 3108–3116 CrossRef CAS.
- R. G. Winkler and A. G. Cherstvy, in Polyelectrolyte Complexes in the Dispersed and Solid State I: Principles and Theory, ed. M. Müller, Springer, Berlin, 2014, pp. 1–56 Search PubMed.
- M. Ogris, S. Brunner, S. Schüller, R. Kircheis and E. Wagner, Gene Ther., 1999, 6, 595–605 CrossRef CAS PubMed.
- J. V. D. Gucht, E. Spruijt, M. Lemmers and M. A. Cohen Stuart, J. Colloid Interface Sci., 2011, 361, 407–422 CrossRef PubMed.
- D. Priftis and M. Tirrell, Soft Matter, 2012, 8, 9396–9405 RSC.
- E. Lai and J. H. van Zanten, Biophys. J., 2001, 80, 864–873 CrossRef CAS PubMed.
- A. Dinari, T. T. Moghadam, M. Abdollahi and M. Sadeghizadeh, Sci. Rep., 2018, 8, 8112 CrossRef.
- F. Lotfipour, S. Hallaj-Nezhadi, H. Valizadeh, S. Dastmalchi, B. Baradaran, M. B. Jalali and F. Dobakhti, J. Pharm. Pharm. Sci., 2011, 14, 181–195 Search PubMed.
- A. K. Blakney, G. Yilmaz, P. F. McKay, C. R. Becer and R. J. Shattock, Biomacromolecules, 2018, 19, 2870–2879 CrossRef CAS PubMed.
- A. Ahmad, F. Khan, R. K. Mishra and R. Khan, J. Med. Chem., 2019, 62, 10475–10496 CrossRef CAS PubMed.
- K.-K. H. Kuo, P.-J. Hsiao, W.-T. Chang, S.-C. Chuang, Y.-H. Yang, K. Wuputra, C.-C. Ku, J.-B. Pan, C.-P. Li, K. Kato, C.-J. Liu, D.-C. Wu and K. K. Yokoyama, Cancers, 2021, 13, 3920 CrossRef CAS PubMed.
- ClinicalTrials.gov. National Library of Medicine (U.S.)., Effect of intratumoral injection of gene therapy for locally advanced pancreatic cancer (THERGAP-02), Identifier NCT02806687, Retrieved October 05, 2021 from https://clinicaltrials.gov/ct2/show/study/NCT02806687.
- W. R. Gombotz and D. K. Pettit, Bioconjugate Chem., 1995, 6, 332–351 CrossRef CAS PubMed.
- K. A. Mislick and J. D. Baldeschwieler, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 12349–12354 CrossRef CAS PubMed.
- A. Akinc, M. Thomas, A. M. Klibanov and R. Langer, J. Gene Med., 2005, 7, 657–663 CrossRef CAS PubMed.
- T. Bus, A. Traeger and U. S. Schubert, J. Mater. Chem. B, 2018, 6, 6904–6918 RSC.
- E. C. Freeman, L. M. Weiland and W. S. Meng, J. Biomater. Sci., Polym. Ed., 2013, 24, 398–416 CrossRef CAS PubMed.
- H. Chang, J. Lv, X. Gao, X. Wang, H. Wang, H. Chen, X. He, L. Li and Y. Cheng, Nano Lett., 2017, 17, 1678–1684 CrossRef CAS.
- V. Postupalenko, D. Desplancq, I. Orlov, Y. Arntz, D. Spehner, Y. Mely, B. P. Klaholz, P. Schultz, E. Weiss and G. Zuber, Angew. Chem., Int. Ed., 2015, 54, 10583–10586 CrossRef CAS PubMed.
- J.-G. Piao, J.-J. Yan, M.-Z. Wang, D.-C. Wu and Y.-Z. You, Biomater. Sci., 2014, 2, 390–398 RSC.
- X. Q. Liu and C. Picart, Adv. Mater., 2016, 28, 1295–1301 CrossRef CAS PubMed.
- P. T. Hammond, Adv. Mater., 2004, 16, 1271–1293 CrossRef CAS.
- J. J. Richardson, M. Björnmalm and F. Caruso, Science, 2015, 348, aaa2491 CrossRef PubMed.
- J. J. Richardson, J. Cui, M. Björnmalm, J. A. Braunger, H. Ejima and F. Caruso, Chem. Rev., 2016, 116, 14828–14867 CrossRef CAS PubMed.
- A. M. T. San Juan, T. Rodgers, C. Bedolla, F. Noriega and G. Romero, J. Appl. Polym. Sci., 2020, 137, 49377 CrossRef CAS.
- Y. Yan, M. Björnmalm and F. Caruso, Chem. Mater., 2014, 26, 452–460 CrossRef CAS.
- S. Zhao, F. Caruso, L. Dähne, G. Decher, B. G. D. Geest, J. Fan, N. Feliu, Y. Gogotsi, P. T. Hammond, M. C. Hersam, A. Khademhosseini, N. Kotov, S. Leporatti, Y. Li, F. Lisdat, L. M. Liz-Marzán, S. Moya, P. Mulvaney, A. L. Rogach, S. Roy, D. G. Shchukin, A. G. Skirtach, M. M. Stevens, G. B. Sukhorukov, P. S. Weiss, Z. Yue, D. Zhu and W. J. Parak, ACS Nano, 2019, 13, 6151–6169 CrossRef CAS PubMed.
- F. Caruso and C. Schüler, Langmuir, 2000, 16, 9595–9603 CrossRef CAS.
- A. Küchler, M. Yoshimoto, S. Luginbühl, F. Mavelli and P. Walde, Nat. Nanotechnol., 2016, 11, 409–420 CrossRef PubMed.
- R. Yang, J. Xu, L. Xu, X. Sun, Q. Chen, Y. Zhao, R. Peng and Z. Liu, ACS Nano, 2018, 12, 5121–5129 CrossRef CAS PubMed.
- M. D. Shin, S. Shukla, Y. H. Chung, V. Beiss, S. K. Chan, O. A. Ortega-Rivera, D. M. Wirth, A. Chen, M. Sack, J. K. Pokorski and N. F. Steinmetz, Nat. Nanotechnol., 2020, 15, 646–655 CrossRef CAS.
- T. Tang, T. Weng, H. Jia, S. Luo, Y. Xu, L. Li and P. Zhang, Biomater. Sci., 2019, 7, 715–732 RSC.
- F.-X. Xiao, M. Pagliaro, Y.-J. Xu and B. Liu, Chem. Soc. Rev., 2016, 45, 3088–3121 RSC.
- A. M. Jhaveri and V. P. Torchilin, Front. Pharmacol., 2014, 5, 77 Search PubMed.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 8572–8610 RSC.
- E. B. Zhulina and O. V. Borisov, Macromolecules, 2012, 45, 4429–4440 CrossRef CAS.
- S. Yorulmaz Avsar, M. Kyropoulou, S. Di Leone, C.-A. Schoenenberger, W. P. Meier and C. G. Palivan, Front. Chem., 2019, 6, 645 CrossRef PubMed.
- J. S. Lee and J. Feijen, J. Controlled Release, 2012, 161, 473–483 CrossRef CAS PubMed.
- C. LoPresti, H. Lomas, M. Massignani, T. Smart and G. Battaglia, J. Mater. Chem., 2009, 19, 3576–3590 RSC.
- M.-C. Jones and J.-C. Leroux, Eur. J. Pharm. Biopharm., 1999, 48, 101–111 CrossRef CAS.
- K. Kita-Tokarczyk, J. Grumelard, T. Haefele and W. Meier, Polymer, 2005, 46, 3540–3563 CrossRef CAS.
- Q. Chen, H. Zhao, T. Ming, J. Wang and C. Wu, J. Am. Chem. Soc., 2009, 131, 16650–16651 CrossRef CAS PubMed.
- L. Caire da Silva, S. Cao and K. Landfester, ACS Macro Lett., 2021, 10, 401–405 CrossRef CAS.
- A. Harada and K. Kataoka, Science, 1999, 283, 65 CrossRef CAS PubMed.
- H. Cabral, K. Miyata, K. Osada and K. Kataoka, Chem. Rev., 2018, 118, 6844–6892 CrossRef CAS.
- P. L. Chariou, O. A. Ortega-Rivera and N. F. Steinmetz, ACS Nano, 2020, 14, 2678–2701 CrossRef CAS.
- S. Iqbal, M. Blenner, A. Alexander-Bryant and J. Larsen, Biomacromolecules, 2020, 21, 1327–1350 CrossRef CAS PubMed.
- S. Rameez, H. Alosta and A. F. Palmer, Bioconjugate Chem., 2008, 19, 1025–1032 CrossRef CAS PubMed.
- A. Kishimura, A. Koide, K. Osada, Y. Yamasaki and K. Kataoka, Angew. Chem., Int. Ed., 2007, 46, 6085–6088 CrossRef CAS PubMed.
- Y. Jiang, J. Zhang, F. Meng and Z. Zhong, ACS Nano, 2018, 12, 11070–11079 CrossRef CAS PubMed.
- T. Einfalt, R. Goers, I. A. Dinu, A. Najer, M. Spulber, O. Onaca-Fischer and C. G. Palivan, Nano Lett., 2015, 15, 7596–7603 CrossRef CAS PubMed.
- L. Messager, J. R. Burns, J. Kim, D. Cecchin, J. Hindley, A. L. B. Pyne, J. Gaitzsch, G. Battaglia and S. Howorka, Angew. Chem., Int. Ed., 2016, 55, 11106–11109 CrossRef CAS PubMed.
- M. Lomora, F. Itel, I. A. Dinu and C. G. Palivan, Phys. Chem. Chem. Phys., 2015, 17, 15538–15546 RSC.
- M. Kumar, M. Grzelakowski, J. Zilles, M. Clark and W. Meier, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20719 CrossRef CAS.
- J. R. Burns, E. Stulz and S. Howorka, Nano Lett., 2013, 13, 2351–2356 CrossRef CAS PubMed.
- I. Neamtu, A. G. Rusu, A. Diaconu, L. E. Nita and A. P. Chiriac, Drug Delivery, 2017, 24, 539–557 CrossRef CAS PubMed.
- E. Mauri, G. Perale and F. Rossi, ACS Appl. Mater. Interfaces, 2018, 1, 6525–6541 CAS.
- H.-Q. Wu and C.-C. Wang, Langmuir, 2016, 32, 6211–6225 CrossRef CAS PubMed.
- K. Landfester, Angew. Chem., Int. Ed., 2009, 48, 4488–4507 CrossRef CAS PubMed.
- L.-P. Lv, Y. Zhao, N. Vilbrandt, M. Gallei, A. Vimalanandan, M. Rohwerder, K. Landfester and D. Crespy, J. Am. Chem. Soc., 2013, 135, 14198–14205 CrossRef CAS.
- K. Maruya-Li, C. Shetty, A. M. Jazani, N. Arezi and J. K. Oh, ACS Omega, 2020, 5, 3734–3742 CrossRef CAS PubMed.
- K. Kolouchova, O. Sedlacek, D. Jirak, D. Babuka, J. Blahut, J. Kotek, M. Vit, J. Trousil, R. Konefał, O. Janouskova, B. Podhorska, M. Slouf and M. Hruby, Biomacromolecules, 2018, 19, 3515–3524 CrossRef PubMed.
- D.-H. N. Tran, T. H. Nguyen, T. N. N. Vo, L. P. T. Pham, D. M. H. Vo, C. K. Nguyen, L. G. Bach and D. H. Nguyen, J. Appl. Polym. Sci., 2019, 136, 47544 CrossRef.
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC.
- K. Landfester, in Colloid Chemistry II, ed. M. Antonietti, Springer, Berlin, 2003, pp. 75–123 Search PubMed.
- A. E. Ekkelenkamp, M. R. Elzes, J. F. J. Engbersen and J. M. J. Paulusse, J. Mater. Chem. B, 2018, 6, 210–235 RSC.
- M. S. Alkanawati, F. R. Wurm, H. Thérien-Aubin and K. Landfester, Macromol. Mater. Eng., 2018, 303, 1700505 CrossRef.
- K. Piradashvili, E. M. Alexandrino, F. R. Wurm and K. Landfester, Chem. Rev., 2015, 116, 2141–2169 CrossRef PubMed.
- K. Landfester and C. K. Weiss, Adv. Polym. Sci., 2010, 229, 1–49 CrossRef CAS.
- S.-M. Jo, S. Jiang, R. Graf, F. R. Wurm and K. Landfester, Nanoscale, 2020, 12, 24266–24272 RSC.
- R. Thiramanas, M. Li, S. Jiang, K. Landfester and V. Mailänder, Cells, 2020, 9, 2043 CrossRef CAS PubMed.
- S.-M. Jo, F. R. Wurm and K. Landfester, Nano Lett., 2020, 20, 526–533 CrossRef CAS PubMed.
- S.-M. Jo, F. R. Wurm and K. Landfester, ACS Appl. Mater. Interfaces, 2018, 10, 34230–34237 CrossRef CAS PubMed.
- Y. Zhao, L.-P. Lv, S. Jiang, K. Landfester and D. Crespy, Polym. Chem., 2015, 6, 4197–4205 RSC.
- O. S. Abu Abed, C. Chaw, L. Williams and A. A. Elkordy, Sci. Rep., 2018, 8, 13158 CrossRef PubMed.
- A. V. Kabanov and S. V. Vinogradov, Angew. Chem., Int. Ed., 2009, 48, 5418–5429 CrossRef CAS PubMed.
- Q. Zhu, X. Chen, X. Xu, Y. Zhang, C. Zhang and R. Mo, Adv. Funct. Mater., 2018, 28, 1707371 CrossRef.
- J. K. Oh, R. Drumright, D. J. Siegwart and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 448–477 CrossRef CAS.
- M. Machtakova, S. Wirsching, S. Gehring, K. Landfester and H. Thérien-Aubin, J. Mater. Chem. B, 2021, 9, 8389–8398 RSC.
- M. Fichter, K. Piradashvili, A. Pietrzak-Nguyen, L. Pretsch, G. Kuhn, S. Strand, M. Knuf, F. Zepp, F. R. Wurm, V. Mailänder, K. Landfester and S. Gehring, Biomaterials, 2016, 108, 1–12 CrossRef CAS PubMed.
- D. Chenthamara, S. Subramaniam, S. G. Ramakrishnan, S. Krishnaswamy, M. M. Essa, F.-H. Lin and M. W. Qoronfleh, Biomater. Res., 2019, 23, 20 CrossRef CAS PubMed.
- J. W. Hickey, J. L. Santos, J.-M. Williford and H.-Q. Mao, J. Controlled Release, 2015, 219, 536–547 CrossRef CAS PubMed.
- S. Chen, Y. Zhong, W. Fan, J. Xiang, G. Wang, Q. Zhou, J. Wang, Y. Geng, R. Sun, Z. Zhang, Y. Piao, J. Wang, J. Zhuo, H. Cong, H. Jiang, J. Ling, Z. Li, D. Yang, X. Yao, X. Xu, Z. Zhou, J. Tang and Y. Shen, Nat. Biomed. Eng., 2021, 5, 1019–1037 CrossRef CAS PubMed.
- D. V. Haute and J. M. Berlin, Ther. Delivery, 2017, 8, 763–774 CrossRef.
- S. M. Moghimi and H. M. Patel, Adv. Drug Delivery Rev., 1998, 32, 45–60 CrossRef CAS.
- X. Duan and Y. Li, Small, 2013, 9, 1521–1532 CrossRef CAS PubMed.
- X. Xie, Q. Tao, Y. Zou, F. Zhang, M. Guo, Y. Wang, H. Wang, Q. Zhou and S. Yu, J. Agric. Food Chem., 2011, 59, 9280–9289 CrossRef CAS PubMed.
- B. Wang, T. Yuan, L. Zha, Y. Liu, W. Chen, C. Zhang, Y. Bao and Q. Dong, Mol. Pharmaceutics, 2021, 18, 1470–1479 CrossRef CAS PubMed.
- S. M. S. Shahriar, J. M. An, M. N. Hasan, S. S. Surwase, Y.-C. Kim, D. Y. Lee, S. Cho and Y.-K. Lee, Nano Lett., 2021, 21, 4666–4675 CrossRef CAS PubMed.
- J. C. Sung, B. L. Pulliam and D. A. Edwards, Trends Biotechnol., 2007, 25, 563–570 CrossRef CAS PubMed.
- C. Xu, H. Tian, P. Wang, Y. Wang and X. Chen, Biomater. Sci., 2016, 4, 1646–1654 RSC.
- Q. Zhang, Z. Shen and T. Nagai, Int. J. Pharm., 2001, 218, 75–80 CrossRef CAS.
- M. M. A. Abdel-Mottaleb, D. Neumann and A. Lamprecht, Eur. J. Pharm. Biopharm., 2011, 79, 36–42 CrossRef CAS PubMed.
- B. Iqbal, J. Ali and S. Baboota, Int. J. Dermatol., 2018, 57, 646–660 CrossRef PubMed.
- R. Alvarez-Román, A. Naik, Y. N. Kalia, R. H. Guy and H. Fessi, J. Controlled Release, 2004, 99, 53–62 CrossRef PubMed.
- L. Busch, Y. Avlasevich, P. Zwicker, G. Thiede, K. Landfester, C. M. Keck, M. C. Meinke, M. E. Darvin, A. Kramer, G. Müller, M. Kerscher, J. Lademann and A. Patzelt, Int. J. Pharm., 2021, 597, 120339 CrossRef CAS PubMed.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- T. Lima, K. Bernfur, M. Vilanova and T. Cedervall, Sci. Rep., 2020, 10, 1129 CrossRef CAS PubMed.
- A. Zielińska, F. Carreiró, A. M. Oliveira, A. Neves, B. Pires, D. N. Venkatesh, A. Durazzo, M. Lucarini, P. Eder, A. M. Silva, A. Santini and E. B. Souto, Molecules, 2020, 25, 3731 CrossRef PubMed.
- M. Faria, M. Björnmalm, K. J. Thurecht, S. J. Kent, R. G. Parton, M. Kavallaris, A. P. R. Johnston, J. J. Gooding, S. R. Corrie, B. J. Boyd, P. Thordarson, A. K. Whittaker, M. M. Stevens, C. A. Prestidge, C. J. H. Porter, W. J. Parak, T. P. Davis, E. J. Crampin and F. Caruso, Nat. Nanotechnol., 2018, 13, 777–785 CrossRef CAS PubMed.
- S. Jesus, M. Schmutz, C. Som, G. Borchard, P. Wick and O. Borges, Front. Bioeng. Biotechnol., 2019, 7, 261 CrossRef PubMed.
- A. Kermanizadeh, N. R. Jacobsen, M. Roursgaard, S. Loft and P. Møller, Heliyon, 2017, 3, e00458 CrossRef PubMed.
- Y. Liu, M. Kong, C. Feng, K. K. Yang, Y. Li, J. Su, X. J. Cheng, H. J. Park and X. G. Chen, Colloids Surf., B, 2013, 103, 345–353 CrossRef CAS PubMed.
- S. K. Jena and A. T. Sangamwar, Carbohydr. Polym., 2016, 151, 1162–1174 CrossRef CAS PubMed.
- K. Sonaje, Y.-H. Lin, J.-H. Juang, S.-P. Wey, C.-T. Chen and H.-W. Sung, Biomaterials, 2009, 30, 2329–2339 CrossRef CAS PubMed.
- S. E.-S. Radwan, M. S. Sokar, D. A. Abdelmonsif and A. H. El-Kamel, Int. J. Pharm., 2017, 526, 366–379 CrossRef CAS PubMed.
- M. Sharma, S. Sharma, V. Sharma, K. Sharma, S. K. Yadav, P. Dwivedi, S. Agrawal, S. K. Paliwal, A. K. Dwivedi, J. P. Maikhuri, G. Gupta, P. R. Mishra and A. K. S. Rawat, Int. J. Biol. Macromol., 2017, 104, 1345–1358 CrossRef CAS PubMed.
- K. Tahara, T. Sakai, H. Yamamoto, H. Takeuchi, N. Hirashima and Y. Kawashima, Int. J. Pharm., 2009, 382, 198–204 CrossRef CAS PubMed.
- M. Kolter, M. Ott, C. Hauer, I. Reimold and G. Fricker, J. Controlled Release, 2015, 197, 165–179 CrossRef CAS.
- G. Romero, I. Estrela-Lopis, J. Zhou, E. Rojas, A. Franco, C. S. Espinel, A. G. Fernández, C. Gao, E. Donath and S. E. Moya, Biomacromolecules, 2010, 11, 2993–2999 CrossRef CAS PubMed.
- S. Uchida and K. Kataoka, J. Biomed. Mater. Res., Part A, 2019, 107, 978–990 CrossRef CAS PubMed.
- S. M. Moghimi, P. Symonds, J. C. Murray, A. C. Hunter, G. Debska and A. Szewczyk, Mol. Ther., 2005, 11, 990–995 CrossRef CAS PubMed.
- M. A. Gosselin, W. Guo and R. J. Lee, Bioconjugate Chem., 2001, 12, 989–994 CrossRef CAS PubMed.
- V. Knorr, V. Russ, L. Allmendinger, M. Ogris and E. Wagner, Bioconjugate Chem., 2008, 19, 1625–1634 CrossRef CAS PubMed.
- S. Sanati, S. Taghavi, K. Abnous, S. M. Taghdisi, M. Babaei, M. Ramezani and M. Alibolandi, Gene Ther., 2021 DOI:10.1038/s41434-021-00234-0.
- D. Liu, F. Yang, F. Xiong and N. Gu, Theranostics, 2016, 6, 1306–1323 CrossRef CAS PubMed.
- K. Smietana, M. Siatkowski and M. Møller, Nat. Rev. Drug Discovery, 2016, 15, 379–380 CrossRef CAS PubMed.
- K. W. Hunter, Nat. Rev. Cancer, 2012, 12, 144–149 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |