
K2S2O8-induced site-selective phenoxazination/phenothiazination of electron-rich anilines†
He
Zhang‡
a,
Shengchun
Wang‡
a,
Xiaoyu
Wang
b,
Pengjie
Wang
a,
Hong
Yi
a,
Heng
Zhang
 *a and
Aiwen
Lei
*ab
*a and
Aiwen
Lei
*ab
aThe Institute for Advanced Studies (IAS), College of Chemistry and Molecular Sciences, Wuhan University, Wuhan, 430072, P.R. China. E-mail: aiwenlei@whu.edu.cn; hengzhang@whu.edu.cn
bNational Research Center for Carbohydrate Synthesis, Jiangxi Normal University, Nanchang, 330022, P.R. China
First published on 4th December 2021
Abstract
By just using cheap K2S2O8 as the oxidant at room temperature in the air, the phenoxazination/phenothiazination of electron-rich anilines to construct or modify triarylamine derivatives has been established. This method demonstrates complete para-selective amination under catalyst-free conditions and its simplicity and efficiency lead to good performance in flow-chemistry synthesis.
10-Phenylphenothiazine derivatives are important structural units in photocatalysts,1 dye-sensitized solar cells2 and OLEDs (organic light-emitting diodes)3 (Scheme 1). Traditionally, these derivatives were constructed via transition metal-catalyzed reactions,4 such as Buchwald–Hartwig, Ullmann and Chan–Lam cross-couplings. Although these methods demonstrate efficient site-selective aromatic amination, the utilization of active substrates, such as aryl-halides or aryl-organoborons, is normally essential in those transformations. As an atom-economical strategy, undirected aromatic amination via Csp2–H/N–H cross-coupling has attracted much attention from chemists.5 To date, photoredox6 and electrosynthetic7 chemistry have advanced the state-of-the-art in this research area. Despite being advanced conceptually, it is still challenging to realize these strategies in industry, partly due to the high demands of the reaction device. Thus, an easy-to-handle, scalable and green method to synthesize 10-phenylphenothiazine derivatives is highly desirable.
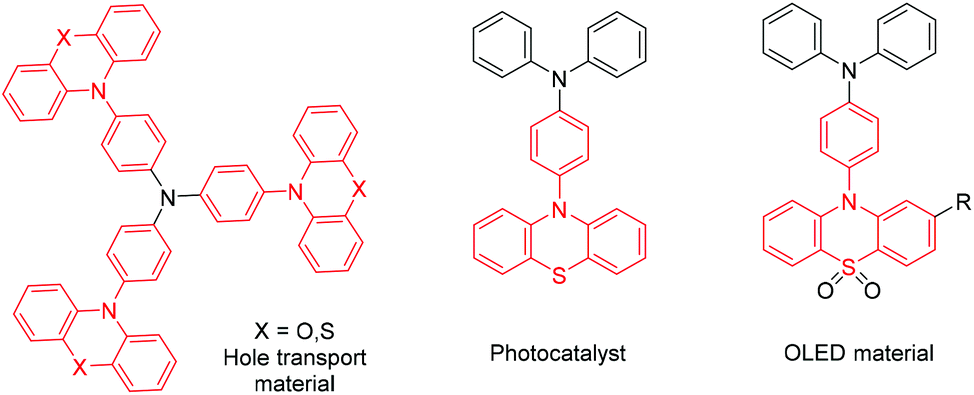 | ||
| Scheme 1 Examples of important functional small molecules. | ||
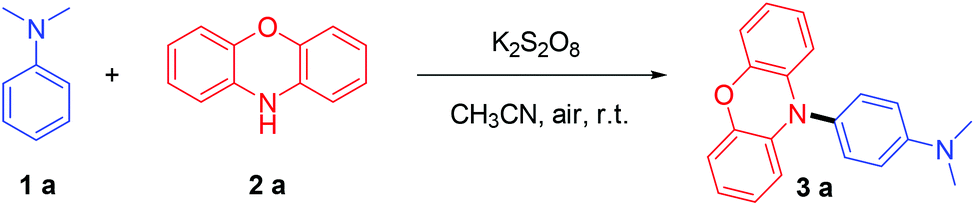
To find an efficient way to achieve this goal, we re-examined the classic Boyland–Sims oxidation reaction.8 Although the product yields are typically low to moderate, the simplicity of the reactions means that they are frequently used (Scheme 2A). In particular, in non-aqueous media, the oxidative coupling of an arylamine leads to the formation of the corresponding dimer or high oligomers and has been widely applied in the synthesis of polyaniline (PANI)9 (Scheme 2B). Thus, we envision that aniline amination via a similar procedure might enable low-cost and powerful applications. However, we are still faced with two challenges: (i) the control of site-selective amination; (ii) how to avoid the polymerization of aniline. These two issues limit the development of efficient amination protocols for anilines. According to our previous electrochemistry and EPR study,7a compared with N,N-dimethylaniline, phenothiazines are easier to be oxidised to stable nitrogen-centered radical intermediates, and these radicals could selectively react with aniline radical cations to construct C–N bonds in high regioselectivity. With such mechanistic insight, herein, we developed an easy-to-handle and efficient method for the preparation of triarylamine derivatives using cheap K2S2O8 as an oxidant. Phenoxazine was used as the amination reagent so that triarylamine blocks were also constructed. Compared with the electrochemical protocol, this procedure is cheaper, easier and does not require special reaction devices. This procedure is more accessible to those researchers who don't have an electrochemical synthesis device. Furthermore, this reaction works well in flow-chemistry reactors, which means that this reaction has potential to be useful for industrial applications.
 | ||
| Scheme 2 Schematic summary of previous work and this work. | ||
In this study, we simply mixed N,N-dimethylaniline (1a, 0.2 mmol), phenoxazine (2a, 0.3 mmol) and K2S2O8 (0.4 mmol) in CH3CN (3 mL). After stirring for 1 h under air at room temperature, a 92% isolated yield of 3a could be obtained (Table 1, entry 1). Good reactivity could still be achieved using other solvents (Table 1, entries 2–4), such as MeOH, DMF or CH3CN & H2O mixed solvent. Notably, the good tolerance toward water means that anhydrous operation is not needed, which is beneficial for the simplification of the experimental procedures. The effect of the amount of 2a was also explored (Table 1, entries 5–7). The results demonstrated that more 2a leads to better yields, because 2a could be overoxidised or polymerized5f under oxidising conditions. Subsequently, other kinds of persulfate were also examined (Table 1, entries 8–10), lower yields were obtained when K2S2O8 was replaced by Na2S2O8, (NH4)2S2O8 or oxone, indicating that K2S2O8 was the best among the tested persulfates. No desired product was isolated when K2S2O8 was omitted (Table 1, entry 11), and a slightly decreased yield was obtained under the N2 atmosphere (Table 1, entry 12).
|
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Yield [%] |
| a Reaction conditions: 1a (0.2 mmol), 2a (1.5 equiv.), K2S2O8 (2.0 equiv.), CH3CN (3.0 mL), room temperature, air, 1 h. Isolated yields. N.D. = not detected. | ||
| 1 | NONE | 92 |
| 2 | MeOH (3.0 mL) instead of CH3CN | 83 |
| 3 | DMF (3.0 mL) instead of CH3CN | 79 |
| 4 | CH3CN/H2O (1.5 mL/1.5 mL) instead of CH3CN | 66 |
| 5 | 1.0 equiv. 2a was added | 76 |
| 6 | 1.25 equiv. 2a was added | 86 |
| 7 | 2.0 equiv. 2a was added | 94 |
| 8 | Na2S2O8 instead of K2S2O8 | 89 |
| 9 | (NH4)2S2O8 instead of K2S2O8 | 78 |
| 10 | Oxone instead of K2S2O8 | 71 |
| 11 | No K2S2O8 | N.D. |
| 12 | Reaction performed in N2 | 88 |
The scope of the direct para C–H amination of aniline derivatives was then explored. As shown in Table 2, the effect of N-substituted groups was firstly investigated. N,N′-Dialkyl substituted and N-alkyl-N-arene substituted anilines were well tolerated in this protocol (Table 2. 3a, 3b, 3e & 3o). However, only a trace amount of product could be detected when triphenylamine was applied (3c), and a large amount of triphenylamine remained, which was been evidenced by GC-MS. As for unsubstituted or N-methyl-N-acetyl aniline, no desired product was observed (3d, 3f); it was speculated that the anilines were completely over oxidised or polymerized because no starting materials could be detected by GC-MS. Then, phenothiazines were also explored; 3g could be afforded in 67% yield, but only 37% yield could be achieved for 2-Cl-phenthiazine. When phenothiazines containing electron-withdrawing substituents were tested, 50–61% isolated yields could be finally obtained (3i, 3j & 3k) after extending the reaction time from 1 to 4 hours. Unfortunately, 2-ethylthiophenothiazine was not compatible with this transformation. Next, we further tested some bioactive molecules, N-methyl-1,2,3,4-tetrahydroquninoline and 4-methyl-3,4-dihydro-2H-benzo[b][1,4]oxazine, which selectively furnished the para amination products with moderate yields (3n, 3q). Notably, the reaction with 2-phenylindole afforded the selective amination product in the 3-position in 96% isolated yield (3p) and a 33% yield could be obtained when 1,2-dimethlyindole (3r) was selected as the substrate. As for the anilines with electron-withdrawing groups (3s, 3t & 3u), no desired products were detected.

Then, the model reaction was carried out on a 5 mmol scale. Due to the existence of side reactions caused by uneven stirring (see ESI Fig. S3†), only a 51% isolated yield of 3a could be obtained by increasing the amount of solvent and substrates (Scheme 3a). Thus, we carried out this reaction in a flow-chemistry reactor (Scheme 3c), so that the substrates can be better mixed. This transformation is so efficient and fast that a 73% isolated yield of 3a could be obtained in a 1 gram (5 mmol) scale reaction after 1 hour. Furthermore, when using phenothiazine as the substrate, 3g could be obtained quickly in 61% (62%) GC yield (15 g scale). These results show the potential for further industrialization of this strategy.
 | ||
| Scheme 3 Scale-up reaction [a]: reaction conditions 1a (5.0 mmol), 2a (1.5 equiv.), K2S2O8 (2.0 equiv., add step by step in 4 h), CH3CN (50 mL), room temperature, air, 6 h. [b] Flow-chemistry reactor. [c and d] Reaction performed in flow-chemistry reactor, a: the reaction was carried out in reactor B; b: the reaction was carried out in reactor C, see ESI† for more details. | ||
Triphenylamine derivatives have been reported to serve as potential optoelectronic materials.3 However, direct modification of triphenylamine remains a challenge because of its good chemical stability. To our delight, when we selected 4,4′-dimethlytriphenyamine and 2-cyano-phenothiazine as the substrates, changed the solvent to a mixture of CH3CN and acetic acid, and extended the reaction time to 24 hours, direct modification of triphenylamine could also be achieved via this procedure, while it could not be achieved via electrochemistry. Then, the substituent effect was studied (Table 3). Triphenylamine only afforded the single amination product 5b in 31% yield, while 4,4′-dimethoxytriphenyamine afforded 5c in 67% yield. As for phenothiazine derivatives, 4-chloride or 4-trifluoromethyl phenothiazine only gave the amination product in low yields (5d, 5e), while 4-acetylphenothiazine gave a moderate yield (4f). Thus, we could conclude that electron-rich triphenylamines and electron-deficient phenothiazines were well-matched in this reaction system, which leads to a moderate yield. Otherwise, low yield or only trace amounts of product would be obtained.
| a Reaction conditions: 4 (1.0 equiv., 0.2 mmol), 2 (1.5 equiv., 0.3 mmol), K2S2O8 (2.0 equiv., 0.4 mmol), CH3CN/AcOH (1.5 mL/1.5 mL), room temperature, air, 24 h. Yields of isolated products are shown. |
|---|

|
Having established the method, our attention turned to understanding the mechanism. As a powerful method, EPR (electron paramagnetic resonance) was used to study the radical pathway. Obviously, a N-centered radical was detected by EPR (Scheme 4, AN = 8.0 g, AH = 3.8 g, AH = 3.8 g, AH = 3.8 g, AH = 3.8 g), when K2S2O8 and phenoxazine were mixed. However, when K2S2O8 and N,N-dimethyl aniline were mixed, no signal could be detected. This result was consistent with the Boyland–Sims reaction, in which a zwitterionic intermediate was formed instead of a radical. N-centered radicals mainly existed when three compounds were mixed together and it proved that phenoxazine was easier to be oxidized by K2S2O8 than N,N-dimethyl aniline.
 | ||
| Scheme 4 Electron paramagnetic resonance (EPR) results; the substrates were mixed in MeOH. | ||
To further verify the reaction mechanism, DFT calculations was carried out. As shown in Scheme 5, the reaction initiates from the single electron transfer between phenoxazine and K2S2O8. The free energy barrier for this single electron process is estimated to be 18.8 kcal mol−1 (see the ESI† for details). The following deprotonation of the phenoxazine radical cation would yield the phenoxazine N radical. The single electron transfer between N,N-dimethylaniline and the KSO4 radical is strongly exergonic (−20.2 kcal mol−1). The following cross-coupling between the phenoxazine N radical and N,N-dimethylaniline radical cation only has a free energy barrier of 8.2 kcal mol−1. The subsequent deprotonation is also strongly exergonic (−34.8 kcal mol−1), with the concomitant formation of the target product. DFT calculations proved that radical/radical cross-coupling is feasible.
 | ||
| Scheme 5 Free energy profiles for the proposed mechanism. Relative energies in kcal mol−1. | ||
Based on these results, a plausible mechanism between 1a and 2a was proposed, as shown in Scheme 6. Firstly, 2a was oxidised by K2S2O8 to generate the the nitrogen radical 6a. At the same time, the radical cation 6b was formed via single electron transfer. The C–N bond was likely to be formed through the selective radical/radical cross-coupling between nitrogen radical 6a and the radical cation 6b. This unique reaction pathway might explain the exclusive para-selectivity for this oxidative C–H/N–H cross-coupling.
 | ||
| Scheme 6 Plausible mechanism. | ||
Conclusions
In summary, we have developed a practical and site-selective amination reaction which is compatible with air and water. Combined with flow-chemistry, this reaction was successfully carried out on a 50 mmol scale, proving its potential for industrialization. Furthermore, the direct modification of triphenylamines, which had seldom been reported, could also be achieved by this novel method. Mechanistic studies proposed that this reaction proceeds via a radical/radical cation cross-coupling.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China No. 2021YFA1500100), National Natural Science Foundation of China (22031008) and Science Foundation of Wuhan (2020010601012192).Notes and references
- (a) K. Targos, O. P. Williams and Z. K. Wickens, J. Am. Chem. Soc., 2021, 143, 4125–4132 CrossRef CAS PubMed; (b) N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. Read de Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS PubMed; (c) E. H. Discekici, N. J. Treat, S. O. Poelma, K. M. Mattson, Z. M. Hudson, Y. Luo, C. J. Hawker and J. R. de Alaniz, Chem. Commun., 2015, 51, 11705–11708 RSC; (d) S. Shibutani, T. Kodo, M. Takeda, K. Nagao, N. Tokunaga, Y. Sasaki and H. Ohmiya, J. Am. Chem. Soc., 2020, 142, 1211–1216 CrossRef CAS PubMed; (e) F. Speck, D. Rombach and H.-A. Wagenknecht, Beilstein J. Org. Chem., 2019, 15, 52–59 CrossRef CAS PubMed; (f) L. Mayer, L. May and T. J. J. Müller, Org. Chem. Front., 2020, 7, 1206–1217 RSC.
- (a) P. Agarwala and D. Kabra, J. Mater. Chem. A, 2017, 5, 1348–1373 RSC; (b) Z. Fang, V. Chellappan, R. D. Webster, L. Ke, T. Zhang, B. Liu and Y.-H. Lai, J. Mater. Chem., 2012, 22, 15397–15404 RSC; (c) A. Higuchi, H. Inada, T. Kobata and Y. Shirota, Adv. Mater., 1991, 3, 549–550 CrossRef CAS; (d) X. Liu, J. Long, G. Wang, Y. Pei, B. Zhao and S. Tan, Dyes Pigm., 2015, 121, 118–127 CrossRef CAS.
- J. Su, H.-J. Jiang, J. Zhang, Y. Tao and R. Chen, New J. Chem., 2013, 37, 977–985 RSC.
- (a) J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534–1544 CrossRef CAS PubMed; (b) C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC; (c) D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338–6361 CrossRef CAS PubMed; (d) M. J. West, J. W. B. Fyfe, J. C. Vantourout and A. J. B. Watson, Chem. Rev., 2019, 119, 12491–12523 CrossRef CAS PubMed.
- For some review papers: (a) K. Murakami, G. J. P. Perry and K. Itami, Org. Biomol. Chem., 2017, 15, 6071–6075 RSC; (b) Y. Yang, D. Zhang and E. Vessally, Top. Curr. Chem., 2020, 378, 37 CrossRef CAS PubMed. Some examples for C–H amination: (c) L. Bering, L. D'Ottavio, G. Sirvinskaite and A. P. Antonchick, Chem. Commun., 2018, 54, 13022–13025 RSC; (d) M.-L. Louillat-Habermeyer, R. Jin and F. W. Patureau, Angew. Chem., Int. Ed., 2015, 54, 4102–4104 CrossRef CAS PubMed; (e) P. Y. Vemuri, Y. Wang and F. W. Patureau, Org. Lett., 2019, 21, 9856–9859 CrossRef CAS PubMed; (f) F. W. Patureau, ChemCatChem, 2019, 11, 5227–5231 CrossRef CAS.
- For some photocatalyst C–H amination: (a) E. Ito, T. Fukushima, T. Kawakami, K. Murakami and K. Itami, Chem, 2017, 2, 383–392 CrossRef CAS; (b) L. Niu, H. Yi, S. Wang, T. Liu, J. Liu and A. Lei, Nat. Commun., 2017, 8, 14226 CrossRef PubMed; (c) N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Science, 2015, 349, 1326–1330 CrossRef CAS PubMed; (d) L. Zhang, L. Liardet, J. Luo, D. Ren, M. Grätzel and X. Hu, Nat. Catal., 2019, 2, 366–373 CrossRef CAS PubMed; (e) Y. Zhao, B. Huang, C. Yang and W. Xia, Org. Lett., 2016, 18, 3326–3329 CrossRef CAS PubMed; (f) Y.-W. Zheng, B. Chen, P. Ye, K. Feng, W. Wang, Q.-Y. Meng, L.-Z. Wu and C.-H. Tung, J. Am. Chem. Soc., 2016, 138, 10080–10083 CrossRef CAS PubMed.
- For some electrochemical C–H amination: (a) K. Liu, S. Tang, T. Wu, S. Wang, M. Zou, H. Cong and A. Lei, Nat. Commun., 2019, 10, 639 CrossRef PubMed; (b) S. Tang, S. Wang, Y. Liu, H. Cong and A. Lei, Angew. Chem., Int. Ed., 2018, 57, 4737–4741 CrossRef CAS PubMed; (c) Y.-C. Wu, S.-S. Jiang, R.-J. Song and J.-H. Li, Chem. Commun., 2019, 55, 4371–4374 RSC.
- (a) E. J. Behrman, Beilstein J. Org. Chem., 2006, 2, 22 CAS; (b) E. J. Behrman, J. Phys. Chem. A, 2011, 115, 7863–7864 CrossRef CAS PubMed; (c) B. Marjanović, I. Juranić and G. Ćirić-Marjanović, J. Phys. Chem. A, 2011, 115, 3536–3550 CrossRef PubMed; (d) B. Marjanović, I. Juranić and G. Ćirić-Marjanović, J. Phys. Chem. A, 2011, 115, 7865–7868 CrossRef; (e) E. J. Behrman, J. Am. Chem. Soc., 1967, 89, 2424 CrossRef CAS.
- (a) A. Janošević, G. Ćirić-Marjanović, B. Š. Paunković, I. Pašti, S. Trifunović, B. Marjanović and J. Stejskal, Synth. Met., 2012, 162, 843–856 CrossRef; (b) E. P. Koval'chuk, S. Whittingham, O. M. Skolozdra, P. Y. Zavalij, I. Y. Zavaliy, O. V. Reshetnyak and M. Seledets, Mater. Chem. Phys., 2001, 69, 154–162 CrossRef; (c) Y. Park, C. Park, J. S. Kim, T. K. Ahn, H. S. Lim, T.-W. Lee and W. Kwon, Adv. Opt. Mater., 2018, 6, 1800577 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc03896f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |