
Tracking high-valent surface iron species in the oxygen evolution reaction on cobalt iron (oxy)hydroxides†
Seunghwa
Lee
 a,
Aliki
Moysiadou
a,
You-Chiuan
Chu
b,
Hao Ming
Chen
b and
Xile
Hu
*a
a,
Aliki
Moysiadou
a,
You-Chiuan
Chu
b,
Hao Ming
Chen
b and
Xile
Hu
*a
aLaboratory of Inorganic Synthesis and Catalysis, Institute of Chemical Sciences and Engineering, ISIC-LSCI, École Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: xile.hu@epfl.ch
bDepartment of Chemistry, National Taiwan University, Taipei 10617, Taiwan
First published on 23rd November 2021
Abstract
The oxygen evolution reaction (OER) is the bottleneck reaction of water splitting, which can be used to generate green hydrogen from renewable electricity. Cobalt iron oxyhydroxides (CoFeOxHy) are among the most active OER catalysts in alkaline medium. However, the active sites of these catalysts remain unclear. Here we use operando ultraviolet-visible (UV-Vis), X-ray absorption, and Raman spectroscopy to reveal oxidations of both Fe and Co ions in CoFeOxHy during the OER. By analyzing samples with different Fe contents and thickness, we find that the concentration of Fe4+ species at the surface, but not the concentration of Co4+ in the bulk, scales with the catalytic activity. These results indicate an Fe4+-containing active site in CoFeOxHy.
Broader contextThe oxygen evolution reaction (OER) is considered as the bottleneck reaction for water splitting, which can potentially be used to convert renewable energies from the sun and wind into the hydrogen fuel. Although great efforts have been devoted to the development of catalysts composed of only Earth-abundant elements, the understanding of these catalysts remains poor. Take cobalt iron oxyhydroxides (CoFeOxHy) as an example. This catalyst has one of the highest intrinsic activity in alkaline medium, but there was no consensus on whether Co or Fe sites were the active sites. In this study, by using a multi-fold of operando spectroscopic tools, we are able to identify various Co and Fe species that are present during OER. Our analysis reveals that surface Fe4+ species rather than bulk Co4+ species are the active sites for OER on CoFeOxHy. This work enhances the understanding of mechanism for a benchmark OER catalyst. The methods presented here might be applied to study the mechanisms of other OER catalysts. |
Introduction
Hydrogen is a promising energy carrier for future sustainable energy systems.1,2 Water electrolyzers can produce green hydrogen using renewable electricity, but a substantial overpotential is required for the oxygen evolution reaction (OER) at the anodes of these devices, leading to energy loss. A tremendous amount of efforts have been devoted to the development of OER catalysts composed of Earth-abundant elements.3–6 Among them are cobalt (Co)-based materials including Co phosphate composites, (oxy)hydroxides, a variety of oxides and chalcogenides.7–12 It has been shown that iron (Fe)-incorporated Co (oxy)hydroxides are among the best OER catalysts.6,8,9Despite considerable efforts to investigate the roles of Co and Fe ions in CoFe oxides and (oxy)hydroxides,3,4,8,9,12–21 conflicting experimental observations and explanations remain. Some studies reported Co3+ species16,17,20 while others reported Co4+ species15,22 as the resting species at OER potentials; some studies showed evidence for Fe3+ to Fe4+ oxidation17,18 while others indicated no oxidation of Fe3+ in the OER.16,20 These discrepancies make it difficult to understand the mechanism of Co- and Fe-containing OER catalysts. Most of these studies employed X-ray absorption spectroscopy (XAS) to probe the oxidation states of Co and Fe ions. Because XAS is a bulk technique and gives averaged information over the entire sample, it might not reveal minority species at the surface of a material which are responsible for the catalytic activity. Therefore, it is necessary to study the catalysts using techniques complementary to XAS. Moreover, measurements should be made to catalysts of different size to probe both surface and bulk species.
Herein, we combine operando ultraviolet-visible (UV-Vis), X-ray absorption and Raman spectroscopy to probe the nature of catalytic active sites in CoFe (oxy)hydroxides (CoFeOxHy). We obtained spectral features corresponding to both Fe and Co oxidations, and unraveled their different relevance to catalysis. By analyzing samples with different Fe contents and thickness, we identified oxidized Fe species at the surface that are essential to the activity of the catalyst. The correlation of surface Fe species with catalytic activity enhances the understanding of this promising OER catalyst.
Results and discussion
CoFeOxHy films with different Fe contents were prepared on fluorine doped tin oxide (FTO) glass by anodic electrodeposition.23 The Fe contents varied from 0% to 42% (see Table S1, ESI†), confirmed by inductively coupled plasma optical emission spectrometry (ICP-OES). By controlling the deposition time the total amount of metal ions was set to be similar for all samples (Table S1, ESI†). Scanning electron microscope (SEM) images (Fig. 1b and Fig. S1, ESI†) indicate that the thicknesses of CoOxHy and CoFeOxHy (Fe 30%) films deposited for 3 min were around 23.8 nm and 12.6 nm, respectively. The thicknesses of the different Fe-containing samples (deposited for 40–70 s at the same anodic current density of 55.6 μA cm−2, Table S1 and Fig. S2, ESI†) subjected to subsequent electrochemical tests (Fig. 1c and Fig. S3, ESI†) were estimated to be 4–5 nm. | ||
| Fig. 1 (a) A schematic presentation of Co100−xFexOyHz on a FTO glass. (b) A representative SEM image of CoFeOxHy film (30% Fe). (c) Linear-sweep voltammograms of the samples with various Fe contents recorded at a scan rate of 1 mV s−1 in Fe-free 0.1 M KOH. | ||
The OER activity of the samples containing 0% to 42% Fe was measured by linear sweep voltammetry (LSV) at a scan rate of 1 mV s−1 in Fe-free 0.1 M KOH (Fig. 1c). As the Fe content increased up to 30%, the OER activity increased, consistent with earlier studies.8,16,18,24 The addition of Fe had also a significant effect on the Tafel slopes (Fig. S3, ESI†). The slope of Fe-free CoOxHy was about 52 mV dec−1. The slope rapidly decreased to about 36 mV dec−1 at an Fe content of 7.2%, and only slightly changed at higher Fe content. The dramatic change in Tafel slope upon addition of Fe suggests a change of mechanism in the OER.
Operando UV-Vis spectroscopy (Fig. S4, ESI†) was employed to track changes in the oxidation states of Co and Fe sites. The spectra were obtained from open circuit potential (OCP) to 1.75 V with an interval of 0.05 V (Fig. S5, ESI†). Control experiments identified spectral features of a bare FTO substrate and an Fe oxyhydroxide film (FeOOH) in the OER potential region (Fig. S6, ESI†). The potential-dependent spectra of two representative samples, CoOxHy and CoFeOxHy (Fe 30%) are depicted in Fig. 2a and b. When the potential increased from OCP up to 1.20 V, a peak appeared at around 420 nm for both samples, which is attributed to the oxidation of Co2+ to Co3+.25 At 1.30 V and above, a broad spectral feature at around 525 nm was visible. A similar peak was reported in an earlier study, which assigned it to the oxidation of Co3+ to Co4+.25 If this is the case, CoOxHy is transformed to CoO2.22,23,26 Into the OER relevant potential region (>1.4 V), distinctive peaks were observed at 450 nm for CoOxHy and 425 nm for CoFeOxHy (Fe 30%), respectively. We attribute the peaks to the accumulation of high-valent Co (450 nm) and Fe (425 nm) species (see below).
 | ||
| Fig. 2 Operando UV-Vis spectra for (a) CoOxHy and (b) CoFeOxHy (Fe 30%) obtained from OCP to 1.70 V with an interval of 0.1 V. (c) Operando UV-Vis spectra for the samples containing 0%, 3.3%, 7.2%, 15%, 23%, 30% and 42% Fe obtained at 1.75 V (vs. RHE). The reference spectrum of FeOOH is shown for comparison. (d) Operando Fe K-edge XANES spectra of CoFeOxHy (Fe 30%) at various potentials compared to reference samples such as Fe foil, FeO, Fe3O4 and Fe2O3. (e) Correlation of edge energy with Fe oxidation state. | ||
Comparison of the spectra for the samples at different Fe (Fig. 2c) content under OER helps further clarify the spectral features. The peak at 525 nm (Co3+ to Co4+) was visible in all samples, suggesting the formation of Co4+ in both Fe-free and Fe-containing catalysts. Appropriate doping of Fe3+ ions was previously reported to promote the oxidation of Co3+ to Co4+,19 while addition of a large amount of Fe3+ ions led to formation of phase-separated FeOOH.8,19,27 Consistent with these reports and assuming the peak intensity on a per Co basis reflects the degree of oxidation to Co4+, the UV-Vis data (Fig. S7a, ESI†) indicate a higher oxidation state of Co under oxidative conditions at a higher Fe content, up to 30% Fe. At an even higher Fe content, the peak intensity decreased due to the formation of inactive FeOOH species. The Fe-dependent formation of Co4+ is further supported by operando Raman spectra (Fig. S8, ESI†) and cyclic voltammograms (Fig. S9, ESI†) of CoOxHy and CoFeOxHy (15% and 30% Fe). The formation of the Co(Fe)O2 phase occurred at earlier potentials upon Fe doping.
The peak at 425 nm in the UV-Vis spectra (Fig. 2c) was observed to grow with a higher Fe content, while the peak at 450 nm disappeared upon addition of Fe. Thus, the peak at 450 nm is due to the oxidation of Co species. This result is consistent with our previous study which showed that during OER on CoOOH the Co4+–OH–Co4+ species was further oxidized to Co4+–O˙–Co4+ in a pre-equilibrium step.23 Meanwhile, the UV-Vis spectrum of FeOOH at 1.75 V indicates that the 425 nm peak is due to the oxidation of Fe at higher potentials (>1.4 V). The intensity ratio of the peaks at 425 nm and 525 nm, denoted as A425nm/A525nm (Fig. S7b, ESI†), should correlate to the portion of oxidized Fe species relative to Co4+ in CoFeOxHy during OER. This ratio increased with an increasing Fe content (Fig. 2c and Fig. S7b, ESI†). An abrupt increase of the ratio at 42% Fe is due to the formation of FeOOH. The correlation of A425nm/A525nm with activity up to 30% Fe suggests the oxidized Fe species is involved in OER.
Operando XAS was also performed to track the oxidation states of Co and Fe sites in the CoFeOxHy (Fe 30%) during OER. Fig. 2d displays the X-ray absorption near edge structure (XANES) for the Fe K-edge. A similar absorption energy to Fe2(III)O3 indicates the initial oxidation state of Fe ion as approximately +3. Upon immersion in the electrolyte, the Fe absorption edge was shifted to a higher energy, indicating that the Fe sites were slightly oxidized by the OH− electrolyte. As the applied potential increased, the Fe absorption energy increases. This increase was quite distinctive at 1.45 V and above where OER begins to take place (see the inset in Fig. 2d). To semi-quantitatively evaluate the Fe oxidation state during OER, the photon energies measured at half height at given potentials were extracted for linear fit with reference points (Fig. 2e).23,28,29 With higher applied potentials, the oxidation state of Fe ions gradually increased.17 Their values were higher than +3.25 at 1.45 V and above, implying the presence of Fe4+ species under OER. This result agrees with the analysis of operando UV-Vis data. XAS also confirmed enhanced oxidation of Co3+ ions in CoFeOxHy (Fe 30%) compared with that in CoOxHy XAS (Fig. S10c, ESI). The absorption edge of CoFeOxHy kept shifting to a higher energy with increasing the applied potential, suggesting that the oxidation state of Co sites increased continuously. At 1.35 V, the corresponding oxidation state of Co in CoFeOxHy was about +3.7, and it increased to about +3.9 at 1.55 V. For comparison, the oxidation state of Co ions in CoOxHy was about +3.4 at 1.55 V according to our previous work.23
Recently, it has been proposed that for certain OER catalysts the activation free energy decreases linearly with the amount of oxidative charge stored, which leads to a linear dependence of log(current density) on the surface coverage of active species.30 We plotted log(current density) versus normalized absorbance of the UV-Vis peaks at 450 nm for CoOxHy and 425 nm for CoFeOxHy (Fe 30%) (Fig. 3). These peaks are assumed to represent the accumulation of high-valent Co and Fe species, respectively. We also plotted log(current density) versus normalized absorbance of the 525 nm, which represents Co4+ species. There are linear correlations between the log(current density) and the normalized absorbance of the UV-Vis peaks at 450 nm (for CoOxHy) and 425 nm (for CoFeOxHy), but not for the peak at 525 nm (for both CoOxHy and CoFeOxHy). The results might be explained by the following considerations: the Co4+ species in CoOxHy is further oxidized in OER and the concentration of the resulting species scales with activity. This result agrees with our previous study which showed that Co4+–O˙–Co4+, formed by oxidation of Co4+–O–Co4+, was the resting species in the OER catalyzed by CoOOH.23 Some bulk Co4+ are not oxidized30–34 so that their concentration does not scale with activity. Upon Fe incorporation (>3.3%), a more active site containing Fe (and likely also Co) is formed. The Fe3+ ions are oxidized and the concentration of the Fe4+ species scales with activity.
 | ||
| Fig. 3 Tafel plots of (a) CoOxHy and (b) CoFeOxHy (Fe 30%), overlaid with normalized absorbance at 450 and 525 nm for CoOxHy and 425 and 525 nm for CoFeOxHy. The features corresponding to the accumulation of high-valent Co (450 nm) and Fe (425 nm) follow the OER activity. | ||
We hypothesize that the Fe-containing active sites in CoFeOxHy are only located at the surface of the catalyst whereas Co4+ species are formed in the entire bulk (see Appendix 1). To verify the hypothesis, we studied CoFeOxHy (30% Fe) with different thickness. The samples were prepared by varying the deposition time from 1 min to 6 min (Fig. S11, ESI†). Similar elemental compositions were confirmed by ICP-OES (Table S2, ESI†). To estimate the electrochemical surface areas of the samples, their double-layer capacitance (Cdl) was acquired by cyclic voltammetry (CV) scans (Fig. S12 and S13, ESI†). The capacitance gradually deviated from a linear correlation with the amount of materials (deposition time), indicating that as the film grows thicker, an increasing portion of the bulk is not electrochemically active.
The above CoFeOxHy (Fe 30%) films were subjected to operando UV-Vis spectroscopy (Fig. 4 and Fig. S14, S15, ESI†) at applied potentials of 1.15 V to 1.75 V with an interval of 0.05 V in 0.1 M Fe-free KOH solutions. When the thickness of the film increased, the peaks corresponding to the Fe4+ and Co4+ species were shifted to longer wavelengths, from 417 nm to 467 nm and from 525 nm to 562 nm, respectively. These changes were attributed to thickness-related optical properties.35–37 More importantly, the ratio of the absorption intensities for the Fe4+ and Co4+ (e.g., A417nm/A525nm for the 1 min-deposited sample) at 1.75 V gradually decrease from 1.14 to 0.91 upon increasing thickness (Fig. S16, ESI†). This result supports the hypothesis that only Fe species at the surface are oxidized to +4 and the Co4+ species are distributed throughout a large part (if not all) of the bulk, as the proportion of the surface sites decreases when the film thickness increases. We studied CoOxHy films in different thicknesses, which again confirms the peak at higher wavelength (e.g., 525 nm for the 1 min-deposited sample) corresponds to the Co4+ species in the entire bulk (Fig. S17, ESI†).
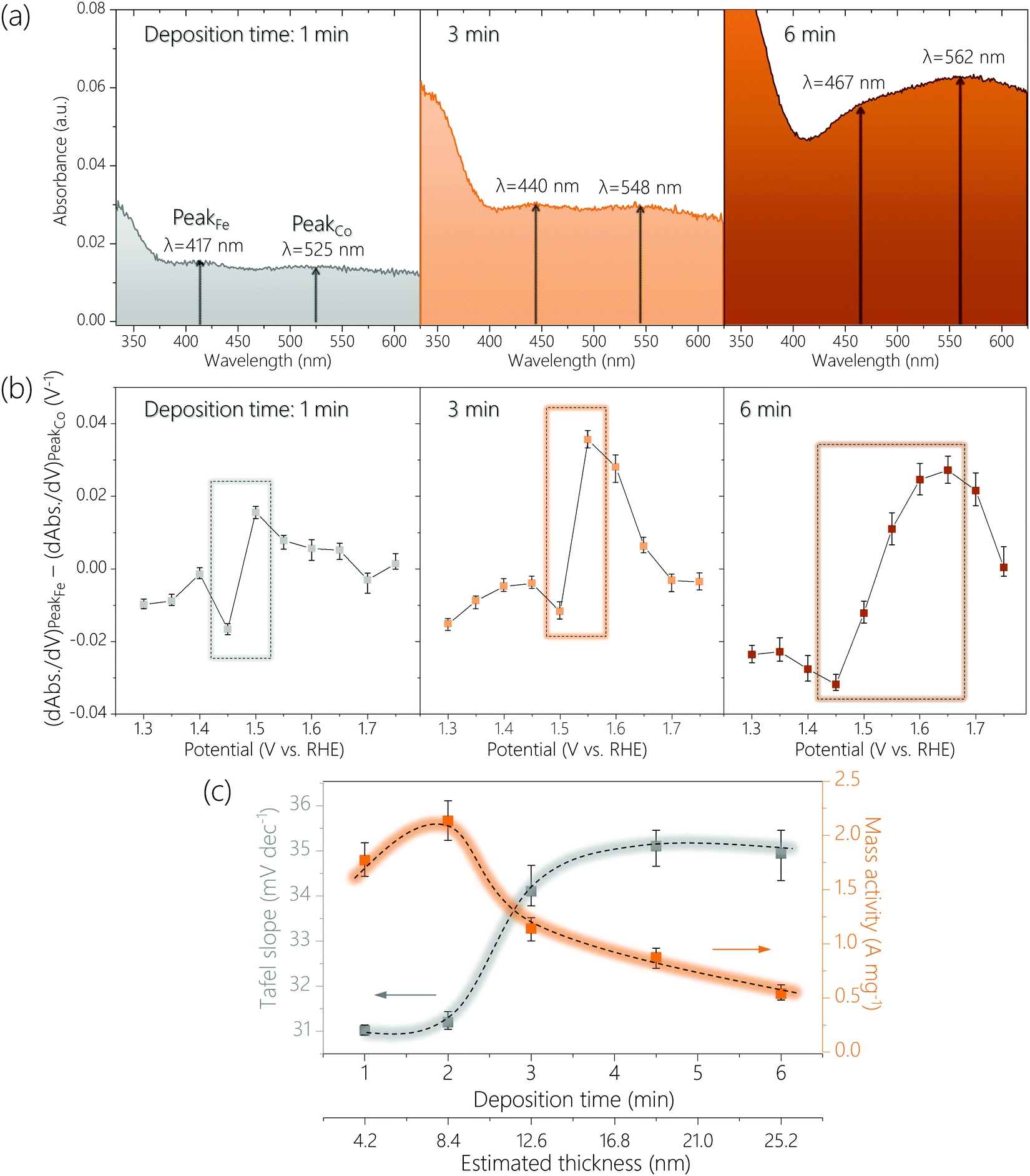 | ||
| Fig. 4 (a) Operando UV-Vis spectra of CoFeOxHy (Fe 30%) films prepared at various deposition durations (1, 3 and 6 min) at 1.75 V (vs. RHE). (b) Difference in differential absorbance at 417, 440, 467 nm (for Fe4+) and 525, 548, 562 nm (for Co4+) for samples deposited at 1, 3 and 6 min as function of potential. (c) Comparison of Tafel slope and mass activity (at an overpotential of 340 mV) as function of deposition time and estimated film thickness. | ||
To further compare the potential-dependent concentration of Fe4+ and Co4+ species, we plotted the differences in differential absorbance at the wavelength where these species absorb as function of potential according to a previously reported procedure (see Experimental methods) (Fig. 4b and Fig. S18, ESI†).32 An upward spike was observed between 1.4 to 1.5 V for all samples, which coincided with the potential region where OER started to occur. This result is consistent with that catalytically relevant Fe4+ species are generated during OER but Co4+ species exist already before OER occurs. The thicker the film was, the higher the potential where the difference (dAbs./dV for Fe4+-dAbs./dV for Co4+) is at the maximum (see rectangular boxes in Fig. 4b and Fig. S18, ESI†). This result agrees with that Fe4+ species are at the surface and Co4+ species are at the bulk.
We analyzed the thickness dependence of electrocatalytic activity (Fig. 4c). The Tafel slope was about 31 mV dec−1 for films of 4.2 nm and 8.4 nm, which increased to about 34 mV dec−1 for a film of 12.6 nm and increased further for thicker films. Meanwhile, total metal mass-based activity followed an opposite trend: best activity for films of 4.2 nm and 8.4 nm followed by continuous decrease at larger thickness. These results again support that the active sites are at the surface.
A previous study showed the optimal mass activity for OER on NiFe nanoparticles was obtained with samples of 5.4 nm.33 Another work reported that the active species in sputtered films of NiOx was accumulated only at the near surface (≤5 nm).31 Even though these previous experimental results are from Ni-based materials it would be worth considering an optimal thickness of approximately 5–6 nm. To probe whether the active surface depth of CoFeOxHy was also 5–6 nm, we prepared a CoFeOxHy (Fe 30%) sample with a thickness of about 6.3 nm (Fig. S19 and S20, ESI†). This sample exhibited similar Tafel slope and spectroelectrochemical behavior but higher mass activity than samples of 4.2 and 8.4 nm. This result suggests the optimal thickness for electrodeposited metal oxyhydroxides is indeed about 5–6 nm.
Co oxides are reported for OER: spinel Co oxides13,38–42 and layered Co (oxy)hydroxides.16–18,22,23 The Co ions in these two types of compounds have different geometries and coordination environments, so the mechanisms of OER are expected to be different as well. Previous studies of spinel Co oxides indicate that the resting state of Co is Co3+, and doped Fe ions are not oxidized during OER.16,38–41 The role of Fe might be the promotion of Co active sites.16,19,41 The electrochemical activity (e.g., Tafel slopes) and spectral features of spinel Co oxides are distinct from those of the Co (oxy)hydroxides reported here. These differences are understandable given their different structures.16,17,19,22,23,38–42 Note that spinel Co oxides might be unstable and transformed into Co (oxy)hydroxides during OER.40,43
We compare our results to those of previous studies of electrochemically deposited CoFe OER catalysts. The active forms of these catalysts are likely CoFe (oxy)hydroxides, although exceptions might exist. The group of Yeo found only Co but not Fe oxidation in Fe-doped CoOx by operando XAS.20 Because the same spectral features but different activity are found for Co in CoOx, CoOx + Fe3+, and Fe48Co52Ox, they suggested that Co was not the active site for Fe-containing catalysts. Based on the lowering of coordination number from 6 to 5.3, they proposed that the unsaturated Fe species are the active sites. Because the Fe ions were adsorbed on CoOx by CV measurements of the latter in Fe-containing solutions, multiple Fe species might form. This possibility, coupled with the thickness of CoOx being about 10 nm, might explain why no Fe oxidation was observed in their study.
In contrast, the group of Boettcher observed XAS spectral features corresponding to Fe oxidation and Fe–O bond shortening but not those corresponding to Co oxidation during the OER on electrochemically deposited thin-film CoFe (oxy)hydroxides.17 Their observation of Fe oxidation agrees with our finding, but the lack of Co oxidation in their study contrasts with our data. On the other hand, many other studies of CoOOH-type catalysts reported the oxidation of Co3+ to Co4+ similar to our work.22,23,25,26 The group of Nocera detected Fe4+ species in CoFeOx films employing ex situ zero-field 57Fe Mössbauer spectroscopy and XAS.18 It is not clear whether the active form of the catalyst has a spinel or layered structure, and whether Co3+ or Co4+ was the main Co species. The detection of Fe4+ species ex situ (meaning at ocp) indicates such species are spectators instead of active sites in OER. The authors suggest these Fe4+ serve as a redox-cooperative center to promote a nearby Co active site. We suspect their materials to be quite different from ours because a significant change of Tafel slopes occurred only at high Fe content (>20%) in their samples, whereas in our samples an Fe content of about 7% already led to a dramatic decrease of Tafel slope. We note that the catalysts employed in the above studies were made by cathodic electrodeposition of Co nitrates, where reduction of nitrate gave OH− ions that reacted with Co ions to form Co(OH)2 species. These films might be quite heterogeneous in composition. On the other hand, our catalyst is deposited by anodic deposition, which is known to give ultrathin films.44
Dionigi et al. applied operando XAS and X-ray scattering spectroscopy as well as density functional theory calculations to study the mechanism of NiFe and CoFe layered double hydroxides (LDHs), which are crystalline reference for the amorphous mixed metal oxyhydroxides made by electrodeposition.15 They provided the first experimental evidence of the OER-active γ-phase of the materials, transformed from the as prepared α-phase of LDHs. According to the calculations, the Ni and Co ions should be in the mixed +3 and +4 formal oxidation state, and the Fe ions should be in the +4 oxidation state or higher. This result agrees with our data. However, due to electrochemically inaccessible nano domains in catalyst films, phase transitions are typically incomplete under OER potentials. This incompleteness results in various weighted averages of +2, +3, and +4 for the oxidation states of metal ions, which are reflected in the different results discussed above. By using ultrathin catalyst films and multiple operando spectroscopy, in this study we are able to differentiate the roles of various surface and bulk species.
Concerning the actual active site for CoFeOxHy, two possibilities are consistent with the spectral data: (i) Fe is the active site which is mostly likely connected to a Co site by a bridging O or OH ligand.45,46 According to the DFT calculations of Dionigi et al.,15 the active site of CoFe LDH is Fe4+–OH–Co4+, and the OER occurs by deprotonation of the bridged OH with concomitant Fe oxidation. This mechanism is consistent with our data, so are catalytic cycles involving an Fe4+![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. (ii) The active site is still Fe–OH–Co, but Co serves as the oxygen evolving center while Fe serves as the redox support. In this scenario, Fe replaces the role of a redox-active O ligand as proposed previously for the OER on Fe-free CoOOH.23 It is difficult to experimentally differentiate the two possibilities, but DFT calculations can be used to compare their energetics. Most DFT computations seem to favor an Fe-based oxygen evolving site.14,15,17,24,47,48
O. (ii) The active site is still Fe–OH–Co, but Co serves as the oxygen evolving center while Fe serves as the redox support. In this scenario, Fe replaces the role of a redox-active O ligand as proposed previously for the OER on Fe-free CoOOH.23 It is difficult to experimentally differentiate the two possibilities, but DFT calculations can be used to compare their energetics. Most DFT computations seem to favor an Fe-based oxygen evolving site.14,15,17,24,47,48
Conclusion
In summary, we applied operando UV-Vis, XAS, and Raman spectroscopy to investigate the oxidation of metal ions during the OER on Co and CoFe (oxy)hydroxides (CoOxHy and CoFeOxHy). For both catalysts the oxidation of Co3+ to Co4+ occurs before the onset of the OER. For CoOxHy, a Co-based oxidation was observed at the onset of the OER. This oxidation is absent in the OER on CoFeOxHy; instead, an Fe3+ to Fe4+ oxidation concomitant to the OER was identified for the latter. The Fe4+ species are only formed at the surface layer of CoFeOxHy while the Co4+ species are present in a large part of the bulk material. The concentration of Fe4+ but not Co4+ correlates with the OER activity. Thus, the role of Fe is not only to promote the oxidation of Co3+ to Co4+, but more importantly, to form a new active site at the surface that has higher activity. The work suggests ultra-thin nanoscale metal oxyhydroxides of about 5 nm thickness will have optimal mass-based activity because all metal ions are accessible for the OER.Experimental
Materials
Cobalt nitrate (Co(NO3)2·6H2O, ≥98%, Sigma-Aldrich), iron nitrate (Fe(NO3)3·9H2O, ≥98%, Sigma-Aldrich) and sodium acetate (NaCH3CO2, anhydrous, ≥99%, Sigma-Aldrich) were used for electrode preparation. Deionized water (18 MΩ) from a Milli-Q® Ultrapure Water System (Millipore, Bedford, USA) was used in this work. Potassium hydroxide solutions (1 N KOH standard solution, Merck KGaA) were diluted for experiments. Fluorine-doped tin oxide (FTO) coated glass (2.3 mm thick and surface resistivity ∼13 Ω sq.−1, Sigma-Aldrich) served as a conductive substrate.Electrode preparation
CoOxHy and CoFeOxHy films were prepared on FTO glass by anodic electrodeposition at a constant current density of 55.6 μA cm−2.23 Prior to deposition, the FTO glass was washed in a mixture of ethanol and acetone (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) using a sonicator. The deposition bath for cobalt (oxy)hydroxide contained 0.01 M Co(NO3)2·6H2O and 0.1 M NaCH3CO2. For the deposition of cobalt–iron (oxy)hydroxide films, a solution containing 0.1 M NaCH3CO2, Co(NO3)2·6H2O and Fe(NO3)3·9H2O with different compositions (total 0.01 M) was used.
:1) using a sonicator. The deposition bath for cobalt (oxy)hydroxide contained 0.01 M Co(NO3)2·6H2O and 0.1 M NaCH3CO2. For the deposition of cobalt–iron (oxy)hydroxide films, a solution containing 0.1 M NaCH3CO2, Co(NO3)2·6H2O and Fe(NO3)3·9H2O with different compositions (total 0.01 M) was used.
Characterization
Electrode compositions were determined by inductively coupled plasma optical emission spectrometry (ICP-OES, Nexlon 350, PerkinElmer). The catalyst-deposited FTO glass was immersed in 2% nitric acid (HNO3) diluted from 65% HNO3 (Merck) such that the catalyst film could be dissolved. The resulting HNO3 solution was analyzed to reveal the amounts of Co and Fe ions. Film thickness was probed with a Gemini field-emission scanning electron microscope operating at 1–3 kV accelerating voltage. Secondary electrons were collected with an in-lens detector. Side-view images were taken to measure the thickness by viewing a cross-section of the catalyst layer on FTO glass.Electrochemical tests
All electrochemical experiments were carried out in KOH solutions purified by a previously reported method to remove Fe impurity.8,23 First, 0.5–1.0 g of Co(NO3)2·6H2O were dissolved in 5 mL of ultrapure water and then 20 mL of 0.1 M KOH was added to this solution. Co(OH)2 precipitate formed, which was subsequently washed with ultrapure water three times via centrifugation and decanting. The resulting Co(OH)2 precipitate was suspended in 50 mL of 1.0 M KOH. Afterward, the suspension was mechanically agitated overnight to absorb Fe impurities from the 1 M KOH solution. Electrochemical measurements were performed with a multichannel potentiostat/galvanostat (VSP, Bio-Logic) in a three-electrode system consisting of a Pt wire counter electrode and a homemade double junction Ag/AgCl electrode (saturated KCl, E(Ag/AgCl) = 0.197 V vs. NHE, normal hydrogen electrode) as reference electrode. IR drop was compensated at a rate 90% by default through a Bio-Logic EC-Lab software. The measurements were carried out in a polypropylene beaker or a Teflon cell to avoid influences of the trace Fe impurity from surface of glass beaker. Unless otherwise stated, the acquired electrochemical data were referred to reversible hydrogen electrode (RHE) scale by following equation:| E (V vs. RHE) = E (V vs. Ag/AgCl) + 0.197 V + 0.0592 × pH | (1) |
Operando ultraviolet-visible (UV-Vis) spectroscopy
The operando UV-Vis measurements were conducted with a Cary 60 UV-Vis spectrophotometer (Agilent Technologies) using a three-electrode system in a custom-made electrochemical Teflon cell. Prior to use, the Teflon cell was washed with an acid solution to remove Fe impurities and other dirt. The catalyst-deposited FTO glass was assembled to the electrochemical cell and then placed in the sample compartment of the UV-Vis spectrometer such that the sample can be irradiated properly. At the OCP, a spectrum as a blank was recorded for automatic baseline correction. With that, subsequent operando spectroscopic measurements were performed at each constant potential. All the spectra were collected in the transmittance mode.Following a previous study,32 differential changes in absorbance (dAbs./dV) were obtained at the wavelengths assigned to Fe4+ and Co4+ (e.g., 417 nm for Fe4+ and 525 nm for Co4+ in the sample deposited during 1 min as shown in Fig. 4a) for the CoFeOxHy (Fe 30%) samples prepared at various deposition times. The dAbs. represents the difference in the absorbance at two consecutive potentials separated by 0.05 V. Hence, dV is 0.05 V. In the potential-dependent plots (dAbs. for Fe4+/dV − dAbs. for Co4+/dV), the positive peaks at OER relevant potentials (1.5–1.65 V) indicate the increases of Fe4+versus Co4+.
Operando X-ray absorption spectroscopy (XAS)
For operando XAS measurements, all of the experiments were carried out at BL 01C, NSRRC, Taiwan. Each spectrum was measured after the calibration of either Co or Fe foil to prevent the deviation of incident energy. The operando XAS measurements were conducted in the customized electrochemical cell composed of Kapton window, a working electrode, a Pt counter electrode, and an Ag/AgCl reference electrode as reported in our previous work.23 In order to analyze the XANES results, the obtained spectra were processed in Athena software to perform standard normalization to exclude the influence of thickness, concentration and amplifier settings.Operando surface enhanced Raman spectroscopy (SERS)
The experiments were carried out in a customized electrochemical Teflon cell using a Raman microscope (in Via Raman microscope, Renishaw).49 Prior to experiments, the Teflon cell was rigorously washed with an acid solution to remove impurities and then washed with a mixture of acetone and ethanol. Consistent with the set-up for operando UV-Vis spectroscopy, a three electrode system was used, in which a Pt wire and a homemade double junction Ag/AgCl electrode served as counter and reference electrode respectively. A 63× water-immersed objective (Leica-Microsystems) was applied to operando spectroscopic measurements. For a strong surface-enhanced effect, the 785 nm laser was used with the laser power of ∼1% at the grating of 1200 l mm−1. Spectrum was calibrated prior to use based on the wavenumber of silicon, 520 ± 0.5 cm−1. During experiments, each spectrum was collected through 15 consecutive scans with 1 s exposure time per scan. We used an electrochemically roughened gold (Au) as a substrate to obtain the surface-enhanced effect.23 First, the Au was mechanically polished with alumina powder (1 μm) and then sonicated several times in 1:1 mixture of ethanol and acetone and then in ultrapure water. The clean Au was subjected to 20 oxidation–reduction cycling between −0.28 and 1.22 V vs. Ag/AgCl in 0.1 M KCl solution, in which the potential was held for 10 s at the negative limit and 5 s at the positive limit. After the potential cycling, the Au surface was reduced for 5 min at a constant potential of −0.3 V. Finally, the resulting brownish Au surface was thoroughly rinsed with ultrapure water. CoOxHy and CoFeOxHy films were deposited on the roughened Au as described above for operando Raman spectroscopy experiments.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the European Research Council (No. 681292) and the Marie Skłodowska-Curie Fellowship (No. 838367) under the European Union's Horizon 2020 research. This study is also supported by the Ministry of Science and Technology, Taiwan (MOST 109-2628-M-002-001-RSP). The work is part of NCCR Catalysis, a National Centre of Competence in Research funded by the Swiss Naitonal Science Foundation.Notes and references
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum and S. Ardo, Energy Environ. Sci., 2013, 6, 1983–2002 RSC.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- F. Song, L. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. Hu, J. Am. Chem. Soc., 2018, 140, 7748–7759 CrossRef CAS PubMed.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116, 14120–14136 CrossRef CAS PubMed.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed.
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Adv. Mater., 2016, 28, 215–230 CrossRef CAS PubMed.
- J. Huang, J. Chen, T. Yao, J. He, S. Jiang, Z. Sun, Q. Liu, W. Cheng, F. Hu and Y. Jiang, Angew. Chem., 2015, 127, 8846–8851 CrossRef.
- Y. Liu, H. Cheng, M. Lyu, S. Fan, Q. Liu, W. Zhang, Y. Zhi, C. Wang, C. Xiao and S. Wei, J. Am. Chem. Soc., 2014, 136, 15670–15675 CrossRef CAS PubMed.
- N. H. Chou, P. N. Ross, A. T. Bell and T. D. Tilley, ChemSusChem, 2011, 4, 1566–1569 CrossRef CAS PubMed.
- S.-F. Hung, Y.-T. Chan, C.-C. Chang, M.-K. Tsai, Y.-F. Liao, N. Hiraoka, C.-S. Hsu and H. M. Chen, J. Am. Chem. Soc., 2018, 140, 17263–17270 CrossRef CAS PubMed.
- C. Feng, M. B. Faheem, J. Fu, Y. Xiao, C. Li and Y. Li, ACS Catal., 2020, 10, 4019–4047 CrossRef CAS.
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. B. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik, D. Fan, A. Bergmann, J. Drnec, J. F. Araujo, M. Gilech, D. Teschner, J. Zhu, W.-X. Li, J. Greely, B. R. Cuenya and P. Strasser, Nat. Commun., 2020, 11, 1–10 Search PubMed.
- R. D. Smith, C. Pasquini, S. Loos, P. Chernev, K. Klingan, P. Kubella, M. R. Mohammadi, D. Gonzalez-Flores and H. Dau, Nat. Commun., 2017, 8, 1–8 CrossRef CAS PubMed.
- L. J. Enman, M. B. Stevens, M. H. Dahan, M. R. Nellist, M. C. Toroker and S. W. Boettcher, Angew. Chem., Int. Ed., 2018, 57, 12840–12844 CrossRef CAS PubMed.
- N. Li, R. G. Hadt, D. Hayes, L. X. Chen and D. G. Nocera, Nat. Commun., 2021, 12, 1–6 CrossRef PubMed.
- L. Zhuang, L. Ge, Y. Yang, M. Li, Y. Jia, X. Yao and Z. Zhu, Adv. Mater., 2017, 29, 1606793 CrossRef PubMed.
- L. Gong, X. Y. E. Chng, Y. Du, S. Xi and B. S. Yeo, ACS Catal., 2018, 8, 807–814 CrossRef CAS.
- F. Yang, K. Sliozberg, I. Sinev, H. Antoni, A. Bähr, K. Ollegott, W. Xia, J. Masa, W. Grünert, B. R. Cuenya, W. Schuhmann and M. Muhler, ChemSusChem, 2017, 10, 156–165 CrossRef CAS PubMed.
- J. Zhou, Y. Wang, X. Su, S. Gu, R. Liu, Y. Huang, S. Yan, J. Li and S. Zhang, Energy Environ. Sci., 2019, 12, 739–746 RSC.
- A. Moysiadou, S. Lee, C.-S. Hsu, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2020, 142, 11901–11914 CrossRef CAS PubMed.
- S. H. Ye, Z. X. Shi, J. X. Feng, Y. X. Tong and G. R. Li, Angew. Chem., Int. Ed., 2018, 57, 2672–2676 CrossRef CAS PubMed.
- M. Risch, F. Ringleb, M. Kohlhoff, P. Bogdanoff, P. Chernev, I. Zaharieva and H. Dau, Energy Environ. Sci., 2015, 8, 661–674 RSC.
- D. Friebel, M. Bajdich, B. S. Yeo, M. W. Louie, D. J. Miller, H. S. Casalongue, F. Mbuga, T.-C. Weng, D. Nordlund, D. Sokaras, R. Alonso-Mori, A. T. Bell and A. Nilsson, Phys. Chem. Chem. Phys., 2013, 15, 17460–17467 RSC.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M. J. Cheng, D. Sokaras, T. C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Norskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
- X. Yang, K. Takada, M. Itose, Y. Ebina, R. Ma, K. Fukuda and T. Sasaki, Chem. Mater., 2008, 20, 479–485 CrossRef CAS.
- D. K. Bediako, B. Lassalle-Kaiser, Y. Surendranath, J. Yano, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2012, 134, 6801–6809 CrossRef CAS PubMed.
- H. N. Nong, L. J. Falling, A. Bergmann, M. Klingenhof, H. P. Tran, C. Spöri, R. Mom, J. Timoshenko, G. Zichittella, A. Knop-Gericke, S. Piccinin, J. Pérez-Ramírez, B. R. Cuenya, R. Schlögl, P. Strasser, D. Teschner and T. E. Jones, Nature, 2020, 587, 408–413 CrossRef CAS PubMed.
- S. Corby, M.-G. Tecedor, S. Tengeler, C. Steinert, B. Moss, C. A. Mesa, H. F. Heiba, A. A. Wilson, B. Kaiser, W. Jaegermann, L. Francàs, S. Gimenez and J. R. Durrant, Sustainable Energy Fuels, 2020, 4, 5024–5030 RSC.
- C. Baeumer, J. Li, Q. Lu, A. Y.-L. Liang, L. Jin, H. P. Martins, T. Duchoň, M. Glöβ, S. M. Gericke, M. A. Wohlgemuth, M. Giesen, E. E. Penn, R. Dittmann, F. Gunkel, R. Waser, M. Bajdich, S. Nemsak, J. T. Mefford and W. C. Chueh, Nat. Mater., 2021, 20, 674–682 CrossRef CAS PubMed.
- C. Roy, B. Sebok, S. B. Scott, E. M. Fiordaliso, J. E. Sørensen, A. Bodin, D. B. Trimarco, C. D. Damsgaard, P. C. K. Vesborg, O. Hansen, I. E. L. Stephens, J. Kibsgaard and I. Chorkendorff, Nat. Catal., 2018, 1, 820–829 CrossRef CAS.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef CAS PubMed.
- S. Agnihotri, S. Mukherji and S. Mukherji, RSC Adv., 2014, 4, 3974–3983 RSC.
- A. Shafiqa, A. A. Aziz and B. Mehrdel, J. Phys.: Conf. Ser., 2018, 1083, 012040 CrossRef.
- G. A. López-Muñoz, J. A. Pescador-Rojas, J. Ortega-Lopez, J. S. Salazar and J. A. Balderas-López, Nanoscale Res. Lett., 2012, 7, 1–6 CrossRef PubMed.
- H.-Y. Wang, S.-F. Hung, H.-Y. Chen, T.-S. Chan, H. M. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 36–39 CrossRef CAS PubMed.
- H.-Y. Wang, S.-F. Hung, Y.-Y. Hsu, L. Zhang, J. Miao, T.-S. Chan, Q. Xiong and B. Liu, J. Phys. Chem. Lett., 2016, 7, 4847–4853 CrossRef CAS PubMed.
- L. Calvillo, F. Carraro, O. Vozniuk, V. Celorrio, L. Nodari, A. Russell, D. Debellis, D. Fermin, F. Cavani and S. Agnoli, J. Mater. Chem. A, 2018, 6, 7034–7041 RSC.
- S. F. Hung, Y. Y. Hsu, C. J. Chang, C. S. Hsu, N. T. Suen, T. S. Chan and H. M. Chen, Adv. Energy Mater., 2018, 8, 1701686 CrossRef.
- A. Bergmann, E. Martinez-Moreno, D. Teschner, P. Chernev, M. Gliech, J. F. De Araújo, T. Reier, H. Dau and P. Strasser, Nat. Commun., 2015, 6, 1–9 Search PubMed.
- A. Bergmann, T. E. Jones, E. M. Moreno, D. Teschner, P. Chernev, M. Gliech, T. Reier, H. Dau and P. Strasser, Nat. Catal., 2018, 1, 711–719 CrossRef CAS.
- C. G. Morales-Guio, L. Liardet and X. Hu, J. Am. Chem. Soc., 2016, 138, 8946–8957 CrossRef CAS PubMed.
- J. Wang, X. Ge, Z. Liu, L. Thia, Y. Yan, W. Xiao and X. Wang, J. Am. Chem. Soc., 2017, 139, 1878–1884 CrossRef CAS PubMed.
- J. Wang, L. Gan, W. Zhang, Y. Peng, H. Yu, Q. Yan, X. Xia and X. Wang, Sci. Adv., 2018, 4, eaap7970 CrossRef PubMed.
- X. Han, C. Yu, J. Yang, X. Song, C. Zhao, S. Li, Y. Zhang, H. Huang, Z. Liu and H. Huang, Small, 2019, 15, 1901015 CrossRef PubMed.
- M. B. Stevens, L. J. Enman, E. H. Korkus, J. Zaffran, C. D. Trang, J. Asbury, M. G. Kast, M. C. Toroker and S. W. Boettcher, Nano Res., 2019, 12, 2288–2295 CrossRef CAS.
- S. Lee, K. Banjac, M. Lingenfelder and X. Hu, Angew. Chem., Int. Ed., 2019, 58, 10295–10299 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ee02999a |
| This journal is © The Royal Society of Chemistry 2022 |