
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe surface composition of amino acid – halide salt solutions is pH-dependent†
Geethanjali
Gopakumar‡
 a,
Isaak
Unger‡
*a,
Clara-Magdalena
Saak
ab,
Gunnar
Öhrwall
c,
Arnaldo
Naves de Brito
d,
Tulio Costa
Rizuti da Rocha
e,
Christophe
Nicolas
f,
Carl
Caleman
*ag and
Olle
Björneholm
*a
a,
Isaak
Unger‡
*a,
Clara-Magdalena
Saak
ab,
Gunnar
Öhrwall
c,
Arnaldo
Naves de Brito
d,
Tulio Costa
Rizuti da Rocha
e,
Christophe
Nicolas
f,
Carl
Caleman
*ag and
Olle
Björneholm
*a
aDepartment of Physics and Astronomy, Uppsala University, Box 516, SE-75120 Uppsala, Sweden. E-mail: isaak.unger@physics.uu.se; carl.caleman@physics.uu.se; olle.bjorneholm@physics.uu.se
bUniversity of Vienna, Department of Physical Chemistry, Währingerstrasse 42, 1090 Vienna, Austria
cMAX IV Laboratory, Lund University, PO Box 118, SE-22100 Lund, Sweden
dUniversity of Campinas, Rua Sergio Buarque de Holanda, 777, Cidade Universitária, 13083-970 Campinas, SP, Brazil
eBrazilian Synchrotron Light Laboratory (LNLS), Brazilian Center for Research in Energy and Materials (CNPEM), 13083-970 Campinas, Sao Paulo, Brazil
fSynchrotron SOLEIL, L'Orme des Merisiers, Saint-Aubin – BP 48, 91192 Gif-sur-Yvette, France
gCenter for Free-Electron Laser Science, DESY, Notkestrasse 85, DE-22607 Hamburg, Germany
First published on 8th March 2022
Abstract
In atmospheric aerosol particles, the chemical surface composition governs both heterogenous chemical reactions with gas-phase species and the ability to act as nuclei for cloud droplets. The pH in aerosol particles is expected to affect these properties, but it is very challenging to measure the pH in individual droplets, precluding the investigation of its influence on the particle's surface composition. In this work, we use photoelectron spectroscopy to explore how the surface composition of aqueous solutions containing inorganic salt and amino acids changes as a function of pH. We observe a change by a factor of 4–5 of the relative distribution of inorganic ions at the surface of a liquid water jet, as a function of solution pH and type of amino acid in the solution. The driving forces for the surface enhancement or depletion are ion pairing and the formation of charged layers close to the aqueous surface.
Environmental significanceLiquid–gas interfaces are commonplace in the atmosphere in the atmosphere, mostly as the surface of aerosol. Depending on the origin and type of aerosol, this creates droplets with a wide variety of compositions. Their chemical surface composition determines the role of these particles in the atmosphere, e.g. as a substrate for chemical reactions with gas-phase species and as cloud condensation nuclei. One of the largest groups of non-anthropogenic aerosol is sea spray aerosol. These particles are already liquid during their generation and contain a mixture of inorganic salt (mostly NaCl) and organic compounds, many of which are surfactants. Sea water, the medium from which these particles generated, is slightly basic, yet measurements find that sea spray aerosol is often acidic, so the pH in aerosol changes. Moreover, aerosol ageing due to chemical reactions with gases might also contribute. Due to the small size of aerosol, measuring the pH inside of them is very challenging, let alone the determination of the pH at the particle surface. Very recently, a few groups succeeded to conduct such measurements and we begin to understand the complexity of the interplay between charge states, pH, and the interactions between ions (often mediated by water). Our present work expands this knowledge by using the strengths of photoemission spectroscopy (XPS) applied to aqueous surfaces: high surface resolution combined with chemical sensitivity. Therefore, XPS complements other techniques already being used in the field such as sum frequency generation and surface tension measurements. In brief, our data suggest that the surface propensity of halides is connected to the pH of a solution/within the aerosol particle if specific organic surfactants are present. In the context of atmospheric chemistry, this could mean that the pH of a particle introduces “pH breaking points” at which the surface concentration of halides increases. These “breaking points” are the pKa of common functional groups in the organic surfactants. An increase/decrease of such ions would shift equilibria for chemical reactions at the particle surface (e.g. reaction of Cl− in solution and OH radicals in the gas phase). In the context of cloud nucleation, these “breaking points” may affect the hygroscopicity of these particles, again due to changes in the surface composition. Whether these “breaking points” really exist in natural aerosol, containing a mixture of organic species, or whether it is more of an incremental change of the particle properties can only be answered by dedicated research on more realistic systems. Our fundamental study suggests the possibility of their existence. |
Introduction
Water is omnipresent in the earth's atmosphere as vapour, rain droplets or in aerosol particles of various origin. Depending on the source of the aerosol, their size and composition vary and they can pick up water while they are suspended in the atmosphere. Two of their most important functions in the atmosphere are their role as cloud condensation nuclei1–3 and as substrates for chemical reactions at the particle-gas interface.4 Both of these functions strongly depend on their surface composition. A chemical reaction at the particle surface can only take place if reactants are present, and the ability to act as cloud condensation nuclei is characterized by the hygroscopicity of the particles, which is also dependent on the chemical surface composition.1,5 As they are exposed to chemical reactions and radiation, the total composition of aerosol particles in the atmosphere changes over time, what is often referred to as ageing of aerosol. The longer aerosol particles remain suspended in the atmosphere, the more sulphuric and nitric acid they pick up, which results in changes of their composition.6,7Sea spray aerosol in particular is produced from slightly basic sea water, and one might expect the particles to exhibit a basic pH, measurements, however, indicate that they are acidic.8–10 Measuring pH in aqueous aerosol is challenging since they are much smaller than the available probes. Only recently, some groups have begun to utilize spectroscopic techniques to measure the pH of aerosol.11–13 Wei et al. even found a pH gradient within basic aerosol particles.14
Variations of the pH in the droplets cause changes in the protonation state of functional groups of solvated organic molecules and thus in their charge state. Many of these molecules are surfactants and interact with inorganic ions present in the solution through their functional groups,15–20 thus they potentially link the presence of inorganic ions at the surface to the pH of the particle. This correlation has implications for chemical reactions at the particle surface, which involve inorganic ions. Prominent examples for such a reaction are the production of Cl2 from 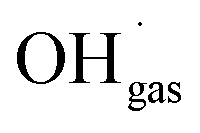 and Clsurface−,21 the ozone depletion through a reaction with iodide and bromide22,23 or the reaction between N2O5(g) and Br−(aq).24
and Clsurface−,21 the ozone depletion through a reaction with iodide and bromide22,23 or the reaction between N2O5(g) and Br−(aq).24
The main concern of this work is to learn about the interactions at aqueous surfaces, and thus we used photoelectron spectroscopy (XPS) for our investigation. This method combines chemical sensitivity with a very high surface sensitivity due to its electron energy dependent probing depth. We estimate the probing depth in our experiments to about 2 nm. This covers what we consider as the ‘surface’ of the solution – a volume, which is affected by the surfactants and surface-induced effect but can vary depending on the properties and contents of the solution.
The interplay of organic surfactants and pH on the surface composition of mixed ionic solutions has been addressed only recently.18–20,25 Our work attempts to further fill this gap and expand our knowledge to the role amino acids play. We chose to use amino acids as organic component in our study as a surrogate for organic solutes in aerosol for several reasons. On one hand, amino acids are present in aerosol particles,26 and on the other hand, they possess two functional groups, whose protonation characteristics behave oppositely to changes of the pH. Moreover, the surface propensity of different amino acids varies. By carefully selecting amino acids we aim to mimic a variety of other organic species present in aerosol with different surface propensity and charge. Amino acids at aqueous surfaces have been simulated using molecular dynamics (MD). These simulations show that amino acids containing hydrophilic side chains, such as glycine (GLY), serine and alanine prefer to be in the bulk. Hydrophobic and amphiphilic amino acids, like valine (VAL), methionine (MET) and phenylalanine (PHE) strive towards the surface.27 The six amino acids included in this study are glycine, valine, phenylalanine, proline (PRO), cysteine (CYS) and methionine. The structures of these amino acids and the bulk pKa values of their functional groups are shown in Fig. 1a. Due to the amino acids possessing different surface propensities, our measurements required us to use solutions of varying amino acid concentration in order to ensure a sufficiently high signal intensity. Though we avoided using concentrations close to the solubility limit, this resulted the amino acid concentrations to differ by up to one order of magnitude.
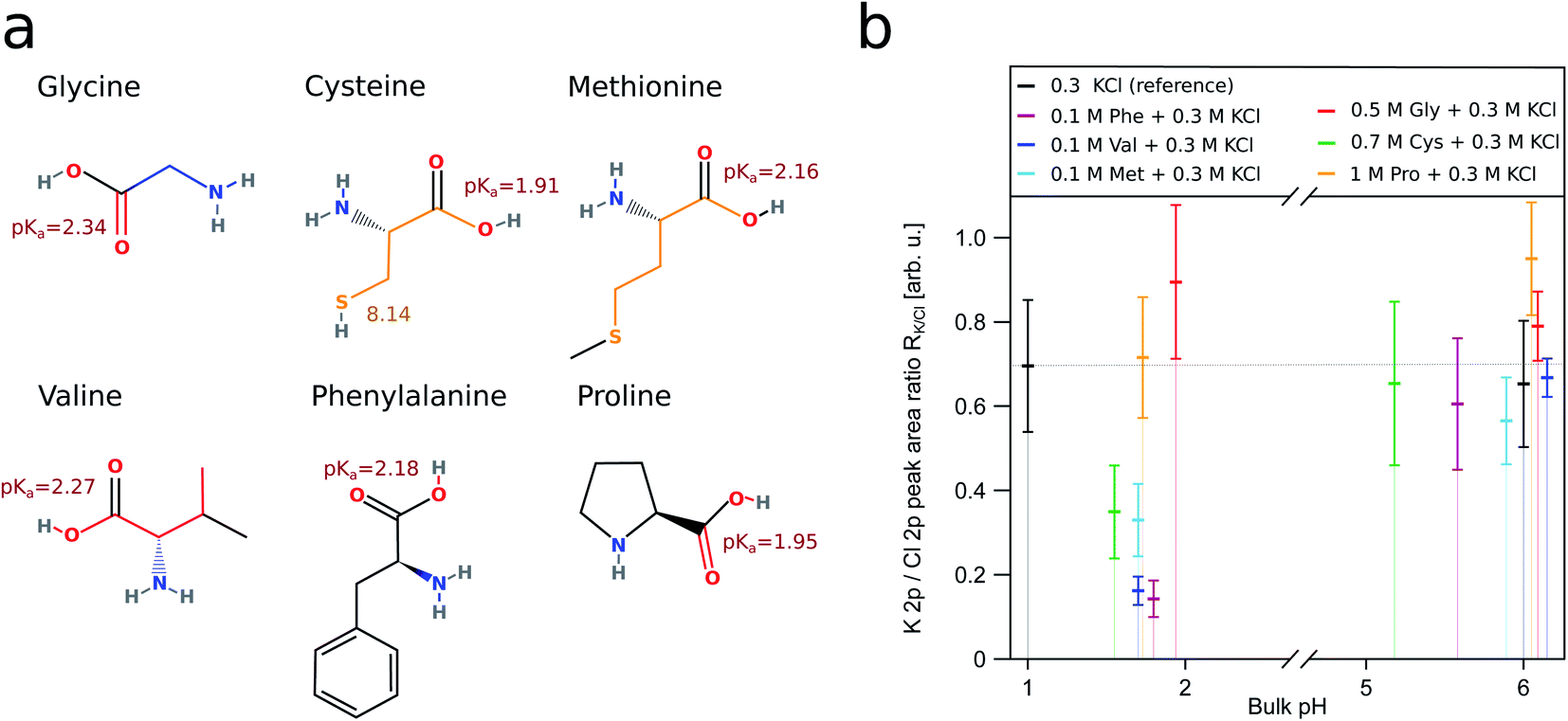 | ||
| Fig. 1 (a) Displays the structure of the amino acids used in this study. The bulk pKa values of their functional groups are depicted next to the respective groups.51 The K 2p/Cl 2p ratio (RK/Cl) for the respective aqueous solutions is shown in (b). All of the ratios have been derived from fits to the K 2p and Cl 2p levels recorded with XPS. We consider a solution containing only 0.3 M KCl (black) as a reference for the behaviour of RK/Cl without the influence of surfactants. The reference solution has RK/Cl ≈ 0.7 indicated by the thin dotted line. The behaviour of RK/Cl in the presence of a surface enriched amino acid is best represented by the solution containing phenylalanine (purple). Due to the hydrophobic side chain, PHE is always enriched at the surface. The large vertical colored bars indicate the error bars; we describe in detail how we obtain these in the ESI.† | ||
This way we could both study amino acids with hydrophobic side chains (VAL, PHE, MET) and without (GLY, CYS, PRO). Amongst these, GLY is expected not to be surface enriched, independent on the pH,28,29 whereas for PHE and VAL the surface enrichment is affected by the charge state of the molecule.28 CYS and MET are included in the study since these are the two amino acids containing sulfur and CYS has the ability to lose both the carboxylic hydrogen and the thiol hydrogen. Overall, we are interested in the impact of amino acids with varied side chains, which is reflected in our choice of the samples. Our focus is on the behaviour of chloride, and we chose to use KCl in our study due to technical reasons (further discussed in the ESI†). NaCl would be a more natural fit in the context of atmospheric science. Though KCl can be found at high concentrations in certain kinds of atmospheric aerosol, these do not originate from sea spray.30,31
Methods
Sets of X-ray photoelectron spectroscopy (XPS) measurements were carried out at the plane grating monochromator (PGM) U11 beamline of the Brazilian synchrotron light laboratory (LNLS),32 with the SOL3 endstation33 at the beamline U49-1 PGM1 at the synchrotron light source BESSY II,34 Berlin, Germany, and at the beamline PLÉIADES35 of the synchrotron SOLEIL in France, using a liquid microjet set up at all three end stations.The experimental setups at LNLS and at BESSY II are similar in design and have been described in previous publications in more detail.33,36 Both machines utilize a High Pressure Liquid Chromatography (HPLC) pump to push the liquid solution from a reservoir through a quartz glass nozzle with inner diameter between 22–25 μm. Pump flow rates vary between 0.6–1 ml min−1 and a thin, liquid filament with laminar flow is created inside the experimental chamber. The synchrotron radiation perpendicularly intersects and ionizes the sample about 1–2 mm downstream from tip of the nozzle, before the jet breaks up into droplets. The emitted electrons pass through a skimmer, which is mounted ∼2 mm away from the liquid surface, perpendicular to the direction of flow of the liquid jet and the direction of the synchrotron radiation. The kinetic energy of the electrons is measured using a hemispherical electron energy analyzer (Scienta Omicron R-4000 at LNLS, Scienta Omicron R-4000 HIPP-2 at BESSY II). The temperature of the jet is kept at 6 °C before entering the vacuum chamber.
At SOLEIL, the photoelectron spectra were recorded using a wide-angle lens Scienta Omicron R-4000 electron spectrometer mounted in a 90° angle relative to the polarization of the X-rays. The configuration of the microjet source coupled to the spectrometer is very similar to the one described by Céolin et al.37 Instead of a cold trap, a heated catcher of inner diameter about 500 μm is employed to collect the used sample inside the vacuum chamber. The catcher is placed about 6 mm away from the glass nozzle and the liquid is pumped into a container outside the experimental chamber. Here, the sample is delivered into the experimental chamber through a glass nozzle of 40 μm inner diameter using a HPLC pump at a flow rate of 1.4 ml min−1.
The sample solutions are prepared by dissolving commercially purchased potassium chloride (KCl) (Alfa Aesar, purity, >98%) and high purity amino acids (phenylalanine and cysteine from Sigma Aldrich with purity, >98%; valine, glycine, proline and methionine from Alfa Aesar, purity, >98%) in MilliQ (18.2 MΩ cm−1) water. The pH of the sample solutions was adjusted with concentrated HCl such that the bulk solution pH is below the pKa of the carboxyl functional group of the respective amino acid. Note that the deprotonation behaviour of organic acids at the liquid–gas interface can differ from the bulk and depends on the presence of co-solvated cations18,19,38 and the concentration of the organic molecules.39 Measurements were also taken from samples without any further pH adjustments, usually resulting in a pH around 6. The concentration of the amino acids and the pH values for the individual measurements are tabulated in the ESI.†
To determine the influence of amino acids on the surface propensity of ions, a reference sample of aqueous 0.3 M KCl solution was also measured. The surface enrichment of K+ and Cl− in different amino acid solutions are monitored by measuring the signal from 2p levels of potassium (binding energy (BE) ≃ 300.8 eV, 298 eV) and chloride (BE ≃ 204.6 eV, 203 eV) ion along with carbon 1 s signal.
The incident X-ray photon energy is kept constant for each set of measurements in order to avoid changes in photon flux due to movements of the beamline optics. This ensures that the intensities within one data set are comparable. This procedure results in varying electron kinetic energies for electrons originating from different atomic levels. Consequently, the electron mean free path of electrons originating e.g. from a K 2p level and a Cl 2p level differs slightly, however not to a degree that this effect invalidates our data.40 The range of the electron mean free path in our experiments is about 1–2 nm and a table of electron kinetic energies and the resulting estimates of the electron mean free path in liquid water is given in the ESI.† The energy calibration of the spectra is done against the BE of 1b1 (highest occupied molecular orbital) of liquid water (BE = 11.16 eV)41 to account for the generally unknown work function of the sample solution42 and the streaming potential of the jet.43
The analysis of the spectra is carried out using Spectrum Analysis by Curve Fitting (SPANCF)44 macro package for Igor Pro (WaveMetrics, Inc., Lake Oswego, USA). All the peaks are fitted using a Voigt profile where the Lorentzian width was set to 0.1 eV and the Gaussian profile was free to vary but kept same for the spin–orbit components of the peaks. Areas under the photoelectron (PE) peaks were determined from peak fits. The peak areas, except for the water 1b1 level, have further been normalized by molarity, photon flux and photoionization cross section. Data obtained from LNLS have also been normalized to the synchrotron ring current, as LNLS had been operated in decay mode, whereas the BESSY II and SOLEIL ran in top up mode. Data shown in Fig. 1b has further been normalized to the variation of the asymmetry factors (β) of K 2p and Cl 2p at different photon energies using the data base provided by ref. 45 (these data base on ref. 46 and 47).
Error bars shown in the figures have been obtained from the fits to the measured data. A detailed description of how we obtain these, including example figures of the intermediate steps, is provided in the ESI.† Our raw data is available at https://doi.org/10.5281/zenodo.4062156.
Before we delve into the discussion of our results, we have to clarify what we mean by “surface” in the context of our work. This is important since we use XPS in the kinetic energy range below 500 eV, which results in a probing depth of only about 2 nm.40 Commonly, such a probing depth is considered to already probe both the surface and the bulk of a solution. We consider the “surface” to be the volume of the liquid, which is appreciably influenced by the presence of the amino acid layer on top, including possible charge compensation layers.48 Note, that it is in general very difficult to give an exact number on how deep the surface reaches into the liquid, since several effects have an influence, e.g. the composition of a solution or even the surface microstructure.
Results and discussion
To quantify the relative concentration of ions, we use the parameter RK/Cl derived as the ratio of the normalized peak areas of the photoelectron spectra measured for the 2p level of the respective ions. RK/Cl in Fig. 1b shows the relative surface distribution of the K+ and Cl− of all samples as a function of the bulk pH value. The ratio for the reference solution is about ∼0.7 and reflects the slight surface enrichment of chloride ions. We expect this ratio to change due to the presence of organic surfactants.49,50 Solutions containing amino acids deviate from the reference value at acidic pH where the functional groups of amino acids are protonated (–COOH and –NH3+). Here, the RK/Cl is lower than the reference value for all amino acids but GLY and PRO. Around pH 6 the K 2p/Cl 2p peak area ratio is similar to the reference within the error limits for all samples except PRO.R K/Cl is very useful to capture of the overall trend of the interaction between organic surfactants and pH on the surface composition of the ions. In order to ascertain the reasons behind the deviations from the reference, we employ a different parameter: RH2O, which is also derived from the peak area (AK2p/Cl2p/1b1) of the ions, but here we use the 1b1 level of water as reference value i.e. we are concerned with the ratios Cl 2p/1b1 and K 2p/1b1. As we are interested in changes at acidic pH, we further only consider the relative change of these ratios relative to the ratio at pH ∼ 6:
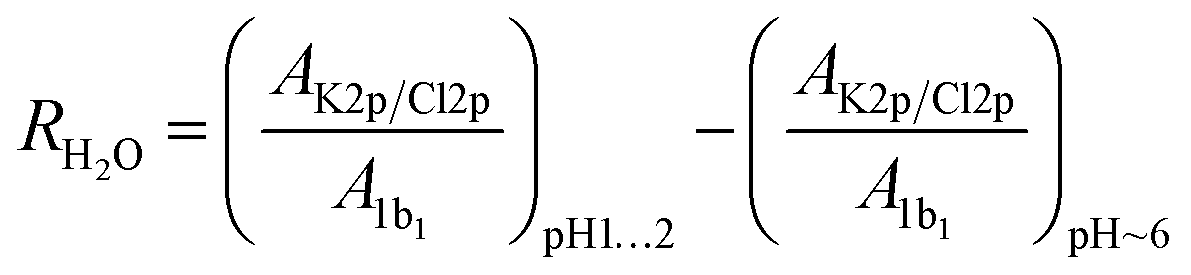 | (1) |
The RH2O can be found in Fig. 2. In essence, a value above 0 indicates that an ion is surface enriched under acidic conditions compared to a solution at pH ∼ 6.
 | ||
| Fig. 2 The RH2O is the ratio between the peak area of Cl 2p (K 2p) levels and the water 1b1 level relative to the same ratio at pH around 6 for each solution. The first column depicts RH2O for the reference solution (0.3 M KCl), and shows values around 0. This indicates that the ion concentration with respect to water is similar at all pH values. The presence of some amino acids changes the concentration of K+ and Cl− at the liquid surface, which is reflected in changes of the RH2O. | ||
We begin our discussion of the results with PHE, which we consider the clearest case. Phenylalanine has a very high surface propensity due to the large hydrophobic side chain and thus PHE is surface enriched independently of the pH. The changing charge of the functional groups of PHE therefore gives rise to the formation of a charged layer at the liquid–gas interface. In acidic conditions, the net charge on PHE is positive due to the protonated amino group, which results in a positively charged surface layer. Hence, Cl− is attracted to the surface, and RK/Cl drops below the reference value. RH2O in Fig. 2 shows that this change is due to an increased surface concentration of Cl−, not due to K+ being repelled from the surface, despite the positively charged surface layer. At pH 5.58 PHE is a zwitterion without net charge, and RK/Cl is about the same as that of the reference solution. Neither of the inorganic ions is particularly attracted to the surface beyond a level observable in the reference solution.
The behaviour of RK/Cl in PHE is shared by MET, VAL and CYS (Fig. 1b), accordingly, we assume that these amino acids are also enriched at the aqueous surface in acidic conditions and structure the K+ and Cl− concentration through the same effects as PHE. Taking a closer look at the RH2O of these four amino acids, though, it becomes apparent that the shared general trend of RK/Cl has different reasons for different amino acids. While PHE predominantly affects Cl−, VAL additionally influences K+. MET and CYS have a much smaller effect on the RH2O than the two other amino acids, but affect the relative surface concentration of both types of ions (RK/Cl), too. RK/Cl of GLY and PRO shows a much different behaviour from the rest. Both of these amino acids are much more soluble than the other samples except CYS. Glycine in particular could be seen as a plausibility check, since it should not be surface enriched.29,52 Neither GLY nor PRO show any profound impact on RK/Cl in acidic solutions, which suggests that none of these two amino acids is surface enriched. Around pH 6, the RK/Cl in PRO solutions exceeds the value of the reference, although by a very small margin. A possible explanation for this behaviour could be found in the protonated NH group in the pyrrolidine ring side chain. The positive excess charge upon protonation can be screened by π electrons in the ring, and as a result the probability of the –NH2+ interacting with Cl− in acidic PRO solutions is lower as the –NH3+⋯Cl− interaction in the other amino acids. What remains is the interaction between the carboxylate group and K+, leading to a small surface excess of K+ at pH ∼ 6. We want to stress, though, that the effect in the our data is so small that it might be an artifact.
Another rather surprising, yet small effect is the RH2O of GLY (Fig. 2). Our data suggest, that both K+ as well as Cl− are reduced in acidic conditions compared to pH 6. Since both ions exhibit the same trend at a similar magnitude, this behaviour is not expressed in RK/Cl. Simulations and experiments have shown that GLY remains mostly in the bulk,29,52 which is corroborated by the RH2O of the carbon signal of glycine (figure inset in 2). Therefore, the simultaneous surface depletion of both inorganic ions in acidic solutions cannot stem from a surface enrichment of GLY. Presently, we can only speculate if the inorganic ions are drawn into the bulk solution by the highly soluble GLY cations or if collective ion–ion interactions could be the cause of the simultaneous depletion of the inorganic ions. Moreover, this effect is also quite small. A dedicated study, combining several experimental techniques with calculations would certainly offer more insight.
Conclusions
The pH-dependent surface propensity of K+ and Cl− in presence of amino acids is mediated by the charge state of the organic molecules if they reside at the surface of the solution. The K+ and Cl− surface propensity in a co-solution of PHE and KCl can be considered as a typical example. Due to the charge state of the amino acid, a positively charged layer forms at the surface of the liquid, which attracts Cl− in acidic conditions. At neutral pH, none of the inorganic ions is surface enriched with respect to a pure KCl solution. Co-solutions of KCl with either methionine, valine and cysteine exhibit the same general behaviour. Co-solutions with glycine and proline apparently have no impact on the K+/Cl− ratio at the surface, most probably because these amino acids are highly soluble and reside predominantly in the bulk, even in their cationic form.It is remarkable, that the effect of the K+-repulsion seems to be much weaker than the attraction of Cl− to cationic amino acids. This might be due to a stronger attractive interaction between the NH3+ and Cl−, but based on our current data, we only have indirect evidence for this. For the interaction of the other functional group of amino acids, COO−, with cations, ion pairing has been found.18,53 We expect the interaction between COO− and cations to be different from the interaction of NH3+ and Cl− due to the different orientation of solvating water molecules in the solvation shells of the ions. This underscores the necessity to improve our understanding of the driving forces behind the surface enrichment of ions, in particular since ion pairing can modify the surface propensity of organic species.19
The potential implications of our findings for aqueous aerosol is the pH-dependence of the surface composition, and hence its potential importance for atmospheric chemistry. In particular reactions involving halide ions at the particle surface will be affected. We have chosen to conduct our study using KCl, where Cl− is a representative for halide ions. It is the most abundant of the three halides common in atmospheric aerosol (I−, Br− and Cl−) but it is also the one that is expected to be the least surface enriched, and therefore it is particularly relevant to focus on how organic ions affect the surface enrichment of Cl−. Recent work by Moreno et al. on the ozone depletion reaction with I− proposed that Cl− might act as a catalyst but their modelling efforts were hampered by a severe lack of knowledge about the surface concentrations of halides in aerosol particles.22,54 This calls for a dedicated study of the competition of halides for the surface of aqueous solutions in the presence of organic species.
Our understanding of other processes sensitive to the surface composition of aerosol particles, like hygroscopic growth, will also benefit from our findings. Connected to hygroscopic growth is the ability of aerosol particles to act as cloud condensation nuclei. Modified surface propensities due to the interaction of ions and organic species can affect the surface composition of solutions. Therefore, the impact on hygroscopicity of the particles may differ from what one would infer from their total chemical composition. Moreover, as the pH in aqueous aerosol particles can change over time, so can the surface propensity of ions and parameters dependent on them. If the functional groups of a majority of surfactants in aerosol have similar pKa values, one might speculate that the pH-dependence we observe introduces pH breaking points for particle properties dependent on the chemical surface composition. If such breaking points really exist in natural aerosol remains questionable until found, given their very complex composition. Motivated by similar considerations, a few other groups have recently begun to investigate the connection between organic molecules, ionorganic ions and pH at aqueous surfaces and our findings here can be considered to be an extension of their results, despite using a different experimental method.18–20,38
Studies such as ours should be expanded to alkaline solutions, divalent cations and anions such as Ca2+ and SO42−, and other organic species. We have used amino acids as a proxy for several kinds of organic species here, since they exhibit two polar moieties with opposite protonation behaviour. This serves the purpose of obtaining a general understanding, but for the direct application for models, similar experiments such as ours with more relevant organics in an atmospheric context would be desirable. Expanding the experiments to basic solutions would likely support findings from other groups conducted with surface tension measurements19,38 and sum frequency generation.15,16,18 The advantage XPS offers in contrast to the other methods is that one obtains signal directly from the inorganic ions and simultaneously gathers information about the chemical surface composition on a nm-scale. A combination of several experimental methods and calculations would certainly prove to be very powerful and greatly advance our knowledge about the physical chemistry of aqueous surfaces.
Though we stress the ramifications of our findings for atmospheric chemistry here, the implications of our results are broader. Any aqueous interface at which organic molecules interact with inorganic ions can potentially exhibit similar pH-driven effects.
Author contributions
The authors contributed as follows to this work; conceptualization: Isaak Unger, Clara-Magdalena Saak, Olle Björneholm, data curation: Geethanjali Gopakumar, Isaak Unger; formal analysis: Geethanjali Gopakumar, Isaak Unger; funding acquisition: Carl Caleman, Olle Björneholm; investigation: Geethanjali Gopakumar, Isaak Unger, Clara-Magdalena Saak, Gunnar Öhrwall, Arnaldo Naves de Brito, Tulio Costa Rizuti da Rocha, Christophe Nicolas; project administration: Geethanjali Gopakumar, Isaak Unger, Clara-Magdalena Saak, Olle Björneholm; supervision: Isaak Unger, Olle Björneholm, Carl Caleman; visualization: Geethanjali Gopakumar, Isaak Unger; writing the original draft: Geethanjali Gopakumar, Isaak Unger; all authors were involved in reviewing and editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the beamtime granted (proposal ID 20180323 & 20181440) at the synchrotron facilities SOLEIL, BESSY and LNLS. I. U. acknowledges the support from the Carl Tryggers Foundation and thanks Stephan Thürmer for his data analysis routines, which have been used for the data analysis. CMS acknowledges funding from the EU Horizon 2020 program under the Marie Skłodowska-Curie grant agreement No. 847693 through the REWIRE program at the University of Vienna. ANB acknowledges support from FAPESP (the Sao Paulo Research Foundation, Process number 2017/11986-5) and Shell and the strategic importance of the support given by ANP (Brazil's National Oil, Natural Gas and Biofuels Agency) through the R&D levy regulation, and National Council for Scientific and Technological Development CNPq 401581/2016-0. We acknowledge support from the Swedish-Brazilian collaboration STINT-CAPES (9805/2014-01). C. C. acknowledges the Swedish Research Council (grant 2018-00740) and the Helmholtz Association through the Center for Free-Electron Laser Science at DESY. O. B. acknowledges the support by the Swedish Research Council (grant 2017-04162). Furthermore, we want to acknowledge the support of Johannes Kirschner, Christina Vantaraki, Rebecka Linnéa Lexelius, Hebatallah Ali, Aaron Ghrist, Florian Trinter, Flávio C. Vicentin and Bernd Winter during the beamtimes.References
- O. Väisänen, A. Ruuskanen, A. Ylisirniö, P. Miettinen, H. Portin, L. Hao, A. Leskinen, M. Komppula, S. Romakkaniemi, K. E. J. Lehtinen and A. Virtanen, In-cloud measurements highlight the role of aerosol hygroscopicity in cloud droplet formation, Atmos. Chem. Phys., 2016, 16, 10385–10398 CrossRef.
- M. W. Christensen and G. L. Stephens, Microphysical and macrophysical responses of marine stratocumulus polluted by underlying ships: Evidence of cloud deepening, J. Geophys. Res., 2011, 116, D03201 CrossRef.
- M. W. Christensen and G. L. Stephens, Microphysical and macrophysical responses of marine stratocumulus polluted by underlying ships: 2. Impacts of haze on precipitating clouds, Atmos. Chem. Phys., 2012, 117, D11203 Search PubMed.
- B. J. Finlayson-Pitts, Reactions at surfaces in the atmosphere: integration of experiments and theory as necessary (but not necessarily sufficient) for predicting the physical chemistry of aerosols, Phys. Chem. Chem. Phys., 2009, 11, 7760 RSC.
- P. Zieger, O. Väisänen, J. C. Corbin, D. G. Partridge, S. Bastelberger, M. Mousavi-Fard, B. Rosati, M. Gysel, U. K. Krieger and C. Leck, et al., Revising the hygroscopicity of inorganic sea salt particles, Nat. Commun., 2017, 8, 15883 CrossRef CAS PubMed.
- A. P. Ault, T. L. Guasco, O. S. Ryder, J. Baltrusaitis, L. A. Cuadra-Rodriguez, D. B. Collins, M. J. Ruppel, T. H. Bertram, K. A. Prather and V. H. Grassian, Inside versus Outside: Ion Redistribution in Nitric Acid Reacted Sea Spray Aerosol Particles as Determined by Single Particle Analysis, J. Am. Chem. Soc., 2013, 135, 14528–14531 CrossRef CAS PubMed.
- J. W. Chi, W. J. Li, D. Z. Zhang, J. C. Zhang, Y. T. Lin, X. J. Shen, J. Y. Sun, J. M. Chen, X. Y. Zhang, Y. M. Zhang and W. X. Wang, Sea salt aerosols as a reactive surface for inorganic and organic acidic gases in the Arctic troposphere, Atmos. Chem. Phys., 2015, 15, 11341–11353 CrossRef CAS.
- A. Fridlind and M. Z. Jacobson, A study of gas-aerosol equilibrium and aerosol pH in the remote marine boundary layer during the First Aerosol Characterization Experiment (ACE 1), J. Geophys. Res., 2000, 105, 17325–17340 CrossRef CAS.
- W. C. Keene, A. A. P. Pszenny, J. R. Maben and R. Sander, Variation of marine aerosol acidity with particle size, Geophys. Res. Lett., 2002, 29, 1101 CrossRef.
- W. C. Keene, A. A. P. Pszenny, J. R. Maben, E. Stevenson and A. Wall, Closure evaluation of size-resolved aerosol pH in the New England coastal atmosphere during summer, J. Geophys. Res., 2004, 109, D23307 CrossRef.
- R. L. Craig, L. Nandy, J. L. Axson, C. S. Dutcher and A. P. Ault, Spectroscopic Determination of Aerosol pH from Acid-Base Equilibria in Inorganic, Organic, and Mixed Systems, J. Phys. Chem. A, 2017, 121, 5690–5699 CrossRef CAS PubMed.
- E. M. Coddens, K. J. Angle and V. H. Grassian, Titration of Aerosol pH through Droplet Coalescence, J. Phys. Chem. Lett., 2019, 10, 4476–4483 CrossRef CAS PubMed.
- H. C. Boyer, K. Gorkowski and R. C. Sullivan, In Situ pH Measurements of Individual Levitated Microdroplets Using Aerosol Optical Tweezers, Anal. Chem., 2020, 92, 1089–1096 CrossRef CAS PubMed.
- H. Wei, E. P. Vejerano, Q. Leng, W. Huang, M. R. Willner, L. C. Marr and P. J. Vikesland, Aerosol microdroplets exhibit a stable pH gradient, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7272–7277 CrossRef CAS PubMed.
- C. Y. Tang, Z. Huang and H. C. Allen, Interfacial Water Structure and Effects of Mg2+ and Ca2+ Binding to the COOH Headgroup of a Palmitic Acid Monolayer Studied by Sum Frequency Spectroscopy, J. Phys. Chem. B, 2011, 115, 34–40 CrossRef CAS PubMed.
- W. Hua, D. Verreault and H. C. Allen, Solvation of Calcium–Phosphate Headgroup Complexes at the DPPC/Aqueous Interface, ChemPhysChem, 2015, 16, 3910–3915 CrossRef CAS PubMed.
- C. B. Casper, D. Verreault, E. M. Adams, W. Hua and H. C. Allen, Surface Potential of DPPC Monolayers on Concentrated Aqueous Salt Solutions, J. Phys. Chem. B, 2016, 120, 2043–2052 CrossRef CAS PubMed.
- E. M. Adams, B. A. Wellen, R. Thiraux, S. K. Reddy, A. S. Vidalis, F. Paesani and H. C. Allen, Sodium–carboxylate contact ion pair formation induces stabilization of palmitic acid monolayers at high pH, Phys. Chem. Chem. Phys., 2017, 19, 10481–10490 RSC.
- M. Luo, N. A. Wauer, K. J. Angle, A. C. Dommer, M. Song, C. M. Nowak, R. E. Amaro and V. H. Grassian, Insights into the behavior of nonanoic acid and its conjugate base at the air/water interface through a combined experimental and theoretical approach, Chem. Sci., 2020, 11, 10647–10656 RSC.
- J. F. Neal, M. M. Rogers, K. A. Smeltzer, A. Morgan, K. A. Carter-Fenk, A. J. Grooms, M. M. Zerkle and H. C. Allen, Sodium Drives Interfacial Equilibria for Semi-Soluble Phosphoric and Phosphonic Acids of Model Sea Spray Aerosol Surfaces, ACS Earth Space Chem., 2020, 4, 1549–1557 CrossRef CAS.
- A. Laskin, H. Wang, W. H. Robertson, J. P. Cowin, M. J. Ezell and B. J. Finlayson-Pitts, A New Approach to Determining Gas-Particle Reaction Probabilities and Application to the Heterogeneous Reaction of Deliquesced Sodium Chloride Particles with Gas-Phase Hydroxyl Radicals, J. Phys. Chem. A, 2006, 110, 10619–10627 CrossRef CAS PubMed.
- C. G. Moreno, O. Gálvez, V. López-Arza Moreno, E. M. Espildora-García and M. T. Baeza-Romero, A revisit of the interaction of gaseous ozone with aqueous iodide. Estimating the contributions of the surface and bulk reactions, Phys. Chem. Chem. Phys., 2018, 20, 27571–27584 RSC.
- S. Chen, L. Artiglia, F. Orlando, J. Edebeli, X. Kong, H. Yang, A. Boucly, P. Corral Arroyo, N. Prisle and M. Ammann, Impact of Tetrabutylammonium on the Oxidation of Bromide by Ozone, ACS Earth Space Chem., 2021, 5, 3008–3021 CrossRef CAS PubMed.
- H. Sobyra, T. B. Pliszka, T. H. Bertram and G. M. Nathanson, Production of Br2 from N2O5 and Br− in Salty and Surfactant-Coated Water Microjets, J. Phys. Chem. A, 2019, 123, 8942–8953 CrossRef PubMed.
- J. Werner, I. Persson, O. Björneholm, D. Kawecki, C.-M. Saak, M.-M. Walz, V. Ekholm, I. Unger, C. Valtl and C. Caleman, et al., Shifted equilibria of organic acids and bases in the aqueous surface region, Phys. Chem. Chem. Phys., 2018, 20, 23281–23293 RSC.
- M. A. Wedyan and M. R. Preston, The coupling of surface seawater organic nitrogen and the marine aerosol as inferred from enantiomer-specific amino acid analysis, Atmos. Environ., 2008, 42, 8698–8705 CrossRef CAS.
- X. Li, T. Hede, Y. Tu, C. Leck and H. Å gren, Cloud droplet activation mechanisms of amino acid aerosol particles: insight from molecular dynamics simulations, Tellus B, 2013, 65, 20476 CrossRef.
- R. Herboth, G. Gopakumar, C. Caleman and M. Wohlert, Charge State Dependence of Amino Acid Propensity at Water Surface: Mechanisms Elucidated by Molecular Dynamics Simulations, J. Phys. Chem. A, 2021, 125, 4705–4714 CrossRef CAS PubMed.
- A. Mocellin, A. Herbert de Abreu Gomes, O. C. Araújo, A. Naves de Brito and O. Björneholm, Surface propensity of atmospherically relevant amino acids studied by XPS, J. Phys. Chem. B, 2017, 121, 4220–4225 CrossRef CAS PubMed.
- M. Pósfai, R. Simonics, J. Li, P. V. Hobbs and P. R. Buseck, Individual aerosol particles from biomass burning in southern Africa: 1. Compositions and size distributions of carbonaceous particles, J. Geophys. Res.: Atmos., 2003, 108, 8483 CrossRef.
- J. Rissler, J. Pagels, E. Swietlicki, A. Wierzbicka, M. Strand, L. Lillieblad, M. Sanati and M. Bohgard, Hygroscopic Behavior of Aerosol Particles Emitted from Biomass Fired Grate Boilers, Aerosol Sci. Technol., 2005, 39, 919–930 CrossRef CAS.
- J. Cezar, P. Fonseca, G. Rodrigues, A. De Castro, R. Neuenschwander, F. Rodrigues, B. Meyer, L. Ribeiro, A. Moreira and J. Piton, et al., The U11 PGM beam line at the brazilian national synchrotron light laboratory, J. Phys.: Conf. Ser., 2013, 072015 CrossRef CAS.
- R. Seidel, M. N. Pohl, H. Ali and E. F. Aziz, Advances in liquid phase soft-X-ray photoemission spectroscopy: a new experimental setup at BESSY II, Rev. Sci. Instrum., 2017, 88, 073107 CrossRef PubMed.
- T. Kachel, The plane grating monochromator beamline U49/2 PGM1 at BESSY II, JLSRF, 2016, 2, A72 CrossRef.
- A. Lindblad, J. Söderström, C. Nicolas, E. Robert and C. Miron, A multi purpose source chamber at the PLEIADES beamline at SOLEIL for spectroscopic studies of isolated species: cold molecules, clusters, and nanoparticles, Rev. Sci. Instrum., 2013, 84, 113105 CrossRef PubMed.
- H. Bergersen, R. Marinho, W. Pokapanich, A. Lindblad, O. Björneholm, L. Sæthre and G. Öhrwall, A photoelectron spectroscopic study of aqueous tetrabutylammonium iodide, J. Phys.: Condens. Matter, 2007, 19, 326101 CrossRef.
- D. Céolin, N. Kryzhevoi, C. Nicolas, W. Pokapanich, S. Choksakulporn, P. Songsiriritthigul, T. Saisopa, Y. Rattanachai, Y. Utsumi and J. Palaudoux, et al., Ultrafast Charge Transfer Processes Accompanying KLL Auger Decay in Aqueous KCl Solution, Phys. Rev. Lett., 2017, 119, 263003 CrossRef PubMed.
- B. A. Wellen, E. A. Lach and H. C. Allen, Surface pKa of octanoic, nonanoic, and decanoic fatty acids at the air–water interface: applications to atmospheric aerosol chemistry, Phys. Chem. Chem. Phys., 2017, 19, 26551–26558 RSC.
- J. R. Kanicky and D. O. Shah, Effect of Premicellar Aggregation on the pKa of Fatty Acid Soap Solutions, Langmuir, 2003, 19, 2034–2038 CrossRef CAS.
- S. Thürmer, R. Seidel, M. Faubel, W. Eberhardt, J. C. Hemminger, S. E. Bradforth and B. Winter, Photoelectron Angular Distributions from Liquid Water: Effects of Electron Scattering, Phys. Rev. Lett., 2013, 111, 173005 CrossRef PubMed.
- B. Winter, R. Weber, W. Widdra, M. Dittmar, M. Faubel and I. Hertel, Full valence band photoemission from liquid water using EUV synchrotron radiation, J. Phys. Chem. A, 2004, 108, 2625–2632 CrossRef CAS.
- G. Olivieri, A. Goel, A. Kleibert, D. Cvetko and M. A. Brown, Quantitative ionization energies and work functions of aqueous solutions, Phys. Chem. Chem. Phys., 2016, 18, 29506–29515 RSC.
- N. Preissler, F. Buchner, T. Schultz and A. Lübcke, Electrokinetic Charging and Evidence for Charge Evaporation in Liquid Microjets of Aqueous Salt Solution, J. Phys. Chem. B, 2013, 117, 2422–2428 CrossRef CAS PubMed.
- E. Kukk, Spectrum Analysis by Curve Fitting (SPANCF)-macro Package for Igor Pro, 2012 Search PubMed.
- Elettra Web Cross Sections, https://vuo.elettra.eu/services/elements/WebElements.html, 2020, accessed: 2020-09-10 Search PubMed.
- J. J. Yeh and I. Lindau, Atomic subshell photoionization cross sections and asymmetry parameters: 1 ≤ Z ≤ 103, At. Data Nucl. Data Tables, 1985, 32, 1–155 CrossRef CAS.
- J. J. Yeh, Atomic Calculation of Photoionization Cross-Sections and Asymmetry Parameters, Gordon and Breach Science Publishers, Langhorne, PE (USA), 1993 Search PubMed.
- P. Jungwirth and D. J. Tobias, Molecular Structure of Salt Solutions: A New View of the Interface with Implications for Heterogeneous Atmospheric Chemistry, J. Phys. Chem. B, 2001, 105, 10468–10472 CrossRef CAS.
- M. J. Krisch, R. Raffaella D'Auria, M. A. Brown, D. J. Tobias, J. C. Hemminger, M. Ammann, D. E. Starr and H. Bluhm, The Effect of an Organic Surfactant on the Liquid-Vapor Interface of an Electrolyte Solution, J. Phys. Chem. C, 2007, 111, 13497–13509 CrossRef CAS.
- X. Xianyuan Zhao, G. M. Nathanson and G. G. Andersson, Competing Segregation of Br− and Cl− to a Surface Coated with a Cationic Surfactant: Direct Measurements of Ion and Solvent Depth Profiles, J. Phys. Chem. A, 2020, 124, 11102–11110 CrossRef PubMed.
- D. R. Lide, CRC Handbook of Chemistry and Physics, Internet Version, CRC Press, Boca Raton, FL, 2005 Search PubMed.
- X. Li, T. Hede, Y. Tu, C. Leck and H. Ågren, Cloud droplet activation mechanisms of amino acid aerosol particles: insight from molecular dynamics simulations, Tellus B, 2013, 65, 20476 CrossRef.
- M.-T. Lee, F. Orlando, M. Khabiri, M. Roeselova, M. A. Brown and M. Ammann, The opposing effect of butanol and butyric acid on the abundance of bromide and iodide at the aqueous solution–air interface, Phys. Chem. Chem. Phys., 2019, 21, 8418 RSC.
- C. Moreno and M. T. Baeza-Romero, A kinetic model for ozone uptake by solutions and aqueous particles containing I− and Br−, including seawater and sea-salt aerosol, Phys. Chem. Chem. Phys., 2019, 21, 19835–19856 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ea00104c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |