
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isocyanate deoxygenation by a molecular magnesium silanide†
Bibian
Okokhere-Edeghoghon
 ,
Samuel E.
Neale
,
Michael S.
Hill
*,
Mary F.
Mahon
and
Claire L.
McMullin
*
,
Samuel E.
Neale
,
Michael S.
Hill
*,
Mary F.
Mahon
and
Claire L.
McMullin
*
Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: msh27@bath.ac.uk; cm2025@bath.ac.uk
First published on 29th November 2021
Abstract
The stoichiometric reactivity of a β-diketiminato (BDI) magnesium silanide towards a variety of organic isocyanates has been assessed. While the primary outcome of reactions of t-BuNCO, DippNCO and CyNCO was the production of β-diketiminato magnesium siloxide adducts of the isonitrile resulting from isocyanate deoxygenation, analogous treatment with i-PrNCO led to multiple products, four of which have been positively identified. Although the specificity of this latter reaction was hampered by competitive isocycanate addition at the γ-methine carbon of the BDI supporting ligand, the identification of [{i-PrNCO}CH{(Me)CNDipp}Mg{Me2PhSi}C(O)Ni-Pr]6 provided corroborative evidence for the likely generation of sila-amidate intermediates in all four reactions under study. The formation of [{Me2PhSi}C(O)NR]− anions as the most likely initial species formed en route to isonitrile and siloxide formation was, therefore, validated by a computational density functional theory (DFT) study.
Introduction
Almost four decades have elapsed since Baldwin and co-workers’ report that the deoxygenation of an organic isocyanate may be accomplished using t-butyldiphenylsilyl lithium to provide the corresponding isonitrile and the lithium siloxide (Scheme 1a).1,2 Despite the potential utility of this transformation, comparable well-defined molecular examples of deoxygenative isocyanate reduction remain a rarity. As part of a study of the ability of the dialkylsilylene (1) to effect the reduction of carbon dioxide to carbon monoxide, Kira and co-workers observed that treatment of 1 with two equivalents of the isoelectronic heterocumulene, DippNCO (Dipp = 2,6-di-isopropylphenyl), gave the dioxasiletane (2) and the corresponding aryl isonitrile (Scheme 1b).3 Similarly, through a combined synthetic and computational study, Roesky and co-workers reported that the cleavage of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of i-PrNCO is mediated by the germylene/borane species (3) to yield the heterocyclic species (4) and (5), most likely through a cooperative activation of the CO bond by the intramolecular Ge/B Lewis pair (Scheme 1c).4
O bond of i-PrNCO is mediated by the germylene/borane species (3) to yield the heterocyclic species (4) and (5), most likely through a cooperative activation of the CO bond by the intramolecular Ge/B Lewis pair (Scheme 1c).4
 | ||
| Scheme 1 (a)–(c) Previously described examples of isocyanate deoxygenation; (d) reactivity of compound 6 with carbodiimdes. | ||
As part of a broader study of the reactivity of alkaline earth derivatives of group 13- and 14-centred anions,5–12 we have recently reported that the β-diketiminato magnesium silanide, [(BDI)MgSiMe2Ph] (6, BDI = HC{(Me)CNDipp}2) reacts with carbodiimides as a potent triorganosilicon nucleophile to provide β-diketiminato magnesium sila-amidinate complexes.10 Dependent on the identity of the N-alkyl or -aryl substituents, this chemistry provided a significant diversity of outcome, including multiple carbodiimide insertion and C–C bond formation at the γ-methine carbon centre of the β-diketiminate ligand (Scheme 1d). Although these variations were ascribed to the relative electrophilicity of the two-coordinate carbodiimide carbon centre, no fragmentation of the {NCN} unit was observed. In this contribution, we report that similar reactions performed with t-BuNCO and DippNCO result in the complete and selective rupture of the CO bond. In contrast, a decrease in the isocyanate steric demands results in reduced kinetic discrimination and reactivity at the BDI γ-methine carbon, albeit this process remains competitive with RNCO/Mg–Si insertion and subsequent deoxygenation.
Results and discussion
As described in our earlier report,10 compound 6 was most conveniently generated in situ by reaction of the organomagnesium reagent, [(BDI)Mgn-Bu] (5), with the commercially available silaborane, pinB-SiMe2Ph (pin = pinacolato). Analysis by 1H NMR spectroscopy of benzene solutions of compound 6 prepared in this manner, with subsequent addition of either t-BuNCO or DippNCO and heating at 60 °C, indicated conversion to the respective new β-diketiminate-containing compounds, 7 and 8 (Scheme 2). After 48 hours the 1H NMR spectra of both compounds displayed resonances that could be assigned to an equimolar ratio of the BDI ligand, the triorganosilyl substituent and the hydrocarbon residue of the isocyanate substrate. The iso-propyl methine resonances of the BDI ligand of compound 8 were separated into two broad (2H by relative integration) signals at δ 3.58 and 3.26 ppm and a series of four (6H) doublet resonances, while the appearance of two discriminated (3H) singlet signals at δ 0.83 and 0.82 ppm were consistent with a diastereotopic disposition of the silylmethyl substituents. In contrast, compound 7 presented a less complex 1H NMR spectrum indicative of a reduced steric influence imposed by the N-tert-butyl residue. A characteristic signal observed at δ 165.4 ppm in the 13C{1H} NMR spectrum of 8, which was revealed through HMBC analysis not to correlate with any proton, was comparable to that assigned to the C-donor carbon centre of Lappert and co-workers’ [Mg{CH(SiMe3)2}2·(CN-C6H3Me2-2,6)2] (9, δ 156.4 ppm), the only previous magnesium adduct of an aryl isonitrile to have been definitively characterised.13 Whereas no comparable resonances were observed in the 13C{1H} NMR spectrum of 7, possibly due to the greater chemical shift anisotropy incurred by a more labile coordination geometry, the identity of both compounds as magnesium siloxide isonitrile adducts was confirmed by X-ray diffraction analysis. | ||
| Scheme 2 Synthesis of compounds 7 and 8. | ||
Colourless block single crystals of compounds 7 and 8 were isolated after removal of the benzene solvent and crystallisation by cooling of pentane solutions to −30 °C. Crystallographic analysis confirmed both structures of compounds 7 and 8 to comprise magnesium siloxides in which the remaining sites of the 4-coordinate alkaline earth centres are occupied by the β-diketiminate ligand and the respective N-tert-butyl- and Dipp-substituted isonitriles. The structures are illustrated in Fig. 1 and selected bond length and angle data are provided in the figure caption. The Mg–O bond lengths observed in compounds 7 and 8 [7 1.8360(14); 8 1.8269(12) Å] lie in the range established [1.814(3)–1.851(2) Å] for the limited number of reported terminal magnesium siloxides in which the alkaline earth centre is similarly 4-coordinate.14–19 The formation of the isonitrile is also confirmed by the clearly distinguishable sp-hybridisation of the donor carbon centres and the short C–N bond lengths of both Mg-coordinated C-donor molecules [7: N(3)–C(38) 1.145(3); 8: N(3)–C(30) 1.151(2) Å]. The Mg–C bond lengths provided by these latter ligands [7 2.2534(19), 8 2.2781(15) Å] are slightly shorter than those observed in Lappert's pseudo-tetrahedral adduct [9 2.306(9) and 2.307(10) Å],13 most likely as a consequence of the more strongly electron donating alkyl co-ligands in the previously reported species.
 | ||
| Fig. 1 ORTEPs (30% probability) of (a) compound 7 and (b) compound 8. Hydrogen atoms, BDI iso-propyl groups and minor disordered components are removed for clarity. Selected bond lengths (Å) and angles (°): (7) Mg1–O1 1.8360(14), Mg1–N1 2.0477(15), Mg1–N2 2.0354(16), Mg1–C38 2.2534(19), Si1–O1 1.5756(14), N3–C38 1.145(3), N1–Mg1–N2 92.66(6), O1–Mg1–N1 121.99(7), O1–Mg1–N2 117.12(7), O1–Mg1–C38 98.18(7), N1–Mg1–C38 113.59(7), N1–Mg1–N2 92.66(6), N2–Mg1–C38 114.59(7), Si1–O1–Mg1 147.32(10), N3–C38–Mg1 165.25(16): (8) Mg1–O1 1.8269(12), Mg1–N1 2.0269(12), Mg1–N2 2.0339(12), Mg1–C30 2.2781(15), Si1–O1 1.5797(12), N3–C30 1.151(2), N1–Mg1–N2 94.87(5), O1–Mg1–N1 121.76(6), O1–Mg1–N2 123.40(6), O1–Mg1–C30 104.25(6), Si1–O1–Mg1 166.19(10), N3–C30–Mg1 174.02(13). | ||
Treatment of 6 with CyNCO resulted in a similar course of reaction to that which yielded compounds 7 and 8 but provided a mixture of two new β-diketiminate species. Compounds 10a and 10b could be discriminated in the resultant 1H NMR spectrum in an approximate 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio by integration of their respective γ-CH methine resonances at δ 4.79 and 4.77 ppm and the corresponding (6H) signals arising from their Si–Me residues, which resonated at δ 0.09 and 0.17 ppm. While neither compound could be crystallised in analytically pure form, the origin of these observations was illuminated by an X-ray diffraction analysis of a co-crystal of both species isolated from a hexane solution at room temperature. Although the data could not be definitively resolved, they were assigned as resulting from a ca. 1:1 co-crystal of the respective CyNC-coordinated magnesium siloxide (10a) and magnesium silanide products, (10b) (Scheme 3). Although the lower selectivity of this reaction and the issue presented by their co-crystallisation precludes the full characterisation of either species, their coincident formation is informative and necessarily indicative of the dissociation of the nitrile moiety subsequent to the deoxygenation process common to the formation of both 7 and 8.
:1 ratio by integration of their respective γ-CH methine resonances at δ 4.79 and 4.77 ppm and the corresponding (6H) signals arising from their Si–Me residues, which resonated at δ 0.09 and 0.17 ppm. While neither compound could be crystallised in analytically pure form, the origin of these observations was illuminated by an X-ray diffraction analysis of a co-crystal of both species isolated from a hexane solution at room temperature. Although the data could not be definitively resolved, they were assigned as resulting from a ca. 1:1 co-crystal of the respective CyNC-coordinated magnesium siloxide (10a) and magnesium silanide products, (10b) (Scheme 3). Although the lower selectivity of this reaction and the issue presented by their co-crystallisation precludes the full characterisation of either species, their coincident formation is informative and necessarily indicative of the dissociation of the nitrile moiety subsequent to the deoxygenation process common to the formation of both 7 and 8.
 | ||
| Scheme 3 Synthesis of compounds 10a and 10b. | ||
Irrespective of the contrasting outcome of the reaction with CyNCO, the formation of 7, 8 and 10a/b is indicative of the operation of a common pathway involving activation of the isocyanate reagents at the Mg–Si bond of 6. In contrast to these straightforward deoxygenation processes, monitoring of an analogous reaction between compound 6 and isopropyl isocyanate by 1H and 13C{1H} NMR spectroscopy (Fig. S10 and S11†) evidenced a more complex mixture of reaction products (Scheme 4). Although an apparent BDI γ-CH methine signal at δ 4.86 and a Si–Me resonance at δ 0.15 ppm were indicative of a predominant reaction product, the simultaneous emergence of up to seven further species was evident by examination of both spectral regions. While an isonitrile carbon resonance at δ 133.8 ppm could be tentatively assigned in the corresponding 13C{1H} NMR spectrum, the identity of these species could not be definitively resolved by NMR spectroscopy. Insight into the nature of these competitive processes was, therefore, reliant on fractional crystallisation of the reaction mixture. Although an initial X-ray study of a crystal obtained from the reaction mixture indicated the possible formation of a mixture of magnesium siloxide and silyl species analogous to the components identified during the analysis of compounds 10a/b, no adequate model could be resolved. Further efforts to isolate a pure bulk sample, however, yielded three further compounds, 11–13, using the same equimolar reaction stoichiometry and experimental conditions. These reactions invariably resulted in the formation of a variety of unassignable species by NMR analysis, and several apparent singlet BDI γ-methine resonances were again observed in the 1H NMR spectrum of the crude reaction mixtures.
 | ||
| Scheme 4 Compounds 11–13 identified from the reaction of compound 6 and i-PrNCO. | ||
Further efforts to identify the products of these reactions entailed the mechanical separation of single crystals of compound 11, X-ray diffraction analysis of which confirmed its constitution as a magnesium complex ligated by two identical chelating N,O-chelating β-ketoiminate ligands (Fig. 2a). This compound, therefore, has evidently been formed by a combination of Schlenk-type ligand redistribution and C–C bond formation between the isocyanate i-PrNCO residues and the γ-methine carbon of the BDI anions. Subsequent tautomerisation of the γ-CH proton then provides a secondary amide unit, the i-PrN-H proton of which interacts strongly with the exocyclic, and now imine-like, N-Dipp residues [N(2)–C(4) 1.292(4); N(5)–C(35) 1.292(4) Å]. The resultant anions are bound to Mg1 in a κ2-N,O-bidentate coordination mode provided by the oxygen atom and one of the N-Dipp residues of the former BDI ligands. A similar C–C bond forming process, albeit without C–H to N–H tautomerisation, has been observed during the reaction of CO2 and the BDI ligand of [(BDI)Mg{HB(C6F5)3}],20 and is reminiscent of the reactions between (6) and p-tolylcarbodiimide (Scheme 1d).10 The atoms comprising the chelate {NCCCOMg} structures of both ligands in 11 are close to planar and provide magnesium with a distorted tetrahedral coordination environment. The various C–C bond lengths within the core of the ligands suggest significant delocalisation around the coupled isocyanate units such that both the central carbon of the former isocyanate and the previous BDI γ-methine carbon atom now display planar geometries.
 | ||
| Fig. 2 ORTEPs (30% probability) of (a) compound 11 and (b) compound 12. Hydrogen atoms, apart from those attached to N3 and N7, isopropyl groups attached to Dipp substituents and the second components of disordered atoms are removed for clarity. Selected bond lengths (Å) and angles (°): (11) Mg1–O1 1.909(2), Mg1–O2 1.909(2), Mg1–N1 2.061(3), Mg1–N4 2.057(3), O1–C30 1.265(4), O2–C63 1.272(4), N1–C2 1.315(4), N1–C18 1.443(4), N2–C4 1.292(4), N2–C6 1.428(4), N3–C30 1.336(4), N3–C32 1.458(4), N4–C37 1.317(4), N4–C39 1.444(4) N5–C35 1.293(4), N5–C51 1.415(4), N6–C63 1.332(4), N6–C64 1.459(4), O1–Mg1–N1 87.86(11), O1–Mg1–N4 112.98(11), O2–Mg1–O1 130.22(11), O2–Mg1–N1 110.70(10), O2–Mg1–N4 88.01(10), N4–Mg1–N1 132.92(11); (12) Mg1–O1 1.951(3), Mg1–O2 1.951(3), Mg1–N1 2.132(3), Mg1–N5 2.130(3), Mg1–C30 2.310(7), O1–C67 1.277(4), O2–C63 1.276(4), N1–C2 1.314(4), N1–C18 1.435(5), N2–C4 1.305(5), N2–C6 1.418(4), N3–C67 1.333(5), N3–C68 1.460(5), N5–C35 1.311(4), N5–C51 1.438(5), N6–C37 1.300(5), N6–C39 1.425(4), N7–C63 1.342(5), N7–C64 1.467(5), N4–C30 1.115(14), N4–C31 1.459(6), O1–Mg1–O2 141.83(9), O1–Mg1–N1 84.18(12), O1–Mg1–N5 94.78(12), O1–Mg1–C30 106.0(8), O2–Mg1–N1 95.17(12), O2–Mg1–N5 84.70(12), O2–Mg1–C30 112.1(8), N1–Mg1–C30 90.4(8), N5–Mg1–N1 178.17(10), N5–Mg1–C30 91.3(8). | ||
With its definitive solid-state identity in hand, a diagnostic downfield doublet observed in the 1H NMR spectrum of the complex mixture of reaction products at δ 11.05 ppm was assigned to the NH-i-Pr protons of compound 11. A further distinctive resonance at 175.2 in the 13C{1H} NMR spectrum was also assigned as the former BDI methine carbon which has undergone C–C coupling with the isocyanate. Further scrutiny of the 1H NMR spectroscopic data revealed the formation of small amount of an additional reaction product (12), which manifested as a multiplet resonance at 3.79 ppm that integrated with half the intensity of a pair of differentiated N-Dipp iso-propyl methine signals at δ 3.19 and 3.12 ppm. The source of this latter observation was resolved, as ever, by single-crystal X-ray diffraction analysis of 12, which was identified to contain an identical magnesium bis(κ2-N,O-β-ketoiminate) constitution to compound 11 but with the addition of a molecule of i-PrNC [C30–N4 1.145(3) Å] coordinated to magnesium (Fig. 2b). The almost linear N(1)–Mg(1)–N(5) [178.17(10)°] and obtuse O(1)–Mg(1)–O(2) [141.83(9)°] bond angles ensure that the coordination geometry at Mg(1) is best described as comprising the isonitrile at the apex of a basally distorted square pyramid (τ5 = 0.61).21,22 Consistent with the increased coordination number of magnesium, the Mg–CNi-Pr bond length [Mg(1)–C(30) 2.310(7) Å] of 12 is slightly elongated in comparison to the analogous measurements in both the 4-coordinate adducts, 7 and 8. The Mg–N and Mg–O bond lengths [Mg(1)–N(1) 2.132(3), Mg(1)–N(5) 2.130(3), Mg(1)–O(1) 1.951(3), Mg(1)–O(2) 1.951(3) Å] and the relevant bond angles [O(1)–Mg(1)–N(1) 84.18(12), O(1)–Mg(1)–N(5) 94.78(12)°] are, however, similar to those observed in compound 11.
Although no complementary siloxide generation could be confirmed, the identification of the isonitrile units in compound 12 is indicative of a similar deoxygenative process to that operating during the syntheses of 7 and 8. In a final attempt to ascertain the fate of the silyl residues during the formation of compounds 11 and 12, therefore, a further reaction between an equimolar quantity of 6 and isopropyl isocyanate was conducted. Although the resultant 1H NMR spectrum of the crude sample again afforded a complex series of aliphatic resonances, a sample of a further compound (13) was provided by fractional crystallisation of the reaction mixture. Single-crystal X-ray diffraction analysis revealed compound 13 (Fig. 3) to be a hexanuclear aggregate of a heteroleptic magnesium species, [{i-PrNCO}CH{(Me)CNDipp}Mg{i-PrNCO(SiMe2Ph)}]6, comprising three unique, four-coordinate, magnesium centres. As was the case in both compounds 11 and 12, each magnesium is ligated by an anion derived from a C–C bond forming reaction between the γ-methine carbon of the β-diketiminate ligand of 2 and a molecule of the isopropyl isocyanate. Unlike 11 and 12, however, this γ-C centre displays a pseudo-tetrahedral geometry such that both N-Dipp residues display C–N bond lengths (ca. 1.27 Å) consistent with their identification as imine-like functions. As a result, each of these anions binds exclusively to magnesium in a κ2-N,O-coordination mode through the oxygen and nitrogen donor atoms of the amidate units. More significantly, insight into the fate of the silyl function during these reactions was provided by the sila-amidate anions, [{Me2PhSi}C(O)Ni-Pr]−, which were also bound to each magnesium in compound 13. Although a number of carbamoyl silanes have been described,23,24 these anions appear to be unprecedented when bound to a metal centre. Their formation, however, is readily rationalised to be a consequence of RNCO insertion into the Mg–Si bond of compound 6, in a process strongly reminiscent of its reactivity with a variety of N,N′-di-alkyl carbodiimides (Scheme 1d).10 In each case these silamidate ligands bridge the magnesium centres via their nitrogen and oxygen atoms, which facilitates the propagation of the heteroleptic units into a hexanuclear aggregate. Of particular note, the various Mg–O interactions [Mg1–O2 1.8671(13) Å, Mg2–O3 1.8718(13) Å, Mg3–O4 1.8691(13) Å] are similar to those within Chisholm's terminal β-diketiminato magnesium tert-butoxide, [(BDI)MgOt-Bu(THF)] [1.844(2) Å],25 and, in conjunction with the correspondingly short C–N bonds [N4–C34 1.304(2), N5–C46 1.308(2), N6–C58 1.309(2) Å], are indicative of localised C–O and CN bonding across the bridging sila-amidate anions.
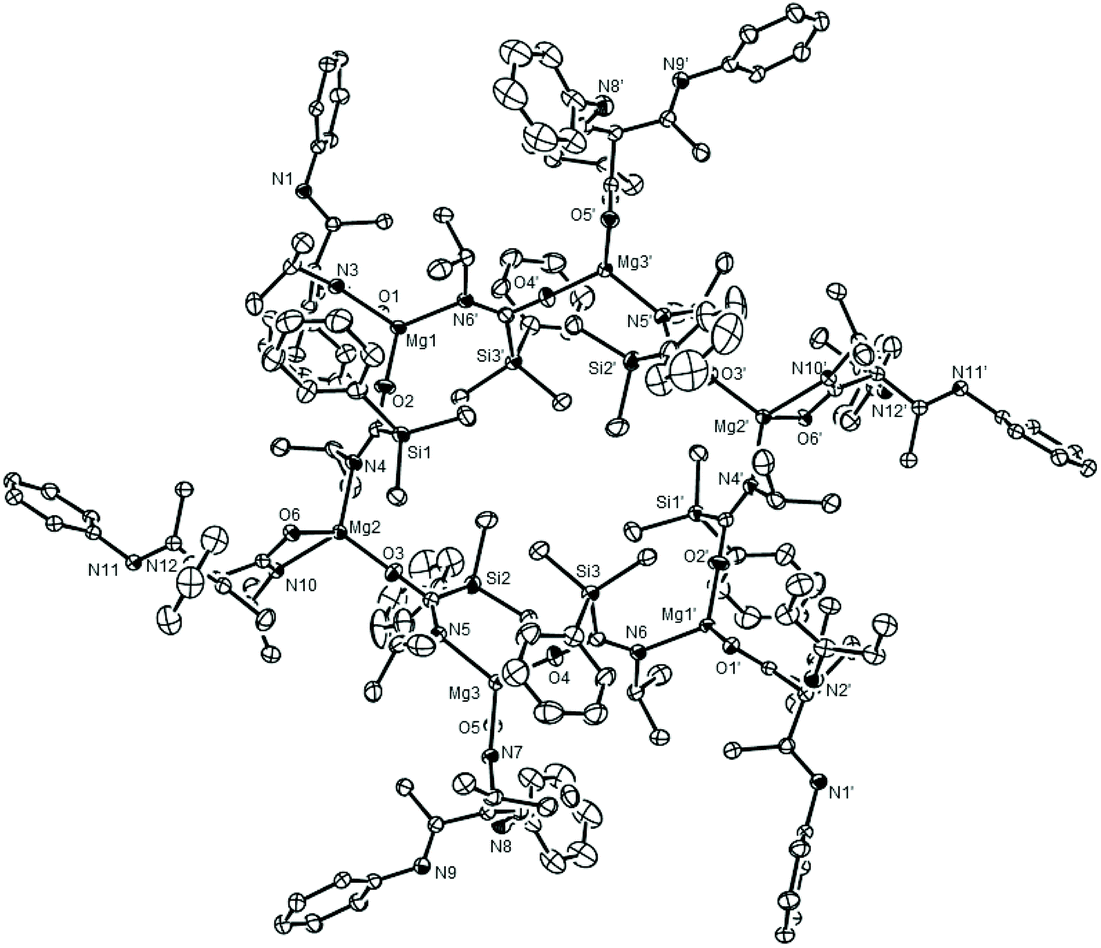 | ||
| Fig. 3 ORTEP (30% probability) of compound 13. Hydrogen atoms, isopropyl groups attached to Dipp substituents and the second components of disordered atoms are removed for clarity. Selected bond lengths (Å) and angles (°): Si(1)–C(34) 1.9094(19), Si(2)–C(46) 1.901(2), Si(3)–C(58) 1.9063(19), Mg(1)–O(1) 2.0106(12), Mg(1)–O(2) 1.8671(13), Mg(1)–N(3) 2.0980(14), Mg(2)–O(3) 1.8718(13), Mg(2)–O(6) 2.0059(14), Mg(2)–N(4) 2.0555(15), Mg(2)–N(10) 2.0930(14), Mg(3)–O(4) 1.8691(13), Mg(3)–O(5) 2.0046(13), Mg(3)–N(5) 2.0599(15) Mg(3)–N(7) 2.0898(14), O(1)–C(30) 1.2969(19), N(3)–C(30) 1.297(2), N(7)–C(70) 1.297(2); C(38)–Si(1)–C(34) 116.68(8), C(38)–Si(1)–C(39) 108.80(9), O(1)–Mg(1)–N(3) 65.31(5), O(2)–Mg(1)–O(1) 121.81(6) O(2)–Mg(1)–N(3) 111.94(6), O(3)–Mg(2)–O(6) 122.26(6), O(3)–Mg(2)–N(4) 113.57(6), O(3)–Mg(2)–N(10) 111.47(6), O(6)–Mg(2)–N(4) 110.61(6), O(6)–Mg(2)–N(10) 65.64(5), N(4)–Mg(3)–N(10) 125.88(6). Symmetry operations to generate primed (‘) labelled atoms; 1 − x, −y, 1 − z. | ||
Computational analysis
The identification of the sila-amidate units of compound 13 strongly indicates that the intermediacy of analogous species also leads to isocyanate deoxygenation and the ultimate generation of the siloxide derivatives; compounds 7, 8 and 10a. Computational studies were, thus, performed to characterise the mechanism of isonitrile formation from t-BuNCO and the silanide species (6) (ΔG = −17.8 kcal mol−1 relative to [(BDI)MgnBu], 5, and corresponding reactants) (Fig. 4). Initial efforts focused on the characterisation of a direct Si–O coupling process to yield the siloxide group. While a saddle point was located in which isocyanate coordination (via the Nt-Bu group) is followed by Si–O coupling viaTS(6-I)B (ΔΔG‡ = +22.5 kcal mol−1, relative to 6), a more accessible process was identified in which Si–C coupling takes place viaTS(6-I) (ΔΔG‡ = +7.9 kcal mol−1) to form an α-silylamidatomagnesium species I (−32.0 kcal mol−1). An alternative Si–C coupling process in which the isocyanate oxygen coordinates to Mg was also located but found to be disfavoured (viaTS(6-I)C, ΔΔG‡ = +14.1 kcal mol−1, Fig. S14†), likely a result of the increased steric pressure of the Nt-Bu next to the silyl substituents rather than the carbonyl O in TS(6-I). The kinetic favourability of Si–C coupling is unsurprising given prior understanding via NBO analysis of the nucleophilicity of the silanide substituent in 6,10 and that isocyanates are significantly electrophilic at the sp-hybridized carbon centre. Moreover, the relatively short contact between Mg and one of the Si–Me bonds in I (where Mg–C = 2.66 Å, Mg–Si = 3.32 Å), along with an elongation of the Si–Me bond itself relative to the other in the –SiMe2Ph group (1.94 Å, vs. 1.90 Å), suggest a δ-agostic Si–C interaction.26 From I, the SiMe2Ph group appears to be more electrophilic in nature compared to the nucleophilicity of the silyl group in 6, as supported by the atomic contributions in the newly formed Si–C bond via NBO (34% on Si vs. 66% on C), and Natural Atomic charges (qSi = +1.43 in Ivs. +0.81 in 6). The –SiMe2Ph unit can then undergo rate-limiting migration to the oxygen of the newly formed α-silylamidato group viaTS(I-II) (−9.1 kcal mol−1) and, in reasonable agreement with the non-optimised experimental reaction conditions, an energetic span of +22.9 kcal mol−1 to afford II (−26.3 kcal mol−1). This process is followed by exergonic C–O cleavage viaTS(II-7) (−13.0 kcal mol−1) to ultimately afford the isocyanide-coordinated magnesium siloxide 7 (−54.6 kcal mol−1). | ||
| Fig. 4 Computed free energy profile (BP86-D3BJ, toluene/BS2//BP86/BS1, in kcal mol−1) for the reaction of silanide species 6 with OCNt-Bu to yield the magnesium siloxide t-BuNC adduct, 7. Free energies are relative to species 5 ([Mg]-nBu) and corresponding reactants. | ||
Conclusion
In conclusion, the stoichiometric reactivity of a β-diketiminato magnesium silanide (6) towards a variety of organic isocyanates has been assessed. Reactions of t-BuNCO, DippNCO and CyNCO were found to yield β-diketiminato magnesium siloxide adducts of the isonitrile resulting from isocyanate deoxygenation as the predominant reaction products. In contrast, analogous treatment of 6 with i-PrNCO led to the identification of four different reaction products, reactivity which is most readily attributed to the decreased kinetic discrimination resulting from the reduced steric demands of the N-alkyl isocyanate substituent. Although the specificity of this reaction was also hampered by competitive isocycanate addition at the γ-methine carbon of the BDI supporting ligand, the identification of compound 13 provided corroborative evidence for the generation of sila-amidate intermediates. The formation of [{Me2PhSi}C(O)NR]− anions as the most likely initial species formed en route to isonitrile and siloxide formation was validated by a computational study. We are continuing to study the reactivity of these conveniently generated sources of nucleophilic silicon to develop this and related deoxygenative processes to a catalytic regime.Experimental section
All reactions of air- and moisture-sensitive compounds were carried out using standard Schlenk line and glovebox techniques under an inert atmosphere of argon. NMR experiments involving air-sensitive compounds were conducted in J. Young tap NMR tubes made up and sealed in a glovebox under argon. NMR spectra were recorded on a Bruker AV300 Ultrashield instrument for 1H (300.2 MHz), a Bruker 400 Ultrashield instrument for 29Si (79.5 MHz) or an Agilent ProPulse instrument for 1H (500 MHz), 13C (126 MHz) and 29Si (99 MHz) spectra at room temperature. The 1H/13C NMR spectra were referenced relative to residual solvent resonances, while 29Si NMR spectra were referenced to an external standard (Me4Si). Solvents (toluene, pentane and hexane) were dried using an MBraun solvent purification system and stored over 4 Å molecular sieves under argon. THF for use in air- and moisture-sensitive reactions was dried over sodium or potassium/benzophenone and distilled before use. C6D6 was purchased from Sigma-Aldrich and dried over a potassium mirror, vacuum transferred into a sealed ampoule and stored in a glovebox under argon. Di-n-butylmagnesium (Mgn-Bu2 1.0 M solution in n-heptane) and carbodiimides were purchased from Sigma-Aldrich and used without further purification. The β-diketiminato magnesium alkyl complex, [(BDI)MgnBu] (BDI = CH{C(Me)NDipp}2, Dipp = 2,6-i-Pr2C6H3) (5), and dimethylphenylsilyl boronic acid pinacol ester (pinBSiMe2Ph, pin = pinacolato) were synthesized by literature procedures.27,28 Elemental analysis was performed by Elemental Microanalysis, Okehampton, UK. For details of the X-ray studies, computational analyses and coordinates of calculated species see the ESI.†Synthesis of [(BDI)Mg{CNt-Bu}OSiMe2Ph] (7)
A solution of 5 (50 mg, 0.10 mmol) and pinBSiMe2Ph (26 mg, 0.10 mmol) in C6D6 (0.5 mL) was added via pipette to a J. Young NMR tube. Complete conversion to compound 6 was obtained after approximately 12 hours at room temperature. tert-Butylisocyanate (11.4 μL, 0.10 mmol) was added to the reaction mixture and complete conversion to product 7 was obtained after 2 days at 60 °C. Crystals suitable for single crystal X-ray diffraction analysis of 7 were obtained by cooling a pentane solution to −30 °C. 1H NMR (500 MHz, C6D6) δ 7.20 (dd, J = 7.6, 1.8 Hz, 2H, o-(C6H5)Si), 7.16–7.12 (m, 6H, Dipp-Ar), 7.09 (br, 1H, m-(C6H5)Si), 7.07 (m, 1H, p-(C6H5)Si), 7.05 (m, 1H, m-(C6H5)Si), 4.78 (s, 1H, CH{C(CH3)NDipp}2), 3.40–3.32 (hept, J = 6.8 Hz, 4H, Dipp-CH(CH3)2), 1.63 (s, 6H, CH{C(CH3)NDipp}2), 1.31 (d, J = 6.8 Hz, 12H, Dipp-CH(CH3)2), 1.20 (d, J = 7.0 Hz, 12H, Dipp-CH(CH3)2), 0.63 (s, 9H, C(CH3)3), 0.09 (s, 6H Si(CH3)2Ph) ppm. 13C{1H} NMR (126 MHz, C6D6) δ 168.6 (CH{C(CH3)NDipp}2), 147.8 (i-(C6H5)Si), 145.3 (i-Dipp-Ar), 142.6 (o-Dipp-Ar), 142.2 (m,p-Dipp-Ar), 133.8 (CNt-Bu), 127.4 (o-(C6H5)Si), 127.2 (m-(C6H5)Si), 125.6 (p-(C6H5)Si), 123.9 (C(CH Dipp)), 94.1 (CH{C(CH3)NDipp}2), 29.1 (C(CH3)3), 28.3 (Dipp CH(CH3)2), 25.7 (Dipp CH(CH3)2), 24.4 (Dipp CH(CH3)2), 24.1 (C(CH3)3), 3.6 (Si(CH3)2Ph) ppm. 29Si{1H} NMR (99 MHz, C6D6, 298 K) δ −16.8 ppm. Elemental analysis, calculated for C42H61MgN3OSi: C, 74.58; H, 9.09; N, 6.21%. Found: C, 74.46; H, 8.77; N, 6.19%.Synthesis of [(BDI)Mg{CNDipp}OSiMe2Ph] (8)
A solution of 5 (50 mg, 0.10 mmol) and pinBSiMe2Ph (26 mg, 0.10 mmol) in C6D6 (0.5 mL) was added via pipette to a J. Young NMR tube. Complete conversion to the product 6 was obtained after approximately 12 hours at room temperature. 2,6-Di-isopropyl-isocyanate (21.4 μL, 0.10 mmol) was added to the reaction mixture and complete conversion to product 8 was obtained after 3 days at 60 °C. Crystals suitable for single crystal X-ray diffraction analysis of 8 were obtained by cooling a pentane solution to −30 °C. 1H NMR (500 MHz, C6D6) δ 7.16–7.05 (m, 9H, Dipp-Ar), 7.05–6.94 (m, 5H, (C6H5)Si), 5.16 (m, J = 6.4 Hz, 1H, Dipp-CH(CH3)2), 4.97 (s, 1H, CH{C(CH3)NDipp}2), 3.58 (br. m, 2H, Dipp-CH(CH3)2), 3.34–3.10 (br. m, 2H, Dipp-CH(CH3)2), 2.32–2.27 (m, 1H, Dipp-CH(CH3)2), 1.64 (s, 6H, CH{C(CH3)NDipp}2), 1.39 (br., 6H, Dipp-CH(CH3)2), 1.15 (d, J = 6.8 Hz, 12H, Dipp-CH(CH3)2), 1.05 (d, J = 6.8 Hz, 6H, Dipp-CH(CH3)2), 1.01 (d, J = 6.2 Hz, 6H, Dipp-CH(CH3)2) ppm. 13C{1H} NMR (126 MHz, C6D6) δ 170.2 (CH{C(CH3)NDipp}2), 165.4 (CN), 144.7 (i-Dipp-Ar), 143.6 (i-Dipp-Ar), 139.5 (Dipp-Ar), 129.3 (i-(C6H5)Si), 126.0 (Dipp-Ar), 124.9 (Dipp-Ar), 124.4 (Dipp-Ar), 122.8 ((C6H5)Si), 95.2 (CH{C(CH3)NDipp}2), 70.4 (Dipp-CH(CH3)2), 28.3 (Dipp-CH(CH3)2), 25.8, (Dipp-CH(CH3)2), 25.1 (Dipp-CH(CH3)2), 24.2 (s, NC(H)(CH3)2), 24.0 (Dipp-CH(CH3)2), 22.4 (Si(CH3)2Ph) ppm. 29Si{1H} NMR (99 MHz, C6D6, 298 K) δ −16.5 ppm. Despite multiple attempts, an accurate microanalysis could not be obtained for this compound.Synthesis of [(BDI)Mg{CNCy}OSiMe2Ph] (10a) and [(BDI)Mg{CNCy}SiMe2Ph] (10b)
A solution of 5 (50 mg, 0.10 mmol) and pinBSiMe2Ph (26 mg, 0.10 mmol) in C6D6 (0.5 mL) was added via pipette to a J. Young NMR tube. Complete conversion to the product 6 was obtained after approximately 12 hours at room temperature. Cyclohexyl isocyanate (12.8 μL, 0.10 mmol) was added to the reaction mixture and conversion to an approximate 3:1 mixture of compounds 10a/b was achieved after 48 hours at room temperature. 1H NMR (500 MHz, C6D6: integrals are quoted relative to the major species 10a) δ 7.23–7.20 (m, 2H, o-(C6H5)Si), 7.15–7.09 (m, 3H, Dipp-Ar), 7.07 (s, 3H, Dipp-Ar), 7.06–7.01 (m, 3H, m,p-(C6H5)Si), 4.78 (s, 1H, CH{C(CH3)NDipp}210a), 4.77 (s, CH{C(CH3)NDipp}210b) 3.37 (m, J = 6.6 Hz, 4H, Dipp-CH(CH3)210a), 3.27 (m, J = 6.6 Hz, Dipp-CH(CH3)210b), 2.58 (m, 1H, (CH2)5CH-NC), 1.64 (s, 6H, CH{C(CH3)NDipp}210a), 1.64 (s, CH{C(CH3)NDipp}210b), 1.32 (d, J = 6.8 Hz, 12H, Dipp-CH(CH3)210a), 1.21 (d, J = 6.8 Hz, 12H, Dipp-CH(CH3)210a/b), 1.18 (d, J = 6.8 Hz, Dipp-CH(CH3)210b), 1.14–1.09 (m, 5H, CH2), 1.00–0.90 (m, 3H, CH2), 0.66–0.57 (m, 3H, CH2), 0.17 (s, Si(CH3)2Ph, 10b), 0.09 (s, 6H, Si(CH3)2Ph, 10a). 13C{1H} NMR (126 MHz, C6D6) δ 168.6 (CH{C(CH3)NDipp}2), 147.8 (SiC), 145.2 (i-Dipp-Ar), 142.3 (C-Dipp-Ar), 133.8 (CN), 134.3 (CNCH), 127.4 (o-Dipp-Ar), 125.4 ((C6H5)Si), 124.0 (m-Dipp-Ar), 123.9 (p-Dipp-Ar), 94.1 (CH{C(CH3)NDipp}2), 31.6 (CH2), 28.4 (Dipp-CH(CH3)2), 25.6 (Dipp-CH(CH3)2), 24.5 (Dipp-CH(CH3)2), 24.1 (CH{C(CH3)NDipp}2), 22.7 (s, CH2), 3.5 (Si(CH3)2Ph) ppm. 29Si{1H} NMR (99 MHz, C6D6, 298 K) δ −16.7 ppm. Elemental analysis, calculated for C44H63MgN3OSi: C, 75.24; H, 9.04; N, 5.98%. Found: C, 74.95; H, 9.26; N, 5.63%.
Isolation of compounds 11–13
Reactions between compound 6 and i-PrNCO were performed as described for the synthesis of compounds 7, 8 and 10a/b. Irrespective of whether the reactions were performed with and without heating to 60 °C, analysis by 1H NMR spectroscopy was indicative of the formation of a complex mixture of products. Single crystals of compounds 11–13 suitable for X-ray diffraction analysis were grown by slow evaporation of hexane solutions of the reaction products and subsequent mechanical separation.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the Petroleum Technology Development Fund (PTDF) of Nigeria for the provision of a PhD scholarship for BO-E. This research made use of the Balena High Performance Computing (HPC) Service at the University of Bath.References
- J. E. Baldwin, J. C. Bottaro, P. D. Riordan and A. E. Derome, J. Chem. Soc., Chem. Commun., 1982, 942–943 RSC
.
- J. E. Baldwin, A. E. Derome and P. D. Riordan, Tetrahedron, 1983, 39, 2989–2994 CrossRef CAS
- X. P. Liu, X. Q. Xiao, Z. Xu, X. M. Yang, Z. F. Li, Z. W. Dong, C. T. Yan, G. Q. Lai and M. Kira, Organometallics, 2014, 33, 5434–5439 CrossRef CAS
-
(a) J. C. Li, B. Li, R. Liu, L. Y. Jiang, H. P. Zhu, H. W. Roesky, S. Dutta, D. Koley, W. P. Liu and Q. S. Ye, Chem. – Eur. J., 2016, 22, 14499–14503 CrossRef CAS
- A. F. Pécharman, A. L. Colebatch, M. S. Hill, C. L. McMullin, M. F. Mahon and C. Weetman, Nat. Commun., 2017, 8, 15022 CrossRef
- A. F. Pécharman, M. S. Hill and M. F. Mahon, Dalton Trans., 2018, 47, 7300–7305 RSC
- A. F. Pécharman, N. A. Rajabi, M. S. Hill, C. L. McMullin and M. F. Mahon, Chem. Commun., 2019, 55, 9035–9038 RSC
- R. J. Schwamm, M. P. Coles, M. S. Hill, M. F. Mahon, C. L. McMullin, N. A. Rajabi and A. S. S. Wilson, Angew. Chem., Int. Ed., 2020, 59, 3928–3932 CrossRef CAS PubMed
-
(a) D. J. Liptrot, M. Arrowsmith, A. L. Colebatch, T. J. Hadlington, M. S. Hill, G. Kociok-Köhn and M. F. Mahon, Angew. Chem., Int. Ed., 2015, 54, 15280–15283 CrossRef CAS
- B. Okokhere-Edeghoghon, M. Dehmel, M. S. Hill, R. Kretschmer, M. F. Mahon, C. L. McMullin, L. J. Morris and N. A. Rajabi, Inorg. Chem., 2020, 59, 13679–13689 CrossRef CAS
- L. J. Morris, M. S. Hill, I. Manners, C. L. McMullin, M. F. Mahon and N. A. Rajabi, Chem. Commun., 2019, 55, 12964–12967 RSC
- L. J. Morris, N. A. Rajabi, M. F. Mahon, I. Manners, C. L. McMullin and M. S. Hill, Dalton Trans., 2020, 49, 10523–10534 RSC
- C. F. Caro, P. B. Hitchcock, M. F. Lappert and M. Layh, Chem. Commun., 1998, 1297–1298 RSC
- P. Sobota, J. Utko, J. Ejfler and L. B. Jerzykiewicz, Organometallics, 2000, 19, 4929–4931 CrossRef CAS
- C. A. Zechmann, T. J. Boyle, M. A. Rodriguez and R. A. Kemp, Inorg. Chim. Acta, 2001, 319, 137–146 CrossRef CAS
- P. Sobota, S. Przybylak, J. Ejfler, M. Kobyłka and L. B. Jerzykiewicz, Inorg. Chim. Acta, 2002, 334, 159–164 CrossRef CAS
- A. Lennartson and M. Håkansson, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2008, 64, m8–m9 CrossRef CAS
- S. Banerjee, Ankur, A. P. Andrews, B. Vargheseb and A. Venugopal, Dalton Trans., 2019, 48, 7313–7319 RSC
- T. Chlupatý, M. Bílek, J. Merna, J. Brus, Z. Růžičková, T. Strassner and A. Růžička, Dalton Trans., 2019, 48, 5335–5342 RSC
- M. D. Anker, M. Arrowsmith, P. Bellham, M. S. Hill, G. Kociok-Köhn, D. J. Liptrot, M. F. Mahon and C. Weetman, Chem. Sci., 2014, 5, 2826–2830 RSC
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC
- A. G. Blackman, E. B. Schenk, R. E. Jelley, E. H. Krensked and L. R. Gahan, Dalton Trans., 2020, 49, 14798–14806 RSC
- S. S. Al-Juaid, Y. Derouiche, P. B. Hitchcock, P. D. Lickiss and A. G. Brook, J. Organomet. Chem., 1991, 403, 293–298 CrossRef CAS
- R. F. Cunico and A. R. Motta, J. Organomet. Chem., 2006, 691, 3109–3112 CrossRef CAS
- M. H. Chisholm, J. C. Huffman and K. Phomphrai, Dalton Trans., 2001, 222 RSC
- Although such an interaction is ill-defined in the literature with respect to Mg centers, previous computational studies have unveiled possible agostic Si–C interactions in lanthanide complexes. For details, see: L. Perrin, L. Maron, O. Eisenstein and M. F. Lappert, New J. Chem., 2003, 27, 121–127 RSC
- A. P. Dove, V. C. Gibson, P. Hormnirun, E. L. Marshall, J. A. Segal, A. J. P. White and D. J. Williams, Dalton Trans., 2003, 3088–3097 RSC
- M. Suginome, T. Matsuda and Y. Ito, Organometallics, 2000, 19, 4647–4649 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra and details of the computational and single crystal X-ray diffraction analysis. CCDC 2119900, 2119901, 2125516, 2125517 and 2119902. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03775g |
| This journal is © The Royal Society of Chemistry 2022 |