
Selective hydrogenation of furfural using a membrane reactor†
Roxanna S.
Delima
 ab,
Mia D.
Stankovic
c,
Benjamin P.
MacLeod
bc,
Arthur G.
Fink
c,
Michael B.
Rooney
c,
Aoxue
Huang
c,
Ryan P.
Jansonius
c,
David J.
Dvorak
b and
Curtis P.
Berlinguette
*abcd
ab,
Mia D.
Stankovic
c,
Benjamin P.
MacLeod
bc,
Arthur G.
Fink
c,
Michael B.
Rooney
c,
Aoxue
Huang
c,
Ryan P.
Jansonius
c,
David J.
Dvorak
b and
Curtis P.
Berlinguette
*abcd
aDepartment of Chemical and Biological Engineering, The University of British Columbia, 2360 East Mall, Vancouver, British Columbia V6Y 1Z3, Canada
bStewart Blusson Quantum Matter Institute, The University of British Columbia, 2355 East Mall, Vancouver, British Columbia V6T 1Z4, Canada
cDepartment of Chemistry, The University of British Columbia, 2036 Main Mall, Vancouver, British Columbia V6T 1Z1, Canada
dCanadian Institute for Advanced Research (CIFAR), MaRS Centre, West Tower, 661 University Avenue, Toronto, Ontario M5G 1M1, Canada. E-mail: cberling@chem.ubc.ca
First published on 3rd November 2021
Abstract
Electrocatalytic palladium membrane reactors (ePMRs) use electricity and water to drive hydrogenation reactions without forming H2 gas. In these reactors, a hydrogen-permeable palladium foil physically separates electrochemical proton generation in aqueous media from chemical hydrogenation in organic media. We report herein the use of the ePMR to electrolytically hydrogenate furfural, an important biomass derivative. This system was proven to convert furfural into furfuryl alcohol and tetrahydrofurfuryl alcohol with 84% and 98% selectivities, respectively. To reach these high selectivities, we designed and built an ePMR for high-throughput testing. Using this apparatus, we tested how different solvents, catalysts, and applied currents impacted furfural hydrogenation. We found that bulky solvents with weak nucleophilicities suppressed the formation of side products. Notably, these types of solvents are not compatible with standard electrochemical hydrogenation architectures where electrolysis and hydrogenation occur in the same reaction chamber. This work highlights the utility of the ePMR for selective furfural hydrogenation without H2 gas, and presents a possible pathway for helping to decarbonize the hydrogenation industry.
Broader contextHydrogenation reactions are responsible for ∼4% of global CO2 emissions. This large carbon footprint arises because industrial hydrogenation reactors use H2 gas derived from methane and require harsh reaction conditions. These challenges motivated us to study alternative electrochemical methods that source hydrogen from water and renewable electricity at ambient conditions, and without H2 gas. We identified the electrocatalytic palladium membrane reactor (ePMR) as a technology solution. Unlike standard electrochemical methods, the ePMR physically separates electrochemical hydrogen generation from chemical hydrogenation. In this study, we used the ePMR to hydrogenate furfural, a biomass-derived chemical that is produced on a large scale (350![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons per year). We show herein that the ePMR enables selective furfural hydrogenation to furfuryl alcohol (FA) and tetrahydrofurfuryl alcohol (THFA), which together make up >65% of the furfural market. The solvent in the reactor governs product selectivities. The best solvent we tested was tert-butanol, a solvent that is not compatible with conventional electrochemical systems. This work demonstrates how membrane reactors could decarbonize the hydrogenation industry that is responsible for emitting 1.4 Gt CO2/year. 000 tons per year). We show herein that the ePMR enables selective furfural hydrogenation to furfuryl alcohol (FA) and tetrahydrofurfuryl alcohol (THFA), which together make up >65% of the furfural market. The solvent in the reactor governs product selectivities. The best solvent we tested was tert-butanol, a solvent that is not compatible with conventional electrochemical systems. This work demonstrates how membrane reactors could decarbonize the hydrogenation industry that is responsible for emitting 1.4 Gt CO2/year.
|
Introduction
The conversion of biomass feedstocks into fuels and chemicals provides a route to decarbonize chemical manufacturing.1,2 Furfural is an appealing chemical feedstock to target because it can be derived from low-value agricultural biomass,3 and upgraded to value-added chemicals through the process of hydrogenation.4,5 Hydrogenation of furfural can produce a variety of products, including furfuryl alcohol (FA), tetrahydrofurfuryl alcohol (THFA), and other furan-containing derivatives (Fig. 1a). FA accounts for >65% of the ∼$800 M furfural market,6 and is widely used in foundry binders, lubricating oils, fragrances, and as a chemical intermediate for pharmaceuticals.7 THFA is a green solvent used in paint-stripping agents, industrial cleaners, epoxy resins, herbicides, dyes, and inks.7 The high miscibility and biodegradability of THFA make it a promising candidate to displace conventional polar solvents (e.g., alcohols) for a wide range of applications. These furfural derivatives are also important building blocks for the emerging bioplastics market.8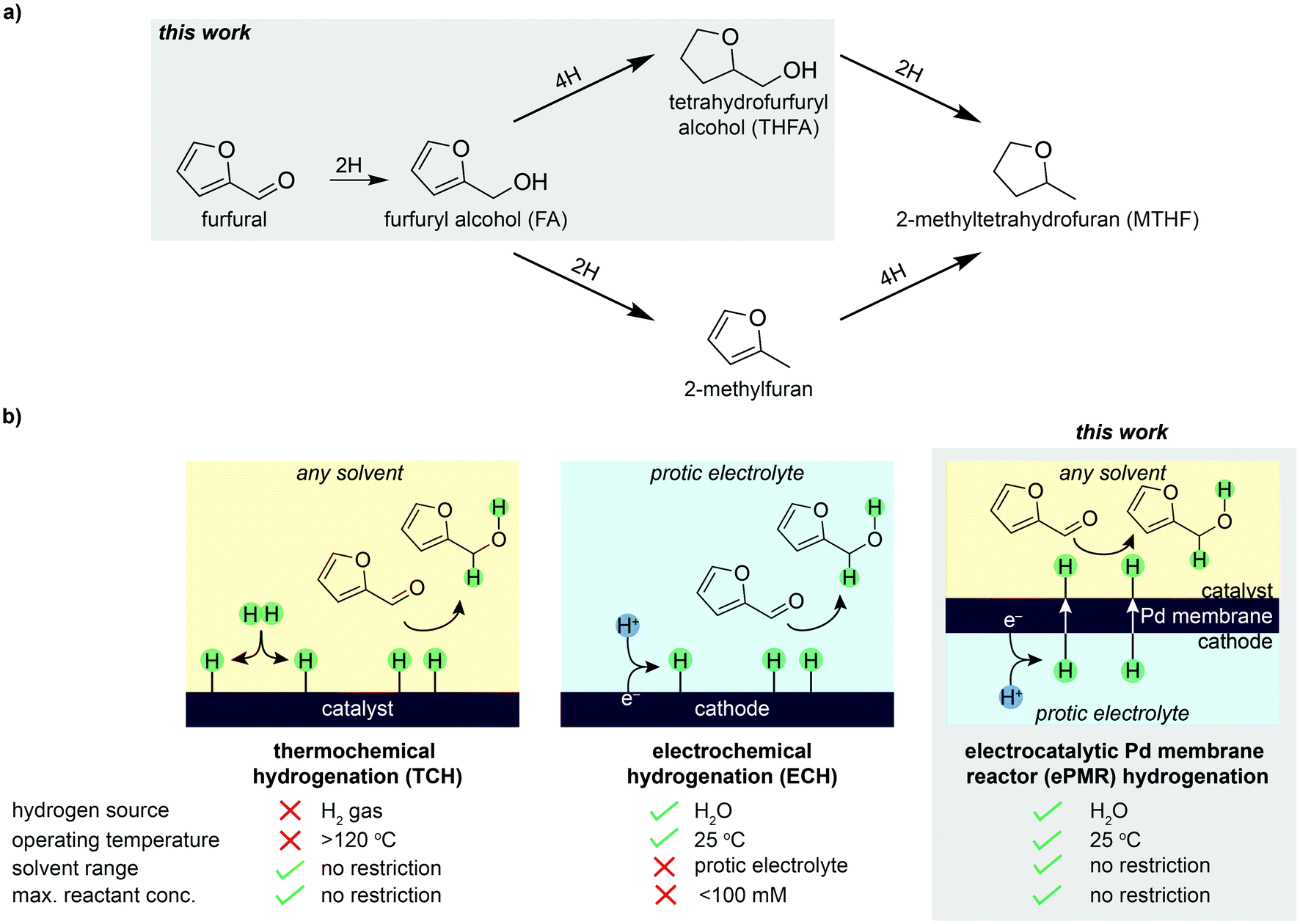 | ||
| Fig. 1 Furfural hydrogenation pathways and methods. (a) Reaction pathways of furfural hydrogenation. (b) Comparison of thermochemical hydrogenation (TCH), electrochemical hydrogenation (ECH), and electrocatalytic Pd membrane reactor (ePMR) hydrogenation. TCH is performed with H2 gas that is dissociated to surface-adsorbed hydrogen atoms (H) that react with furfural to form hydrogenated products. ECH is performed with protons (H+) derived from a protic electrolyte that are reduced to H and react with furfural to form hydrogenated products. Hydrogenation in an ePMR is performed with H+ derived from a protic electrolyte that are reduced to H, permeate the Pd membrane, and resurface on the opposing side to react with furfural to form hydrogenated products. Note: reaction pathways in (a) and main products differ for the three methods. | ||
Furfural provides the opportunity to make industrial chemicals renewably, yet the current method for producing furfural derivatives, thermochemical hydrogenation (TCH), requires high temperatures and H2 gas derived from fossil fuels (Fig. 1b).7 In addition to the high carbon intensity, H2 gas is difficult to handle and store, particularly in the remote locations where biomass is plentiful and inexpensive.9
The electrochemical hydrogenation (ECH) of furfural is an alternative method for hydrogenating furfural. In this case, hydrogen is derived from protic electrolyte rather than H2 gas (Fig. 1b).10–17 In ECH, protons are produced electrolytically at an anode and migrate to a cathode where they are reduced to surface-adsorbed hydrogen atoms (denoted herein as “H atoms”). These H atoms then react with furfural to form hydrogenated products. Electrochemical hydrogenation is appealing because it uses water and electricity to form reactive hydrogen at low temperatures and pressures to hydrogenate furfural. However, there are several fundamental challenges that need to be addressed before ECH can be commercialized. For example, ECH reactions require a protic electrolyte to enable the electrochemical production of H atoms and subsequent hydrogenation of furfural to occur simultaneously in the same reaction chamber (Fig. 1b). This requirement limits the scope of solvents that can be used for the reaction, and leads to additional challenges such as product separation from solution14,18 and byproduct formation (e.g., furfural products reacting with electrolyte to form polymerized byproducts).13,19 Finally, the low furfural concentrations (≤100 mM) that are recommended to prevent oligomer formation and electrode fouling are not conducive to commercial reactors.19
These challenges prompted us to explore alternative ways to utilize electrochemistry to source hydrogen. This search converged on the use of the electrocatalytic palladium membrane reactor (ePMR), which uses a membrane to physically separate the electrochemical hydrogen production from the hydrogenation reaction (Fig. 1b).20–27 The defining feature of an ePMR is that a palladium foil acts as: (i) a cathode for reducing protons (produced from water electrolysis) to form reactive H atoms; (ii) a hydrogen-selective membrane to transport H atoms to an isolated reaction vessel; and (iii) a catalyst for hydrogenation.20–22 In this architecture, protons produced at a platinum anode migrate to the palladium cathode, where they are reduced to surface-adsorbed H atoms. H atoms subsequently absorb into interstitial octahedral sites of the palladium face-centered cubic (fcc) lattice,28 permeate across the membrane, and resurface on the opposite face of the membrane where they react with unsaturated bonds of organic molecules to form hydrogenated products. The palladium membrane therefore acts as a physical barrier to separate the electrochemical production of H atoms from the hydrogenation reaction (Fig. 1b). Consequently, hydrogenation can be performed in any solvent (including protic and organic solvents) and at any concentration of furfural (i.e., not constrained to ≤100 mM).
We report here the first demonstration of hydrogenation of furfural using an ePMR. These experiments were performed successfully with furfural dissolved in n-BuOH in the hydrogenation compartment and 1 M H2SO4 in the electrochemical compartment. While this result was important, the reaction was not selective for a single hydrogenation product. This issue is common to furfural hydrogenation chemistries where the large number of products (>10) that can form are sensitive to reaction conditions (e.g., temperature, pressure, catalyst, H2 flow rate, solvent, etc.).5 The challenge we faced is that the extensive literature linking reaction conditions to furfural selectivity does not translate effectively to the ePMR: the ePMR sources monoatomic hydrogen from water, and hydrogenation occurs through a non-faradaic process that is fundamentally different from both TCH (which involves the dissociation of H2 gas at the catalyst surface) and ECH (which involves faradaic processes). We needed to empirically build a new database for reaction chemistry in the ePMR.
We therefore designed and built a customized high throughput ePMR platform to rapidly screen furfural hydrogenation reaction chemistries. This platform, which we call “MultiThor”, enabled a ∼60-fold faster testing of furfural hydrogenation conditions (e.g., H atom supply at the surface, solvent, and catalyst). These accelerated experiments revealed to us that, under the reaction conditions tested, the reaction chemistry in the ePMR occurs in two successive steps: (i) the carbonyl bond is first hydrogenated to form FA (rate-limiting step); and (ii) the furan ring is subsequently hydrogenated to form THFA. We also found that THFA could be further hydrodeoxygenated to form 2-methyltetrahydrofuran (MTHF). The outcome of these experiments is the identification of reaction conditions necessary to convert furfural to FA and THFA with selectivities of 84% and 98%, respectively. Although the production of FA and THFA generally requires an entirely different set of conditions for each product,7 we showed that the ePMR can selectively produce both without changing the system. This study highlights the opportunity for using the ePMR to selectively hydrogenate furfural without carbon-intensive H2 gas.
Experimental
Materials
Wafer bars (1 oz) of Pd (99.95%) were obtained from Silver Gold Bull. PdCl2 (99.9%) was purchased from Strem Chemicals. DCM (≥99.8%), H2SO4, HNO3, H2O2 solution (30 wt% in H2O), MeOH (≥99.8%), EtOH (95%), n-BuOH (99.4%), i-PrOH (99.5%), t-BuOH (≥99.7%), 2-methylfuran (99%), and 2-methyltetrahydrofuran (≥99%) were purchased from Sigma-Aldrich. Furfural (98%), furfuryl alcohol (98%), Pt gauze (52 mesh, 99.9%), and Pt wire (0.5 mm, 99.95%) were obtained from Alfa Aesar. Ag/AgCl reference electrodes (RE5B) were purchased from BASi. Viton gasket material (⅛′′ thick), quick turn polycarbonate plastic couplings (¼-28′′ thread), M3 and M4 socket head 18–8 stainless steel bolts, 304 stainless sheet metal (⅜′′ thick), acrylic sheet (⅜′′ thick), and M5 dowel pins were purchased from McMaster Carr. Kapton (500 HN) substrates and tape were purchased from American Durafilm. Copper tape (¼′′ thick) manufactured by 3M was purchased from Digikey. A low-flow chemical metering pump (part no. 4049K55) was purchased from McMaster-Carr.MultiThor design
The MultiThor design consisted of: (i) an 8 mL electrochemical compartment; (ii) six 2 mL isolated hydrogenation “wells”; and (iii) a 30 μm thick Pd foil membrane that separated the hydrogenation wells from the electrochemical compartment (Fig. 2 and Fig. S1, S2, ESI†). Inert fluorosilicone gaskets were used to seal the interfaces between compartments. The electrochemical compartment contained a 1 cm2 Pt mesh anode that acted as the counter electrode against the Pd foil cathode/working electrode. The Pd foil membrane had an exposed geometric surface area of 4 cm2 at the electrochemical compartment interface and 0.43 cm2 at each hydrogenation well interface. A high surface area Pd catalyst was electrodeposited on the Pd foil membrane (Pd/Pd membrane) facing the hydrogenation side of the reactor with electrochemical surface area measurements showing a ∼218-fold increase in catalytic surface area (Fig. S3, ESI†).22 An additional 10 to 50 nm layer of metal catalyst (Ni, Cu, Ag, Ir, Pt, or Au) was sputter-deposited on top of the Pd/Pd membrane based on previously reported procedures to enhance reactivity for carbonyl functionalities (Fig. S4, ESI†).29 | ||
| Fig. 2 MultiThor architecture. (a) A rendering of the MultiThor design. The six hydrogenation wells and an electrochemical compartment are separated by a Pd foil cathode/membrane with electrodeposited Pd catalyst. A Pt anode is used as a counter electrode in the electrochemical compartment, Cu tape is attached to the Pd foil to provide electrical contact, and fluorosilicone gaskets are used to seal intercompartmental interfaces. (b) Illustration of hydrogen pathway through MultiThor. An applied current across the Pd cathode and Pt anode results in water oxidation to form protons (H+). These H+ are reduced to surface-adsorbed hydrogen atoms (H) at the Pd surface, H permeate through the Pd membrane, and resurface in the isolated hydrogenation wells where they are poised to react on the high surface area Pd catalyst. (c) External MultiThor setup showing pump, electrochemical reservoir, and electrode leads that are connected to a potentiostat. | ||
Electrochemistry
All experiments were performed with a Metrohm Autolab PGSTAT302N potentiostat coupled with MultiThor, with the exception of the initial proof-of-concept reaction where an H-cell was used (ESI† Experimental methods). Experiments were chronopotentiometric, where a reductive current ranging from 75 to 300 mA (18.75 to 75 mA cm−2) was applied across the Pd foil cathode and Pt anode, and the cell potential was measured. For all experiments performed with MultiThor, the electrochemical compartment was filled with 25 mL of 1 M H2SO4 and operated under flow conditions (250 mL min−1) with inlet tubing directing electrolyte to the Pd membrane to remove H2 bubbles formed during electrolysis (Fig. S2, ESI†). A small hole (2 mm diameter) over each hydrogenation well enabled excess H2 bubbles to escape and provided access for aliquots to be taken periodically during the reaction. Upon completion of each experiment, an oxidative potential (chronoamperometric, +0.2 V) was applied across the cell for 30 min to draw out any H atoms that remained in the Pd lattice.Pd membrane preparation
Pd foils were rolled from a 1 oz Pd wafer bar using an MTI MR-100A electric rolling mill. The bar was rolled to achieve a thickness of 30 μm determined by a Mitutoyo digital micrometer. The resulting Pd foil was cut into ∼3 × 3 cm2 squares and annealed at 850 °C for 1.5 h under N2 gas. The foils were then cleaned using 1:2:1 HNO3:H2O:H2O2 v/v ratio solution until vigorous bubbling subsided (∼40 min), rinsed with acetone and Milli-Q water, and dried with N2 gas.
Pd catalyst was electrodeposited on Pd foils using a one-compartment electrochemical cell. A Pd foil was clamped into the cell with an exposed geometric surface area of 2 × 2 cm2. The Pd foil served as a working electrode in reference to a Ag/AgCl reference electrode and Pt mesh counter electrode. The compartment was filled with 15 mL of 15.9 mM PdCl2 in 1 M HCl electrolyte. A voltage of −0.2 V vs. Ag/AgCl was applied to the Pd foil working electrode to reduce Pd ions in solution. The electrodeposition ended when a charge of 30 C had been passed, corresponding to a catalyst loading of approximately 7.5 C cm−2, similar to a previously reported procedure.22 This Pd layer increased the catalytic surface area of the hydrogenation side of the Pd membrane by ∼218-fold (Fig. S3, ESI†).
An additional layer of catalyst was deposited on the Pd/Pd membranes by sputter-deposition (M/Pd/Pd membranes). A Kapton mask with 6 cut-outs (0.5 cm2 each) that aligned with the 6 wells of MultiThor was used to enable customized deposition of catalysts in each well. A Leica EM MED020 coating system was used to sputter-deposit Pt, Ir, Cu, Ag, and Au and a Univex 250 RF magnetron sputtering system with an Onyx-2 IC Mag II cathode was used to deposit Ni. More details on sputter-deposition conditions can be found in ESI† Experimental methods. The catalyst coated Pd/Pd membranes containing up to 6 sputter-deposited catalysts were used for hydrogen permeation and hydrogenation experiments without any further processing.
Hydrogen permeation
Hydrogen permeation experiments were conducted in MultiThor with 1 M H2SO4 flowing through the electrochemical compartment and solvent without any furfural in all hydrogenation wells. All compartments were closed to air. The potentiostat was used to apply a constant applied current (75 to 300 mA). The production of gaseous H2 (2 m/z current ratio over time; 10 mL min−1 flow rate) in the hydrogenation wells and electrochemical compartment was monitored by an ESS CatalySys atmospheric–mass spectrometer (atm–MS). H2 evolution measurements were taken by alternating between one hydrogenation well and the electrochemical compartment every 2 s with a purge time of 5 s between measurements for 30 min for each well. Current was applied for 60 min prior to each set of measurements and the ion current value was taken once the signal had equilibrated. The equilibrated ion current was used to determine the ratio of H2 evolution in the hydrogenation:electrochemical compartments.
Product quantification
Gas chromatography–mass spectrometry (GC–MS) was used to quantify products for the hydrogenation of furfural. GC–MS measurements were conducted on an Agilent GC–MS using a HP–5 ms column and electron ionization. Aliquots of 30 μL diluted in 1 mL DCM were taken at time intervals for up to 24 h of reaction. The prepared samples were run on an autosampler with a 1 μL injection volume and a split ratio of 20:1. The oven temperature began at 40 °C for 1 min and ramped to 80 °C at 10 °C min−1 then to 200 °C at 25 °C min−1. A solvent delay of 2 min was employed. Furfural, FA, THFA, 2-methylfuran, and 2-methyltetrahydrofuran peaks were identified by searching the National Institute of Standards and Technology (NIST) database for matching mass spectra and confirmed with standards of each of the compounds.
Results
Proof-of-principle reactions
For the first stage of this study, we set out to demonstrate that furfural could be hydrogenated in the ePMR. This proof-of-concept reaction was performed in an H-cell architecture (Fig. S5a; see ESI† for details).22–24,26,27 The hydrogenation compartment was filled with 30 mL of 0.25 M furfural dissolved in n-BuOH, the electrochemical compartment with 30 mL of 1 M H2SO4, and a 45 mA current (37 mA cm−2) was applied to drive the reaction. Reaction progress monitored by GC–MS after 24 h of reaction showed that furfural was hydrogenated to form FA and THFA, as well as >6 other side products with higher boiling points (Fig. S5b, ESI†). Although this proof-of-concept study demonstrated furfural hydrogenation in the ePMR, the large number of products formed and the slow reaction rates (6.3 μmol h−1) highlighted the shortcomings of the H-cell architecture for studying furfural hydrogenation.To address these challenges, we constructed MultiThor (Fig. 2) in order to enable 6 reactions to be performed in parallel and at ∼10-fold faster furfural hydrogenation rates than the H-cell (64.8 μmol h−1 compared to 6.3 μmol h−1, Table S1, ESI†). This faster hydrogenation rate was made possible by using a lower-volume hydrogenation compartment with a 10-fold higher catalyst surface area exposed to the furfural solution (Table S1, ESI†). MultiThor was also designed such that a bulk water electrolysis chamber (i.e., electrochemical compartment) provided H atoms to all six isolated wells (Fig. 2b). Control experiments showed that hydrogen permeation and hydrogenation were evenly distributed across the six wells at the reaction conditions tested (Fig. S6 and S7; see ESI† for details). These results confirmed that MultiThor could be used to study furfural hydrogenation at a ∼60-fold faster rate of testing.
Effect of solvent on selectivity
We used MultiThor to study the effect of different solvents on furfural selectivity and hydrogenation rate. For each experiment, furfural was dissolved into 1 mL of solvent (CHCl3, t-BuOH, n-BuOH, i-PrOH, EtOH, MeOH; 0.25 M) and a current of 150 mA was applied for 2 h (Fig. 3a). The production of FA, THFA, and any side products that were formed during reaction (labelled “other”) for each solvent are plotted in Fig. 3b. These results showed that solvents with lower nucleophilicity generally produced fewer side products (<15%), while more nucleophilic solvents (i.e., EtOH and MeOH) resulted in the formation of >40% side products (particularly 2-furaldehyde diethyl acetal and 2-furaldehyde dimethyl acetal, respectively; Fig. S8 and S9, ESI†). This finding is consistent with previous studies showing the formation of 74% acetal products during furfural hydrogenation, which was ascribed to EtOH and MeOH nucleophilic attack of the solvent on furfural.30 For reactions where furfural was not consumed by the solvent, furfural consumption rates decreased as follows: n-BuOH ∼ i-PrOH > t-BuOH > CHCl3 (Fig. 3c). This trend is similar to those found in literature that correlate hydrogenation rate to H-donating ability of the alcohol solvent.31 For all experiments hereafter, t-BuOH was used as the solvent because of the effectiveness of side product suppression (Fig. 3b and Fig. S8, ESI†).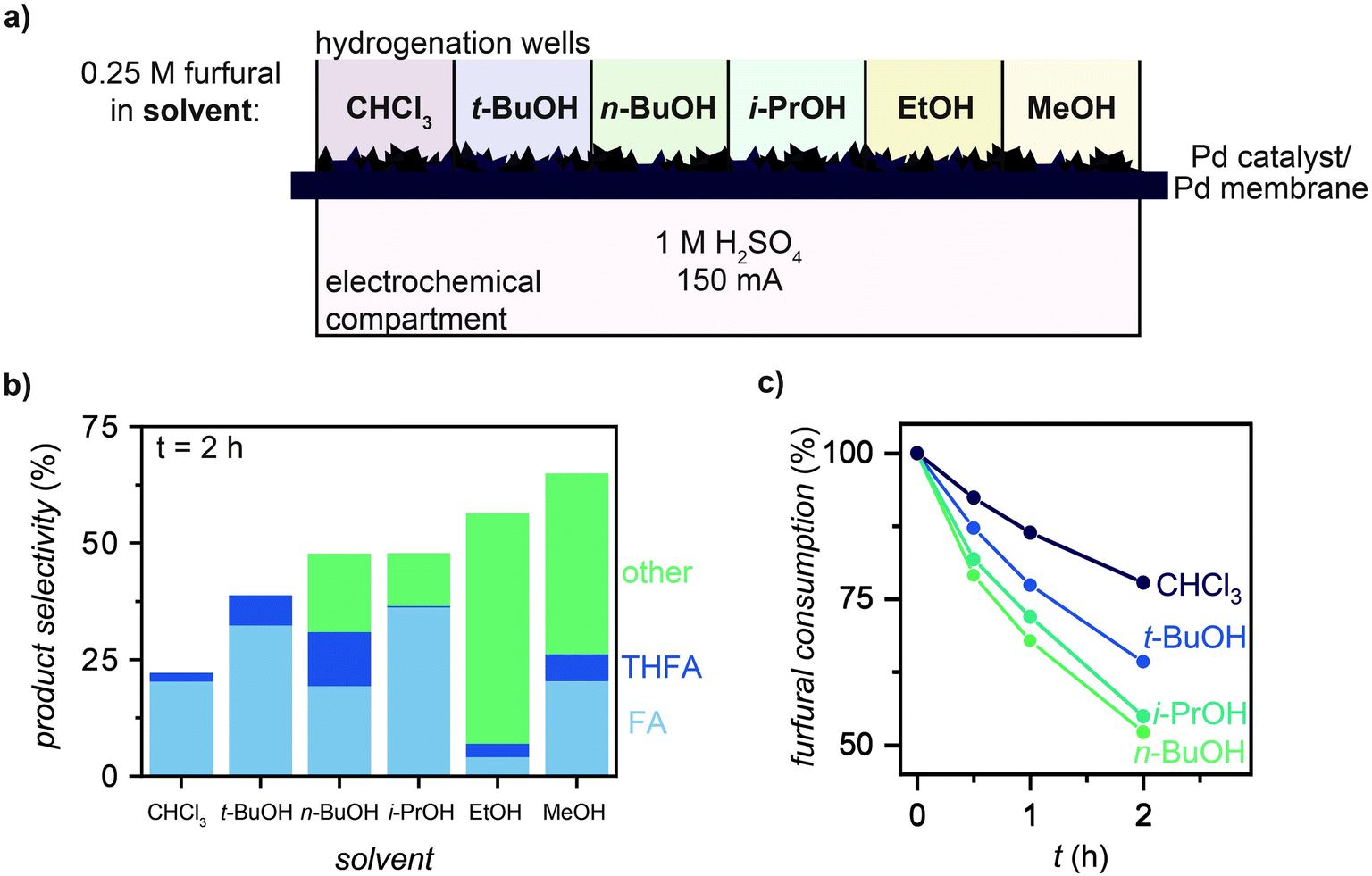 | ||
| Fig. 3 Effect of solvent on furfural hydrogenation selectivity. (a) MultiThor setup for solvent measurements. (b) Product selectivity of furfural hydrogenation to furfuryl alcohol (FA), tetrahydrofurfuryl alcohol (THFA), and any other products formed during reaction (other) in CHCl3, t-BuOH, n-BuOH, i-PrOH, EtOH, and MeOH after 2 h of reaction at 150 mA. (c) Furfural consumption during hydrogenation in CHCl3, t-BuOH, i-PrOH, and n-BuOH. Note: MeOH and EtOH are not shown in (c) due to solvent attack on furfural to form 2-furaldehyde diethyl acetal and 2-furaldehyde dimethyl acetal (Fig. S9, ESI†). | ||
Effect of catalyst and current on selectivity
The selective hydrogenation of furfural in t-BuOH was tested for six different catalysts to examine the effect of metal identity on FA and THFA selectivity. For each experiment, a Pd/Pd membrane was prepared with six sputter-deposited catalysts (10 nm; Ni, Cu, Ag, Ir, Pt, and Au) using a masking technique such that one catalyst was deposited in each well (see Experimental for details). The M/Pd/Pd membrane was secured in MultiThor, 1 mL of 0.25 M furfural in t-BuOH was placed into each well, and a current of 75, 150, or 225 mA was applied across the electrochemical compartment (Fig. 4a). Product selectivity measured after 8 h of reaction showed that FA selectivity increased with decreasing current (i.e., highest selectivity of 67% was achieved with Pt at 75 mA), while THFA selectivity increased with increasing current (i.e., highest selectivity of 98% achieved with Pt at 225 mA).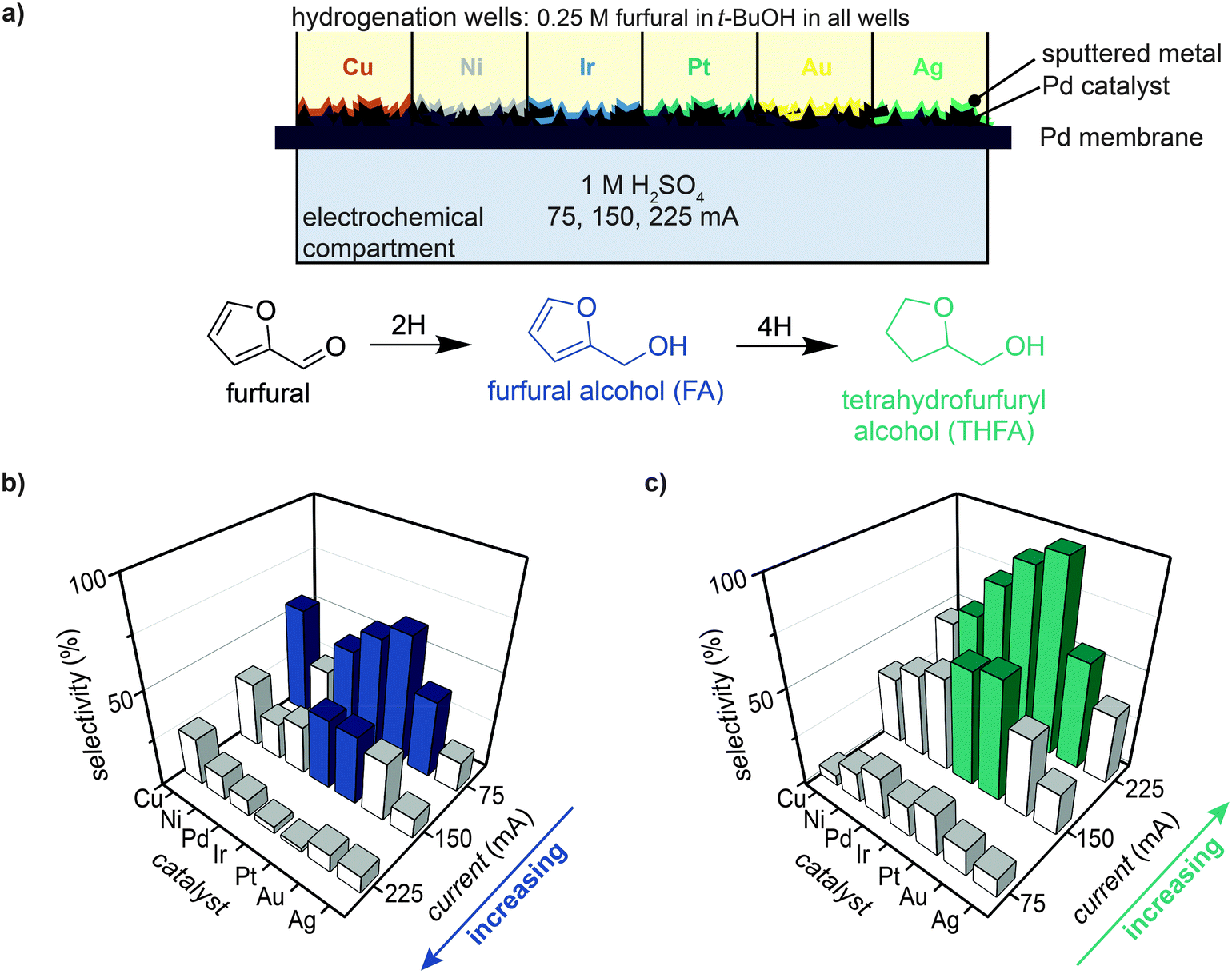 | ||
| Fig. 4 Effect of catalyst identity and applied current on furfural selectivity. (a) MultiThor architecture for testing how different sputtered metals affect furfural hydrogenation. Selectivity of (b) furfuryl alcohol (FA) and (c) tetrahydrofurfuryl alcohol (THFA) for 75, 150, and 225 mA applied current with M/Pd/Pd membrane (10 nm; M = Cu, Ni, Pd, Ir, Pt, Au, and Ag). Coloured bars represent selectivity of >50% and white bars represent selectivity of <50%. Samples were taken after 8 h of applied current. | ||
The trend for highest to lowest selectivity for FA production at 75 mA was Pt > Ir ∼ Cu > Pd ∼ Au > Ni > Ag, while for THFA at 225 mA the trend was Pt ∼ Ir > Pd > Au ∼ Ni > Cu > Ag (Fig. 4b, c and Fig. S10–S12, ESI†). These data highlight that reactivity can be enhanced by the addition of a catalyst layer in an ePMR, and show that reactivity for FA and THFA generally follow a similar catalyst trend (e.g., Pt and Ir enabled the highest reactivity and Ag the lowest). Importantly, a selectivity of 98% THFA was achieved with a 10 nm Pt/Pd/Pd membrane, which demonstrates the ability to perform selective furfural hydrogenation in the ePMR.
The effect of applied current was further investigated by comparing FA and THFA production at equivalent charge passed (Fig. S13, ESI†). A current of 75 mA applied for 8 h is equivalent (in terms of charge passed) to a current of 225 mA applied for 2.7 h. The observed FA selectivity at these two applied currents varied by at least 2- to 6-fold depending on the catalyst tested (Fig. S13a, ESI†). These results show that applied current (and not just charge passed) influences reaction selectivity. A second experiment performed for a longer amount of time at each applied current (e.g., 75 mA for 24 h and 225 mA for 8 h) showed that THFA selectivity varied by merely ∼10% at the two applied currents because almost all furfural and FA are consumed over this time period with our apparatus (Fig. S13b, ESI†). It is worth noting that ∼14% MTHF was observed after 24 h of reaction at 225 mA (Fig. S10d, ESI†), indicating that THFA can be further hydrodeoxygenated to form MTHF.32
Effect of catalyst thickness selectivity
We then tested the effect of catalyst thickness on furfural selectivity (Fig. 5 and Fig. S14–S16, ESI†). We chose different thicknesses of Pt and Ir because these catalysts enabled the highest selectivity towards FA in our initial catalyst experiments (Fig. 4b). A 10 to 50 nm thick layer of Pt and Ir was sputter-deposited on the Pd/Pd membrane (using the same masking technique as before), t-BuOH was used as the solvent in all hydrogenation wells, and a current of 75 mA was applied to drive the hydrogenation reaction. Furfural consumption rates were calculated using the slopes of the initial 2 h of the reactions (μmol h−1; Fig. S14, ESI†). For both the Pt and Ir catalysts, thicknesses of 20 nm enabled a >30% increase in furfural consumption rates (Fig. 5a). Moreover, samples recorded after 8 h of reaction showed a 17% increase in FA selectivity when the catalyst thicknesses were increased from 10 to 50 nm (Fig. 5b). Notably, SEM images and ECSA measurements for all thicknesses demonstrate that there are negligible changes in catalytic surface area, suggesting that the rough morphology of the underlying Pd is retained during sputter-deposition (Fig. 5c and Fig. S3, ESI†). FA selectivity was highest for Pt/Pd/Pd membranes, which showed a selectivity of 84% at 50 nm. These results highlight how reaction selectivity can be adjusted by catalyst thickness. | ||
| Fig. 5 Effects of catalyst thickness on furfural selectivity. (a) Furfural hydrogenation rate calculated for the first 2 h of reaction for Pt and Ir with different sputtered catalyst thicknesses. (b) Furfural to furfuryl alcohol (FA) selectivity for 10 to 50 nm of Pt and Ir sputter-deposited on a Pd/Pd membrane measured after 8 h of reaction. (c) SEM images of 10, 20, and 50 nm of Ir on a Pd/Pd membrane. (d) Relative rates of consumption of starting material for experiments that start with furfural or furfuryl alcohol. | ||
We also performed a control experiment using 0.25 M FA in t-BuOH as the starting material instead of 0.25 M furfural to define the reaction mechanisms that govern furfural hydrogenation in the ePMR. We observed a 4-fold faster reaction rate when FA was used as the starting material instead of furfural (Fig. 5d). This result points to furfural hydrogenation being the rate-determining step (RDS), and the subsequent hydrogenation of FA to THFA being a relatively fast step (eqn (1)).
 | (1) |
Discussion
Furfural hydrogenation to FA and THFA is typically performed using a series of reactors to enable: (i) H2 production; (ii) H2 purification; (iii) hydrogenation of FA; and (iv) hydrogenation of THFA.7 These reactors require expensive infrastructure to ensure safe handling of H2 gas.33 The H2 gas is first produced by steam-methane reforming (SMR), a process that requires large amounts of heat and fossil fuels to reduce water and methane into H2 gas and CO.34 The H2 gas is then purified to 99.999% to ensure that the H2 feed is not contaminated with CO which is known to poison many hydrogenation catalysts.35 Large amounts of purified H2 gas are then transported into a reactor containing furfural to produce FA at ∼120 °C in the presence of Cu-based catalysts.7 This CO2-intensive process produces FA at high yields of >95% in a solvent-free environment. FA is further hydrogenated into THFA using a separate reactor vessel in the presence of H2 gas and Ni-based catalysts at ∼100 °C.7 These reactions are well-established; however, the high infrastructure costs and large carbon footprint associated with conventional TCH,36 as well as the expanding furfural market6 have resulted in a growing interest in furfural hydrogenation using renewable methods.Electrochemical hydrogenation achieves this goal by sourcing hydrogen from renewable electricity and water (or protic media), but there are limitations to performing electrolysis and hydrogenation in the same environment (Fig. 1b). Consider, for example, that the two most common products of ECH of furfural (furfuryl alcohol and 2-methylfuran) undergo side reactions in acidic environments that can result in product polymerization.13 Moreover, 2-methylfuran, which has a low boiling point (64 °C) and high hydrophobicity relative to the surrounding protic solution, can evaporate during ECH of furfural.13 This evaporation can be suppressed with cosolvents, but these cosolvents increase the cell potential and can also trap furfural to reduce the amount of available reactant.18 Low furfural concentrations (≤100 mM) are also required to prevent oligomer formation and electrode fouling,19 a constraint that does not exist for ePMR hydrogenation. Moreover, an ePMR can enable operation at significantly lower voltages (∼1 V) than ECH.24
These factors led us to hypothesize that the ePMR could be used to hydrogenate furfural using electricity and water without requiring H2 gas. We first tested this hypothesis by performing electrochemical furfural hydrogenation in a proof-of-concept ePMR “H-cell” architecture (Fig. S5a, ESI†). We validated our hypothesis by observing the successful conversion of furfural to FA and THFA during these experiments, which proceeded without an external H2 gas supply (Fig. S5b, ESI†). Notably, the reaction was performed in n-BuOH, a solvent that is not compatible with ECH.
These positive results notwithstanding, reactivity was slow (6.3 μmol h−1) and there were >6 side products formed (Fig. S5b, ESI†). A powerful feature of the ePMR is that multiple experimental parameters can be varied to optimize for reaction rate and selectivity (e.g., solvents, current densities, electrolytes, co-additives, catalysts). In order to accelerate the testing of myriad experimental variables, we designed and constructed a high throughput testing platform that we named “MultiThor”. This platform enabled a 10-fold faster hydrogenation rate (64.8 μmol h−1) and a ∼60-fold higher throughput of testing reaction conditions than the H-cell (Table S1, ESI†). For this study, we tested furfural hydrogenation using 6 different solvents and 7 catalyst formulations (two of which were selected for testing at 5 different thicknesses) at 3 different applied electrical currents.
We observed the highest reaction rates and selectivities when using the bulky weakly nucleophilic solvent, t-BuOH (Fig. 3 and Fig. S8, ESI†). Solvents of high nucleophilicities (e.g., EtOH, MeOH) reacted immediately with furfural to form undesirable acetal side products, while non-bulky weakly nucleophilic solvents (e.g., n-BuOH, i-PrOH) reacted with hydrogenation products FA and THFA (Fig. 3 and Fig. S8, S9, ESI†). In contrast, no side products were observed when the reaction was run in t-BuOH (Fig. S8 and S10, ESI†). We attribute this observation to the steric bulk of the solvent inhibiting undesirable reaction chemistry.37 In terms of reaction rates, the observed trend tracks the H-donating ability of the solvent (n-BuOH ∼ i-PrOH > t-BuOH > CHCl3; Fig. 3c). This observation is consistent with the TCH literature, where H-donating solvents are known to accelerate the rate of furfural hydrogenation by affecting the hydrogen transfer process or by activating the carbonyl of furfural.38,39 For the parameter space we tested, t-BuOH was identified as the optimal solvent for both reaction rate and selectivity. Importantly, this solvent is not compatible with ECH reactors.
We also observed that applied current, charge passed, and catalyst identity influence selectivity and rate of furfural hydrogenation (Fig. 4 and Fig. S10–S12, ESI†). Lower applied currents mediated faster FA formation (maximum selectivity of 67% at 75 mA), while higher currents resulted in higher THFA production (maximum selectivity of 98% at 225 mA). Samples taken after 24 h at 225 mA demonstrated the production of ∼14% MTHF (Fig. S10d, ESI†). These results suggest that the –OH bond of THFA can be further hydrogenated to produce MTHF, however, this step occurs much slower than the FA and THFA production steps. Moreover, plots of equivalent charge passed at applied currents of 75 mA and 225 mA (measured at 8 h and 2.7 h, respectively) for FA selectivity demonstrated two different trends (Fig. S13a, ESI†), while measurements taken after 24 h and 8 h, respectively for THFA selectivity showed two similar trends (Fig. S13b, ESI†). These results suggest that applied current (and not only charge passed) influences selectivity, particularly for intermediate products such as FA.
We ascribe the observed trend for FA selectivity to a change in effective pressure across the palladium membrane mediated by changing the applied current. This observation is supported by studies that have shown that a small applied voltage of −0.2 to 0.3 V can lead to a large effective pressure of 1000 to 10000 atm.40 This feature is made even more pronounced by applying the current directly to the palladium membrane (as opposed to a producing hydrogen at a secondary Pd cathode and then fed through a Pd membrane). Hydrogenation rates were 6-fold faster when hydrogen was fed through the palladium membrane directly (Fig. S17, ESI†). These results highlight a stark advantage of the ePMR for direct hydrogenation compared to producing H2 gas using conventional water electrolysis and then using that H2 gas to perform hydrogenation.
The FA production rates at 75 mA for different catalysts followed the trend Pt > Ir ∼ Cu > Pd > Au > Ni > Ag. The rate of THFA production at 225 mA followed the trend Pt ∼ Ir > Pd > Au > Ni > Cu > Ag. The rate of FA production (i.e., hydrogenation of the furfural C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond) tracks the substrate-metal adsorption energies,41 but the trend for THFA production (i.e., hydrogenation of the FA CC bond) did not.42
O bond) tracks the substrate-metal adsorption energies,41 but the trend for THFA production (i.e., hydrogenation of the FA CC bond) did not.42
This result points to furfural hydrogenation being rate-limiting, and the subsequent hydrogenation of FA to THFA being a relatively fast step (eqn (1)). This hypothesis was supported by an experiment with 0.25 M FA (instead of furfural) in t-BuOH as the starting material occurring at a rate that was ∼4-fold faster (Fig. 5d). A higher activation energy for furfural hydrogenation relative to FA hydrogenation is also supported by activation energy calculations.32 Our data demonstrates that FA hydrogenation rates decreased with increasing Pt thicknesses because Pt can passivate the superior catalytic properties of Pd hydrogenating CC bonds (Fig. S15, ESI†).
These results taught us that adding a thicker layer of sputter-deposited catalyst would slow down the FA to THFA hydrogenation reaction and increase selectivity towards FA. We measured the effects of 10–50 nm thicknesses of Pt and Ir (i.e., catalysts that enabled the highest reactivity to FA). We found that 50 nm Pt improved FA by 17% (Fig. 5a). Furfural hydrogenation rates also increased at 20 nm catalyst thicknesses, indicating that the 10 nm catalyst thicknesses used for much of this study were not optimal (Fig. 5b).
The thickness of the catalyst layer influences two variables: (i) hydrogen permeation; and (ii) furfural reactivity. Hydrogen permeation experiments with 10–50 nm of Ir at 75 mA showed >96% hydrogen permeation for the thinner films but only ∼78% hydrogen permeation for the 50 nm film (Fig. S16, ESI†). The thicker 50 nm films reduce furfural hydrogenation rates and increase FA selectivity by limiting the amount of hydrogen permeating through the membrane. These findings highlight the important role of catalyst thickness on catalytic rates and substrates in the ePMR.
Conclusion
We demonstrated that the ePMR can hydrogenate furfural with high selectivities and activities, using water as the H atom source. This system is driven by electricity rather than thermal energy, and enables electrolysis at high current densities to be combined with furfural chemistry in organic media. The ePMR made it possible for us to optimize product selectivity by adjusting solvent, current, catalyst identity, and catalyst thickness in operando without modifying the system. We also designed and demonstrated a high-throughput ePMR platform to quickly identify the industrially relevant conditions for furfural hydrogenation.Author contributions
R. S. D. and B. P. M. developed the “MultiThor” concept. M. B. R. designed and built the MultiThor reactor used in this study. R. S. D designed the experimental plan. R. S. D. and M. D. S. performed all experiments. R. S. D., A. G. F., and A. H. performed product characterization and data analysis. D. J. D. and M. D. S. performed sputter-deposition and membrane characterization. C. P. B. supervised the project. The draft manuscript was written by R. S. D., and all authors contributed to the construction of the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Ben Herring at the UBC Shared Instrument Facility for assistance with the GC–MS instrument (without which this research would not have been possible during the COVID-19 pandemic), Fraser Parlane for help with XRF spectroscopy, and Camden Hunt for help with ECSA measurements. We are grateful to the Stewart Blusson Quantum Matter Institute's Quantum Electronic Science and Technology Initiative, Canadian Natural Science and Engineering Council (RGPIN-2018-06748), Canadian Foundation for Innovation (229288), Canadian Institute for Advanced Research (BSE-BERL-162173), Canada Research Chairs, and the Canada First Research Excellence Fund for financial support. SEM imaging and sputter-deposition were performed at the UBC Centre for High-Throughput Phenogenomics, a facility supported by the Canada Foundation for Innovation, British Columbia Knowledge Development Foundation, and the UBC Faculty of Dentistry.References
- B. Kamm, P. R. Gruber and M. Kamm, Biorefineries - Industrial Processes and Products: Status Quo and Future Directions, Wiley, 2010 Search PubMed.
- D. Tilman, R. Socolow, J. A. Foley, J. Hill, E. Larson, L. Lynd, S. Pacala, J. Reilly, T. Searchinger, C. Somerville and R. Williams, Science, 2009, 325, 270–271 CrossRef CAS PubMed.
- T. Werpy and G. Petersen, Top value added chemicals from biomass: Volume I – results of screening for potential candidates from sugars and synthesis gas, National Renewable Energy Lab., Golden, CO (US), 2004 Search PubMed.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. López Granados, Energy Environ. Sci., 2016, 9, 1144–1189 RSC.
- Y. Wang, D. Zhao, D. Rodríguez-Padrón and C. Len, Catalysts, 2019, 9, 796 CrossRef.
- Furfural Market Size & Share, https://www.grandviewresearch.com/industry-analysis/furfural-market, (accessed 18 February 2021).
- H. E. Hoydonckx, Wiley-VCH Verlag GmbH & Co. KGaA, Ullmann's Encyclopedia of Industrial Chemistry, Weinheim, Germany, 2000, vol. 66, p. 733 Search PubMed.
- N. Thomopoulos, Global markets for renewable chemicals manufacturing, https://www.bccresearch.com/market-research/environment/renewable-chemicals-manufacturing-markets-report.html, (accessed 9 March 2021).
- C. E. G. Padro and V. Putsche, Survey of the economics of hydrogen technologies, National Renewable Energy Lab., Golden, CO (US), 1999 Search PubMed.
- W. C. Albert and A. Lowy, Trans. Electrochem. Soc., 1939, 75, 367 CrossRef.
- P. Nilges and U. Schröder, Energy Environ. Sci., 2013, 6, 2925–2931 RSC.
- S. Jung and E. J. Biddinger, Energy Technol., 2018, 6, 1370–1379 CrossRef CAS.
- S. Jung and E. J. Biddinger, ACS Sustainable Chem. Eng., 2016, 4, 6500–6508 CrossRef CAS.
- C. H. Lam, W. Deng, L. Lang, X. Jin, X. Hu and Y. Wang, Energy Fuels, 2020, 34, 7915–7928 CrossRef CAS.
- S. A. Akhade, N. Singh, O. Y. Gutiérrez, J. Lopez-Ruiz, H. Wang, J. D. Holladay, Y. Liu, A. Karkamkar, R. S. Weber, A. B. Padmaperuma, M.-S. Lee, G. A. Whyatt, M. Elliott, J. E. Holladay, J. L. Male, J. A. Lercher, R. Rousseau and V.-A. Glezakou, Chem. Rev., 2020, 120, 11370–11419 CrossRef CAS PubMed.
- Y. Kwon, K. J. P. Schouten, J. C. van der Waals, E. de Jong and M. T. M. Koper, ACS Catal., 2016, 6, 6704–6717 CrossRef CAS.
- L. Liu, H. Liu, W. Huang, Y. He, W. Zhang, C. Wang and H. Lin, J. Electroanal. Chem., 2017, 804, 248–253 CrossRef CAS.
- Z. Li, S. Kelkar, C. H. Lam, K. Luczek, J. E. Jackson, D. J. Miller and C. M. Saffron, Electrochim. Acta, 2012, 64, 87–93 CrossRef CAS.
- A. S. May and E. J. Biddinger, ACS Catal., 2020, 10, 3212–3221 CrossRef CAS.
- H. Inoue, T. Abe and C. Iwakura, Chem. Commun., 1996, 55–56 RSC.
- T. Sato, S. Sato and N. Itoh, Int. J. Hydrogen Energy, 2016, 41, 5419–5427 CrossRef CAS.
- R. S. Sherbo, R. S. Delima, V. A. Chiykowski, B. P. MacLeod and C. P. Berlinguette, Nat. Catal., 2018, 1, 501–507 CrossRef CAS.
- R. S. Delima, R. S. Sherbo, D. J. Dvorak, A. Kurimoto and C. P. Berlinguette, J. Mater. Chem. A, 2019, 7, 26586–26595 RSC.
- R. S. Sherbo, A. Kurimoto, C. M. Brown and C. P. Berlinguette, J. Am. Chem. Soc., 2019, 141(19), 7815–7821 CrossRef CAS PubMed.
- R. P. Jansonius, A. Kurimoto, A. M. Marelli, A. Huang, R. S. Sherbo and C. P. Berlinguette, Cell Rep. Phys. Sci., 2020, 1, 100105 CrossRef.
- A. Kurimoto, R. S. Sherbo, Y. Cao, N. W. X. Loo and C. P. Berlinguette, Nat. Catal., 2020, 1–8 Search PubMed.
- A. Huang, Y. Cao, R. S. Delima, T. Ji, R. P. Jansonius, N. J. J. Johnson, C. Hunt, J. He, A. Kurimoto, Z. Zhang and C. P. Berlinguette, J. Am. Chem. Soc., 2021, 1(3), 336–343 CAS.
- E. Wicke, H. Brodowsky and H. Züchner, Top. Appl. Phys., 1978, 73–155 CrossRef CAS.
- A. Kurimoto, R. P. Jansonius, A. Huang, A. M. Marelli, D. J. Dvorak, C. Hunt and C. P. Berlinguette, Angew. Chem., Int. Ed., 2021, 60, 11937–11942 CrossRef CAS PubMed.
- M. J. Taylor, L. J. Durndell, M. A. Isaacs, C. M. A. Parlett, K. Wilson, A. F. Lee and G. Kyriakou, Appl. Catal., B, 2016, 180, 580–585 CrossRef CAS.
- Y. Deng, R. Gao, L. Lin, T. Liu, X.-D. Wen, S. Wang and D. Ma, J. Am. Chem. Soc., 2018, 140, 14481–14489 CrossRef CAS PubMed.
- A. Banerjee and S. H. Mushrif, ChemCatChem, 2017, 9, 2828–2838 CrossRef CAS.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- Hydrogen Production: Natural Gas Reforming, https://www.energy.gov/eere/fuelcells/hydrogen-production-natural-gas-reforming, (accessed 21 February 2020).
- I. Tkatchenko, Comprehensive Organometallic Chemistry, G. Wilkinson, F.G.A. Stone, E.W. Abel ed., 1982 Search PubMed.
- IEA, Technology Roadmap Energy and GHG Reductions in the Chemical Industry via Catalytic Processes, 2013.
- L. Peng, M. Wang, H. Li, J. Wang, J. Zhang and L. He, Green Chem., 2020, 22, 5656–5665 RSC.
- X. Gao, S. Tian, Y. Jin, X. Wan, C. Zhou, R. Chen, Y. Dai and Y. Yang, ACS Sustainable Chem. Eng., 2020, 8, 12722–12730 CrossRef CAS.
- L. Bendiaf, I. Bahadur, A. Negadi, P. Naidoo, D. Ramjugernath and L. Negadi, Thermochim. Acta, 2015, 599, 13–22 CrossRef CAS.
- T. Maoka and M. Enyo, Electrochim. Acta, 1981, 26, 607–614 CrossRef CAS.
- J. Hagen, Industrial Catalysis: A Practical Approach, John Wiley & Sons, 2015 Search PubMed.
- X. Li, X. Lan and T. Wang, Green Chem., 2016, 18, 638–642 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information on MultiThor design, sputter-deposition procedure, ECSA measurements, XRF spectroscopy, SEM imaging, and proof-of-concept experiments including Fig. S1–S17 and Table S1. See DOI: 10.1039/d1ee02818a |
| This journal is © The Royal Society of Chemistry 2022 |