
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbene chemistry of arsenic, antimony, and bismuth: origin, evolution and future prospects
Rajesh
Deka
and
Andreas
Orthaber
 *
*
Synthetic Molecular Chemistry, Department of Chemistry – Ångström laboratories, Uppsala University, Box 523, 75120 Uppsala, Sweden. E-mail: andreas.orthaber@kemi.uu.se
First published on 9th May 2022
Abstract
The discovery of the first isolable N-heterocyclic carbene in 1991 ushered in a new era in coordination chemistry. The remarkable bonding properties of carbenes have led to their rapid proliferation as auxiliary ligands for a wide range of transition metals and main group elements. In the case of group 15, while carbene-stabilized nitrogen and phosphorus compounds are extensively studied, the scope of research has shrunk significantly from arsenic to bismuth. This is essentially attributed to the decrease in stability of the C–E bond upon descending the group. Even so, modulating the carbene backbone or introducing alternative synthetic strategies not only alleviates the stability issues but also offers promising results in terms of the bonding and reactivities of these compounds. The purpose of the present perspective is to provide a comprehensive overview of the origins and development of carbene chemistry of arsenic, antimony, and bismuth, as well as to highlight the future prospects of this field.
Introduction
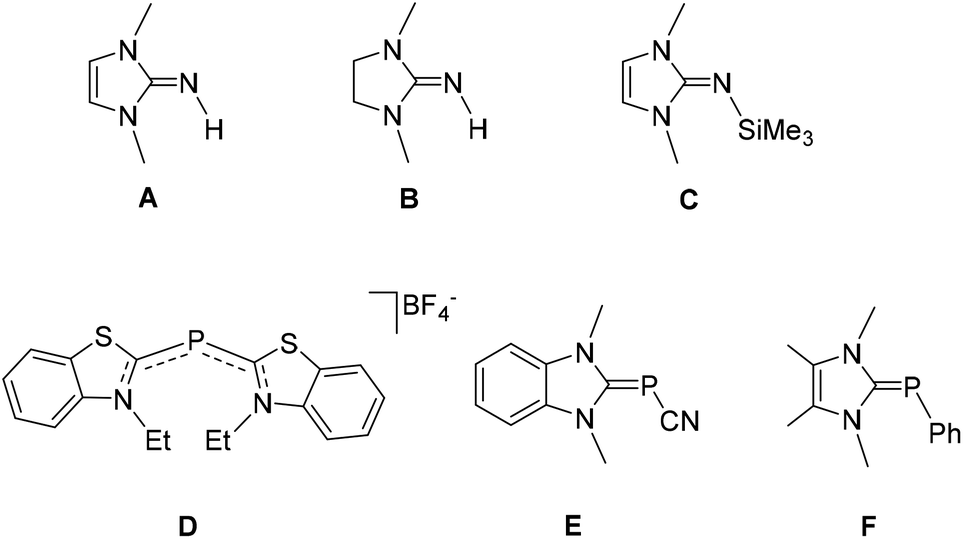
Carbenes, with their strong σ-donor and variable π-acceptor properties, have gradually overtaken the cyclopentadienyl and phosphine-based coordination chemistry, and unequivocally evolved as one of the key players in contemporary synthetic chemistry.1 Described as a neutral species with a divalent carbon atom bearing a six-electron valence shell, carbenes were regarded as highly reactive, which in turn caused their progress to stagnate for many years. In fact, despite being known for a long time, it was not until 1968 when carbenes found their first practical significance as a spectator ligand by Öfele2 and Wanzlick.3 After several years, based on the seminal work by Bertrand and coworkers on stable [bis-(diisopropylamino)phosphino](trimethylsilyl)carbene,4 followed by Arduengo's report on the first isolable, free N-heterocyclic carbene (NHC) species,5 the carbene-based chemistry has experienced a dramatic rise in research interest. With the ease of fine-tuning the electronic and steric properties, NHCs have eventually evolved into an organometallic tool with absolute practical significance and its coordination complexes with transition metals cover almost all aspects of modern science ranging from organic transformations (particularly catalysis),6 medicinal chemistry7 to material science.7c,8 As the chemistry of carbene-ligated transition metal complexes flourished, considerable attention has also been drawn to synthesizing carbene complexes with main-group elements. In particular, the unique ability of NHCs to stabilize a broad range of low-valent and low-coordinate species by their strong σ-donating ability makes them very promising ligand motifs in main-group chemistry.9 Following the success of NHC in main-group compounds, a wide variety of non-NHC derivatives including mesoionic (abnormal) and remote NHCs have also been explored, which show promising results regarding both fundamental bonding studies and various synthetic applications.10 This has led to the publication of a large number of reviews over the years on the carbene-based chemistry of main group compounds.9c,dAmong the main group elements, the carbene chemistry of lighter group 15 elements (N and P) has been known for a long time. The research on carbene-stabilized nitrogen adduct dates back to 1977, long before the report on Arduengo's isolable carbene. Using creatinine derivatives for the reaction with LiAlH4, Rapoport and his coworker synthesized 1,3-dimethylimidazolin-2-imine, A, and its 4,5-dihydro derivative, B, which can be regarded as the first NHC-stabilized nitrogen adducts (Fig. 1).11 In 2004, Kuhn and coworkers reported an alternative synthetic protocol for A with better yield, which in turn accelerated the chemistry of NHC-stabilized nitrogen derivatives.12 Tamm and coworkers reported the synthesis of N-silylated 2-iminoimidazoline, C, which has found profound interest as a precursor for various carbene-stabilized nitrogen adducts.13 Following these reports, a large number of carbene-stabilized nitrogen adducts have been studied with regard to their intriguing structural and bonding features as well as their diverse reactivity.9c,d,14
 | ||
| Fig. 1 Early examples of carbene-stabilized nitrogen and phosphorus species. | ||
Similarly in 1964, even before the genesis of carbene chemistry, Dimroth and coworker, reported a cationic phosphacyanine compound D, which can be regarded as the first carbene adduct of phosphorus.15 In 1980, Schmidpeter reported carbene-stabilized cyanophosphinidene species E by the treatment of N-ethyl-2-chlorobenzothiazoliumfluoroborate with tris-(hydroxymethy1)phosphine.16 In 1991, Arduengo and coworkers made the first deliberate synthesis of carbene–phosphinidene adduct F by the treatment of free carbene with pentaphenylpentaphosphorus.17 Since then, carbene-stabilized phosphorus adducts spark profound interest in the literature and offer rich chemistry.9c–f,18
Unlike the highly developed chemistry of carbene-stabilized nitrogen and phosphorus compounds, the chemistry of heavier analogs is still largely unexplored, with the scope of the research dwindling significantly from As to Bi. Nevertheless, with the contributions of several researchers in the last few years, the carbene chemistry of heavier group 15 elements is making noteworthy headway towards becoming a highly valuable research field. We intend to provide an up-to-date review of the chemistry of heavier group 15 compounds (As, Sb and Bi) stabilized by heterocyclic carbenes (NHCs, CAACs, etc.), with particular emphasis on their synthesis and bonding aspects.
Carbene adducts with arsenic
Low coordinate and low valent adducts
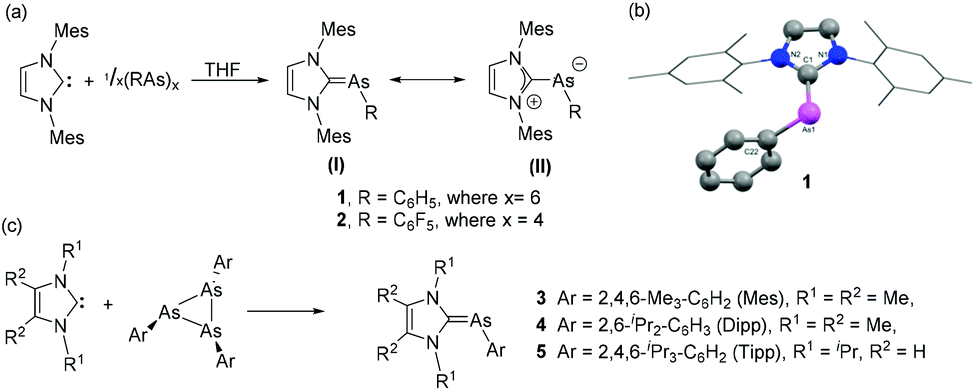
Arduengo and coworkers have introduced the chemistry of N-heterocyclic carbene (NHC) in the field of heavier group 15 elements by synthesizing a series of NHC-supported arsinidene adducts exploring the ability of cyclopnictanes to act as simple “pnictinidene” transfer reagents.19 The syntheses of NHC–arsinidene adducts, 1 and 2 were accomplished by the treatment of 1,3-bis(2,4,6-trimethylphenyl)imidazolin-2-ylidene (IMes) with hexaphenyl cyclohexaarsane, and tetrakis(pentafluorophenyl) cyclotetraarsane, respectively (Fig. 2a). In the molecular structures of 1 and 2, the As centre adopts a bent geometry with CNHC–As–CAryl bond angles of 97.3(1)° and 99.8(3)°, respectively (Fig. 2b). Both these bond angles deviate significantly from the ideal sp2 centre (120°). The corresponding As–CNHC bond distances in 1 and 2 are 1.899(3) Å and 1.902(7) Å, respectively. These bond distances are shorter than As–C single bond [∑rcov (C, As) = 1.96 Å], indicating the presence of double bond character and/or attractive ionic contributions to the bond. In both compounds, the As–CAryl bond is twisted out of the imidazole plane [torsion angle C–As–CNHC–N = 29.6(3)° (1) and 30.6(7)° (2)], implying a weak Cpп–Aspп interaction between the imidazole rings and the unsaturated As centres. In solution NMR data suggest a free rotation around the formal As![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. Thus, both the solid and solution-state structures suggest a strongly polarized pπ–pπ interaction in the As–CNHC bonds, thereby imparting an ‘ylide’ character to these compounds represented by resonance structure (II). In contrast to the lighter P-derivatives, where 31P NMR chemical shifts are prime indicators for the ylene/ylide character, for the heavier analogs metrical parameters, solution behavior and of late computational studies are of utmost importance to describe the bonding situations.
C double bond. Thus, both the solid and solution-state structures suggest a strongly polarized pπ–pπ interaction in the As–CNHC bonds, thereby imparting an ‘ylide’ character to these compounds represented by resonance structure (II). In contrast to the lighter P-derivatives, where 31P NMR chemical shifts are prime indicators for the ylene/ylide character, for the heavier analogs metrical parameters, solution behavior and of late computational studies are of utmost importance to describe the bonding situations.
 | ||
| Fig. 2 (a) Synthesis of NHC–arsinidene adducts 1–2; ylene (I) and ylide (II) resonance structures; (b) solid-state structure of 1; (c) synthesis of NHC–arsinidene adducts 3–5 from triarsiranes. | ||
Recently, Hering-Junghans and co-workers, have reported that triarsiranes with sterically demanding aromatic substituents (ArAs)3 are also excellent arsinidene transfer agents and readily react with NHC at room temperature to afford NHC–arsinidene adducts, 3–5 (Fig. 2c).20 The solid-state structures of 3–5 are isostructural with 1–2, and the As–CNHC bond lengths and related bond angles are in good agreement with 1–2. Theoretical studies are carried out to elucidate the electronic and structural properties of the synthesized NHC–arsinidene adducts. In detail, the NBO analysis suggests an As–CNHC π orbital, which is strongly polarized towards the As centre, giving an overall Wiberg Bond Index (WBI) of ca. 1.2 indicative of more pronounced ylene character.
Driess and coworkers have reported that NHC-stabilized germylidenylarsinidene 6 can be isolated by the reaction of β-diketiminato chlorogermylene with NHC 1,3-di(isopropyl)imidazolin-2-ylidene (IPr) using sodium arsaethynolate, [Na(AsCO)(dioxane)x] (x = 2.3–3.3) as single atom arsenic source.21 During the reaction, the intermediate arsaketenyl germylene undergoes a transition metal-like ligand substitution of CO by the NHC to afford the desired germylidenylarsinidene, 6 (Fig. 3a). Importantly, in absence of a carbene, the release of CO results upon dimerization in a 1,3-digerma,2,4-diarsabutadiene derivative. In the solid-state structure of 6 (Fig. 3b), the As–CNHC distance is 1.946(2) Å, which is significantly longer than the values observed in 1–5. The corresponding Ge–As bond distance is 2.4283(2) Å, significantly shorter compared to an analogous phosphine stabilized system indicating a strong influence of the NHC on the Ge–As bonding situation. The NBO analysis reveals that the σ(Ge–As) orbital in 6 is partly delocalized towards the π*(C–N) orbital of the carbene ring. This results in a WBI of 1.11 for the Ge–As bond. A similar weak delocalization of As lone pair to the π*(C–N) orbital is observed in the As–CNHC bond, resulting in a WBI 0.99, i.e. significantly lower than that observed in the adducts 3–5, rationalizing the elongated As–CNHC bond.
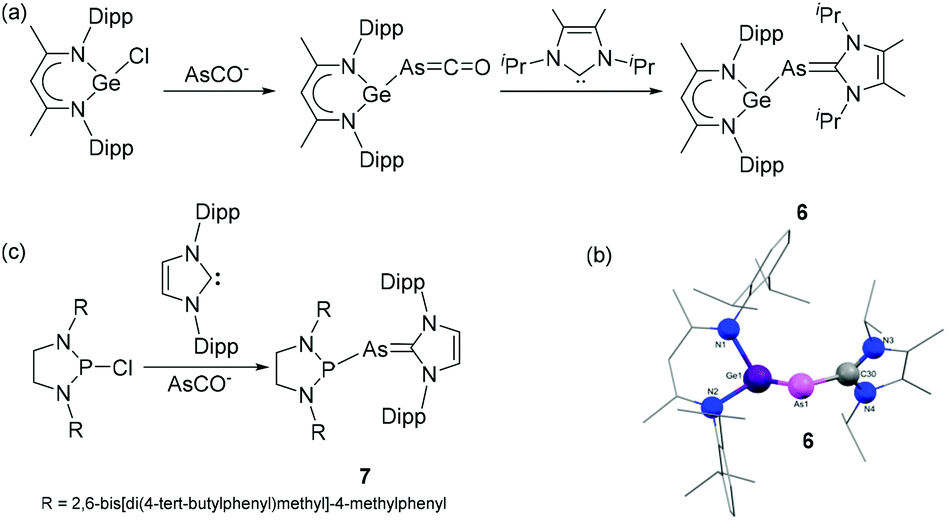 | ||
| Fig. 3 (a) Synthesis of NHC-stabilized germylidenylarsinidene 6; (b) solid-state structure of 6; (c) synthesis of NHC-stabilized phosphino–arsinidene adduct 7. | ||
Goicoechea and coworkers utilized sodium arsaethynolate for the isolation of an NHC-stabilized phosphino–arsinidene adduct.22 The reaction of chlorodiazaphospholidine (ArNCH2)2PCl (Ar = 2,6-bis[di-(4-tert-butylphenyl)methyl]-4-methylphenyl) with 1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene (IDipp) in presence of [Na(dioxane)3.3][AsCO] afforded phosphino–arsinidene adduct 7 (Fig. 3c). In the solid-state structure of 7, the As–CNHC distance is 1.919(3) Å, which is in good agreement with that of 1–2. The P–As bond distance is 2.3532(2) Å well in the expected range of P–As single bonds. A sharp 31P NMR resonance at 198.3 ppm for the phosphine is indicative of an electron deficient P-centre. This reactivity contrasts the observed cleavage of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O bond and formation of a PO and an arsaallenyl fragment, when a cyclic alkyl amino carbene (CAAC) ligand is used instead.
O bond and formation of a PO and an arsaallenyl fragment, when a cyclic alkyl amino carbene (CAAC) ligand is used instead.
Over the past decade, several studies reported synthetic approaches towards NHC–arsinidene adducts and detailed theoretical studies which provided a better understanding of their bonding and electronic structures. However the parent carbene–arsinidene (AsH) adduct remained elusive for a long time. These highly reactive arsinidene species, which bears 6 valence electrons, are often stabilized through sterically demanding substituent (vide infra) or complexation. Consequently, detection and characterization of arsinidene (AsH) species was long limited to only spectroscopic and theoretical studies. In fact, the solid-state structure containing the primary arsinidene motif (As–H) bond was precluded until recently, when Liddle and coworkers reported the first structurally characterized uranium arsinidene complex, [U(TrenTIPS)(AsH)]−, [TrenTIPS = N(CH2CH2NSiiPr3)3].23
Only a couple of years later, Tamm and coworkers have reported the first example of NHC-stabilized discrete arsinidene species by using two different synthetic routes (Fig. 4a).24 In one protocol, the reaction of As(SiMe3)3 with N,N′-disubstituted-2,2-difluoroimidazoline afforded NHC–As(SiMe3) adducts via elimination of TMS-F. Further desilylation of the adduct using excess anhydrous methanol afforded the first example of discrete NHC–arsinidene species 8 without inducing further decomposition. Alternatively, 8 was synthesized by reaction of [Na(dioxane)3.31][AsCO] with the corresponding imidazolium chlorides via a formal salt elimination, C–H bond insertion and decarbonylation reaction. In the solid-state structure of 8, the geometry around the As centre is distorted V-shaped with CNHC–As–H bond angles are in the range of 90(2) to 98(1)° for the IDipp and IMes adducts, respectively (Fig. 4b). This indicates a high degree of p-character for the arsenic-element bonds, and large s-character of the lone pair. The corresponding As–CNHC bond distances fall within the range of 1.883(2)–1.896(2) Å, which are shorter than those observed in NHC–arsinidene adducts. Nonetheless VT-NMR studies of the IMes-derivative suggest that the rotational barrier of the As–H fragment about the CNHC–As bond is only 10 kcal mol−1. This is in line with natural resonance theory (NRT) analysis showing a 33% and 38% contribution of the double bonded and zwitterionic resonance forms, respectively.
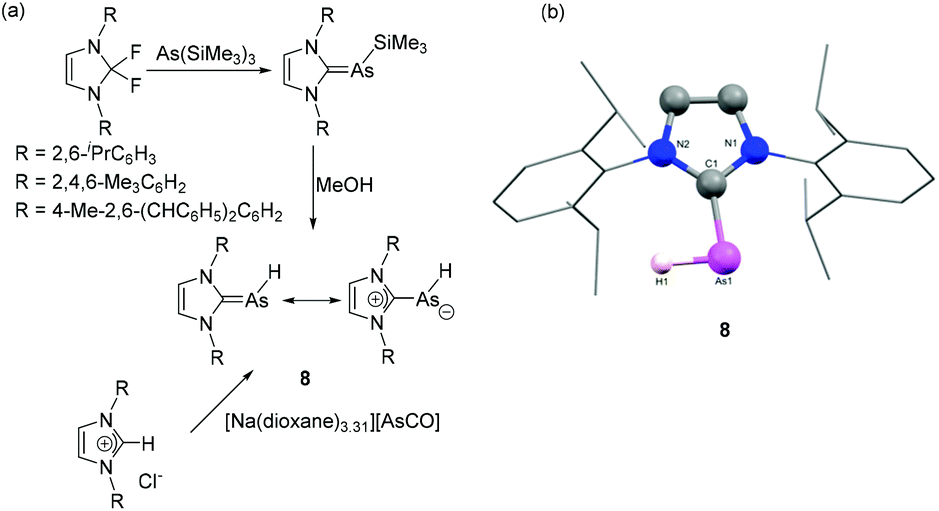 | ||
| Fig. 4 (a) Synthesis of NHC–arsinidene 8 starting from [AsCO]− and As(TMS)3; (b) solid-state structure of 8. | ||
In a collaborative work by the Goicoechea and Wolf groups, the reactivity of sodium arsaethynolate to a carbene stabilized transition metal was studied. Reacting (η5-C5H5)Ni(IMes) with [Na(dioxane)3.0][(AsCO)], the reaction afforded a carbene-arsinidenyl-bridged Nickel-dimer, (μ2-CO)[μ2-As-(IMes)]Ni2(IMes)(η5-C5H5), 9 (Fig. 5).25 Complex 9 represents the first example of any NHC–arsenidenide complex with an anionic arsinidenyl [As(IMes)]− moiety. Here, [As(IMes)]− acts as a formal 4-electron ligand bridging two Ni(I) centres with As–Ni bonds distances of 2.389(1) and 2.282(1) Å. Analogous to the reported phosphorus derivative, the As–CNHC bond length of 1.955(3) Å is at the longer end of the spectrum for an AsC double bond, though significantly shorter than an As–C single bond.
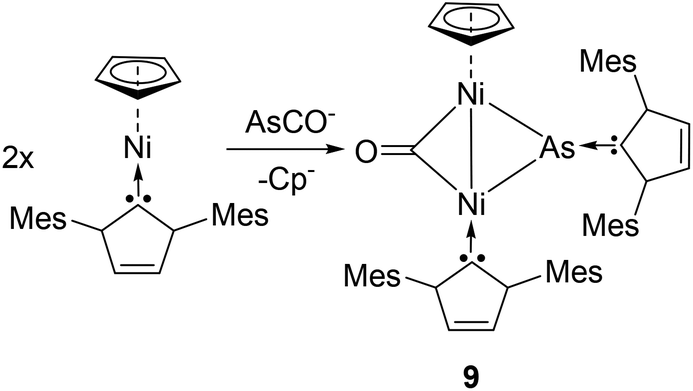 | ||
| Fig. 5 Synthesis of NHC-stabilized arsinidenyl-bridged Ni complex 9. | ||
The carbene adducts obtained from reactions with trivalent As species are often used as starting points towards the synthesis of low valent derivatives by means of reduction, or elimination reactions. Moreover, halogenated precursors can be subjected to various dehalogenation protocols yielding cationic derivatives. Of late, the synthesis of diatomic main-group allotropes in a formal zero oxidation state has gained significant interest, owing to their promising structural, electronic and photophysical properties in comparison to the naturally occurring allotropes and mononuclear main-group species. In this regard, NHCs have been widely used as an ideal ligand system for stabilizing various, diatomic group 13–14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9h and phosphorus allotropes.26 In a seminal contribution, Robinson and coworkers have reported the synthesis of first example of diarsenic allotrope stabilized by an NHC. To achieve that, the NHC adduct of arsenic trichloride 10 was synthesized by the treatment of IDipp with AsCl3 (Fig. 6a).27 In the solid-state structure of 10, the geometry around the four coordinated As is a distorted see-saw arrangement with an As–CNHC distance of 2.018(3) Å. The reduction of the 10 with potassium graphite results in the formation of the first Lewis base (LB) stabilized diarsenic compound, 11.28 The compound adopts a trans-bent geometry around the As–As bond (Fig. 6b). Each As–CNHC bond distance is 1.881(2) Å, which is significantly shorter than the As–CNHC bond distance in the precursor 10, indicative of a partial AsCNHC double bond character in 11. The As–As bond distance is 2.442(1) Å, which is in good agreement with a single bond [∑rcov (As, As) = 2.42 Å]. The partial double bond character of the AsCNHC fragment was further corroborated by NBO-NLMO analysis, which indicates that the As–CNHC is twofold: a σ-donation from: CNHC to As and a p–π back donation of the As lone pair to the empty p-orbital of CNHC. Furthermore, Robinson and coworkers have explored the oxidation reaction of 11 towards Lewis acid (LA) GaCl3. In particular, the reaction of 11 with 2 equivalents of GaCl3 afforded a monocationic diarsenic radical [IDipp:AsAs:IDipp]˙+([GaCl4]−)2, 12, while the reaction of 11 with 4 equivalents of gallium trichloride afforded a dicationic diarsene [IDipp:AsAs:IDipp]2+([GaCl4]−)2, 13.29 The As–As bond distances in the solid-state structures of 12 and 13 are 2.332(3) and 2.2803(5) Å, respectively. Both these distances are significantly shorter compared to that in 11 and indicate a stepwise increase of AsAs double character concomitant with an elongation of the AsCNHC bond upon oxidation. EPR studies at room temperature of the radical cation 13 show a septet originating from hyperfine coupling to two equivalent 75As nuclei (I = 3/2).
9h and phosphorus allotropes.26 In a seminal contribution, Robinson and coworkers have reported the synthesis of first example of diarsenic allotrope stabilized by an NHC. To achieve that, the NHC adduct of arsenic trichloride 10 was synthesized by the treatment of IDipp with AsCl3 (Fig. 6a).27 In the solid-state structure of 10, the geometry around the four coordinated As is a distorted see-saw arrangement with an As–CNHC distance of 2.018(3) Å. The reduction of the 10 with potassium graphite results in the formation of the first Lewis base (LB) stabilized diarsenic compound, 11.28 The compound adopts a trans-bent geometry around the As–As bond (Fig. 6b). Each As–CNHC bond distance is 1.881(2) Å, which is significantly shorter than the As–CNHC bond distance in the precursor 10, indicative of a partial AsCNHC double bond character in 11. The As–As bond distance is 2.442(1) Å, which is in good agreement with a single bond [∑rcov (As, As) = 2.42 Å]. The partial double bond character of the AsCNHC fragment was further corroborated by NBO-NLMO analysis, which indicates that the As–CNHC is twofold: a σ-donation from: CNHC to As and a p–π back donation of the As lone pair to the empty p-orbital of CNHC. Furthermore, Robinson and coworkers have explored the oxidation reaction of 11 towards Lewis acid (LA) GaCl3. In particular, the reaction of 11 with 2 equivalents of GaCl3 afforded a monocationic diarsenic radical [IDipp:AsAs:IDipp]˙+([GaCl4]−)2, 12, while the reaction of 11 with 4 equivalents of gallium trichloride afforded a dicationic diarsene [IDipp:AsAs:IDipp]2+([GaCl4]−)2, 13.29 The As–As bond distances in the solid-state structures of 12 and 13 are 2.332(3) and 2.2803(5) Å, respectively. Both these distances are significantly shorter compared to that in 11 and indicate a stepwise increase of AsAs double character concomitant with an elongation of the AsCNHC bond upon oxidation. EPR studies at room temperature of the radical cation 13 show a septet originating from hyperfine coupling to two equivalent 75As nuclei (I = 3/2).
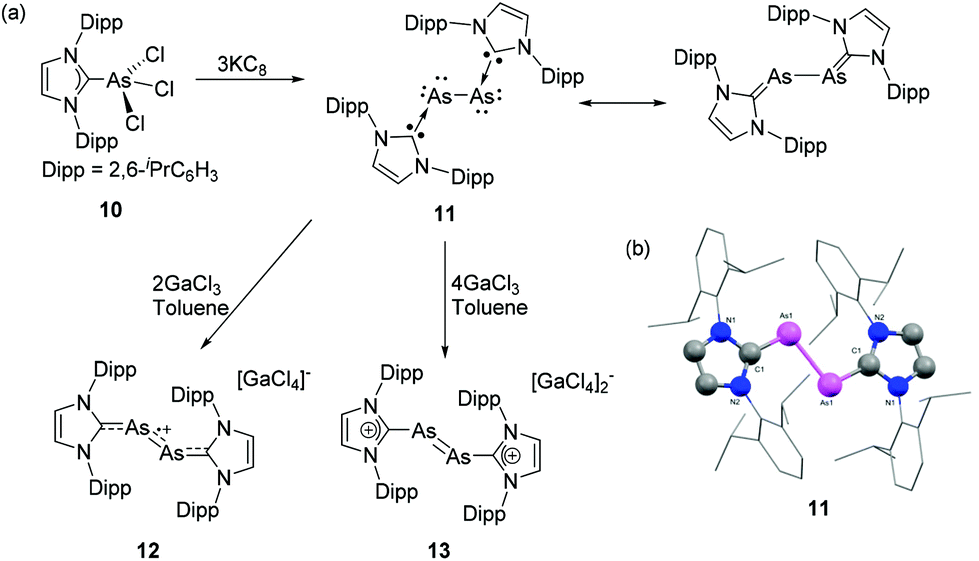 | ||
| Fig. 6 (a) Syntheses of NHC-supported diarsenic, 11, diarsenic radical cation 12, and diarsene dication 13; (b) solid-state structure of 11. | ||
In order to explore the chemistry of diarsenic species bearing an anionic NHC motif, Tamm and coworkers have synthesized a dichloroarsane (WCA-NHC)AsCl2, 14, where the NHC ligand was appended with a weakly coordinating anionic (WCA) trishexafluorophenyl borate moiety.3014 was synthesized by salt metathesis reaction of AsCl3 with (WCA-NHC)Li˙-toluene (Fig. 7a). In the solid-state structure of 14, the geometry around the As centre is trigonal pyramidal with CNHC–As–Cl bond angles of 97.47(7)° and 98.19(7)° and a Cl–As–Cl angle of 98.80(3)° (Fig. 7b). The As–CNHC bond distance is 1.978(2) Å and the As–Cl bond distances are 2.1679(8) and 2.1626(8) Å. Particular interesting about the solid-state structure of 14 is that an As⋯arene interaction is observed, as indicated by an As–Cipso distance of 3.080(2) Å between the As atom and the ipso-carbon atom of the NHC adjacent to the borate moiety. Such pnictogen⋯arene interactions, i.e. Menshutkin complexes,31 are much more prominent in the heavier pnictogen derivatives, e.g.73 (vide infra) owing to the further increased Lewis acidity. When 14 was reacted with 1,3-bis(trimethylsilyl)-1,4-dihydropyrazine or magnesium powder, the reaction afforded a diarsenic species 15 with a formal AsAs double bond. Similar to 11, compound 15 exhibits a trans-bent geometry around the central diarsenic motif. The As–As bond distance is 2.2882(8) Å, which is in good agreement with the bond lengths observed in [(IDipp):AsAs:(IDipp)]2+([GaCl4]−)2, 13. Evidently, the use of such anionic carbene ligands has a positive effect on the stabilization of unsaturated arsenic species. In order to assess the role of London dispersion interactions in the thermodynamic stabilization of 15, Tamm and coworkers have carried out the local energy decomposition (LED) analysis. It is observed that in addition to the stabilizing effects of the WCA, the London dispersion interactions arising from the iso-propyl substituent of the NHC ligand play a crucial role in the overall stability of the diarsenic species.
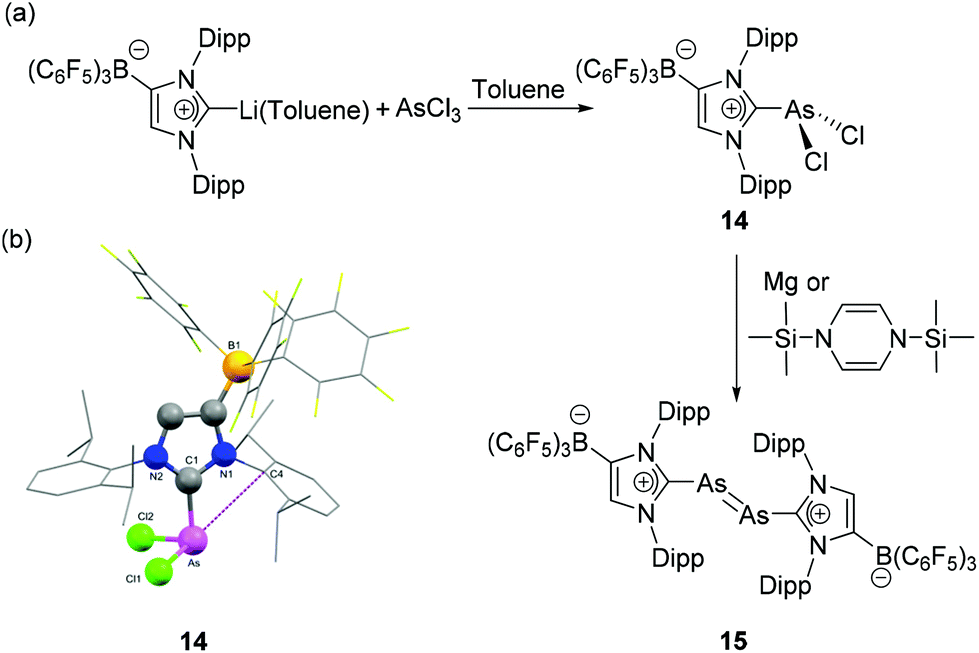 | ||
| Fig. 7 (a) Synthesis of diarsene 15 from NHC-stabilized dichloroarsane, 14; (b) solid-state structure of 14 showing As–Cipso interactionds. | ||
Following the report on diarsenic species stabilized by anionic NHC, Tamm and coworkers have synthesized the NHC-stabilized heteronuclear dipnictogen species, namely arsenic monophosphide and its derivatives.32 To synthesize arsenic monophosphide species, (IDipp)PSiMe3 was treated with (IMes)AsCl3 to afford [(IMes)As(Cl)P(IDipp)]Cl, 16 (Fig. 8a). In the 31P NMR spectrum, the chemical shifts at 132.2, 20.3, 16.5, and 1.8 ppm are attributed to isomers with different chlorine binding modes. The subsequent reaction of 16 with KC8 afforded heteroleptic dipnictogen compound 17, which shows a single 31P NMR resonance at −60.6 ppm, which is attributed to the PCNHC bond. While the reaction of 17 with ferrocenium hexaflourophosphate afforded monocationic species 18, the chloride abstraction of 16 by GaCl3 afforded dicationic dipnictogen species 19. In the solid-state structures, compounds 17–19 adopt a trans-bent geometry around the As–P bond (Fig. 8b). Interestingly, the P–As–CNHC angles in 17–19 are only marginally smaller compared to the As–P–CNHC angles. The As–P bond distances in 17–19 are 2.3149(8) Å, 2.2379(4)/2.2416(4) Å, and 2.1610(8) Å, respectively, indicating a step-wise increase of double bond character of the As–P bond upon oxidation. The corresponding Wiberg Bond Index increases from 1.02 in 17, 1.22 in 18 to 1.63 in 19. This trend was further rationalized by the analysis of frontier molecular orbitals, which shows a π*(As–P) based HOMO for 17. After subsequent oxidation, the π*(As–P) orbital transformed to the singly occupied molecular orbital in the case of 18 and the lowest unoccupied molecular orbital in 19. EPR studies of 18 suggest an equal distribution of the radical density over the As (0.39e) and P (0.31e) sites and only minor contributions from the N atoms (0.04e).
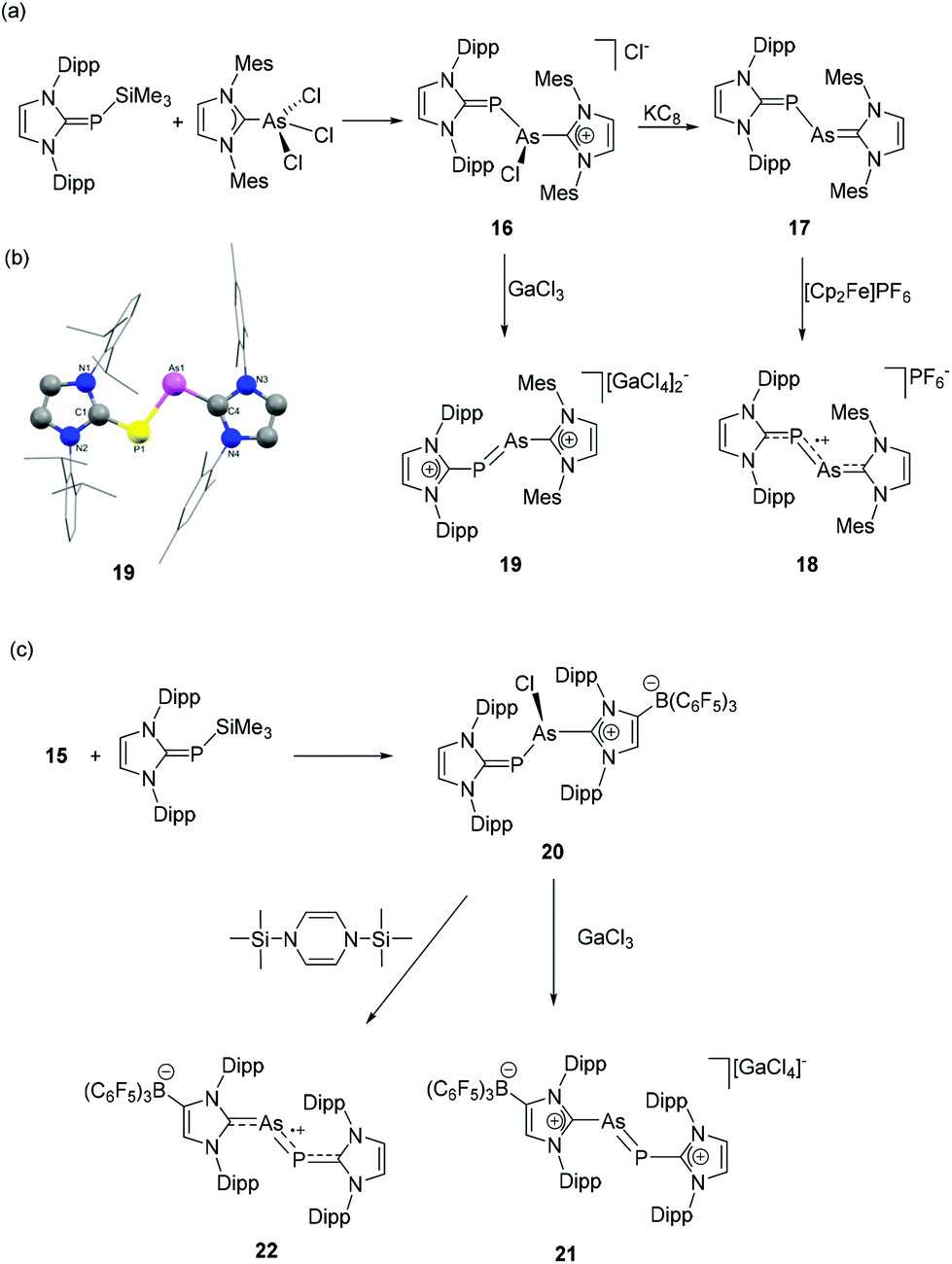 | ||
| Fig. 8 Syntheses of heteroleptic, NHC-supported arsenic monophosphide species 17–19 (a) and 21–22 (c); (b) solid-state structure of 19. | ||
These synthetic protocols were further extended to heteroleptic NHC ligands, i.e. combining anionic and neutral NHCs. The reaction of (WCA-NHC)AsCl2 with (IDipp)PSiMe3 afforded the heteroleptic dicarbene adduct, (WCA-NHC)As(Cl)P(IDipp), 20 (Fig. 8c).33 Similar to previous observations, the 31P NMR spectrum shows four signals at 156, 121, −1, and −31 ppm, indicating the presence of two isomers with variable binding of Cl atom to the pnictogens. The reaction of 20 with GaCl3 and 1,4-bis(trimethylsilyl)-1,4-dihydropyrazine afforded heteroleptic, cationic arsaphosphene, 21, and neutral dipnictene radical, 22, respectively. In the 31P NMR spectrum of compound 21, the resonance at 456 ppm is ascribed to the P atom in a low-valent arsaphosphene moiety, which is further supported by the solid-state structure analysis, showing a P–As bond distance of 2.1577(7) Å. Expectedly, the P–As bond order is reduced in line with a slightly longer bond distance of 2.2385(13) Å in the case of the zwitterionic derivative 22.
To continue exploring the synthesis of NHC-stabilized heteroleptic diatomic pnictogen allotropes, Tamm and coworkers have recently reported NHC-stabilized arsenic mononitride species. The synthetic approach had to be altered, initially reacting [(IDipp)NSiMe3] with AsCl3 to afford the dichloroarsine [(IDipp)NAsCl2], 23 (Fig. 9a) followed by the addition of one equivalent IMes, which gave the heteroleptic, cationic dicarbene adduct [(IDipp)NAs(Cl)(IMes)]Cl, 24 as a highly reactive intermediate which is prone to decomposition.34 The in situ generated 24, in reduction with KC8 afforded the desired heteroleptic NHC-stabilized arsenic mononitride, 25. Expectedly, compound 25 adopts a trans-bent geometry, and the As–N distance of 1.8803(16) Å indicates an As–N single bond (Fig. 9b). In the P-analogue the CNHC–Pn–N angle is somewhat larger (25: 95.8(1)°, where PnAs, compared to 104.0(1)°, where PnP), while the corresponding Pn–N–CNHC angles are close to 120° (for PnP: 118.9(1)° and PnAs: 123.3(1)°). Further reactivity studies of these heteroleptic systems containing the lightest pnictogen atom were prevented by the significantly reduced stability compared to 11–21.
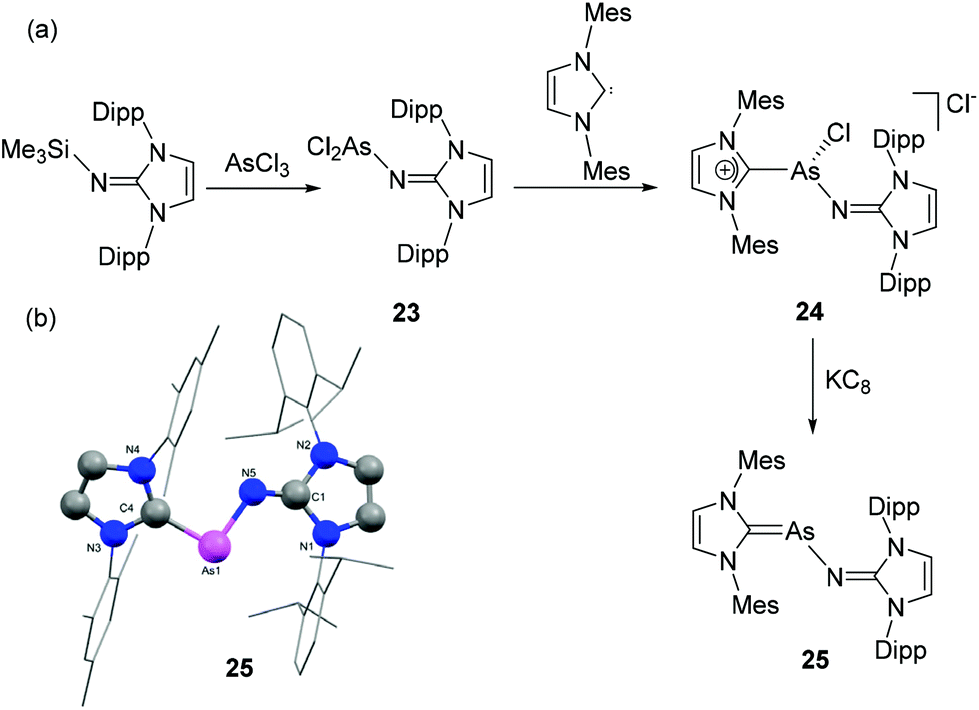 | ||
| Fig. 9 (a) Synthesis of heteroleptic NHC-stabilized arsenic mononitride, 25; (b) solid-state structure of 25. | ||
Arsenium and arsonium carbene adducts
In contrast to the highly developed chemistry of NHC adducts of cationic phosphorus species, analogous arsenic species are not as widely studied. Weigand and coworkers have reported the NHC-stabilized monocationic dichloroarsane, 26 obtained by the treatment of 2-trimethylsilylimidazolium triflate with AsCl3 (Fig. 10a).35 Particularly interesting about the synthetic protocol is that the silylimidazolium cation acts as an imidazoliumyl-transfer reagent and prohibits the tendency of As(III) derivative for the reductive formation of As(I) species in presence of Lewis Base. The cation 26 on treatment with Me3SiCN and Me3SiN3 afforded NHC-stabilized dicyanoarsenium triflate 27 and diazidoarsenium triflate 28, respectively. In the solid-state structure of 26, there is a strong cation–anion interaction with As⋯Otriflate distance of 2.882(1) Å, which is significantly shorter than the sum of the van der Waals radii of the two elements [∑rvdw (O, As) = 3.37 Å]. As such, the geometry around As cation can be considered distorted bisphenoidal, i.e., having coordination number of 4 (Fig. 10b). The As–CNHC bond distance is 1.974(1) Å, which is expectedly longer than its phosphorus analogue (Å). The As–Cl bond distances are 2.1705(5) and 2.2036(5) Å. The Cl1–As–Cl2 bond angle is 97.25(2)°, which is closer to 90° than that of the corresponding phosphorus analog. This is attributed to the higher p-character in the bonds of the arsenium cation.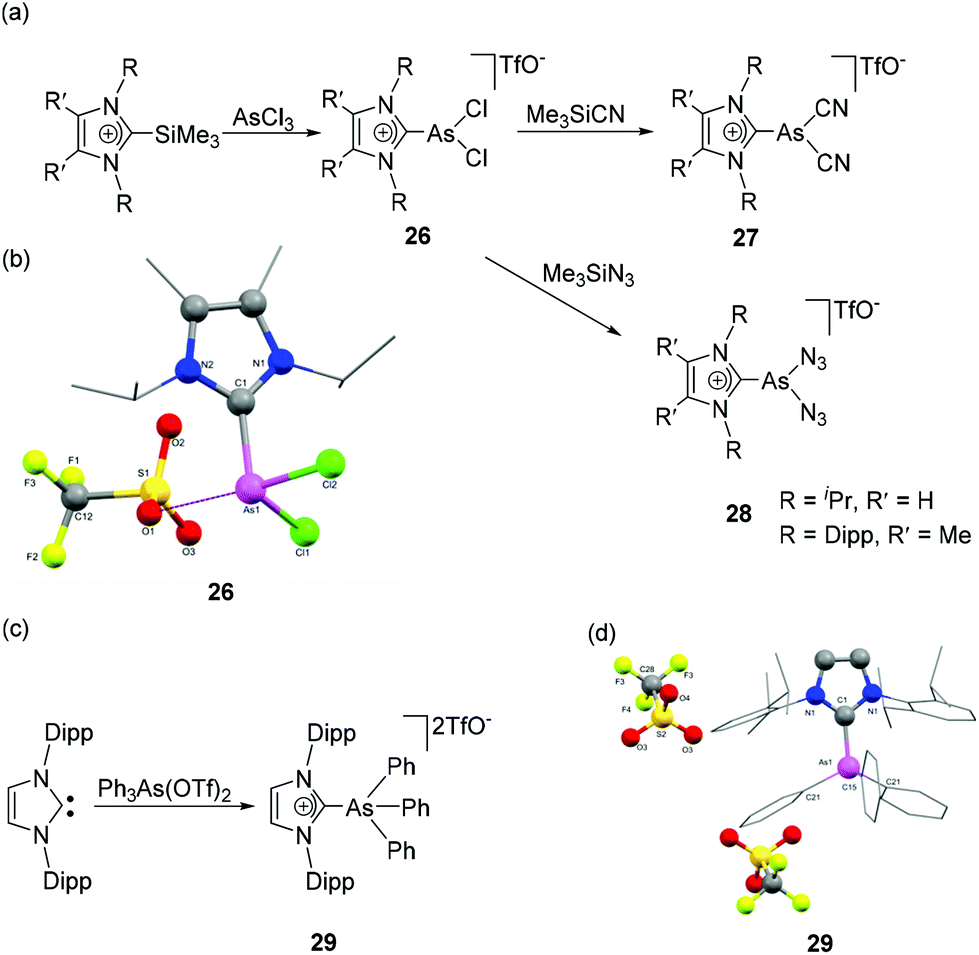 | ||
| Fig. 10 Syntheses of NHC-stabilized cationic arsenic species 26–29 (a) and (c); solid-state structures of 26 (b) and 29 (d). | ||
Weigand and coworkers have also reported the synthesis of NHC-supported dicationic tetraorganoarsenium species, 29 by the treatment of Ph2As(OTf)2 with IDipp (Fig. 10c).36 During the reaction, the pentacoordinated As(V) species, being a highly reactive Lewis acid, undergoes triflate anion displacement with a concomitant change in coordination number from five to four and results in a dicationic NHC adduct, 29. In the solid-state structure, the As adopts a tetrahedral geometry with the CNHC–As–CPh bond angles falling within the range of 105.98(6)° to 113.36(10)° (Fig. 10d). The As–CNHC bond distance is 1.948(2) Å. Interestingly, the As–CPh bond distances are slightly shorter and lie in the range of 1.906(2)–1.9103(17) Å. The lengthening of CPh–As bond distances in comparison to the As–CNHC bond distance might be attributed to the relative hindrance adhered by the flanking mesitylene arm of the imidazole moiety. A similar trend in the As–C bond distances was also observed in the structure of NHC-stabilized α-cationic arsine reported by Alcarazo and coworkers.37 The cationic triorganoarsine was synthesized by the treatment of IMes with Ph2AsI to afford [IMesAsPh2]I, which on treatment with AgOTf resulted in [IMesAsPh2]OTf, 30 (Fig. 11a). The geometry around As is distorted trigonal-pyramidal (Fig. 11b). Similar to 29, the As–CNHC bond distance is 1.9858(8) Å, which is substantially longer than the As–CPh distances [1.9468(9) and 1.9461(9) Å]. The reaction of 30 with [AuCl(Me2S)] afforded a rare example of an Au(I) complex of any cationic arsenium species, 31. While the molecular structure of 31 was not known, it is observed in a related cationic arsine-based metal complex that due to the higher electrophilic character of As centre there is a minute but perceptible shortening of As–CNHC bond distances in comparison to the parent cation. Furthermore, a significant increase in the CNHC–As–C bond angles is also observed in the metal complex indicating an almost ideal sp3 hybridization at the arsenic centre.
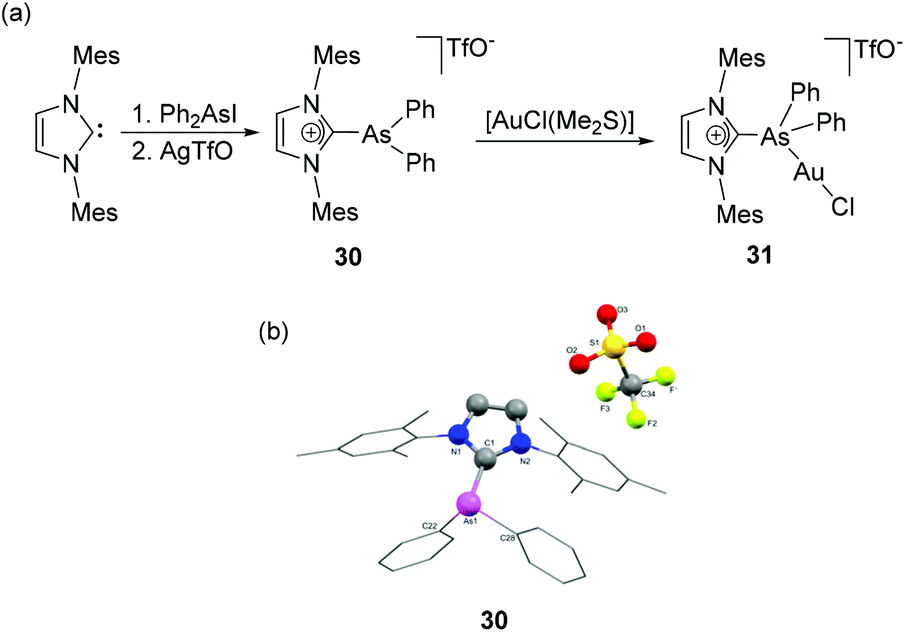 | ||
| Fig. 11 (a) Syntheses of NHC-stabilized α-cationic arsine, 30 and Au(I) complex, 31; (b) solid-state structure of 30. | ||
In a recent report, Scheer and coworkers have reported the serendipitous synthesis of the first structurally characterized NHC-stabilized As(I) cation.38 It was observed that when a triple-decker sandwich complex, namely [(Cp*Mo)2(μ,η6:6-As6)] was treated with NHC, a rare type of NHC-induced ring contraction takes place along with the formation of NHC-stabilized diorganoarsenic(I) cation 32 (Fig. 12a). In the solid-state structure, the As(I) centre adopts a bent geometry with a C–As–C bond angle of 94.5(1)° (Fig. 12b). The corresponding As–C bond distances are 1.924(3) and 1.927(3) Å, i.e. shorter than those observed in cationic 30.
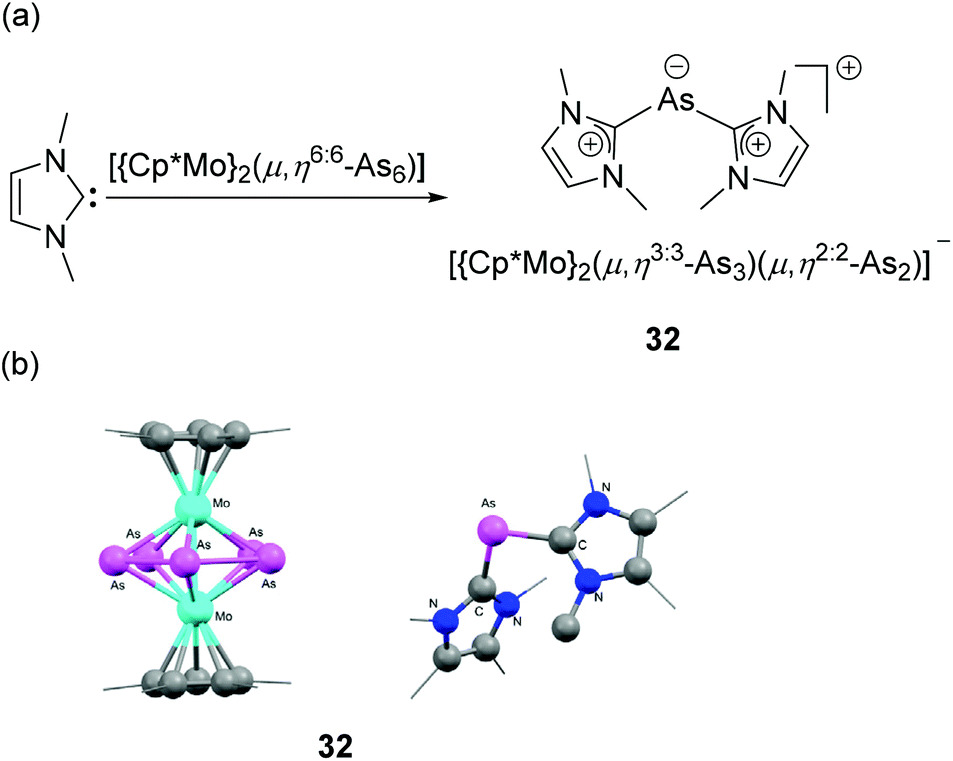 | ||
| Fig. 12 (a) Synthesis of NHC-stabilized As(I) cation, 32; (b) solid-state structure of 32. | ||
Adducts with coordination number three and higher
Arduengo et al., reported the hypervalent adduct of arsenic pentafluoride with an NHC. Compound 33 is obtained by the treatment of 4,5-dichloro-1,3-dimesitylimidazol-2-ylidene (IMes2Cl2) with AsF5 in 1,3-bis(trifluoromethyl)benzene as a solvent (Fig. 13a).39 The octahedral geometry around the As is only slightly distorted with the As atom sitting on a plane made by four equatorial fluorine atoms and average C–As–F and F–As–F cis angles being 89.7° and 90.2°, respectively (Fig. 13b). The As–CNHC bond distance of 1.991(6) Å in hypervalent 33 is longer than that observed for NHC–arsinidene adducts and are even beyond that of a covalent single bond [∑rcov (C, As) = 1.96 Å]. Solution 19F NMR data allow to differentiate two F-environments with geminal F–F couplings of 55.2 Hz and resonance similar to those found for the phosphorus congener.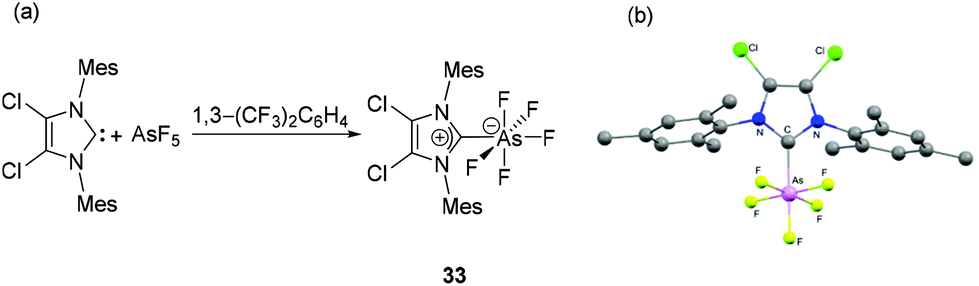 | ||
| Fig. 13 (a) Synthesis of NHC-stabilized hypervalent arsenic species 33; (b) solid-state structure of 33. | ||
Carbene adducts with antimony
Even less is known about the chemistry of carbene–Sb adducts. By necessity of an increased acceptor character to stabilize these heavier derivatives, a more diverse range of carbenes, e.g. NHCs and CAACs, have been explored for this class of compounds. The increased Lewis acidity of Sb compared to its lighter congeners becomes evident and is illustrated in several examples.Adducts with coordination numbers of three and higher
In 1999, Arduengo and coworkers reported the first NHC-stabilized antimony compound, a hypervalent antimony species (34) obtained by the nucleophilic addition of IMes2Cl2 to Sb(CF3)3 (Fig. 14). In the solid-state structure, the Sb centre adopts a see-saw geometry, with the carbene ligand and one CF3 group occupying the axial (linear) positions. The occupation of an axial position by the IMes2Cl2 ligand suggests that the effective electronegativity of the imidazoline-2-ylidine moiety is higher than that of the CF3 groups. The NHC appears to be weakly coordinated to the Sb centre with Sb–CNHC bond distance of 2.821(5) Å beyond ∑rcov(C, Sb) of 2.15 Å. This weak interaction between NHC and Sb was further corroborated by the NMR spectroscopic studies, which indicates a facile dissociation of the carbene fragment from the antimony centre.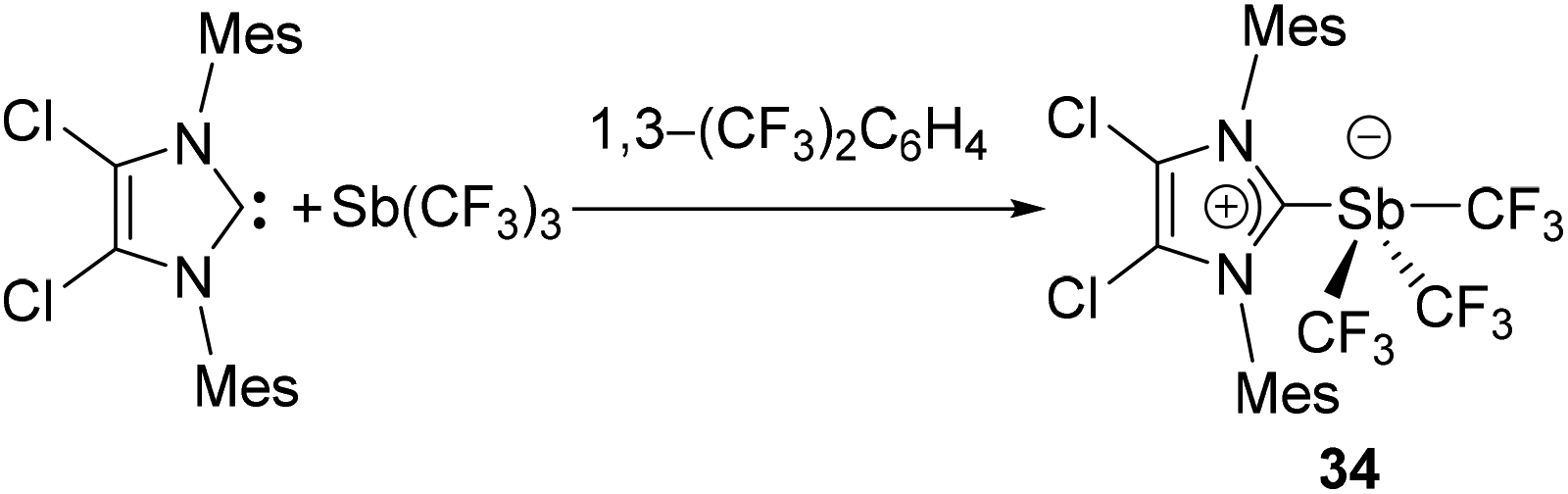 | ||
| Fig. 14 Synthesis of NHC-stabilized antimony adduct 34. | ||
As in the case of carbene-ligated As species, most of the antimony analogs are synthesized from reactions of the corresponding carbene-stabilized antimony halide adducts. As such, significant research has been devoted to the design and synthesis of a wide range of carbene-ligated antimony halide adducts, in view of their subsequent reactivity studies (vide supra). Radius and coworkers have synthesized a series of twelve NHC- and CAAC-stabilized Sb(III) adducts by the treatment of corresponding carbene ligands with SbCl3 or RSbCl2 (R = Ph, Mes) (Fig. 15a).40 The solid-state structures of 35–47 are essentially isostructural and adopt a distorted see-saw geometry at antimony, a feature that is commonly found in these carbene–pnictogen adducts (Fig. 15b). In all these structures, two Cl atoms occupy the axial positions, whereas the carbene ligand and the third Cl/C atom occupy the equatorial positions. This is in contrast to the tris(trifluoromethyl) antimony adduct 34, which is explained by the positioning of the more electronegative substituents in such hypervalent pseudo-trigonal bipyramidal compounds. In case of 48, due to steric strain associated with the Mes groups and the carbene ancillary arms, both the Cl atoms sit cis to each other. The Cl–Sb–Clax. bond angles for compound 35–47 range from 164.772(19)° to 173.064(15)°, while the Cl–Sb–Cleq./CAryl bond angles lie in the range of 96.70(5)° to 104.5(2)°. The corresponding Cl–Sb–Clcis angle in 48 is 83.77(8)°. The Sb–Clax. bond distances in 35–47 are in the range of 2.4990(6) to 2.6351(5) Å. These bond distances are significantly shorter than the corresponding Sb–Cleq. distances, which range from 2.3506(6) to 2.3858(5) Å. The elongation of Sb–Clax. bonds over the Sb–Cleq. bonds could be attributed to the donation of electron pair from the carbene centre to the σ*-orbital of the Sb–Clax. bonds along with the typical 4-electron-3-centre bonding of the axial positions. The Sb–Ccarbene bond distances in 35–48 range from 2.193(6) to 2.367(6) Å. In the cases of 41–48, the corresponding Sb–Caryl bond distances are slightly shorter and fall within the narrow range of 2.159(3) to 2.184(3) Å.
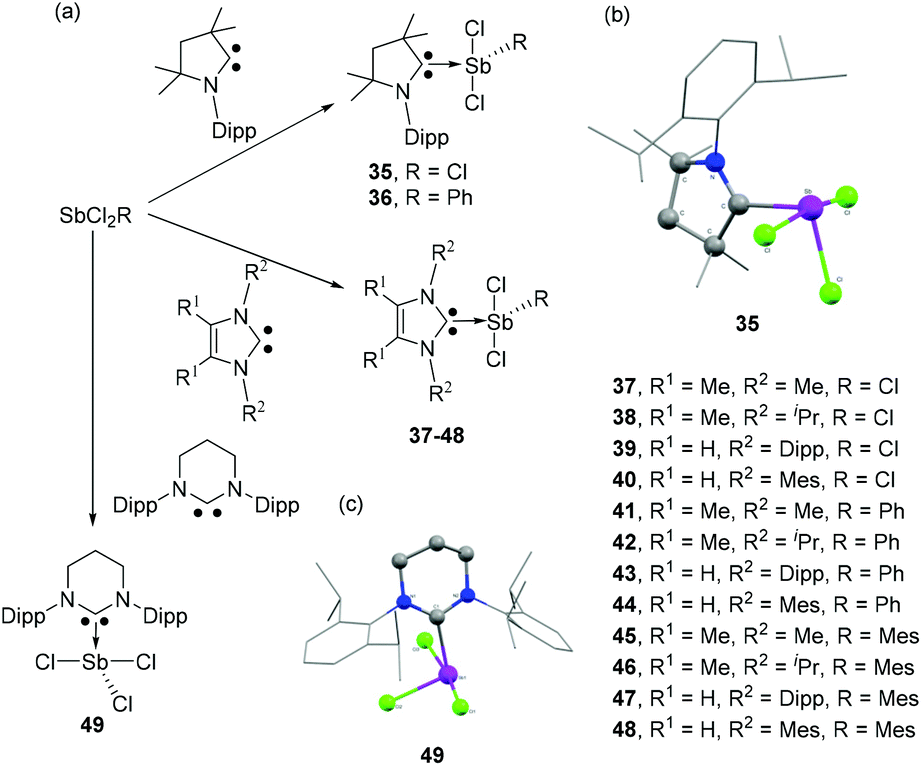 | ||
| Fig. 15 (a) Syntheses of carbene-stabilized antimony adducts 35–49; solid-state structures of 35 (b) and 49 (c). | ||
Jones and coworkers synthesized NHC-stabilized antimony trichloride adduct 49 using one of the expanded NHCs, namely 6-Dipp (6-Dipp = 1,3-bis(2,6-di-iso-propylphenyl)-4,5,6-hexahydropyirimidine-2-ylidine), which should provide increased steric shielding and nucleophilicity, along with a reduced π-acidity compared to CAAC (Fig. 15a).41 Similar to compounds 35–47, the Sb centre adopts a see-saw geometry with two Cl atoms occupying the axial positions, while the carbene ligand and the third Cl atom occupy the equatorial positions (Fig. 15c). However, in the 1H and 13C NMR spectra, 49 shows a symmetrical structure as indicated by one methine and two methyl resonances. This observation is essentially attributed to the Berry-pseudorotation in the solution state. The Sb–CNHC bond distance is 2.288(2) Å, which is significantly shorter than that observed in 34 but is in good agreement with the values observed for 35–47. Attempts had been made to reduce 49 for the synthesis of low oxidation state Sb species. However, the treatment of 49 resulted in a complex mixture of products, which infers that either the low electrophilicity of the expanded NHC or the increased steric demand precludes stabilization of Sb species in a low oxidation state.
Arduengo and coworkers have reported NHC-stabilized antimony pentafluoride, 50 by the treatment of IMes2Cl2 with SbF5 (Fig. 16).39 Similar to its lighter analog 33, the Sb(V) centre adopts a distorted Oh geometry, sitting on the plane made by the four fluoride ligands. The Sb–CNHC bond distance of 2.176(5) Å, which is shorter than that observed in 34. Detailed NMR spectroscopic studies were however precluded due to peak broadening by quadrupolar coupling of the 121Sb centre with 13C and 19F nuclei.
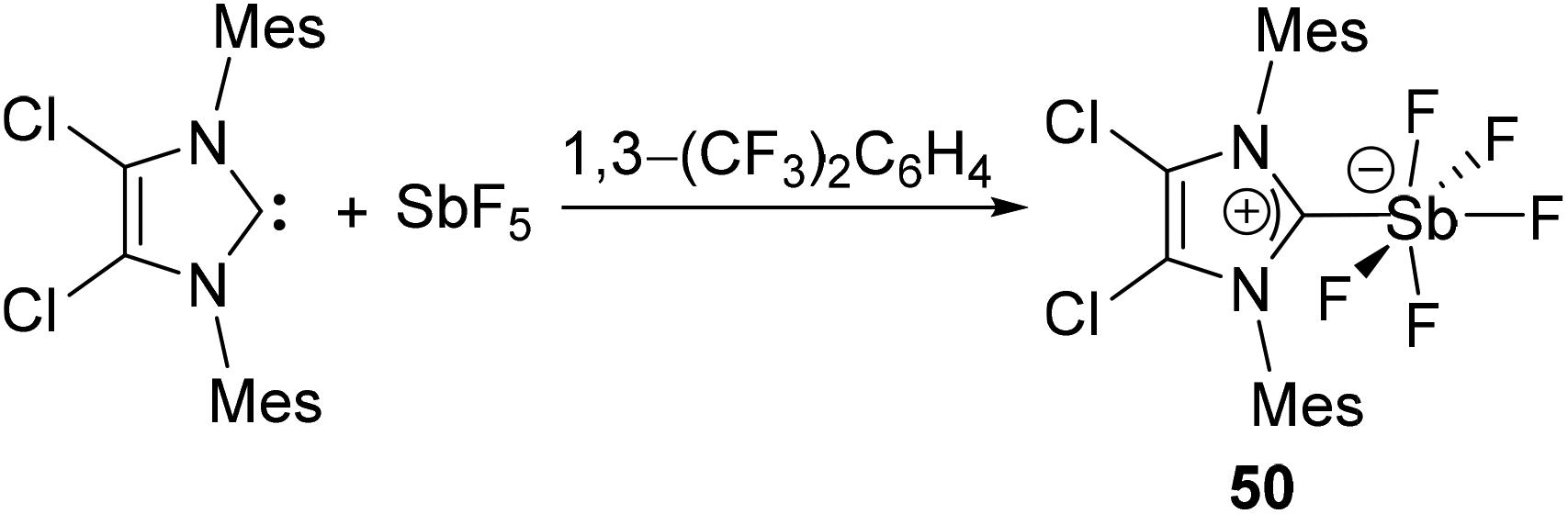 | ||
| Fig. 16 Synthesis of hypervalent Sb-carbene adduct 50. | ||
Low coordinate and low valent adducts
To overcome this stability limitation, Bertrand and coworkers have used the more electrophilic cyclic alkyl amino carbenes (CAAC) and reported upon reduction a carbene stabilized diantimony allotropes. Expectedly, the reaction of CAAC with SbCl3 afforded CAAC-stabilized adduct 51.42 The step-wise reduction of 51 with one, two, and three equivalent of KC8 afforded the neutral antimony centred Sb(II) radical species 52, a CAAC-stabilized chlorostibinidene(II) species 53, and a diantimony species 54 stabilized by two CAACs, respectively (Fig. 17a). Compound 51 is isostructural with 49 and adopts a similar see-saw geometry. The Sb–CCAAC bond distance of 2.223(3) Å is in good agreement with that of 49. DFT calculations of 52 reveal two possible conformations; with one having most spin density located at the antimony centre (90.7%), while the other is characterized by larger carbon (58.7%) and nitrogen (22.1%) spin localizations. Experimental and simulated EPR data clearly support the antimony based radical description, which is at odds with most of the carbene-stabilized main group radical species wherein a carbene-centred spin density is prevalent. In the solid-state structure compound 53 adopts a bent geometry, with a CCAAC–Sb–Cl bond angle of 100.8(1)° and a short Sb–CCAAC bond distance of 2.082(5) Å. The WBI for the Sb–CNHC bond is 1.282 also indicates only a partial double bond character. NLMO study illustrate that the Sb–CCAAC σ-bond is attributed to the donation of carbene lone pair to Sb, while the Sb lone pair, which is predominantly p-character, is involved in π-back donation to an empty p-orbital of the carbene. Similar to arsenic analogs, both the carbene moieties in 54 adopt a mutually trans disposition with respect to the Sb–Sb bond (Fig. 17b). The C–Sb–Sb bond angles are 104.1 (3)° and 105.6(2)°. The Sb–CCAAC bond distances of 2.084(11) and 2.088(10) Å match well with the chlorostibinidene 53. The WBI for Sb–CCAAC bond is 1.234 Å, which is lower than that of the NHC-stabilized arsenic analog (1.341). This indicates a reduced p-character of Sb–CNHC bond in compound 54 in comparison to NHC stabilized diarsenic derivative 14. The Sb–Sb bond distance is 2.8125 Å, which is slightly beyond the sum of their covalent radii [∑rcov(Sb,Sb) = 2.78 Å].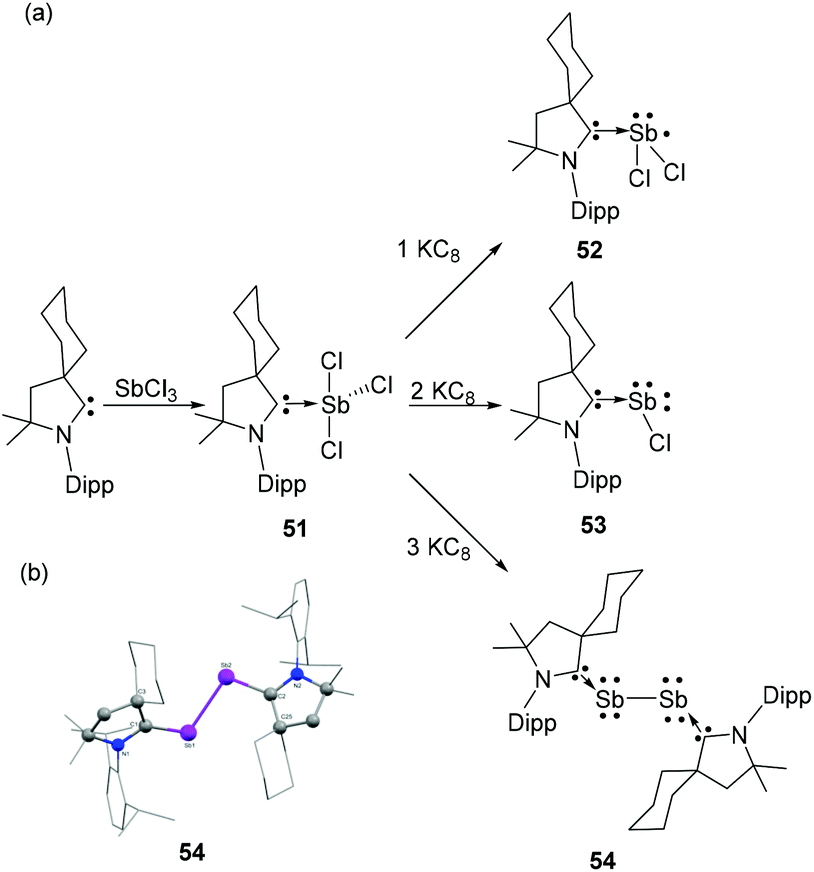 | ||
| Fig. 17 (a) Syntheses of CAAC-stabilized antimony adducts 51 and 52, chlorostibinidene 53, and diantimony species 54; (b) solid-state structure of 54. | ||
Due to the high reactivity of Sb(I) species, most of the stibinidenes are trapped and stabilized by complexation with transition metals, or obtained as (cyclic) oligomers/polymers. To attenuate the electron-rich environment around the Sb(I) centre, Hudnall and co-workers have utilized an electrophilic diamidocarbene (DAC) ligand and stabilized a monomeric stibinidene species.43 In detail, DAC upon reaction with PhSbCl2 afforded carbene-stabilized Sb(III) adduct 55 (Fig. 18a). Subsequent reduction of 55 with two equivalents of Mg afforded the stibinidene species 56. Similar to its lighter analogs, compound 56 adopts a bent geometry with CDAC–Sb–CPh bond angle of 105.5(2)° (Fig. 18b). The corresponding Sb–CDAC bond distance is 2.086(7) Å, which is significantly shorter than that observed in the Sb(III) precursor 55 [2.326(4) Å]. Both these metrical parameters are indicative of a formal CDACSb bond in 56 with a hindered rotation about this bond according to NMR studies. Similar to 54, compound 56 is stabilized by the π-back bonding interaction from the Sb(I) centre to the electron-deficient DAC moiety. In 56, the sum of WBI around the Sb centre is 2.842, in line with NBO orbital analysis showing both a σ- and π bonds of the CDAC–Sb fragment. The HOMO in 56 is a π-bonding orbital with 57.5% and 42.5% localization at Sb and CDAC, respectively, while the LUMO is an antibonding π-orbital distributed over the entire Sb–C and DAC fragment. However, comparing with the similar DAC-stabilized phosphinidene, the extent of π-back donation is less pronounced in compound 56, attributed to the poor overlap between the C(2p)-orbital at the DAC with the Sb(5p) orbital.
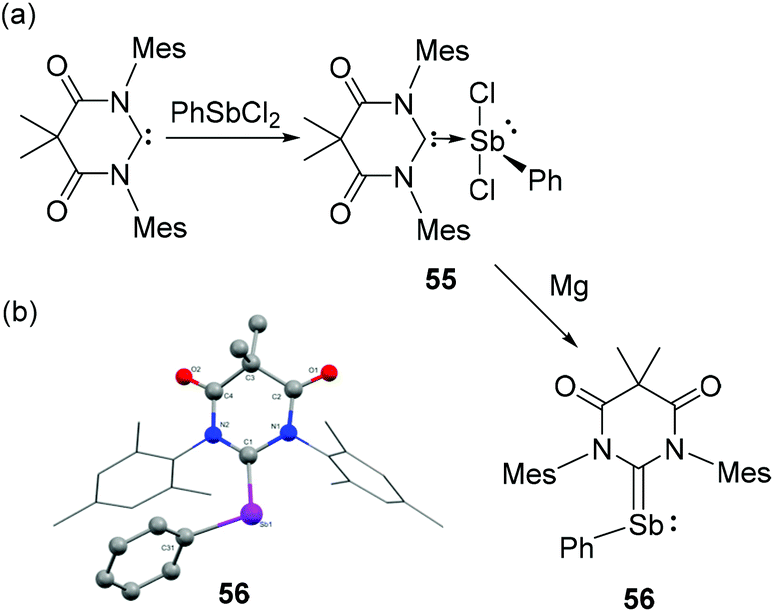 | ||
| Fig. 18 (a) Syntheses of carbene-stabilized antimony adduct 55 and stibinidene 56; (b) solid-state structure of 56. | ||
Recently, Schulz and coworkers have reported the synthesis of carbene-stabilized Ga-substituted stibinidenes. The gallane diyl LGa {L = HC[C(Me)N(Dip)]2} is treated with half equivalent of SbX3 (X = Cl, Br) to afford disubstituted halostibane [L(X)Ga]2SbX.44 Subsequent reaction of [L(X)Ga]2SbX with one equivalent of NHC/CAAC afforded carbene-stabilized Ga-substituted stibinidenes, 57–60 (Fig. 19a). In the solid-state structures, the Ga–Sb bond lengths in CAAC-stabilized stibinidenes 57 (Fig. 19b) and 58 are 2.605(1) Å and 2.599(2) Å, respectively. These bond distances are somewhat longer than the values observed in corresponding NHC-stabilized stibinidenes 59 [2.571(1) Å] and 60 [2.562(1) Å]. The corresponding C–Sb–Ga bond for 57 [107.2(2)°] and 58 [107.2(3)°] are slightly smaller than that of 59 [112.0(1)°] and 60 [114.3(2)°]. An inverse trend is seen for the Sb–Ccarbene bond distances with 57 [2.083(4) Å] and 58 [2.089(1) Å] being substantially shorter than those observed in 59 [2.228(1) Å] and 60 [2.378(1) Å]. Notably, the heavier halide substituent (Br vs. Cl) has an elongating effect on the Sb–Ccarbene bonds, which are in good agreement with the value observed in DAC-stabilized stibinidene 56. Interestingly, the WBI for Sb–Ccarbene bonds in 57 and 58 are 1.19 and 1.18, respectively, indicating the partial double bond character to the bond. However, relatively low WBI in 59 (0.97) and 60 (0.96) suggest Sb–Ccarbene single bonds in agreement with the observed bond distances. Similar to 56, the σ-bond in 57–60 stems from the donation of carbene lone pair to the empty p orbital of the Sb centre. In the cases of 57 and 58, significant π-back bonding interactions were observed from the Sb lone pair orbital to the empty p orbital of the CAAC. The corresponding HOMOs in 57 and 58 were expanded along the Sb–Ccarbene bond, while such expansion of HOMOs was not observed in 59 and 60. The π-back bonding interactions also attribute a balanced partial charge between the Sb and Ccarbene atoms in 57 and 58. Contrastingly, in absence of π-back donation, the Sb atoms in 59 and 60 exhibit a pronounced partial negative and the CNHC atoms a partial positive charge hinting at the description of these systems as zwitterionic stibinidene ylides rather than ylene-like stibaalkenes. Besides the clear differences originating from the nature of the carbene, it is important to state that in all the Ga-substituted stibinidenes, 57–60, London dispersion plays a crucial role in the thermodynamic stabilization of these compounds.
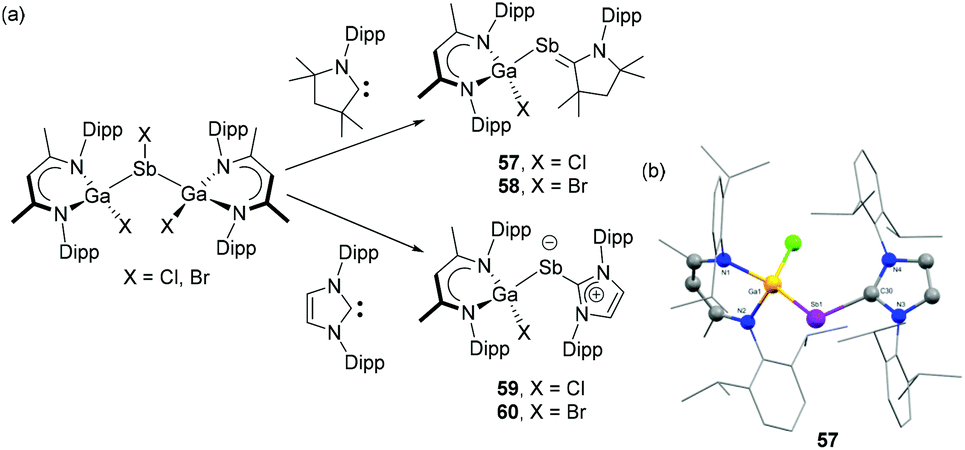 | ||
| Fig. 19 (a) Syntheses of carbene-stabilized Ga-substituted stibinidenes 57–60; (b) solid-state structure of 57. | ||
Tamm and coworkers have expanded the use of their WCA-NHC ligand, where the carbene centre bears a formal positive charge towards antimony.30 The synthetic protocol was identical to that of its lighter arsenic congener (vide supra), wherein the salt metathesis reaction of SbCl3 with (WCA-NHC)Li˙− toluene salt afforded dichlorostibane 61. Subsequent reduction of 61 with 1,3-bis(trimethylsilyl)-1,4-dihydropyrazine or magnesium powder afforded the diantimony compound 62 (Fig. 20a). Expectedly, compound 62 adopts a trans-bent geometry around the SbSb bond, with a coplanar arrangement of the carbene ligands, as evident from the C–Sb–Sb–C torsion angle of 180° (Fig. 20b). The C–Sb and SbSb single and double bond show distances of 2.194(2) and 2.882(8) Å, respectively, which are in good agreement with the values observed in CAAC stabilized diantimony species, 54. The increased s character of the Sb lone pair results in a further decrease of the C–Sb–Sb angle to 96.81(7)°.
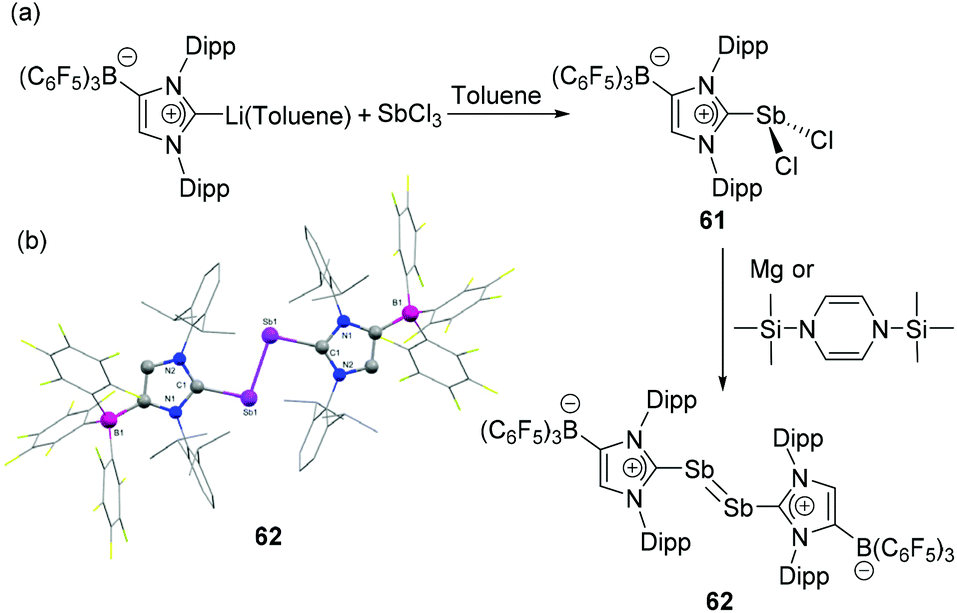 | ||
| Fig. 20 (a) Synthesis of diantimony compound 62 from NHC-stabilized dichlorostibane, 61; (b) solid-state structure of 62. | ||
Cationic Sb-derivatives
NHC-stabilized dichlorostibenium cation 63, the first carbene-ligated cationic antimony compound was synthesized by Weigand and coworkers.3563, similar to its arsenic analog 26, was synthesized by the treatment of 2-trimethylsillyimidazolium triflate with SbCl3 (Fig. 21a). Similar to 26, a strong Sb⋯Otriflate interaction was observed in 63, which result in a distorted bisphenoidal geometry around the Sb centre (Fig. 21b). The Sb–CNHC bond distance is 2.212(3) Å, and the Sb–Cl bond distances are 2.4022(6) and 2.212(3) Å. The steric demand of the carbene is crucial and using bulky groups, allows exclusive isolation of the monocationic species, irrespective of the nature of the pnictogen centre.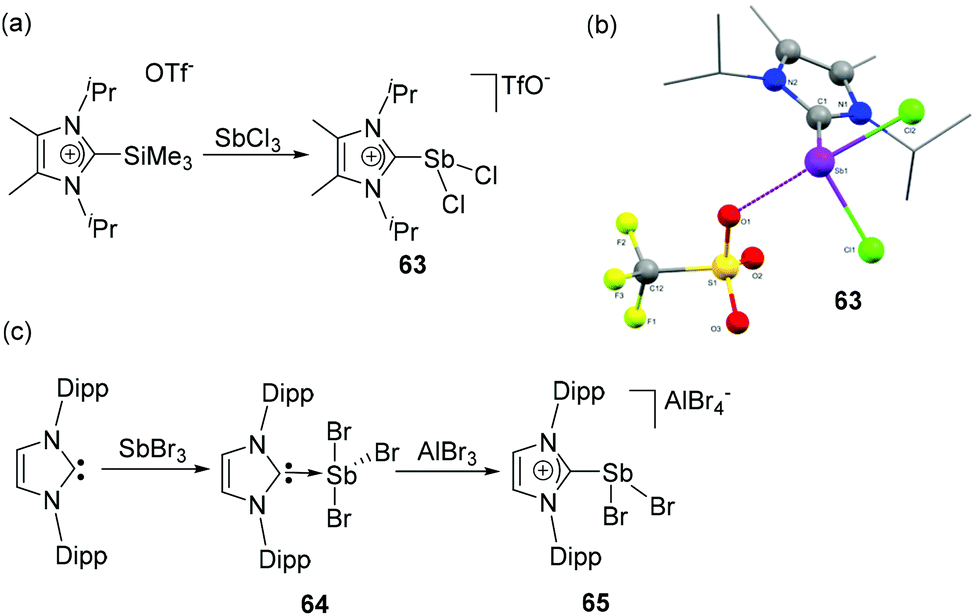 | ||
| Fig. 21 Syntheses of NHC-stabilized cationic dichlorostibane 63 (a) and 65 (c); solid state structure of 63 (b). | ||
Goicoechea and coworkers have synthesized a related NHC-stabilized dichlorostibenium cation, 65, however with a different synthetic strategy.45 When IDipp was treated with SbBr3, the reaction afforded NHC-stabilized adduct 64 in a classical see-saw geometry. Additionally, a coordinating THF molecule is located trans to the carbene ligand. As a consequence of electron donation from the carbene centre, the Sb–Brax. bond distances in 64 are longer [2.727(1) and 2.729(1) Å] than the corresponding Sb–Breq. bond distance [2.508(1) Å]. Taking into consideration of this relative labialization of the Sb–Br bonds, when 64 was treated with AlBr3, a bromide abstraction took place, leading to the formation of dichlorostibenium cation, 65 (Fig. 21c). As observed in 63, due to the close contact between the Sb centre and one of the bromide ligands of the anion, 65 adopts a distorted bisphenoidal geometry. The Sb–CNHC distance of 2.223(4) Å is in good agreement with that of cation 63. Interestingly, excess of IDip during the syntheses triggered a rearrangement to the zwitterionic aIDip(SbBr3), which is observed as a (μ-Br)2-bridged dimer, which in itself shows a rich chemistry.
In a recent development, Roesky and coworkers have reported the CAAC-stabilized Sb(I) cation, 66 by the reduction of SbCl3 with KC8 in the presence of CAAC, i.e. a heavier group 15 isoelectronic analog to carbones (Fig. 22a). The Sb(I) cation is obtained upon anion exchange to triflate and adopts a bent geometry (Fig. 22c) in the solid-state as observed in its lighter As(I) analog, 32. Noteworthily, no direct anion–Sb interactions are observed. The C–Sb–C bond angle is 111.87(7)° and the Sb–C bond distances are 2.145(2) and 2.1498(18) Å. DFT calculations suggest that the bonding in 66 can be described in terms of a CAAC: → Sb ← :CAAC dative bond, wherein the neutral carbene ligands donate electron pairs to the Sb(I) centres and the Sb(I) centre is involved in π-back donation to the vacant π-orbital of the carbene carbon, exploiting the outstanding π-acceptor and σ-donating character of CAACs.
 | ||
| Fig. 22 Syntheses of Sb(I) cation 66 (a) and Sb(III) cation 67 (b) stabilized by two CAACs and NHCs, respectively; solid-state structures of 66 (c) and 67 (d). | ||
Tavčar and coworkers have reported that when soluble SbF3(tmeda) [(tmeda = N,N,N′,N′-tetramethylethane-1,2-diamine ] was treated with IDipp, it initially afforded an NHC-stabilized trifluouro antimony intermediate, IDippSbF3. Thermal treatment of the antimony adduct led to an NHC-mediated SbF3 auto-ionization to afford a cationic species, 67 (Fig. 22b). Interestingly, one of the IDipp ligands also undergoes a proton shuffling between the carbene carbon and alkenic double bond to rearrange to a mesionic carbene ligand (aIDipp) and coordinate to the Sb centre via C4 position. Such a mesionic NHC rearrangement is not prevalent in an Sb–F system. In the solid-state structure, compound 67 adopts a bisphenoidal geometry, where the two carbene moieties occupy equatorial positions and the F− ligands occupy the axial positions (Fig. 22d). The Sb–CIDIPP and Sb–CaIDipp bond distances are 2.197(3) and 2.165(3) Å, respectively. The SbF4− counter ion shows no direct interactions with the stibenium centre.
Carbene adducts with bismuth
Similar to the situation of antimony, carbene adducts of the heaviest pnictogen are very little explored. The larger atomic size of bismuth causes it to have poorer orbital overlap with the carbene centre than its lighter congeners. Additionally, due to its metallic nature, organo-bismuth compounds exhibit significantly different reactivity and stability. Therefore, the synthesis of carbene-stabilized bismuth species is even more challenging.Adducts with coordination numbers of three and higher
As such, carbene–bismuth compounds were long considered elusive until Dutton and coworkers first reported NHC-stabilized bismuth adducts 68–69 in 2014 (Fig. 23a).46 These adducts were obtained in a clean reaction by the treatment of an NHC (IDipp and IiPr) with an equimolar amount of bismuth(III)-chloride. The reaction of 68 with Me3SiOTf resulted in the formation of carbene-stabilized adduct 70. In the solid-state, compound 68 exists as a dimer with a crystallographically imposed centre of inversion, featuring a Bi2Cl2 core formed by the μ2-bridging chlorine atoms (Fig. 23c). The intramolecular Bi⋯Cl interactions [Bi⋯Cl 3.129(2) Å], donating electron density to the σ*(Bi–Cl) bond of the neighboring molecule, result in a longer Bi–Cl2 bond [2.702(2) Å] in comparison to the other Bi–Cl1 [2.611(2) Å] and Bi–Cl3 bond distances [2.438(2) Å]. Overall the geometry around each Bi centre is distorted square-pyramidal. As with 68, 70 also exists as a dimer in which two triflate moieties act as bridging ligands. Each bismuth atom is coordinated to a THF molecule of solvation, resulting in a distorted octahedral geometry around the Bi centre. The Bi–CNHC bond distance in 70 [2.37(1) Å] is in good agreement with that observed in 69 [2.389(8) Å], but substantially longer than in BiPh3 (2.24–2.27 Å). Similar to the arsenic analogs, attempts to reduce 68 and 70 with KC8 or Mg0 were unsuccessful which is ascribed to the poor electrophilicity of NHCs, and the increased propensity of bismuth to from metallic Bi. | ||
| Fig. 23 Synthesis of NHC-stabilized bismuth adducts 68–72 (a) and (b); solid-state structures of 68 (c) and 72 (d). | ||
A related NHC-stabilized bismuth tribromide adduct 71 was reported by Goicoechea and coworkers by the treatment of IDipp with BiBr3 (Fig. 23b).45 The bromide abstraction of 71 by AlBr3 led to the isolation dichlorobismuthenium cation 72. Similar to 68, 71 also exists as a dimer, featuring intermolecular Bi⋯Br interactions, while its lighter IDipp·SbBr3 (64) analog is stabilized as a THF adduct. Interestingly, there is a significant deviation of Bi2Br2 core from planarity in comparison to that observed in 68. The deviation is attributed to the additional intermolecular Bi⋯Br interactions of the Bi atom with a neighboring Br atom with a distance of 3.389(1) Å, which falls significantly below the sum of van der Waals radii (3.70 Å). Mirroring the same trend in the Bi–Br bond distances with that of 68, the bridging bromide atom makes a longer bond with the Bi atom [2.850(1) Å] in comparison to that with the terminal bromide atoms [2.792(1) and 2.619(1) Å]. The Bi–CNHC bond distances in 71 are 2.418(6) and 2.400(7) Å, which are slightly longer in comparison to that observed in 68. The solid-state structure of 72 is isostructural with its Sb congener 65, wherein it adopts a similar bisphenoidal geometry with a cation⋯anion interaction distance of 3.3354(1) Å (Fig. 23d). The Bi–CNHC bond distance in 72 is 2.355(5) Å, which is significantly shorter in comparison to its precursor 71 eluding to an increased bonding between the carbene and the cationic fragment.
Low coordinate and low valent adducts
Tamm and coworkers have extended the use of a WCA-NHC ligand for the synthesis of dichlorobismuthane 73 (Fig. 24a).47 The reduction of 73 with 1,3-bis(trimethylsilyl)-1,4-dihydropyrazine afforded the first example of NHC-supported dibismuthene 74, featuring a bismuth–bismuth double bond. The Lewis acidic character of Bi is again illustrated in the solid-state structure of 73, wherein the benzene molecule of solvation makes an η6-coordination to the Bi atom, resulting in a distorted tetrahedral geometry around the Bi centre (Fig. 24b), which is often referred to as a Bi⋯arene “Menshutkin complex” (vide infra). The corresponding Bi–Ccentroid distance is 3.107(4) Å. The Bi–CNHC bond distance is 2.311(4) Å, which is significantly shorter in comparison to that observed in dimeric adducts 68 and 70. The solid-state structure of 74 is isostructural with its antimony analog, wherein it adopts a trans-bent geometry with Bi′–Bi–C bond angle of 94.11(8)°. Both the carbene ligands adopt a coplanar arrangement as indicated by the C–Bi–Bi–C torsion angle of 180°. There are no notable changes of the Bi–CNHC distance of 74 [2.313(3) Å] compared to its precursor 73.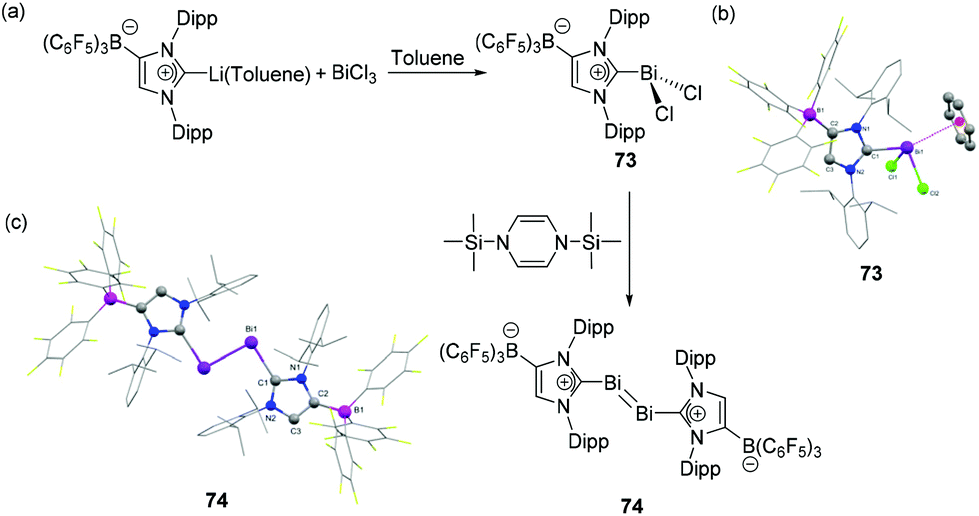 | ||
| Fig. 24 (a) Syntheses of NHC-stabilized bismuth adduct 73, and dibismuthene compound 74; solid-state structures of 73 (b) and 74 (c). | ||
Initial attempts of Gilliard and coworkers to react CAACs with BiCl3 often resulted in unstable species and metallic bismuth, as also observed by Dutton et al. for the corresponding NHCs. In order to stabilize the desired Bi(III) adducts, PhBiCl2 was explored, which should provide additional stabilization via the phenyl substituent.48 Accordingly, the reactions of Et2CAAC and CyCAAC with phenylbismuth dichloride resulted in the formation of the first examples of CAAC-stabilized bismuth adducts 75 and 76 (Fig. 25a). Both compounds were stable below −20° in solution, however increased stability of 76 over 75 is observed in the solid state. In the solid-state structures, both 75 and 76 exist as dimers (Fig. 25b), via intramolecular Bi⋯Cl interactions with each penta-coordinated bismuth centre adopting a distorted square pyramidal geometry. The Bi–CCAAC distances in 75 and 76 are significantly longer with values of 2.4566(15) and 2.4123(19) Å, respectively, indicating a weak dative nature of the CAAC → Bi interactions. The covalent Bi–CPh bonds are much shorter with distances of 2.2732(16) and 2.267(2) Å, respectively and similar to those in BiPh3. To investigate the ligand effect on the electronic structures of the adducts, Gilliard and coworkers have synthesized analogous bismuth adducts of NHC. The reaction of SIPr (SIPr = 1,3-bis(2,6-diisopropylphenyl)-4,5-dihydroimidazole-2-ylidene) and IPr with phenylbismuth dichloride afforded NHC-stabilized bismuth adducts 77 and 78, respectively in ∼25% yields. Only for 78, when kept in solution at −37 °C, isomerization took place to afford an abnormal carbene adduct 79. The solid-state structure of 77 reveals that the compound was unequivocally stabilized as a THF solvate. The THF molecule forms a dative-type bond with the Bi centre at a bond distance of 2.814(2) Å, which results in a distorted square pyramidal geometry around the Bi centre. In case of 78, a similar square pyramidal geometry of the Bi centre was achieved by a weak interaction of the Bi with the phenyl ring of the neighboring NHC ligand, i.e. a Menshutkin like stabilization. The corresponding Bi–Ccentroid distance is 3.500 Å. The Bi–CNHC distance [2.428(3) Å] in 77 matches well with the CAAC stabilized bismuth adducts 75 and 76. The corresponding Bi–CNHC distances in 78 are slightly shorter with values of 2.342(6) and 2.367(6) Å.
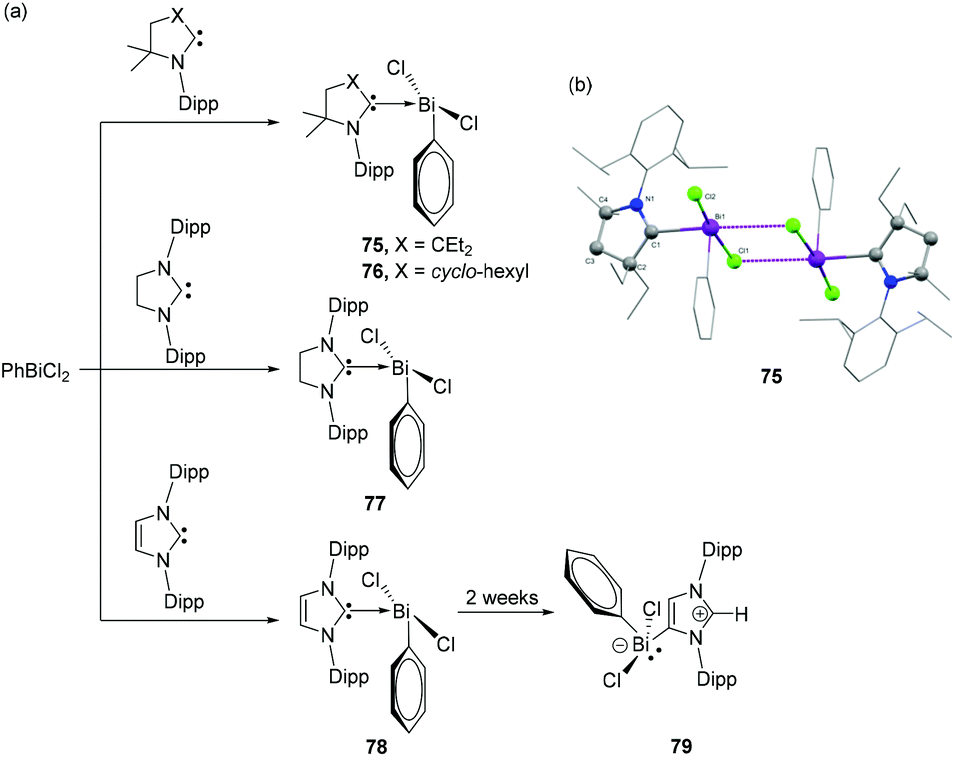 | ||
| Fig. 25 (a) Syntheses of CAAC-stabilized bismuth adducts 75–79; (b) solid-state structure of 75. | ||
Adduct 75 was further utilized for the synthesis of the first example of carbene-stabilized bismuthinidene compound (Fig. 26a).49 The subvalent bismuth compound 80 was synthesized by using a CAAC-stabilized Be(0) compound as reducing agent. Alternatively, when the CAAC-stabilized Be(0) compound was treated with PhBiCl2(THF), a simultaneous ligand transfer and reduction resulted in the same bismuthinidene compound. In contrast, attempted reduction e.g. with KC8 was unsuccessful. In the solid-state structure, compound 80 adopts a bent geometry around the Bi centre (Fig. 26b). The Bi–Ccarbene distance is 2.1992(2) Å, which is significantly shorter than the Bi–CPh bond distance [2.278(2) Å], and the Bi–Ccarbene distance observed in 75. Thus, the bond between Bi and the carbene carbon can be considered to have partial double bond character, which was further corroborated by detailed DFT calculations.
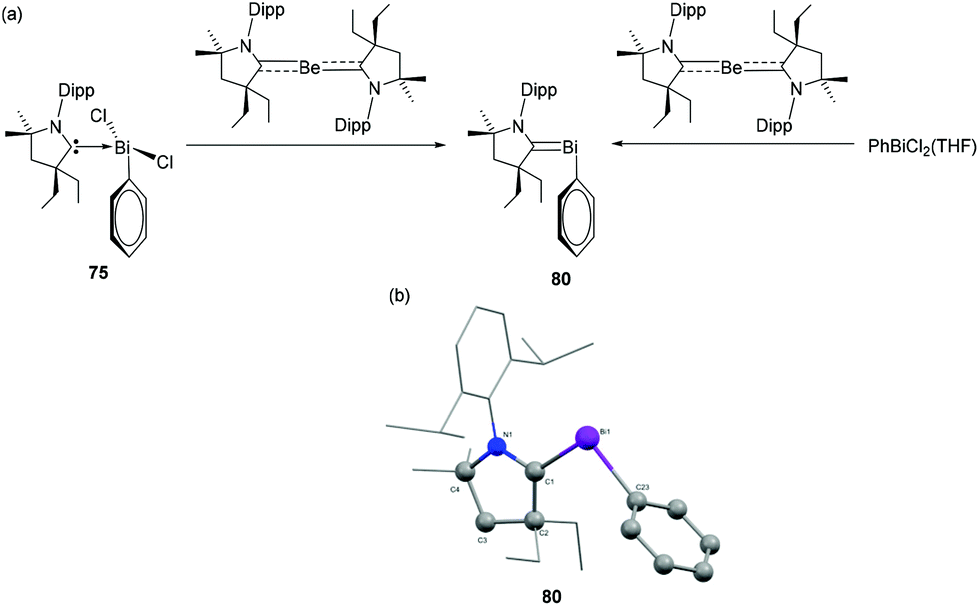 | ||
| Fig. 26 (a) Synthesis of carbene-stabilized bismuthinidene compound 80, (b) solid-state structure of 80. | ||
An attempt to synthesize cationic bismuth species failed when NHC-stabilized diphenyl bismuth chloride adduct 81 was treated with Ag[SbF6]. Interestingly, when 81 was recrystallized from THF, a solvent-promoted cationization took place to afford the desired NHC-supported bismuthenium cation 82 (Fig. 27a) with a dichloro bismuthate anion. Alternatively, similar cationic species 83 and 84 could be obtained by the direct reaction of NHC and CAAC, respectively with [Ph2Bi(μ-Cl)]2 illustrating the subtle effects originating from the different carbene ligands (Fig. 27b). Compounds 82–84 adopt bisphenoidal geometry wherein one of the chloride atoms of the bismuthate moiety coordinates to the bismuthenium centre (Fig. 27c). The Bi–CCarbene bond distances are in the range of 2.383(3)–2.39(1) Å.
 | ||
| Fig. 27 Syntheses of carbene-stabilized bismuth cations 82–84 (a) and (b); (b) solid-state structure of 82. | ||
Gilliard and coworkers have hypothesized that CDC, being a stronger electron donor in comparison to NHCs, would facilitate halide abstraction in chloro bismuthanes. To study the effects of CDC and NHC on the labilization of Bi–halogen bond, NHC and CDC were reacted with PhBiCl2(THF) (Fig. 28a). While in the case of NHC a dimeric adduct 85 was obtained, CDC more effectively attenuated the electron deficiency around the bismuth centre and stabilized the Bi centre as a monomer (86, Fig. 28b). Sequential chloride abstraction from 86 with silver salts led to the formation of mono- and dipositive bismuthenium cations 87 and 88, respectively. In the solid-state structures, both cations showed weak interactions with the [SbF6]− anion with Bi⋯F distances ranging from (2.603(8) to 2.90482) Å (∑rvdW = 3.77 Å; Fig. 28c). With the increase in electrophilicity of the Bi centre, the π-donation from the ligand increase, and consequently, the Bi–CCDC distances decrease in the order of 86 [2.249(6) Å] > 87 [2.226(3) Å] > 88 [2.157(11) Å]. Notably, the Bi–C bond distance in 88 is even shorter than that observed in carbene–bismuthinidene compound 80. As such, compound 88 is regarded as a dicationic bismaalkene species.
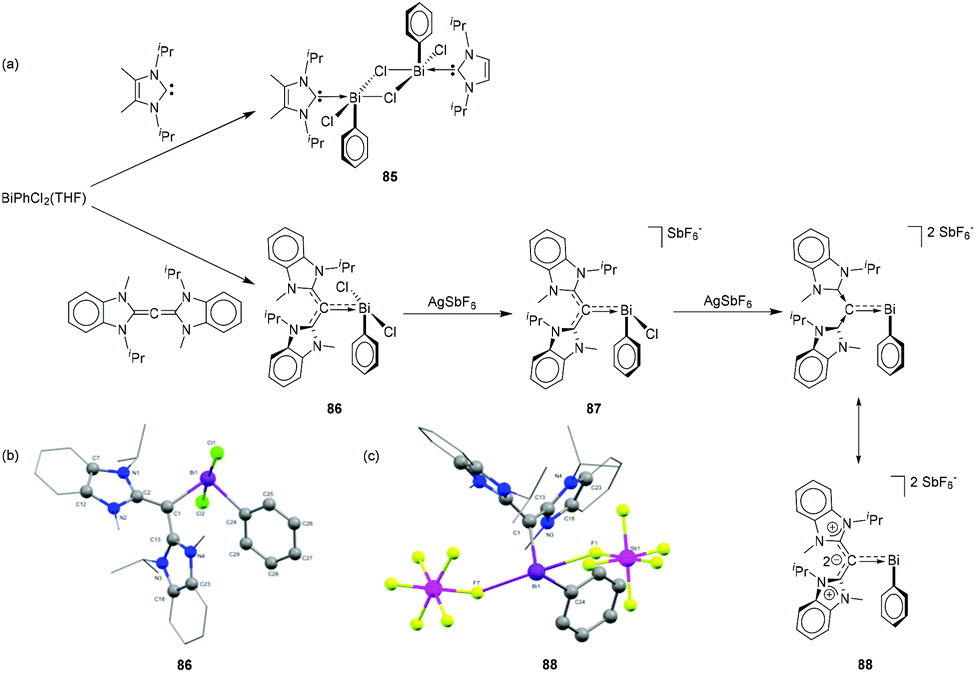 | ||
| Fig. 28 (a) Syntheses of NHC- and CDC-stabilized bismuth adduct 85, and cationic species 86–88; solid-state structures of 86 (b)and 88 (c). | ||
To target bismuthenium cations having no contacts to their anions, CDC was reacted with 2 equivalents of BiBr3 to afford dimeric adduct 89 (Fig. 29a) as a suitable starting material. The Bi–C bond of 2.292(6) Å in 89 is significantly shorter than that observed in 86. The Bi–Br bond distances are in good agreement with the values observed in related NHC-stabilized dimer 71, with two Bi–Br bond distances [2.8390(12) and 2.9196(12) Å] being significantly longer than the third one [2.6629(16) Å]. When 89 was treated in dichloromethane with 2 equivalents of AgSbF6, one bromide abstraction took place to afford monocationic bismuth species 90. Deliberate addition of THF suppresses anion interactions and gives two coordinated THF molecules. Under similar conditions, the reaction of 89 with four equivalents of silver (bis-trifluoromethylsulfonyl)imide (AgNTf2) afforded dicationic bismuth species, 91 with four coordinating THF molecules. Interestingly when compound 89 was treated with six equivalents of AgNTf2 in the presence of free CDC ligand, the strong electron donating effect of the free carbene ligands made the third Bi–Br bond more labile, leading to the formation of tricationic species 92. In the solid-state structures, no cation–anion interaction was observed for 89–92 (Fig. 29b). The additional THF coordination to the bismuth centre increases the coordination number from four (e.g.87 and 88) to five for 89 and 91. While there is no significant difference in the Bi–Ccarbene bond distance of 90 [2.226(12) Å] with the related monocation 87 [2.226(3) Å], the corresponding Bi–C bond distance in 91 [2.199(5) Å] is significantly longer than that observed in 88 [2.157(11) Å]. The Bi–Ccarbene bond distances in trication 92 are 2.1666(2) and 2.197(2) Å. Theoretical studies revealed that dications 88 and 91, and trications 92 exhibit double dative bonds (CCDC ⇉ Bi) consisting of a strong σ- and a significantly weaker π-component.
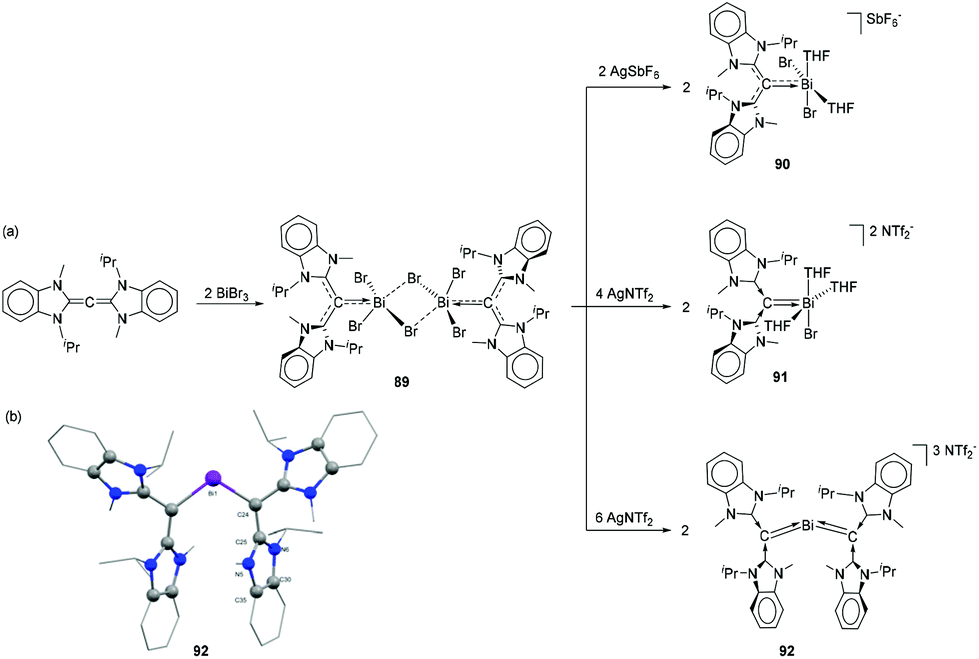 | ||
| Fig. 29 (a) Syntheses of CDC-stabilized bismuth adducts 89, and cationic species 90–92; (b) solid-state structures of 92. | ||
Conclusion and outlook
Over the past decade carbene adducts of arsenic, antimony and bismuth have become an increasingly popular study subject and massive progress has already been made in the directed synthesis of trivalent carbene adducts as well as cationic derivatives which are often obtained by simple halide abstraction protocols. Carbene-EX3 adducts are widely available for almost all halides and carbenes irrespective of the pnictogen centre; see for example Dipp-ECl3, 10 (As), 39 (Sb), and 68 (Bi). The synthesis of low valent pnictinidene–carbene adducts or carbene stabilized dipnictogens followed established routes for related phosphorus derivatives. However, the reducing agents often needed to be tailored to the pnictogen–carbene adduct to achieve the targeted low valent compound. Notably, the increased difficulty to stabilize the heaviest (Sb and Bi) carbene adducts required targeted strategies and the exploration of different carbene reagents to circumvent prevailing stability issues. In particular, the use of strongly π-accepting carbenes and sterically demanding substituents (both acting as shielding groups but also giving rise to attractive London dispersion) has allowed stabilization and isolation of unprecedented motifs as seen in examples of the (WCA-NHC)E = E(WCA-NHC), where E = As (15), Sb (62), Bi (74) and the CAAC stabilized distibene 54. This contrasts regular NHCs, which are only reported to stabilize the diarsene (e.g. Dipp-AsAs-Dipp 15), but insufficient to stabilize any of the heavier congeners. With the increased size and poorer C–Pn orbital overlap, the description of the bonding situation is an important aspect of ongoing research. Detailed theoretical investigations and judicious choice of the carbene fragment have already resulted in a large variety of bonding motifs spanning ylidic/ylenic, donor–acceptor, and ionic systems. In fact, despite all the challenges, dedicated synthetic efforts resulted in a plethora of carbene (NHC, CAC, and DCC) pnictogen adducts of As, Sb, and Bi. In particular, the use of anionic and strongly accepting carbene ligands has proven very useful in this respect.
The low valent pnictaalkene/pnictinidene–carbene and dipnictene derivatives are also prone to a rich redox-chemistry forming radical species. While the potential of these reactions has already been explored for many of the phosphorus derivatives and it has been tapped into with respect to some arsenic derivatives almost nothing is known about the Sb- and Bi-derivatives.
So far, the reported adducts have mainly been limited to mono and dinuclear pnictogen centres stabilized by one or two carbenes. As exemplified by recent literature, carbenes are also excellent ligands to stabilize a variety of poly-phosphorus clusters, such as adducts of the P4-tetrahedron, P8 and P12 clusters, but also being able to trigger fascinating P4-core rearrangements.50 Even the closest related system, yellow arsenic,51 has not been studied yet, which provides a massive opportunity for future work. The propensity of all pnictogens to chains, clusters, Zintl ions, etc. could open up multiple opportunities to study the interplay of these larger systems with carbenes.
While NHC–phosphorus adducts have been reacted to a variety of boranes similar attempts with the heavier derivatives have not been explored. In contrast to the halide abstraction protocols using Lewis acids (LAs), other acid–base adduct chemistry with LAs are lacking for all heavier pnictogen–carbene adducts. While the increased Lewis acidity and significantly reduced Lewis basicity of the heaviest pnictogen derivatives may preclude rich chemistry with LAs, this might provide ample opportunities to explore the Lewis acidic character as indicated by numerous solvent, anion, and arene interactions (vide supra, e.g.73). Coordination towards transition metals is, with the exception of the arsinidenyl–nickel complex, virtually unknown for the heavier pnictogen derivatives.25 Based on the fascinating complexes using phosphorus–carbene adducts as ligands,52 this presents itself as another field of exploration for neutral, cationic, and anionic motifs as novel ligands towards (transition) metals with exceptional electronic properties.
Current investigations are mainly focused on the synthetic aspects and fundamental (solid-state) structural characterizations of these heavier group 15 carbene adducts. Surprisingly, the reaction of pnictogen–carbene adducts with chalcogens giving unusual oxides/sulfides (e.g. NHC–P(O)–P(O)–NCH), and trapping of elusive low valent pnictogen chalcogen species such as phosphinidene sulphide, i.e. Mes*P(S)IDipp,53 is limited to phosphorus. To the best of our knowledge none of this reactivity has been reported for any of the heavier pnictogens. In general, the reactivity of said heavier pnictogen adducts with small molecules, in stochiometric or (auto)catalytic processes will remain the target of future investigations. In contrast to other main group elements the use of more elaborate, multidentate, or multiple carbene motifs to stabilize uncommon and highly reactive species is still very limited for the pnictogens. Currently, investigations are mostly focused on simple 1:1 adducts, rather than geometrically constrained and tailored multidentate ligands.
The particular redox properties of pnictogens have recently received increased attention,54 for example exploiting the P(III/V),55 Bi(I/III)56 redox couple for catalysis, as well as the Bi(II/III) couple in photocatalytic processes.57 However, these efforts have not yet been explored for carbene stabilized pnictogens.
Similar opportunities for studying the optical and electronic properties will arise within this maturing field of research. First reports on the characterization of arsenic-based radicals (e.g. diarsenic radical cation 12) show the great potential and future work will continue to reveal unprecedented and unexpected bonding situations and opto-/electronic properties.
We hope this perspective will augment basic understanding of the carbene chemistry of arsenic, antimony, and bismuth, as well as inspire the readers to design new synthetic strategies, enabling the synthesis of hitherto unknown carbene species with these elements, and exploring their intriguing chemistry.
Author contributions
R. D. and A. O. both contributed to the writing of the perspective.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Swedish research council (Vetenskapsrådet) the Olle-Engkvist foundation (Olle-Engkvists stiftelse) and the Wenner-Gren foundation (Wenner-Gren stiftelserna) for their financial support.Notes and references
- (a) P. Bellotti, M. Koy, M. N. Hopkinson and F. Glorius, Nat. Chem. Rev., 2021, 5, 711–725 CrossRef CAS; (b) H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS PubMed; (c) S. Díez-González and S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874–883 CrossRef; (d) D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC; (e) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- K. Öfele, J. Organomet. Chem., 1968, 12, P42–P43 CrossRef.
- H.-W. Wanzlick and H.-J. Schönherr, Angew. Chem., Int. Ed. Engl., 1968, 7, 141–142 CrossRef CAS.
- A. Igau, H. Grutzmacher, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 1988, 110, 6463–6466 CrossRef CAS.
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- (a) D. Zhang and G. Zi, Chem. Soc. Rev., 2015, 44, 1898–1921 RSC; (b) E. Lu, Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2021, DOI:10.1016/B978-0-12-820206-7.00015-9; (c) Q. Zhao, G. Meng, S. P. Nolan and M. Szostak, Chem. Rev., 2020, 120, 1981–2048 CrossRef CAS PubMed; (d) P. L. Arnold and I. J. Casely, Chem. Rev., 2009, 109, 3599–3611 CrossRef CAS PubMed; (e) J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561–3598 CrossRef CAS; (f) N. Wang, J. Xu and J. K. Lee, Org. Biomol. Chem., 2018, 16, 8230–8244 RSC; (g) Z. Wang, L. Jiang, D. K. B. Mohamed, J. Zhao and T. S. A. Hor, Coord. Chem. Rev., 2015, 293–294, 292–326 CrossRef CAS; (h) C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247–2273 CrossRef CAS; (i) D. J. Cardin, B. Cetinkaya and M. F. Lappert, Chem. Rev., 1972, 72, 545–574 CrossRef CAS; (j) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed; (k) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS; (l) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed; (m) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed; (n) W. A. Herrmann, M. Elison, J. Fischer, C. Köcher and G. R. J. Artus, Angew. Chem., Int. Ed. Engl., 1995, 34, 2371–2374 CrossRef CAS.
- (a) K. O. Marichev, S. A. Patil, S. A. Patil, H. M. Heras Martinez and A. Bugarin, Expert Opin. Ther. Pat., 2022, 32, 47–61 CrossRef CAS PubMed; (b) W. Liu and R. Gust, Coord. Chem. Rev., 2016, 329, 191–213 CrossRef CAS; (c) L. Mercs and M. Albrecht, Chem. Soc. Rev., 2010, 39, 1903–1912 RSC; (d) K. M. Hindi, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Chem. Rev., 2009, 109, 3859–3884 CrossRef CAS PubMed; (e) S. Nayak and S. L. Gaonkar, ChemMedChem, 2021, 16, 1360–1390 CrossRef CAS PubMed.
- (a) S. Kang, S. E. Byeon and H. J. Yoon, Bull. Korean Chem. Soc., 2021, 42, 712–723 CrossRef CAS; (b) R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC.
- (a) Y. Wang and G. H. Robinson, Dalton Trans., 2012, 41, 337–345 RSC; (b) R. Deb, P. Balakrishna and M. Majumdar, Chem. – Asian J., 2022, 17, e202101133 CrossRef CAS PubMed; (c) A. Doddi, M. Peters and M. Tamm, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS PubMed; (d) V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed; (e) Y. Wang and G. H. Robinson, Inorg. Chem., 2014, 53, 11815–11832 CrossRef CAS PubMed; (f) C. D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2013, 4, 3020–3030 RSC; (g) D. P. Curran, A. Solovyev, M. Makhlouf Brahmi, L. Fensterbank, M. Malacria and E. Lacôte, Angew. Chem., Int. Ed., 2011, 50, 10294–10317 CrossRef CAS PubMed; (h) R. Yadav, S. Sinhababu, R. Yadav and S. Kundu, Dalton Trans., 2022, 51, 2170–2202 RSC.
- (a) S. Roy, K. C. Mondal and H. W. Roesky, Acc. Chem. Res., 2016, 49, 357–369 CrossRef CAS PubMed; (b) O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445–3478 CrossRef CAS PubMed; (c) R. H. Crabtree, Coord. Chem. Rev., 2013, 257, 755–766 CrossRef CAS; (d) P. L. Arnold and S. Pearson, Coord. Chem. Rev., 2007, 251, 596–609 CrossRef CAS; (e) Á. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493–9586 CrossRef; (f) A. Krüger and M. Albrecht, Aust. J. Chem., 2011, 64, 1113–1117 CrossRef; (g) S. C. Sau, P. K. Hota, S. K. Mandal, M. Soleilhavoup and G. Bertrand, Chem. Soc. Rev., 2020, 49, 1233–1252 RSC; (h) R. Jazzar, M. Soleilhavoup and G. Bertrand, Chem. Rev., 2020, 120, 4141–4168 CrossRef CAS PubMed; (i) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed.
- J. F. Stearns and H. Rapoport, J. Org. Chem., 1977, 42, 3608–3614 CrossRef CAS.
- N. Kuhn, R. Fawzi, M. Steimann, J. Wiethoff, D. Bläser and R. Boese, Z. Naturforsch., B: Chem. Sci., 1995, 50b, 1779–1784 CrossRef.
- M. Tamm, S. Randoll, T. Bannenberg and E. Herdtweck, Chem. Commun., 2004, 876–877, 10.1039/B401041H.
- X. Wu and M. Tamm, Coord. Chem. Rev., 2014, 260, 116–138 CrossRef CAS.
- K. Dimroth and P. Hoffmann, Angew. Chem., Int. Ed. Engl., 1964, 3, 384–384 Search PubMed.
- A. Schmidpeter, W. Gebler, F. Zwaschka and W. S. Sheldrick, Angew. Chem., Int. Ed. Engl., 1980, 19, 722–723 CrossRef.
- I. A. J. Arduengo, H. V. Rasika Dias and J. C. Calabrese, Chem. Lett., 1997, 26, 143–144 CrossRef.
- T. Krachko and J. C. Slootweg, Eur. J. Inorg. Chem., 2018, 2018, 2734–2754 CrossRef CAS.
- A. J. Arduengo, J. C. Calabrese, A. H. Cowley, H. V. R. Dias, J. R. Goerlich, W. J. Marshall and B. Riegel, Inorg. Chem., 1997, 36, 2151–2158 CrossRef CAS PubMed.
- A. Schumann, J. Bresien, M. Fischer and C. Hering-Junghans, Chem. Commun., 2021, 57, 1014–1017 RSC.
- S. Yao, Y. Grossheim, A. Kostenko, E. Ballestero-Martínez, S. Schutte, M. Bispinghoff, H. Grützmacher and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 7465–7469 CrossRef CAS PubMed.
- A. Hinz, M. M. Hansmann, G. Bertrand and J. M. Goicoechea, Chem. – Eur. J., 2018, 24, 9514–9519 CrossRef CAS PubMed.
- B. M. Gardner, G. Balázs, M. Scheer, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2015, 7, 582–590 CrossRef CAS PubMed.
- A. Doddi, M. Weinhart, A. Hinz, D. Bockfeld, J. M. Goicoechea, M. Scheer and M. Tamm, Chem. Commun., 2017, 53, 6069–6072 RSC.
- G. Hierlmeier, A. Hinz, R. Wolf and J. M. Goicoechea, Angew. Chem., Int. Ed., 2018, 57, 431–436 CrossRef CAS PubMed.
- Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2008, 130, 14970–14971 CrossRef CAS PubMed.
- Y. Wang and G. H. Robinson, Inorg. Chem., 2011, 50, 12326–12337 CrossRef CAS PubMed.
- M. Y. Abraham, Y. Wang, Y. Xie, P. Wei, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, Chem. – Eur. J., 2010, 16, 432–435 CrossRef CAS PubMed.
- M. Y. Abraham, Y. Wang, Y. Xie, R. J. Gilliard, P. Wei, B. J. Vaccaro, M. K. Johnson, H. F. Schaefer, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2013, 135, 2486–2488 CrossRef CAS PubMed.
- L. P. Ho, A. Nasr, P. G. Jones, A. Altun, F. Neese, G. Bistoni and M. Tamm, Chem. – Eur. J., 2018, 24, 18922–18932 CrossRef CAS PubMed.
- W. Smith, J. Chem. Soc., Trans., 1879, 35, 309–311 RSC.
- A. Doddi, D. Bockfeld, M.-K. Zaretzke, T. Bannenberg and M. Tamm, Chem. – Eur. J., 2019, 25, 13119–13123 CrossRef CAS PubMed.
- L. P. Ho, M.-K. Zaretzke, T. Bannenberg and M. Tamm, Chem. Commun., 2019, 55, 10709–10712 RSC.
- D. Bockfeld and M. Tamm, Z. Anorg. Allg. Chem., 2020, 646, 866–872 CrossRef CAS.
- F. D. Henne, A. T. Dickschat, F. Hennersdorf, K. O. Feldmann and J. J. Weigand, Inorg. Chem., 2015, 54, 6849–6861 CrossRef CAS PubMed.
- M. Donath, M. Bodensteiner and J. J. Weigand, Chem. – Eur. J., 2014, 20, 17306–17310 CrossRef CAS PubMed.
- J. W. Dube, Y. Zheng, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2016, 138, 6869–6877 CrossRef CAS PubMed.
- M. Piesch, S. Reichl, M. Seidl, G. Balázs and M. Scheer, Angew. Chem., Int. Ed., 2019, 58, 16563–16568 CrossRef CAS PubMed.
- A. J. Arduengo, F. Davidson, R. Krafczyk, W. J. Marshall and R. Schmutzler, Monatsh. Chem., 2000, 131, 251–265 CrossRef CAS.
- M. S. M. Philipp, M. J. Krahfuss, K. Radacki and U. Radius, Eur. J. Inorg. Chem., 2021, 2021, 4007–4019 CrossRef CAS.
- A. Sidiropoulos, B. Osborne, A. N. Simonov, D. Dange, A. M. Bond, A. Stasch and C. Jones, Dalton Trans., 2014, 43, 14858–14864 RSC.
- R. Kretschmer, D. A. Ruiz, C. E. Moore, A. L. Rheingold and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 8176–8179 CrossRef CAS PubMed.
- C. L. Dorsey, R. M. Mushinski and T. W. Hudnall, Chem. – Eur. J., 2014, 20, 8914–8917 CAS.
- J. Krüger, C. Wölper, L. John, L. Song, P. R. Schreiner and S. Schulz, Eur. J. Inorg. Chem., 2019, 2019, 1669–1678 CrossRef.
- J. B. Waters, Q. Chen, T. A. Everitt and J. M. Goicoechea, Dalton Trans., 2017, 46, 12053–12066 RSC.
- A. Aprile, R. Corbo, K. Vin Tan, D. J. D. Wilson and J. L. Dutton, Dalton Trans., 2014, 43, 764–768 RSC.
- L. P. Ho and M. Tamm, Dalton Trans., 2021, 50, 1202–1205 RSC.
- G. Wang, L. A. Freeman, D. A. Dickie, R. Mokrai, Z. Benkő and R. J. Gilliard, Inorg. Chem., 2018, 57, 11687–11695 CrossRef CAS PubMed.
- G. Wang, L. A. Freeman, D. A. Dickie, R. Mokrai, Z. Benkő and R. J. Gilliard Jr., Chem. – Eur. J., 2019, 25, 4335–4339 CrossRef CAS PubMed.
- (a) M. H. Holthausen, S. K. Surmiak, P. Jerabek, G. Frenking and J. J. Weigand, Angew. Chem., Int. Ed., 2013, 52, 11078–11082 CrossRef CAS PubMed; (b) J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2007, 129, 14180–14181 CrossRef CAS PubMed.
- M. Seidl, G. Balázs and M. Scheer, Chem. Rev., 2019, 119, 8406–8434 CrossRef CAS PubMed.
- (a) C. L. B. Macdonald, J. F. Binder, A. a. Swidan, J. H. Nguyen, S. C. Kosnik and B. D. Ellis, Inorg. Chem., 2016, 55, 7152–7166 CrossRef CAS PubMed; (b) J. F. Binder, S. C. Kosnik and C. L. B. Macdonald, Chem. – Eur. J., 2018, 24, 3556–3565 CrossRef CAS PubMed.
- C. M. E. Graham, T. E. Pritchard, P. D. Boyle, J. Valjus, H. M. Tuononen and P. J. Ragogna, Angew. Chem., Int. Ed., 2017, 56, 6236–6240 CrossRef CAS PubMed.
- J. M. Lipshultz, G. Li and A. T. Radosevich, J. Am. Chem. Soc., 2021, 143, 1699–1721 CrossRef CAS PubMed.
- N. L. Dunn, M. Ha and A. T. Radosevich, J. Am. Chem. Soc., 2012, 134, 11330–11333 CrossRef CAS PubMed.
- H. W. Moon and J. Cornella, ACS Catal., 2022, 12, 1382–1393 CrossRef CAS PubMed.
- J. Ramler, J. Poater, F. Hirsch, B. Ritschel, I. Fischer, F. M. Bickelhaupt and C. Lichtenberg, Chem. Sci., 2019, 10, 4169–4176 RSC.
| This journal is © The Royal Society of Chemistry 2022 |