
A recyclable metal-free catalytic system for the cationic ring-opening polymerization of glycidol under ambient conditions†
Si Eun
Kim
ab,
Hyun Ji
Yang
a,
Soonyoung
Choi
a,
Eunbyul
Hwang
 a,
Minseong
Kim
cd,
Hyun-Jong
Paik
b,
Ji-Eun
Jeong
a,
Young Il
Park
a,
Jin Chul
Kim
*a,
Byeong-Su
Kim
*c and
Sang-Ho
Lee
*a
a,
Minseong
Kim
cd,
Hyun-Jong
Paik
b,
Ji-Eun
Jeong
a,
Young Il
Park
a,
Jin Chul
Kim
*a,
Byeong-Su
Kim
*c and
Sang-Ho
Lee
*a
aCenter for Advanced Specialty Chemicals, Korea Research Institute of Chemical Technology, Ulsan 44412, Republic of Korea. E-mail: slee@krict.re.kr; jckim81@krict.re.kr
bDepartment of Polymer Science and Engineering, Pusan National University, Busan 46241, Republic of Korea
cDepartment of Chemistry, Yonsei University, Seoul 03722, Republic of Korea. E-mail: bskim19@yonsei.ac.kr
dDepartment of Chemistry, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea
First published on 9th December 2021
Abstract
A recyclable catalytic system for ionic polymerization under ambient conditions is still undoubtedly a challenging issue that needs to be addressed for industrial production. In this study, a metal-free cationic ring-opening polymerization of glycidol (GD) using tris(pentafluorophenyl)borane (B(C6F5)3, BCF) as a catalyst affords a well-controlled branched cyclic polyglycidol (BC-PGD) structure and a recycling polymerization process was achieved using unpurified reagents and ambient conditions. Although homogeneous catalysts cannot usually be readily recycled during polymerization, the growing PGD chains in nonpolar solvents induces self-precipitation in catalyst solutions with increasing molecular weight and hydrophilicity, causing a phase separation of PGD with a uniform molecular weight distribution. Specifically, the recycling polymerization process is successfully performed by repeating the simple sequence of decantation and addition of the unpurified monomer. The unique structure of the obtained PGDs was confirmed by 1H NMR, inverse-gated 13C NMR analyses, SEC, and MALDI-ToF-MS. Based on the green and recyclable BCF-catalyzed cationic ring-opening polymerization, more intriguing examples with simple and well-reproducible polymerization techniques are anticipated for challenging industrial applications.
Introduction
In the past few years, efforts have been devoted to recycling catalytic systems for organic synthesis1–3 and polymerization4,5 to reduce the cost of industrial production and improve the purity of the final products. Employing an immiscible phase separation phenomenon after the reaction is a simple strategy for reusable synthesis, which affords a simple production procedure as well as purification.6–9 Although this synthetic strategy provides various advantages, the reported examples are not applicable to the ionic polymerization field due to concerns about the decreased reactivity of active species and tedious purification steps.10,11 An ideal recyclable catalytic system for ionic polymerization under ambient conditions needs to satisfy the following three factors: (i) high air and humidity tolerance of the catalyst; (ii) easy isolation between the polymer product and catalyst solution via phase separation; and (iii) scalable polymerization and high reproducibility for uniform production.Hyperbranched polyglycidols (hb-PGDs) possess several remarkable features, including a highly flexible aliphatic polyether backbone, multiple hydrophilic groups, and excellent biocompatibility.12 In general, for preparing hb-PGDs, anionic ring-opening polymerization13 and cationic ring-opening polymerization (CROP)14,15 of glycidol (GD) are usually employed, providing well-controlled molecular weights and polymer structures; however, harsh reaction conditions and sufficient purification of reagents are required. Recently, new synthetic approaches have been proposed to control the polymer architecture using a buffer solution or Lewis acid catalyst. For example, Haag and coworkers employed citric acid as the proton donor and activating reagent for a CROP system to prepare hb-PGDs.11 Moreover, Kim et al. obtained hb-PGDs using a double metal cyanide catalyst, which promotes the active chain end (ACE) mechanism by coordination between the glycidol (GD) monomer and catalyst.16 In contrast, Harth et al. developed semibranched PGDs with a low branching density using phosphate buffered saline in aqueous media.17 Furthermore, efforts have been directed to fabricating a macrocyclic polymer structure using tris(pentafluorophenyl)borane (B(C6F5)3, BCF), which are provided by the zwitterionic ring-expansion polymerization (ZREP) of glycidyl ether-based monomers.18–20 In addition, in the presence of water, the ZREP system yields a linear polymer structure due to the protonation of the active chain end. Despite the successful synthesis of topologically controlled PGDs, the synthetic methodologies are still limited because of the molecular weight control of polymers and the tedious purification steps prior to the polymerization.
To increase the interest in recyclable catalytic polymerization systems and overcome the sensitivity of metal catalysts under ambient conditions, we examined a recyclable metal-free catalytic system for the CROP of GD using the BCF catalyst. In this context, the BCF catalyst has high air and humidity tolerance, which enables CROP with unpurified reagents under ambient conditions.21,22 Interestingly, while homogeneous catalysts cannot generally be easily recycled, the growing PGD chains in nonpolar solvents, such as toluene, might be automatically separated from the catalyst solution upon the growth of polymer chains with an increased molecular weight and hydrophilicity, which should lead to phase separation and a uniform molecular weight (Scheme 1). Obviously, the BCF-catalyzed ring-opening polymerization forms zwitterionic intermediates with the GD monomer in the initiation step, and a cyclic topology is generated via ring-expansion polymerization.18 Simultaneously, branched structures are afforded from an AB2-type GD monomer,23 leading to the formation of branched cyclic PGDs (BC-PGDs) via two competing propagation mechanisms: the ACE and activated monomer (AM) mechanisms (Scheme 2). Herein, based on this CROP system, green and recyclable catalysis for CROP under ambient conditions is established for producing BC-PGDs with high reproducibility.
 | ||
| Scheme 1 Schematic of the recyclable metal-free catalytic system for the cationic ring-opening polymerization of the glycidol monomer. | ||
 | ||
| Scheme 2 Mechanism of the metal-free cationic ring-opening polymerization of the glycidol monomer. | ||
Experimental
Materials
Tris(pentafluorophenyl)borane (BCF, Sigma-Aldrich; purity >95%), dimethyl sulfoxide (DMSO) (Wako; 99%), anisole (Sigma-Aldrich; 99.7%), ethyl acetate (Sigma-Aldrich; 99.8%), methyl ethyl ketone (Sigma-Aldrich; ≥99%), and diethyl ether were used as received. Glycidol (GD, Sigma-Aldrich; purity >96%) and tetralin (Sigma-Aldrich, 99%) were dried overnight over calcium hydride and purified by distillation over calcium hydride before use. Toluene (Sigma-Aldrich; ≥99.9%) was purified by passing it through purification columns (JCM, JCM-3SPS-SA-6) and bubbling with dry nitrogen gas for more than 15 min immediately before use.Measurements
The Mn and Mw/Mn of the polymers were measured by size-exclusion chromatography (SEC) at 45 °C using dimethylformamide (DMF) as the eluent. For DMF-SEC, three polystyrene-gel columns [KD-802 (from Shodex): pore size, 150 Å; 8 mm i.d. × 300 mm; KD-803 (from Shodex): pore size, 500 Å; 8 mm i.d. × 300 mm; and KD-804 (from Shodex): pore size, 1500 Å; 8 mm i.d. × 300 mm] were connected to a PU-4180 pump, a RI-4030 refractive-index detector, and a UV-4075 ultraviolet detector (JASCO). The flow rate was set to 1.0 mL min−1. For the obtained PGD samples, the columns were calibrated against 13 standard poly(ethylene glycol)/poly(ethylene oxide) samples (Agilent Technologies; Mp = 980–811![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500; Mw/Mn = 1.03–1.11). 1H NMR spectra were recorded on a Bruker Ultrashield spectrometer operating at 300 MHz. 1H NMR (400 MHz), 13C NMR (100 MHz), and 19F NMR (376 MHz) spectra were obtained using an AVANCE III HD NMR spectrometer (Bruker). All spectra were recorded in ppm units with the deuterated solvent DMSO-d6 at room temperature. Furthermore, matrix-assisted laser desorption/ionization time-of-flight (MALDI-ToF) analysis was performed using an Ultraflex III MALDI mass spectrometer; 2,5-dihydroxybenzoic acid was used as the matrix. UV–vis spectra were obtained using a Jasco V-770 UV–vis spectrometer.
500; Mw/Mn = 1.03–1.11). 1H NMR spectra were recorded on a Bruker Ultrashield spectrometer operating at 300 MHz. 1H NMR (400 MHz), 13C NMR (100 MHz), and 19F NMR (376 MHz) spectra were obtained using an AVANCE III HD NMR spectrometer (Bruker). All spectra were recorded in ppm units with the deuterated solvent DMSO-d6 at room temperature. Furthermore, matrix-assisted laser desorption/ionization time-of-flight (MALDI-ToF) analysis was performed using an Ultraflex III MALDI mass spectrometer; 2,5-dihydroxybenzoic acid was used as the matrix. UV–vis spectra were obtained using a Jasco V-770 UV–vis spectrometer.
Metal-free cationic ring-opening polymerization
Polymerization was performed using the syringe technique in dry argon-baked glass tubes equipped with a three-way stopcock or in sealed glass vials. The typical procedure for GD with BCF is described as follows. In a round-bottom flask (50 mL) filled with argon, GD (0.67 mL, 10.0 mmol), toluene (2.23 mL), and tetralin (0.30 mL) were sequentially added. The reaction mixture was cooled to 0 °C, and a BCF catalyst solution (0.80 mL, 25 mM in toluene) was added to the polymerization mixture; the total volume was set as 4.00 mL. Immediately after that, the reaction mixture was placed in a reaction bath at 25 °C. Monomer conversion was determined from the residual monomer concentration measured by 1H NMR with tetralin as the internal standard. Then, the quenched solution was decanted and dissolved in DMSO. The polymer solution was precipitated in cold diethyl ether to remove oligomers and the catalyst. The final product was dried overnight under vacuum at 30 °C (68.3 mg, 92% yield) and characterized by SEC (Mn = 3800, Mw/Mn = 1.17).Recycling process for the metal-free cationic ring-opening polymerization of glycidol
Polymerization was performed under air using unpurified reagents in a vial. In a vial, the BCF catalyst (0.077 g, 0.15 mmol) and toluene (29.0 mL) were sequentially added. Then, the reaction mixture was cooled to 0 °C and GD (0.997 mL, 15 mmol) was introduced into the polymerization mixture; the total volume was set as 30.0 mL. The reaction mixture was placed in a reaction bath at 25 °C for 10 min. Thereafter, the catalyst solution (top layer) was separated from the polymer (bottom layer) and placed in a new 50.0 mL vial by decantation. Additional GD (0.997 mL, 15 mmol) was added to the recycled solution, and the solution was maintained at 25 °C for 10 min. The above sequence of decantation and monomer feeding was repeated till the fifth polymerization (volume loss <4% for each cycle and 83.3% material retention after 5 cycles). Exceptionally, the reaction time of the fifth polymerization was 30 min. Furthermore, the monomer conversion was determined from the residual monomer concentration measured using the 1H NMR spectra. The obtained PGDs were dried for several days under vacuum at 60 °C after decantation and characterized using SEC.Results and discussion
BCF was used as a Lewis acid catalyst for the metal-free CROP of GD to afford BC-PGDs comprising dendritic (D), linear (L), and terminal (T) units (Scheme 1). In the initiation step of the BCF-catalyzed ring-opening polymerization, zwitterionic intermediates initially formed between BCF and the GD monomer; then, propagation occurred via the nucleophilic attack of GD through the ACE and AM mechanisms (Scheme 2). Generally, in the cationic polymerization of GD, the ACE mechanism generates only primary alcohols, such as L1,3 and T2 units. On the other hand, the AM mechanism yields branched polymer chains via the nucleophilic attack of the hydroxyl group of GD or the polymer chain. As is known, the AM mechanism can afford all possible structural composition units (i.e., L1,3, L1,4, D, T1, and T2). However, the most significant difference between the AM and ACE mechanisms is that the AM mechanism promotes secondary alcohol formation, whereas the ACE mechanism favors the formation of primary alcohols. Thus, the combination of these two propagation mechanisms could afford the BC-PGD structure.24–26BCF-catalyzed ring-opening polymerization is highly exothermic, and the reaction can be violent if performed at high temperatures. Therefore, the reaction parameters, such as the monomer and catalyst concentrations, polymerization temperature, and solvent polarity, were carefully optimized (Table 1). Briefly, reactions were conducted with varying monomer and BCF catalyst concentrations at −40 °C in toluene for 3 h; a high conversion with BC-PGDs of similar molecular weights was obtained (P1–P8 in Table 1 and Fig. S1–S5†). The high concentration of the Lewis acid, used as a catalyst, in the polymerization affected the monomer conversion rate, and the concentration of the monomer influenced the molecular weight distributions of the obtained polymers.
| Polymer code | [GD]0 (mM) | [B(C6F5)3]0 (mM) | Solvent | Temperature (°C) | Time | Conversiona (%) |
M
nb |
M
w/Mnb |
DBc |
|---|---|---|---|---|---|---|---|---|---|
| a Conversion was determined by 1H NMR with tetralin as an internal standard. b Measured by SEC calibrated with PEO standards in DMF (50 mM LiBr, 45 °C, and a flow rate of 1.0 mL min−1). c The degree of branching (DB) was calculated by inverse-gated 13C NMR using the following equation: (D + T)/(D + T + L) (D = dendritic unit; T = terminal unit; and L = linear unit). d Not determined. e The polymerization was performed under air using an unpurified monomer and solvent. f Large-scale polymerization was performed under air using an unpurified 200 g-scale monomer and solvent. | |||||||||
| P1 | 2500 | 1 | Toluene | −40 | 3 h | 83.3 | 1600 | 2.16 | 0.36 |
| P2 | 2500 | 3 | Toluene | −40 | 3 h | 93.7 | 3300 | 1.29 | 0.38 |
| P3 | 2500 | 5 | Toluene | −40 | 3 h | 99.1 | 3500 | 1.25 | 0.34 |
| P4 | 2500 | 10 | Toluene | −40 | 3 h | >99.9 | 3700 | 1.43 | 0.36 |
| P5 | 500 | 5 | Toluene | −40 | 3 h | >99.9 | 3500 | 2.18 | 0.41 |
| P6 | 1000 | 5 | Toluene | −40 | 3 h | 96.3 | 3850 | 2.19 | 0.43 |
| P7 | 5000 | 5 | Toluene | −40 | 3 h | 95.2 | 3400 | 1.37 | 0.42 |
| P8 | 10000 |
5 | Toluene | −40 | 3 h | 98.9 | 3300 | 1.54 | 0.42 |
| P9 | 2500 | 5 | Toluene | 0 | 15 min | 94.7 | 3600 | 1.28 | 0.39 |
| P10 | 2500 | 5 | Toluene | 25 | <1 min | >99.9 | 3800 | 1.17 | 0.41 |
| P11 | 2500 | 5 | Chloroform | 25 | 15 min | 99.4 | 2200 | 1.52 | 0.41 |
| P12 | 2500 | 5 | DMSO | 25 | 6 h | <2.0 | n/dd | n/dd | n/dd |
| P13e | 2500 | 5 | Anisole | 25 | 10 min | >99.9 | 2360 | 1.39 | 0.42 |
| P14e | 2500 | 5 | Ethyl acetate | 25 | 24 h | >99.9 | 2190 | 1.08 | 0.43 |
| P15e | 2500 | 5 | MEK | 25 | 24 h | 42.3 | 850 | 1.28 | n/dd |
| P16f | 2500 | 5 | Toluene | 25 | <1 min | >99.9 | 2450 | 1.53 | 0.41 |
However, the molecular weights of the obtained branched cyclic polymers were independent of the catalyst and monomer concentrations despite the high monomer conversion.
Three polymerization temperatures (i.e., −40, 0, and 25 °C) were employed to investigate the effect of temperature on the polymerization kinetics and the degree of branching (DB) of the polymer chain (P3 and P9–P10 in Table 1 and Fig. S6–S7A†). Obviously, the polymerization rate increased with the reaction temperature because of the self-acceleration due to the exothermic nature of the BCF-catalyzed CROP. Particularly, the metal-free CROP of GD at room temperature was completed within 1 min and yielded the most controlled molecular weight dispersity (P10 in Table 1, conversion >99.9%, Mn,SEC = 3800, Mw/Mn = 1.17). Moreover, the high reaction temperature slightly increased the DB of the polymer, as expected (Table S1† and Fig. 1A).27 Interestingly, although the DB values of P9 and P10 were relatively greater than that of P3, the composition of the L1,3 unit on the branched cyclic polymer chain was constant, indicating that only the L1,4 unit participated in the formation of additional T and D units. This difference is likely due to the different reactivities of the secondary hydroxyl group in the L1,4 unit and the primary hydroxyl group in the L1,3 unit. Furthermore, the composition of the L1,4 unit decreased to the same extent that the D unit increased, suggesting that no other secondary alcohol from the T1 unit in the propane-1,2-diol group participated in the formation of the additional T and D units.
 | ||
| Fig. 1 Structural analyses of (A) inverse-gated 13C NMR spectra (DMSO-d6, 25 °C) and (B) representative MALDI-ToF spectra of the P10 and P16 samples in the mass region from 2300 to 2500 Da. | ||
The careful selection of the solvent polarity for cationic polymerization is essential for achieving successful ionic polymerization as it influences the formation and stabilization of ion pairs. In particular, a polar cyclic solvent (e.g., tetrahydrofuran or dioxane) not only affects the polymerization system by, for example, stabilizing the initiating or propagating ion pair, but also acts as a comonomer for CROP.15 Thus, based on their polarity, toluene, chloroform, and dimethyl sulfoxide (DMSO) were employed as solvents to analyze the effects of solvent polarity on the metal-free CROP of GD (P10–P12 in Table 1 and Fig. S7–S9†). The polymerization in toluene completed within 1 min, resulting in well-controlled branched cyclic polymers (P10 in Table 1; conversion >99.9%, Mn,SEC = 3800, Mw/Mn = 1.17). Similarly, GD was consumed with a high conversion and afforded a comparable DB value in chloroform (P11 in Table 1; conversion = 99.4%, Mn,SEC = 2200, Mw/Mn = 1.52). However, the polymerization rate decreased, and the polymer exhibited broad dispersity. In contrast, no GD polymerization occurred in DMSO over a period of 6 h (P12 in Table 1; conversion <2.0%). This result is likely due to the decrease in the reactivity of the BCF catalyst due to the formation of a highly stable complex with DMSO or the stabilization of the zwitterionic intermediate by the polar solvent.
Next, our effort was then directed towards expanding the solvent scope to improve the green credentials of the process.28 For the choice of a green solvent, there was a limitation that the catalyst should not be affected by solvents such as alcohols. Thus, based on the polarity, anisole, ethyl acetate (EA), and methyl ethyl ketone (MEK) were employed as green solvents for the metal-free CROP of GD (P13–P15 in Table 1 and Fig. S10A†). The polymerizations successfully occurred within 10 min in anisole and 24 h in EA, resulting in fairly controlled PGDs (P13 in Table 1; conversion >99.9%, Mn,SEC = 2360, Mw/Mn = 1.39 and P14 in Table 1; conversion >99.9%, Mn,SEC = 2190, Mw/Mn = 1.08). However, the polymerization of GD in MEK showed a relatively low monomer conversion after 24 h (P15 in Table 1; conversion >42.3%, Mn,SEC = 850, Mw/Mn = 1.28). It is likely due to the higher polarity of MEK than the other solvents, which might affect the zwitterionic intermediate during propagation.
Typical cationic polymerization requires low polymerization temperatures and the rigorous purification of the monomers and solvents. However, encouraged by the high catalytic efficiency and good tolerance of the BCF catalyst toward its environment, we performed the polymerization of GD under air using the non-purified, as-received GD and solvent (P16 in Table 1 and Fig. 1).
Surprisingly, BC-PGD was obtained by large-scale (200 g-scale monomer) polymerization within 1 min (conversion >99.9%, Mn,SEC = 2450, Mw/Mn = 1.53). The dispersity of the obtained BC-PGD was slightly broader than that of the BC-PGD prepared under inert conditions (P10 in Table 1, conversion >99.9%, Mn,SEC = 3800, Mw/Mn = 1.17). Moreover, the structural changes of P16, such as changes in the composition of D, L, and T units and in the ratio between BC-PGD and ring-opened BC-PGD due to humidity (i.e., hb-PGD), were not observed, as confirmed by inverse-gated 13C NMR and MALDI-ToF MS (Table S1 and Fig. 1, S11 and S12†). In general, while MALDI-ToF MS can be regarded to be limited for quantitative analysis, there are many studies on the applications of MALDI-ToF for the evaluation of the absolute molecular weight and end group analyses for polymers as well as the confirmation of polymer topologies (e.g. cyclic or linear).18,29 As such, here we assessed the relative content of the prepared polymer with different structures in a quantitative manner. For example, in the MALDI-ToF spectra, the major series (blue circles in Fig. 1B) were observed, comprising regularly spaced peaks corresponding to the GD mass interval (74.04). Moreover, the absolute m/z value of 2392.26 corresponds to a BC-PGD chain with 32 GD units and Na+ as a countercation. Additionally, two minor series (red squares and green stars in Fig. 1B) were observed, indicating the presence of hb-PGD (observed m/z: 2410.34 for P10 and 2410.37 for P16; [32 GD units + H2O + Na+]) and dehydrated BC-PGD, respectively, which possibly stemmed from the dehydration of the water molecules from the numerous alcohol groups on the polymer chain upon strong laser irradiation during the measurement (observed m/z: 2374.37 for P10 and 2374.28 for P16; [32 GD units − H2O + Na+]).18,29 Interestingly, although P16 was synthesized using non-purified GD and solvent under air, the fraction of hb-PGD was nearly as small as that of P10. Thus, although the air and humidity in the non-purified GD and toluene might apparently affect the polymerization, the fast polymerization rate of the exothermic reaction and phase separation between the hydrophilic PGD and nonpolar solvent after the reaction prevent the side reactions.
Generally, recyclable catalysis systems have not been employed for ionic polymerization due to the concerns about the decreased reactivity of active species and the tedious purification steps. As discussed in the preceding section, however, the BCF catalyst exhibits high air and humidity tolerance, affording successful polymerization with unpurified reagents under ambient conditions. Moreover, the growing PGD chains in toluene were automatically isolated from the catalyst solution with an increasing molecular weight and hydrophilicity. Thus, our effort was then directed to a recyclable polymerized catalysis system using a decanted BCF solution.
In a typical run, the first polymerization was performed using unpurified reagents under air, successfully forming BC-PGDs within 10 min at room temperature (Fig. 2, S13 and Table S2;† conversion >99.9%, Mn,SEC = 2040, Mw/Mn = 1.53, DB = 0.40).
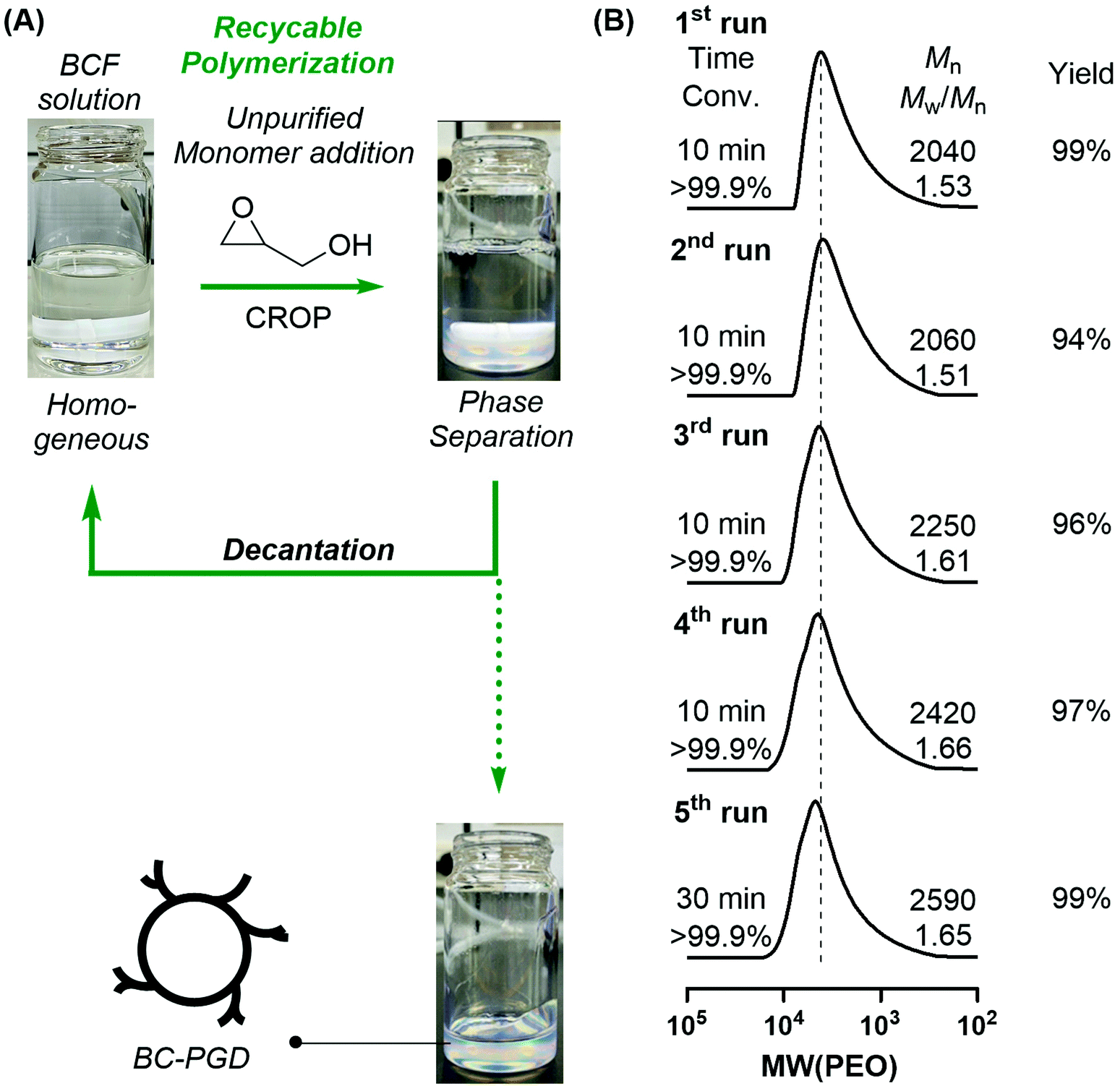 | ||
| Fig. 2 (A) Recyclable metal-free catalytic system for the CROP of GD, (B) SEC curves for the obtained PGDs calibrated with PEO standards in DMF (1st polymerization: [GD]0 = 500 mM; [B(C6F5)3]0 = 5 mM in toluene; 2nd–5th polymerization: [GD]add = 500 mM). | ||
After the first polymerization, the solution was decanted for reuse as the polymerization catalyst solution. The second to fifth polymerizations were then successively conducted by adding identical amounts of the unpurified GD monomer and performing decantation after the phase separation. Interestingly, the polymers obtained from the recycling process exhibited similar molecular weights and DB values to those of the first polymerization (2nd run: Mn,SEC = 2060, Mw/Mn = 1.51, DB = 0.39; 3rd run: Mn,SEC = 2250, Mw/Mn = 1.61, DB = 0.39; 4th run: Mn,SEC = 2420, Mw/Mn = 1.66, DB = 0.37; 5th run: Mn,SEC = 2590, Mw/Mn = 1.65, DB = 0.39). The reaction time increased with the number of polymerizations, which were performed for 10–30 min; this was probably due to the loss of the BCF catalyst because of trapping on the polymer phase.
Thus, the catalyst residues were confirmed in the obtained PGDs using the absorption spectra (Fig. S14 and S15†). Furthermore, to investigate the quantitative catalyst residue in the polymer phase depending on the number of reuse, the absorption spectrum of the PGDs obtained after polymerization was obtained (10 mg mL−1). After the first polymerization, the BCF absorbance at 204 nm was compared with the calibration curve according to the BCF concentration, showing BCF trapping of 7.2 mg (∼10% of the initial amount) in 1.0 g of PGD. The amount of the trapped BCF was similar even after the fifth polymerization (6.2 mg in 1.0 g of PGD), denoting that a certain amount of BCF (∼10% of the initial amount) was trapped in the polymer phase for every polymerization. Moreover, the BCF concentration in the supernatant gradually decreased with increasing polymerization time; however, the polymerization could be completed within 30 min even after five repetitions in the presence of a recycled catalyst.
The samples were then further investigated for the reusability of the BCF catalyst by 19F NMR spectroscopy (200 mg mL−1) using sodium trifluoroacetate (Na-TFA) as the standard. As a result, from the third polymerization, the peak splitting was observed indicating that the BCF catalyst was affected by humidity and air. However, the major peaks in the spectra were still from the BCF catalyst, indicating the BCF catalyst can be reusable (Fig. S16†).
Similarly, anisole was used as an alternative green solvent to determine the recyclability as compared to toluene. After repeated polymerizations, BC-PGDs were successfully obtained in anisole indicating that recyclable polymerization is applicable in the green solvent (Fig. S10B†).
The study results show that controlling the polymerization parameters, such as the monomer and catalyst concentrations, reaction temperature, and solvent polarity, is important for fabricating BC-PGDs. Notably, the ambient conditions with unpurified reagents, comprised air and humidity, did not clearly affect the polymerization even in the recycling process (e.g., propagation disturbance, formation of hb-PGD, and structural changes). Therefore, this recyclable metal-free catalytic system has great potential as a green CROP system, and its application can be expanded to various industrial fields due to its environmental and economic impacts.
Conclusions
BC-PGDs were successfully synthesized via the metal-free CROP of GD as an AB2-type monomer, forming branches on the cyclic PGD with numerous hydroxyl end groups. The BCF-catalyzed CROP afforded a well-controlled BC-PGD structure by large-scale polymerization even when using unpurified reagents and under ambient conditions. Particularly, recyclable polymerization was successfully achieved by repeating the simple sequence of decantation and addition of the unpurified monomer. The interesting structure of the obtained PGDs was confirmed by 1H NMR, inverse-gated 13C NMR analyses, MALDI-ToF, and size-exclusion chromatography (SEC). We believe that the green and recyclable BCF-catalyzed CROP with the simple and well-reproducible polymerization technique will afford intriguing examples of challenging industrial applications.Author contributions
The manuscript was written through the contribution of all authors. All authors have given approval to the final version of the manuscript. S. E. K.: experimentation, analysis, and writing; H. J. Y., S. C., E. H., and M. K.: experimentation and analysis; H.-J. P., J.-E. J., and Y. I. P.: analysis; J. C. K.: conceptualization and supervision; B.-S. K. and S.-H. L.: conceptualization, supervision, and editing.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This study was supported by the Ministry of Trade, Industry & Energy (MOTIE, Korea) under the Industrial Technology Innovation Program (No. 20011123), a National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2020R1C1C1005569), and the Korea Research Institute of Chemical Technology (KRICT) (No. KS2041-00). This work was also supported by the NRF (NRF-2021R1A2C3004978).References
- V. K. Dioumaev and R. M. Bullock, Nature, 2003, 424, 530–532 CrossRef CAS.
- S. M. Mercer, T. Robert, D. V. Dixon and P. G. Jessop, Catal. Sci. Technol., 2012, 2, 1315–1318 RSC.
- J. Steinbauer, L. Longwitz, M. Frank, J. Epping, U. Kragl and T. Werner, Green Chem., 2017, 19, 4435–4445 RSC.
- C.-G. Wang, J. J. Chang, E. Y. J. Foo, H. Niino, S. Chatani, S. Y. Hsu and A. Goto, Macromolecules, 2020, 53, 51–58 CrossRef CAS.
- A. Kanazawa, S. Kanaoka and S. Aoshima, J. Am. Chem. Soc., 2007, 129, 2420–2421 CrossRef CAS.
- D.-H. Chang, D.-Y. Lee, B.-S. Hong, J.-H. Choi and C.-H. Jun, J. Am. Chem. Soc., 2004, 126, 424–425 CrossRef CAS PubMed.
- Y.-H. Fu, C. Perales, T. Eliason and D. E. Bergbreiter, Ind. Eng. Chem. Res., 2019, 58, 14579–14587 CrossRef CAS.
- T. J. Malinski, H. S. Bazzi and D. E. Bergbreiter, ChemSusChem, 2019, 12, 416–419 CrossRef CAS.
- D. Chouikhi, I. Kulai, D. E. Bergbreiter, M. Al-Hashimi and H. S. Bazzi, Appl. Sci., 2019, 9, 120 CrossRef CAS.
- S. Boileau and N. Illy, Prog. Polym. Sci., 2011, 36, 1132–1151 CrossRef CAS.
- E. Mohammadifar, A. Bodaghi, A. Dadkhahtehrani, A. Nemati Kharat, M. Adeli and R. Haag, ACS Macro Lett., 2017, 6, 35–40 CrossRef CAS.
- D. Wilms, S.-E. Stiriba and H. Frey, Acc. Chem. Res., 2010, 43, 129–141 CrossRef CAS PubMed.
- J. Song, L. Palanikumar, Y. Choi, I. Kim, T.-Y. Heo, E. Ahn, S.-H. Choi, E. Lee, Y. Shibasaki, J.-H. Ryu and B.-S. Kim, Polym. Chem., 2017, 8, 7119–7132 RSC.
- A. T. Royappa, M. L. Vogt and V. Sharma, J. Appl. Polym. Sci., 2004, 91, 1344–1351 CrossRef CAS.
- P. Kubisa, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier, Amsterdam, 2012, vol. 4, pp. 141–164 Search PubMed.
- C. H. Tran, M. W. Lee, S. A. Kim, H. B. Jang and I. Kim, Macromolecules, 2020, 53, 2051–2060 CrossRef CAS.
- B. R. Spears, M. A. Marin, J. R. Montenegro-Burke, B. C. Evans, J. McLean and E. Harth, Macromolecules, 2016, 49, 2022–2027 CrossRef CAS.
- I. Asenjo-Sanz, A. Veloso, J. I. Miranda, J. A. Pomposo and F. Barroso-Bujans, Polym. Chem., 2014, 5, 6905–6908 RSC.
- I. Asenjo-Sanz, A. Veloso, J. I. Miranda, A. Alegría, J. A. Pomposo and F. Barroso-Bujans, Macromolecules, 2015, 48, 1664–1672 CrossRef CAS.
- F. M. Haque, C. M. Schexnayder, J. M. Matxain, F. Barroso-Bujans and S. M. Grayson, Macromolecules, 2019, 52, 6369–6381 CrossRef CAS.
- J. R. Lawson and R. L. Melen, Inorg. Chem., 2017, 56, 8627–8643 CrossRef CAS PubMed.
- L. A. Körte, J. Schwabedissen, M. Soffner, S. Blomeyer, C. G. Reuter, Y. V. Vishnevskiy, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2017, 56, 8578–8582 CrossRef.
- D. Hölter, A. Burgath and H. Frey, Acta Polym., 1997, 48, 30–35 CrossRef.
- R. Tokar, P. Kubisa, S. Penczek and A. Dworak, Macromolecules, 1994, 27, 320–322 CrossRef CAS.
- A. Dworak, W. Walach and B. Trzebicka, Macromol. Chem. Phys., 1995, 196, 1963–1970 CrossRef CAS.
- M. Gosecki, M. Gadzinowski, M. Gosecka, T. Basinska and S. Slomkowski, Polymer, 2016, 8, 227 Search PubMed.
- S.-E. Stiriba, H. Kautz and H. Frey, J. Am. Chem. Soc., 2002, 124, 9698–9699 CrossRef CAS PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- J. Liu, R. S. Loewe and R. D. McCullough, Macromolecules, 1999, 32, 5777–5785 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc03063a |
| This journal is © The Royal Society of Chemistry 2022 |