
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigation of the preparation and reactivity of metal–organic frameworks of cerium and pyridine-2,4,6-tricarboxylate†
Juliana
Fonseca de Lima
 a,
Fernanda V. S.
Moreno
a,
Bruno A. T.
Menezes
a,
Jader
da Silva Barbosa
b,
Matthew C.
Waddington
c,
Siân A.
Franklin
c,
Guy J.
Clarkson
c,
Marc
Walker
d,
Osvaldo A.
Serra
b and
Richard I.
Walton
*c
a,
Fernanda V. S.
Moreno
a,
Bruno A. T.
Menezes
a,
Jader
da Silva Barbosa
b,
Matthew C.
Waddington
c,
Siân A.
Franklin
c,
Guy J.
Clarkson
c,
Marc
Walker
d,
Osvaldo A.
Serra
b and
Richard I.
Walton
*c
aInstituto de Química, Universidade do Estado do Rio de Janeiro, 20550-900 Rio de Janeiro, Brazil
bDepartamento de Química, Faculdade de Filosofia, Ciências e Letras de Ribeirão Preto, Universidade de São Paulo, Avenida dos Bandeirantes, 3900, 14040-901, Ribeirão Preto, SP, Brazil
cDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: r.i.walton@warwick.ac.uk
dDepartment of Physics, University of Warwick, Coventry, CV4 7AL, UK
First published on 29th November 2021
Abstract
The synthesis of three coordination polymers of cerium(III) and the ligand pyridine-2,4,6-tricarboxylate (PTC) is reported. Two of the materials crystallise under hydrothermal conditions at 180 °C, with [Ce(PTC)(H2O)2]·1.5H2O, (1), being formed on extended periods of reaction time, 3 days or longer, and Ce(PTC)(H2O)3, (2), crystallising after 1 day. Both phases contain Ce(III) but are prepared using the Ce(IV) salt Ce(SO4)2·4H2O as reagent. Under solvothermal conditions (mixed water-N,N-dimethylformamide (DMF)), the phase [Ce(PTC)(H2O)(DMF)]·H2O (3) is crystallised. The structures of the three materials are resolved by single crystal X-ray diffraction, with the phase purity of the samples determined by powder X-ray diffraction and thermogravimetric analysis. (1) is constructed from helical chains cross-linked by the PTC linkers to give a three-dimensional structure that contains clusters of water molecules in channels that are hydrogen-bonded to each other and to additional waters that are coordinated to cerium. (2) also contains nine-coordinate cerium but these are linked to give a dense framework, in which water is directly coordinated to cerium. (3) contains corner-shared nine-coordinate cerium centres, linked to give a framework in which Ce-coordinated DMF fills space. Upon heating the material (1) in air all water is irreversibly lost to give a poorly crystalline anhydrous phase Ce(PTC), as deduced from X-ray thermodiffractometry and thermogravimetric analysis. The material (1), however, is hydrothermally stable, and is also stable under oxidising conditions, where immersion in 30% H2O2 gives no loss in crystallinity. Oxidation of around 50% of surface Ce to the +4 oxidation state is thus possible, as evidenced by X-ray photoelectron spectroscopy, which is accompanied by a colour change from yellow to orange. Photocatalytic activity of (1) is screened and the material shows effective degradation of methyl orange.
Introduction
Metal–organic frameworks of cerium have attracted significant interest in the past few years.1 Part of this focus lies in the fact that cerium is one of the few lanthanide elements that adopts two stable oxidation states that can interconvert readily. The redox chemistry associated with Ce(IV)/Ce(III) finds uses ranging from oxidants in organic synthesis,2 to heterogeneous catalysis,3 and integrating these properties into MOFs is an interesting proposition for development of new functional solids that might have practical applications. In the case of MOFs, a variety of Ce(III) materials have been produced and these are often analogues of structures that contain other larger lanthanides, such as those of La(III) or Nd(III). As expected, high coordination numbers are observed, often with irregular geometry and this can give unique structures, not found for metal cations from other parts of the Periodic Table. In the case of Ce(IV) the first MOFs formed were analogues of Zr(IV) materials, or mixed-metal variants where solid-solutions of Ce and Zr are found in an isostructural series.4 It is noteworthy that the chemistry of Ce(IV) MOFs is largely associated with the hexameric oxy-clusters seen in zirconium chemistry, whereas for Ce(III), a variety of coordination geometries and connectivity of the primary polyhedra is seen, depending on the ligands used to construct the material.1aA variety of carboxylate linkers have been used for the preparation of cerium MOFs, each with inherent structural directing connectivity, depending on the number of connection points, and also with the possibility of structural flexibility, depending on the organic backbone of the ligand.1a Carboxylates provide versatile ligands for construction of MOFs based on lanthanide cations because the variety of potential bonding modes of the carboxylate function allows the high coordination number of the cation to be satisfied,5 as well as the strong electrostatic contribution to bonding leading to thermally, and potentially hydrothermally, robust materials.
In terms of properties, the MOFs reported for cerium have been studied for a range of potential applications. The reported works span catalysis, photocatalysis and sensing, as well as gas adsorption that would be associated with many other families of MOFs. For example, the cerium(IV) analogue of UiO-66 was used as an oxidation catalyst, proven with the model reaction of conversion of benzyl alcohol to benzaldehyde, and good conversion was reported even though the reaction was found to occur only at the surface of the material.6 Homochiral Ce(III) MOFs produced using methylenediisophthalate as linker were found to give enantiomeric selectivity in the asymmetric cyanosilylation of carbonyl-containing substrates, and were easily recovered by filtration and reused without loss of efficiency.7 Application in photocatalysis were predicted by computational simulation for Ce(IV)-UiO-66, which showed that only the Ce analogue of material could allow ligand-to-metal charge transfer to separate photogenerated charges.8 Photo-oxidation of benzylic alcohols to benzaldaldehydes under near-ultraviolet light irradiation was independently proven for the material.9 In sensing applications a chromogen has been used in combination with Ce-MOFs to detect species such as ascorbic acid, using a mixed-valent Ce(III)/Ce(IV) MOF,10 and Hg2+ using Ce(IV)-UiO-66.11 The emerging applications of Ce-MOFs have recently been reviewed.1
In this paper we describe an exploration of the synthesis of metal–organic frameworks of cerium using the linker pyridine-2,4,6-tricarboxylate (PTC). This linker has been used for a number of metal organic frameworks,12 but is worthy of further investigation since tridentate binding by the nitrogen and two neighbouring carboxylate oxygens can potentially provide robust materials, with the third carboxylate moiety offering the possibility of bridging to further metal centres to create three-dimensionally extended structures. Das et al. reported eleven possible bonding bonds for the PTC ligand.12 Here, we report three distinct Ce(III)-PTC materials and for one of the materials, prepared in water with extended crystallisation times, we study its stability and explore possible applications. A structural comparison is made with previously reported lanthanide pyridine-2,4,6-tricarboxylate materials.
Experimental section
The synthesis of the ligand precursor pyridine-2,4,6-tricarboxylic acid, H3PTC, was carried out by oxidation of 2,4,6-trimethylpyridine and recrystallisation via formation of the methyl ester of the tricarboxylic acid. 2,4,6-Trimethylpyridine (6 mL, 43 mmol) was added to concentrated H2SO4 (200 mL) at 0 °C followed by small amounts of CrO3 (39 g, 0.39 mmol, in total) with the temperature maintained at 0 °C. The mixture was heated to 75 °C for 24 h, then placed into ice water (0.5 mL) and kept at 4 °C. After 3 days the mixture was filtered and the filtrate was placed in 40 mL of methanol and 0.7 mL of H2SO4. After 12 h of reflux, the solid methyl ester of pyridine-2,4,6-tricarboxylic acid was recovered by filtration with a yield of 60%. Hydrolysis of the ester was performed in 30 mL of water and 30 mL of 38% hydrochloric acid, and the solution was heated until complete dissolution (1 h). At room temperature the pH of the solution was adjusted to 2 and the precipitated H3PTC crystals were filtered and washed with ice-cold H2O (50 mL) to give a yield of 80%.The cerium precursors Ce(SO4)2·4H2O and Ce(NO3)3·4H2O were used as provided by chemical suppliers, with the water content verified using thermogravimetric analysis. [Ce(PTC)(H2O)2]·1.5H2O (1) was isolated as single crystals suitable for structure determination in a hydrothermal reaction. Ce(SO4)2·4H2O (0.101 g, 0.25 mmol) and H3PTC (0.106 g, 0.5 mmol) were dispersed in 5 mL H2O with rapid stirring and 4 drops (∼0.2 mL) of triethylamine were added to aid deprotonation of the tricarboxylic acid. The mixture was sealed in a 23 mL PTFE-lined steel autoclaved and heated in a fan-assisted oven 180 °C for 10 days. The solid product was recovered by suction filtration and allowed to dry under ambient conditions. Ce(PTC)(H2O)3 (2) was formed using identical conditions to (1), except the time of reaction was shortened to 1 day. [Ce(PTC)(H2O)(DMF)]·H2O (3) was prepared at room temperature in a mixed solvent system. 0.0211 g of H3PTC was dissolved in 5 mL of 1 M aqueous NaOH and the 5 mL DMF (N,N-dimethylformamide) was added. Separately, 5 mL 0.02 M aqueous Ce(NO3)3·4H2O was diluted with a further 5 mL water. The second solution was added dropwise to the first one and the mixture allowed to stand. After 30 days crystals suitable for single-crystal diffraction were isolated.
Single crystal X-ray diffraction was performed on suitable crystals that were selected and mounted on glass fibres with Fromblin oil and placed on a Rigaku Oxford Diffraction SuperNova diffractometer with a dual source (Cu at zero) equipped with an AtlasS2 CCD area detector. The crystals were kept at 150(2) K during data collection. Using Olex2,13 the structures were solved with the ShelXS structure solution program14 using the Patterson Method and refined with the ShelXL refinement package15 using least squares minimisation. The ESI† contains further details of the structure solution and crystal structure data.
Powder XRD data for phase identification were recorded using a Siemens D5000 X-ray diffractometer operating with Cu Kα1/2 radiation. Data were recorded in Bragg–Brentano geometry from finely-ground samples pressed into silicon holders.
Thermogravimetric analysis was performed using a Mettler-Toledo TGA/DSC 1 Thermogravimetric Analyser. Approximately 10 mg of powdered sample was placed in an alumina pan and its mass monitored in static air upon heating to 1000 °C at a rate of 10 °C min−1.
Powder X-ray thermodiffractometry was performed using a Bruker D8 instrument with Cu Kα1/2 radiation, fitted with an Anton Paar XRK 900 chamber and a VÅNTEC solid-state detector Samples were heated from room temperature to 900 °C in flowing air and XRD patterns were recorded at intervals of 50 °C.
Diffuse reflectance UV-Vis spectroscopy was performed on powder samples using a Shimadzu UV-2600i UV-Vis spectrophotometer. A barium sulfate standard was used as a baseline for the measurements.
X-ray photoelectron spectroscopy (XPS) was performed on a Kratos Axis Ultra DLD using monochromatic Al-Kα excitation (1486.7 eV) with detection at 90° with respect to the surface. A charge neutraliser was used during the experiments in order to prevent surface charging, with the binding energy scale subsequently referenced to the C–C/C–H component of the C 1s spectrum at 285.0 eV during data analysis. Data were analysed using the CasaXPS package applying Voigt peaks and Shirley backgrounds for all regions. The Ce 3d region was interpreted using previous literature reports on cerium oxides as guidance.16
Photocatalytic testing was carried out using a Hg vapour lamp (250 W) with 2 mg solid catalyst and 10 mL of a 4.0 × 10−5 M solution of methyl orange at pH = 3.0. The concentration of dye was monitored at 505 nm using UV-Vis spectroscopy as a function of time on a Hewlett Packard HP 8453 spectrophotometer.
Results and discussion
The synthesis of two of the cerium organic frameworks (1) and (2) was carried out under hydrothermal conditions, with the only difference being the extended reaction time to form (1). The metal–organic framework structure of (1) is the same as reported for Nd3+ and Pr3+ materials,17 and already found for Ce3+,18 but (1) contains a different amount of occluded water than the previously reported structures. The previously reported structure of the cerium material by Zhao et al. has chemical formula [Ce(PTC)(H2O)2]·H2O.18 For the Nd analogue a chemical formula of [Nd(PTC)(H2O)2]·2H2O was reported by Ghosh and Bharadwaj,17a whereas for Pr, isostructural [Pr(PTC)(H2O)2]·2H2O16a and [Pr(PTC)(H2O)2]·1.5H2O17b materials have been reported (Table S2†). This leads us to conclude that variable water content is possible. The structure of (1) is constructed from 9-coordinate Ce centres created from coordination of the PTC ligand and water molecules, Fig. 1a. Three of these coordinating atoms are from tridentate coordination of the PTC ligand via nitrogen and two oxygens from neighbouring carboxylates, four are oxygens of other carboxylate, and two are oxygens of water molecules. Each PTC ligand thus connects five Ce centres, Fig. 1b. This bonding mode PTC is Type VII according to the classification by Das et al. (ESI†).12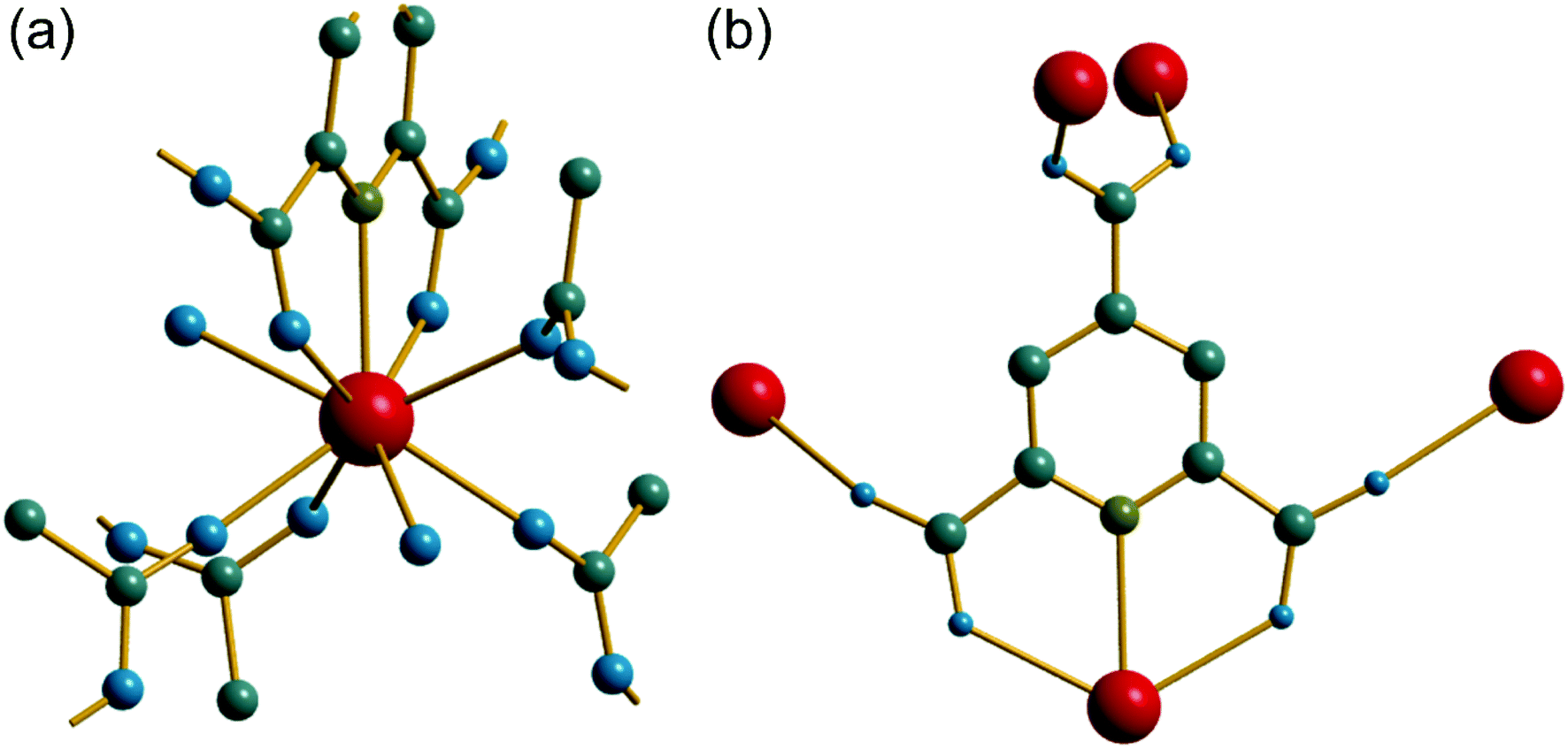 | ||
| Fig. 1 (a) The local environment of Ce in (1) with cut-off bonds showing connection to neighbouring atoms. (b) The connectivity of the PTC ligand in (1). In both cases, red spheres are cerium, grey are carbon, blue are oxygen, olive are nitrogen and hydrogens are not shown. | ||
The 9-coordinate Ce centres are not directly corner shared in (1) but are most closely linked via Z,Z-μ2-η1:η1 bridging carboxylate oxygens to form helical chains that run parallel to b, Fig. 2a. There are chains of each handedness in the crystal structure. The chains are cross-linked in both directions by to give a three-dimensionally connected framework, Fig. 2b. Running parallel to a are channels that contain occluded water, as well as that directly coordinated to cerium. The pores present can be defined by closest distances between the water oxygens directly bound to cerium of 3.29 Å, and in this confined space the occluded water is held by a network of hydrogen bonding, as evidenced by short O⋯O distances (Fig. 2c and Table S7†), which includes interactions between the occluded water and the water that is coordinated to the cerium directly.
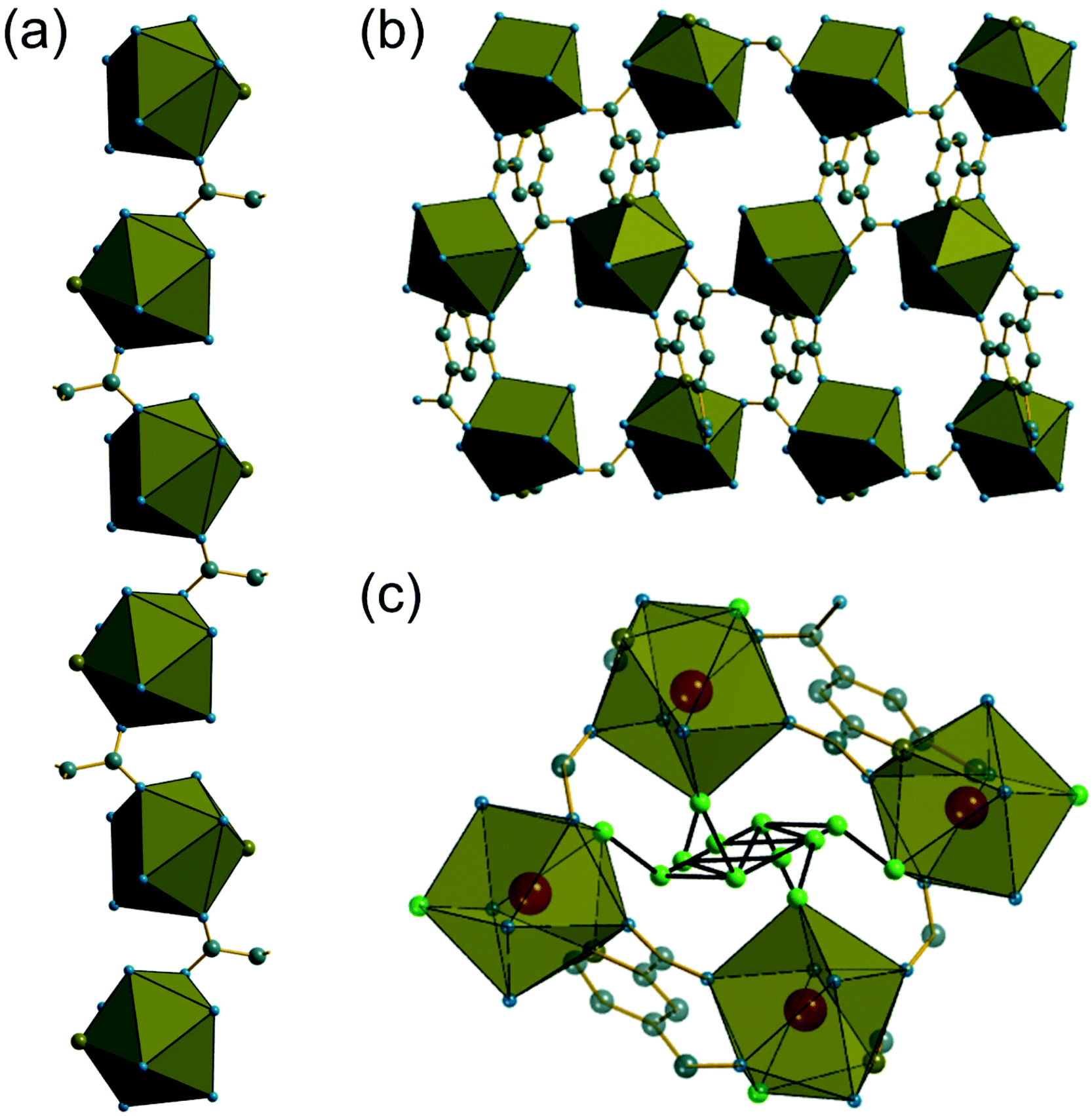 | ||
| Fig. 2 (a) Chains of nine-coordinate Ce centres found in (1) running parallel to b. (b) Projection of the structure in the ab plane showing connection of chains to yield an open structure (extra-framework water molecules not shown). (c) The local environment of water in (1) with short oxygen–oxygen distances (2.5 to 3 Å) indicative of hydrogen bonding shown in black. Note that the extra framework water sites are not fully occupied so not all will be simultaneously populated. The cerium environment is represented by the polyhedra, and grey spheres are carbon, blue are oxygen, olive are nitrogen and hydrogens are not shown. In (c) the green spheres are the oxygens of water molecules. | ||
The material (2) contains two crystallographically distinct Ce sites, both of which have coordination number nine and the same set of coordinating atoms, Fig. 3a. These consist of one tridentate PTC ligand bound through a nitrogen and two neighbouring carboxylate oxygens, three further carboxylate oxygens from distinct PTC ligands and three water molecules. Each PTC ligand, of two crystallographically distinct types, connects four Ce centres in (2), as shown in Fig. 3b, such that one of the carboxylate oxygens is terminal: this is a mode of bonding not described in the Das et al. notation.12
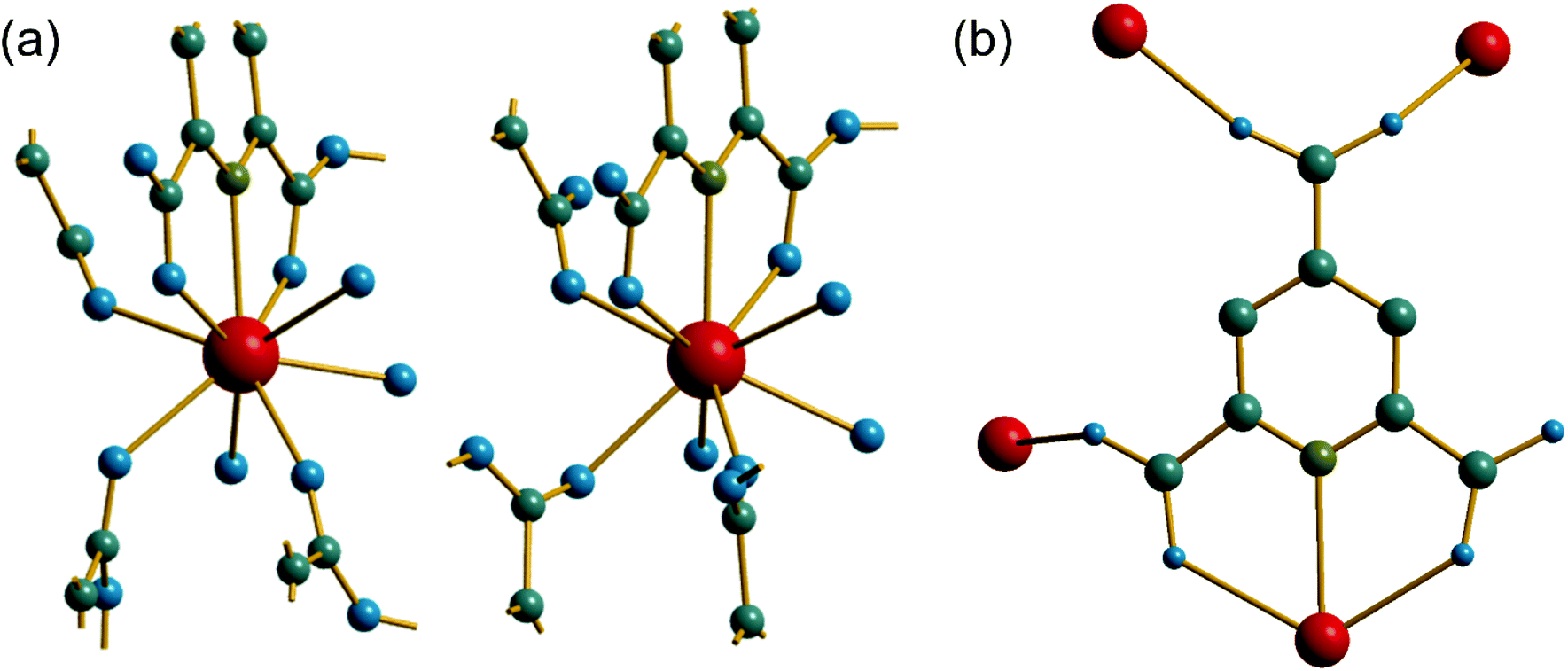 | ||
| Fig. 3 (a) The local environment of the two Ce sites in (2) with cut-off bonds showing connection to neighbouring atoms. (b) The connectivity of one of the PTC ligands in (2); the second PTC ligand has identical connectivity and is not shown. In both cases, red spheres are cerium, grey are carbon, blue are oxygen, olive are nitrogen and hydrogen are not shown. | ||
As in (1), the 9-coordinate Ce(III) centres in (2) are not connected by directly shared corners, and here are linked to the neighbouring cerium centres via Z,Z-μ2-η1:η1 bridging carboxylates to give a three-dimensionally connected array. Each Ce centre is thus connected to four other centres and this occurs in a strictly alternating manner with one of the two crystallographically distinct cerium centres surrounded only by the second type and vice versa, as seen when the structure is viewed along b, Fig. 4a. The structure is more densely packed than (1) but the terminal carboxylate oxygens project into small voids, seen when the structure is viewed along a, Fig. 4b. These oxygens lie within 3 Å of oxygens of water molecules connected directly to more distant cerium centres, meaning that there is the no possibility of permanent porosity in (2), indeed there is no occluded water that might be removed to access any free pore space.
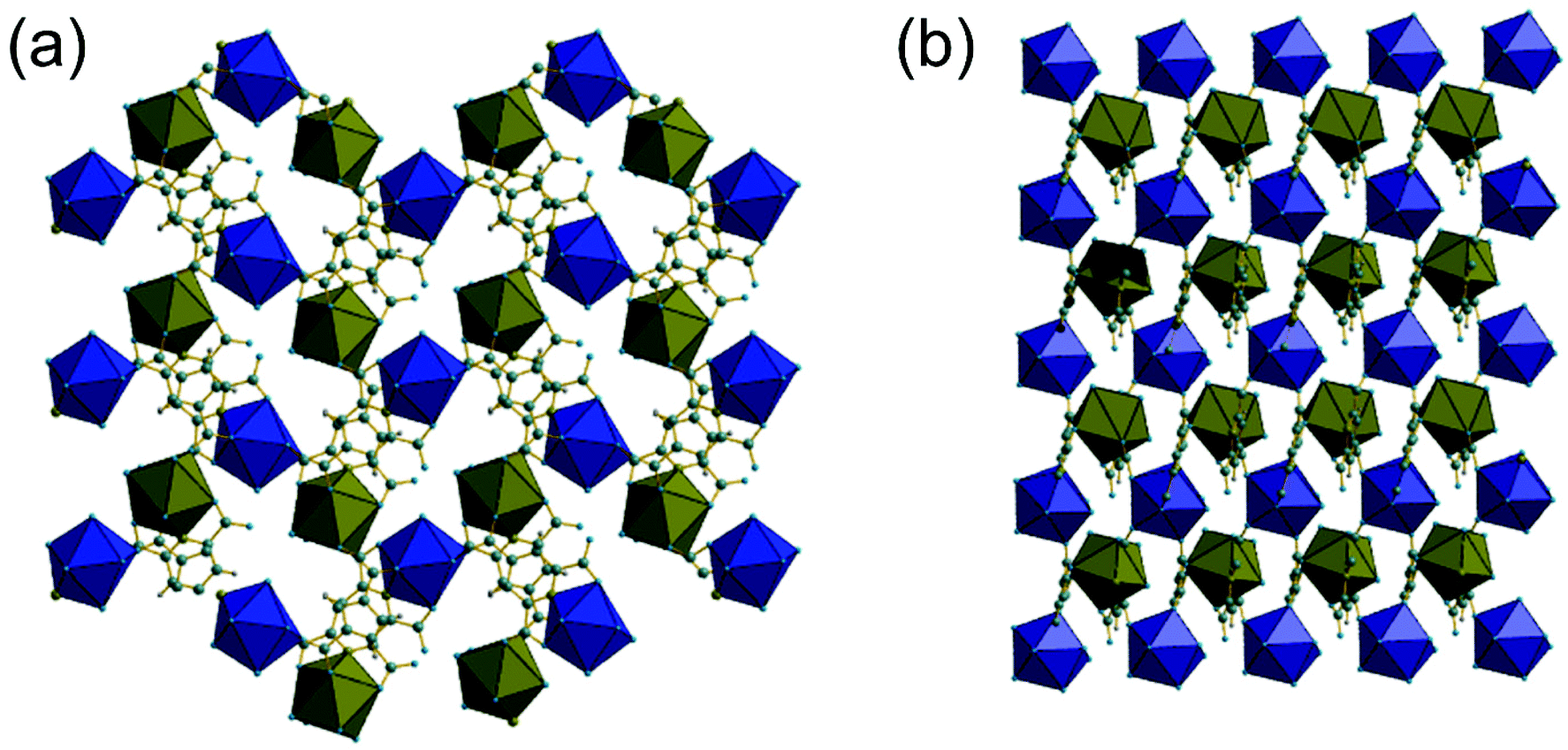 | ||
| Fig. 4 (a) View of (2) along b with the two distinct types of cerium shown in different coloured polyhedra (Ce1 blue and Ce2 olive green). (b) View of (2) along a showing the dense packing of the structure. Grey spheres are carbon, blue are oxygen, olive are nitrogen and hydrogens are not shown. | ||
The phase purity of (1) and (2) was determined by powder XRD, Fig. 5a. This showed that the bulk sample of (2) is contaminated with just a small quantity of (1). Powder XRD patterns of materials prepared at intermediate reaction times are also shown on Fig. 5, which reveals an increasing fraction of (1) over (2) with increasing time of synthesis. At 72 hours the sample is phase pure (1) (note that the crystal studied for structure solution was grown using an even more extended reaction time of 10 days).
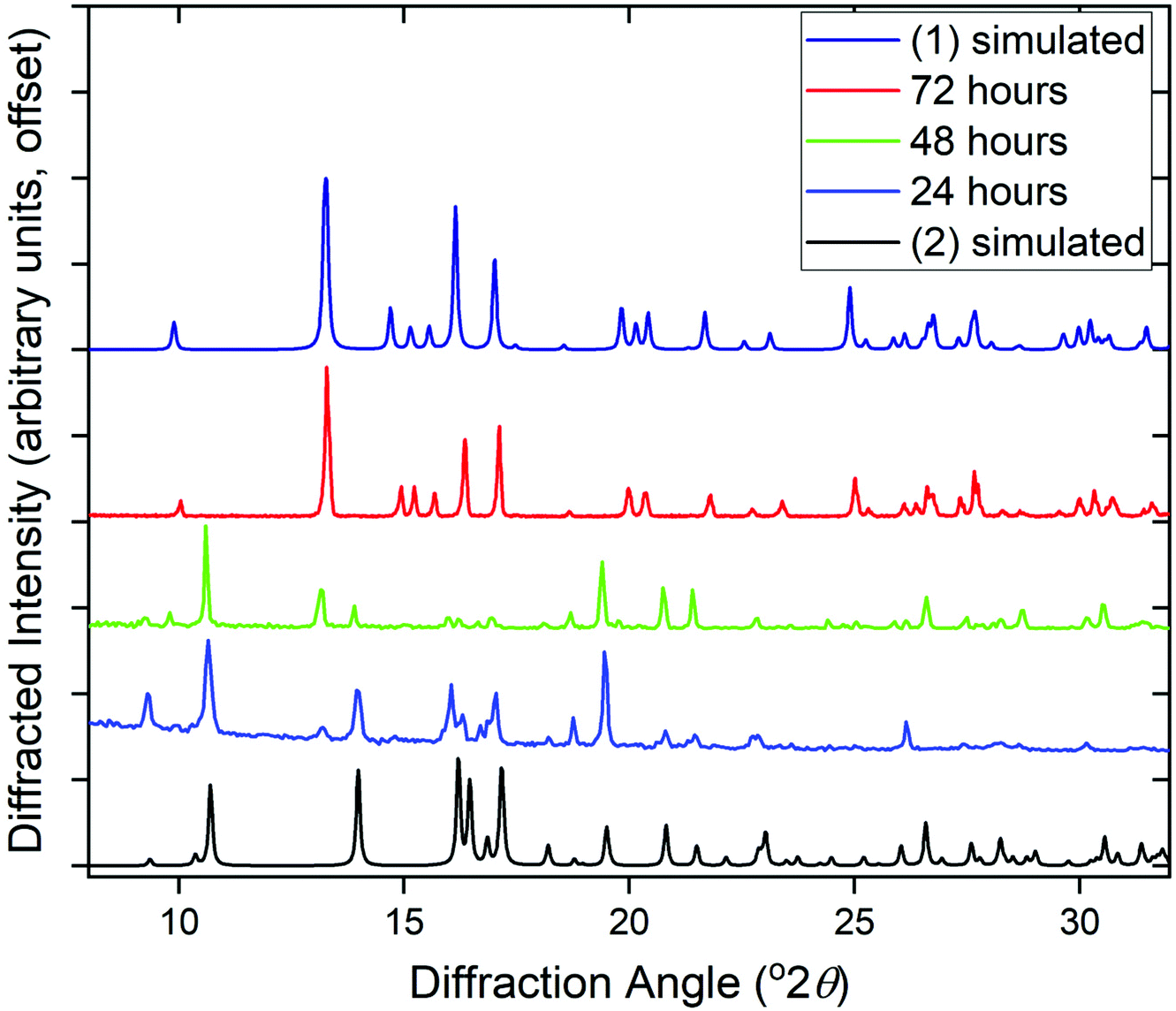 | ||
| Fig. 5 Powder XRD patterns of cerium pyridine-2,4,6-tricarboxylates prepared at different reaction times compared to simulated patterns of (1) and (2), showing the evolution of the product formed from (2) to (1). | ||
Given that the two structures bear no obvious relationship in atomic connectivity, it is likely that the successive crystallisation of (2) followed by (1) occurs via dissolution of the initially formed (2). In other MOF chemistry, the formation of kinetic phases has been observed, that may have no structural relationship to the final crystalline product.19 The presence of Ce3+ in (1) and (2) is assigned to achieve charge balance, but also verified using bond valence sums,20 which give an oxidation state of 3.23 for the single unique cerium in (1) and 3.14 and 3.27 for Ce1 and Ce2, respectively, in (2). The reduction of the Ce4+ precursor used for both materials must take place readily in solution. It has previously been noted that Ce4+ MOFs are only produced under solvothermal reaction conditions from (NH4)2[Ce(NO3)6] in water/DMF mixtures at low reaction temperatures ≤100 °C using short reaction times (t ≤ 30 min).1a In other cases where Ce(IV) precursors have been used, reduction to Ce(III) occurs readily in situ to yield materials that contain solely the lower oxidation state. For example, in the case of porphyrin-based MOFS, the use of (NH4)2[Ce(NO3)6] lead to Ce(III) materials, which was ascribed to the reducing properties of the ligand precursor.21 We also attempted reactions using NaOH as base but this yielded only crystals of the sodium salt Na3PTC, as verified by single crystal diffraction (see ESI†).
The material (3) is formed at room temperature from a mixed DMF-water solvent using a Ce3+ containing precursor. The crystal structure contains one unique Ce centre, which is directly coordinated to nine ligand atoms, Fig. 6a. Three of these are from the tridentate coordination of a PTC ligand via its nitrogen and neighbouring oxygens of adjacent carboxylate groups. A further PTC ligand is connected via two oxygens in an η2 binding mode, while a third PTC ligand is connected by a single oxygen atom. Two directly coordinated water molecules and an O-connected DMF molecule complete the coordination environment. The single unique PTC ligand thus connects a total of four different Ce centres, as shown on Fig. 6b, such that one of the carboxylate oxygens is terminal, as in (2). This mode of binding is not one reported in the Das et al. classification.12
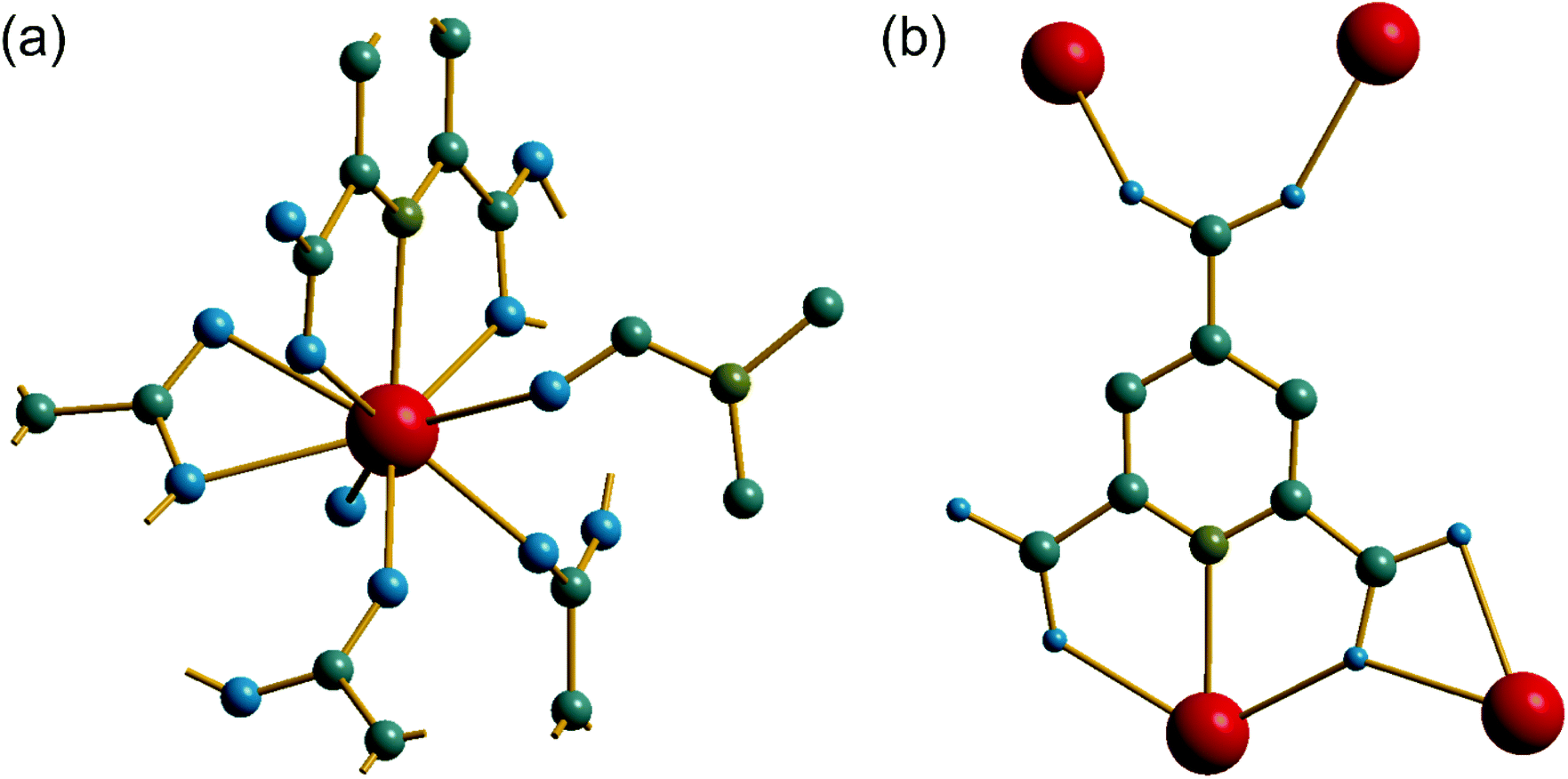 | ||
| Fig. 6 (a) The local environment of Ce in (3) with cut-off bonds showing connection to neighbouring atoms. (b) The connectivity of the PTC ligand in (3). In both cases, red spheres are cerium, grey are carbon, blue are oxygen, olive are nitrogen and hydrogens are not shown. | ||
Unlike compounds (1) and (2), the cerium centres in (3) directly share a common oxygen via a μ2-η2:η1 connected PTC ligand, using one of the carboxylates adjacent to the nitrogen of the pyridine ring. This gives infinite chains running parallel to c, Fig. 7a. The carboxylate in the 4-position of the pyridine ring then cross links the chains by connection to two ceriums on a neighbouring chain in a Z,Z-μ2-η1:η1 fashion. The Ce–O–Ce bond angle along the chain is 138° and so the chains are not linear: there is a zig-zag arrangement of cerium centres that repeats every second cerium, evident in a projection parallel to c, Fig. 7b, which shows the connection of the chains to each other. The DMF molecules, coordinated to the Ce centres, fill any void space that may otherwise be present, and are found to be disordered, modelled as being present in two different orientations, with occupancy of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30.
:30.
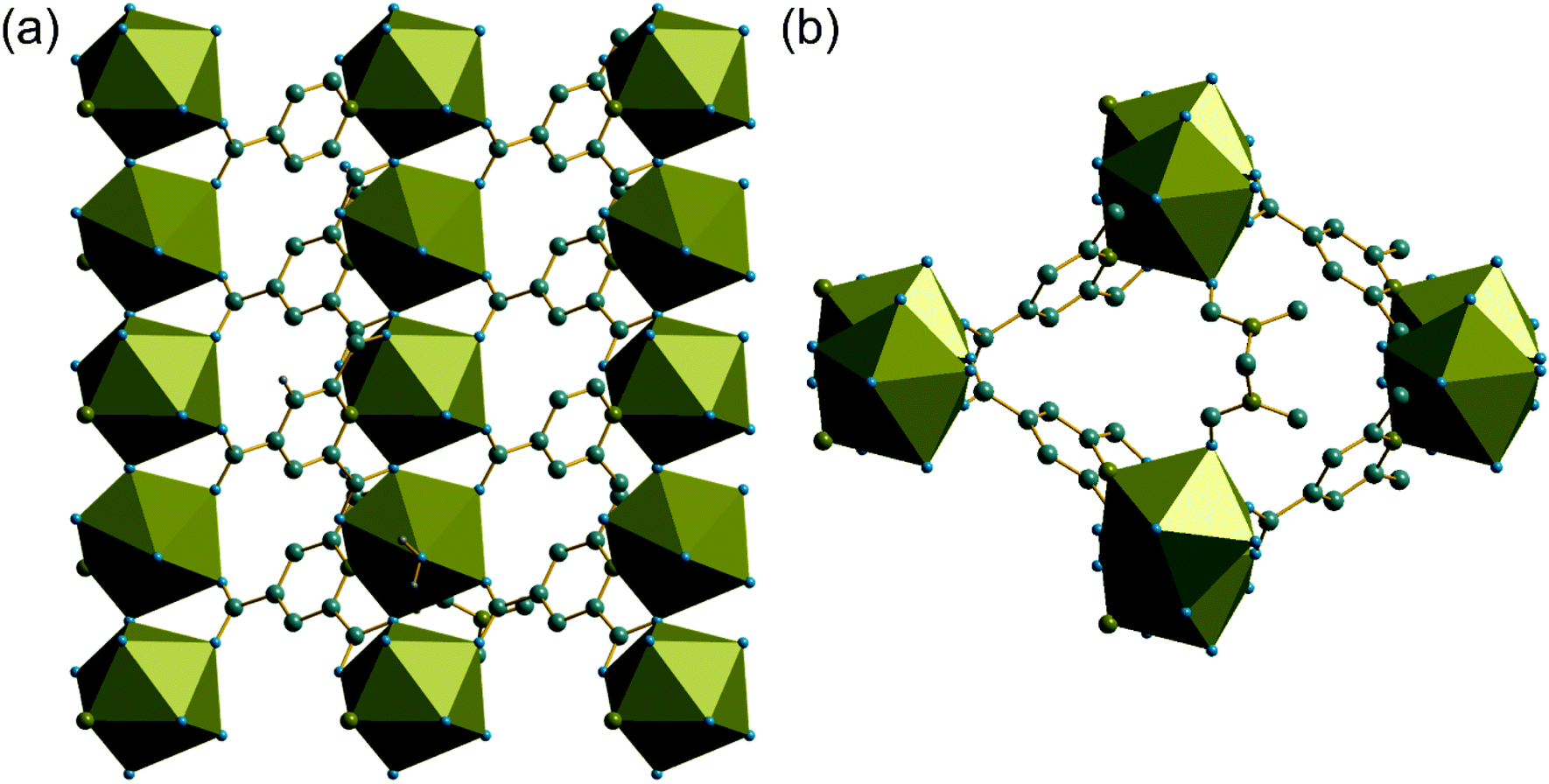 | ||
| Fig. 7 (a) View of (3) along b showing cross-linked chains of corner-shared cerium-centred polyhedra. (b) Projection of (3) along c showing coordinated DMF (one orientation shown) filling extra-framework space. The cerium environment is represented by the polyhedra, and grey spheres are carbon, blue are oxygen, olive are nitrogen and hydrogens are not shown. | ||
Two other cerium(III) pyridine-2,4,6-tricarboxylates have already been reported in the literature. [Ce(PTC)(H2O)3]·H2O was reported by Sharif et al. and contains a single unique Ce coordinated by 9 ligand atoms, but has a different local coordination environment to our materials, since two of the ligand positions around Ce are formed from η2 binding mode from a single PTC ligand, which is coordinated in Type XI mode using the Das et al. notation. This material was prepared in a hydrothermal reaction in the presence of trifluoroacetic acid. The material [Ce(PTC)(H2O)5]·4H2O, also reported by Sharif et al., is much more water-rich and contains five directly coordinated water molecules to each nine-coordinate Ce centre.22 The PTC ligand is bound in a manner not classified by Das et al. where two cerium centres are connected by each PTC ligand (see ESI†). This material was produced in water–methanol mixture at room temperature. Other lanthanide pyridine-2,4,6-tricarboxylates are known, and for the later lanthanides the smaller size of the Ln3+ cations with increasing atomic number may result in lower coordination numbers, even for an isostructural series, where the water content may be variable.23 We also note that PTC has been used as a ligand in combination with a second ligand to form extended coordination networks with cerium, such as with oxalate or glycine,24 but structural comparison with these materials is not straightforward due to the influence of the second ligand on the architecture of the structure.
For each of the materials studied here, thermogravimetric analysis, in addition to powder XRD, confirmed phase purity and showed stepwise loss of occluded water, coordinated water (or DMF where present), before final combustion of the PTC ligand (ESI†). Since (1) is the material produced under hydrothermal conditions for extended periods of heating, it represents the most stable phase in the this set of materials and we further studied its properties. Thermodiffractometry shows the material maintains crystallinity upon heating to 100 °C, with no evident shift in Bragg peak positions or broadening of Bragg peaks, Fig. 8. Above 110 °C a significant change in the powder XRD pattern is seen with evidence of a phase transition, but also a broadening of all Bragg peaks indicating a loss of crystallinity. The phase produced is stable to 310 °C. Looking at the TGA analysis, the phase formed between 110 °C and 310 °C would correspond to fully dehydrated Ce(PTC), where loss of occluded water and directly bound water has occurred. The TGA shows no evidence that the two types of water can be removed separately, implying that the extra-framework water present plays a significant role in stabilising the open structure through its hydrogen bonds. It is noteworthy that the poorly crystalline Ce(PTC) phase found between 110 and 310 °C, shows no low-angle diffraction features (ESI†), implying that the structure is more dense than (1). Its BET surface area is also small, ∼7 m2 g−1, consistent with the formation of a dense structure. In addition, the anhydrous phase does not convert back to (1) on standing in ambient air or immersion in water, consistent with significant structural rearrangement having taken place upon dehydration. Attempts to remove the water by vacuum treatment were also unsuccessful and the same collapsed Ce(PTC) phase was observed by powder XRD (ESI†).
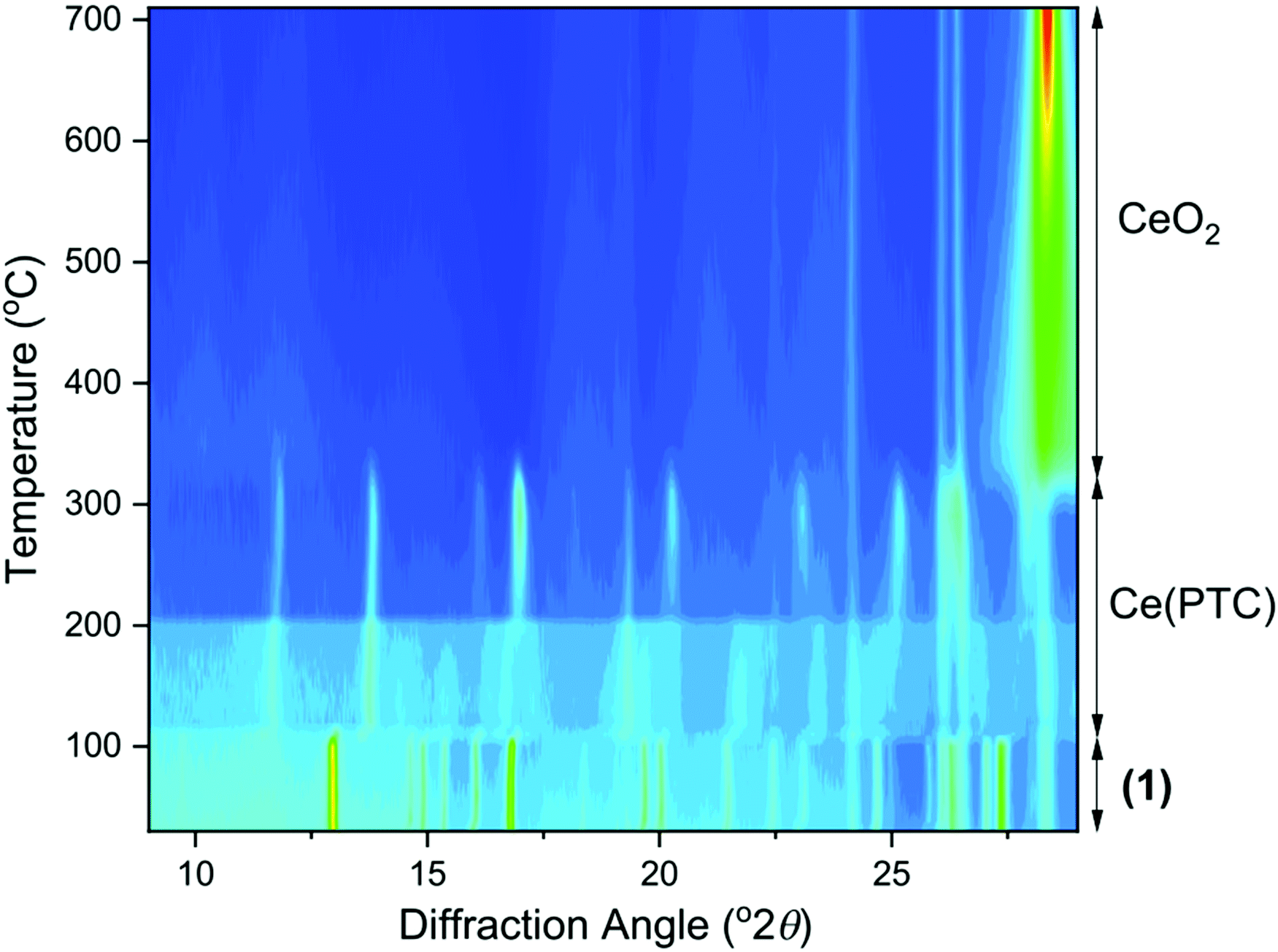 | ||
| Fig. 8 Thermodiffractometry measured for (1) on heating in air, with the phases present indicated. | ||
Upon heating above 310 °C, further structural collapse is evident, with the first appearance of the (111) Bragg peak of CeO2 that subsequently increases in intensity and sharpens on continued heating. Thus above 310 °C, combustion of the organic ligand commences, which corresponds with the TGA analysis.
The possibility of Ce3+/Ce4+ redox is useful to explore in coordination polymers. We thus assessed the stability and behaviour of (1) towards hydrogen peroxide. After immersion of (1) in 30 v/v H2O2 solution for 2 hours, the powder XRD pattern shows the same characteristic signature, with no changes in Bragg peaks positions and only small changes in relative peak intensities, Fig. 9a. The colour of the sample, however, is notably different, with a change from pale yellow to orange. This colour change was quantified using diffuse-reflectance UV-Vis spectroscopy, which indicates a small decrease in band gap from ∼2.0 to ∼1.9 eV, Fig. 9b. XPS was used to examine the surface oxidation state of the material before and after oxidation, Fig. 9c and d. Quantification of the signal shows that around 50% of the Ce3+ present at the surface is oxidised to Ce4+ after oxidation. Given that the powder XRD shows little change and that XPS has a probe depth of a few nanometres, it is probable that the reduction of cerium is associated with the surface of the sample, and in turn responsible for the bulk colour change observed. TGA also shows no significant differences after the oxidation treatment, which again implies only the surface is modified (see ESI†). Xiong et al. previously studied a cerium(III) benzenetricarboxylate material that could be partially oxidised by treatment with H2O2,25 and thus (1) behaves in a similar manner.
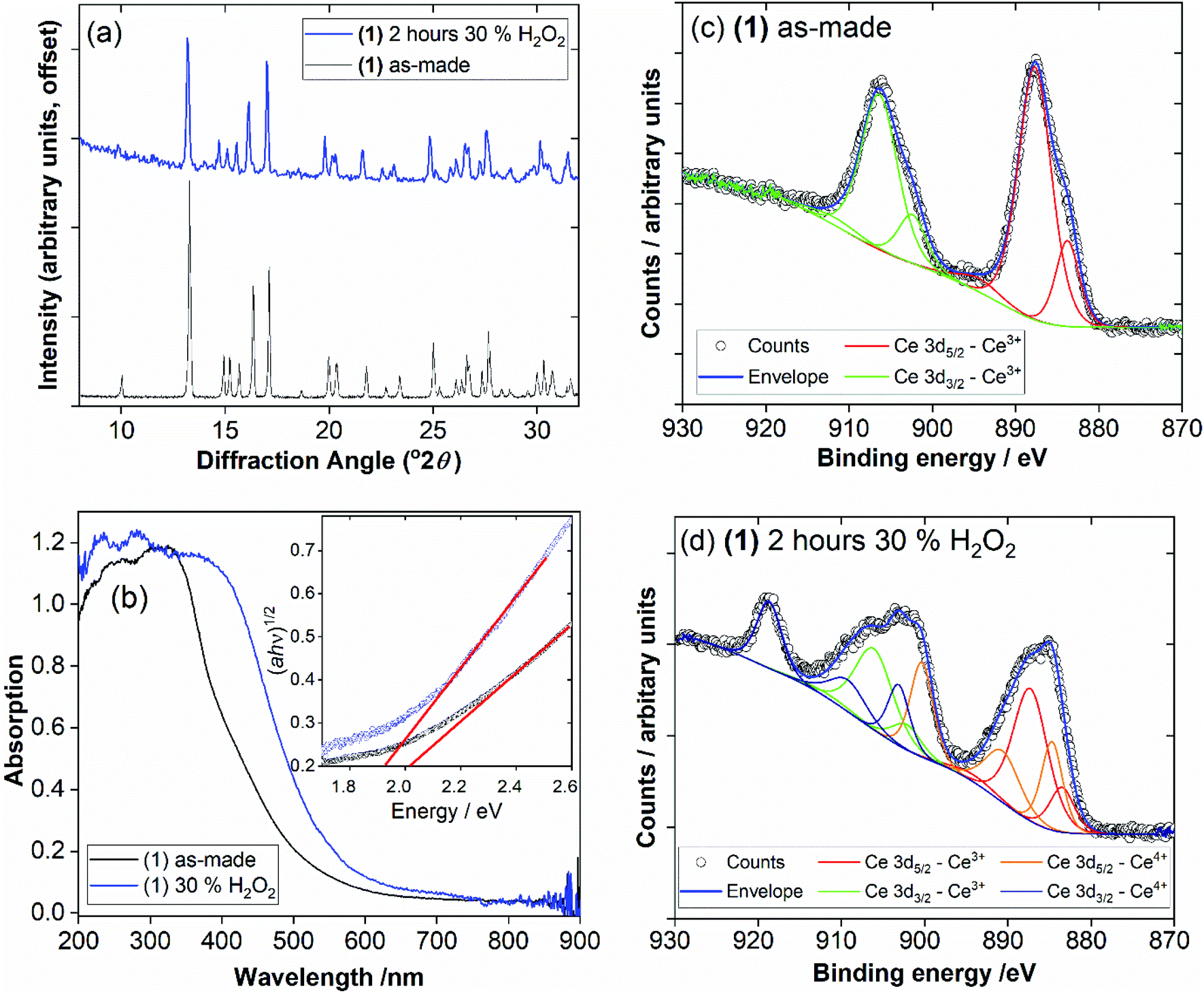 | ||
| Fig. 9 Characterisation of (1) upon application of oxidising conditions: (a) powder XRD measured before and after exposure to 30% H2O2 solution, (b) UV-Vis of the two solids, with Tauc plots inset, where red lines showing the linear extrapolation to estimate band gap, and (c) and (d) XPS of the materials, with the deconvolution and assignment of signals indicated. | ||
One interesting application of cerium–organic frameworks is in photocatalysis, and we assessed the possibility of (1) for this purpose, given its stability in aqueous conditions. The degradation of methyl orange was studied using a 250 W mercury lamp as UV source. This showed an effective decomposition of the dye in the presence of (1) alone, which was enhanced with added H2O2, Fig. 10a. We also verified the decomposition of dye in the presence of (1) that had been previously oxidised. Adsorption effects were found to be negligible (below 0.2%), as shown by a background measurement in the absence of light (ESI†). A decrease in the intensity of the absorption band (∼505 nm) relative to –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– double bond in the azo dye and the decolourisation of the solution indicate an efficient photodegradation (see ESI†). Also, for photocatalysis in the presence of our catalyst a new band around 280 nm appears and becomes stronger over reaction time, indicating the cleavage of the azo bond and the formation of structures with –NH2 groups.26
N– double bond in the azo dye and the decolourisation of the solution indicate an efficient photodegradation (see ESI†). Also, for photocatalysis in the presence of our catalyst a new band around 280 nm appears and becomes stronger over reaction time, indicating the cleavage of the azo bond and the formation of structures with –NH2 groups.26
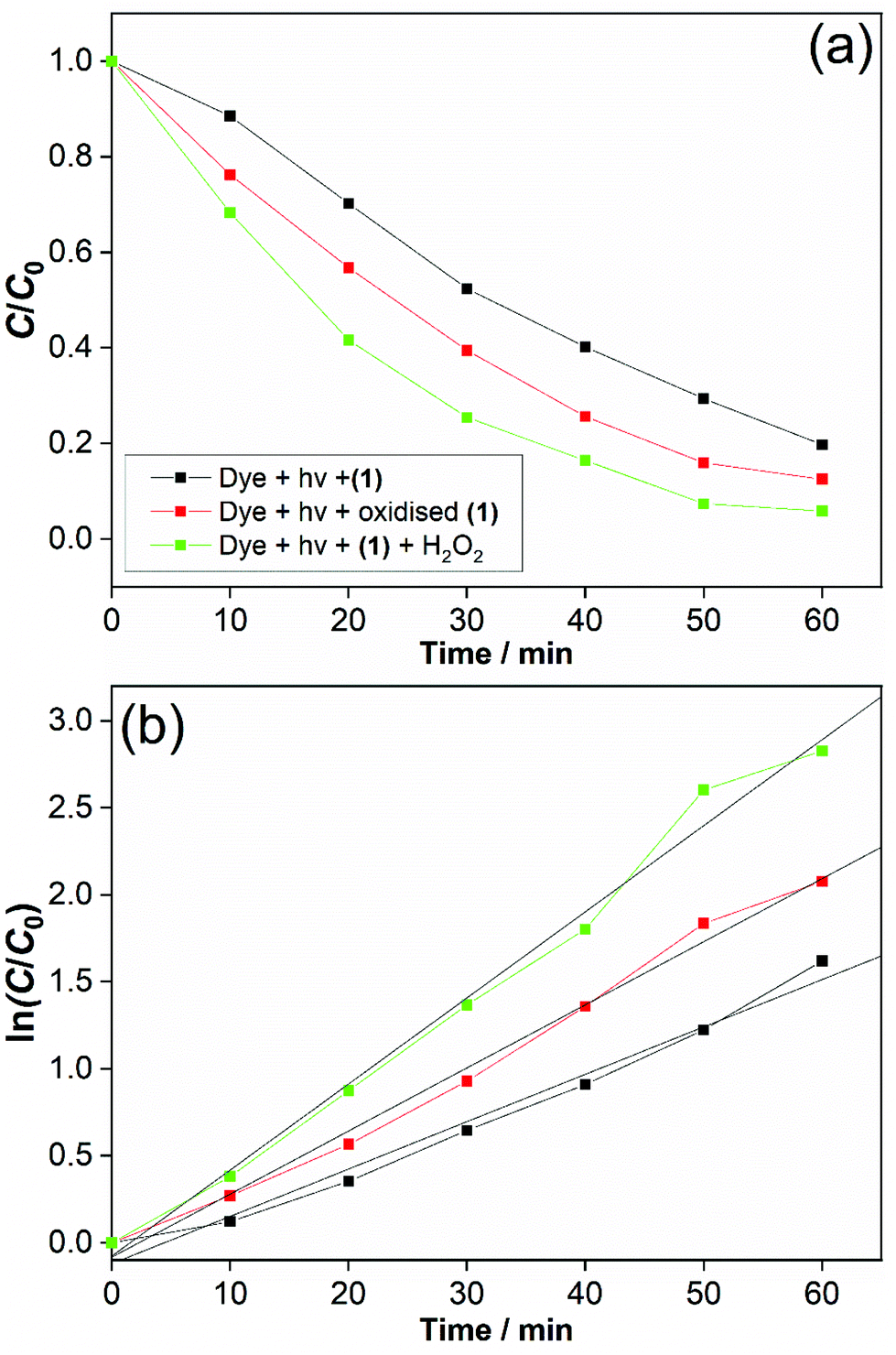 | ||
| Fig. 10 (a) Photodegradation of methyl orange at catalysts, 25 °C, UV light (250 W). (b) Kinetic fit of the time trace and linear plots (pseudo first-order) of the UV–VIS band at λmax = 505 nm, R2 = 0.982 (black line), 0.992 (red line) and 0.987 (green line). The legend in (b) is the same as in (a). | ||
After 60 min, the degradation rates were 80%, 87% and 94% for (1) alone, previously oxidised (1) and (1) + H2O2. Degradation rates of methyl orange were estimated by dye decolourisation following eqn (1):
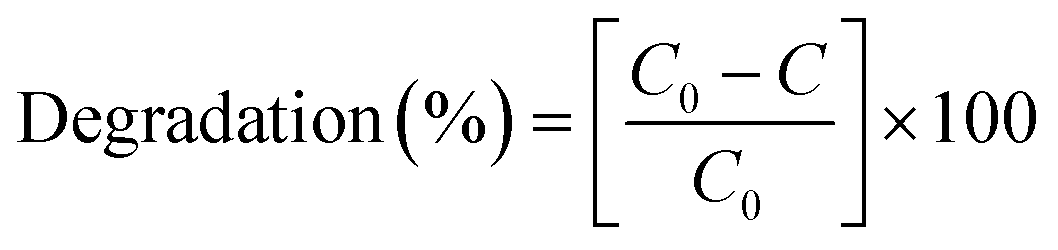 | (1) |
The photodegradation follows pseudo first-order kinetics, Fig. 10b, as expected for heterogeneous photocatalysis.27 The corresponding rate constants kobs (min−1) for methyl orange were determined by pseudo first-order kinetic expression (eqn (2)). Fitted linear equation (R2 = 0.982–0.992) for all photocatalytic experimental conditions is showed in Fig. 10b.
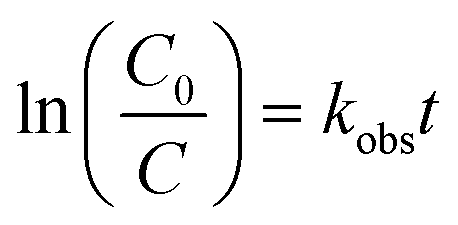 | (2) |
The photocatalytic performance of cerium-PTC based MOFs was significantly affected by the presence of surface Ce3+/Ce4+. Simultaneous presence of these species is known to favour redox reactions in the photocatalytic mechanisms.28 The photodegradation of methyl orange was conducted in the presence of catalyst (1) and H2O2 (0.01 M), simulating a Fenton-like process.29 The photocatalysis was improved and best conversion was achieved with methyl orange almost completely degraded after 60 min. For this photodegradation the effect of hydrogen peroxide on the dye must be considered (ESI†). Although the photocatalysis was enhanced with H2O2 addition, the performance of (1) alone and previously oxidised (1) must be highlighted. Very low quantities of catalysts were employed in our experiments and good conversion rates were obtained. The reported works for dye degradation in the presence of metal–organic frameworks have typically described the use more than three-fold excess of catalyst per dye concentration.27b,30 While CeO2 has been employed as catalyst with similar results for photodegradation,28,31 it is noteworthy that water-stable and oxidatively-stable cerium organic frameworks provide effective photocatalysts.
Conclusions
We have explored the hydrothermal chemistry of cerium salts in combination with a polydentate ligand that includes a strongly binding tridentate binding motif, via a pyridine nitrogen and two adjacent carboxylate motifs. Despite using cerium(IV) precursors, the crystalline product are Ce(III) frameworks. It can be noted that the previous synthesis of the material isostructural to (1) used a Ce(III) precursor, illustrating the strong tendency of cerium to form Ce(III) carboxylates. The variety of bonding modes of pyridine-2,4,6-tricarboxylate is illustrated by the materials we have crystallised, which add to a wider family of lanthanide pyridine-2,4,6-tricarboxylates already reported in the literature. The material (1) is an example of a hydrothermally-stable metal–organic framework, which is also stable under oxidative conditions, where evidence is found for oxidation of a large proportion of cerium at the surface to the +4 oxidation state.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank FAPESP for awarded of a postdoctoral fellowship to JFL, and CNPq for funding this work, including award of a Pesquisador Visitante Especial position at USP Ribeirão Preto for RIW. We are grateful for James Crosland for measuring the UV-Vis spectra and David Hammond for measuring thermogravimetric data. CCDC 2110894–2110897† contain the supplementary crystallographic data for this paper. Some the equipment used in this research was provided by the University of Warwick's Research Technology Platforms.References
- (a) J. Jacobsen, A. Ienco, R. D'Amato, F. Costantino and N. Stock, Dalton Trans., 2020, 49, 16551–16586 RSC; (b) Z. Hu, Y. Wang and D. Zhao, Chem. Soc. Rev., 2021, 50, 4629–4683 RSC.
- V. Sridharan and J. C. Menéndez, Chem. Rev., 2010, 110, 3805–3849 CrossRef CAS PubMed.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- K. A. Lomachenko, J. Jacobsen, A. L. Bugaev, C. Atzori, F. Bonino, S. Bordiga, N. Stock and C. Lamberti, J. Am. Chem. Soc., 2018, 140, 17379–17383 CrossRef CAS PubMed.
- S. R. Batten, S. Neville and D. R. Turner, Coordination Polymers: Design, Analysis and Application, Royal Society of Chemistry, Cambridge, UK, 2009 Search PubMed.
- M. Lammert, M. T. Wharmby, S. Smolders, B. Bueken, A. Lieb, K. A. Lomachenko, D. D. Vos and N. Stock, Chem. Commun., 2015, 51, 12578–12581 RSC.
- D. Dang, P. Wu, C. He, Z. Xie and C. Duan, J. Am. Chem. Soc., 2010, 132, 14321–14323 CrossRef CAS PubMed.
- X.-P. Wu, L. Gagliardi and D. G. Truhlar, J. Am. Chem. Soc., 2018, 140, 7904–7912 CrossRef CAS PubMed.
- M. Campanelli, T. Del Giacco, F. De Angelis, E. Mosconi, M. Taddei, F. Marmottini, R. D'Amato and F. Costantino, ACS Appl. Mater. Interfaces, 2019, 11, 45031–45037 CrossRef CAS.
- L. Luo, L. Huang, X. Liu, W. Zhang, X. Yao, L. Dou, X. Zhang, Y. Nian, J. Sun and J. Wang, Inorg. Chem., 2019, 58, 11382–11388 CrossRef CAS.
- Y. Zhang, X. Zeng, X. Jiang, H. Chen and Z. Long, Microchem. J., 2019, 149, 103967 CrossRef CAS.
- M. C. Das, S. K. Ghosh, E. C. Sañudo and P. K. Bharadwaj, Dalton Trans., 2009, 1644–1658 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- (a) T. Hasegawa, S. M. F. Shahed, Y. Sainoo, A. Beniya, N. Isomura, Y. Watanabe and T. Komeda, J. Chem. Phys., 2014, 140, 044711 CrossRef; (b) M. N. Revoy, R. W. J. Scott and A. P. Grosvenor, J. Phys. Chem. C, 2013, 117, 10095–10105 CrossRef CAS; (c) E. Paparazzo, Mater. Res. Bull., 2011, 46, 323–326 CrossRef CAS.
- (a) S. K. Ghosh and P. K. Bharadwaj, Eur. J. Inorg. Chem., 2005, 2005, 4886–4889 CrossRef; (b) H.-L. Gao, L. Yi, B. Ding, H.-S. Wang, P. Cheng, D.-Z. Liao and S.-P. Yan, Inorg. Chem., 2006, 45, 481–483 CrossRef CAS PubMed.
- C.-Q. Zhao, Y.-M. Ou, Z.-Y. Zhao, X.-J. Yin and Y.-M. Jiang, Z. Kristallogr., 2013, 228, 3–4 CAS.
- (a) P. M. Forster, A. R. Burbank, C. Livage, G. Férey and A. K. Cheetham, Chem. Commun., 2004, 368–369 RSC; (b) F. Millange, M. I. Medina, N. Guillou, G. Férey, K. M. Golden and R. I. Walton, Angew. Chem., Int. Ed., 2010, 49, 763–766 CrossRef CAS; (c) H. H.-M. Yeung, Y. Wu, S. Henke, A. K. Cheetham, D. O'Hare and R. I. Walton, Angew. Chem., Int. Ed., 2016, 55, 2012–2016 CrossRef CAS PubMed.
- I. D. Brown, The Chemical Bond in Inorganic Chemistry: The Bond Valence Model, Oxford University Press, Oxford, 2002 CAS.
- T. Rhauderwiek, N. Heidenreich, H. Reinsch, S. Øien-Ødegaard, K. A. Lomachenko, U. Rütt, A. V. Soldatov, K. P. Lillerud and N. Stock, Cryst. Growth Des., 2017, 17, 3462–3474 CrossRef.
- S. Sharif, I. U. Khan, S. Zaheer and S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m624–m625 CrossRef CAS.
- C.-J. Li, M.-X. Peng, J.-D. Leng, M.-M. Yang, Z. Lin and M.-L. Tong, CrystEngComm, 2008, 10, 1645–1652 RSC.
- I. U. Khan, S. Sharif and O. Sahin, J. Coord. Chem., 2013, 66, 3113–3125 CrossRef CAS.
- Y. Xiong, S. Chen, F. Ye, L. Su, C. Zhang, S. Shen and S. Zhao, Chem. Commun., 2015, 51, 4635–4638 RSC.
- (a) Y. Sha, I. Mathew, Q. Cui, M. Clay, F. Gao, X. J. Zhang and Z. Gu, Chemosphere, 2016, 144, 1530–1535 CrossRef CAS; (b) Z. T. Ildefonso, P. B. José de Jesús, T. L. Celeste Yunueth, L. R. Luis, M. L. Maria Luisa and M. V. Yunny, RSC Adv., 2016, 6, 76167–76173 RSC.
- (a) N. El-Wakiel and H. El-Ghamry, Int. J. Chem. Kinet., 2017, 49, 464–476 CrossRef CAS; (b) D. Zhao and C. Cai, Dyes Pigm., 2021, 185, 108957 CrossRef CAS.
- X. Tao, W. Cong, L. Huang and D. Xu, J. Alloys Compd., 2019, 805, 1060–1070 CrossRef CAS.
- S. S. F. Carvalho, A. C. C. Rodrigues, J. F. Lima and N. M. F. Carvalho, Inorg. Chim. Acta, 2020, 512, 119924 CrossRef CAS.
- R. Paz, H. Viltres, N. K. Gupta and C. Leyva, J. Mol. Liq., 2021, 337, 116578 CrossRef CAS.
- S. Zhao, S. Li, Y. Long, X. Shen, Z. Zhao, Q. Wei, S. Wang, Z. Zhang, X. Zhang and Z. Zhang, Chemosphere, 2021, 280, 130637 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2110894–2110897. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03514b |
| This journal is © The Royal Society of Chemistry 2022 |