
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Expanding plastics recycling technologies: chemical aspects, technology status and challenges†
Houqian
Li
 a,
Horacio A.
Aguirre-Villegas
b,
Robert D.
Allen
cd,
Xianglan
Bai
e,
Craig H.
Benson
f,
Gregg T.
Beckham
cd,
Sabrina L.
Bradshaw
f,
Jessica L.
Brown
eg,
Robert C.
Brown
eg,
Victor S.
Cecon
h,
Julia B.
Curley
cd,
Greg W.
Curtzwiler
h,
Son
Dong
i,
Soumika
Gaddameedi
j,
John E.
García
a,
Ive
Hermans
ai,
Min Soo
Kim
a,
Jiaze
Ma
a,
Lesli O.
Mark
i,
Manos
Mavrikakis
a,
Olumide O.
Olafasakin
e,
Tim A.
Osswald
k,
Konstantinos G.
Papanikolaou
a,
Harish
Radhakrishnan
e,
Marco Antonio
Sanchez Castillo
l,
Kevin L.
Sánchez-Rivera
a,
Khairun N.
Tumu
h,
Reid C.
Van Lehn
a,
Keith L.
Vorst
h,
Mark M.
Wright
e,
Jiayang
Wu
a,
Victor M.
Zavala
a,
Panzheng
Zhou
a and
George W.
Huber
*a
a,
Horacio A.
Aguirre-Villegas
b,
Robert D.
Allen
cd,
Xianglan
Bai
e,
Craig H.
Benson
f,
Gregg T.
Beckham
cd,
Sabrina L.
Bradshaw
f,
Jessica L.
Brown
eg,
Robert C.
Brown
eg,
Victor S.
Cecon
h,
Julia B.
Curley
cd,
Greg W.
Curtzwiler
h,
Son
Dong
i,
Soumika
Gaddameedi
j,
John E.
García
a,
Ive
Hermans
ai,
Min Soo
Kim
a,
Jiaze
Ma
a,
Lesli O.
Mark
i,
Manos
Mavrikakis
a,
Olumide O.
Olafasakin
e,
Tim A.
Osswald
k,
Konstantinos G.
Papanikolaou
a,
Harish
Radhakrishnan
e,
Marco Antonio
Sanchez Castillo
l,
Kevin L.
Sánchez-Rivera
a,
Khairun N.
Tumu
h,
Reid C.
Van Lehn
a,
Keith L.
Vorst
h,
Mark M.
Wright
e,
Jiayang
Wu
a,
Victor M.
Zavala
a,
Panzheng
Zhou
a and
George W.
Huber
*a
aDepartment of Chemical and Biological Engineering, University of Wisconsin-Madison, Madison, WI 53706, USA. E-mail: gwhuber@wisc.edu
bBiological Systems Engineering, University of Wisconsin-Madison, Madison, WI 53706, USA
cRenewable Resources and Enabling Sciences Center, National Renewable Energy Laboratory, Golden, CO 80401, USA
dBOTTLE Consortium, National Renewable Energy Laboratory, Golden, CO 80401, USA
eDepartment of Mechanical Engineering, Iowa State University, Ames, IA 50011, USA
fDepartment of Civil and Environmental Engineering, University of Wisconsin-Madison, Madison, WI 53706, USA
gBioeconomy Institute, Iowa State University, Ames, IA 50011, USA
hPolymer and Food Protection Consortium, Department of Food Science and Human Nutrition, Iowa State University, Ames, IA 50011, USA
iDepartment of Chemistry, University of Wisconsin-Madison, Madison, WI, 53706 USA
jZoology and Environmental Studies, University of Wisconsin-Madison, Madison, WI 53706, USA
kDepartment of Mechanical Engineering, University of Wisconsin-Madison, Madison, WI 53706, USA
lFacultad de Ciencias Químicas, Universidad Autónoma de San Luis Potosí, San Luis Potosí 78210, Mexico
First published on 14th September 2022
Abstract
Less than 10% of the plastics generated globally are recycled, while the rest are incinerated, accumulated in landfills, or leak into the environment. New technologies are emerging to chemically recycle waste plastics that are receiving tremendous interest from academia and industry. Chemists and chemical engineers need to understand the fundamentals of these technologies to design improved systems for chemical recycling and upcycling of waste plastics. In this paper, we review the entire life cycle of plastics and options for the management of plastic waste to address barriers to industrial chemical recycling and further provide perceptions on possible opportunities with such materials. Knowledge and insights to enhance plastic recycling beyond its current scale are provided. Outstanding research problems and where researchers in the field should focus their efforts in the future are also discussed.
 Houqian Li | Houqian Li is currently a postdoctoral research associate at University of Wisconsin Madison in Chemical and Biological Engineering and Chemical Upcycling of Waste Plastics (CUWP) center working with Professor George Huber. His research focuses on chemical methods to recycle/upcycle biomass and waste plastics and also the mechanistic study of sustainable catalysis. He received his PhD in Chemical and Biological Engineering from Washington State University under the supervision of Prof. Yong Wang. |
 Jessica L. Brown | Jessica Brown received her Bachelor's Degree in chemical engineering in 2019 from Iowa State University. She is currently pursuing her Ph.D. at Iowa State University in mechanical engineering under Dr Robert Brown. Her research interests are upcycling plastic wastes using thermochemical deconstruction and hybrid processing. |
 Victor S. Cecon | Victor is a doctoral student and graduate research assistant in Food Science and Technology at Iowa State University (Ames, IA, USA) and is affiliated with the Polymer and Food Protection Consortium. His major professor is Dr Keith Vorst. He received his B.S. in Chemical Engineering from the Federal University of Sao Carlos (UFSCar, Brazil) in 2019, being a visiting scholar at the University of British Columbia (UBC, Canada) for one year. His current research is focused on the sorting, processing, and characterization of post-industrial and post-consumer plastic waste, including landfill-diverted waste streams from Materials Recovery Facilities (MRFs). |
 Julia B. Curley | Julia Curley received her PhD in inorganic chemistry from Yale University under the supervision of Prof. Nilay Hazari in September 2021. Her graduate research included synthetic, catalytic, and mechanistic studies of iron and ruthenium pincer complexes used in reactions relevant to renewable energy storage. She began her postdoc at the National Renewable Energy Laboratory in October 2021 under the guidance of Dr Gregg Beckham and Dr Robert Allen. Her current work focuses on the development of efficient and scalable systems for polyester chemical recycling. |
 Harish Radhakrishnan | Harish Radhakrishnan, born in 1996, obtained his master's degree in Mechanical Engineering from Iowa State University and has been working on his Ph.D. dissertation for the past three years. His dissertation concerns the thermochemical and plasma-based upcycling of post-consumer plastic wastes into fuels and chemicals for downstream processes. His research activities include pyrolyzing and liquefaction of polymers, modeling polymer pyrolysis and plasma-based deconstruction chemistries, and plasma reactor design. He also performs catalytic and non-catalytic experiments with gas–solid, liquid–solid, and gas–gas plasma interactions for various applications, including natural and synthetic polymer deconstruction, as well as CO2 conversion and NOx production. |
 Kevin L. Sánchez-Rivera | Kevin L. Sánchez-Rivera graduated from the University of Puerto Rico-Mayagüez in 2019 and joined Prof. George Huber's group at the University of Wisconsin–Madison in the Chemical and Biological Engineering Department. His current work focuses on developing the Solvent-Targeted Recovery and Precipitation (STRAP) technology to recycle different types of multilayer plastics, as part of the efforts of the Chemical Upcycling of Waste Plastics (CUWP) center. |
1. Introduction
Plastics have revolutionized our society, allowing us to inexpensively make a variety of materials that have a myriad of uses throughout industry. The first three synthetic plastics were polystyrene (PS) in 1839, polyvinyl chloride (PVC) in 1835 and Bakelite in 1907.1 Industrial plastic production really began to expand in about 1950 when global plastic production reached 2 million tons per year.2 Current global plastic production is around 380 million tons per year and it is estimated that humankind will have produced more than 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 million tons of plastics by the end of 2050.2 The growth in plastic production is occurring for three likely reasons:2–4 (1) new plastic materials (made by both changing the structure and processing conditions of the plastics) allow for the use of plastics in new applications, (2) the developing world is using more plastics as their economy improves, and (3) the decreasing cost of plastic production due to cheap fossil feedstocks.
000 million tons of plastics by the end of 2050.2 The growth in plastic production is occurring for three likely reasons:2–4 (1) new plastic materials (made by both changing the structure and processing conditions of the plastics) allow for the use of plastics in new applications, (2) the developing world is using more plastics as their economy improves, and (3) the decreasing cost of plastic production due to cheap fossil feedstocks.
Fig. 1 shows a Sankey diagram for the production, utilization and recycling of packaging plastics adapted from the Ellen McArthur Foundation.5 This shows that 32 percent of all plastics is mismanaged (littered or inadequately disposed).6 Between two to five percent of plastics are transported into the ocean through riverways.6 Any plastic waste on land has the potential to eventually reach the ocean as the ocean is the ultimate sink of the earth.7 In the environment, plastics will slowly degrade into microplastics, which are plastic fragments less than 5 mm in diameter. Recently, microplastics were even found in the troposphere.8 Microplastics can also enter the environment from the degradation of plastic clothing into wastewater.9 Degradation mechanisms include weathering, leaching, fragmentation and potentially assimilation and mineralization. Plastic leaks into the environment through improper disposal, transport of plastics from collection sites to a central facility, and leakage from the solid waste facilities. Ocean plastics have two well-studied impacts on marine and freshwater animals: waste ingestion–egestion of plastics and entanglement in plastic waste.7
 | ||
| Fig. 1 Sankey Diagram showing the life cycle of plastic packaging.5 | ||
Forty percent of plastics is landfilled. Developed countries often have landfills with thick plastic covering which prevents leakage into the environment.10 However, less developed countries often have open landfills where plastic can easily leak into the environment. In the US, which has a modern solid waste disposal infrastructure, it has been estimated that 0.98–1.26 million metric tons of plastic (or 2.33–2.99% of plastic waste generated) leak into the environment.11 Large amounts of plastics wastes are also released into the environment during natural disasters. For example, in 2011 the Tohoku earthquake and tsunami in Japan deposited an estimated 5 million tons of debris into the ocean. By 2030, 58.4 million tons of plastics will be added to the oceans each year around the world.7
Fourteen percent of plastics is burned for energy recovery. While this does not create a solid waste problem, it does generate greenhouse gases. Fourteen percent of plastics are “recycled” with 8% being “cascaded recycled” to lower quality materials and only 2% of plastics used in closed-loop recycling where virgin plastics can be used. The amount of plastics recycled varies by countries, with the US recycling 10% and the EU 31% of plastics consumed.4 As will be discussed in this review, the low amount of recycled plastics is due to several reasons including: (1) plastics have a wide variety of compositions, (2) the high cost of collecting and sorting different types of plastics, especially the removal of contamination, and (3) the high capital costs and technological uncertainties of more “advanced recycling options”. Recent changes in legislation along with the desire from consumers for more sustainable products has created a dramatic push by industry for more advanced plastic recycling technologies. IHS Markit has projected that $300 billion of capital spending that is earmarked for new plastic production capacity should be redirected to plastic recycling technologies to meet circular economy goals.12
Fig. 2 shows the current and future infrastructure for plastic recycling technologies. The first step in plastic recycling is the sorting of plastic wastes into different streams through the material recovery facility (MRF). The MRF sorts out the glass, metals, carboard, and plastics, bails them, and then sells them to a downstream recycler. Plastic wastes can be divided up into four areas: post-industrial waste (PIW), post-consumer waste (PCW), plastics that are present in municipal solid wastes (MSW), and ocean plastics. PIW is typically of more uniform composition and lower in contaminants than the other types of plastic wastes. Oftentimes industry will recycle PIW in closed-loop recycling processes. However, PIW often contain multi-layer films which cannot be recycled by mechanical recycling due to material incompatibility. The current plastic recycling infrastructure is primarily focused on mechanical recycling of rigid #1 and #2 plastics. MRFs do not collect flexible films or textiles as this will clog the equipment. Ocean plastic waste is not currently collected at any large scale. MSW is primarily sent to landfills although some “dirty MRFs” can process these streams as well.
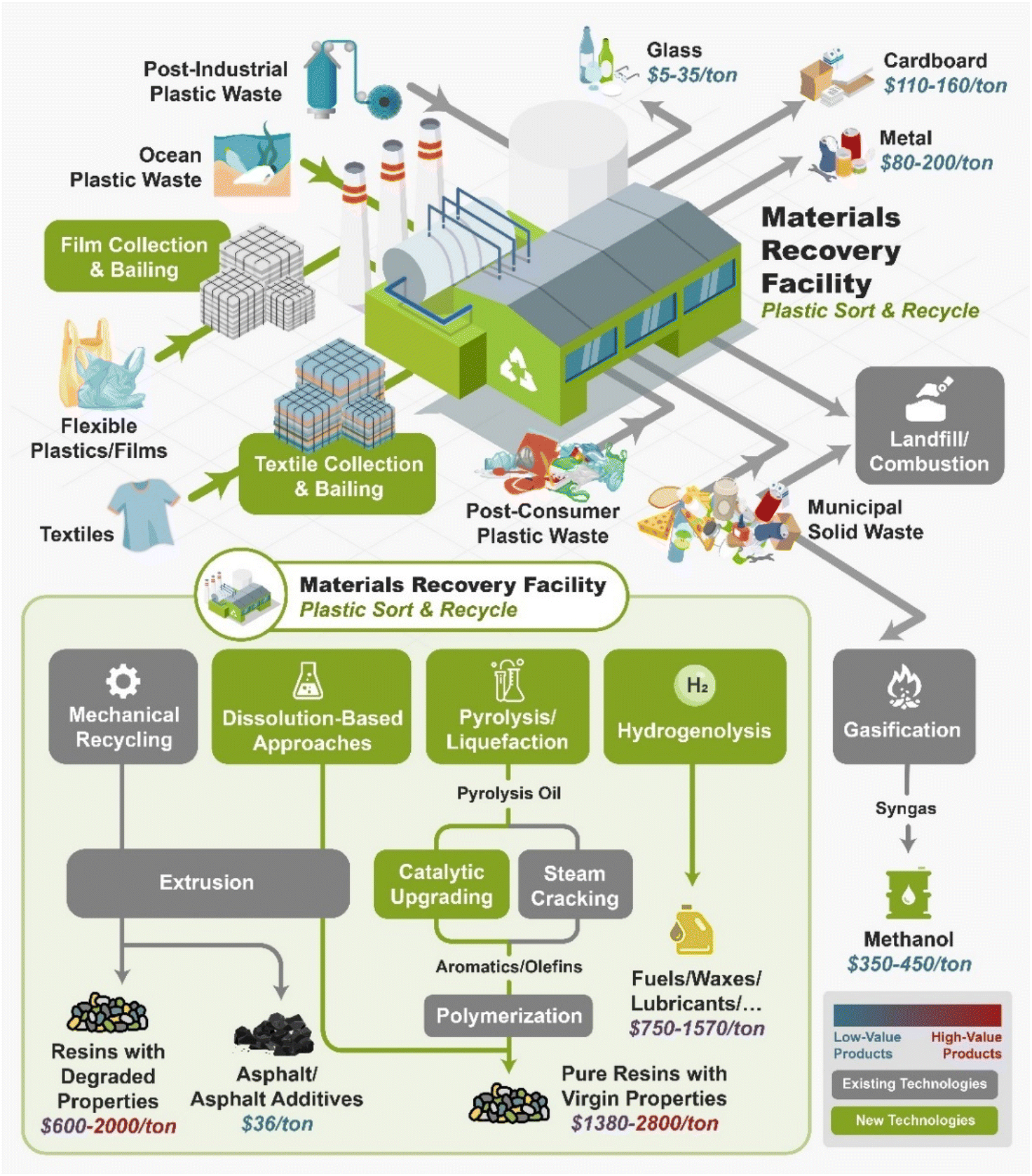 | ||
| Fig. 2 Summary of current management system of waste plastics. | ||
As shown in Fig. 2, several technologies currently exist to chemically recycle plastics. Plastics can undergo thermal degradation to produce a liquid known as pyrolysis oil. Pyrolysis oil can be converted into aromatics and olefins by either steam cracking or using a form of catalytic upgrading as discussed in sections 5 and 6. This produces aromatics and olefins which can then be re-used to make new recycled plastics that have the same properties as the virgin plastics. Multilayer-plastics can be processed via dissolution-based recycling approaches generating pure plastic flakes which can then be re-extruded into recycled plastic resins. Polyesters and polycarbonates can be chemically or enzymatically (section 9) converted into their monomers by methanolysis and other technologies. These monomers can then be re-used to remake the polymers. Plastics can also be gasified to synthesis gas which can be used to make methanol or transportation fuels as discussed in section 7. Methanol can then be converted into aromatics and olefins.13 Other approaches to recycle plastics, primarily being studied in academia at present, include hydrogenolysis to produce lubricants and oils, functionalization of plastics, and the creation of plastic alloys as discussed in section 10.
Sorting, recycling, disposal, and product redesign in the management of waste plastics have been reviewed.14–22 The objective of this review is to first describe current plastic recycling infrastructure followed by a discussion of the chemistry, engineering, and technology of advanced plastic recycling. We first review the manufacture of plastics (section 2), landfilling of waste plastics and MSW sortation (section 3), and mechanical recycling of waste plastics (section 4). We then summarize the technologies that are being used or developed including pyrolysis (section 5), liquefaction (section 6), gasification (section 7), dissolution-based approaches (section 8), PET conversion to monomers (section 9), followed by other catalytic processes for plastics conversion (section 10). Conclusions and outlook are provided in section 11.
2 Manufacture of plastics and the environmental footprint of plastics disposal
2.1 Plastic industry supply chain
 | ||
| Fig. 3 Supply chain of the manufacture of #1–#6 plastics and their volume of production in 2019.19 | ||
The chemical process of polymerization is not the same for all plastics, since the reaction is dependent on monomer chemistry and controlled by several conditions such as temperature, pressure, monomer concentration, and desired end use as well as by structure-controlling additives including catalysts, activators, accelerators, initiators and inhibitors.23 There are two basic classifications that encompass most polymerization schemes: step growth (condensation) and chain growth (addition) polymerization. Linear and non-linear step growth polymerizations are processes in which the polymerization occurs through the reaction of more than one molecular species. On the other hand, chain polymerization processes occur with monomers that have an unsaturated group. For example, polyethylene (PE) is produced from the polymerization of ethylene (or ethene, olefin) monomers through polyaddition either from the radical polymerization of ethylene monomers or by using Ziegler–Natta or metallocene catalysts. Table 1 lists the various polymerization mechanisms and some examples of their polymer products.
| Classification | Polymerization | Examples |
|---|---|---|
| Step linear | Polycondensation | Polyamides (PA) |
| Polycarbonate (PC) | ||
| Polyesteramide (PEA) | ||
| Polyetherimide (PEI) | ||
| Polyaddition | Polyethylene (PE) | |
| Step non-linear | Network polymers | Epoxy resins (EP) |
| Melamine formaldehyde (MF) | ||
| Chain | Free radical | Polymethylmethacrylate (PMMA) |
| Polyvinyl acetate (PVAC) | ||
| Polystyrene (PS) | ||
| Polyvinyl chloride (PVC) | ||
| Cationic | Polyisobutene (PIB) | |
| Anionic | Butadiene rubber (BR) | |
| Polymethylmethacrylate (PMMA) | ||
| Ring opening | Polyamide from e-caprolactam (PA6) | |
| Polycaprolactone (PCL) | ||
| Polyethylene oxide (PEO) | ||
| Polypropylene Oxide (PPO) | ||
| Ziegler–Natta | Polyethylene (PE) | |
| Metallocene | Polypropylene (PP) |
PP, the most widely used thermoplastic globally, is exceptional due to its production cost-effectiveness and wide applications, such as flexible barrier film pouches, caps, closures, and containers. LDPE, the second most widely used resin, is typically used to make plastic films, and injection- or blow-molded products, such as wash bottles and computer hardware covers and packaging. HDPE is typically used as fibers for industrial plastics, pipes and tubing, and consumer products, such as containers. These polyolefins account for almost 48% of global plastic demand.24
Copolymerization is used to manipulate the properties of manufactured plastics to meet specific requirements and industrial needs, allowing for the improvement of mechanical and chemical properties.25 Through manipulation of monomer chemistry, more customized materials like copolymers have been formed and successfully introduced commercially, such as acrylonitrile–butadiene–styrene (ABS), styrene–butadiene copolymer (SBR), nitrile rubber (NR), styrene–acrylonitrile (SAN), styrene–isoprene–styrene (SIS), and ethylene–vinyl acetate (EVA), all of which are formed through chain growth polymerization. Linear low-density polyethylene (LLDPE) is produced by polymerizing ethylene with 1-butene and smaller amounts of 1-hexene and 1-octene, leading to a product that is structurally similar to LDPE but with short, uniform branches, leading to improvements to the polymer's mechanical and chemical properties. Step growth polymerization has been used to produce most polyamides, e.g., nylon-12 and 66 polymers, as well as the PET family. These polymers consist of at least two types of constituents (monomers, e.g., dicarboxylic acids and dialcohols) and can be classified depending on the repeat unit structure.
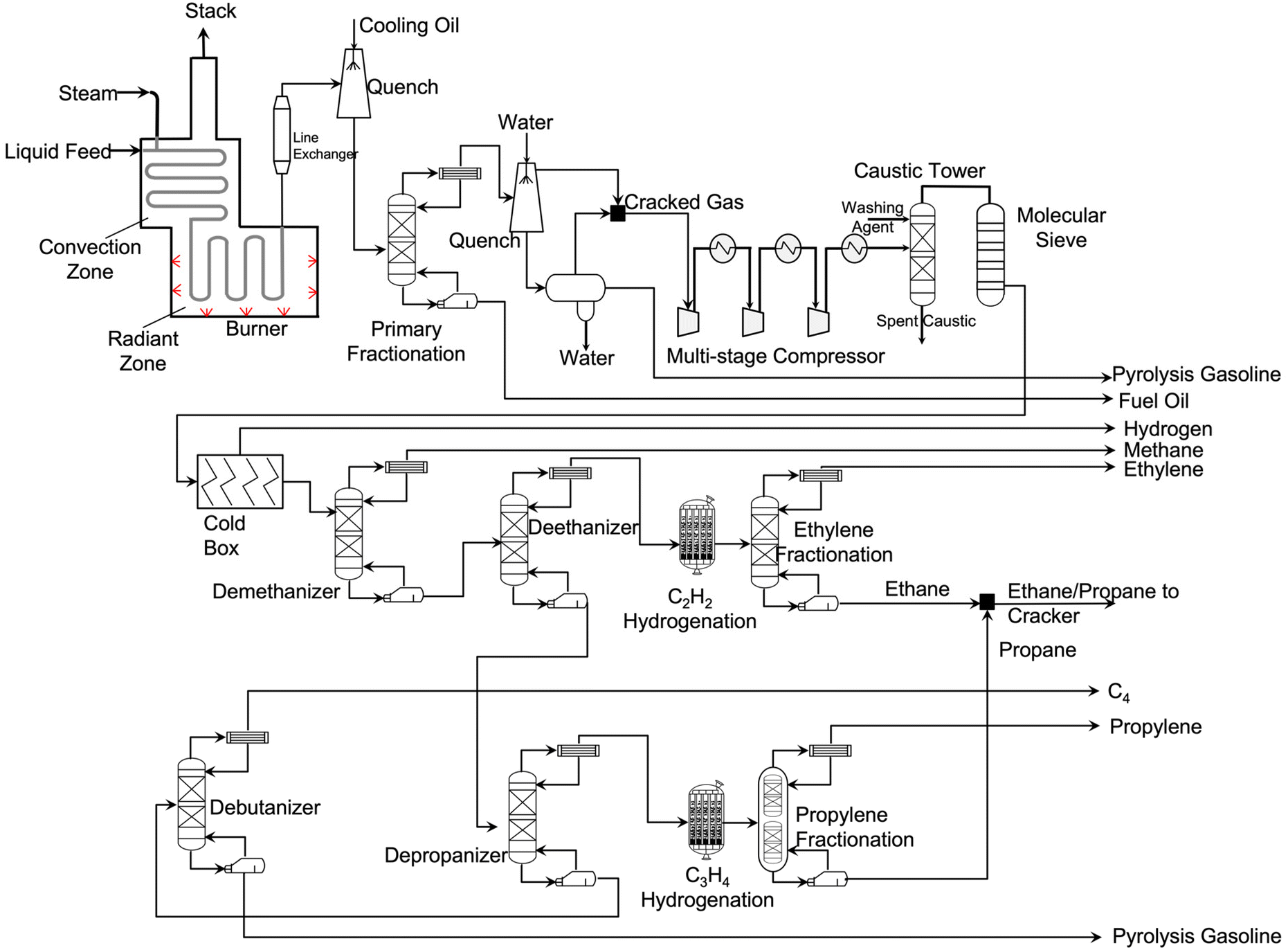 | ||
| Fig. 4 A simplified process flowsheet diagram of steam cracking.27–29 | ||
When the feedstock is naphtha, a similar free radical reaction mechanism occurs. Adding steam to the steam cracker facilitates the control over residence time, prevents coke formation, and reduces hydrocarbon partial pressure. Steam does not participate in the free radical reaction mechanism. Its main role is to react with coke to form CO and CO2. Since the cracking process is highly endothermic, energy is input from burners in the radiant zone.
The effluent stream from the cracking furnaces is cooled by a transfer line exchanger (TLE) where feed water is vaporized, and high-pressure steam is generated. Then the cracked gas is further cooled by contact with cooled oil in a quench tower. The resulting stream is sent to a primary fractionation column where fuel oil is obtained at the bottom. The stream on the top is further cooled down using a quench water tower. On the bottom of this quench tower, a three-phase separator splits water, liquid, and gas hydrocarbons. The liquid hydrocarbons are collected as gasoline-ranged products that is heavy in aromatics. These aromatics are also used to produce other types of plastics. On the top of the quench water tower, gas effluent is obtained and mixed with gas effluent of the three-phase separator. This stream contains light hydrocarbons, including ethylene and propylene.
After compressing the cracked gas with a multi-stage compressor, the gas is sent to an acid–gas removal unit where CO2 and H2S are removed. After drying the purified gas with a molecular sieve dryer, the cracked gas is sent to the cold box where it is cooled down to −156 °C. Under these conditions, hydrogen is recovered for use in hydrogenation of C3H4. The separation of hydrocarbons starts from a demethanizer where methane is removed. This methane stream is supplied to the cracker as fuel gas for providing the reaction heat. The bottom stream of the demethanizer is sent to a deethanizer where ethane or lighter components are obtained. To convert acetylene into ethylene, this C2 stream is hydrogenated and fractionated cryogenically. The ethylene fractionator column splits ethylene and ethane. The ethane is mixed with propane and recycled back to the cracker. On the bottom of deethanizer, C3 and heavier components are obtained and sent to a depropanizer where C3 and C4/heavy fractions are separated.
After hydrogenating methylacetylene and propadiene, the C3 stream is split into propylene and propane. The propane is recycled back to the cracker. Meanwhile, the C4 and heavier components stream is sent to a debutanizer where butylene/butane and gasoline-ranged products are obtained. When ethane/propane are fed, no primary fractionator is needed. The gas cleaning and separation processes are very similar to the above one when feeding the cracker with ethane only. Information related to steam cracking facilities in the US can be found in SI 1.0 in ESI†.
2.2 Plastics processing
The conversion of raw or virgin plastics into finished products involves a series of plastic manufacturing processes. These manufacturing processes will vary depending on the type of plastic (thermoplastic, thermosetting, or elastomeric) and the final product desired. Plastic manufacturing can be commonly divided into three stages. The first stage is the compounding of the raw plastic with additives to achieve the desired material properties. For example, PE and PP films often stick together, which makes them difficult to separate, as when opening a PE bag. Anti-blocking agents (such as limestone and talc) can be added to lower the blocking force between the film layers. Table 2 lists the most common additives and their classifications. The compounding process is highly complex and involves taking into consideration the effect of all the additives utilized on the final properties of the plastic product. More often, a plastic will contain several additives to aid the processing of the material, modify color, and achieve the required mechanical properties. These formulations tend to be proprietary and will vary depending on the manufacturer.| Additive classification | Common agents |
|---|---|
| Anti-blocking agents | Limestone |
| Synthetic silica gel | |
| Natural silica | |
| Talc | |
| Slip additives | Erucamide |
| Oleamide | |
| Stearamide | |
| Plasticizers | Dioctylphthalate (DOP) |
| Antioxidants | Hydrogen donors – phenols |
| Hydroperoxide decomposers – phosphites and phosphonites | |
| Alkyl radical scavengers – hindered amine stabilizers and hydroxyl amines | |
| Metal deactivators | |
| Flame retardants | Aluminum hydroxide/oxalate |
| Zinc borate/chloride/sulfide | |
| Alumina trihydrate | |
| UV stabilizers | Sterically hindered amines (HALS) |
| Carbon black | |
| Antistatic agents | Fatty acid esters |
| Ethoxylated alkylamines | |
| Antimicrobial agents | 10,10′-Oxybisphenoxarsine (OBPA) |
| Antifogging agents | Glycerol esters |
| Polyglycerol esters | |
| Alcohol ethoxylates | |
| Chemical blowing agents | Azodicarbonamide |
| Carbonates | |
| Semi-carbazides | |
| Colorants | White – titanium dioxide, zinc oxide |
| Black – spinel black, iron oxide black | |
| Yellow-orange – iron oxide yellow, zinc ferrite | |
| Brown – chrome iron brown, rutil brown | |
| Red – iron oxide red, cadmium orange | |
| Green – chrome oxide green, cobalt spinel green | |
| Blue – cobalt blue, ultramarine blue | |
| Metallic – aluminum, copper | |
| Fluorescent whitening | Bis-benzoxazoles |
| Agents | Phenylcoumarins |
| Fillers | Glass |
| Talc | |
| Ceramic | |
| Porcelain clay |
The second stage, called forming, involves imparting the desired shape on the material. An inherent requirement of this step is that the material must be able to maintain this shape after the step is completed. Synthetic fibers, typically polymerized from petroleum-based chemicals, are formed through extruding a plastic solution or melt through a spinneret, a process known as spinning. This liquid-to-fiber formation process can be compared to the spinning of cotton candy. Depending on the type of spinneret used, either solid or hollow fibers can be formed. Synthetic fibers account for about half of all fiber usage, with applications in every field of fiber and textile technology. Nylon, polyester, acrylic, and polyolefin-based synthetic plastics dominate the market with these four accounting for approximately 98% of synthetic fiber production. Polyester alone accounts for around 60%.38 The final stage involves finishing the piece, which includes machining, decoration, and assembly. Plastics processing and manufacturing has greater opportunities for cost-savings through minimization of finishing processes as compared to the manufacture of more traditional materials.30Table 3 lists common methods attributed to the three stages of plastics manufacturing.
| Stage | Methods |
|---|---|
| Compounding | Powder, melt, dispersion, solution |
| Forming | 2-D: extrusion, calendering, coating, spinning |
| 3-D: thermoforming, molding (compression, transfer, injection, blow) | |
| Finishing | Machining, decoration, assembly |
For polymeric materials, it is typical for a particular product to require multiple forming processes for ease of transfer and shipping (such as extruding a tube, then pelletizing for shipping and further use) or to carry out a specific chemical process (such as crosslinking or vulcanization). These forming processes typically involve the softened stage of the polymer, which is achieved by heating the polymer material. Each forming process will impact a thermal history and, in the case of reactive polymeric systems, will impact a degree of cure. These repeated forming processes will influence the properties of the material, either through the degradation of the material or unwanted side reactions. Setting the shape of the product is typically achieved either by cooling (which can cause crystallization, further impacting material properties) or a chemical process (crosslinking) to achieve dimensional stability. Table 4 demonstrates the most common plastics processing techniques used to form and set material shapes.
| Processing Method | Description |
|---|---|
| Extrusion | Thermoplastic granules are forced through a heated barrel and the plastic melt is squeezed through a die with the desired profile for the extruded component. The extruded material is cooled as it leaves the die, then cut to the desired length |
| Spinning | A specialized form of extrusion that utilizes a spinneret to form multiple continuous filaments. Extruded liquid plastic filament is continuously drawn and simultaneously solidified to form a continuous synthetic fiber |
| Blow molding | Compressed air is introduced underneath a warmed sheet of thermoplastic material, forcing the material into a cavity or allowing it to expand freely |
| Vacuum forming | A sheet of plastic is set over a mold, where it is warmed and softened. Air is drawn from under the softened plastic sheet, which forces it over or into a mold by atmospheric pressure |
| Extrusion blow molding | Plastic material is first extruded as a tube shape into an open die. The die is then closed to seal the ends of the tube and air is blown in, forcing the plastic tube to take the shape of the die cavity |
| Injection molding | Granulated plastic is heated until a melt is formed. The melt is then injected into a closed mold using high pressures and cooled inside the mold |
| Rotational molding | Plastic is placed in a closed mold that is heated and rotated slowly around a vertical and horizontal axis. The plastic material melts and sticks to the hot mold surface, building up the required thickness. The mold is gradually cooled while rotating |
| Foaming | The base plastic is mixed with foaming/blowing agents. When heated, these agents release gas which form bubbles in the plastic. A separate method is to inject compressed nitrogen gas into molten plastics during the molding process |
| Calendering | A thermoplastic melt is extruded onto heated rotating rollers which squeeze the material into a continuous sheet or film |
| Compression molding | A thermosetting plastic charge is placed into a preheated lower mold cavity. The mold is then closed and subjected to further heat and pressure. The combined pressure and heat caused polymerization and the flow of the plasticized material within the mold |
2.3 Mechanical and thermal properties
Most plastics produced are specifically designed to serve very particular applications. Fig. 5 demonstrates material properties and pricing of thermoplastics, aluminum, and ceramics compared to steel. During the forming of the product, it is standard for manufacturers to use additives to manipulate the properties of the raw material to achieve specific characteristics or homogenize the characteristics of their material batches as shown in Table 2. These additives influence the mechanical and thermal properties of the plastics, either by manipulating the glass transition temperature, imparting higher chemical resistance or increasing impact strength and elasticity, among other things.31Fig. 5 demonstrates average mechanical properties and pricing of common commercial plastics. Many commercial plastics will have different characteristics from those of the base plastic, even though they may have the same chemistry. In general, it is important to understand that many plastics are technically combinations of several materials. As a result, mechanical and thermal properties are influenced by each component. Often, manufacturers change additives or blend ratios due to environmental concerns, to enhance specific properties, or simply for more economical processing. These subtle changes and introduction of new materials and compounding may drastically change one or more engineering properties; because of this, it is of utmost importance to continually test and evaluate commercial plastics and their specifications. Table 5 contains thermal and mechanical properties for common commercial plastics. It is of note that these properties are averages and specific material formulations can manipulate these properties for specific applications.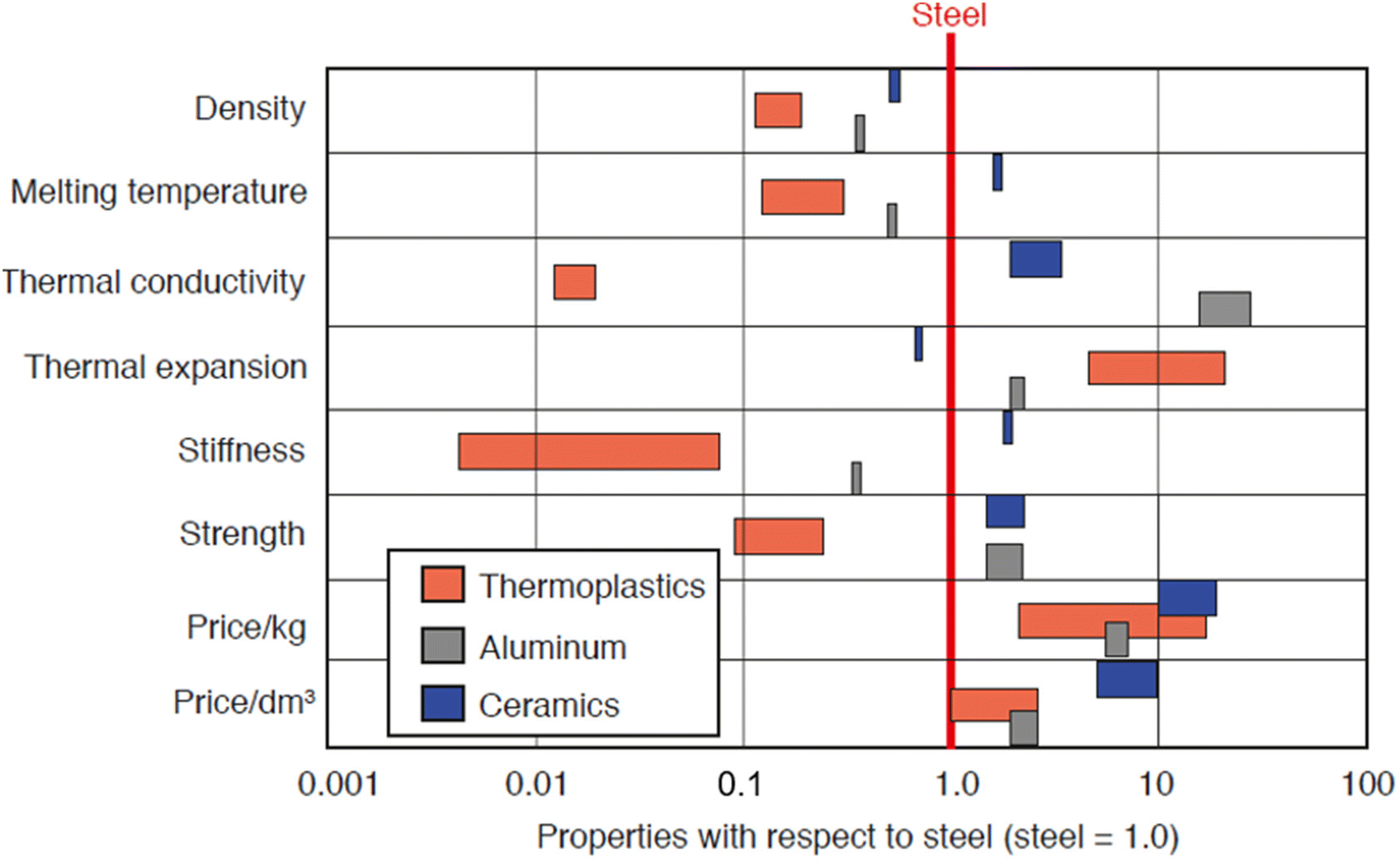 | ||
| Fig. 5 Properties of thermoplastics, aluminum, and ceramics with respect to steel. Reproduced with permission from ref. 32 from Carl Hanser Verlag, Munich, copyright 2012. | ||
| Material | Morphology | Density [g cm−3] | Glass transition temperature [°C] | Melting temperature [°C] | Initial degradation temperature [°C] | Tensile strength (MPa) | Tensile modulus (Mpa) | Strain at break (%) |
|---|---|---|---|---|---|---|---|---|
| ABS | Amorphous | 1.03–1.07 | −85/95–105/125 | — | 420–428 | 32–45 | 1300–2700 | 15–30 |
| EVA | Semi-crystalline | 0.92–0.95 | −40–20 | 30–110 | 345–360/470–480 | 18–35 | 7–120 | 600–900 |
| LDPE | Semi-crystalline (40%–55%) | 0.91–0.93 | −130 to −100/−30 to −10 | 100–115 | 487–498 | 8–23 | 200–500 | 300–1000 |
| LLDPE | Semi-crystalline | 0.91–0.94 | −130 to −100/−70 to −25 | 122–127 | 475–485 | — | — | — |
| HDPE | Semi-crystalline (60%–80%) | 0.94–0.96 | −130 to −100 | 125–135 | 480–498 | 18–35 | 700–1400 | 100–1000 |
| UHMWPE | Semi-crystalline | 0.93–0.94 | −130 to −100 | 130–145 | 480–490 | — | — | — |
| PLA | Semi-crystalline | 1.21–1.43 | 45–65 | 150–160 | 350–375 | — | — | — |
| PP | Semi-crystalline | 0.90–0.91 | −20–20 | 160–165 | 450–470 | 21–37 | 1100–1300 | 20–800 |
| PS | Amorphous or Semi-crystalline | 1.05 | 80–105 | — | 415–425 | 45–65 | 3200–3250 | 3–4 |
| PVAL | Semi-crystalline | 1.21 | 70–100 | 220–260 | 260–320/420–450 | — | — | — |
| PVC-Plasticized | Amorphous | 1.16–1.35 | −50–80 | — | 290–315/460–475 | 10–25 | — | 170–400 |
| PVC-unplasticized | Amorphous | 1.38–1.55 | 80–90 | — | 285–315/460–475 | 50–75 | 1000–3500 | 10–50 |
| PA6 | Semi-crystalline (30%–40%) | 1.12–1.15 | 45–80 | 225–235 | 445–460 | 70–85 | 1400 | 200–300 |
| PA66 | Semi-crystalline (35%–45%) | 1.13–1.16 | 65–90 | 225–265 | 430–473 | 77–84 | 2000 | 150–300 |
| PC | Amorphous | 1.20–1.24 | 140–150 | — | 480–535 | 56–67 | 2100–2400 | 100–130 |
| PET | Semi-crystalline | 1.33–1.45 | 70–85 | 245–260 | 425–445 | 47 | 3100 | 50–300 |
| PUR | Thermoset | 1.10–1.70 | 10–180 | — | 240–350 | 70–80 | 4000 | 3–6 |
| SBR | Rubber with hard and soft segments | 0.94 | −55 to −35 | −20 | 435–470 | 26–38 | 1800–2500 | 25–60 |
Temperature can have varying effects on polymer products, from melting to degradation to morphological transitions.33 Temperature can also affect mechanical properties due to enhanced polymer chain movement and free volume. Two key temperatures describe these effects: the glass transition temperature and melting temperature. The glass transition temperature is a property of the amorphous region of the plastic, whereas the melting point is a property of the crystalline regions. At temperatures below the glass transition temperature, the molecules in the amorphous region exist in a frozen state where movement is minimized and the polymer chains can vibrate slightly. This is typically referred to as the plastic's glassy state. The rubbery state occurs when the plastic is heated above the glass transition temperature, allowing the polymer chains to move and shift around each other, which in turn allows the plastic to become soft and flexible. Glassy (amorphous) plastics exposed to heat eventually soften and flow into a viscous liquid.
Melting transitions in some plastics refer to the transition between a crystalline solid and a liquid. When a semi-crystalline plastic is heated above the melting temperature, it forms a viscous liquid and flows. Amorphous plastics do not experience melting since there are no crystal regions to melt. For semi-crystalline plastics, crystallinity is dependent on the type and structure of the plastic. Crystallinity is typically induced by cooling a melt, although strain-induced crystallization may occur due to the molecular chains disentangling, unfolding, and straightening above its yield point. Molecular weight plays a large role in crystallinity and most polymeric properties. A plastic's tacticity will also influence its crystallinity, with isotactic plastics demonstrating higher crystallinity than syndiotactic plastics, and atactic plastics being considered uncrystallizable due to the lack of chain regularity. Strong intermolecular forces and a stiff chain backbone favor the formation of crystals, while bulky side groups and branching have the opposite effect, due to the difficulty in folding and aligning the chains along the crystal growth direction. Crystallinity highly influences the thermal, mechanical, and physical properties since it impacts strength, toughness, and opaqueness depending on the degree of crystallinity. These properties are further varied by the plastic's chemistry, composition, and compounding, which in turn influence the molecular weight of the plastic. Fig. 6 demonstrates the key characteristics of various amorphous and semi-crystalline plastics based on their performance and applications.
 | ||
| Fig. 6 International Association of Plastics Distribution Thermoplastics Rectangle. Reproduced from ref. 34 with permission from International Association of Plastics Distribution, copyright 2021. | ||
The mechanical properties of plastics are highly varied, although there are specific relations between plastic structure and their properties. A plastic's strength (tensile, compressional, flexural, torsional, and so on) depends on the molecular weight, presence of crosslinking, crystallinity, and structure. In general, a linear plastic has less strength than a branched plastic, followed by cross-linked plastics having greater strength, and network plastics exhibiting the highest strength. Low molecular weight plastics exhibit low strength, regardless of crystallinity, due to weak van der Waals forces and the ease of movement between chains, while high molecular weight plastics exhibit a greater degree of entanglement, which greatly increases the plastic's strength. Crosslinking increases the strength of the plastic by restricting chain motion, while crystallinity increases strength due to the significant intermolecular bonding. These properties are highly influenced by environmental and application factors such as loading, temperature, humidity, weather conditions, exposure, and time.
2.4 Environmental assessment on plastic production systems
Fig. 7 presents the supply chain greenhouse gas emission (GHG) arising in the production of major types of plastic. PET has the largest carbon footprint of these plastics. The equivalent GHG emissions for producing one kg of PET film is up to 4.5 kg, where 60% of the emissions result from the usage of processing fuel and 28% of emissions are from electricity usage. General-purpose (GP) HDPE has the lowest GHG emissions among the plastics listed. The production of injected molded (IM) LDPE tends to have a high emission associated with electricity usage (1.8 kg equivalent CO2 per kg of plastic). In comparison with the GHG emissions of electricity and processing fuel, the emission associated with transportation is relatively low, and only corresponds to 9 to 18% of the emission of the plastic supply chain.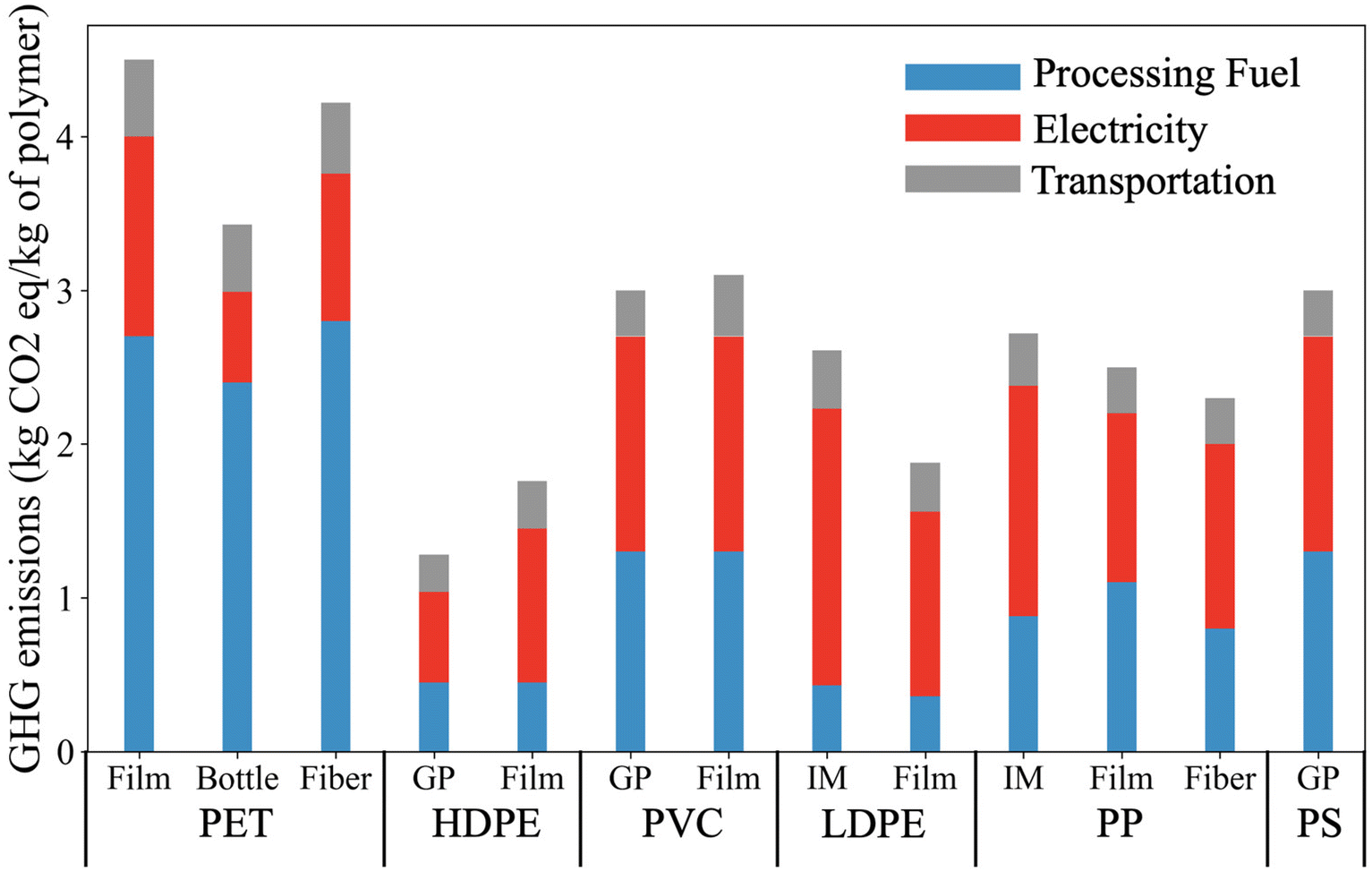 | ||
| Fig. 7 Supply chain GHG emissions arising in the production of different plastics. Reproduced from ref. 14 with permission from Cell Press, copyright 2021. LCA concepts were employed to estimate the cradle-to-gate energy requirement and GHG emissions. The GHG emissions associated with processing fuel and electricity correspond to the total energy consumption of the plastic supply chain. This includes emission associated with raw materials extraction, intermediate materials productions (e.g., monomer), and plastic production. | ||
2.5 LCA on plastic recycling technologies
In the past 17 years, 13 major life cycle assessment (LCA)/environmental assessment studies on recycling technologies and plastic waste disposal options have been conducted.35–47 These recycling/disposal options could be classified into four types: mechanical recycling, chemical recycling, incineration, and landfilling. These studies explored the environmental impact of each technology from GHG emissions, water/air pollution, human health, and resource depletion perspectives. From these publications, some general conclusions can be made:(a) Climate change: without considering the electricity credit, incineration of plastics has the highest GHG. The GHG emission of incineration ranges from 1.8–3 kg CO2,eq per kg plastic wastes.43,48 However, accounting for the electricity credit lowers the GHGs of incineration to around 1.4 kg CO2,eq per kg plastic wastes. Meanwhile, the chemical recycling technologies (e.g., pyrolysis, gasification) also have higher GHGs than the GHGs of mechanical recycling methods, due to the direct emission and electricity consumption.44
(b) Acidification: chemical recycling technologies tend to have high acidification potential. For example, gasifying one ton of plastic waste results in 0.49 kg emission of NOx and 0.09 kg emission of SOx.48 However, after accounting for credit to avoid incineration of plastic waste, the acidification potential of chemical recycling options is lower than the acidification potential of producing virgin plastic from fossil fuel.49
(c) Air pollution: incineration contributes to air pollution. The air emission of organic compounds is ten times higher than that of the air emission of pyrolysis (1.42 g emission of organic compounds per kg of plastic processed). Mechanical recycling has the lowest air emission among plastic disposal options.
(d) Resource depletion: it is estimated that producing 1 kg of plastic requires 2 kg of crude oil, which is lost in cases where plastics are landfilled.48 Meanwhile, incineration also has high resource consumption, since the plastic is combusted for generating electricity.39 Among all the disposal options, mechanical recycling has the advantage in terms of natural resource depletion. After accounting for the credit of avoided landfill or incineration, the amount of resource consumption of mechanical recycling (crude oil/gas) is even negative.39
Incineration causes air pollution, CO2 emission, and acidification of the disposal of plastic wastes. Although landfills tend to have low air pollution, they can cause soil and groundwater pollution. The available space for landfills is limited, even if parts of these areas can be reused in some cases.49 Therefore, landfills are not a sustainable option. In comparison with chemical recycling, mechanical recycling has a lower energy consumption and a lower carbon emission. However, as will be discussed in section 4, only a portion of plastic wastes can be mechanically recycled (e.g., PET and HDPE bottles). For plastics that cannot be mechanically recycled (e.g., plastic films), chemical methods are the most environmentally friendly approach.
3. Landfills, disposal, and sorting of post-consumer waste plastics
3.1 Landfill and incineration
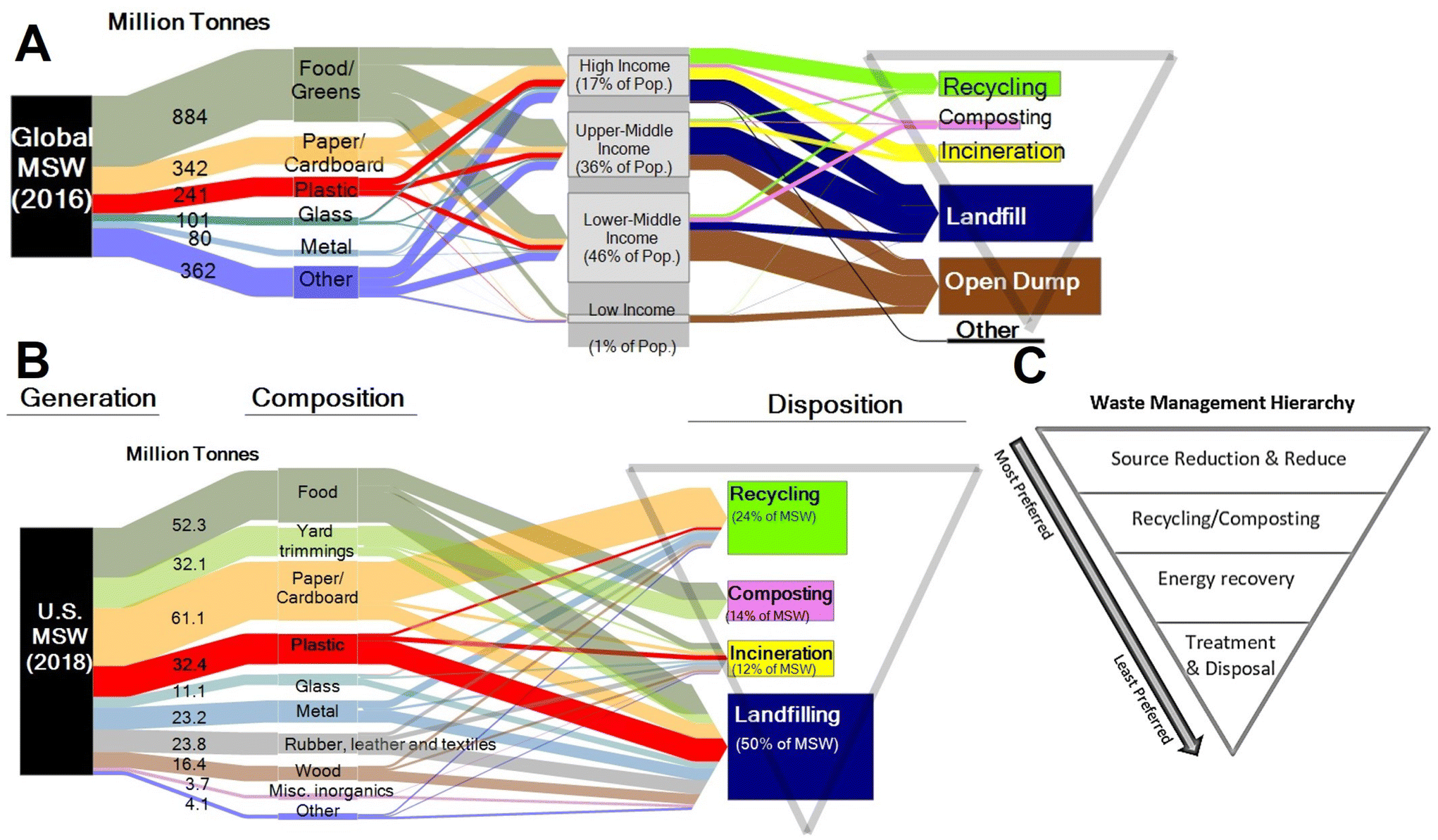 | ||
| Fig. 8 Municipal solid waste (MSW) generation and final disposition: (A) global generation in 2016 with fractions associated with national income, (B) US generation in 2018, and (C) waste management hierarchy. Inserted triangle illustrating how disposition pathways fit with preferred waste management hierarchy. | ||
Plastics currently comprise 12% of all MSW generated globally.50 In the US, only 9% of plastics are recovered through recycling, 16% incinerated, and 76% landfilled.51 The EU recycles 33% of plastics, with 43% incinerated, and 25% landfilled.52 Estimates for the cumulative global fate of plastics indicate 79% of plastics produced have been landfilled or left in the open environment, whereas 12% have been incinerated, and 9% recovered for recycling.6
The UN Environment Programme and US EPA53,54 have established waste hierarchies focused on shifting from a philosophy of waste management to a philosophy of resource management in the context of a circular economy. The recommended hierarchy (highest to lowest priority) consists of waste prevention, waste minimization, reuse, recycling, energy recovery, and disposal (Fig. 8). Most waste today, in the US and globally, is managed by disposal in landfills – the lowest priority in the hierarchy.
A modern engineered landfill consists of an engineered containment system situated in an earthen depression (Fig. 9). The base is comprised of a multicomponent liner overlain by a leachate collection system.10 Waste is placed in layers approximately 3–4 m thick (called “lifts”) that are compacted with heavy machinery. A gas collection system is installed in the waste to collect landfill gas (LFG) for treatment or production of renewable energy. Interim cover (soil or synthetic material) is placed over the waste during filling to reduce ingress of precipitation, control emissions and odors, and limit vectors. Once final elevations are met, a final cover is placed to limit the ingress of precipitation to de minimis amounts, facilitate gas collection, and provide separation from the surrounding environment for as long as the waste poses a threat. As with the liner, the final cover includes a multilayer hydraulic barrier overlain by a drainage layer. The surface is covered with vegetated cover soils for protection and aesthetics.10
 | ||
| Fig. 9 MSW landfill with liner, cover, leachate collection, leachate recirculation, and gas collection. | ||
Leachate is generated when water percolating through the landfill contacts the waste.59 Leachate contains a variety of inorganic and organic contaminants, and therefore is treated before being discharged. Leachates are pumped from the leachate collection system, and generally are transported to a public wastewater treatment plant (a publicly owned treatment works, or POTW) where they are treated along with other wastewater (e.g., sewage from a community, etc.). In some cases, leachates are treated on site or are recirculated in the waste to stimulate waste decomposition to promote more rapid waste stabilization and greater gas production for renewable energy.55,56
Landfills operate under anaerobic conditions where the microbial community decomposes the degradable organic fraction and generates gas comprised of approximately equal parts methane (CH4) and carbon dioxide (CO2).56 MSW landfills are the third largest source of anthropogenic CH4 in the US57 and contribute approximately 3% of global GHG emissions.60 LFG also contains a variety of other contaminants in small quantities. Consequently, LFG must be treated before being released. Thermal treatment is most common, either through a flare or through combustion to create renewable energy (e.g., LFG to electricity). In recent years, landfills have also become a significant source of renewable natural gas (RNG), which is produced by treating and compressing LFG extracted from the landfill.56
Incineration is the combustion of MSW at temperatures at around 750–1100 °C, with the heat being used to produce steam for generating electricity and/or heating.70,71 Plants producing steam for heating and electricity can have efficiencies on the order of 80%; whereas the efficiency is on the order of 20% for plants producing only electricity.61 Most MSW incineration is conducted as mass burn (bulk MSW incinerated in a single chamber) or as refuse derived fuel (RDF) where screened and shredded MSW is combusted.72 RDF is common in the Portland cement industry and can represent up to 80% of the thermal requirements in cement plants.61 Nearly 15% of all MSW is incinerated globally. Incineration rates vary significantly, with incineration more common in developed land-constrained countries and islands,70 less common in developed countries without land constraints, and uncommon (<1%) in lower-middle and low-income countries. For example, Japan has significant land constraints and incinerates 74% of its MSW,73 whereas the US has virtually no land constraints and incinerates only 12% of MSW. The effect of income is evident in the location of incinerators; 80% of MSW incinerators globally are located in North America, Europe, and Asia Pacific.70 US incinerators manage 22% of discarded food and 16% of discarded plastics.74 Most WtE incinerators in the US (77%) only generate electricity, 19% produce electricity and heat, and 4% export steam to local users.75
Incineration reduces the mass and volume of MSW that is landfilled (70–85% by mass and 75–90% by volume)71 while generating energy for use or sale. Despite these advantages, incinerators are difficult to implement, especially in the US. Emissions from MSW incinerators can contain dioxins and furans, which are toxic persistent organic pollutants and human carcinogens.76,77 These pollutants are often of great concern to surrounding communities even when plants are in compliance with emissions criteria in the Clean Air Act. MSW incineration also generates ashes that need to be managed,75 generally through landfill disposal. Incinerator ashes tend to have higher levels of contaminants per unit mass than MSW, resulting in leachate with higher concentrations that requires more treatment. In some cases, ashes can be reactive, leading to heat generation that is problematic in landfills.78
3.2 Material recovery facilities (MRFs)
MRFs (pronounced “murfs”) receive waste material and separate (or bail) these materials. Their origin can be explained when terms such as MSW and waste management began to be established in the 1950–60s, with the increased waste generation linked to economic growth and popularization of petrochemical derivatives. In the US, the first MRF was established in Groton, Connecticut in the early 1980s,79 with the number of facilities multiplying in the following years. There were 40 MRFs in operation or under planning in 1991, 166 in 1993, and 307 in 1995.80MRFs are classified based on the input waste material they receive: single-stream, dual-stream, mixed-waste, or pre-sorted, with the first being a typical design in the US.42 Single and mixed-waste MRFs (containing more organic contaminants from MSW) have similar inbound composition, receiving a single stream with paper, plastic, glass, and metals, usually from automatic or semi-automatic vehicles. Dual-stream MRFs have a two-category stream line: fibers, composed of paper and cardboard, and containers, composed of plastic, metal, and glass, whereas pre-sorted MRFs receive source-separated material streams.42,81Fig. 10 is a block flow diagram for single-, mixed-, and dual-stream MRF frameworks (differences between operation modes are shown; otherwise, the process is the same for all) based on the work of Pressley et al.42 who drew on common MRF designs around the US. The design of a MRF is unique to each location, with different specifications required and associated with inbound composition, investment capacity, and ownership and operation type.
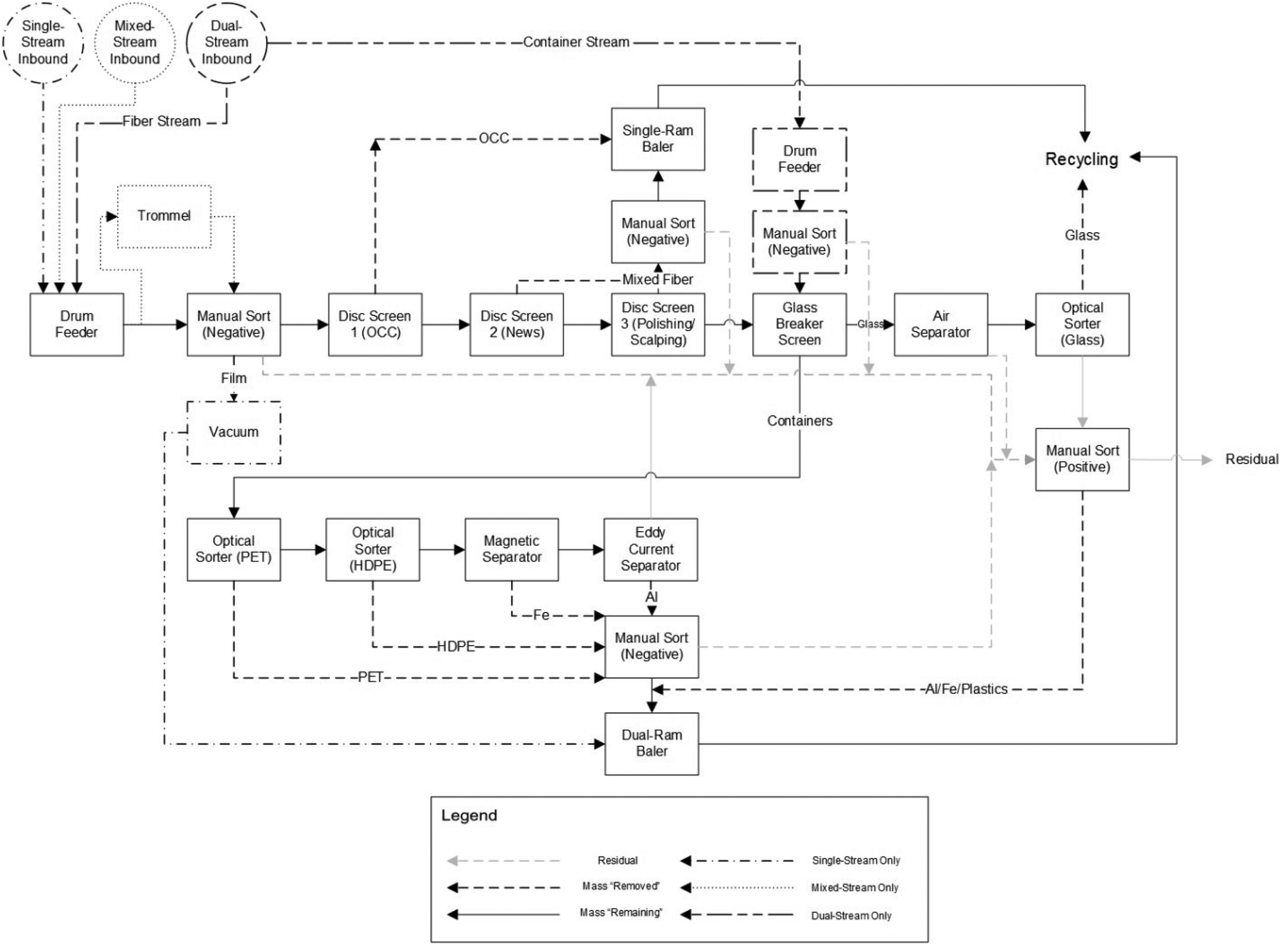 | ||
| Fig. 10 Block Flow Diagram of a single-, mixed-, and dual-stream MRF. Reproduced from ref. 42 with permission from Elsevier, copyright 2015. | ||
Single and dual-stream frameworks have their advantages and disadvantages. Single-stream benefits from waste collection with larger volumes and lower costs but with increased difficulty and costs for processing at the MRF, in addition to an increased contamination rate.82 For example, during conversion from dual to single-stream, a MRF from Brookhaven (New York, US) observed a 25% increase in volume of input recycling streams, which led to a diversion of recyclable material from landfills from 50000 to 35000 tons, along with an increase in non-recyclables in the single-stream.83 Another study that investigated GHG emissions observed benefits from the conversion of three medium-size MRFs from dual-stream to single-stream systems, avoiding the emission of 711 kg-CO2-equivalent per metric ton.84 The advantages of switching from a dual to single stream were not observed on the economic side, as observed in a study conducted using data from 223 municipalities in the province of Ontario, Canada. For example, it was found that single-stream recycling incurs 48.7% higher processing costs and 9.6% lower values of a recyclable saleable stream compared to multi-stream recycling systems, with only a 3% reduction in the collection costs.85
Specifications were determined in the US in an effort to standardize the inbound recycling streams at MRFs, such as those stipulated by the Institute of Scrap Recycling Industries (ISRI).86Table 6 describes the materials accepted, prohibited, and the possible contaminants in the majority of US MRFs. It should be noted that plastic bags and films are considered contamination due to the possibility of issues during sorting and are removed early in the process. A pilot study conducted by Resource Recycling Systems (RRF) for the Materials Recovery for the Future (MRFF) on an existing MRF in Birdsboro (Pennsylvania, US) looked at how to modify an MRF design to accept flexible packaging from residential single-stream inbound waste. It demanded additional equipment for separation, with a processing cost of $2.41 per ton and a breakeven point compared with the sorted polypropylene market value.87
| Allowed materials | |||
|---|---|---|---|
| Paper (single stream) | Plastic (single stream) | Metal (single stream) | Glass (single stream) |
| Paper (dual stream–separated) | Containers (dual stream – not mixed with paper) | ||
| Uncoated white office, printing, and writing paper | Empty PET (#1), HDPE (#2), PP (#5) plastic bottles, jugs, jars, and containers | Aluminum, tin, steel beverage and product containers | Bottles and jars of any color |
| Colored paper | Bulky rigid plastics such as crates, buckets, totes, and baskets | ||
| Newspaper and magazines | Tubes and lids | ||
| Phonebooks and junk mail | Cartons | ||
| Tissue boxes/rolls, paper towel and toilet paper rolls | |||
| Paperboard and Cardboard | |||
| Brown or kraft paper | |||
| Paper boxes/cartons, Pizza boxes (without food/liner) | |||
| Contamination (materials which will be sent to landfill) | |||
| Shredded paper | Flexible plastics (plastic bags and plastic film) | All metal except for those allowed | All glass except for those allowed |
| Containers coated with wax, plastic or other materials | Expanded polystyrene foam (#6) | Aerosol cans that are under pressure or partly filled | Drinking glasses, crystal, or tableware |
| Wallpaper | Multi-layered juice pouches | Mirrors | |
| Material with excess food residue or other organic material | Plastic form electronics | Non-container glass | |
| Plastic marked as biodegradable or compostable | Light Bulbs | ||
| Windows | |||
| Prohibited materials | |||
| Sharps and needles, batteries, radioactive materials, hazardous materials, corrosives, medical waste, pesticides, poisons, biohazards, compressed gas cylinders, refrigerants, polychlorinated biphenyl (PCB)-containing capacitors, transformers, and ballast, asbestos, wax, electronics, wood, ceramics, food waste, rock, dirt, asphalt, concrete, regulated materials, materials that can damage the equipment | |||
Two types of conveyors are usually used: rubber and chain belt conveyors. Rubber belts are divided into two categories: slider beds, where the belt moves through a flat or shaped trough and is ideal for sorting with the material more spread-out, and idler conveyors, which have a belt over idler rollers perpendicular to the material movement, better suited for fine or gritty material. Chain belt conveyors are often employed when there is a need to move material through increased angles, such as feeding other equipment or weighty loads due to chain-link drive systems being more robust than rubber conveyors. The top speed for these conveyors is considerably slower.88
After entering the MRF environment, the waste stream passes through multiple separation equipment and operations. The simplest method is known as manual sorting, a labor-intensive step in the MRF operation and a source of potential health and occupational hazards,89 including physical, chemical, and biological concerns. The sorting can be divided into a negative sort when undesired materials are removed from the waste stream and a positive sort that looks for recyclables and placement into the correct bin.90 For the separation involving multiple material sizes into a specific range, different types of screens are commonly employed, such as vibratory, disc, trommel, ballistic, and, more recently, auger screens, as described in Table 7.
| Types | Description | Illustration |
|---|---|---|
| Vibratory (shaking) | The screen can be horizontal or inclined, with the waste stream cascading through a series of finger elements, perforated plates, or meshes, promoting material separation. Presents better performance when operating with dry and cleaner waste, as it can be easily clogged |

|
| Disc | Rotating discs are positioned in shafts, moving the waste stream throughout the screen in horizontal or angled positions. The distance between discs (interface opening) is associated with the screened material size. Disc rolling might help by partially breaking the material on the screen. A specialized version of a disc screen that separates 2D (paper or film) from 3D materials (containers) can be called a polishing or planar screen |

|
| Trommel (revolving) | Cylinder-mounted rollers in an inclined position contain holes in the sides and might have baffles. They are used for primary or final size screening. Factors that affect the input and separation efficiency include screen openings, trommel diameter, rotational speed, type and number of baffles, and cylinder inclination. Tumbling motion can be cascading, cataracting, or centrifuging |

|
| Ballistic | Combines parallel paddles that vibrate (similar to a vibratory screen) with a 2D/3D screen framework. Three flows are collected: a fines stream with material that passed through the paddles, a 2D stream with paper, cardboard, and films, and a 3D stream mainly consisting of containers |

|
| Auger | One of the newest types of equipment launched to the market consists of multiple cantilevered augers in corkscrewing motion, which prevents the wrap of plastic films and hoses. Mainly employed as a pre-sort step for removing large flat fractions such as (old corrugated containers) OCC from smaller objects |

|
Air separation exploiting different densities of materials in mixed waste streams is another option at MRFs. A heavy fraction composed of metal, glass, and stones falls to the bottom of the equipment while a lighter fraction of paper, plastic, and dry organics is carried away by the air stream, requiring the use of a cyclone, for example, for the recycling of the air stream.90 Equipment in this category includes zig-zag classifiers, air-drum or rotary air, aspirator air or suction hood, cross-current air, and airbed classifiers.88,90
Another crucial piece of equipment typically used for material sorting is the optical sorter. The use of this type of equipment can substitute for manual labor and lead to high sortation efficiencies; however, a significant capital cost is required for its installation, which hinders small and locally owned MRFs from selling bales in a market with low tolerance to contamination. An estimate using data from the United Kingdom by Eule showed that a MRF with a single operation shift could take up to 3.14 more years to break even after installing an optical sorter compared to manual labor.92
Optical sorters use color-sensitive cameras and near-infrared (NIR) and ultraviolet (UV) spectroscopy sensors, or a combination of them, to identify different materials that pass through a conveyor belt. Once the material desired to be positively sorted is identified, air jets are activated to eject the piece to a different chute.90,92 Spectrometric techniques exploit the unique wavelength signature of different kinds of materials.88 NIR optical sorters are most commonly used for plastic separation as a non-destructive technique with high-speed measurement, penetration depth and signal-to-noise ratio.91 NIR optical sorter use has been growing for sorting fibers and cartons.93 Another advance in sorting technology is the use of hyperspectral imaging (HSI), which combines imaging from a digital camera with spectrometric analysis, to obtain a discrete spectrum for every pixel collected. This is then computationally analyzed using algorithms such as principal component analysis94,95 for the sorting of more complex plastic streams containing low and high density polyethylene, for example.96 Machine learning algorithms are being studied to overcome some of the challenges associated with NIR sorters,97 such as the difficulty of correctly sorting black plastics98 or avoiding the still high costs of HSI technology.99
For the sortation of ferrous materials, magnetic separators are used to sort materials such as steel from the remaining waste stream. Multiple factors are considered to determine the system's efficiency, including the distance between the conveyor carrying the waste stream and the magnet, the magnetic force employed, the conveyor speed, and the burden depth on the conveyor belt. Common types of setup include a drum holding magnet, a belt holding magnet, and a suspended type magnetic separator.100 For non-ferrous materials, such as aluminum and copper, eddy current separators are used, often after the magnetic separator. The sorting is based on the eddy currents formed when conductive objects are close to a magnetic field (created using rotating magnets), with another magnetic field of opposite direction. According to Lenz's law, this creates a force that expels these objects from the initial magnetic field, giving them a different trajectory. Considering that less dense and conductive objects demand a lesser force, aluminum has the greatest potential for separation through this equipment since it has one of the highest conductivity/density ratios compared to other metals, such as lead, copper, zinc, and tin.90
At the end of the sorting process, each material stream is compacted or densified to reduce its volume to minimize storage space requirements and increase payload for recyclable material and residue transportation, significantly reducing costs.90 Balers are commonly used to reduce the bulk density of the material and can be divided into two different types: vertical balers, an option with reduced output but low costs, or horizontal balers, which are the most utilized in MRFs.101 Horizontal balers are usually single-ram or dual-ram (two-ram), with the first mainly used for fiber, paper, and OCC output lines, while the latter is considered a multipurpose equipment utilized for plastic and aluminum outputs due to its capacity to produce more heavy and uniformly packed bales.88,102 Single-ram balers operate with the sorted material being pushed from a collection chamber into a narrow hopper, with the densified material moving continuously as more material is fed and tied with wires. On the other side, a dual-ram baler (Fig. 11) has the main ram that loads the material into a chamber until the desired amount is achieved, while a secondary ram is responsible for moving the bale to a tying process.88
 | ||
| Fig. 11 Two-ram Baler from Machinex Recycling. Reproduced from ref. 104 with permission from Machinex Technologies, copyright 2022. | ||
The Association of Plastic Recyclers (APR) has developed some specifications to standardize plastic bales and facilitate the sales between MRFs and recycled plastic reprocessors, brokers, or companies that can use them in their process. For rigid plastic containers bales, including natural and colored HDPE, PP, and #1–7 plastics, it is expected to have a bale density between 15 and 20 lb ft−3 (240 and 320 kg m−3), whereas, for a PET bottle bale, the targeted bale density is between 15 and 18 lb ft−3 (240 and 288 kg m−3), all using non-corrosive galvanized bale wires of 10–12 gauge (2.6–3.2 mm).103 The main products of a MRF are the bales of sorted material with the ultimate goal of producing saleable recycled material and the least amount of non-recyclable residues. In the optimal scenario, these residues can be used as fuel for waste-to-energy conversion, in anaerobic digestion or, in the worst case, be an expense for discard in a landfill. For plastic bales, the ISRI Scrap Specifications Circular defines standards for some of the most common types of bales from a MRF in terms of composition, contaminants, and prohibited items, as shown in Tables 8 and 9. The historical prices for some plastic bales are listed in Table 10.
| Bale type | Material description | Main product | Allowed contamination |
|---|---|---|---|
| #1–7 bottles and small rigid plastic | Rigid plastics, consisting of at least 65% bottles, bulky rigid containers greater than 5 gallons should be avoided | Bottle and non-bottle containers | 5 wt% total, with less than 2 wt% paper/cardboard and 1 wt% of metal/plastic bags/liquid/other residues |
| #3–7 bottles and small rigid plastic | Rigid plastics without PET (#1) and (#2), bulky rigid containers greater than 5 gallons should be avoided | Bottle and non-bottle containers | 5 wt% total, with less than 2 wt% paper/cardboard/metal and 1 wt% of liquid/other residues |
| Mixed bulky rigid (#2 and #5) | Large rigid plastics HDPE (#2) and PP (#5) | Bulky rigid plastic | 15 wt% total, with less than 4 wt% for plastic/packaging items (#1, #3, #6, #7), 2 wt% of metals, glass, wood, liquid/other residues, paper/cardboard, plastic bags/sheets/films |
| Tubes and lids (#2, #4, and #5) | Any whole container of HDPE (#2), LDPE (#4), and PP (#5) | Tubes and lids | 10 wt% total, with less than 2 wt% of metals, liquid/other residues, paper/cardboard, injection-molded HDPE (#2), PET (#1) bottles and thermoforms, plastic/packaging items (#1, #3, #6, and #7) |
| Bale grade | Grade A | Grade B | Grade C | Grade F | |
|---|---|---|---|---|---|
| PET (#1) | Total fraction (wt%) | >94 | 93–83 | 82–73 | <72 |
| Max. contamination (wt%) | 6 | 7–17 | 18–27 | >28 | |
| HDPE (#2) | Total fraction (wt%) | >95 | 94–85 | 84–80 | <79 |
| Max. contamination (wt%) | 5 | 6–15 | 16–20 | 21 |
| Materials | National average price (cents lb−1) | |||||
|---|---|---|---|---|---|---|
| Year | 2017 | 2018 | 2019 | 2020 | 2021 | 2022 |
| Plastics PET (baled) | 11–16 | 14–17 | 9–15 | 6–11 | 7–26 | 18–25 |
| Plastics film (grade A, sorted, 800 + lb bales) | 8–11 | 8–12 | 9–12 | 8–11 | 11–21 | 21 |
| Plastics Natural HDPE (baled) | 24–33 | 30–39 | 20–59 | 33–64 | 56–108 | 55 |
| Plastics colored HDPE (baled) | 13–17 | 14–17 | 10–16 | 4–14 | 17–58 | 20 |
| Plastics commingled (#1–7, baled) | 2–3 | 2–3 | 2 | 1–2 | 1 | 1 |
| Plastics PP post consumer (baled) | 7–9 | 8–11 | 7–13 | 3–12 | 12–39 | 19–23 |
| Plastics polystyrene EPS (baled) | 2 | 2 | 2 | 2 | 2 | 2 |
129 tons.106 The Recycling Partnership conducted another survey with data from Resource Recycling, initially finding 360 MRFs in the United States in 2019, a number that was updated in February 2020 to 367 after a verification conducted by The Last Beach Cleanup (a nonprofit 501(c)3 organization), with a spreadsheet available on their website107 and results published by Greenpeace.108 The distribution of MRFs across the U.S. is shown in Table 11. A list of the largest 75 MRFs in North America was published in 2019 by Recycling Today, with 65 located in the United States and 10 in Canada. The largest MRF was Sims Municipal Recycling's Sunset Park, located in New York, which shipped 241884 tons in 2018.109 In England, data from the last quarter of 2019 totaled 85 regulated material facilities (>1000 tonnes per year recyclables processed and required to submit their data to England's Environmental Agency), resulting in daily input of 9237 tonnes. In Wales, there were 12 facilities in the same year, with daily input of 710.6 tonnes.110 Ali and Courtenay111 observed from data that approximately 60% of United Kingdom MRFs have an annual capacity higher than 50000 tonnes. Italy had 33 facilities for plastic sorting (“centri di selezione” – CSS) in 2020 associated with COREPLA (National Consortium for Collection and Recycling of Plastic Packaging) that receive a pre-sorted waste stream mainly composed of mixed plastics from 996 smaller and local sorting centers (“centri comprensoriali”).112,113 Germany's federal statistics reported 987 waste sorting facilities in 2019, with a total input of 25120700 tonnes.114 In France, it was reported that 399 waste sorting facilities (“centres de tri de déchets”) were operating in 2016, receiving 11.1 million tons.115
| Region | Number of MRFs | |
|---|---|---|
| EPA (2018) | The last beach cleanup/Greenpeace (2020) | |
| Northeast | 128 | 66 |
| South | 142 | 107 |
| Midwest | 139 | 76 |
| West | 123 | 118 |
| Total (U. S.) | 532 | 367 |
In this section we discuss waste pickers in PET recycling in Mexico to illustrate the challenges in Latin America. Mexico is the second largest PET bottle consumer in the world, mainly due to the high consumption of soft drinks and bottled water.120,121 The high consumption of PET bottles, many of them discarded in public places, roads and transport systems and rural areas, combined with a lack of recycling programs caused environmental and health problems, and the clogging of drainage systems in urban areas. Since the mid-1990s, the Mexican government has set voluntary PET recovery and recycling programs that were successful to some extent.121 PET recycling activities involve sorting, washing, re-granulating and pellet production by local enterprises. The pellets are then shipped to domestic Mexican plastic transformation industry (35%) or exported to other markets (65%). In Mexico, recycled PET facilities produce textile fibers, bottles for non-food uses and industrial strapping.
In Mexico, recycling of the MSW is mostly in the hands of waste pickers, middlemen or brokers.122,123 Waste pickers are self-employed, sometimes resorting to collecting plastics from neighborhood garbage containers before they are collected by contracted waste management services. Waste pickers often organize themselves into associations, cooperatives, unions or microenterprises, through which they feel empowered to carry out their activities.116,121 Often local authorities promote and recognize the formation of associations or cooperatives, which are able to negotiate agreements for waste collection and recycling services. However, even with these associations the waste pickers income is not fixed but based on the quantity and quality of recycled materials and the selling price in local markets.
Currently, recycled PET price depends on factors such as impurities (i.e., amount of other plastics or residues), quality (i.e., clear vs. color PET), cleanliness, type of organization the waste pickers belong to, middleman or broker who buys the product, and even the geographical area (i.e., urban vs. rural) where PET is collected and sold. An average price for recycled PET ranges between $0.24–0.48 per kg,105 although it is likely that most waste pickers receive the lowest price. Waste pickers who recycle PET must work long hours to obtain sufficient income to support their families. The official minimum daily wage set by the Mexican federal government was $8.25 per day in January 2022. If an average recycled PET price of $0.36 per kg is considered, a waste picker must collect around 23 kg of PET every day to receive the equivalent of the minimum daily wage. A number of studies highlight the low monthly income ($120–360 per month) by waste pickers in Latin America.124 There is an urgent need to propose business schemes that improve the income of waste pickers, in addition to providing them health, safety, labor and social benefits through formal employment. The development of business models in Latin America that formalize the inclusion of waste pickers in the recycling value chain, and that limit the actions and profits of middlemen, will have social benefits for waste pickers and will bring favorable economic benefits for the Mexican recycled PET industry.
Some successful initiatives show that when waste pickers are organized by non-profit and for-profit organizations and they are formally included in the PET recycling value chain, a number of favorable social and economic benefits occurs.118 Social-oriented programs for waste pickers lead to their recognition and empowerment through the establishment of formal jobs, with fixed salaries and labor and health benefits with local government or private companies. Social-oriented programs may have a profound impact on the education and culture of waste piker's communities. These social-based initiatives are emerging in the MSW management value chain of Mexico, Brazil, Argentina, Colombia and Chile, but the benefits are still limited to a few communities. In the current socio-economic situation in Latin America, the development of technologies and industries that facilitate the processing of plastic waste into products with higher added value in the market, is a critical factor to facilitate the inclusion of waste pickers in the value chain of plastic recycling.
MSW from urban and rural areas is typically processed by public services run by local governments to avoid negative impacts on health and the environment. In countries such as Mexico and Brazil, the management of MSW is regulated by federal and local laws, which establish guidelines for the collection, separation, recycling and final disposal of waste, using strategies that, in principle, contribute to the preservation of the environment and take care of the health and safety of the employees.118,119 Various technological, economic, administrative, social and cultural factors prevent risk and safety management from being carried out properly, especially in rural areas. As a result, it is estimated that around 40 million people (6% of population) in Latin American communities lack trash service. Up to a third of MSW is disposed in open dumps.125–128 Consequently, the prevailing situation is that the MSW management in Latin America is a focus of concern in terms of public health, environmental risks, informal economy, security and social inequality, particularly for the communities where the MSW transfer units and final disposal sites are located.
A license is granted to private companies when MSW cannot be adequately collected by local governments. A growing number of companies in Latin America offer the following benefits in MSW management: a reduction in public spending, selective processing of domestic, commercial, and industrial waste, use of specialized technology and skilled workforce, generation of well-paying jobs, and management projects contributing to sustainable development.129,130 This recycling strategy is very distinctive from the circular economy and it has been identified as a vector contributing to the fulfillment of Sustainable Development Objectives set by the United Nations. For this reason, federal and local governments in Latin American communities have established legal frameworks to trigger waste recycling, as in the case of Brazil, which in 2010 established the National Solid Waste Policy, and Mexico, which in 2003 approved the Law for the Prevention and Integrated Management of Waste, and very recently the General Law of Circular Economy with the aim of promoting efficiency in the use of waste through reuse and recycling.119,131–133
Poverty and the growing market value of recyclable products has induced many people in conditions of social and economic vulnerability to work as waste pickers. It is very common nowadays to find fixed human settlements around waste transfer and final disposal units. Entire families live in these settlements. It is common for waste pickers' children to continue with the family tradition. The situation of waste pickers is similar in other countries that do not have a modern waste management system.134 The development of advanced waste management systems combined with new plastic recycling technologies will drastically improve the quality of life for waste pickers.
4. Mechanical recycling
Mechanical recycling is defined by the U.S. Food and Drug Administration (FDA) as secondary recycling and is the most common approach for recycling plastic solid waste (PSW). This process has an established infrastructure in most countries. In secondary recycling, the post-consumer plastic waste is reprocessed and converted into new materials.135 With the advancement and availability of mechanically recycled PSW, virgin plastics are substituted with post-consumer recycled materials or blends thereof in many industrial and consumer applications.136,137 Mechanical recycling involves collection/segregation, cleaning and drying, chipping/sizing, coloring/agglomeration, pelletization/extrusion, and manufacturing the end product. Both primary and secondary recycling involve similar process steps.138 Primary recycling as defined by the FDA recovers pre-consumer or post-industrial recycled (PIR) waste material and produces new products. Secondary recycling physically reprocesses post-consumer waste materials to produce new materials.139 Primary recycling maintains the same quality as virgin material, whereas downcycling is common in secondary recycling due to some degradation of the plastic's properties.136 Post-consumer materials, in particular polyolefins, can have unpredictable physical properties due to the large range of polymer structures, molecular weights, and additive packages. Such variation and lack of grading can make using these materials more difficult. The variation in color further complicates usage in new products due to the inability to control the final colors. For example, plastic materials having different colors produce undesirable grey color after reprocessing into pellets.1404.1 Processing technologies in mechanical recycling
Mechanical recycling consists of a few general steps: shredding/granulation, contaminant removal, followed by flake segregation. The primary step involves the melting and re-extruding of the plastic into uniform pellets136 which can then be utilized in cast/film extrusion, injection molding, blow molding, etc.136,141 The molecular weight of a plastic is highly influential to all the plastic's innate properties.142–144 In general, an increase in molecular weight increases the glass transition temperature, the melting temperature, plastic strength, elasticity, stiffness, toughness, and viscoelastic properties. The processing residence time and reprocessing of the material also influence the molecular weight of the polymeric materials.32,145–147Fig. 12 demonstrates the change in melt-flow index (MFI) of various plastics subjected to extrusion and injection molding. The repeated exposure to high heat, time, and shear rates can cause degradation of the polymeric material, and in some cases, unwanted side reactions that can either increase or decrease the molecular weight of the final product. These structural modifications lead to changes in both flow and mechanical properties of the material. Manufacturers may be inclined to utilize additives to recover material properties or homogenize the recycled material, although this typically adds cost to the recycled plastic and may further complicate future recycling attempts of the material waste. Different variations of mechanical and thermal processing technologies (Fig. 13) used in recycling are described below. | ||
| Fig. 12 Change in MFI as a function of number of processing events for extrusion (left) and injection molding (right). MFI0 and MFIF are the MFI prior to any processing event and the MFI after the total processing events, respectively. Published from ref. 32 with permission from Carl Hanser Verlag, Munich, copyright 2012. | ||
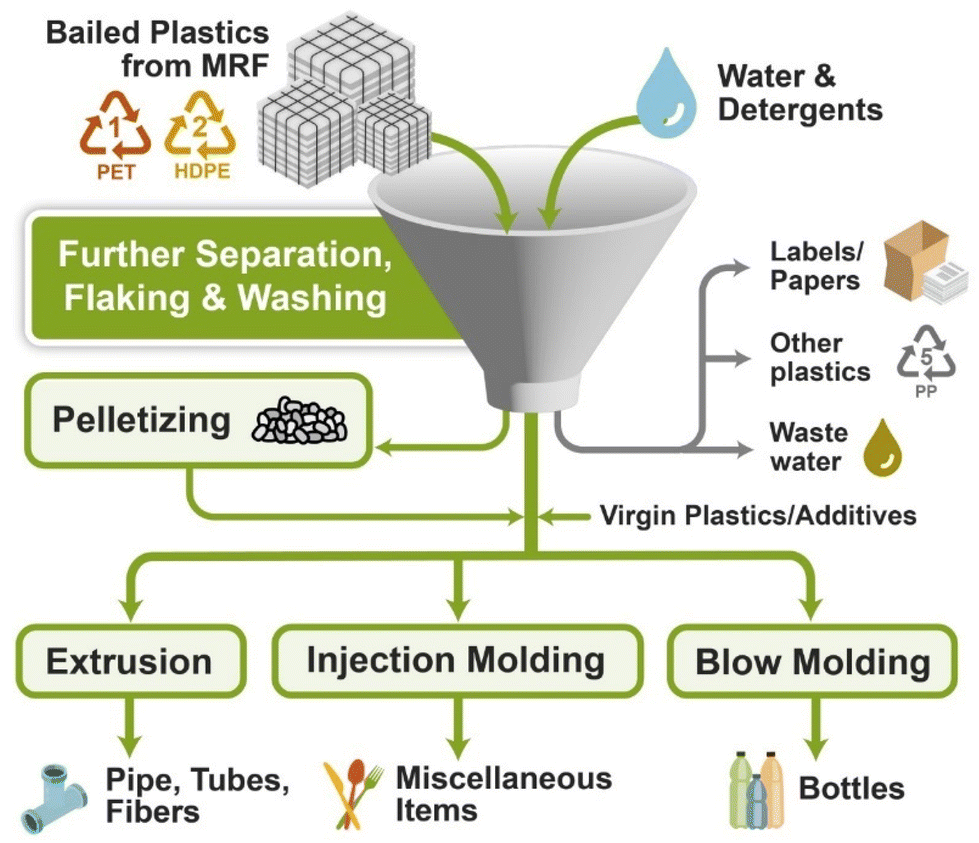 | ||
| Fig. 13 Various approaches for mechanical recycling of PSW (plastic solid waste).136 | ||
4.2 Extrusion molding
In extrusion molding, plastic pellets enter the hopper into the barrel of the extruder in a continuous process. The plastic is fed into the barrel in the screw feed zone, and the screw forces raw material through the hot barrel and causes melting. The constant moving of the plastic through the barrel restricts overheating and limits resonance time, which may degrade the plastic.148 Because of the heating during extrusion, some alteration to the polymer chain occurs. The plastic melt in the barrel passes through screen packs to remove gross contaminants during the extrusion process. The screen pack and breaker plate are found in commercial plastic extrusion between the extruder and the die. Melted plastic is re-shaped using a die.136 The breaker plate ensures the proper mixing in the screw of the plastic by creating backpressure and reorients the plastic creating a stable linear flow before entering the die. Uneven flow of the molten plastic into the die can create undesirable residual stress that can cause warping during the cooling stage.148 The extrudate is cooled with a water bath or cooled air stream. This process is complicated as plastics are good thermal insulators.148 The extrusion process for recycling is similar to a new product manufacturing technique. Details of extrusion molding, injection molding, blow molding, film molding, and fiber processing can be found in SI 2.0 in ESI†.4.3 Mechanical reprocessing of PET
Bailed PET often contains a large number of contaminants including adhesives, sugars, and other plastics. It is important to clean the PET and separate it from other materials before it is melt-processed through extrusion to produce recycled PET pellets (rPET).149Fig. 14 illustrates a typical PET bottle recycling process which is adapted from one of the PET bottle recycling companies in Europe.150 The delivered bottles are sorted depending on the type of material, color, processing method etc. The bottles are then shredded into small flakes followed by washing processes. The flakes go to the extruder after cleaning and drying.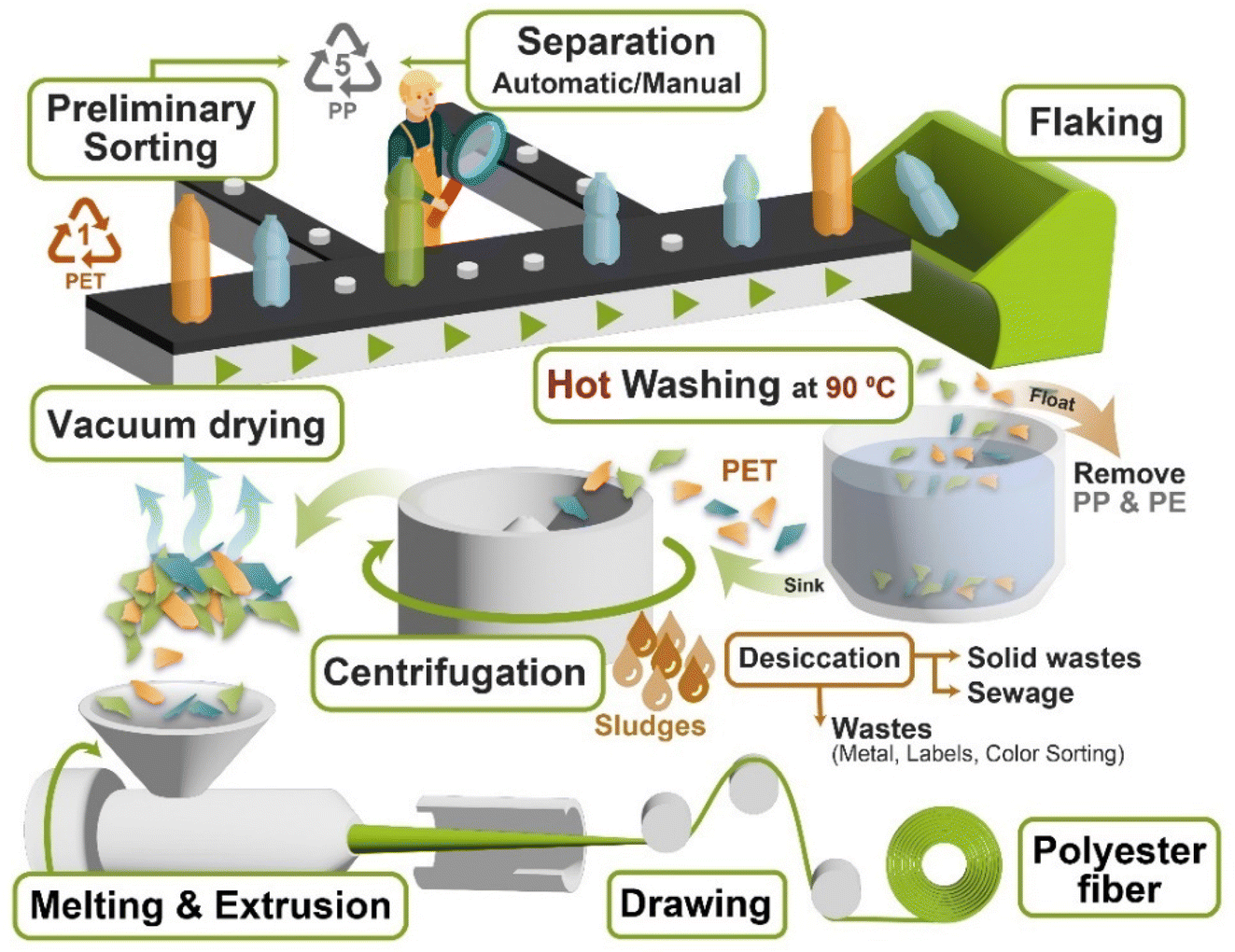 | ||
| Fig. 14 A generalized process flow diagram for a recycling of PET bottles.150 | ||
Melt extrusion of clean washed PET flake or granules is used to manufacture pellets of uniform shape for subsequent manufacturing processes (injection molding, extrusion, injection stretch blow molding, etc.).151 When PET reprocessing uses an established facility, processing becomes more cost-effective, more straightforward, and possesses less environmental impact. Switching from virgin to recycled PET depends on material pricing, secure recycled material supply chains, and the ability to use existing facilities.152 Processing PET at high temperatures may cause chain degradation which influences crystallinity of the plastic.146 Plastic degradation from chain scission causes molar mass plastic decrease, which makes bottle-to-bottle recycling challenging.153
rPET can be used for most applications instead of virgin materials provided that the properties meet the quality criteria. Clarity and yellowing are major property challenges for rPET.137 Like other plastics, PET's quality is also affected by chain degradation because of thermo-oxidation.154,155 Some contaminants (such as PVC and polyvinyl alcohol) accelerate chain scission in PET plastic during melt processing due to acidolysis.156,157 Impurities in PET enhance chain scission reactions, and traces of PVC and polylactic acid (PLA) can accelerate acidolysis or hydrolysis. These reactions alter the plastic's microstructure and the mobile amorphous region is altered causing thickening of the crystalline domains.155 Crosslinking can also occur, resulting in increased viscosity in PET, hindering processability for applications such as injection molding. The multipole extrusion cycles of mechanical recycling increase the rate of chain scission because of thermo-oxidation.
Compounding additives into rPET during reprocessing can protect the plastic from chain scission and improve mechanical properties. To minimize thermo-oxidation of the plastic, metal-based stabilizers are often used.154 The use of radical scavengers, such as organic phosphates, is another option to hinder radical induced oxidation of the terephthalate ring, which is a source of yellowing.155,158 The most common problem in PET reprocessing is viscosity reduction (molecular weight reduction) due to hydrolytic degradation and thermal oxidation. Color change in the post-consumer PET is another vital problem and normally the industry solves the problem by “open-loop recycling” or “semi-closed-loop recycling”.154 The concepts of “open-loop recycling” or “semi-closed-loop recycling” are described in section 4.3.1.1.
If the molecular weight reduction is less severe, the plastic can be subjected to a process called solid state polymerization (SSP) to help rebuild the molecular weight.159 The plastic is heated to a temperature between the melting and glass transition temperatures to induce esterification and transesterification reactions.153 Industries are currently looking for a new stream of PET waste to reprocess, including colored PET.160 For example, in 2018 ‘Indorama Ventures’ partnered with ‘Unilever’ to recycle fifty billion bottles per year by 2025, including colored PET. According to them, traditionally non-recyclable colored PET is subjected to a chemical recycling process. These colored PET are reduced to monomeric species, and then the contaminants removed before being used as raw material for polymerization. This technology produces transparent food-grade PET plastic material from post-consumer PET.161,162 Recycling for food contact packaging requires a very demanding and clean plastic. No Objection Letters (NOLs) by the Food and Drug Administration are often sought by recyclers and manufacturers to demonstrate that their recycling process and materials are suitable for direct food contact applications.163
Continuous improvements in plastic reprocessing is an ongoing effort with new technologies in degassers and filters to facilitate increased plastic melting quality.164 Degassing employs a vacuum vent on the extruder to enable volatile compound stripping from a plastic melt. This stage reduces hydrolysis by removing moisture and adds value by producing a high-quality plastic with maintained molecular weight and decreased contamination. The addition of a filter helps remove unwanted non-volatile contaminants from the plastic and increases the mechanical properties of the recycled plastic.165 When selecting recycling technologies in manufacturing processes, it is important to select the appropriate mechanism to minimize the degradation of the plastic for high-quality new parts.155
4.3.1.1 Closed-loop and open-loop recycling. The recycled raw material supply increases as plastic waste collection increases. These raw materials can be used to produce new material as ‘open loop’ or ‘semi-closed loop’ or ‘closed loop’ recycling.166 Closed-loop recycling means the recycled raw material is used for the same product and fully replaces the virgin material, e.g., PET bottle to bottle recycling. Open-loop recycling means the recycled material is used for a different product and fails to replace the virgin raw material in the original application, also referred to as downcycling. The inherent properties of raw materials for open-loop recycling are often too degraded by the service life and recycling process and the plastic cannot be used in the original application. Semi-closed recycling means the recycled raw materials are used to produce another type of product but cause no change in the inherent properties of the recycled materials.167
4.3.1.2 Recycled PET products. rPET is incorporated to form a wide range of new products, for example, shoes, bags, fibers, boards, mechanical parts, etc., as open-loop recycling. Most rPET is used for fibers.168 Consumers can currently purchase rPET food packaging containers. According to a 2020 NAPCOR (National Association for PET Container Resources) Report post-consumer rPET used in the US and Canada has continuously increased since 2004. It has been predicted that by 2026 the market will reach up to $12.5 billion, representing an ∼8% Compound Annual Growth Rate (CAGR) from 2019 to 2026.168,169 The NAPCOR Chairman mentioned that post-consumer rPET utilization is proceeding towards a circular economy. According to NAPCOR, fiber sectors made up more than 40% of the worldwide rPET market in 2019.170 The fiber market includes manufacturing mattresses, insulators, car equipment, etc. Asia Pacific plastic industries are increasing their dependency on rPET. Colored rPET is used for food and non-food applications for its stability and strength. These have high popularity as they are lightweight and provide strength for a long period.168
4.4 Mechanical reprocessing of polyolefins
Mechanical processing of polyolefins also induces degradation at the molecular level and the formation of aldehydes, ketones, and short-chain hydrocarbons.171–173 The degradation mechanism is different in each type of polyolefin. The general degradation mechanisms begin with the generation of macroradicals within the molecular chain. This radical creates many shorter, branched, or cross-linked polymer chains. When these radicals react with oxygen centered radicals, further degradation occurs.174 Previous studies have demonstrated that HDPE is more prone to crosslinking whereas PP shows a large number of chain scission events. These degradation reactions mainly occur in the amorphous phase below the plastic's melting temperature because oxygen cannot diffuse into the crystalline phase.155As post-consumer plastic films are contaminated with various waste materials, an intensive pre-treatment is crucial before extrusion. The source of raw material in secondary recycling is MSW mixed with other plastics. This needs an initial separation step to separate the contaminants. The mechanical recycling process described here (Fig. 15) is an example of a European processing facility.175 The process starts with comminution of large plastic parts and shredding/sieving to remove as much of the contaminants (metals, glass, paper, etc.) as possible. The contaminant-free plastics are sent for pre-washing then vigorous washing to avoid micro-physical contamination. Afterward, the material enters the hydrocyclone to separate undesirable particles.175 A friction washer is used for washing to consistently remove contaminants from the plastic such as labels, glue, or organic dirt on the surface.176 After washing, the plastics are dried thermally to lower the moisture content before entering the extrusion and pelletizing stage.175 The function of extruder and blowing machines are discussed above. The quality of the recycled plastic film is degraded leading it to be used in applications such as products for constructions, public spaces, and traffic applications after injection molding or extrusion.177
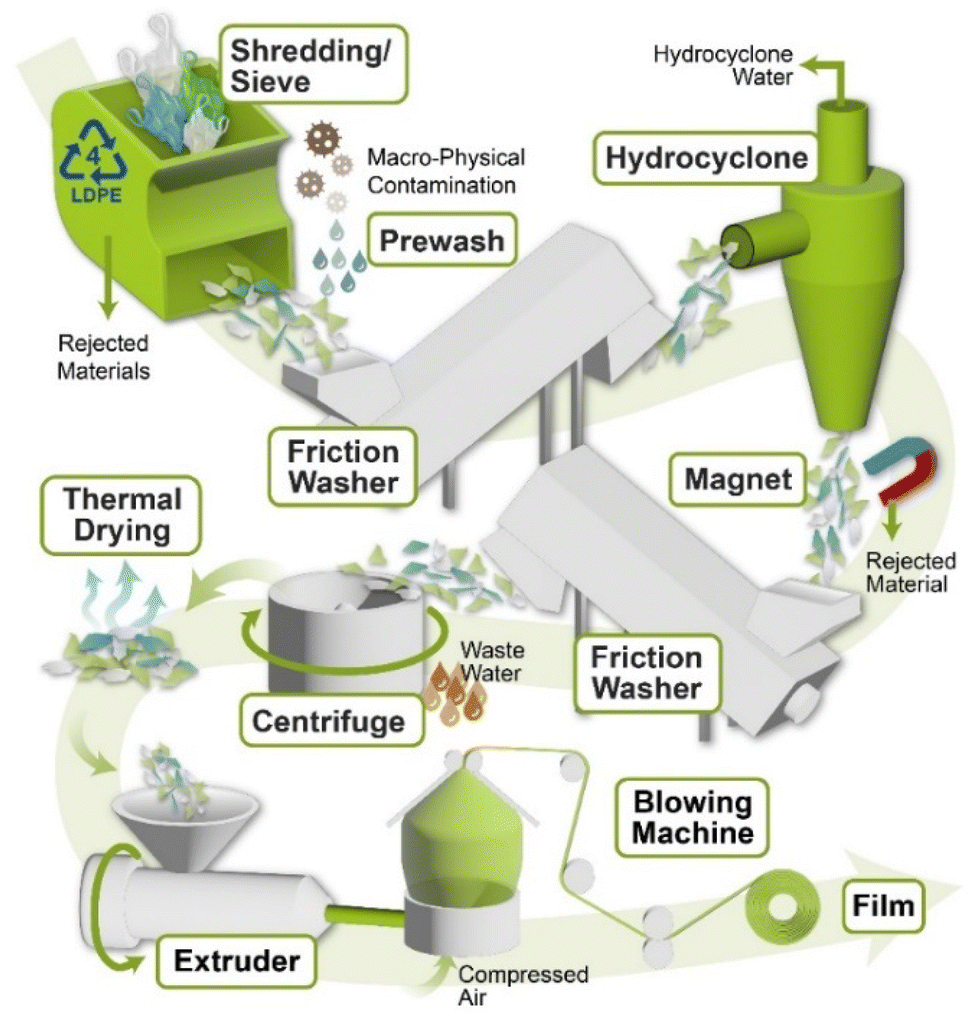 | ||
| Fig. 15 Mechanical recycling process of polyethylene plastic film at an industrial plant.175 | ||
Recycled HDPE and other recycled polyolefins have multiple applications in the food packaging sector and multilayer packaging applications. However, there is a concern with recycled raw material in direct food contact because potentially toxic small molecules (plasticizers, stabilizers, etc.) could diffuse and migrate into food products.180 As a result, recycled polyolefins are often utilized as the middle layer in multilayered packaging, and the surface layers are made of virgin plastics. Recycled HDPE has a large market as the processing cost can be relatively low. Post-consumer HDPE is a feasible option to produce plastic lumber which is a replacement for wooden lumber.
4.5 How legislation changes the scenario
The municipal waste management system and plastic recycling in most European countries has made remarkable advancement. EU countries with landfilling restrictions or bans have higher recycling and waste recovering percentages as shown in Fig. 16. Recycling of HDPE, LDPE, PET, PP, and PS plastics are performed together.181 Post-consumer plastic waste collection was more than 32% in several EU countries which was mostly comprised of mechanical recycling techniques. The highest recycling rate was found in Norway at 45% in comparison to ∼9% in the United States.137,182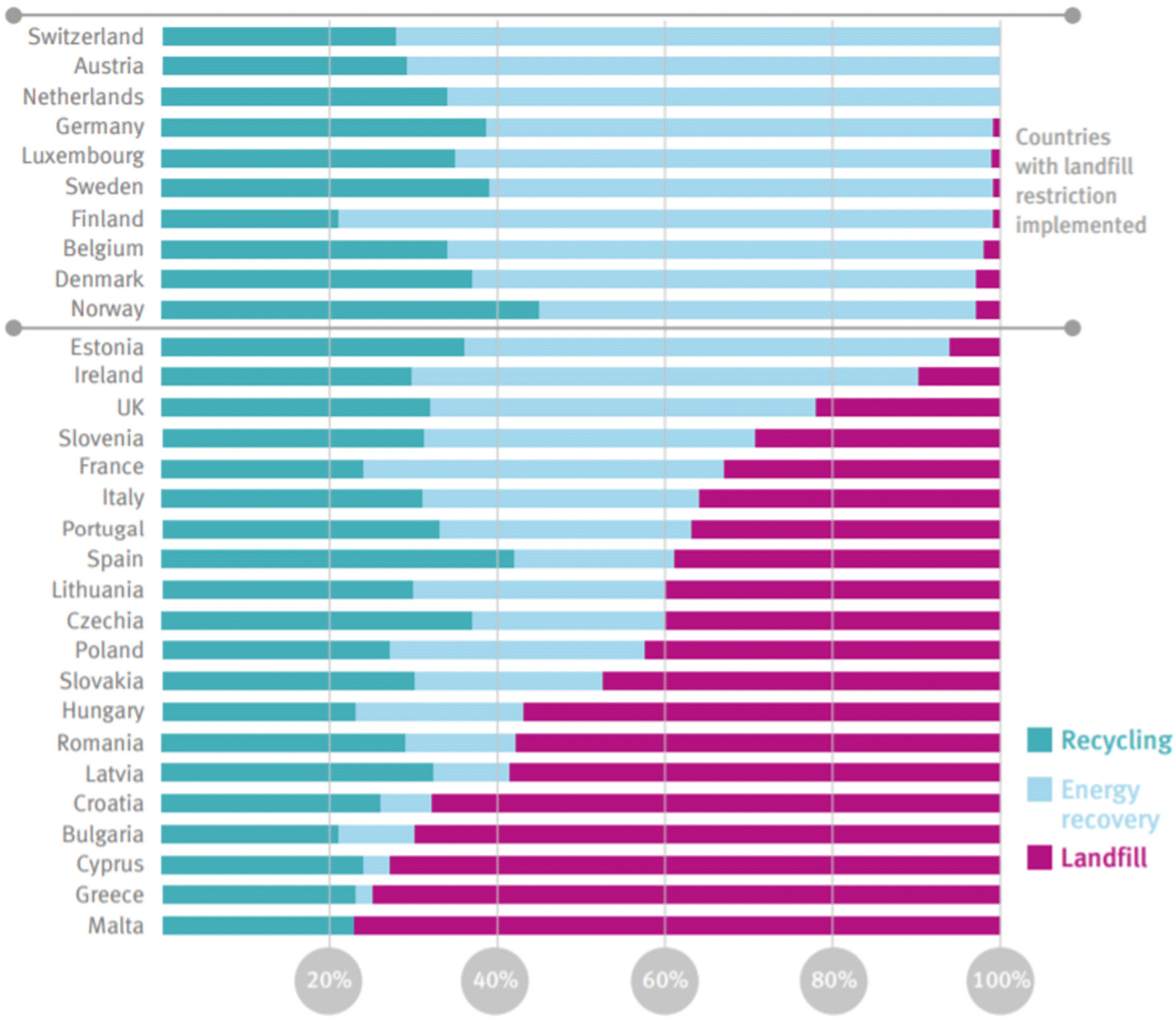 | ||
| Fig. 16 Post-consumer plastic waste recycling rate, energy recovery rate and landfilling rate per European country in 2018. Reproduced from ref. 186 with permission Plastic Europe, copyright 2022. | ||
The number of recycling companies is increasing. There are almost 115 PET recycling and 40 PE recycling facilities in the US. For example, ‘77 recycling' is a plastics (post-industrial plastic scrap) recycling company that accepts most materials (PET, PP, PS, PVC, HDPE, LDPE, etc.).183 China's ban on imports of plastic waste increased the recycling facilities across the US.184 Good communication between product manufacturers and the recycling industries is needed to confirm the recycling of packaging materials. This communication can strengthen the use of recycled raw materials and create a new market for these recycled plastics.185
• When recycled plastic/material is used as FCM, the chance of contaminants in the final product may increase.
• Post-consumer recycled plastic/materials are permissible to use as FCM material.
• The adjuvants (for example petroleum waxes, synthetic petroleum wax, colorants, plastic modifiers188) coming from the post-consumer recycled plastic/material may not be suggested for use in FCMs as the contaminant present in the recycled material can pass on to the new product and migrate into the food matrix.189
• The FDA regulates the plastic recycling processes to ensure that the recycled materials are not contaminated and that the recycling process is suitable for direct food contact.163 For example, in secondary recycling, the physical reprocessing steps, grinding, melting, and reforming, are documented and submitted for evaluation to obtain a favorable opinion letter through the No Objection Letter (NOL) program. For plastic depolymerization, it is necessary to confirm that the monomers are contamination-free and the produced plastic is the same as that from virgin monomers.190
• In the case of multi-layered packaging, sometimes, when proper contamination removal is complex, the recycled layer is used as the most outer layer. For example, for recycled PET, FCM is used as the outer layer in multi-layered containers, and the inner layer in contact with food is made of virgin material.155
Manufacturers of FCMs using post-consumer recycled materials often need special evaluation.163 The NOL determines the assessment of the plastic used for a FCM and whether the recycling process is physical or chemical. The earliest NOL191 was issued in 1990 for a physical recycling process of whole egg cartons.192 Many of the early NOLs that granted favorable opinions were for PET and PS plastics.193 However, in recent years, much of the focus has been on polyolefin materials; utilization of post-consumer polyolefin streams for direct food contact applications is challenging due to a large number of different molecular structures, additive packages, and uses for which consumer abuse can lead to incidental contamination.194 Such challenges are overcome through careful source control of the post-consumer material and challenge surrogate chemical testing to determine if the recycling process can reduce the concentration of different types of compounds, which is often a requirement for the NOL process.
For the surrogate chemical challenge testing, the plastic is soaked in a chemical cocktail containing a volatile polar compound (e.g., chloroform, chlorobenzene, 1,1,1 trichloroethane, diethyl ketone), a non-volatile polar compound (toluene), a heavy metal compound (copper(II) 2-ethylhexanoate), non-volatile polar (benzophenone or methyl salicylate), and a non-volatile non-polar compound (tetracosane, lindane, methyl stearate, phenylcyclohexane, 1-phenyldecane, 2,4,6-trichloroanisole) using an isopropyl/hexane solvent delivery mixture to artificially contaminate the plastic.163 After the artificial contamination soaking of two weeks at 40 °C, the spiked plastic is subjected to recycling. The concentration of the artificial contaminants is quantified in the recovered, recycled plastic and the starting spiked plastic to demonstrate the ability of the recycling process to reduce contamination. These results, along with a description of the recycling procedure and post-consumer material feedstocks, are submitted to the FDA to evaluate suitability for manufacturing direct FCMs. Upon favorable review of the packet, a NOL is issued. As of the end of 2021, 254 NOLs have been issued by the FDA.192
5. Thermal decomposition of plastics
5.1 Non-catalytic thermal decomposition of plastics
Pyrolysis is the thermal decomposition of organic compounds typically in an oxygen-deficient atmosphere. The decomposed products are then condensed and collected as oils, waxes, gases, and char, with distributions dependent on feedstock, reaction conditions, and reactor type. Temperatures in the range of 350–700 °C are typical for plastics pyrolysis,195 with the general trend of higher yields of gas at higher temperatures.196 Pyrolysis is generally classified according to heating rate (slow, fast, or ultra) and environment (catalytic pyrolysis, steam pyrolysis, vacuum pyrolysis, microwave pyrolysis, plasma pyrolysis, oxidative pyrolysis, etc.).Pyrolysis is the most researched chemical recycling method for plastics.195 The main advantage of plastic pyrolysis is the ability to convert plastic into lower molecular weight products to be used as fuels or feedstock for new chemicals or plastics.197 Pyrolysis also produces less emission than plastics incineration.198 Unlike mechanical recycling, pyrolysis can handle highly heterogeneous feedstocks with a higher degree of contamination.197
5.1.1.1 Polyolefins. PE follows a free radical thermal decomposition that takes place through sequential random bond–scissions.199–209 The degradation begins in weak links (mainly peroxide groups formed during preparation and storage of PE) and proceeds in the pristine segments of the plastic.210 Peterson et al.211 employed thermogravimetric analysis to demonstrate that the activation energy depends on the extent of the decomposition reaction and assumes values between 150 kJ mol−1 and 240 kJ mol−1. The latter number corresponds to the random scission process for pristine PE.212 Huber and coworkers reported a similar activation energy (234.8 kJ mol−1), and argued that only models that include two random scission steps are capable of capturing the details of weight loss curves as obtained from thermogravimetric analysis.200 The complexity of the free radical thermal decomposition of PE is substantial, and this is reflected in the large variety of pyrolysis products. The main products are alkenes, alkanes and alkadienes, while Diels–Alder reactions may also lead to the formation of small amounts of cycloalkenes.200
The initiation of PE pyrolysis involves a homolytic C–C cleavage reaction that happens at a random position along the PE chain, and gives two macroradicals (1) – (Scheme 1).213 Each of these radicals may undergo a β-scission to produce ethylene monomer and another primary radical.214 However, this unzipping process is not kinetically favorable at the early stage of PE decomposition (ca. 400 °C), and as a result no or little ethylene is produced in experiments (<1%).215,216 In contrast, radical (1) prefers to undergo a backbiting (often 1,5-backbiting through a six membered transition state) reaction, thereby generating a secondary internal radical (2) (Scheme 1).211,216 Subsequently, a β-scission at (I) gives rise to an alpha–olefin (3), whilst if the same process occurs at (II), a terminally unsaturated polymer residue (5) is produced, as well as a radical that transforms into an alkane (6) after abstracting a hydrogen atom. The formation of alkadienes (8) takes place either via a hydrogen abstraction reaction from (5) (Scheme 1) or via chain scission followed by a backbiting reaction. These processes give rise to (7) that, after a β-scission, is converted into an α,ω-alkadiene (8) (Scheme 1) and a new primary radical (1), which likely undergoes the processes of Scheme 1 anew.
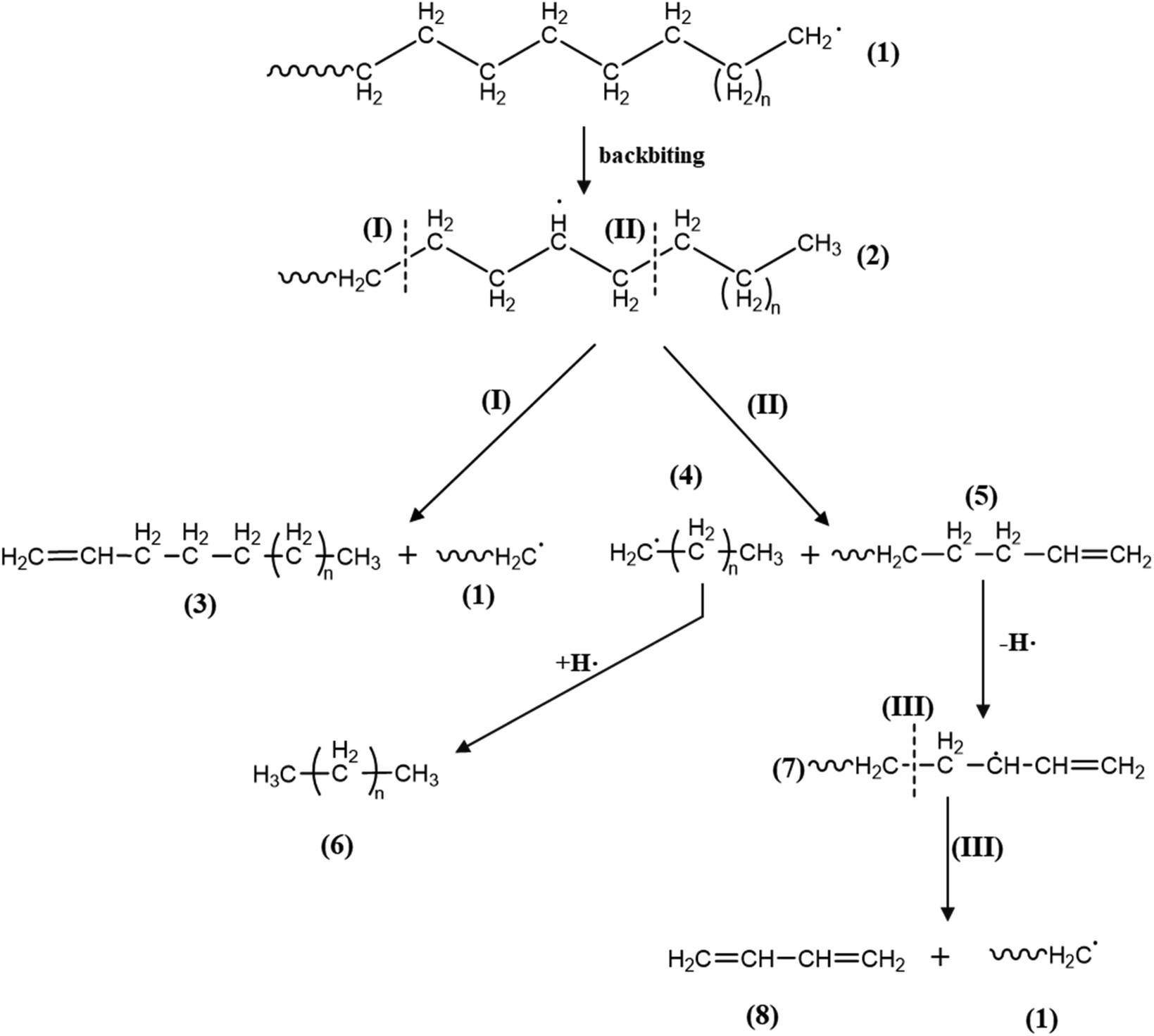 | ||
| Scheme 1 Fundamental reactions for the thermal decomposition of PE leading to alkanes (6), alkenes (5) and alkadienes (8). Arabic numerals indicate chemical species that form during the pyrolysis of PE, while Roman numerals indicate the occurrence and position of the scission of chemical bond. | ||
Similar to PE, the thermal decomposition of PP (9), Scheme 2, proceeds via random scissions along the backbone of the polymer with an activation energy of about 250.0 kJ mol−1.212 Each random scission produces a primary (10) and a secondary (11) radical (Scheme 2), which, in turn, are converted to alkanes,214 alkenes,217 and alkadienes.218 The major decomposition products are alkenes and alkadienes, and their formation takes place in a manner similar to what we described in Scheme 1.
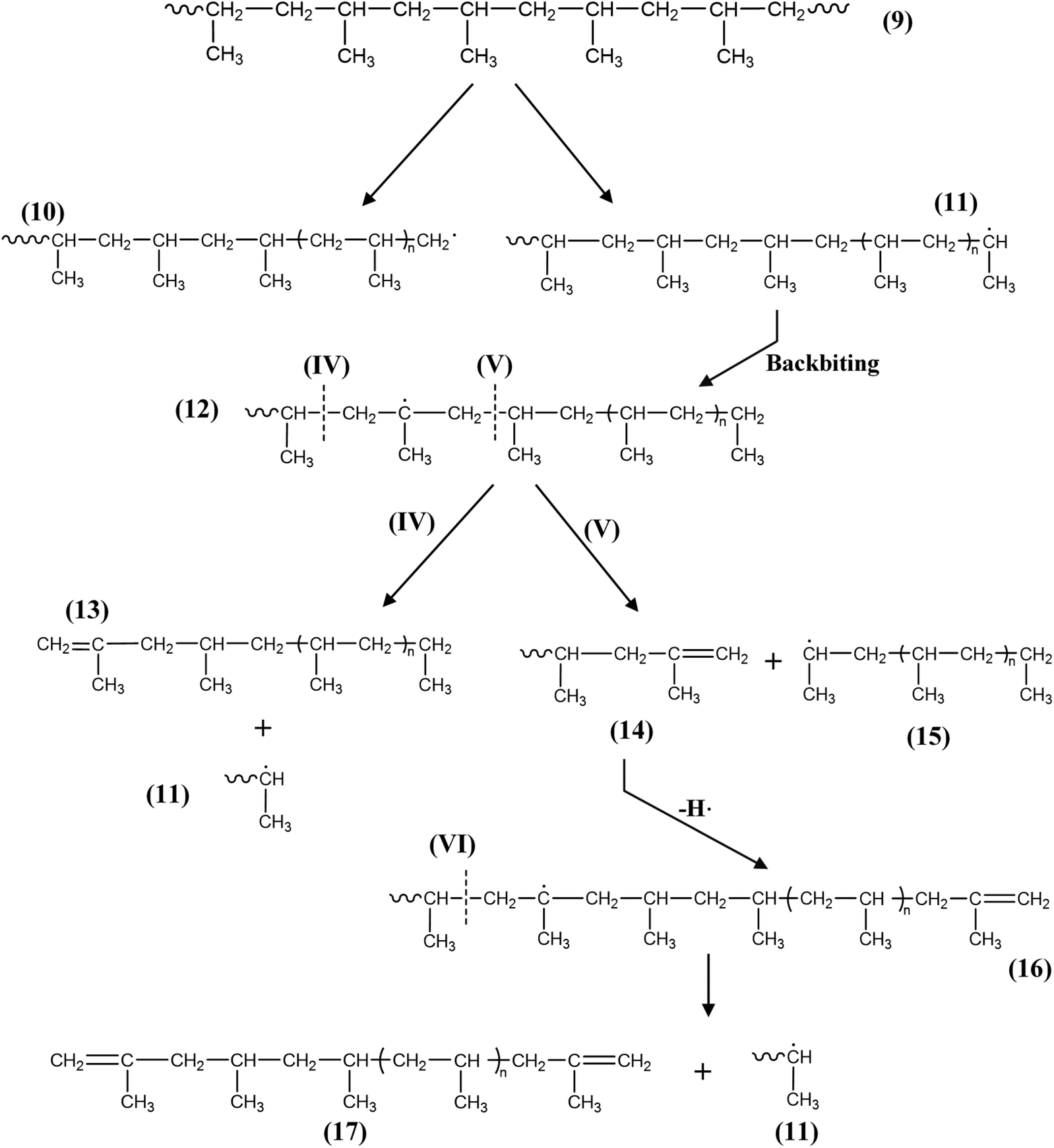 | ||
| Scheme 2 Fundamental reactions for the thermal decomposition of PP leading to alkenes and alkadienes. Arabic numerals indicate chemical species that form during the pyrolysis of PE, while Roman numerals indicate the occurrence and position of the scission of a chemical bond. | ||
The highest products from PP pyrolysis (i.e., 2-methyl-1-pentene, 2,4-dimethyl-1-heptene) are produced via the decomposition of the secondary radical (11), while only minor quantities of products are derived from the primary radical (10).211 On this basis, Scheme 2 focuses on the formation of alkenes and alkadienes via (11), which after a backbiting reaction and a β-scission at (IV) forms an alkene product (13) and another secondary radical (11). The new secondary radical is decomposed following the same sequence of steps. Alternatively, the β-scission may occur at (V), which results in a terminally unsaturated polymer (14). The latter species may undergo an atomic hydrogen abstraction reaction followed by a β-scission at (VI), thereby forming another secondary radical (11) and a series of alkadiene products (17).
5.1.1.2 Polyvinyl chloride. The thermal decomposition of PVC (18), Scheme 3, follows a completely different mechanism compared to polyolefins. According to thermogravimetric analysis studies, PVC (18) starts decomposing at much lower temperatures (<250 °C) than other common polymers, and its decomposition involves two distinct stages.219
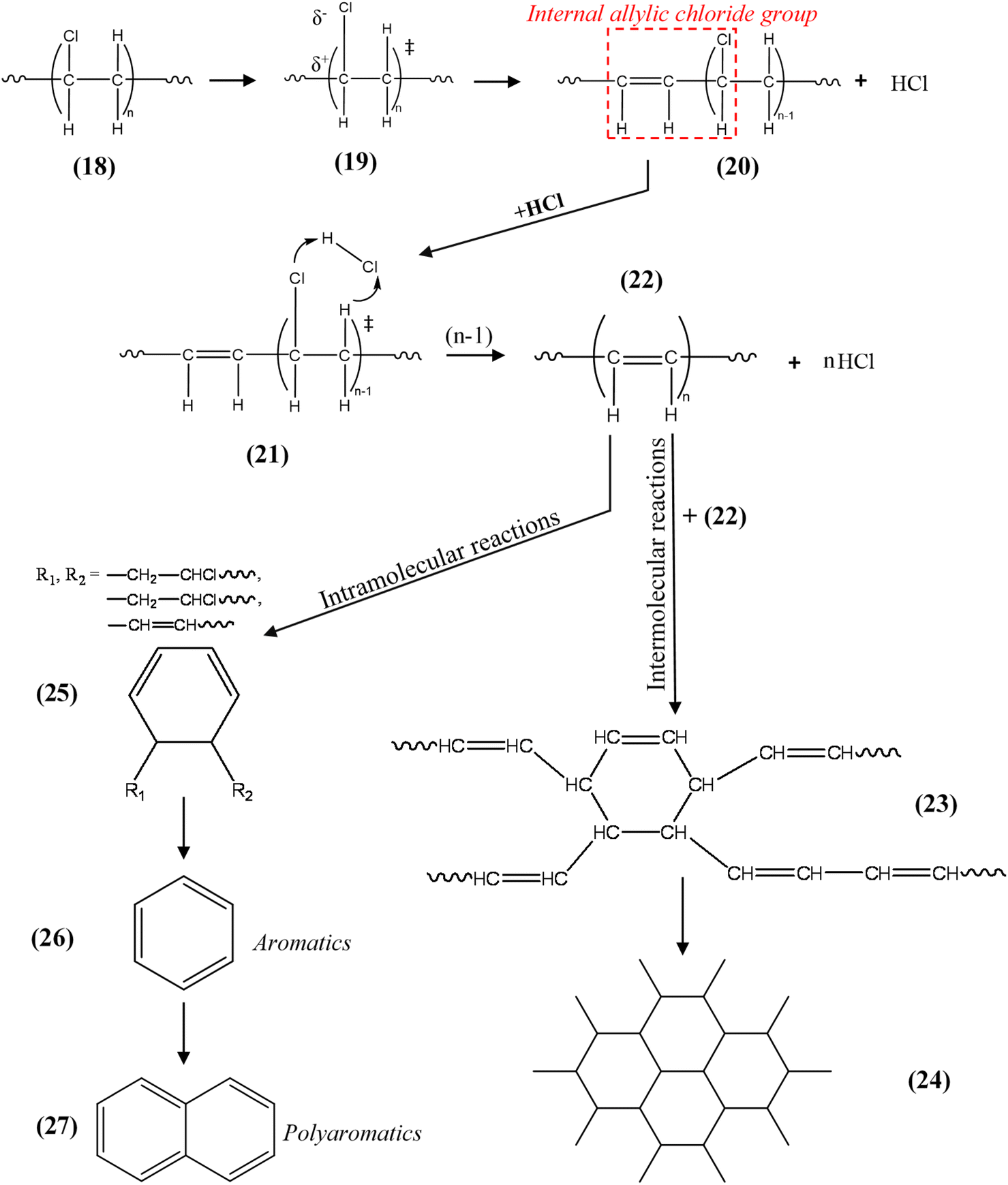 | ||
| Scheme 3 Fundamental reactions for the thermal decomposition of PVC leading to different products. The symbol ‡ indicates a transition state. Arabic numerals indicate chemical species and intermediates that form during the thermal degradation of PVC. | ||
The dominant reaction in the first stage (250 °C–350 °C) is the dehydrochlorination of the backbone that leads to a 60% weight loss.220 The activation energy for this process has been determined in several studies, and values in the range of 112.0 kJ mol−1–150.0 kJ mol−1 have been reported.221,222 It has been conjectured that the initiation occurs in thermally labile defects like butyl branches associated with tertiary chloride.223,224 Once the reaction is initiated, its propagation proceeds via a quasi-ionic mechanism whereby HCl is removed in a concerted manner via a four-center transition state (19).225 Subsequently, the propagation reaction is relatively fast owing to the continuous formation of internal allylic chloride structures [see (20) in Scheme 3], which are more thermally unstable and susceptible to C–Cl heterolysis via HCl catalysis [i.e., autocatalysis – transition state (21) in Scheme 3] than the pristine PVC polymer segments.226,227 Remarkably, first-principles calculations by Yanborisov and Borisevich showed that the HCl-catalyzed dehydrochlorination of small chlorinated molecules (e.g., 2-chlorobutane) can be 60.0 kJ mol−1 more facile than the uncatalyzed process.228
The main dehydrochlorination products are HCl gas and π-conjugated polyene sequences (22). The latter products are highly unstable under pyrolytic conditions and therefore undergo intramolecular and intermolecular reactions as soon as they form.229 The intermolecular reactions require the interaction between two polyene chains (22),230 and give rise to crosslinked polyene chains (23),229 which are the precursor of char (24).231 Char is extensively formed in the second stage of the thermal pyrolysis.231 Alternatively, a polyene chain may undergo an intramolecular electrocyclic reaction, thereby giving rise to π-conjugated 1,3-cyclohexadiene rings (25). The latter are converted to benzene (26), and subsequently polyaromatics (27), after two consecutive homolytic C–C cleavages that occur at temperatures beyond 350 °C having activation energies larger than 220.0 kJ mol−1.232 C–C bond breaking also gives rise to small amounts of non-condensable gases (e.g., CH4 and H2).230 Importantly, we note that the formation of aromatics can be promoted by applying a high-temperature pyrolysis step in the presence of a solid catalyst (e.g., ZSM-5 zeolite).233
5.1.1.3 Polystyrene. The thermal degradation of PS (28) follows a radical chain mechanism (Scheme 4), with styrene (35) being the dominant product.234 By means of thermolysis experiments, Wall et al.235 demonstrated that the thermal stability of PS (28) depends on the polymerization method (i.e., anionic or radical polymerization) used in its synthesis. Preparation through a radical polymerization gives rise to polymer chains with randomly distributed “weak links”, which perhaps are peroxidic in nature and are the sites where the thermal decomposition of PS starts.236 Yet, anionic polymerization yields PS of high thermal stability;234 the general thermal decomposition mechanism for this case is described in Scheme 4 and the following text.
 | ||
| Scheme 4 Fundamental reactions for the thermal decomposition of PS leading to different products. Arabic numerals indicate chemical species, while Roman numerals indicate the occurrence and position of the scission of a chemical bond. | ||
The experimentally measured activation energy for the thermal degradation of PS is 200.0 kJ mol−1 and remains constant regardless of the extent of the reaction.211 This is indicative of a single rate-determine step during the reaction: the β-scission that forms styrene (35) (see Scheme 4).211 According to Guyot and coworkers,234,237 a likely initiation pathway for “anionically” prepared PS is via an end chain scission reaction that forms a benzyl radical (30) and a primary radical (29). The two radicals go through a disproportionation reaction and give toluene (32) and a chain with a thermally unstable unsaturated end (31). In turn, (31) decomposes into α-methylstyrene (33) and a secondary radical (34) that leads the degradation process thereafter. DFT work by Huang et al. revealed that the latter reaction is exothermic by 80.8 kJ mol−1 and its activation energy is 288.2 kJ mol−1.238
The propagation occurs by a β-scission process at (IX), thereby generating styrene (35) and another secondary radical (34). According to DFT calculations on a PS trimer, this reaction has an activation energy of ca. 117.0 kJ mol−1, but it is strongly endothermic (+105.4 kJ mol−1) and therefore not favorable at temperatures below 250 °C.238 Interestingly, the computed activation energy for the unbuttoning reaction (i.e., 117.0 kJ mol−1) is in stark contrast to the experimental value (200.0 kJ mol−1) and this can, to some degree, be justified by the considerably smaller size of the simulated PS trimer as compared to actual PS chains. (34) can either undergo further unbuttoning to give extra monomer (35) or a backbiting reaction followed by another β-scission reaction at (X) and give a dimer (37) along with another secondary radical (34). Based on DFT, such backbiting and β-scission reactions require kinetic barriers of 174.0 kJ mol−1 and 134.5 kJ mol−1, respectively, and the whole process that forms (34) and (37) is endothermic by 94 kJ mol−1.238 We note that the formation of trimer or tetramer species takes place in an analogous manner to that for dimer production (37) (Scheme 4) with the only difference that different tertiary hydrogen atoms are involved in the backbiting reaction.
5.1.1.4 Poly(ethylene terephthalate). PET (38) is a thermally stable polymer thanks to the combination of short aliphatic chains and aromatic rings. Its thermal decomposition follows a molecular mechanism and the initiation reaction involves a scission at the ester linkage (Scheme 5).239–241 This reaction takes place through a six–membered transition state (39), and recent DFT studies predict activation and reaction energies of 184.0 and +48.0 kJ mol−1, respectively.242
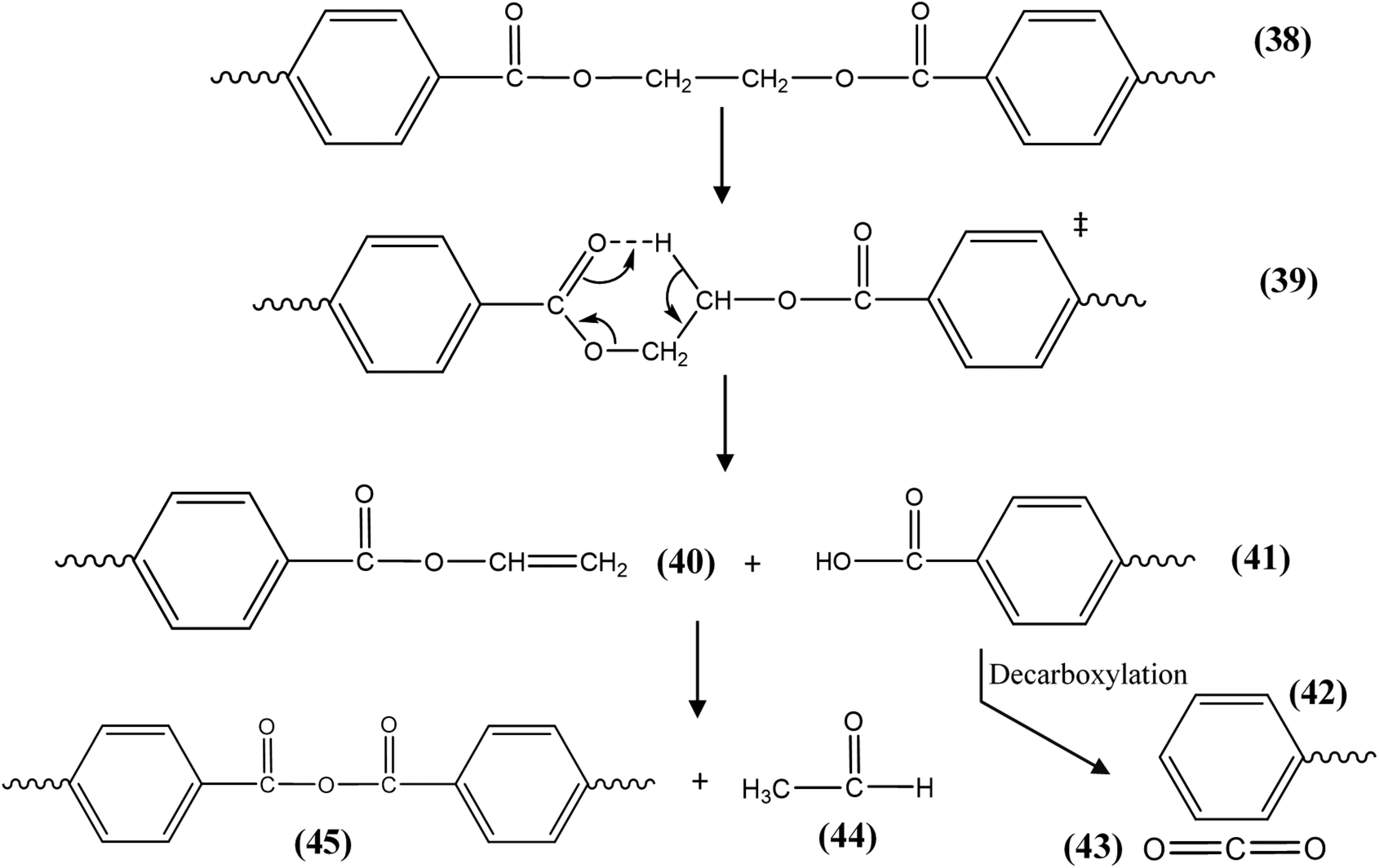 | ||
| Scheme 5 Fundamental reactions for the thermal decomposition of PET. The symbol ‡ indicates a transition state. Arabic numerals indicate chemical species and intermediates formed during the pyrolysis of PET. | ||
The initiation reaction leads to the formation of an oligomer with an olefin end group (40) and another oligomer with a carboxylic end group (41). These two species decompose further to produce a wide spectrum of products with acetaldehyde (44) and CO2 (43) being the most abundant at temperatures around 300 °C.240 The former can be produced directly from (40) and (41) or via the degradation of EG end groups as proposed by Holland and Hay,243 while CO2 is released by decarboxylation of (41). More than 45 wt% of the PET is degraded into CO2 with trace CO.244
• C1–5 = gas
• C6–20 = naphtha or oil, liquid at ambient conditions
• C21+ = wax, solid or highly viscous liquid at ambient conditions
Al-Salem et al.246 published an in-depth review addressing plastic pyrolysis variables and their influences on reaction and product distribution and asserted that the main decomposition behavior of the plastic is controlled by temperature. In PE pyrolysis, early studies showed that only liquid and gas – no wax – are produced above 600 °C.247 Increasing temperature leads to increased aromatic production, as gas is converted to aromatics through a Diels–Alder reaction and unimolecular cyclization reactions,248,249 although a study by Kulas et al.250 showed that the reaction pathway from gas to aromatics has low temperature dependence.
The composition of naphtha is vastly different than the composition of plastic oil (Fig. 17). The main products of naphtha are paraffin with around 10% aromatics, while the main products of plastic pyrolysis oil are olefins and cycloalkanes/alkadienes with a much wider range of carbon numbers.
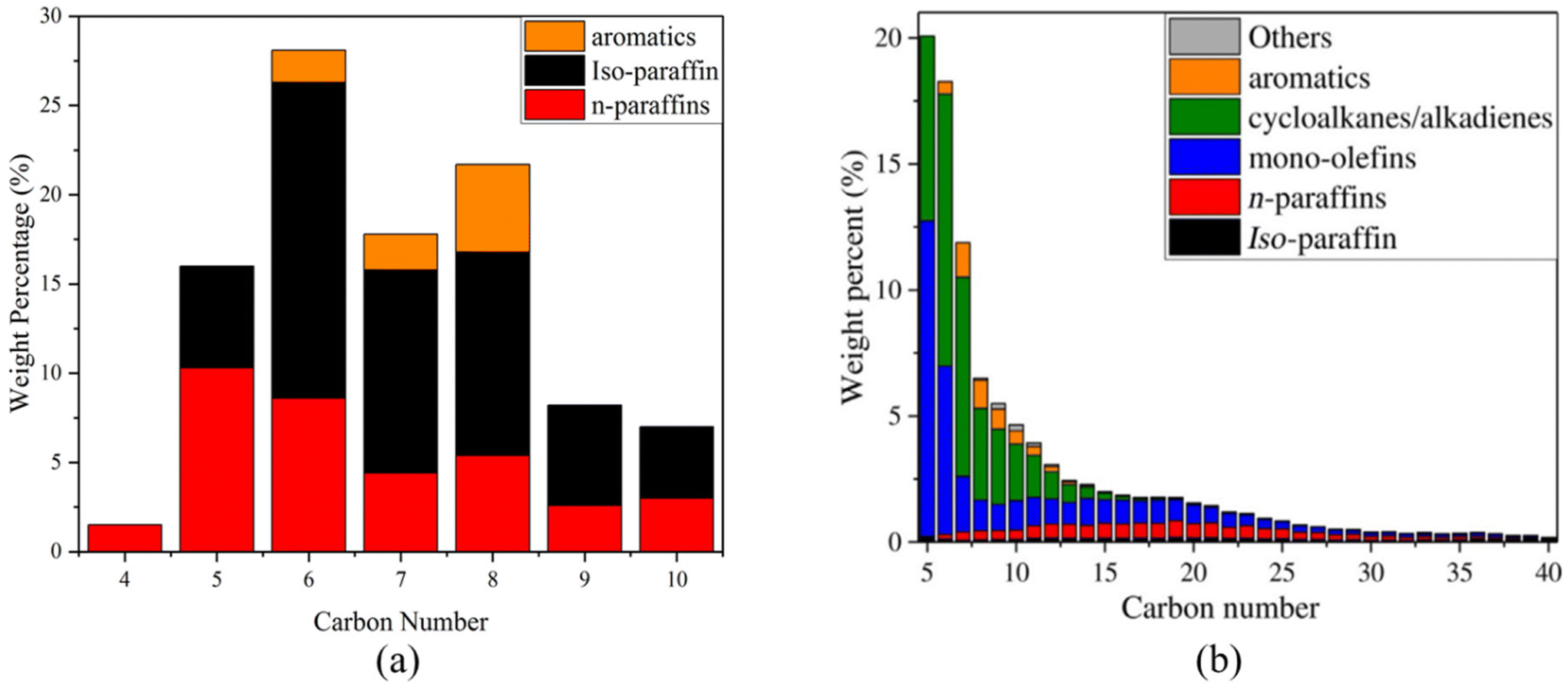 | ||
| Fig. 17 (a) A typical naphtha composition (graph made based on the data obtained from ref. 251, (b) a typical plastic pyrolysis oil composition obtained from PE pyrolysis. Reproduced from ref. 252 with permission from Wiley-VCH, copyright 2020. | ||
Increasing vapor residence time increases the yields of oil and non-condensable gases.248,253 As primary products are exposed to heat for longer periods of time, secondary reactions and further cracking to smaller molecules are encouraged.254 Thus, the product distribution shifts from high molecular weight products to lower molecular weight hydrocarbons (MWHCs).255–257 For polyolefin feedstocks, longer reaction times decreased product olefin concentrations and increased polyaromatic hydrocarbon concentrations.258
Polyolefins have received the most attention in the field of plastics pyrolysis. PE and PP decompose into a distribution of hydrocarbons, with decreasing product molecular weights as reaction temperature and time increase. Waxes and heavy oil olefins dominate product distributions at low temperatures, while gases and light oils are the main products at high temperatures.250 Polyolefins such as HDPE, LDPE, and PP decompose into a distribution of hydrocarbon products that have high concentrations of olefins and dienes (>70%).259,260 Mixed plastic feedstock with high PE content will lead to higher paraffin concentrations.261 Pure PP yields higher amounts of olefins, with lesser amounts of paraffins, naphthenes, and aromatics.262,263 Due to the tertiary carbon present on the backbone of PP, multiple types of secondary radicals are formed, shifting liquid product composition towards branched olefins.261,264
PS decomposes primarily into the monomer styrene and its oligomers265,266 with the polymer undergoing chain scission followed by random scission.266 PS pyrolysis produces liquid products at yields greater than 90 wt% in both batch262,267,268 and continuous247,269,270 reactors. Liu et al.269 pyrolyzed PS in a fluidized bed reactor at 600 °C and achieved 98.7 wt% oil, of which 78.7 wt% was styrene. Secondary reactions and increased reaction temperature decrease the concentration of styrene in the liquid product and increases the formation of coke and gas.269 PS accelerates the pyrolysis of polyolefins due to higher concentrations of free radicals.271 Green Mantra Technologies is commercializing PS pyrolysis to recover styrene.272 A thorough review on the pyrolysis of PS can be found elsewhere.266
The pyrolysis of PET yields oxygenated compounds from the presence of oxygen in the backbone of the polymer chain. Fast pyrolysis of PET produces mostly mixtures of aromatics and the organic acids benzoic and phthalic acid, char, and gas.273 Formation of TPA is particularly problematic due to its high boiling/sublimation temperatures, which clogs reactor and condenser internals.273,274 The presence of phthalic acids in product oils increases viscosity and acidity and lowers calorific value, resulting in low-quality oils.273,275,276 Pyrolysis temperature has little effect on calorific values of PET products.260 PET in mixed plastics increases the CO and CO2 concentrations in product gases from pyrolysis reactions.196,277 While some investigators have concluded that PET is not suitable for plastic pyrolysis,255,278 others advocate for it based on the valorization of product char to activated carbon and product oil to fuel.273 US-based pyrolysis company Agilyx has undertaken pyrolysis of PET with some success,279 but PET is rarely used as a feedstock in commercial pyrolysis processes. Other possible chemical recycling processes of PET are detailed elsewhere and in section 9.1.280–283
Many commercial pyrolysis processes have low tolerance to PVC, which produces significant concentrations of organic chlorine, HCl and a solid residue.275,277 The HCl causes corrosion issues with downstream processing of the products. Thermal decomposition of PVC is initiated by a dechlorination reaction, resulting in the formation of hydrogen chloride and benzene.284,285 The polymer chain then undergoes cyclization, yielding aromatic and alkylaromatic compounds that are often chlorinated.220 Pyrolysis oil containing chlorinated compounds has limited use.286 Most previously reported pyrolysis processes had a specification that the PVC content of the plastic feeds be less than 2 wt%.287
Plastics can also be co-processed with waste tires,288,289 wastepaper,290 and agricultural residues,291,292 which has been shown to enhance products for fuels and value-added chemicals.293 The majority of waste plastics will contain high levels of contaminants. Additives, heavy metals, and pigments are often added to plastic to enhance its physical appearance or performance. Specific problematic elements include nitrogen, oxygen, sulfur, chlorine, iron, sodium, and calcium.294 In low-temperature pyrolysis, sulfur presents in the pyrolytic oil, which reduces its ability to be used as ultra-low sulfur diesel.294 Produced liquid may also contain amines or alcohols if the feedstock contains N, O, or S.295 Siloxanes can be present in the pyrolysis oil as well.296 Dechlorination is possible through the use of sorbents,286,297–301 absorption and neutralization,302 subcritical water treatment,303 catalysts,304 or stepwise pyrolysis,286 but the latter was shown to modify the composition of liquid products.286 Untreated plastic waste pyrolysis oils with high levels of contaminants are not feasible feedstocks for steam cracking unless sufficiently diluted.305 Contamination may lead to catalyst deactivation if catalytic upgrading of products is desired. For example, a municipal plastic waste pyrolysis plant in Idaho, U.S.A. produced an oil only suitable for low-value applications due to severe contamination.306
Thermal oxo-degradation (TOD) involves adding small amounts of oxygen to the pyrolysis process to increase chain cleavage.307 Plastics subjected to TOD are decomposed to oxygenated products and hydrocarbons, with exothermic partial oxidation reactions releasing energy to provide heat back to the process. This process mirrors that of autothermal pyrolysis developed for the process intensification of biomass,308,309 with the possibility to achieve autothermal operation for plastics.310
 | ||
| Fig. 18 Schematics of various plastic pyrolysis reactors including fixed bed, fluidized bed, conical spouted bed, stirred tank, auger, and rotary kiln reactors. | ||
Conical spouted bed reactors are a type of fluidized bed with a cylindrical geometry and a conical base. Cyclic and vigorous movements by sand particles in the bed prevent agglomeration upon melting and subsequent bed defluidization.259,326 Conical spouted beds have been employed in laboratory-scale pyrolysis studies of PS327 and polyolefins,326,328,329 which report high yields of wax. Orozco et al.259 identified stable operating conditions for conical spouted bed reactors for individual plastic feedstocks.
Fixed bed reactors are inexpensive, simple to operate, and have low maintenance requirements. However, these types of reactors would be difficult to operate for solid feedstocks on the industrial scale due to large amounts of solid coke/char formation. Vertical fixed beds use gravity to force products through the reactor. Tubular reactors are very similar to fixed beds, but are usually horizontal with co-current flow of feedstock and carrier gas.330,331 Due to the relatively long times required to devolatilize plastic, the entrance to the tubular reactor can become quickly blocked with melted plastic, limiting feed rates. While commonly used in laboratory settings,332–335 fixed beds are not suitable for industrial applications.275,313 BASF reported the use of a tubular pyrolysis reactor to crack plastic wastes at scale to produce oil and gas, but the feed rates achieved were not documented.312
Auger reactors consist of a rotating screw inside a tubular reactor. The helical screw is used to convey feedstock while also discharging residual solid products. The auger reactor is a simple system that can be easily scaled and is feedstock flexible. Providing enthalpy for pyrolysis through the reactor wall is increasingly difficult as the reactor is made larger. This limitation can be substantially overcome by conveying heated metal shot or granular material into the reactor.336 As outlined by Campuzano et al.,336 potential disadvantages of auger reactors include the risk of blockage, mechanical wear, and higher vapor residence times, which can lead to sometimes undesirable secondary reactions. Auger reactors have been used to pyrolyze plastics to high yields of liquids for use as fuel337 or wax,338 or as the first stage in a multi-staged pyrolysis system to activate molecules for further cracking.321 Brightmark, a US company, is using an auger reactor to process plastic pyrolysis, and the reactor has five heating zones in series to improve the heat transfer. The number of zones of a reactor can be varied depending on different feedstocks.339
The rotary kiln is one of the most common reactors used in industrial pyrolysis processes. The kiln is a slightly inclined cylindrical vessel rotated slowly on its axis to allow feedstock and residual solids to move through the kiln. Because they can handle various feedstock sizes and shapes, rotary kilns have been used to process scrap tires340,341 and municipal wastes.342,343 Heat is supplied to the kiln by internally or externally heated walls,311 although heating rates are so slow as to qualify as slow pyrolysis.314 Residence times of solids and vapors in rotary kilns are highly controllable, depending upon rotational speed and carrier gas flow rates.344 Rotary kilns are considered robust and affordable however they are limited to certain scales due to heat transfer.311
Stirred tank reactors, also known as melting vessels, have been used for pyrolysis of plastics in batch,267,345,346 semi-batch,258 and continuous operation. Commercial examples include the Smuda process,347 the Hitachi Zosen process,275 and the Plastic Energy process.348 Stirred tank reactors are characterized by a simply-designed vessel heated through the reactor wall and a stirrer to promote uniform heat distribution and homogenous reactants. Heat gradients, large infrastructure requirements, and frequent maintenance limit the use of stirred tank reactors.312 Stirred tank reactors are primarily used for batch reactions, which is not economical on a large continuous scale process.
Pyrolysis reactors can be heated by a heat carrier (Fig. 19a) or with an external heat exchanger (Fig. 19b). The heat balance when a heat carrier is used is shown in eqn (1), where  is the mass flow rate of catalyst (or heat carrier), Cp is the heat capacity of the gas, T is the temperature,
is the mass flow rate of catalyst (or heat carrier), Cp is the heat capacity of the gas, T is the temperature, 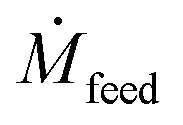 is the mass flow rate of reactant, and ΔHrxn is the net energy of the heat of the pyrolysis reaction. When using a heat carrier, the reactor can be scaled to larger industrial sizes without limitations to the heat transfer area by use of an external fluid. In contrast if an external fluid is used for heating the energy balance is dependent on the heat transfer area as shown in eqn (2) and (3).
is the mass flow rate of reactant, and ΔHrxn is the net energy of the heat of the pyrolysis reaction. When using a heat carrier, the reactor can be scaled to larger industrial sizes without limitations to the heat transfer area by use of an external fluid. In contrast if an external fluid is used for heating the energy balance is dependent on the heat transfer area as shown in eqn (2) and (3).
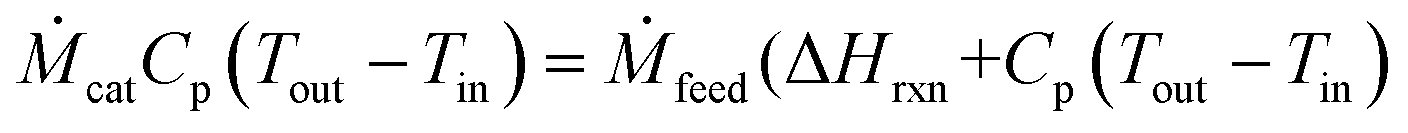 | (1) |
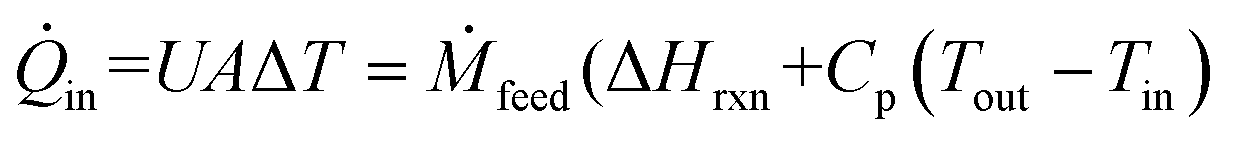 | (2) |
| A = 2πrL | (3) |
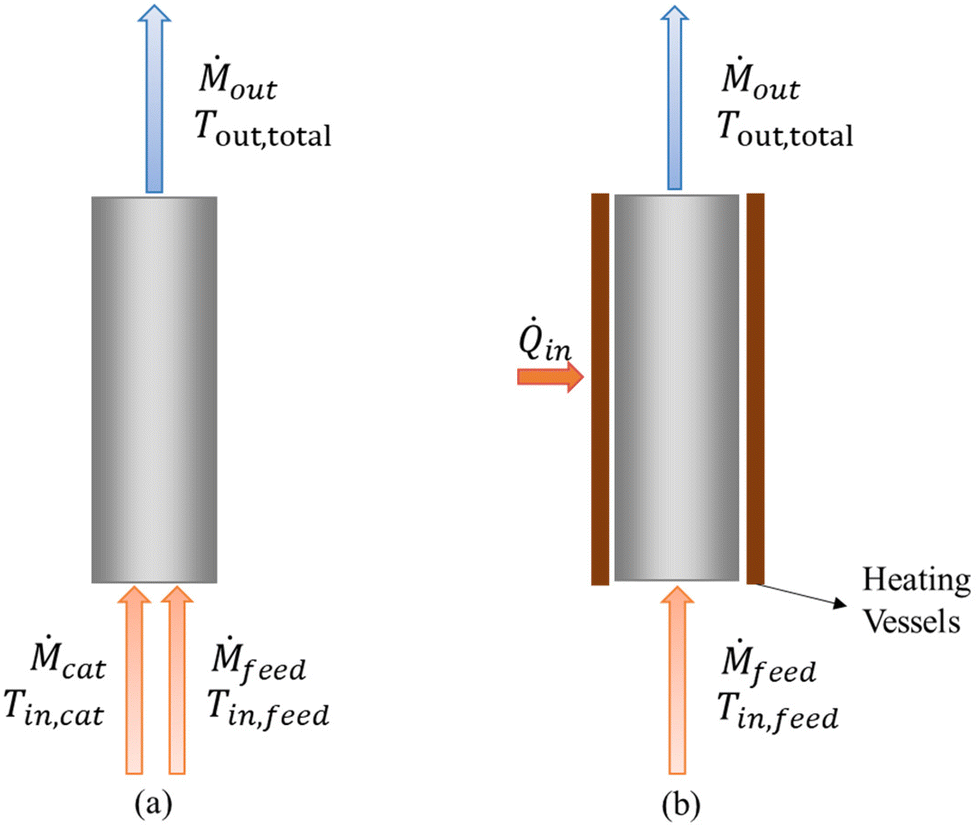 | ||
| Fig. 19 (a). Scheme of reactor with heat carrier (b) Scheme of reactor with external heating vessels. | ||
PE pyrolysis requires around 260 kJ kg−1 of heat.349 In a larger reactor, the heat transfer efficiency decreases due to the larger reactor diameter. Brown et al.350 reported that the maximum diameter of a biomass fast pyrolizer is 9.4 mm, and an analogous calculation for a PE pyrolizer obtained a maximum diameter of 6.4 cm. The reaction rate of PE used for the calculation is obtained from Zhao et al.252 In addition, the overall heat transfer will be affected by the bulky nature of the fed plastic, and the real diameter of the plastic pyrolyzer may be smaller than 6.4 cm. Autothermal pyrolysis can help to reduce the amount of heat required for pyrolysis by adding a certain amount of oxygen to partially oxidize the hydrocarbon feed, which makes the rate of energy release from the oxidation reaction equal the rate of energy consumed by the plastic pyrolysis.350 However, one main problem of the autothermal process is that it causes hot or cold spots due to the heat absorbed or released from the autothermal pyrolysis. The key to eliminating the hot and cold spots is again improving heat transfer within the reactor.
Various studies have shown that the profitability of plastic pyrolysis refineries depends on several factors including feedstock cost, yield rate, product type, and facility scale. Feedstock composition, pyrolysis yield, and product selectivity are critical drivers for process efficiency. Feedstock cost, capital investment, and market prices play a significant role in the profitability of a plastic pyrolysis facility. Table 12 summarizes the key assumptions and findings from several studies on plastic pyrolysis.278,356–358 Fivga & Dimitriou investigated the pyrolysis of plastic waste to produce wax/oil products that can be used as a heavy fuel oil substitute or as raw materials by the petrochemical industry. The study evaluated a base case scenario of a 0.7 kton per year pyrolysis pilot plant built by a recycling company in the United Kingdom. The base case analysis resulted in a capital cost of $1.34 million. The product cost from the base case scenario was $0.98 per kg of fuel produced, which was more than the market price of the heavy fuel oil product of ca.$0.8 per kg. Increasing the pyrolysis scale from 70.1 kton per year to 701 kton per year decreased the product price from 29.4 cents to 3.39 cents per kg.356 The pyrolysis plants can be designed as either closed-loop or open-loop.357 Larrain et al. also emphasized that to make an open-loop and closed-loop pyrolysis of mixed olefins attractive to investors, closed-loop and open-loop plastic pyrolysis plants should be scaled up to at least 70 kton per year and 115 kton per year, respectively.357
| Technology | Feedstock | Major products | Region | Capacity (kton per year) | Capital Cost ($ million) | NPV ($) | Ref. |
|---|---|---|---|---|---|---|---|
| Pyrolysis | PS, PP, PE | Heavy oils petrochemical feedstock | United Kingdom | 0.7–701 | 1.36–77.2 | −0.44 per kg–0.71 per kg | 356 |
| Pyrolysis + upgrading | PS, PP, PE, PET | Hydrocarbon fuel | Korea | 260 | 118 | 0.062 per gal | 278 |
| Pyrolysis | PE, PP, PET | Diesel power char | Australia | 14.6 | 3.76 | 2.03 million | 360 |
| Pyrolysis & heat integration | HDPE | Ethylene propylene | United States | 193 | 118.5 –120.5 | 367.8 million–383million | 359 |
| Fast pyrolysis (open-loop & closed-loop) | Mixed polyolefins mainly LDPE and residual PP | Naphtha | Belgium | 120 | Not disclosed | open loop: 32.5 per ton closed loop: 2.72 per ton | 357 |
| Pyrolysis | Plastic waste (PP, PE, PS) | Light oil heavy oil | Malaysia | 120 | 58.6 | 20.9 million | 358 |
Larrain et al. investigated the economic feasibility of open-loop and closed-loop pyrolysis of mixed polyolefins (mainly LDPE and residual PP) in a 120 kton per year facility in Belgium. In the open-loop system, the process only produces naphtha, while the closed-loop system produces naphtha and wax. The study recorded a base case net present value (NPV) per tonne of plastic waste processed of $32.5 per ton and $2.72 per ton for the open-loop and closed-loop systems, respectively. The market value of the products resulted in a significant difference of $24.75 per ton, thereby emphasizing that the type of products plays a vital role in the facility's profitability. The study went further to consider the uncertainties in oil prices that significantly affect the prices of wax and naphtha in the market and concluded that the open-loop system has about 98% probability of achieving positive returns compared to 58% recorded for the closed-loop system.357
Gracida-Alvarez et al. conducted the economic analysis of a multi-product 193 kton per year pyrolysis-based refinery for the conversion of HDPE to value-added chemicals (ethylene, propylene, and aromatic mixtures) and hydrocarbon mixtures via pyrolysis. In this study, the authors estimated the capital cost, operating cost, and NPVs for a base case where HDPE is converted to the products and a heat integrated case. The authors reported a capital cost of $118.5 million for the base case and $120.5 million for the heat integrated case. The NPV can range from $320 million to $450 milliondepending on the selling price of low MWHCs and the internal rate of return (IRR).359 The study showed that plastic pyrolysis targeting multiple products could be very profitable.
| Name | Plant location | Scale (kton per year) | Status | Reactor type | Feedstocks | Product | Collaborators |
|---|---|---|---|---|---|---|---|
| BP361 | Grange-mouth (Polit plant) | 25 | 1998–2000 (Design only) | Fluidized bed reactor | PP, PE, PET, PS, PVC | Hydrocarbon liquid | VALPAK, and Shanks & McEwan |
| BASF361 | Ludwig-shafen (Polit plant) | 15 | 1994–1996 | Not reported | Not reported | Not reported | Duales System Deutschland |
| Agilyx376 | Tigard (Polit plant) | 3 | Operational since 2018 | Stirred tank reactor | PS | Styrene monomer | Toyo, Exxon Mobil, Braskem, AmSty, Lucite, and NextChem |
| Quantafuel339,367,368 | Skive (Polit plant) | 20 | Operational since 2017 | Fluidized bed rector | HDPE, LDPE, PP, PS, and PET | Liquid oil, non-condensable gas, and carbon rich ash. The liquid oil will be used to produce virgin-quality plastic | BASF, VITOL, and VITTI |
| Amsterdam | 100 | Under construction and plan to operate in 2023–2024 | |||||
| Sunderland | 100 | Under construction and plan to operate in 2024 | |||||
| Pryme353,380 | Rotterdam | 40–60 | Under construction and plan to operate in 2022 | Fluidized bed reactor | PS, PE, and PP | Kerosine, naphtha, and wax followed with steam cracking to obtain petroleum products | Shell |
| Brightmark339,373,381 | Asheley | 100 | Partially operational since 2021 | Auger rector | PET, HDPE, PVC, LDPE, PP, and PS | Ultra-low sulfur diesel, naphtha, and wax | BP, Chevron, clean fuel Partners, and Northeast Indiana Solid Waste Management District |
| Plastic energy354,366 | Seville (Pilot plant) | 5 | Operational since 2017 | Stirred tank reactor | LDPE, HDPE, PP, PS | Diesel and naphtha followed with steam cracking to obtain raw materials to make new plastic | SABIC, ExxonMobil, and freepoint Eco-Systems |
| Le Havre | 25 | Design phase planned to start up in 2023 | |||||
| Almeria | Not reported | Operational since 2014 | |||||
| Freepoint Eco-Systems374 | Houston | 33 | Plan to operate in Mid-2024 | Fluidized bed reactor | LDPE, HDPE, PP, PS | Synthetic oil | Plastic energy, and TotalEnergies |
| Obetz | 90 | Plan to operate in 2023 | |||||
| Shell377 | Pulau Bukom | 50 | Under construction and plan to operate in 2022 | Not reported | Not reported | Not reported | Pryme |
| ExxonMobil374 | Baytown | 30 | Under construction and plan to operate in end-2022 | Fluidized bed reactor | Not reported | Not reported | Cyclyx, agilyx, and plastic energy |
In Europe, there are 3 operating pyrolysis plants owned by Quantafuels and Plastic Energy. Quantafuel's first commercial production facility is in Skive, Denmark. The plant processes 20 kton per year. The liquid product is sent to BASF, who uses the liquid product to produce virgin plastic and other chemicals.362 Plastic Energy currently has two plants in Almeria (started in 2014) and Seville (started in 2017) in Spain. The plant in Seville has the ability to treat 5 kton per year.363
Several companies have announced plans in the past few years to build plastic pyrolysis plants and convert the plastic oils into aromatics/olefins by steam cracking. In Germany, SABIC and BP announced in 2021 plans to build a plastic recycling plant in Gelsenkirche.355 BP will build plants to steam crack the plastic oil obtained from SABIC and forward the cracking materials to SABIC for making new plastics.355 One goal of BP is to achieve 30% of propylene and ethylene produced from recycled material by 2030.364 Shell plans to work with Pryme to build a plastic pyrolysis plant and produce circular chemicals in the Netherlands and Germany with the capacity of 60 kton per year in 2022.365 Plastic Energy announced a collaboration with ExxonMobil in early 2021. Starting in 2023, ExxonMobil will use the plastic oil from Plastic Energy and upgrade the plastic to a naphtha-quality petrochemical feedstock.354 The company has started to construct a recycling plant in France next to ExxonMobil's Notre Dame de Gravenchon petrochemical complex, which will have a capacity of 25 kton per year.366 QuantaFuel plans to build a 100 kton per year plant in Sunderland UK and to work with VITOL and VITTO to build another 100 kton per year plant in Amsterdam.367,368 Fuenix Ecogy raised €4 million ($4.53 million) to scale up its technology to recycle mixed plastic waste, and in a collaboration with Dow to build a plant to pyrolyze plastic in Weert, Netherlands in 2019.369,370 The pyrolysis plastic oil will be sent to Dow Terneuzen, Netherlands, for steam cracking and producing virgin quality plastics.371 The goal for both Dow and Fuenix is to generate 100 kton of the recycled plastics by 2025.372
In the US, Brightmark is planning to build one of the largest plastic pyrolysis plants in the world, with a scale of 100 kton per year, in Ashley, Indiana.373 Freepoint Eco-Systems plan to invest $60 million to construct a plastic pyrolysis plant in Obetz, Ohio, with a capacity of 90 kton per year. The company claims that the plant will be operating in 2023 and will be able to convert 300 tons of waste plastic into 1500 barrels of oil.374,375 TotalEnergies is also working with the Freepoint Eco-Systems to build a 33 kton per year plant in Houston, Texas in mid-2024. The products will be sent to TotalEnergies’ steam cracker to produce monomer and manufacture high-quality plastics.352,376 Agilix and ExxonMobil created a joint venture together called Cyclyx International, which will be focused on creating novel ways to collect and pre-process massive amounts of plastic trash into feedstocks for higher-value products.374
There are also some plastic pyrolysis projects underway in Asia. Shell decided to start producing plastic oil with a capacity of 50 kton per year at Pulau Bukom, Singapore in 2023.377 Plastic Energy collaborates with WWF-Indonesia and aims to help Indonesia to reduce 100 kton of plastic waste by 2025.378 Insights provided by McKinsey showed that pyrolysis could be the most profitable process in the coming years.379 Due to the large amounts of oil required to run the hydrocarbon economy, the use of blends of plastic pyrolysis products and petroleum-derived feedstocks is expected to expand.305
5.2 Catalytic thermal decomposition of plastics
For mesoporous aluminosilicates, the plastic decomposition product distribution was dependent on the acidity of the material. Low-acidic materials (Al-MCM-41, Al-SBA-15) have high resistance to coking and increased cracking activity compared to all-silica materials.392,400,402–404 The additional acidity induced by the presence of alumina sites facilitates plastic cracking and can yield higher selectivity towards lighter hydrocarbons. For example, the product distribution for a PP/PE mixture was shifted towards C6–C12 products for Al-MCM-41, while Al-SBA-15 yielded high-quality diesel oil (high selectivity towards C13–C20 fraction, Fig. 20). However, both Al-MCM-41 and Al-SBA-15 were found to degrade a mixed plastics feed (HDPE, LDPE, PP, PE, and PS) to a similar product distribution of near equal amounts of C1–C5, C6–C12, and C13–C40 hydrocarbons.402,405 The large pore size of mesoporous aluminosilicates allows for improved intercalation of polymer molecules to Brønsted acid sites but prevents the moderately cracked products from undergoing re-adsorption and secondary reactions that result in extensive cracking.406 However, it was found that the promising cracking ability of mesoporous materials was only effective with polyolefin and polyaromatic PSW – polymers with other heteroatoms form coke which causes catalyst deactivation.402
 | ||
| Fig. 20 Distribution of liquid and wax products for thermal pyrolysis and mesoporous aluminosilicates in the conversion of PE/PP (ratio 6:5). Pyrolysis experiments were conducted at 500 °C for 30 minutes using a batch reactor set up, where the catalyst-to-feedstock ratio was 0.03:1. Reproduced from ref. 401 with permission from Wiley-VCH, copyright 2020. | ||
While mesoporous aluminosilicates were found to be capable of producing favorable product distributions with limited amount of coke, zeolites have still been extensively explored for the catalytic pyrolysis of plastic waste.20,382 Zeolite Y was found to have high cracking ability due to its high accessibility to active sites, but the larger micropores and high acidity were found to facilitate condensation and secondary reactions leading to fast deactivation.407–415 Similarly, ZSM-5 has high cracking activity for polyolefins due to its strong acidity.413,416–425 The microporous network within the ZSM-5 structure is ideal for the aromatization of cracked intermediates, however this precludes coke formation and fast deactivation. β-Zeolite yields higher gas fractions at the expense of liquid products because it is highly active for secondary reactions.389,426–431 Additionally, β-zeolites were found to produce high quantities of residue and wax. Natural zeolites were found to have moderate cracking ability with higher selectivity towards the liquid fraction, but its efficiency was strongly correlated to the plastic/catalyst ratio.393,432–442 Given the uncontrolled structure of natural zeolites, the materials contain impurities and there is little consistency amongst the materials. As such, there is high variability in the effectiveness of natural zeolites. Lastly, doped zeolites have been explored to enhance the acidity of the material.436,443–452 Doped zeolites were found to yield high selectivity towards olefinic products and were active for the removal of heteroatoms such as Br. One great challenge of the catalytic pyrolysis of plastic waste with zeolites is catalyst deactivation due to secondary coking reactions and limited diffusion of polymeric molecules into the microporous network.
Clays are another form of aluminosilicate that are of interest due to their weaker acidity, wide availability, and ability to produce medium distillate products such as diesel fuel.409,453–456 Clays are composed of silica and alumina or magnesia (or both) and have a macroporous structure which induces high coke resistance and moderate polymer cracking to heavier products. Structured clays, such as pillared clays (PILC), can include heteromoieties like metal oxides (Al, Fe, Ti, and Zr) to improve the textural properties and increase catalyst acidity.453–456 As such, PILCs have been found to have improved catalytic performance for HDPE and PE, as well as improved regeneration abilities. Clays, however, have lower activity due to the macroporous nature of the material, resulting in reduced contact of the feedstock with the weakly acidic sites within the structure.
Spray dried catalysts (typically Geldart Particle size A) are used in fluidized bed reactors because they are easier to flow in the fluidized bed. Spray dried catalysts (Fig. 21) are a mixture of clays or binders, zeolites, and silica-alumina matrices.457–463 Spray dried catalysts can have a bimodal pore structure with micropores in the zeolite framework and mesopores in the silica-alumina matrix. It was recently determined by Weckhuysen and coworkers that, in the conversion of PP, the fluidized catalytic cracking (FCC) matrix was responsible for aromatization of cracked products and the zeolite domain was responsible for coking.457 Zeolite and silica alumina catalysts can undergo structural changes during the reaction due to water vapor (dealumination) and impurities from the feed building up on the catalyst surface. Thus, in a circulating fluidized bed reactor, such as in fluid catalytic cracking, it is important to study the “equilibrium catalyst”. It is the equilibrium catalyst which is the actual catalyst in the reactor. Studies of the effect of the equilibrium FCC catalyst on PP conversion found the same aromatic content as fresh FCC catalysts with reduced coking. The zeolite domains in an equilibrium catalyst were found to be inaccessible – resulting in reduced coking – and the FCC matrix was found to have enhanced cracking activity due to leached metal deposits. This work by Weckhuysen and co-workers suggests that the strong acidity and microporous structure of the zeolite domain within an FCC catalyst is not necessary for the aromatization of polyolefin feedstocks.457 Further, the presence of zeolite domains may even be detrimental to the effectiveness of FCC catalysts for the catalytic pyrolysis of plastic waste.
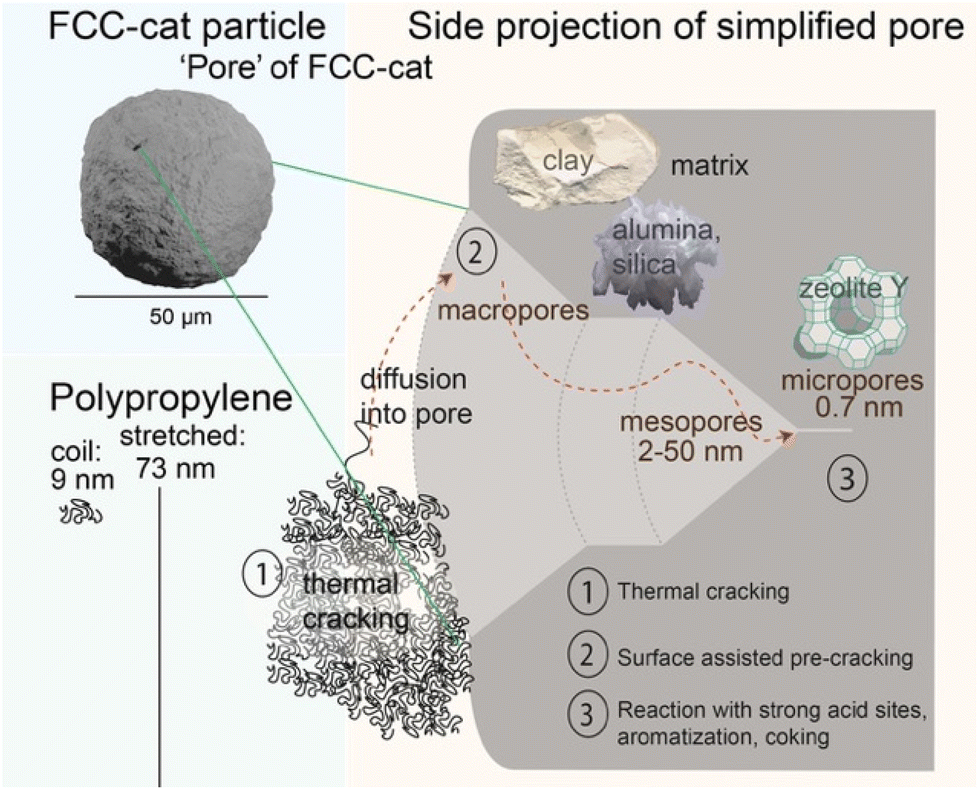 | ||
| Fig. 21 Structure of a spray dried FCC catalyst. Reproduced from ref. 457 with permission from Wiley-VCH, copyright 2021. | ||
Like FCC catalysts, multi-catalyst systems are zeolites mixed with MCM-41, NiO, muds, clays, salts, or metal silicates.396,464–468 The systems are typically formed by either the mechanical mixing of the two separate catalysts via mortar and pestle or by layering the catalysts within the bed. As with other catalytic pyrolysis systems, the presence of the catalysts reduces the required temperature for polymeric degradation as compared to thermal pyrolysis.464,467 The use of muds and clays with zeolites – such as a combination system of zeolite Y, metakaolin, aluminum hydroxide, and sodium silicate – has been found to improve the selectivity toward liquid products for a mixed plastic waste feed stream as compared to thermal pyrolysis.415,469,470 Additionally, red mud and calcium hydroxide was found to reduce chlorine content in the products. The combination of zeolites and MCM-41 has been found to induce synergistic effects on plastic degradation, where the MCM-41 provides enhanced surface area, and the zeolite provides strong acidity.464 For zeolite/MCM-41 systems, diffusional limitations were observed for the direct catalytic pyrolysis of HDPE. In two-stage pyrolytic-catalytic cracking with HZSM-5/MCM-41, a high yield of aromatic gasoline range products was formed.464 Similarly, HY-zeolite/NiO was explored in a two-stage reactor.466 The addition of NiO was found to increase gas selectivity at the expense of liquid products and reduce coke formation as compared to HY-zeolite alone. NiO was speculated to promote primary degradation of larger polymer fragments into radicals preventing coke deposition.466 In total, composite systems can induce favorable synergistic effects that can result in a narrowed carbon number distribution of products as compared to thermal pyrolysis, however the favorable effects are highly dependent on the catalyst mixture ratio. Further, diffusional limitations are increased in composite systems given the physical mixture of varying pore networks and may require two-stage pyrolysis-catalysis.
Like composite systems, hierarchical and core–shell catalysts combine the advantageous properties of various materials into one system. Unlike composite systems, hierarchical and core–shell catalysts are contained within one structure rather than in a heterogeneous mixture or across multiple reactors. Many of the studies to date of hierarchical or core–shell catalysts for plastic waste deconstruction combine the micropores of zeolites with secondary mesoporosity.445,446,449,450,471–481 The goal is to allow for enhanced accessibility where faster intra-crystalline diffusion and lower steric hindrances occur. Catalysts such as these have been found to have higher resistance to coking and deactivation compared to traditional microporous zeolites.479 Further, the secondary porosity of such materials renders improved gas and aromatic hydrocarbon production than their microporous counterpart. Hierarchical and core–shell materials are capable of supporting metals to enhance acidity and therefore activity.446,449,450,471–476,478,479 However, these materials require refined and detailed synthesis – making scale up difficult – and the dual porosity must be well defined otherwise deactivation via coking will occur.
Silica and aluminosilicates have dominated catalytic pyrolysis because of their porous networks and acidity, however other metal oxides such as ZrO2, MgO, and CaO have shown interesting results.482–494 In particular, sulfated zirconia – despite suffering from coke deactivation – has shown high cracking activity and isomerization, esterification, and hydrocracking ability.485,487,492,494 By sulfating the metal oxide, the acidity of the material is enhanced, resulting in improved reactivity. Further, studies have explored promoting sulfated ZrO2 with platinum and found that the material – in a two-step pyrolysis-catalytic hydrocracking process – can enhance the hydrocracking of LDPE into liquid products as compared to the non-doped material.483 Other metal oxides such as MgO and CaO have been shown to reduce activation barriers and enhance selectivity toward phenolic compounds for PC catalytic pyrolysis, as well as reduce production of light gases (H2, CO2, CO, and methane) for PC and LDPE degradation.482,484,488,489,491 It was determined that the basicity of CaO allowed for the reaction of CO2 in the pyrolysis vapors, resulting in the production of CaCO3, while MgO was found to interact with H2 and utilize the molecule for hydrogenation reactions.488,491 However, CaO and MgO produce significant amounts of solid residue, where the carbonaceous char inhibited the movement of free radicals during the cracking reaction, preventing propagation and degradation of the plastic molecules.484
Outside of metal oxide catalysts, activated carbon has been tested for plastic catalytic pyrolysis but has been found to only minimally change product distribution.396,481,495–497 Further, activated carbon has variable acidity – which can be modulated by the addition of different acid groups (–OH, –OOH, and –P–O) – that dictates the product distribution.396,497 Weakly acidic activated carbons have been found to have high selectivity towards jet-fuel range alkanes within the liquid fraction and strongly acidic carbons (with P functionality) have improved selectivity towards jet-fuel range aromatics.481 Additionally, metal-doped carbons have been utilized for PSW decomposition due to the high thermal stability of the material and the cracking ability of transition metals. For example, Pt/C and Pd/C were found to decompose PET into monocyclic compounds within the liquid fraction by improving decyclization activity and promoting thermal free radical cracking mechanisms.498 Pt/C was also found to crack the carbonaceous compounds that formed during catalytic pyrolysis of PET, further inhibiting the production of undesirable products.498
Like many of the catalysts described above, carbonates have been found to increase the rate, reduce operational temperature, and reduce residence time of plastic decomposition. However, carbonates are prone to decomposition under thermal treatment, resulting in the formation of oxide compounds and CO2. MgCO3, CoCO3, and CuCO3 were all found to yield largely diesel fuel components in the liquid fraction for the catalytic pyrolysis of HDPE.499–502 At the same time, gas yields were increased by 10–15% due to the decomposition of the material under reaction conditions.499 The basic carbonates were hypothesized to initiate the plastic cracking mechanism via a different step than the acidic catalysts previously described, where the carbonates will accept a proton from a plastic defect site to create a carbocation chain rather than the catalyst donating the proton to form a carbonium molecule. The carbocation chain will undergo β-scission to form lower carbon number alkanes and alkenes, then further reaction will occur through carbonium ion attacks or interactions with the catalyst basic sites.
Ashes from plastic incineration plants have also proven to be effective in reforming plastic pyrolysis vapor using a pyrolysis-reforming reactor.503 Incineration ashes contain ppm levels of metals due to metal evaporation and adsorption to the surface of ash particles during incineration. When used for in-line upgrading of plastic vapor, flying ash increased the BTX content of liquid product compared to non-catalytic upgrading. The Fe content of ash promoted the formation of monoaromatics while inhibiting the formation of polyaromatics.455,504 Although the use of ash as a catalyst is attractive as it is a waste, the catalytic performance of this material is still inferior compared ZSM-5. Furthermore, pretreatment of ashes is required to remove impurities that prevent access to active sites.
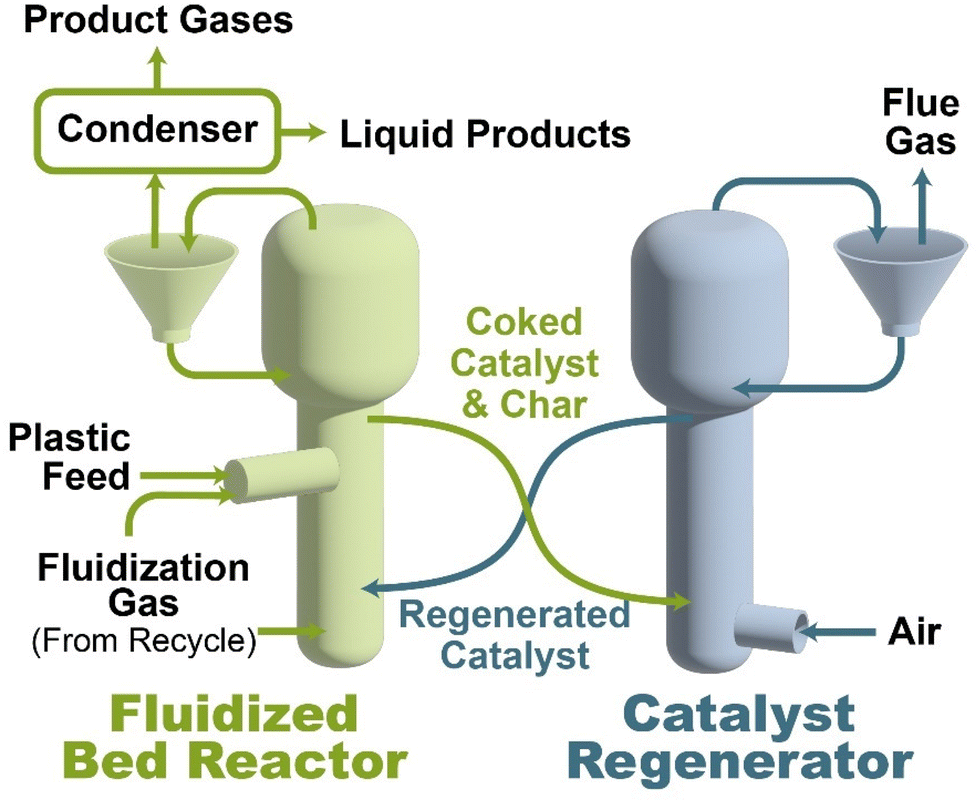 | ||
| Fig. 22 Fluidized bed reactor for plastic pyrolysis.508 | ||
Braskem, the leading biopolymer producer of thermoplastic polyolefins in America, has formed several partnerships to develop new catalysts for plastic catalytic upgrading.510–512 Braskem is working with FCC S.A., the leading catalysts supplier of fluid catalytic cracking in the South American market, to develop new catalysts for plastic upgrading. In addition, Braskem and Encina have formed a long-term partnership to produce recycled PP. Encina development group is focusing on developing a catalytic pyrolysis process to produce BTX from plastics #3 through 7, with the focus on PP.
6. Liquefaction of plastics
Liquefaction typically operates at temperatures between 200–450 °C under high pressure to convert the plastic feedstock into a liquid oil in the liquid phase. Heterogenous or homogenous catalysts are sometimes added.513,514 When liquefaction occurs in a solvent, it is called solvent liquefaction. Fuel liquids or monomers can be obtained from the liquefaction of plastics.513,515–523 During liquefaction, pressurized nitrogen or other gases keep the pressure high, helping maintain the products in the liquid phase. Pressurized hydrogen enhances polymer chain cleavages, caps the chain ends with hydrogen, and produces liquids with higher H/C ratios and/or low boiling point products.521 Various organic solvents, including hydrocarbons, alcohols, and water, are used to enhance the reaction rate and lower the activation energy of plastic decomposition, in addition to reducing unfavored secondary reactions for repolymerization.524–527 Chemically-compatible solvents can facilitate product removal and dissolution, suppressing intermolecular coupling reactions.520 Hydrogen-donating solvents can enhance product dissolution and stabilize intermediate products.518When performed under supercritical states of solvents, solvent liquefaction also takes advantage of drastically increased reaction rates and enhanced solubilization capability of the polar and non-polar products.518,528 In the case of hydrothermal liquefaction (HTL), water is a low-cost green solvent. Table 14 summarizes some of the previous work on plastic liquefaction processes, which are grouped into liquefaction using pressurized gases, HTL, and liquefaction by organic solvents with the majority of this work in stirred-tank batch reactors.
| Type of plastics | Temp. (°C) | Pressure (MPa) | Time (h) | Concentration (%) | Solvent/gas/catalyst | Products | Oil yield (wt%) | Year | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Liquefaction under pressurized gases | |||||||||
| PE, PP, PS (continuous reactor) | 400 | — | — | — | w/or w/o ZSM-5 | Majority aromatics, some aliphatic liquids and gases | 85% (PE, PP); 90% (PS) | 1992 | 515 |
| MDPE, HDPE, PP, PET or mix | 420–450 | 5 | 1 | 66% plastics in tetralin/waste oil | H2; w/or w/o 1 wt% HZSM-5/ferrihydrite catalyst | Hydrocarbon oil and gases | ![[w with combining low line]](https://www.rsc.org/images/entities/char_0077_0332.gif) ![[/ with combining low line]](https://www.rsc.org/images/entities/char_002f_0332.gif) ![[o with combining low line]](https://www.rsc.org/images/entities/char_006f_0332.gif) ![[t with combining low line]](https://www.rsc.org/images/entities/char_0074_0332.gif) ![[i with combining low line]](https://www.rsc.org/images/entities/char_0069_0332.gif) ![[h with combining low line]](https://www.rsc.org/images/entities/char_0068_0332.gif) ![[c with combining low line]](https://www.rsc.org/images/entities/char_0063_0332.gif) ![[a with combining low line]](https://www.rsc.org/images/entities/char_0061_0332.gif) ![[l with combining low line]](https://www.rsc.org/images/entities/char_006c_0332.gif) ![[y with combining low line]](https://www.rsc.org/images/entities/char_0079_0332.gif) ![[s with combining low line]](https://www.rsc.org/images/entities/char_0073_0332.gif) ![[) with combining low line]](https://www.rsc.org/images/entities/char_0029_0332.gif) : 11 to 96% (HDPE); 83 to 98% (PP); 33 to 93% (MDPE) : 11 to 96% (HDPE); 83 to 98% (PP); 33 to 93% (MDPE) |
1994 | 516 |
| Mix of HDPE, LDPE, PET and PS | 400–440 | 5.6 | 0.5–2 | 50% plastics in tetralin, decalin, dodecane, C12–C20 alkanes | H2; 10–20 wt% HZSM-5/FCC catalysts | Hydrocarbon oil and gases | 56.2–75.8% conversion (mixture); 90–100% conversion (individual plastics) | 1996 | 513 |
| PS and SBR | 350–450 | 3.45–17.23 | 0.25–2 | 1–5 wt% Fe2O3/SO42− and ZrO2/SO42− | Aromatics (PS); aromatics and C5–C9 paraffins/cycloparaffins. | 80.3% (PS); 72% (SBR) | 1996 | 517 | |
| LDPE, PET, PVC | 420–440 | 5.5 | 0.25–1 | 70% in tetralin | Hydrogen | C9–C40 hydrocarbons and gases | 59% (LDPE) | 1996 | 518 |
| MDPE, HDPE, PP | >420 | 0.68–5.5 | 0.3–1 | 30–50% in tetralin; H2 or N2; HZSM-5 or Al2O3–SiO2-ferrihydrite | Light, medium and heavy oils | >90% (all plastics) | 1996 | 519 | |
| HDPE, PP, PB | 350–450 | 3.5–13.8 | 0.5–3 | n-Octadecane; H2; 1–2 wt% Fe2O3/SO42− and ZrO2/SO42− | Gasoline range paraffins as major products | > 90% | 1997 | 520 | |
| PE and PP | 500 | 0.79 | 0.5 | Hydrogen | Light and heavy oils | up to 60% | 1998 | 521 | |
| Post-consumer plastic (PCW) mixture | 415–455 | 1.4 | 0.5–1 | H2, 1–5 wt% of HZSM-5 and others | Higher gasoline range oil with catalysts | up to 85% | 1999 | 522 | |
| PE, PP, PS, PVC, and PET (standalone and mixed) | 500 | 1 | 1 | Nitrogen and hydrogen | Hydrocarbon oil and gases with high concentrations of alkanes and single-ring aromatics | ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) ![[u with combining low line]](https://www.rsc.org/images/entities/char_0075_0332.gif) ![[e with combining low line]](https://www.rsc.org/images/entities/char_0065_0332.gif) ![[d with combining low line]](https://www.rsc.org/images/entities/char_0064_0332.gif) ![[m with combining low line]](https://www.rsc.org/images/entities/char_006d_0332.gif) ![[x with combining low line]](https://www.rsc.org/images/entities/char_0078_0332.gif) ![[v with combining low line]](https://www.rsc.org/images/entities/i_char_0076_0332.gif) ![[s with combining low line]](https://www.rsc.org/images/entities/i_char_0073_0332.gif) ![[. with combining low line]](https://www.rsc.org/images/entities/char_002e_0332.gif) ![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif) ![[W with combining low line]](https://www.rsc.org/images/entities/char_0057_0332.gif) ![[( with combining low line]](https://www.rsc.org/images/entities/char_0028_0332.gif) ![[D with combining low line]](https://www.rsc.org/images/entities/char_0044_0332.gif) ![[S with combining low line]](https://www.rsc.org/images/entities/char_0053_0332.gif) ![[F with combining low line]](https://www.rsc.org/images/entities/char_0046_0332.gif) : 72.3% vs. 32.5/64.1% (N2); 75.12% vs. 48.2/70.6% (H2) : 72.3% vs. 32.5/64.1% (N2); 75.12% vs. 48.2/70.6% (H2) |
2007 | 523 | |
| Hydrothermal liquefaction | |||||||||
| HDPE | 425 | Corresp. to T | 0.16–3 | None | Light and medium oil | 90.2% (120 min) | 1999 | 524 | |
| SBR | 450–686 | <1000 | 0.022–0.5 | 5/10% H2O2 | Light and medium HCs | 25.9–66% | 2002 | 530 | |
| Hydrothermal liquefaction | |||||||||
| HDPE | 450–480 | Corresp. to T | 0.016–0.5 | 25–50 | None | Light and medium oil | 91.4% (460 °C, 1 min) | 2004 | 531 |
| PVC | 200–600 | 1.6–55.7 | 1 | 0.1–2 | None | Low-molecular weight aromatic and aliphatic compounds | 179 ppm (300 °C), 396 ppm (400 °C) | 2004 | 532 |
| HDPE (continuous reactor) | 530 | 25 | 0.02–0.07 | — | None | C7–C11, C12–C18, C19–C24, >C24 | 79% | 2007 | 533 |
| Model mix of PE, PP, PS and PVC | 200–400 | 1–5 | 100–200 | Glass powder additive | Chlorine content after NaOH-based dechlorination | 40–120 ppm in oil (negl.) | 2011 | 534 | |
| PBT, PC, PLA, PMMA, POM, PPO, PVA, SB. | 400 | 25 | 0.25 | 10 | None | ![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) ![[% with combining low line]](https://www.rsc.org/images/entities/char_0025_0332.gif) : nil/50.8 (PBT), 99.8/nil (PC), /68.5 (PET), 48/nil (PMMA), 13.7/8.1 (POM), 78.9/8.8 (PPO), 35.4/2.9 (PVA), 80.8/1.2 (SB) : nil/50.8 (PBT), 99.8/nil (PC), /68.5 (PET), 48/nil (PMMA), 13.7/8.1 (POM), 78.9/8.8 (PPO), 35.4/2.9 (PVA), 80.8/1.2 (SB) |
2017 | 535 | |
| High impact PS (HIPS) | 350–550 | 30 | 0.12–1 | 1–9 | None | Ethylbenzene (51.3 wt%), toluene (14 wt%) and other polyaromatics (490 °C per 1 h) | Maximum carbon liquefaction rate of 77 wt% (490 °C, 1 h) | 2019 | 536 |
| PP | 425–450 | 23 | 0.5–4 | — | None | 80% naphtha | 91% (2 h per 425 °C; 1 h per 450 °C) | 2019 | 537 |
| ABS, PA6, PA66, PET, Epoxy, PC, PUR, HDPE, PVC, LDPE, PP, PS | 350 | Corresp. to T | 0.33 | 5.6 | KOH | Bisphenol-A & phenol (PC, epoxy), caprolactam + (PA6, PA66), TE & EG (PET), TDA + (PUR) and no polyolefin-derived products | 2020 | 538 | |
| PP, PS, PC and PET | 350–450 | 25+ | 0.5–1 | 0.06–0.35 | None | 32% (PP, 425 °C, 30 min), 16% (PET, 450 °C, 30 min), 86% (PS, 350 °C, 30 min), and 60% (PC, 425 °C, 30 min). | 2020 | 528 | |
| PC | 350–450 | Corresp. to T | 0.03–1 | 5 | None | IPP, IPrP, phenol, BPA, and other alkylphenols | 57.7 | 2020 | 539 |
| HDPE | 400–450 | Corresp. to T | 0.5–4 | 57.1 | None | Naphtha, heavy oil and heavy waxes | 86–87% (425 °C, 2.5 h or 450 °C, 0.75 h) | 2020 | 540 |
| LDPE, HDPE | 380–450 | Corresp. to T | 0.25–4 | 20 | 1% acetic acid | Alkanes, alkenes, cycloalkanes, aromatics, and negligible alcohols | 85–90% (425–450 °C, 1 h) | 2020 | 514 |
| Hydrothermal liquefaction | |||||||||
| Mix of LLDPE and PP | 400 | 25 | 1 | 16.67 | None | Paraffins, olefins, cyclics and aromatics | 90.7% (mixture); 87.04% (LLDPE); 86.42% (PP) | 2021 | 541 |
| A waste mixture containing polyolefins, PET, nylon, PVAc, and cellulose | 340 | Corresp. to T | 5 | 10 | 2% NaOH | Aromatics, oxygenated compounds, benzoic acid, phenols, caprolactones, fatty acids, and no polyolefin-derived products | ![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) ![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) : oil (7.7%/7.4%), solid (75.1%/65.5%), gas (11%/11%), unreacted & aqueous-phase products (6.2%/16%) : oil (7.7%/7.4%), solid (75.1%/65.5%), gas (11%/11%), unreacted & aqueous-phase products (6.2%/16%) |
2021 | 542 |
| Liquefaction with other supercritical solvents | |||||||||
| HDPE, PP and HDPE/PP mix | 450–470 | Corresp. to T | 1 | 10% | Acetone | Aliphatic hydrocarbons | : 88% (HDPE); 96% (PP); 90% (mix) |
2021 | 525 |
| PS | 370 | Corresp. to T | 1.5 | 7–10% | n-Hexane | Styrene (primary); toluene and ethyl benzene (secondary) | 90% conversion | 2001 | 526 |
| PS | 340–420 | Corresp. to T | 0.25–1 | — | Methanol | Styrene, toluene, ethylbenzene, dimer, and other 2-ringed aromatics | 92% conversion (380 °C/15 min) | 2008 | 527 |
| HDPE; HDPE + spirulina microalgae | 340 | Corresp. to T | — | 6–14% | Ethanol | Aliphatic hydrocarbons | 7.55% (PE oil); ∼50% (HDPE + spirulina) | 2012 | 543 |
| PS | 350 (SC ethanol); 370 (HTL) | Corresp. to T | 0.25–1.25 | 25–400% | Ethanol; water | Alkyls, alkenes, and aromatics (SC ethanol) | : 84.74% (SC ethanol); 78.3% (HTL) |
2020 | 544 |
| HDPE + sugarcane bagasse mix | 240–280 | Corresp. to T | 1–1.5 | 5–10% | Ethanol | Aliphatic hydrocarbons | 32.3% (bio-oil yield) | 2020 | 545 and 546 |
6.1 Products
Solid acid catalysts are often used during the hydrogen liquefaction of plastics. In general, the addition of catalyst lowers reaction temperature, increases conversion, and produces oils with higher light fractions. Shabtai et al. reported that conversion of HDPE during hydrogen liquefaction at 350 °C increased from nearly zero without a catalyst to 30% with a 17 wt% of SO4/ZrO2 catalyst, and 64.7% using a 33% Pt/SO42−/ZrO2 ctalyst.520 Much lower metal loadings were used in other related studies. For example, Zmierczak et al.517 found catalytic hydrogen liquefaction of PS at 375 °C increased the conversion from 59.4% without a catalyst to 70.5% with a 5 wt% Fe2O3/SO42− catalyst and to 80.3% with a 5 wt% ZrO2/SO4 catalyst. Other catalysts were also evaluated for hydrogen liquefaction, as shown in Table 14. Shah et al. converted post-consumer plastic waste using 1 wt% HZSM-5 zeolite, a ZrO2/WO3 catalyst, nanoscale ferrihydrite treated with citric acid, ferrihydrite containing 5% Mo, a SiO2–Al2O3 binary oxide, and two TiO2–SiO2 binary oxides.522 They reported that while there were no significant differences in the oil yields, the presence of ZSM-5 reduced the boiling point distribution of the oil products resulting in the most gasoline-like products. Similar results were also observed by Feng et al.519
Chemically compatible or hydrogen donor solvents have been used to aid the hydrogen liquefaction of plastics. For example, adding n-octadecane during Pt/SO42−/ZrO2 catalyzed hydrogen liquefaction of HDPE at 350 °C increased the conversion from 64.7% without solvent to 92.5% with the solvent.520 The plastic conversion in the solvent without catalyst was only 2%.520 Tetralin has been studied as a hydrogen donor for coal liquefaction. As shown in Table 14, Taghiei et al.516 reported that tetralin-aided hydrogen liquefaction of PE and PP increased the oil yield from 11% without a catalyst to 83% with a catalyst.2 In contrast, the oil yield only increased from 62% to 64% for PET. Since PE and PP-derived products are non-polar hydrocarbons whereas PET-derived products are oxygenated polar products, chemical compatibility between the solvent and plastic decomposition products seemed to play a critical role in their study. Murty et al.518 reported that PVC absorbs tetralin and that this promoted PVC decomposition. Feng et al.519 reported that liquefaction of co-mingled waste plastics in the absence of solvent resulted in higher oil yields than the liquefaction in tetralin or waste motor oil, regardless of whether catalyst was added or not.
The liquefaction of plastics is highly sensitive to reaction temperature. Zmierczak et al.517 reported an increase in the distillable product yield from 71% at 350 °C to 93% at 450 °C during catalytic hydrogen liquefaction of PS. Increasing temperature also increased the formation of single-ring aromatics (i.e., benzene and alkylbenzenes) by further decomposing multi-ring products (e.g., diphenyl or triphenyl alkenes).517 However, increasing the temperature during catalytic liquefaction can also promote the cracking of liquid products to gases. Compared to catalytic liquefaction, reaction temperature seemed to have a greater effect on non-catalytic liquefaction. Feng et al.519 showed that the oil yield from mixed post-consumer plastics at 430 °C was only about 25% with no catalyst and 60–63 wt% with 1 wt% HZSM-5 or Al/Si as the catalyst. However, oil yields of over 90% were obtained at 460 °C both with and without catalysts.519 For all cases, gasoline and kerosine fractions increased with increasing temperature.
In general, increasing reaction time increased both conversion and secondary reactions. Shabtai et al.520 reported that increasing reaction time during catalytic hydrogen liquefaction of HDPE at 450 °C improved the conversion from 82.5% to 100% but also reduced the oil yield from 92.8% to 60.2% due to increased cracking at prolonged reaction times. A similar trend was also observed with PP.520 Increasing the reaction time also enabled the complete depolymerization of PS to single ring aromatics and promoted secondary cyclization of the primary monomers to form more naphthalene and indane as reported by Zmierczak et al.517 While the effect of hydrogen pressure is usually less significant than reaction temperature or time, it can improve both conversion and oil quality.519,520 For example, Murty et al.518 reported that the oil produced from LDPE at 440 °C using 800 psi hydrogen pressure was three times less viscous than the oil obtained using 100 psi hydrogen pressure because of the conversion from heavy oil to light oil at higher pressure. Increasing pressure also promoted single-ring aromatics and suppressed secondary cyclization during catalytic liquefaction of PS.517
Feedstock heterogeneity is the most challenging problem in converting real-world plastic wastes. Williams and Slaney523 evaluated the feedstock compositional variability under comparable liquefaction in this context. They conducted non-catalytic liquefaction of a mixture containing HDPE, PP, PET, and PVC. The product yields obtained from the mixture were then compared with the theoretical product yields calculated using those obtained when their constituent polymers were independently converted under the same condition. The oil yields produced from the mixed plastics were significantly lower than the calculated yields for nitrogen and hydrogen liquefaction. The effect of feedstock preparation methods showed that the PCW prepared in Belgium by Fost Plus via air blowing separation (obtaining a low density fraction of PCW) produced much higher oil yields and lower solid residues than the non-plastic removed PCW prepared by water washing (Waste DSD).523 Plastic wastes from Waste Fost Plus were expected to contain a high fraction of low-density plastics (LDPE, PP, and PS) and a lower fraction of high-density plastics (HDPE, PVC, and PET).523 Unfortunately, the authors investigated neither the product compositions nor the possible interactions among the compositions to elaborate on the crucial results. Contaminant content in mixed plastics also had a significant detrimental impact during hydrogen liquefaction. The catalytic effect was severely hindered during Shah et al.’s study when the dirtier PCW prepared by dry separation was converted.522 The authors found no noticeable changes in either yields or boiling point distributions of the products before and after using 1 wt% of HZSM-5 or other catalysts. In contrast, adding identical amounts of catalysts to a cleaner PCW prepared by water washing significantly increased the gasoline fraction of the oil products.522 The dirtier PCW contained slightly more N and Cl elements than the cleaner PCW (0.67% vs. 0.65% for N, 1.26% vs. 0.03% for Cl), likely from PVC and polyurethane.522 Thus, catalyst deactivation by the heteroatoms Cl and N can be considered among the reasons for the reduced catalyst performance.
Depending on their polymer structure, either monomers or fuel oil products are obtained during the HTL of plastics. Some step-growth polymers formed by condensation polymerization, such as PET, PC, polyamides, and polyurethane (PUR), have ether, ester, or acid amide linkages.528,535,538,539 These functional groups can be hydrolyzed to rapidly depolymerize the plastics with high chemical selectivity and liquefaction efficiencies even under sub-critical conditions.528,538,539 There is pronounced support for hydrogen bonding and ion solvation due to the higher density of the sub-critical water medium.528,538 HTL of PUR mainly promotes monomer production. It was reported that the sub-critical liquefaction of PUR at only 250 °C for 30 min produced diaminotoluene (TDA) at 72% yield along with some other aromatics and polyols.547 Passosa et al.538 reported oligomers as the primary products when PUR was converted at 350 °C using fast heating rates and short residence times, suggesting partial depolymerization under such conditions. HTL of PUR produces NH3. The solubilized NH3 then acts as a base catalyst to promote PUR decomposition.538 As reported by Passosa et al.538 and Goto et al.,548 polyamides (PA including PA6 and PA66) were hydrolyzed during HTL at temperatures above 300 °C to produce ε-aminocaproic acid, which further decomposed to form ε-caprolactam in nearly theoretical yield. When caprolactam was further hydrolyzed over extended reaction time, a range of water-soluble oxygenated platform chemicals were produced.538,548 Passosa et al.538 showed that adding KOH catalyst to the HTL of PA could significantly increase oil yield and promote complete depolymerization in reduced residence times. The presence of the base catalyst also accelerated the production of oxygenated products soluble in water542 and increased the selectivity of polyaromatics and oxygenated compounds derived from plastics like PC and PURs.538
HTL of non-polar plastics (polyolefins and PS) usually requires supercritical or near supercritical conditions to be effective. According to Ciuffi et al.,542 compounds from PE, PP, and PS depolymerization were absent from the liquid products obtained for subcritical HTL of mixed plastic wastes. The lack of heteroatoms (i.e., polar reactive sites) in the polyolefins and PS prevents the plastics from depolymerizing under mild conditions.538,542 As shown in Table 14, HTL of PS has been widely studied. Kwak et al.549 reported the complete and nearly instantaneous conversion of PS at 400 °C. Higher temperatures or prolonged reaction times at supercritical conditions will further decompose liquefied products by promoting secondary reactions. Seshasayee et al.528 reported a decrease in PS-derived oil yield from 86.2% at 350 °C to 38.3% at 450 °C after 30 minutes of treatment. HTP of PS usually produces styrene monomer, ethylbenzene, α-methylstyrene, styrene dimer, styrene trimer, and other dimer/trimer associated products. Toluene and ethylbenzene were the major liquid products under supercritical reaction conditions. Bai et al.536 reported yields for toluene and ethylbenzene at 490 °C to reach 14% and 51.3%, respectively.
HTL of polyolefins was conducted under supercritical conditions to produce an oil similar to naphtha. Several articles have reported 85–90% oil yields from HDPE and LDPE in supercritical HTL.514,540,541 As given in Table 14, oil yields higher than 90% from HDPE were reported by Murty et al.518 and Su et al.533 where HTL was performed in batch reactors.524,531 In comparison, the highest yield obtained using a continuous reactor system was 79% at 530 °C (Table 14).533 Supercritical HTL of polyolefins yields oils with a wide range of molecular weights. A narrower distribution can be obtained by increasing temperature and reaction time.531 HTL of PE produces n-alkanes as the major products, followed by 1-alkenes and alkadienes.514,540
Intramolecular β-scission and intermolecular hydrogen abstraction (H abstraction) are generally considered the primary mechanisms for polyolefins to degrade during HTL. When the PE backbone undergoes homolytic cleavage, the random scission produces free radicals with a random number of carbon atoms. These radical ends can be terminated by hydrogen abstraction from within the molecule (by β-scission) or from other molecules (by H abstraction).550 The ratio between 1-alkenes and n-alkanes in the product can indicate which reaction is prominent.550 During HTL, polymer fragments with radical ends immediately dissolve in SCW, where large amounts of excited H radicals are available. Thus, H abstraction becomes more prominent than β-scission.528 This is also why previous studies frequently report the 1-alkene-to-n-alkane ratio to be lower in HTL of PE than in thermal pyrolysis of PE.524 At extended reaction times, n-alkanes can undergo isomerization, and alkenes can undergo secondary reactions like cyclization.540 Gas production is also increased under these conditions due to increased cracking or recombination of short-chain free radicals.540 Aromatization is suppressed under HTL, thus producing less char from plastics.524 When HTL was conducted in a continuous or a semi-batch reactor system, the radicals were quickly removed from the reaction region. This led to a decreased opportunity for H abstraction, producing more alkenes than n-alkanes.533,551 In addition to promoting chain cleavage, HTL can also oxidize the hydrocarbon products to form alcohols and ketones. Products such as 2-propanol, 2-butanol, 2-propanone, and 2-butanone were found in the aqueous phase products obtained during HTP of polyolefins by Moriya et al.524
As shown in Table 14, oil yields up to 91% from supercritical HTL of PP were reported using reaction temperatures up to 450 °C.537 The oil fraction consisted of olefins, paraffins, and cyclic and aromatic compounds, with 80–90% of these compounds in the naphtha range. The authors also reported that the PP-derived oil is comparable to gasoline in terms of their H/C and O/C ratios, boiling point range, carbon numbers, density, viscosity, and surface tension.537 Similar product composition from HTL of PP was also reported by others,541 presenting an opportunity for PP-derived oil to be directly used as fuel feedstocks and blendstocks without further upgrading. Zhao et al. studied co-conversion of LLDPE and PP by HTL at 400 °C and reported the oil yield from the plastic mixture to be higher than the yield produced by converting LLDPE or PP alone, as shown in Table 14.541 In this study, PE produced more heavy oil than medium or light oils whereas PP produced equal amounts of naphtha (light oil), medium, and heavy oils. In comparison, the mixtures of PP and PE produced more heavy oil than naphtha or light oil. HTL co-conversion also increased the formation of cyclic compounds while reducing paraffin production.541 Thus, relatively lower quality oil was obtained by co-conversion, although the oil yield synergistically increased.
Subcritical HTL of PVC was also investigated because hydrochloric acid, produced from the PVC, is water soluble. PVC was dechlorinated in subcritical water at 200 °C in Takeshita et al.'s work to yield polyenes as solid residue without producing any hazardous organochlorine compounds.532 When the reaction temperature increased to 250–350 °C, a mixture of low-molecular-weight aromatic and aliphatic compounds was produced. As the temperature further increased to 400 °C, the increased production of light alkanes and alkenes was also accompanied by the formation of aliphatic ketones and oxygenated benzene compounds at higher yields.532 Inside the highly acidic solution created by the PVC-derived HCl, the PVC-derived polyene can undergo cyclization, hydrolysis, oxidation, and thermal degradation.532,538 As shown in Table 14, base catalysts, such as NaOH or KOH, were added during HTL of PVC to study their effect on dechlorination. Passosa et al.538 showed that although adding KOH did not affect the oil and solid yields produced from PVC at 350 °C, it caused more chlorine to leave as Cl2 gas rather than remaining in the solution as HCl.
Increasing temperature and treatment time during HTL often facilitated conversion to increase liquid and gas yields.514,524,531,539,540 However, it has been noted that high reaction temperatures and a prolonged residence time can reduce the desired product yield and increase coke formation due to over-cracking of the liquefied products.530,532,536 The feedstock-to-solvent mass ratio also affected the HTL process. Increasing the feedstock mass loading caused the liquefaction reaction rate to first increase and then decrease. Increasing the solid mass loading to beyond the corresponding optimum led to increased heat transfer limitations and decreased mobility of the macromolecular free-radical fragments in the solution.539 Such limitations favored undesired secondary reactions like repolymerization. Jin539et al. also reported that increasing temperature and mass ratio would eventually result in decreased liquefaction efficiency for the same reaction time. When high mass ratios were employed at higher reaction temperatures, increased reaction rate and higher H+/H˙ concentration in the solution caused the monomers and oligomers of the liquified products to undergo further cracking to form gaseous products.539 The effect of pressure during HTL of plastics is generally considered less significant than reaction temperature and time, especially for continuous and semi-batch reactor systems.533 Although the pressure effect was only observed in some studies, an increase in pressure increased the ion products of the water due to increased density, subsequently bolstering the cracking and hydrolysis process.536 An increase in pressure also enables enhanced interaction among different reaction species, promoting cyclization, hydrolysis, oxidation, and thermal degradation all at the same time.532 Depending on plastic types and the availability of reactive sites, any of these reactions may take precedence.
Ethanol achieves its critical states at relatively milder operating conditions, and its low dielectric constant increases the solubility of both polar and non-polar products. Furthermore, ethanol can also act as a hydrogen-donor solvent to stabilize the intermediate products and reduce repolymerization reactions. Breaking alcohol solvent at the critical state produces excessive hydrogen and hydroxyl ions, aiding the hydrogenation reactions. Ahmad et al. reported that the oil yield produced from PS conversion in ethanol at 350 °C was 84.74%, higher than 71% from pyrolysis at 500 °C, and 78.3% obtained using HTL at 370 °C.544 Rather than producing styrene as the primary monomer, the liquid produced using ethanol solvent consisted of alkyls, alkenes, and aromatics.544 Compared to PS, less liquid was produced when PE was converted in ethanol. According to Pei et al., the oil yield of HDPE in 340 °C ethanol was only 7.55%.543 However, co-liquefying PE with microalgae or lignocellulosic biomass in ethanol was found to synergistically increase the liquid yield because the oxygenated feedstock enhanced the thermal decomposition of PE in the solvent probably by hydrogen abstraction from PE by the oxygenated biopolymers.545,546
Methanol is also a hydrogen donor solvent with low critical conditions. Shin and Bae reported 92% conversion for PS by treating with 380 °C methanol for 15 min.527 Their comparative study showed that the activation energy for degrading PS in supercritical methanol was 117.2 kJ mol−1, lower than 132 kJ mol−1 in supercritical n-hexane, 157 kJ mol−1 with supercritical water, and 224 kJ mol−1 in pyrolysis. The authors reported the hydrogenation of unsaturated products to be the predominant reaction in methanol. Besides donating hydrogen, methanol also directly reacted with styrene monomer and α-methylstyrene to produce 3-phenyl propanol and 3-phenyl-1-butanol.527
6.2 Reactor designs
Lab-scale liquefaction studies have predominantly been carried out in batch reactors with few accounts of continuous reactor systems. Industrial-scale processes have incorporated both batch and continuous reactor designs. Stirred or agitated tank reactors are the most employed batch reactor designs at both lab and industry-scale. The different continuous reactor designs adopted for industrial-scale processes are continuous flow stirred tank, rotary kiln, fixed bed, and tubular reactors, with gravity or auger/extruder-based material feeding mechanisms.552,553Fig. 23 shows an example of an industrial-scale continuous flow reactor design.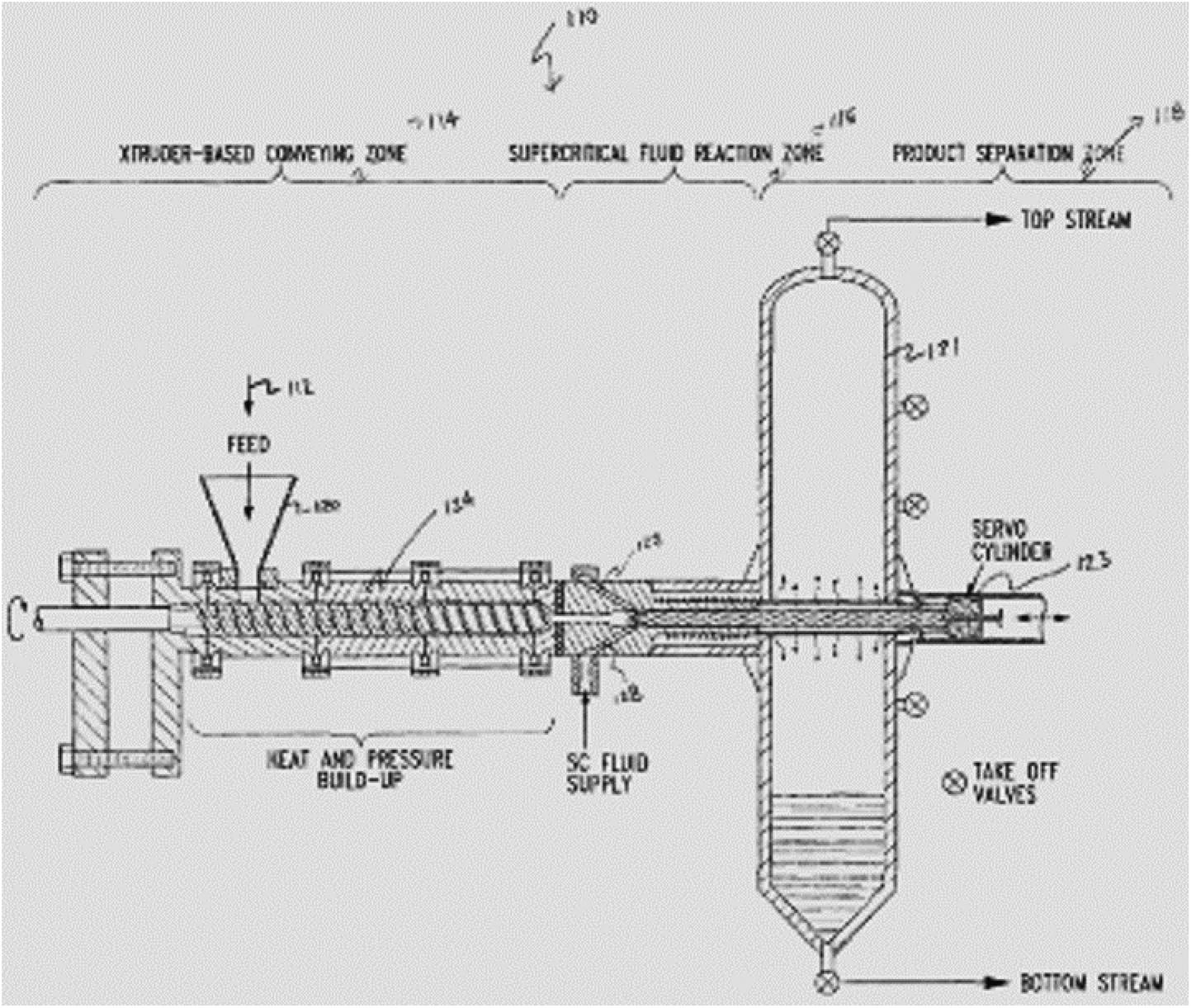 | ||
| Fig. 23 Example of a continuous tubular reactor design for hydrothermal liquefaction of plastics (extracted from the US patent US 8980143B2).554 | ||
6.3 Commercial activities in plastics liquefaction
The Sapporo Plastic Recycle Co. Ltd (SPR) was established in 2000 with the support of PWMI and Sapporo city with an initial capacity of 13.5 kton year which later grew into 14.5 kton per year.553 The SPR plant treated mixed plastics at three stages. The first stage involved pretreatment, sorting and pelletizing, followed by dechlorination, which fed calcium hydroxide into the system. The third and final stage involved thermally decomposing the waste plastics in a rotary kiln reactor at 400 °C and 5 kPa overpressure to produce light, medium, and heavy fractions of oils.553,556 Approximately 65% of the total oil produced was utilized as a process fuel within the plant.553 Due to the tender system governed by the Japan Container and Package Recycling Association (JCPRA), only half of the baled plastics were used in the SPR liquefaction plant. Additionally, the small operation scale of the SPR plant also raised the cost of waste plastics recycling.553 In Mikasa city, another large scale (6 kton per year capacity) liquefaction plant started operation in 2000 but was shut down in early 2004 due to a lack of waste plastic feedstock to process.556 By 2010, all major liquefaction plants in Japan ceased, with the advanced SPR plant being the last of them. A 2019 PWMI report suggested that the need for the crude oil product to be further cracked and refined meant that the process was not commercially viable at that time.557 Furthermore, these facilities are prone to fire accidents due to the risk of an explosion associated with the liquefaction reactors. Since the 2000s, liquefaction research and development have improved direct liquefaction methods to convert waste feedstocks to naphtha and diesel and thus reduced the burden for further refining. Nevertheless, the issues related to the scale of the plants and businesses remain, and any new companies adopting this technology will face difficulties.557
Another company called Aduro Clean Technologies from Canada has also developed an HTL-based commercial process. They call their process Hydrochemolytic Plastic Upcycling where the reactor operates at only 240–390 °C and takes about 25 min to completely convert the plastic mixtures.562,563 According to their claim, their water-based process operates under less severe conditions and allows the manipulation of chemical reactions associated with different types of plastics to form a comprehensive approach for chemical conversion of plastics.563
7. Gasification of waste plastics
7.1 Fundamentals of gasification reactions
Gasification (or partial oxidation) of MSW converts solid waste into synthesis gas or producer gas (CO, H2, CH4) by the reaction shown in eqn (4), where CHqOr represents the feedstock; q and r are the hydrogen and oxygen to carbon ratios, respectively; m and b are the input air and steam to MSW ratios; xi is the mole number of molecular species i.564 Producer gas (CO, H2, N2) is produced when air is used for the gasification while synthesis gas (CO, H2) is produced when oxygen is used for gasification. Synthesis gas can be further processed to produce a range of chemicals and fuels. MSW and mixed plastic waste can be used as the feed for gasification.565 MSW gasification could be more profitable and environmentally sustainable than incineration.566,567 A comparative study estimates that the global warming impacts of waste gasification with a combined cycle powerplant are over 50% lower than incineration.568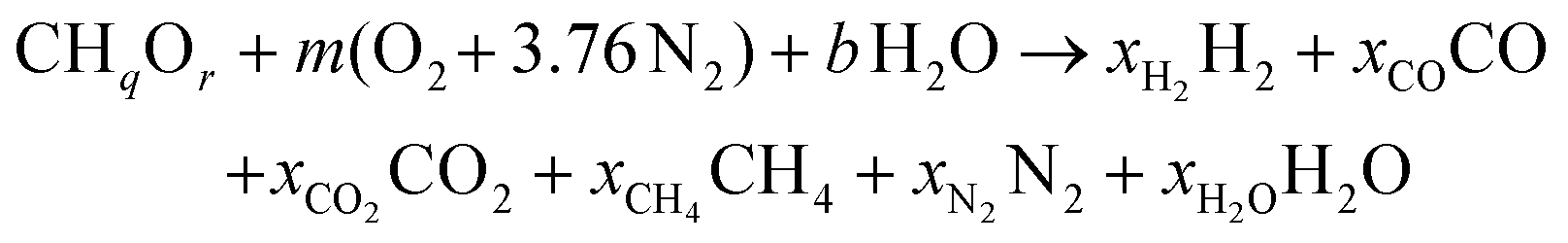 | (4) |
Gasification of MSW occurs above 550 °C.569 The main MSW gasification products are gases including CO, H2, CO2, and CH4. Solid residues (i.e., ash) are also produced. The amount of ash depends on the equivalence ratio (i.e., the ratio of oxygen in the employed oxidant and the oxygen required for complete combustion). Ash can be minimized by applying equivalence ratios between 0.25 and 0.35. Accordingly, equivalence ratios within this range are usually used in commercial reactors. Chemical thermodynamics can be used to predict the gas phase products during gasification.570,571
As shown in Table 15, at lower gasification temperatures (i.e., around 600 °C) there is a significant amount of CO2572 formed from the highly exothermic CO oxidation (eqn (5)), the oxidation of carbon (eqn (6)) and the water–gas shift reaction (eqn (7)). These reactions are thermodynamically unfavorable at higher gasification temperatures (>700 °C) leading to a decrease in the amount of CO2 produced.571,573 Conversely, more CO is formed at these higher temperatures by the steam reforming of CH4 (eqn (8)), steam reforming of hydrocarbons, the steam gasification of char (eqn (9)) and the dry reforming of hydrocarbons (eqn (11)) all of which are highly endothermic reactions. The Boudouard reaction (eqn (10)) is yet another highly endothermic reaction that yields CO.574 Material and operational constraints such as tar condensation and accumulation575 often require maintaining gas temperatures above 700 °C.
| Reaction | ΔH (kJ mol−1) | Name | Eqn |
|---|---|---|---|
| CO + ½O2 → CO2 | −283.7 kJ mol−1 | CO oxidation | (5) |
| C + O2 → CO2 | −394.1 kJ mol−1 | Carbon partial oxidation | (6) |
| CO + H2O ↔ CO2 + H2 | −41.0 kJ mol−1 | Water–gas shift | (7) |
| +CH4 H2O ↔ CO + 3H2 | +206.0 kJ mol−1 | Methane steam reforming | (8) |
| C + H2O ↔ CO + H2 | +131.0 kJ mol−1 | Steam gasification of char | (9) |
| C + CO2 ↔ 2CO | +172.0 kJ mol−1 | Boudouard reaction | (10) |
| CxHy + xCO2 ↔ 2xCO + y/2H2 | Endothermic | Dry reforming | (11) |
7.2 Waste plastic gasification process flow diagram
Fig. 24 shows a general flow diagram for waste plastic gasification processes. The feedstock is fed into a gasifier where gasification takes place. The volatiles enter a combination of physical, thermal, or chemical gas cleaning technologies where char, slags, and ash are removed. The clean syngas is then cooled in a quench unit to the temperature required for a secondary gas cleaning step. The secondary gas cleaning step removes contaminants that can impact catalyst performance, such as sulfur, halides, HCN, NH3, and tar, to levels of less than 0.1 parts per million by volume.576 The cleaned syngas can either go to a turbine for power generation or for catalytic upgrading to other products. Synthetic gas consisting mostly of CO and H2 is the feedstock for many catalytic synthesis systems.577,578 The synthetic gas mixture can be catalytically converted to methanol and other alcohols. Platform chemicals such as ethylene, propylene, and other alkenes can be produced via the methanol-to-olefins prcoess.579 | ||
| Fig. 24 Gasification of waste plastics and upgrading to heat, power, olefins, alcohols, and other fuels. | ||
The design and configuration of waste plastic gasification facilities depend on the facility scale and targeted products. Some gasification facilities combust the synthetic gas and solid by-product streams to produce combinations of heat and power. Other gasification facilities include steam generation, catalytic upgrading, and other product recovery units to improve energy efficiency and target higher-value markets.
7.3 Commercial activities scale
According to the Energy Information Administration (EIA), global waste gasification capacity is estimated to be less than 10000 Megawatts thermal (MWth) compared to more than 200000 MWth of coal gasification capacity.580 Despite the low total capacity, the number of global waste gasifiers in operation is estimated to be about 100 facilities which is similar to the number of biomass and natural gas gasifiers.580 These gasifiers primarily produce electricity and heat.
One of the leaders in gasification of MSW is Enerkem who is currently planning to build four commercial MSW recovery facilities. Enerkem's facility, in Québec, Canada, cost $680 million and will convert 200 kton per year of MSW and forest residues to 125 million liters of biofuels and chemicals starting in approximately 2023. In Alberta, Canada Enerkem recently commissioned a $50 million waste-to-biofuel facility with a capacity of 38 million liters per year.581 In partnership with Shell and the Port of Rotterdam, Enerkem plans to convert about 360 kton per year of MSW to 80 kton of renewable products including aviation fuels. Finally, Enerkem, Agbar, and Repsol plan to build a facility processing 400 kton per year to 220 million liters per year of fuels in Tarragona, Spain pending final investment decisions.582
7.4 Techno-economic analysis
The profitability of waste plastic gasification facilities depends on several process factors and market drivers. Feedstock composition, process yields, and energy efficiency are key drivers for process performance. Feedstock tipping fees, capital costs, and market prices have a significant impact on profitability. Several studies show that waste plastic gasification is more profitable than conventional waste management practices such as incineration and landfilling.58,583,584There are numerous techno-economic analysis studies on MSW gasification, but only a small subset focuses on waste plastic upgrading. For this review, we identified three comparative analyses that investigate the costs of waste plastic gasification as a primary strategy. Table 16 shows a summary of the key assumptions and findings from these studies.
| Technology | Feedstock | Major products | Region | Feed capacity (kton per year) | Capital cost ($ million) | IRR (%) | Ref. |
|---|---|---|---|---|---|---|---|
| Chemical recycling + gasification + catalytic upgrading | MSW | Propylene, ethylene, LPG, others | Germany | 76 | 158.2–271.2 | Not reported | 583 |
| Bubbling fluidized bed gasifier | Package derived fuel | Syngas, electricity | Italy | 4 | 0.00549–0.00841/kWe | 0.5–29.8 | 584 |
| Gasification to hydrocarbons | Waste polypropylene | Propylene, ethylene, aromatics | United States | 100 | 49.09 (annualized) | 16.91 | 58 |
Voss et al. compared the economic performance of chemical recycling and gasification to incineration as municipal waste treatment options in Germany.583 In their study, they assumed that their feedstock consists of “not differentiable mixed municipal waste” and “household waste, household-type commercial waste” as defined by the European Waste Classification Syste.585 Their MSW stream contains 6.9 wt% plastics. Most of Germany's waste streams are incinerated directly via combustion to produce heat and power or indirectly via anaerobic digestion of the organic fraction and the refuse-derived fuel (RDF) combustion. The chemical recycling scenario assumes that 1 tonne of MSW is initially mechanically separated into the organic fraction (750 kg) and RDF (250 kg). The organic fraction undergoes anaerobic digestion followed by a biogas-powered combined heat and power system. The RDF fraction is gasified to produce syngas, and the syngas is catalytically upgraded to methanol and subsequently to olefins using sodium hydroxide as the catalyst. Hydrocarbons including propylene (32 kg), ethylene (30 kg), butadiene (4.2 kg), LPG (4.0 kg), and butane (1.9 kg) are recovered in the recovery stage. The chemical recycling unit has a capital cost of $176.6 million for a 76 kton per year facility. Under the basic scenario, this technology is not profitable. It yields a net present value of −$180.8 million versus $8927 million for direct incineration. A combination of increasing the plant scale, environmental regulations, and favorable market conditions increases the net present value to $497.2 million.
Gregorio and Zaccariello investigated the economic performance of gasifying packaging-derived fuel (PDF) to produce power, heat and power, and district heating in Italy. PDF is a mixture of primarily plastic-derived materials. Their samples contained 53.9 wt% carbon, 7.7 wt% hydrogen, and 26.0 wt% oxygen. The material contains 0.1 wt% sulfur and 0.3 wt% chlorine, among other contaminants. The nominal capacity (4 kton per year) for the plant was 500 kilowatt-electric (kWe). Total plant costs ranged from $5500 for the power system to $8400 for the district heating scenario. The revenue generated from the three scenarios analyzed includes a tariff of $0.07 per kWe delivered to the grid and $0.09 per kWe for the sale of saturated or superheated steam production in the combined heat and power plant. Based on the revenues, the internal rate of return was estimated at 0.5% for power generation, 18.9% for district heating, and 29.8% for combined heat and power generation.584
Bora, Wang, and You compared plastic gasification to other recycling strategies. In their study, the gasifier processes plastic waste containing 84.5 wt% carbon, 13 wt% hydrogen, and 2.5 wt% oxygen. The gasification facility has a plant capacity of 100 kton per year, and an annualized capital cost of $49.1 million. They estimated a 16.91% internal rate of return from the sale of synthetic gases.58
8. Dissolution-based approaches for plastics recycling
Dissolution-based approaches are methods to use solvents to separate and recycle plastics without chemically modifying their structures. The basic workflow of a dissolution-based polymer recycling process can be described as follows: first, the target polymer is selectively dissolved in a suitable solvent or solvent mixture at a defined temperature; second, the mixture is filtered to separate the solid plastic and the liquid phase that contains the solvated polymer; last, the polymer precipitates from the solution by adding an antisolvent (i.e., a solvent in which the polymer is insoluble) and/or decreasing the temperature. Dissolution-based polymer recycling has several advantages over chemical or mechanical recycling methods: it produces high-quality polymers by removing impurities and additives, it can deal with multicomponent plastic mixtures by selectively dissolving a single target polymer; it does not require a high-purity input stream, and it is typically more environmentally friendly than chemical recycling.586–5888.1 Thermodynamics of selective dissolution and solvent screening
The key to a dissolution-based polymer recycling process is the selection of appropriate solvents. In a selective dissolution process, the solvent should dissolve the target polymer with a sufficient solubility without dissolving other unwanted components of the input stream. Since each polymer presents unique interactions that dictate solubility, and the composition of plastic waste is often complicated, theoretical approaches are valuable for systematically identifying suitable solvents. The thermodynamics of polymer-solvent mixing is governed by the free energy of mixing (eqn (12)),589 where ΔGmix is the molar Gibbs free energy of mixing, ΔHmix is the molar enthalpy of mixing, T is temperature, and ΔSmix is the molar entropy of mixing. A polymer will favorably mix with a solvent if ΔGmix is negative.| ΔGmix = ΔHmix − TΔSmix | (12) |
Key contributions to the Gibbs free energy of mixing can be understood using Flory–Huggins theory, a mean-field model for polymer solutions (eqn (13)),590 where kB is the Boltzmann constant, ϕ is the volume fraction of the polymer, N is the degree of polymerization, and χ is the Flory interaction parameter that characterizes interactions in the mixture.
 | (13) |
The χ parameter is the critical parameter that governs how favorable a solvent system is for a selected polymer; smaller values promote mixing. The χ parameter can be estimated by eqn (14), where v0 is the molar volume of a monomer and δi is the Hildebrand solubility parameter of compound i, which is defined as the square root of the cohesive energy density. A polymer is expected to interact more favorably – and exhibit a higher solubility – in a solvent with a similar solubility parameter, reflecting the general axiom of “like dissolves like”. This conceptual framework thus highlights polymer- and solvent-specific solubility parameters as critical to the design of dissolution-based processes.
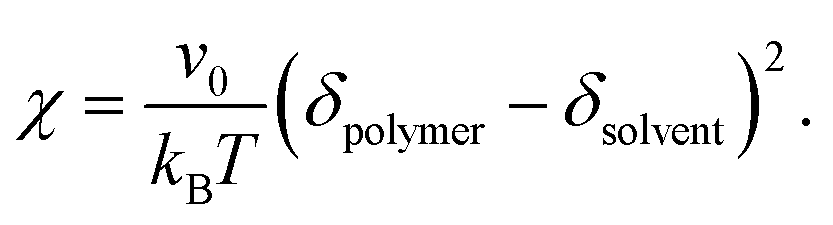 | (14) |
8.2 Origin of polymer dissolution and precipitation processes
The simplicity and relevancy of dissolution/precipitation for the separation and purification of polymeric materials has been demonstrated experimentally and disclosed in patents since the 1930s. For example, the dissolution and purification of condensation products, mainly polyesters, in ethyl alcohol was disclosed in a 1936 patent by Sändig.591 In 1949, Young and Sparks of the Standard Oil Development Company patented a method to purify polyesters via the solvent-mediated separation of high-molecular weight compounds using paraffinic mineral oils.592 These approaches showed the feasibility of using solvents to dissolve and purify polymerization products, giving way to the idea that this could be used to separate dissimilar polymers. In the 1970s, Seymour and Stahl demonstrated the separation of PE, PVC, PS, polyvinyl acetate (PVAC), and polymethyl methacrylate (PMMA) using a series of solvents that included toluene, methanol, and petroleum ether.593 Around the same time, companies like DuPont filed patents for the recovery of solid scrap thermoplastic polymers like PET, pol(hexamethylene adipamide), poly(acrylonitrile), and poly(oxymethylene) by dissolving them in hexafluoroisopropanol.594 In 1977, Monsanto patented a process for recovering solid PET, poly(cyclohexanedimethanol terephthalate), and poly(butylene terephthalate) from fibers using naphthalene as a selective solvent and dimethylformamide (DMF), water, and acetone as quenching mediums (antisolvent).5958.3 Applications of dissolution-based plastic recycling
Building upon these early materials recovery and purification processes, applications of dissolution and precipitation to plastic recycling has gained interest along the years. Nauman et al. proposed an approach to separate plastics in physically comingled solid mixtures by selective dissolution. Single solvents were used to dissolve plastics like PS, PVC, LDPE, HDPE, and PP, at different temperatures and pressures.596,597 The process consisted of shredding the comingled solids, dissolution of the plastic, filtration, flash devolatization, and pelletization of the separated plastic. Model experiments of plastic recycling with the dissolution/precipitation method were performed by Kampouris, Papaspyrides, and collaborators to recover PVC, LDPE, PP and PS.598–602 In one study, Pappa et al. designed a pilot unit for the separation of LDPE/PP mixtures using the dissolution/precipitation method with xylene and 1-propanol as solvent and antisolvent, respectively. They claimed that the costs of the plastics produced from the process were comparable to those of the virgin plastics.603 Regarding the impact of solvent treatment to the plastic properties, early recycling experiments with LDPE, HPDE, PET and PP showed that applying these processes once or twice did not affect the rheological, thermal, or mechanical properties of the final plastic.587,598,602,604 Many of these reports established solvent/antisolvent systems for common plastics, including solvent combinations, as summarized in Table 17.| Polymer | Solvent | Antisolvent | Ref. |
|---|---|---|---|
| PVC | Cyclohexanone | n-Hexane | 601 |
| PVC | Dichloromethane | Methanol | 611 |
| PVC | Toluene | Methanol | 611 |
| PVC | Tetrahydrofuran (THD), xylene | — | 596 |
| PS | Methyl ethyl ketone (MEK) | Methanol, n-hexane | 599 |
| PS | p-Xylene | n-Pentane | 599 |
| PS | Benzene, toluene | Water | 600 |
| PS | Toluene | n-Hexane | 611 |
| PS | Xylene | Methanol | 611 |
| PS | THF, toluene, xylene | — | 596 |
| PS | p-Cymene | 612 | |
| EPS | D-Limonene | — | 611 |
| HDPE | Xylene | n-Hexane, methanol | 611 |
| HDPE | THF, toluene, xylene | — | 596 |
| HDPE, LDPE, PP | Turpentine | n-Hexane, petroleum ether (PetE) | 613 and 614 |
| HDPE, LDPE, PP | Turpentine/PetE | n-Hexane, PetE | 613 and 614 |
| HDPE, LDPE, PP | Turpentine/benzene | n-Hexane, PetE | 613 and 614 |
| LDPE | Xylene | n-Hexane | 611 |
| LDPE | THF, toluene, xylene | — | 596 |
| PE, PP, PS | n-Butane, propane | — | 615 |
| PP | Xylene | n-Hexane | 611 |
| PP | Tetrachloroethylene | — | 616 |
| PP | THF, toluene, xylene | — | 596 |
| PET | THF | — | 596 |
| PET | γ-Valerolactone (GVL) | — | 617 |
| PET | N-Methyl-2-pyrrolidone (NMP) | n-Octane, n-hexane | 604 |
| PET | Diphenyl, diphenyl ether, naphthalene, methylnaphthalene, benzophenone, diphenylmethane | — | 618 |
| PET | Benzyl alcohol | Methanol | 611 |
| PC | Dichloromethane | Acetone | 619 |
| PA 6 | Dimethyl sulfoxide (DMSO) | MEK | 620 |
| PA 6,6 | Formic acid | MEK | 620 |
Recent and ongoing research efforts have developed dissolution/precipitation methods for increasingly complex sources of plastic waste. Researchers at the Illinois Sustainable Technology Center have developed a nontoxic energy-efficient chemical solvent process to recover plastics from blends of electronic waste that are converted to fuel oil by pyrolysis. The process has been demonstrated at the lab scale and can produce plastics with properties similar to virgin resins and will begin a pilot-scale project depending on the performance of the recovered plastics.605–607 Recently, Georgiopoulou et al.608 demonstrated a process to recycle Tetra Pak® packaging materials composed of LDPE, paperboard, and aluminum. Their approach included a hydropulping process to recycle the paper component and a dissolution/precipitation process to recover LDPE, using xylene and i-propanol as the solvent/antisolvent pair. Researchers at the Center for the Chemical Upcycling of Waste Plastics at the University of Wisconsin–Madison have developed a solvent-based method called Solvent-Targeted Recovery and Precipitation (STRAP) that has been demonstrated for the recycling of different post-industrial multilayer films composed of PE, EVOH, PET and EVA.609,610 STRAP combines experiments, computational modeling, and process design tools to develop solvent systems to recycle multilayer plastics via selective dissolution. A technoeconomic analysis showed that STRAP can produce plastics at costs similar to virgin resins and can be more economically feasible when the use of antisolvents is reduced by instead inducing precipitation via temperature.609
Other than targeting specific plastic components in plastic waste, solvents can also be used to extract additives from plastics or to selectively dissolve or decompose adhesives in plastic structures to trigger delamination. While these topics are out of the scope of this discussion, they have been discussed at length in recent publications.621–624
8.4 Industrial demonstrations of dissolution/precipitation processes
A handful of technologies are being commercialized for the recycling of different plastic waste feeds via dissolution methods. Here, we focus primarily on technologies that are currently in the process of commercialization. We omit discussion of past efforts; for example, the Solvay VinyLoop process was developed to separate PVC from a polymer coating using a selective dissolution step in a proprietary solvent,625 but the process ceased operations in 2018.APK AG's NewCycling process is being demonstrated in a 8000 ton per year plant in Germany to produce polyamides (PA) and PE from PIW multilayer plastics.626–628 Their technology is based on dissolving a plastic using a solvent mixture from a group of alkanes, isooctane or cycloalkanes.629 The plastics are recovered from solution and then pelletized by extrusion.626,627 Centrifuges separate the solids from the plastic solution and solvent.630 APK AG claims that PP, PET, PS, PLA and aluminum could also be recovered with this process in the future.627
The Fraunhofer Institute is developing a solvent-based process called CreaSolv that produces plastics with comparable properties to virgin materials, effectively removing contaminants and additives.631 The process is likely based on the dissolution of a target plastic, mainly polyolefins, using a solvent with a Hansen parameter δH between 0.0 to 3.0 MPa1/2, selected from a group of aliphatic hydrocarbons. An antisolvent made out of mono/polyhydroxy hydrocarbons, like 1-propanol or 1,3-propanediol, with a δH between 4.0 and 38.0 MPa1/2 is then used to precipitate the polyolefin from the mixture.632 The institute has also studied the separation of PS633 and is researching the recycling of multilayer food packaging that can consist of PET, PE, PP, EVOH, PA and metalized layers.634 Currently, Unilever is implementing the CreaSolv process in a 1100 ton per year demonstration plant in Indonesia to recover PE from multilayer sachets.635,636 CreaSolv is also reported to have a demonstration plant in Holland to separate EPS from POP, a pilot plant for recycling packaging and automotive compounds, and a demonstration plant in Germany for multilayer films.637
PureCycle Technologies is building a 54000 ton per year facility that uses solvents to produce PP from PIW and PCW in collaboration with P&G.638,639 The method consists of contacting the plastic waste with an alkane at elevated temperatures and pressures to obtain the purified PP.640 The process removes impurities, undesired colors and odors and produces PP with comparable properties to the virgin resin.639 P&G has recently filed patents with methods to separate and purify adhesives, PP, PE, PET, cellulose, and polyacrylic acid (PAA).641 The company also has inventions in the purification of PE using a pressurized solvent consisting of hydrocarbons, primarily n-butane or pentane.642 Additionally, they have a process for the separation of PS, PP, PE and poly(dimethylsiloxane) under similar conditions.615
Polystyvert developed a dissolution process to recycle different types of PS, including expanded PS, extruded PS and high-impact PS. Polystyvert operates at 125 kg per hour with an output of 600 metric tons per year.605 In their technology p-cymene is used as a selective solvent for PS and a non-polar solvent is used as an antisolvent to precipitate the plastic and remove p-cymene.612 Through this approach they can separate and recover PS from other materials, as only PS dissolves in the solvent and other materials float or sink in the system.643
Dissolution-based recycling processes have also been applied to textile waste. For example, Worn Again Technologies has patented a technology to recycle PET into pellets. In their process, the polyesters are dissolved in a solvent system with either benzyl acetate, benzyl benzoate, benzaldehyde, or similar solvents, precipitated by cooling of the solvent system, and then separated via filtration. After the plastic is separated, it is washed and dried and then molded into pellets and/or converted into fibers.644 This process can be applied to cotton textiles, post-consumer PET bottles, and plastic packaging containing PET. Worn Again has also partnered with companies such as H&M and Kering to promote the reduction of textile waste.645 Another start-up company, Ambercycle, has developed a technology to purify and separate polyesters from different garments and is planning a demonstration plant to produce a metric ton of recycled material per day.646,647
Other approaches are being used to recycle multilayer plastics using a combination of mechanical and solvent-mediated steps. For example, Saperatec has introduced a low-energy mechanical recycling process that involves shredding the multilayer film followed by use of surfactant-containing microemulsions to break up and separate the layers.648 The technology separates PE and aluminum film, PP from aluminum, and PE from PP. The technology was originally developed for rigid packaging and is being extended to flexible packaging and will be demonstrated at a 17000 ton per year facility in Germany.649
8.5 Advanced solvent selection for complicated feedstocks
As presented above, several solvents and antisolvents have already been identified for many common plastics, enabling the development of dissolution/precipitation processes for single plastics by selecting solvents from past literature.588,650,651 However, an ongoing challenge is solvent selection for plastic mixtures which are ubiquitous in realistic plastic waste. Multicomponent polymeric materials are often made from two or more plastics, each of which is selected to contribute its own useful properties.652 For example, food packaging boxes are typically multilayer plastic films, which are extremely challenging to recycle because of their complex compositions and the incompatibility of different polymer layers.610 Dissolution-based methods are promising for recycling such materials because selective dissolution enables the separation of different plastics and is tolerant to the additives and impurities present in waste. However, due to the variety of possible plastic combinations, as well as the necessity for choosing appropriate operating temperatures for dissolution and precipitation, solvent selection for plastic mixtures is a difficult task.Based on the thermodynamic framework described above, one method for solvent selection is to compute solubility parameters for target plastics and solvents to predict dissolution. Hildebrand solubility parameters can be applied with reasonable accuracy to predict solubility in systems dominated by non-polar interactions, but are inaccurate for mixtures with strong polar or specific interactions such as hydrogen bonds.590,653 Similar to the Hildebrand solubility parameter, other parameter sets have been applied to identify solvents and antisolvents for plastics; examples include Hansen,654 Kamlet-Taft,655 Gutmann,656 and Swain657 parameters. These parameters assess the chemical similarity between species to predict dissolution behaviors.651 Among them, Hansen solubility parameters (HSP) are the most widely used to select solvents for plastic dissolution.588,651,658 The HSP system assigns each compound three parameters that account for dispersion (δD), polar (δP), and hydrogen-bonding (δH) interactions. Solvent–polymer interactions are measured by their distance Ra in the 3-dimensional HSP space (2δD − δP − δH space) as shown in eqn (15).
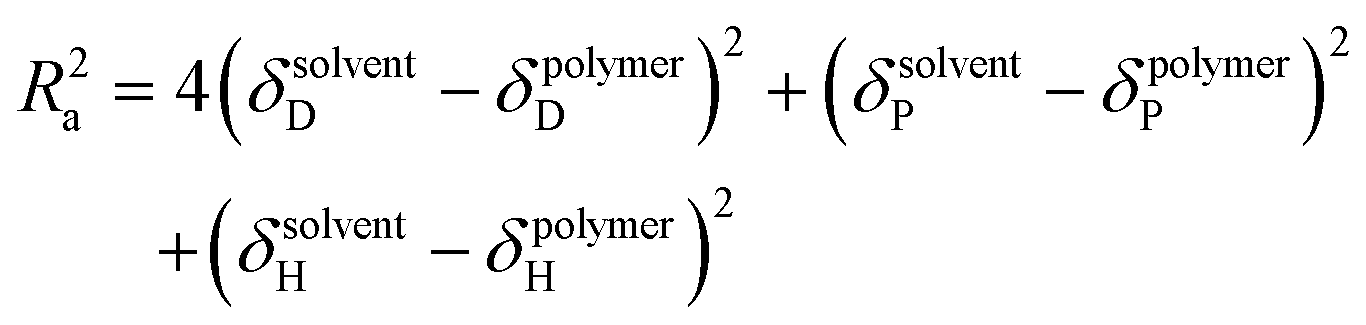 | (15) |
Each polymer has an additional radius parameter, R0, that defines a sphere in the HSP space. Solvents within this sphere (Ra/R0 < 1) are expected to dissolve the polymer, whereas solvents outside of this sphere (Ra/R0 > 1) are not expected to dissolve the polymer. Tabulated HSP and R0 values are available for a large number of polymers and solvents based largely on empirical data and have been used in many successful applications of solvent screening.658,659
Building upon the success of these solubility parameter methods, newer computational approaches for predicting polymer solubilities and screening solvents with minimal experimental effort have emerged in recent years. A recent example was the development of machine learning techniques for the large-scale binary classification of solvents and antisolvents for polymers, thus categorizing solvents into similar categories as HSPs. Specifically, Chandrasekaran et al.660 developed a deep neural network to classify 24 common solvents for over 4500 homopolymers with an accuracy of 93.8%. This example demonstrates the feasibility of applying data-centric methods to rapidly screen potential polymer–solvent combinations for selective dissolution processes, although more data will be required to extend the solvent library.
The preceding techniques primarily focus on qualitatively classifying solvents and antisolvents for target polymers. However, the development of dissolution/precipitation processes requires quantitative analysis of solubilities as a function of temperature in order to assess the feasibility and economics of potential industrial-scale processes. To address this challenge, Zhou et al. developed a computational approach for quantitatively predicting temperature-dependent polymer solubilities using molecular-scale models.661 This approach employs molecular dynamics simulations to sample polymer conformations in representative solvents. These conformations are used as input to the COnductor-like Screening MOdel for Real Solvents (COSMO-RS), a model that used quantum mechanical calculations and statistical thermodynamics methods to predict polymer solubilities through solid–liquid equilibrium calculations.610,661 After initial polymer conformations are available, temperature-dependent solubilities can be obtained rapidly (∼minutes). This approach was applied to screen over 500 solvent combinations for the selective dissolution of PE and EVOH and extended to develop a STRAP process for a commercial multilayer film with four polymer components.609,610,661
8.6 Techno-economic analysis of STRAP
A published techno-economic analysis of the STRAP process has demonstrated that plastics recovered via this technology can be sold at prices that are comparable to virgin resins.610 Due to STRAP being a new technology, the size of the process required to achieve a competitive sell price is important, as large capital costs can make scaling up to an industrial level difficult. At a feed rate of 3000 tonnes per year, plastic recycled by the STRAP process can be sold at a minimum selling price (MSP) of $2.18 per kg, while delivering a discounted return on investment (DROI) of 10% to investors over a 20 year lifetime. The MSP can be further reduced through optimization of STRAP design parameters, as shown in Fig. 25.609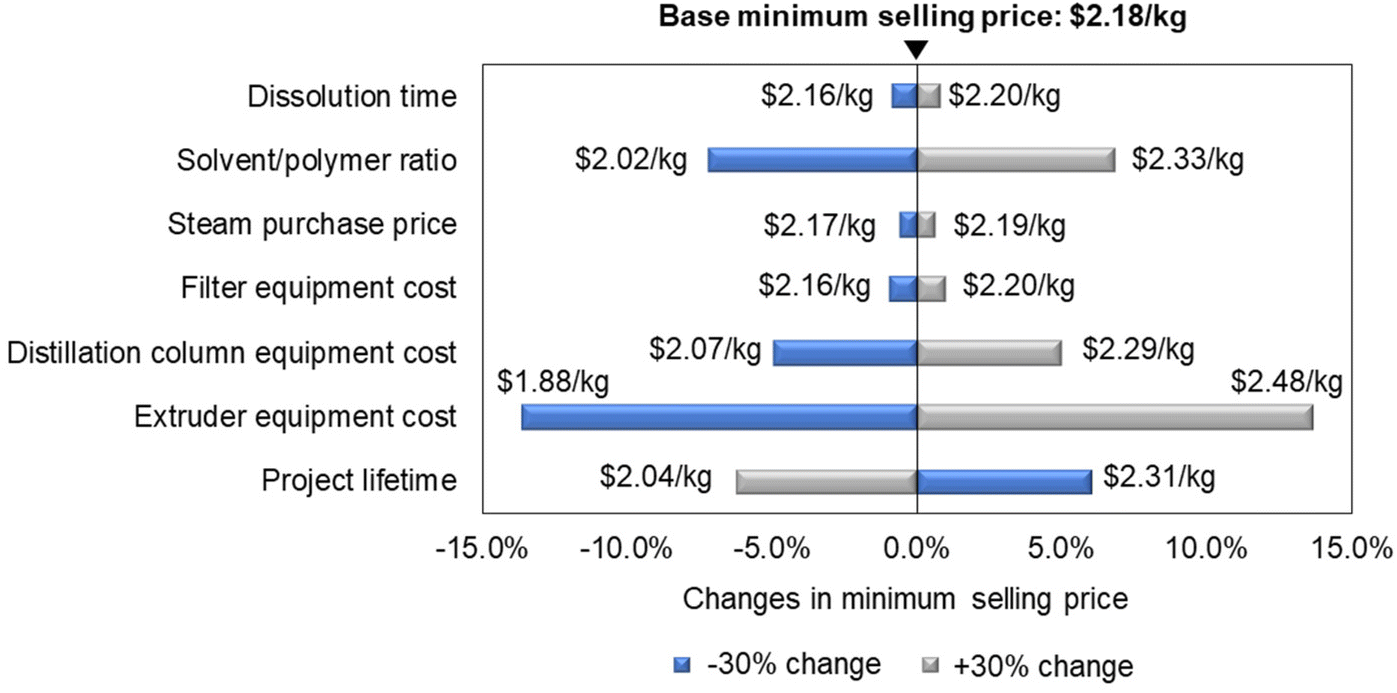 | ||
| Fig. 25 Sensitivity analysis of MSP for the STRAP process. Reproduced from ref. 609 with permission from Wiley-VCH, copyright 2021. | ||
A key feature that drives economic performance is solvent selection. A solvent with a high affinity for a target polymer can decrease the dissolution time and solvent/polymer ratio which can in turn decrease the filter equipment cost by reducing its size. A low-boiling solvent/antisolvent pair with a high relative volatility or solvent that allows for temperature-controlled precipitation can reduce the size of or eliminate the need for distillation columns.609 Using less solvent, an easily separable solvent/antisolvent system, or eliminating the need for an antisolvent then reduces the quality and quantity of steam required. This illustrates that efficient solvent screening and selection is essential for establishing economic as well as technical feasibility of STRAP.
Fig. 26 shows that STRAP is far from the point of diminishing returns at a processing capacity of 3000 tonnes per year. These data indicate that there will be a significant economic incentive to scale STRAP up to larger sizes if the process is shown to be effective at smaller processing capacities. At a feed rate of 15000 tonnes per year, the recycled plastic could be sold for $0.95 per kg to achieve a DROI of 10% or deliver a DROI of approximately 30% if the selling price of $2.18 per kg is maintained while requiring about twice as much capital investment.610 This analysis demonstrates that the process has significant economic potential and can be made even more competitive via process optimization and solvent selection. More broadly, this analysis, along with the commercial demonstrations described previously, supports the economic viability of dissolution-based recycling techniques.
 | ||
| Fig. 26 Capital investment and MSP for the STRAP process as a function of feed capacity (top) and DROI as a function of MSP at three different feed capacities (bottom) Reproduced from ref. 609 with permission from Wiley-VCH, copyright 2021. | ||
9. Chemical recycling of polyester to monomers
9.1 Chemical recycling of PET
The commercial uses of PET span a wide variety of industrial sectors, from fiber for textiles to resin for beverage bottles. In 2021, global PET consumption totaled 89.3 million metric tons (MMT) (Fig. 27). It is the fourth most consumed plastic in the world after PE, PP, and PVC.14,662,663 There are many reviews in the literature summarizing recent progress in chemical plastics recycling, as well as several which focus solely on PET.17,281,663–674 Chemical recycling of PET is an area of intense focus in the literature, and a comprehensive review of every report in this field is thus not attempted here. This section summarizes each strategy for PET depolymerization, with a focus on important and practical advances and their potential for industrial application. This review is organized into sections based on the approach to PET depolymerization: aerobic oxidation, hydrolysis, alcoholysis, glycolysis, aminolysis, hydrogenolysis, and enzymatic depolymerization (Fig. 28). Current commercial activity in chemical PET depolymerization is included at the end of this section, along with comments on the economics of these processes.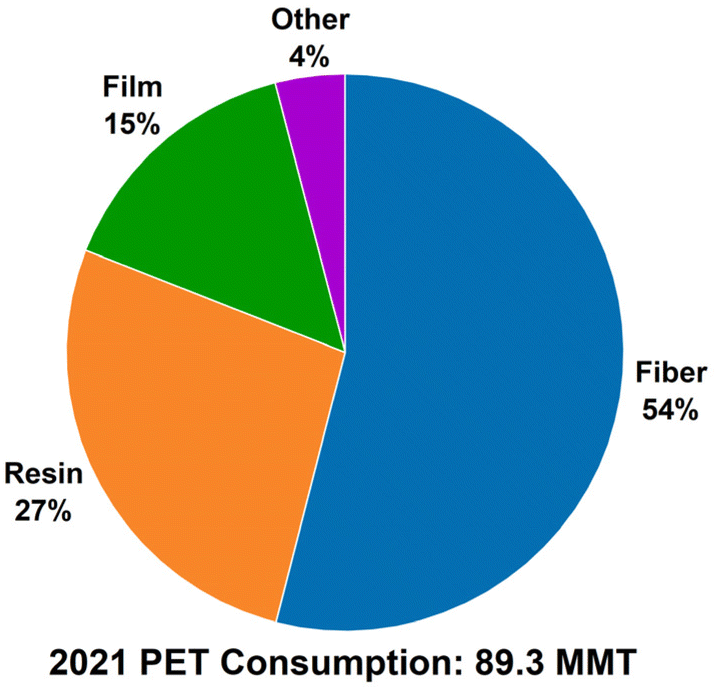 | ||
| Fig. 27 Global PET consumption in 2021. | ||
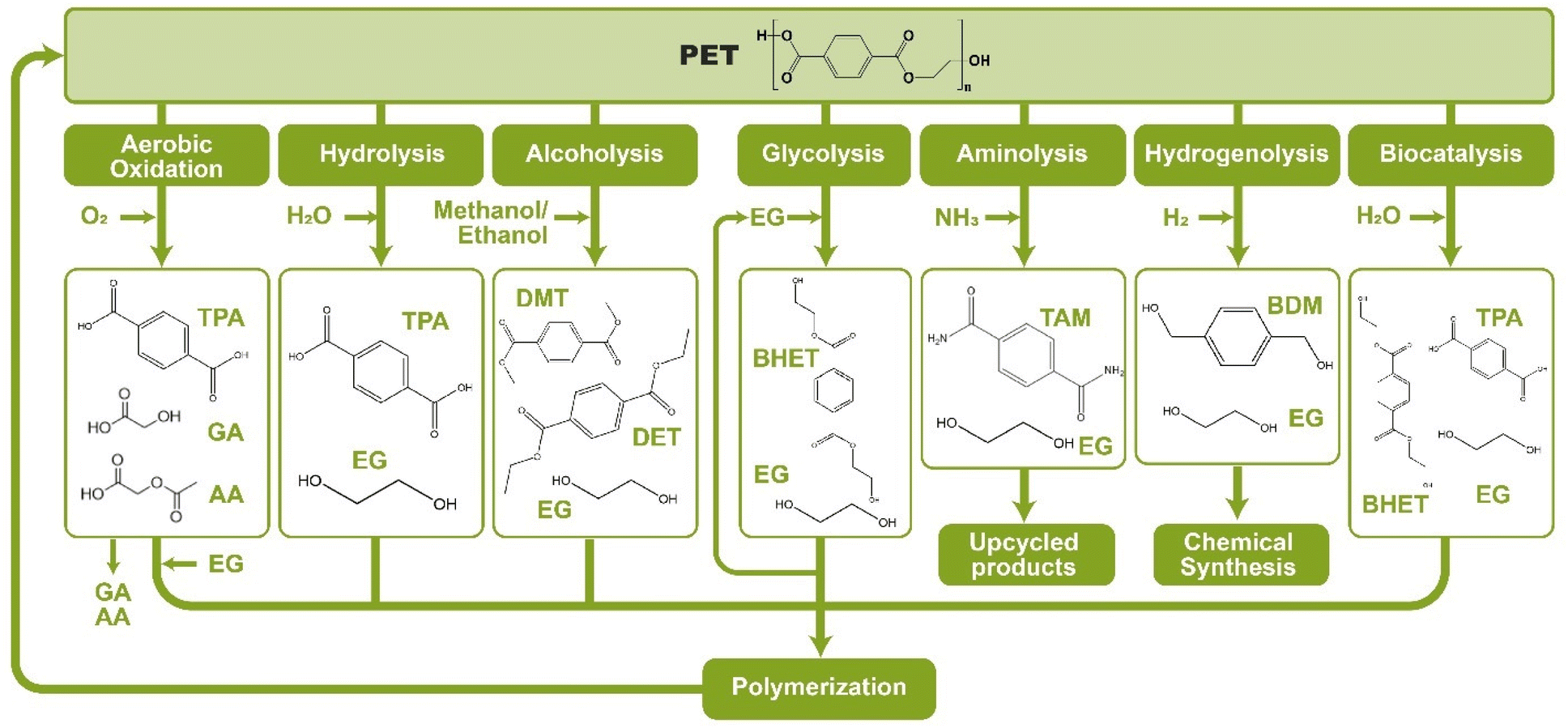 | ||
| Fig. 28 Strategies for PET chemical recycling discussed in this section, and their most common products. (TPA = terephthalic acid, EG = ethylene glycol, DMT = dimethylterephthalate, DET = diethylterephthalate, BHET = bis-2-hydroxyethylterephthalate, BDM = benzenedimethanol, MHET = mono-2-hydroxyethylterephthalate.). | ||
Partenheimer676 received a patent in 2005 for the oxidation of a wide variety of polymers using a range of simple metal salt promoters. PET sourced from blue film, recycled resin, and shirts were depolymerized as 4–20 wt% solutions in acetic acid under 1000 psia air pressure at 150–205 °C. A combination of Co/Mn/Br/Zr in varying ratios was used as a catalyst, and the effect of adding co-oxidants such as toluene and p-xylene was also investigated (Fig. 29). TPA yields of up to 100% were reached, and more efficient conversion was observed at higher temperatures, longer reaction times, higher initial PET concentrations, and in the presence of either co-oxidant. Partenheimer further published that a 56% yield of TPA could be isolated from the oxidation of PET at 190 °C in a water/acetic acid mixture using a similar Co/Mn/Br/Zr catalyst. The TPA yield increased to 100% in the presence of toluene as a co-oxidant.677 This chemistry was based on an extensive review published by Partenheimer about a decade earlier on metal/bromide autoxidation of hydrocarbons.678
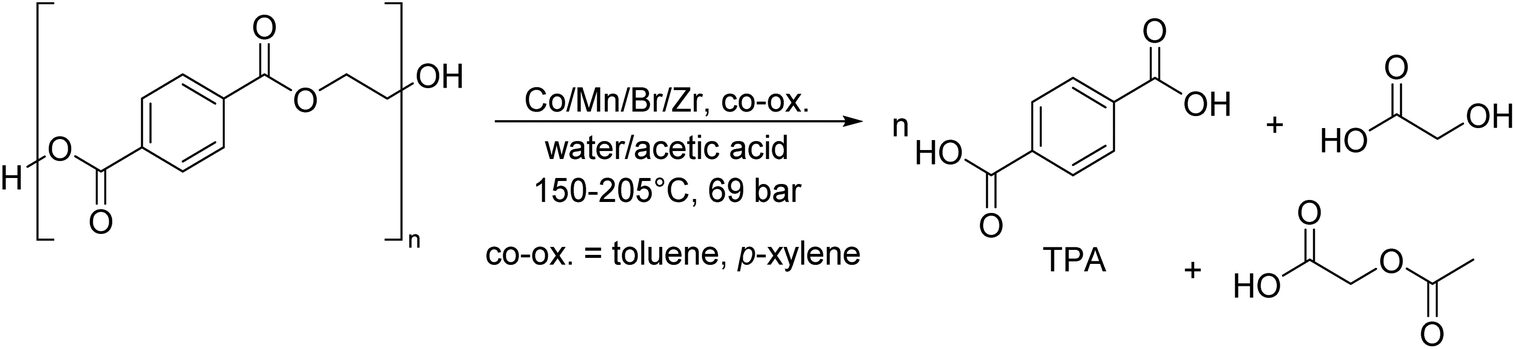 | ||
| Fig. 29 Typical reaction conditions and products for PET aerobic oxidation. | ||
9.1.2.1 Basic hydrolysis. Basic hydrolysis is typically carried out in a 4–20 wt% solution of NaOH or KOH in water. Reaction temperatures average around 200 °C and pressures are usually 15–20 bar (Fig. 30a). The disodium or dipotassium terephthalate product must be protonated with acid after the reaction, generating stoichiometric inorganic salt waste.681 LCA performed on this reaction demonstrated that decreasing the volume of water required for product purification is critical to achieve lower GHG emissions for this recycling process compared to PET incineration.679 Yoshioka and coworkers evaluated a system in which PET was hydrolyzed in concentrated NaOH at 250 °C and 5 MPa O2 pressure for 5 hours. The disodium terephthalate was not oxidized under these conditions and was obtained in quantitative yields. However, EG was oxidized to oxalic acid in roughly 70% yield, generating a value-added product that could be sold to further improve the economics of this recycling strategy.682
 | ||
| Fig. 30 Typical reaction conditions and products for uncatalyzed PET (a) basic, (b) neutral, and (c) acidic hydrolysis. Calculations of enthalpy and Gibbs free energy were performed in the presence of solvent [polarizable continuum model (PCM)]. Exchange-correlation functional: B3LYP. Basis set: 6-31++g(d,p). Neutral and basic environments H2O is the solvent – at small concentration of NaOH/KOH reactant and product species will mainly interact with H2O molecules. Acidic environment was simulated by setting the refractive index equal to 1.3945, which corresponds to 50 wt% sulfuric acid; the result is not very sensitive to the value of dielectric constant and the one for H2O was used (i.e., 78). | ||
A phase transfer catalyst such as a quaternary ammonium salt can be used to lower the temperatures and pressures required for alkaline hydrolysis. Kosmidis and coworkers683 used this strategy to isolate TPA at high purity using aqueous NaOH and catalytic trioctylmethylammonium bromide to depolymerize post-consumer PET soda bottles under atmospheric pressure and at temperatures as low as 70 °C. Similarly, microwave irradiation has been used to lower the required temperature of basic PET hydrolysis.684 Khalaf and Hasan685 isolated TPA from post-consumer soda bottles in almost quantitative yields after only 1 hour of irradiation in the presence of tetrabutylammonium iodide as a phase transfer catalyst. The catalyst could be recovered from the reaction mixture after product extraction, which improves the potential practicality of this method.
Hu et al.686 found that using a mixed solvent system of an ether and an alcohol enabled complete depolymerization of PET with NaOH in 40 minutes at only 60 °C. Pure TPA and EG were easily isolated from the reaction mixture. In 2017, Essaddam687 received a patent for the room temperature hydrolysis of PET. The system consisted of a nonpolar solvent such as CH2Cl2, which swelled the polymer, and an alcoholic solution of NaOH to hydrolyze the PET. High purity TPA could be isolated using this method in up to quantitative yields. Ügdüler and coworkers679 used ethanol as a cosolvent in the alkaline hydrolysis of PET to achieve 95% TPA yield in only 20 minutes at 80 °C. When post-consumer multilayer PET/PE trays were used as a substrate, yields were lower but still reached 45%.
9.1.2.2 Neutral hydrolysis. Neutral hydrolysis is in many ways preferable to basic or acidic strategies, as the solution will not require specialized reactor materials to prevent corrosion and stoichiometric salts are not required or formed in the reaction. However, this also means that impurities and additives present in the PET remain in the recycled monomer, which is not the case in TPA generated from acidic and basic hydrolysis.680,681 Additionally, high temperatures and pressures are required for the reaction to run at an appreciable rate. The temperatures used are 200–300 °C, and often in the upper half of this range so the reaction runs in the melt phase of PET. Pressures of 10–40 bar are typically used, along with a large excess of water or steam (Fig. 30b). The large volume of water required leads to very dilute solutions of EG in the product mixture, which makes isolation more difficult and costly. The reaction proceeds in about 1 hour at 275 °C, and it has been proposed that the TPA formed in the reaction can catalyze the hydrolysis, rendering the reaction autocatalytic.681,688
Campanelli and coworkers689 investigated possible catalytic effects in the neutral hydrolysis of PET but found that zinc salts exhibit only a minute accelerating effect on the reaction. Full conversions were only reached at temperatures above 250 °C. Xylene was utilized as a cosolvent in neutral hydrolysis, which reduced the required temperature, pressure, and volume of water in the reaction. Additionally, this strategy enabled the efficient isolation of EG as a concentrated solution in the organic layer.690
Zhang et al.691 used a dual phase-transfer catalyst [(CH3)3N(C16H33)]3[PW12O40] in the neutral hydrolysis of post-consumer bottle PET at 145 °C for 3 hours to obtain TPA yields of up to 93%. The catalyst allowed the reaction to be run at much lower temperatures and could be separated from the products and recycled at least three times without any observed decrease in efficiency. Stanica-Ezeanu and Matei692 found marine water to be an excellent solvent for neutral PET hydrolysis due to the presence of various metal ions that can act as catalysts. They used simple salts commonly found in the ocean, including NaCl, KHCO3, and CaCl2, as catalysts in the depolymerization of PET at 200 °C and 40 bar to obtain TPA in up to 85% isolated yield. Yields were increased at milder conditions when more than one salt was added or when marine water was used as a catalyst.
9.1.2.3 Acidic hydrolysis. Several systems have been described for the acidic hydrolysis of PET in concentrated (>50 wt%) H2SO4 solutions to generate high purity TPA in high yields at ambient pressures and relatively low temperatures (Fig. 30c). However, these reactions are highly impractical at scale due to the corrosivity of the solution, the need to recycle large volumes of acid, the generation of stoichiometric salt waste, and the difficulty in isolating EG from the reaction.672,681,693 Yoshioka and coworkers694 discovered that they could decrease the H2SO4 concentration by about half and still achieve complete hydrolysis of PET to TPA in 5 hours at 150 °C. The acid was subsequently recovered by dialysis. The same group also reported that 7–13 M HNO3 completely hydrolyzed PET over about 2 days at temperatures below 100 °C. The EG was simultaneously oxidized to oxalic acid, a value-added product.695
Liu et al.696 studied the hydrolysis of PET using the ionic liquid (IL) 1-butyl-3-methylimidazolium (Bmim) chloride as the solvent and an acid-functionalized IL (1-methyl-3-(3-sulfopropyl)-imidazolium hydrogen sulfate) as a catalyst. After 4.5 hours at 170 °C, they isolated TPA in 88% yield, and were able to recycle the ILs at least 8 times with no decrease in yield. Yang and coworkers found that TPA itself was an effective catalyst for acidic PET hydrolysis. They achieved a 95.5% yield of high purity TPA after 3 hours at 220 °C. The only workup required was a simple filtration, and the hydrolysis efficiency was maintained over 8 consecutive reaction cycles.697
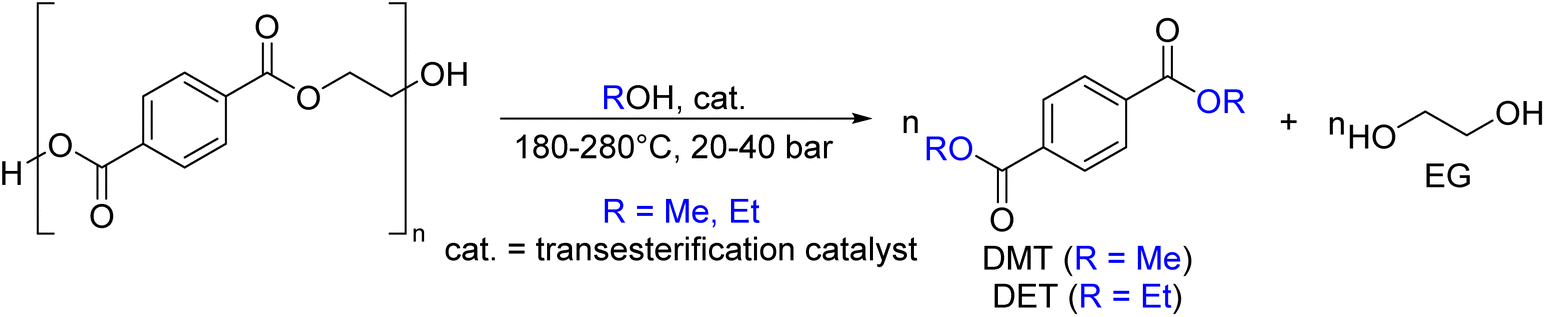 | ||
| Fig. 31 Typical reaction conditions and products for PET alcoholysis. | ||
9.1.3.1 Methanolysis. Methanolysis of PET produces dimethylterephthalate (DMT) and EG. DMT can be used directly in transesterification polymerization to form PET or further hydrolyzed to TPA.672,680,698 However, methanolysis of post-consumer PET leads to high separation and purification costs due to the mixture of glycols, alcohols, and phthalate derivatives produced in the reaction.663 Methanolysis typically operates at temperatures of 180–280 °C and pressures of 20–40 bar. The high pressures ensure that the methanol remains a liquid during the reaction. Transesterification catalysts such as zinc acetate, magnesium acetate, cobalt acetate, or lead dioxide are typically used, and yields of DMT average 80–85%. This reaction can be run in continuous flow, but complex reactors are required to add feedstocks while maintaining the required high reaction pressures. In a typical continuous method, melted PET and preheated methanol are fed into an autoclave with a set residence time to complete the depolymerization, then the mixture is flowed to the bottom of a second pressurized reactor at a lower temperature so that high density impurities can be removed. After leaving the second autoclave the reaction pressure is reduced, the product mixture is further cooled in a mixer, and finally DMT is isolated by precipitation.672,680,699
Sako and coworkers700 reported in 1997 that PET could be completely depolymerized to DMT, EG, and some oligomers with no catalyst in only 30 minutes in supercritical methanol. This was achieved at temperatures above 300 °C and pressures of 110 bar. Above supercritical conditions, the pressure of the reaction was not found to have a significant effect on DMT yield, however the severe conditions make this process too cost intensive for large scale use and too difficult to adapt to continuous flow.680
Kurokawa et al.698 used the transesterification catalyst Al(OiPr)3 to promote the methanolysis of PET from soda bottles under more mild conditions. They achieved 88% DMT and 87% EG yields in 4:1 methanol:toluene at 200 °C and no added pressure. Improved yields in this solvent mixture compared to pure methanol indicate that the rate of methanolysis strongly depends on PET solubility. Hofmann and coworkers701 found that microwave irradiation facilitated the methanolysis of PET under even milder conditions using zinc acetate as a catalyst and CH2Cl2 as a cosolvent to improve solubility. In only 20 minutes at 160 °C, moderate to high yields of DMT were isolated from a variety of post-consumer PET sources, including a dish detergent bottle (46% yield), a soap dispenser with a label (70% yield), and a transparent food container (98% yield).
Pham and Cho702 reported that the inexpensive salt K2CO3 catalyzes PET methanolysis at room temperature. A 93% yield of DMT was obtained after 24 hours in a mixed solvent system of 1:1 methanol:CH2Cl2. Tanaka and coworkers703 developed a new strategy wherein they trapped the EG formed in the reaction as ethylene carbonate. This biased the depolymerization equilibrium towards the monomers and enabled the reaction to run in only 5 hours at ambient temperature. Various simple alkali salts were used as catalysts, and optimized yields of DMT exceeded 90% using 5 mol% LiOMe.
9.1.3.2 Ethanolysis. PET ethanolysis yields diethylterephthalate (DET) and EG. The strategy is similar to methanolysis in many ways but could be particularly advantageous in countries such as Brazil where ethanol from sugarcane is cheap and abundant.704
In 2006, it was reported that PET could be completely depolymerized to its monomers in 5 hours in supercritical ethanol. At 528 °C and pressures of 76–116 bar, DET yields of 65–98% were obtained using post-consumer PET from a variety of sources including green bottles, mechanically recycled fiber, and polyester string.705 Fávaro and coworkers704 studied the ethanolysis of multilayer food packaging containing PET, PE, and aluminum. The material was depolymerized over 2 hours in supercritical ethanol at 255 °C and 116 bar. High purity DET was obtained in 80% yield after precipitation, and metallic aluminum could also be isolated from the reaction. Lozano-Martinez and coworkers found that the product distribution from PET ethanolysis could be changed by altering the reaction conditions. Long reaction times in supercritical ethanol generated DET as the only product, while shorter reaction times at lower temperatures and pressures led to the formation of TPA.706
Nunes et al.707 reported that the addition of catalytic [Bmim][BF4] reduced the required reaction time for PET depolymerization in supercritical ethanol from 6 hours to only 45 minutes. Cobalt and nickel oxides have also been found to be effective catalysts for PET ethanolysis under supercritical conditions, and provide almost quantitative yields of DET. The required reaction time was only 90 minutes with low catalyst loadings.708
:PET ratio on depolymerization kinetics. Higher pressures were reported to enable faster reactions, and at higher EG concentrations BHET was in equilibrium with PET oligomers. At lower EG concentrations, no monomer was observed.
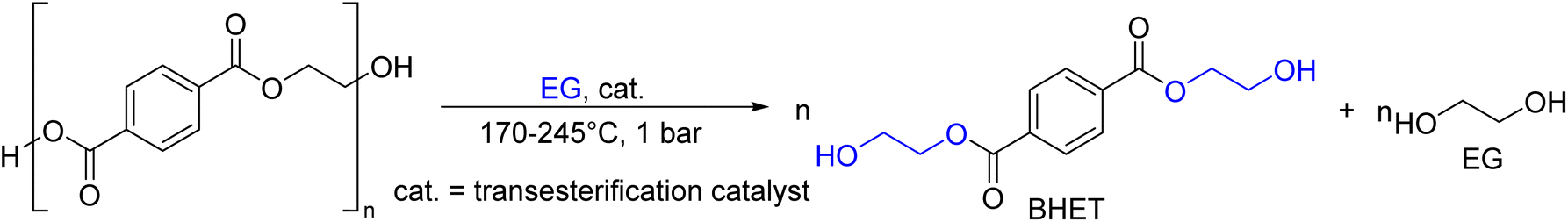 | ||
| Fig. 32 Typical reaction conditions and products for PET glycolysis with EG. | ||
Metal acetates were first reported as PET glycolysis catalysts in 1989 and have been widely studied.709,711,712 Güçlü and coworkers713 used xylene to form a multiphase reaction in zinc acetate-catalyzed PET glycolysis with EG at temperatures of 170–245 °C. BHET was pulled into the xylene layer to bias the equilibrium of the depolymerization and prevent oligomer formation, providing monomer in up to 80% yield. Other diols such as butanediol and triethylene glycol have also been used in PET glycolysis with zinc acetate to generate monomers and dimers.714 Chen found that the optimal PET glycolysis temperature was 190 °C when using manganese acetate as a catalyst. Conversion of PET to BHET and dimer reached ∼100% after 1.5 hours.715 Ghaemy and Mossaddegh716 used various metal acetate catalysts in the glycolysis of PET in refluxing EG. Yields of BHET reached up to 85% from both fiber and flake PET.
Troev and coworkers717 demonstrated that light metal salts could also be used as glycolysis catalysts. They reported that titanium(IV) phosphate catalyzes the depolymerization of PET to BHET in up to 97% selectivity at 190 °C with a reaction time of 2.5 hours. Fang et al.718 utilized polyoxometalates as catalysts for PET glycolysis under mild conditions. BHET was isolated in 85% yield after only 40 minutes at 190 °C and 0.018 mol% catalyst loading, with no loss in activity observed after the catalyst was recycled four times.
ILs have gained interest in the literature as catalysts for PET glycolysis due to the ease of separating the products from the reaction mixture, enabling facile catalyst recycling.719–721 H. Wang et al.719 first published the use of ILs as catalysts for PET glycolysis in 2009. They synthesized and tested several different ILs as catalysts and were able to achieve full conversion of PET to primarily BHET and some oligomers at 180 °C in 8 hours at atmospheric pressure. Higher reaction temperatures led to improved BHET selectivity. A subsequent study found that the IL could be recycled with no loss in activity.722 They then found that an iron-containing IL could catalyze the depolymerization at temperatures as low as 140 °C.723 Q. Wang720 and coworkers studied different metal-containing ILs as PET glycolysis catalysts. They achieved isolated yields of up to 80% high purity BHET after 75 minutes at 175 °C and atmospheric pressure. Other metallic ILs have been shown to be effective catalysts for this reaction under a variety of conditions.724 Yue et al.725 used basic ILs at catalysts in PET glycolysis to isolate BHET in yields of up to 71% after 2 hours at 190 °C.
Deep eutectic solvents (DESs) have also garnered recent interest as catalysts for PET glycolysis due to their low cost, simple synthesis, low toxicity, and other characteristics shared with ILs such as thermostability and tunability.726,727 Q. Wang and coworkers first used DESs as catalysts for PET glycolysis in 2015. Under optimized conditions of 170 °C and only 30 minutes, BHET was obtained in 83% yield. These results are similar to those obtained under supercritical conditions. The high activity is attributed to a synergistic effect between H-bonds and coordination bonds between the DES and EG. Sert et al.728 synthesized five different DESs to evaluate as PET glycolysis catalysts. The best catalyst tested was formed from potassium carbonate and EG, and reached isolated BHET yields of up to 88% after 2 hours at 180 °C.
A variety of different heterogeneous catalysts have also been used in the glycolysis of PET. Shukla and coworkers729 used β- and γ-zeolites as catalysts for PET glycolysis in 2008. BHET yields of over 60% were reached after 8 hours in refluxing EG. Al-Sabagh et al.730 used multiwalled carbon nanotubes doped with Fe3O4 as catalysts for PET glycolysis, and reported quantitative BHET yields after 2 hours at 190 °C. The catalyst was successfully recycled 8 times with no loss in activity. Veregue et al.731 used 3 nm cobalt nanoparticles as catalysts, and isolated pure BHET without additional water in 77% yield after 3 hours at 180 °C. The catalyst could be reused at least 5 times with no drop in activity. Laldinpuii and coworkers732 used bamboo leaf ash as a bio-waste derived catalyst for PET glycolysis. Several metal oxides and other salts were identified in the catalyst, which provided BHET in up to 83% yield after 3 hours at 190 °C. The catalyst could be reused four times with only a slight decrease in yield, and EG was also recovered from the reactions for reuse. Z. Wang et al.733 developed a colloidal catalyst based on graphitic carbon nitride for this reaction. 80% BHET yield was achieved in 30 minutes at 196 °C for five consecutive catalyst cycles. Several different post-consumer PET sources were successfully depolymerized to give BHET in ∼80% yield, including green bottle flake, noise deadening foam, and bottle flake contaminated with PP.
IL catalysts have been tethered to heterogenous supports to enable even more facile catalyst recovery and product isolation. Najafi-Shoa734 and coworkers used a cobalt-containing IL supported on graphene to reach 95% isolated BHET yields after 3 hours at 190 °C and ambient pressure. The catalytic activity was maintained over five cycles. Al-Sabagh et al.735 supported iron-containing ILs on bentonite to reach a maximum 44% BHET yield. Similarly, Cano et al. supported iron-containing ILs on silica-coated, magnetic Fe3O4 nanoparticles. The recoverable catalyst gave quantitative BHET yields for an impressive twelve consecutive 24 hours cycles at 190 °C, and the magnetic particles greatly simplified reaction workup.736
Organocatalytic PET glycolysis was first reported in 2011 by Fukushima et al.737 using the amine base 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD). Isolated BHET yields of up to 78% were reached after 3.5 hours at 190 °C and atmospheric pressure, comparable to some of the most efficient metal-catalyzed reactions. A study of several other nitrogen-based catalysts led to the discovery that 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) was an even more efficient catalyst when EG was used as the glycol. Mechanistic studies revealed that EG and other short chain diols can act as cocatalysts to activate the PET ester group and assist in depolymerization (Fig. 33).738 Jehanno and coworkers739 used a salt derived from TBD and methanesulfonic acid as a catalyst in PET glycolysis. 91% BHET was isolated after 2 hours at 180 °C. The catalyst could be recycled at least five times, and the monomer isolated from these reactions was used to synthesize new PET with similar thermal properties as commercial material.
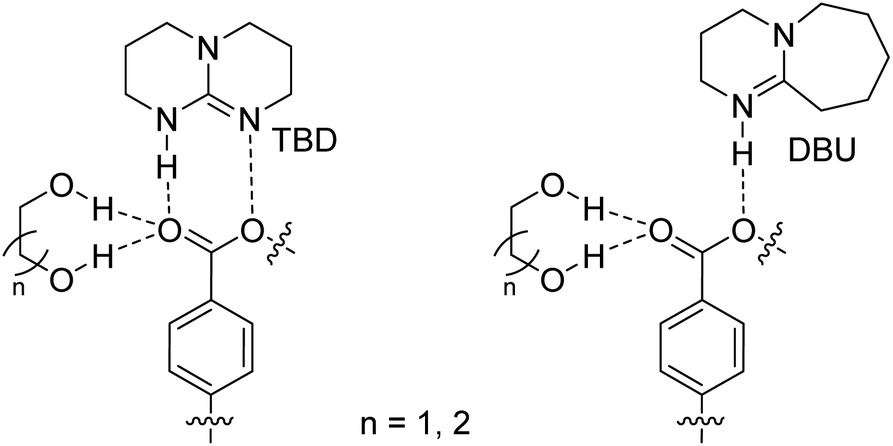 | ||
| Fig. 33 Proposed mechanism of PET activation by the organic bases TBD or DBU and short chain alcohols. Longer chain alcohols cannot participate in this reaction. | ||
Microwave irradiation is another growing strategy for facilitating PET glycolysis at milder conditions than conventional heating. Pingale and Shukla740 first published in 2008 that, under otherwise identical zinc acetate-catalyzed glycolysis conditions, microwave irradiation provided the same yield of BHET in 35 minutes that conventional heating did in 8 hours. This increased efficiency was confirmed by Chaudhary et al.741 Achilias et al.742 studied the microwave glycolysis of PET to oligomers using diethylene glycol. They found that complete degradation was achieved in only 5 minutes at 180 °C, compared to more than 4 hours using conventional heating. Saravari et al.743 glycolyzed PET bottles with propylene glycol under microwave irradiation with zinc acetate as a catalyst. Oligomers obtained from the reaction were reacted with linseed oil and toluene diisocyanate to obtain urethane oil with similar properties to commercially purchased material, showing the potential of this strategy for PET upcycling. Parrott744 received a patent for microwave-assisted PET glycolysis in 2020. BHET yields of up to 94% were achieved in only 5 minutes of irradiation at 250 °C with 0.1 wt% zinc acetate as a catalyst. A variety of simple salt catalysts were investigated, and zinc salts were generally the most effective.
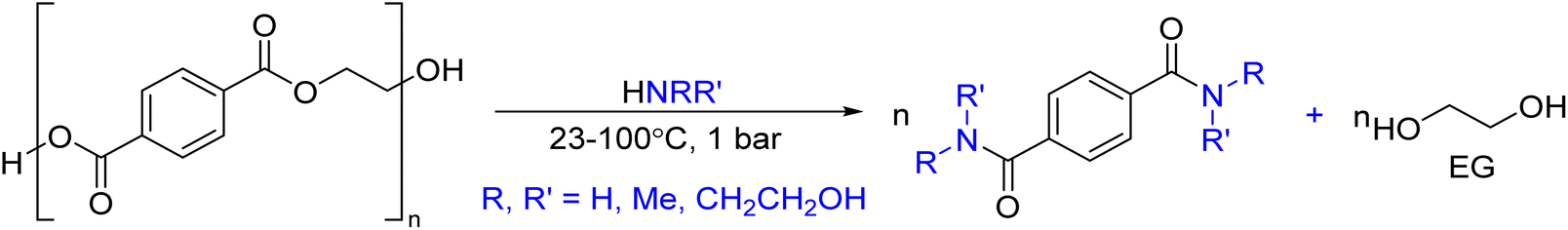 | ||
| Fig. 34 Typical reaction conditions and products for PET aminolysis. | ||
Soni and Singh reported the aminolysis of PET in the presence of aqueous methylamine or ammonia using cetyl ammonium bromide as a catalyst in 2005. They found that the catalyst reduced the required time for complete degradation to N,N′-dimethylterephthalamide to 45 days at 40 °C, while large PET flakes were still visible in the uncatalyzed reaction after the same time.
Ammonolysis of PET was much slower, and full depolymerization was not achieved under any tested conditions.749 In 2006, Shukla and Harad750 reported the aminolysis of PET using excess ethanolamine and simple catalysts including acetic acid, sodium acetate, and potassium sulfate. The product bis(2-hydroxy ethylene)terephthalamide (BHETA) was obtained in yields of up to 91% from post-consumer PET and 83% from bottle PET after 8 hours at reflux. Sodium acetate was found to give the highest yields in all reactions.
Tawfik and Eskander751 used dibutyltin oxide as a catalyst in the aminolysis of PET with ethanolamine at 190 °C and ambient pressure. Pure BHETA was obtained in yields of 49–62% after 1–4 hours. Chan and coworkers752 used zinc acetate as a catalyst to depolymerize post-consumer bottle PET in the presence of tri- and tetraamines. The reactions were complete in 30 minutes at 190 °C, and no purification of the polyamine products was necessary before the authors used them in crosslinking reactions with ethylene glycol diglycidyl ether to form hydrogels with potential uses in removing industrial dyes from water and other environmental and agricultural applications (Fig. 35).
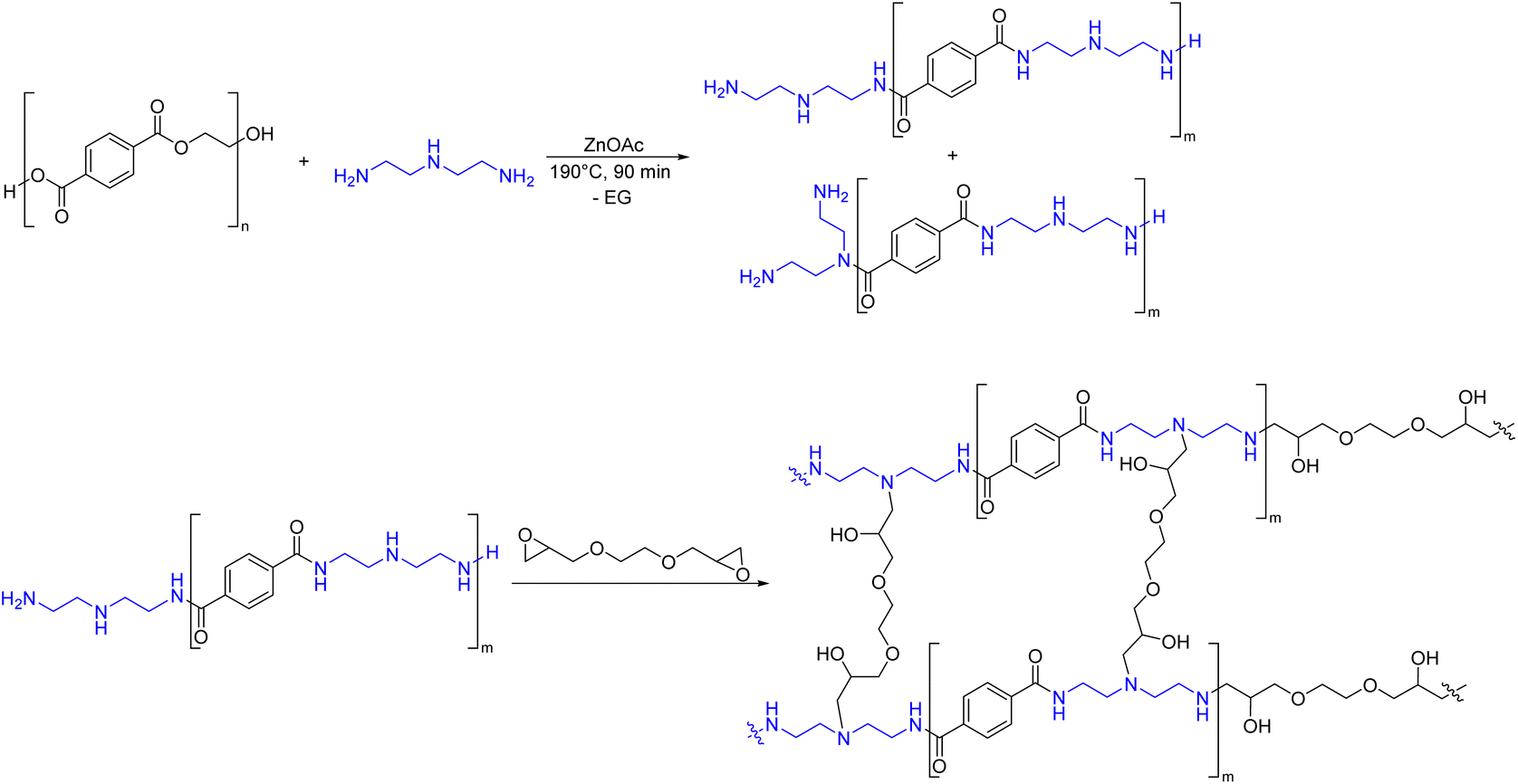 | ||
| Fig. 35 Aminolysis of PET using a polyamine, followed by a crosslinking reaction to form a PET hydrogel. | ||
Fukushima and coworkers745 used TBD as a catalyst in PET aminolysis with a variety of primary amines. The terephthalamide products were obtained in moderate to high yields at reaction temperatures of 110–190 °C in less than 24 hours. The monomers have potential applications as building blocks for high performance materials with desirable thermal and mechanical properties. The same group used this catalytic system to depolymerize PET with o-phenylenediamine and 2-aminophenol to generate bis-benzamidazole and bis-benzoxazole, which can be used in the synthesis of medicines and high-performance polymers.753
Microwave irradiation has also been studied in PET aminolysis. Pingale and Shukla754 reported the microwave-assisted aminolysis of PET using simple sodium salt catalysts and excess ethanolamine in 2009. BHETA yields of up to 90% were obtained in only 5 minutes under microwave irradiation, whereas similar yields required 8 hours using conventional heating. Cleaned post-consumer PET bottles and pristine PET fibers were both effectively depolymerized using this system. Achilias and coworkers755 utilized microwave heating to quantitatively depolymerize PET to BHETA in only 5 minutes at 260 °C without the addition of a catalyst.756 Hoang and Dang reported the uncatalyzed aminolysis of PET to bis(2-aminoethyl)terephthalamide and its oligomers α,ω-aminoligo(ethylene terephthalamide), but Sharma et al.757 found that the same reaction could be carried out in only 10 minutes at 250 °C using microwave irradiation. They then used the obtained amides to synthesize bis-benzoxazines, which can be used as advanced performance thermosets.
The ammonolysis of PET involves the reaction of the monomer with excess ammonia to generate terephthalamide and EG as products. Terephthalamide can be further converted into platform chemicals p-xylenediamine or 1,4-bis(aminoethyl)cyclohexane.17 A patent from 1988 describes the ammonolysis of PET under 20 bar NH3 pressure at temperatures of 120–180 °C for 1–7 hours. A simple filtration and drying workup provided the product in >99% purity and >90% yield. The same patent describes the use of zinc acetate as a catalyst to facilitate the transformation at only 70 °C and provide terephthalamide in 87% yield.17
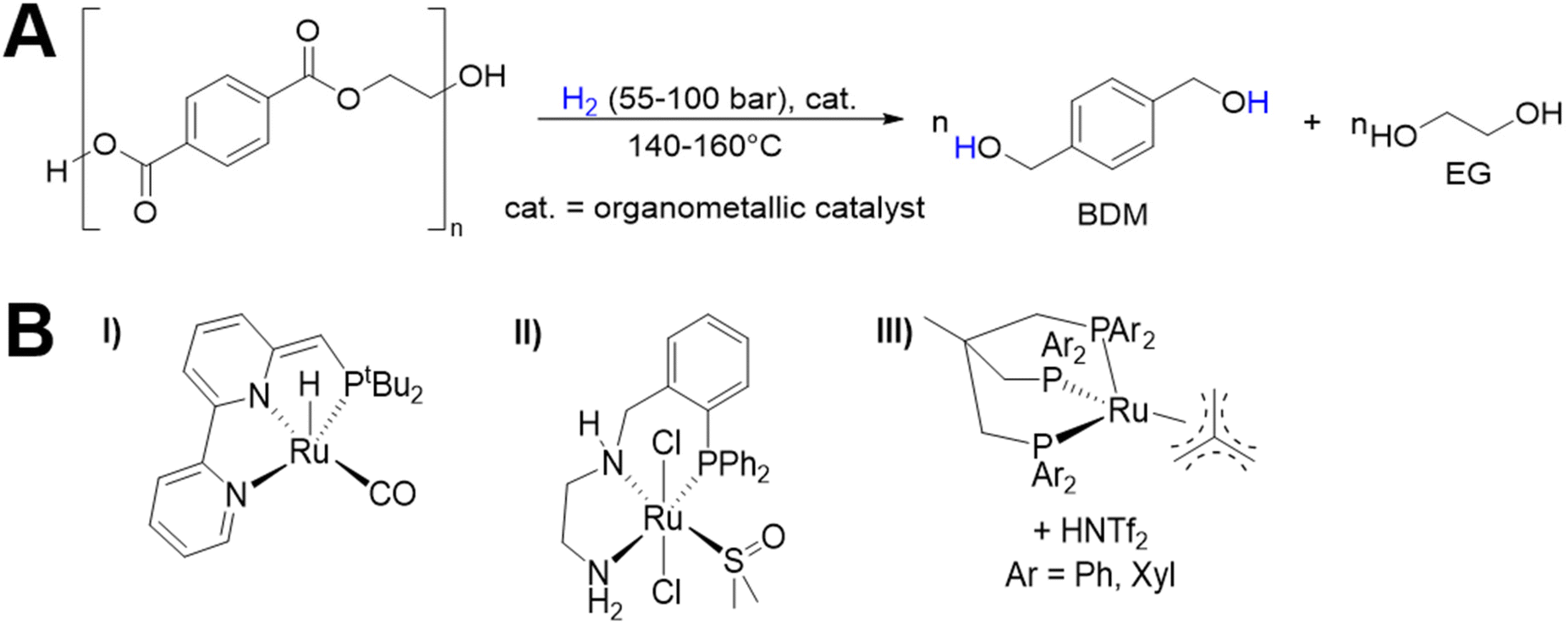 | ||
| Fig. 36 (A) Typical reaction conditions and products for PET hydrogenolysis and (B) organometallic catalysts used for PET hydrogenolysis by (I) Krall et al., (II) Fuentes et al., and (III) Westhues et al. | ||
In 2014, Krall and coworkers761 found that a ruthenium pincer complex developed by Milstein could be used as a catalyst in the hydrogenolysis of PET (Fig. 36B I). At 2 mol% catalyst loading, 55 bar H2, and 160 °C in anisole/THF, quantitative conversion to BDM and EG was observed after 48 hours. Post-consumer PET soda bottles were used in these experiments without pretreatment. Fuentes et al.762 found that a novel PNN pincer complex (Fig. 36B II) catalyzed PET hydrogenolysis with moderate BDM yields, but the catalyst was poisoned by the EG formed in the reaction. This precluded monomer yields higher than 73%. Westhues et al.763 used a ruthenium triphos catalyst (Fig. 36B III) to depolymerize PET from a variety of sources, including water bottles, pillow filling, and a sports jersey. Reaction selectivity for BDM and EG were over 98% with most substrates, but relatively forcing conditions of 100 bar H2 and 140 °C were required. An entire PET water bottle with PP cap and PE label was depolymerized without pretreatment. Quantitative conversion to BDM was observed, with no conversion of PP or PE. The other polymers could be filtered off and subsequently depolymerized.
Supported metal catalysts have also been used in PET hydrogenolysis. In 2020, Kratish et al.764 selectively depolymerized PET to TPA and ethylene using a carbon-supported molybdenum-dioxo catalyst under only 1 bar H2. No solvent was required for the reaction, and TPA was obtained in 85% yield after 24 hours at 260 °C. The air and moisture stable catalyst could be recycled four times with no loss in activity, and the yield of TPA did not change when PET from a post-consumer beverage bottle was used as the substrate. Jing and coworkers765 used a Ru/Nb2O5 catalyst to convert PET to benzene, toluene, and p-xylene in 84% total yield. The reactions were run in octane at 280 °C and 5 bar H2 for 8 hours. The catalyst also successfully depolymerized a mixture of PET, polycarbonate (PC), PS, and polyphenylene oxide to a variety of aromatic monomers in 79% overall yields, showing the viability of this system in mixed waste streams. However, a 1:1 mass ratio of polymer to catalyst was required in this system. Wu et al.766 prepared an N-doped carbon-supported bimetallic catalyst for the conversion of PET to TPA in 91% yield in 10 hours at 260 °C and 1 bar H2. No solvent was used in the reaction, and the catalyst could be reused at least six times with no observed decrease in activity. Almost the same yield of TPA was achieved with PET from bottles and with a mixture of PET and PP from bottles.
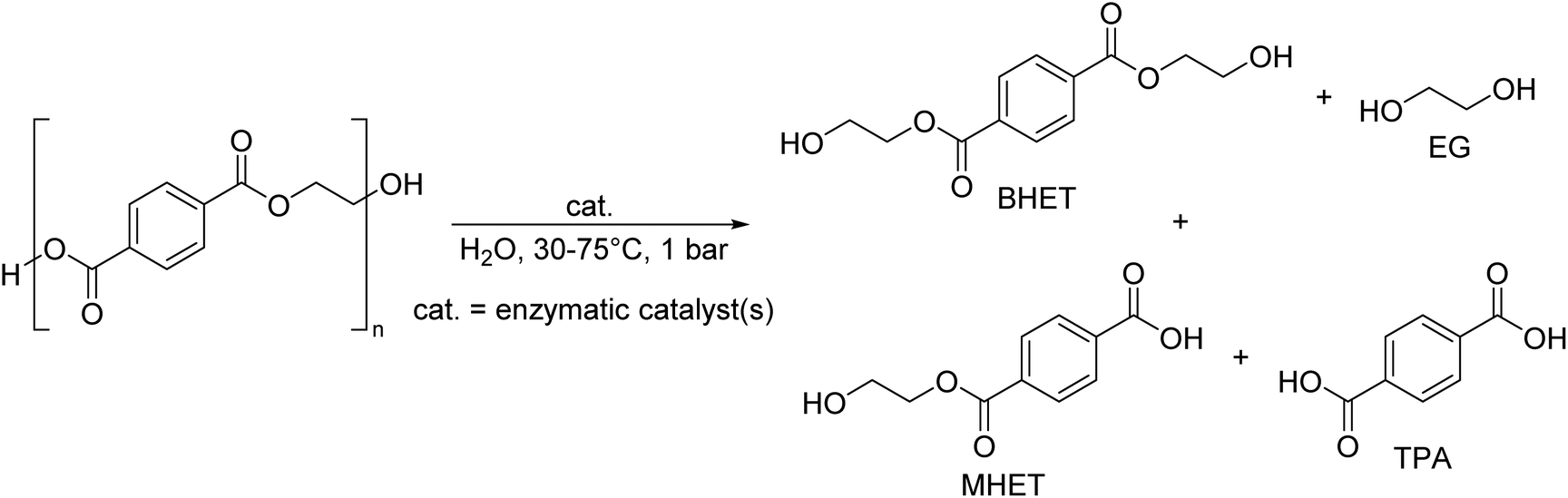 | ||
| Fig. 37 Typical reaction conditions and products for enzymatic PET depolymerization. | ||
The first report of enzymatic hydrolysis of PET was in 2005 from Müller et al.773 wherein they showed appreciable extents of PET conversion over 3 weeks at 55 °C using a cutinase enzyme from the thermophilic bacterium Thermobifida fusca. This study prompted a search in genome and metagenome databases for additional cutinases capable of PET hydrolysis. In addition to the discovery and characterization of homologous enzymes from the genus Thermobifida and closely related bacteria,774–782 Sulaiman et al. reported the discovery of a leaf-branch compost cutinase (LCC) that has been subsequently engineered by Tournier et al. for greater thermal stability and activity, enabling quantitative conversion of amorphous micronized PET substrates in 10 hours at 72 °C and enzyme loadings concomitant with industrial performance.783–785 Shirke et al.786 demonstrated that the addition of glycosylation to the LCC enzyme via expression in Pichia pastoris results in greater stabilization of the enzyme, thus highlighting the potential for post-translational modifications as another means of enzyme stabilization and engineering for industrial applicability. A similar enzyme to LCC was recently reported by Sonnendecker et al.787 that is able to achieve similarly high PET conversion with no substrate pretreatment. Additional studies to diversify the known PET hydrolytic enzyme suite will likely be enabled by computational methods and the massive number of genome and metagenome sequences that are continuously being reported.788,789
The seminal discovery of Ideonella sakaiensis by Yoshida and coworkers790 in 2016 highlighted that natural microbial systems are seemingly responding to the presence of PET in the natural environment. In particular, I. sakaiensis secretes a two-enzyme system, comprising the enzymes PETase and MHETase, to fully depolymerize PET into TPA and EG via the intermediates BHET and MHET. This two-enzyme system has been extremely well studied to date, with structural biology techniques791–797 and many types of protein engineering and evolution approaches used to improve the stability and performance particularly of the PETase enzyme.796,798,799 The I. sakaiensis system also points to the potential utility of an enzyme cocktail for industrial application of an enzymatic recycling approach, as is common in industrial cellulose deconstruction, especially in this case to overcome product inhibition due to the buildup of MHET during enzymatic hydrolysis.783,800
Singh et al. recently conducted a detailed techno-economic, energy, and GHG emissions analysis of enzymatic PET recycling using the patent literature and the work from Tournier et al.785,800 Therein, they estimated that with low-cost PET feedstocks, enzymatic recycling processes could achieve cost parity with virgin PET manufacturing at substantially reduced energy inputs and GHG emissions. This work directly highlighted two areas of prominence that merit further attention in process development for enzymatic PET recycling, which also may translate to other PET chemical recycling approaches discussed herein. Namely, it was estimated that approximately half of the energy input and GHG emissions arise from substrate pretreatment, which was modeled as thermal extrusion and cryo-grinding to yield micronized, amorphous PET. The development of enzymatic systems that can deconstruct crystalline substrates, which has been highlighted by studies as a major need for the field,787,799,801,802 would thus be a major step forward for this approach. Secondly, EG recovery from water was estimated to roughly equate to the other half of energy use and GHG emissions. Advanced approaches to separations for EG that operate in the condensed phase or employ reactive distillation may be of use to further improve this process and will likely be relevant for EG recovery in chemical solvolysis reactions as well.
9.2 Chemical recycling of polycarbonates and other polyester feedstocks
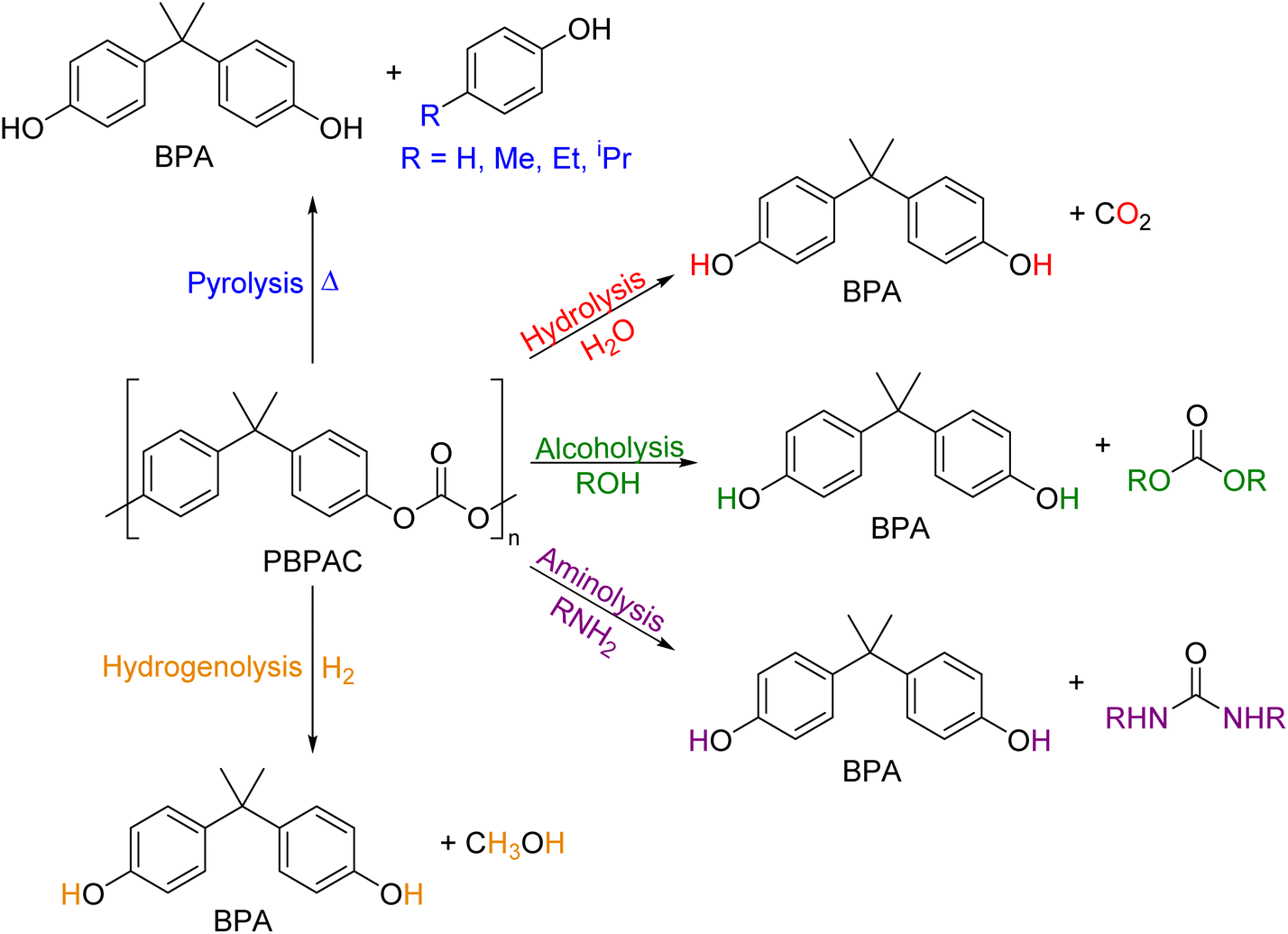 | ||
| Fig. 38 Methods for the chemical depolymerization of PBPAC and their primary monomeric products. | ||
PBPAC pyrolysis produces a large amount of char due to the flame retardancy of the polymer. Uncatalyzed pyrolysis at 300–500 °C leads to the generation of BPA and phenolic compounds as the primary liquid products, leaving behind a residue which accounts for 20–30 wt% of the starting material.804 Metal chloride or oxide catalysts such as CuCl2, FeCl3, ZnCl2, SnCl2, CaO, and MgO have been shown to lower the temperatures required for pyrolysis, narrow the product distribution, and decrease the amount of char waste formed in the reaction.484,488,807,808 Due to the hydrophobicity of PBPAC, hydrolysis of this polymer requires steam or supercritical conditions.805 At temperatures above 240 °C, BPA and CO2 are the primary products of PBPAC hydrolysis.809–811 Alkaline earth metal oxides and hydroxides, alkali hydroxides and carbonates, and rare earth triflates all catalyze PBPAC hydrolysis, enabling milder reaction conditions and higher BPA yields especially when an organic cosolvent such as 1,4-dioxane is used.812–818 Similar to hydrolysis of PET, ionic liquid catalysts819–821 and microwave heating822,823 also facilitate this reaction.
To our knowledge, PBPAC alcoholysis was first reported with methanol, NaOH catalyst, and toluene cosolvent to swell the polymer and obtain BPA and methylene carbonate as products.824 The reaction has since been adapted to continuous flow,825 accelerated using microwave heating,823 and run under supercritical conditions.826–828 Alkali halide, heterogeneous, ionic liquid, and organic base catalysts have also been employed to increase yield and selectivity for BPA under milder conditions.829–837 The aminolysis of PCs provides value-added ureas and BPA as products. The yield of monomeric products is increased when a diamine is used to form a stable cyclic urea.838,839 An ionic liquid/ZnO nanoparticle catalyst system provides excellent BPA and urea yields with a variety of mono- and diamines.840 Ammonolysis of PBPAC in an aqueous ammonia solution provides BPA and urea in high yields.841,842 Finally, many of the catalysts studied in PET hydrogenolysis also successfully depolymerize PCs under similar conditions to generate the corresponding alcohol monomer and methanol in high yields.761,763,765
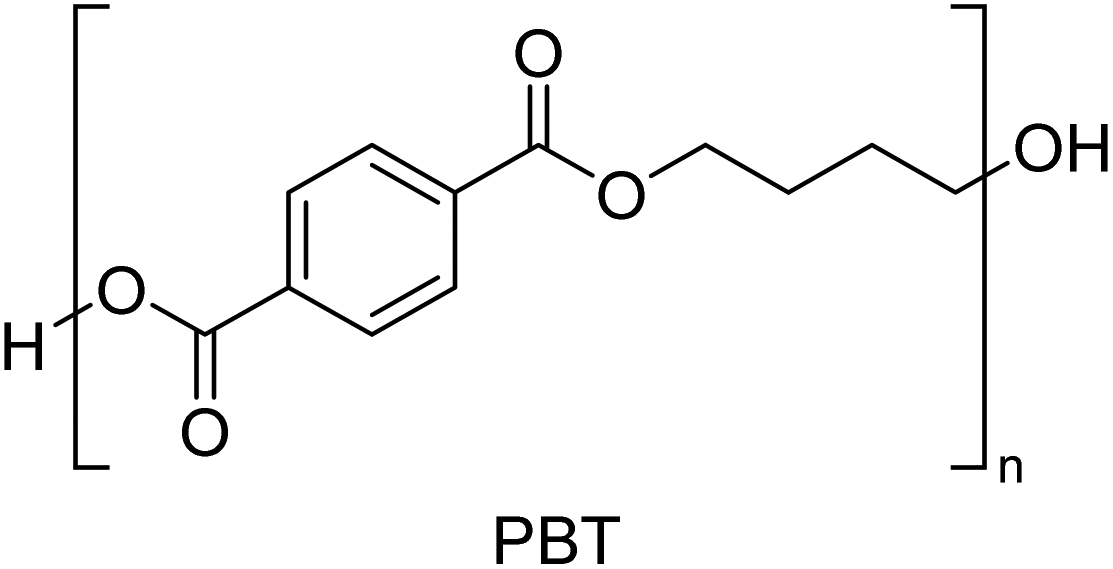 | ||
| Fig. 39 Chemical structure of PBT. | ||
PLA is a widely used, industrially compostable bioplastic derived from the fermentation of renewable feedstocks.668 PLA has good mechanical properties but a low glass transition temperature, narrow temperature-processing window, and undesirable brittleness.667 Depolymerization of PLA to the lactide monomer is not possible due to side reactions including elimination to acrylic acid and epimerization.844,845 The primary method used for the deconstruction of PLA on the lab scale is hydrolysis to lactic acid (Fig. 40).668 Additionally, some catalysts used in the hydrogenolysis of PET are capable of reductively depolymerizing PLA to form propylene glycol in high yields.761,763
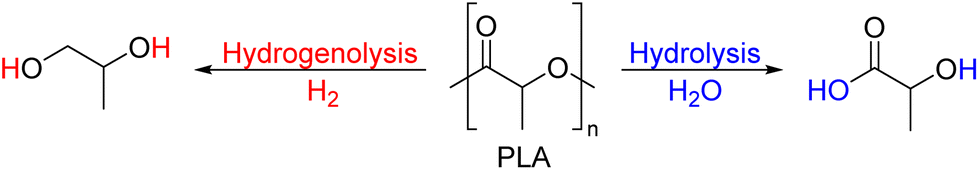 | ||
| Fig. 40 Hydrolysis and hydrogenolysis of PLA to form lactic acid and propylene glycol, respectively. | ||
9.3 Commercial activity and economics
Much of the current commercial progress in PET chemical recycling is not captured in scientific publications, with patents and press announcements typically constituting the public information in this field. A recent review of advanced PET chemical recycling focuses primarily on the development of commercial processes.666 At the time of publication, commercial chemical PET recycling has not yet been realized. However, many commercialization projects exist using different depolymerization chemistries and subsequent downstream purification processes.Mechanical recycling of PET is complementary to chemical recycling processes, and both are needed to achieve a high degree of circularity in PET applications. Chemical recycling feedstocks will comprise primarily colored and/or opaque plastic bottles and polyester-based textiles that are unsuitable for the high purity standards required in mechanical recycling.662,664 The availability of a high volume, low cost feedstock is critical for commercial process viability, which will require processing highly impure PET feedstocks to produce high purity, polymerization-grade monomers at scale. The quality of the output of rPET from chemical recycling is higher than that from mechanically recycled PET. This allows the rPET from chemical recycling facilities to be infinitely recyclable, whereas the rPET produced from mechanical recycling degrades during each cycle.
The commercial activity described below is in the research and development-to-pilot stage unless otherwise indicated. To our knowledge, hydrolysis, methanolysis, glycolysis, and enzymatic depolymerization are the current primary areas of commercial focus, but it is likely that additional technologies will soon emerge on the commercial landscape. The intention of the processes described here is the transformation of waste PET plastic and textiles to monomers for subsequent repolymerization to rPET with physical and mechanical properties identical to those of pristine material.
10. Other catalytic approaches for plastics conversion
10.1 Hydrogenolysis with noble metals
Hydrogenolysis of plastics cleaves the carbon–carbon bonds in the presence of hydrogen converting the polymers into hydrocarbons (ranging from C1 to C50) typically with noble metal (e.g., Ru, Pt etc.) catalysts. In such processes, heterogeneous catalysts are used which enables simple separation and recycling from the reactants and products.Table 18 summarizes different technologies utilized in hydrogenolysis of waste plastics where plastics were converted in batch reactors. Ruthenium supported on metal oxides or carbon has been reported to catalyze the production of alkanes, aromatics, and liquid fuels from polyolefins (i.e., LDPE, HDPE, PP), PS, and PC.863–869 LDPE, HDPE, and PP can be converted to liquid fuel (C5–C21) and lubricants/waxes (C22–C45) at low temperature and low H2 pressure (i.e., 200–250 °C, 20–30 bar) on metal oxides-supported Ru (e.g., Ru/TiO2, Ru/CeO2).863,869 Over multifunctional Ru/Nb2O5, monocyclic aromatics can be selectively produced from single or mixed aromatic plastics (i.e., PET, PC, PS, polyphenylene oxide) at 200–320 °C in the presence of hydrogen and solvent (e.g., water, octane etc.).864 Ru hydrogenation sites with low coordination numbers (i.e., 5–6) prevent the over-hydrogenating of the aromatics, while the surface Lewis acid and Brønsted acid sites on NbOx species act in concert to selectively adsorb, activate C–O bonds, and cleave C–C bonds of the plastics. The support can be adjusted to treat different plastics and obtain varied products.865–868 Over Ru/WO3/ZrO2, LDPE can be converted into higher molecular weight fuels and wax/lubricant base-oils at 250 °C and 50 bar H2.868 While on Ru/FAU, methane (>97%) can be produced from PE, PP, and PS under 50 bar H2 at 300–350 °C.867 Ru nanoparticles can also be supported on carbon (Ru/C) to catalyze the hydrogenolysis of LDPE, HDPE, and PP towards liquid n-alkanes and alkane gases (C1–C6) under mild conditions (200–250 °C, 20–50 bar H2).865,866 In the liquid phase (water, n-hexane etc.), around 90 wt% of HDPE can be converted to liquid hydrocarbons (C8–C38) within 1 h on Ru/C catalysts at 220 °C in the presence of H2 (30 bar).870
| Catalyst | Polymer | Temperature (°C) | Pressure (Bar) | Time (h) | Polymer/catalyst (Mass) | Products | Ref. |
|---|---|---|---|---|---|---|---|
| Ru/CeO2 | LDPE, HDPE, PP | 200–240 | 20–60 | 5–24 | 34 | Liquid fuels, waxes (C5–C45) | 863 |
| Ru/TiO2 | PP | 250 | 30 | 8–16 | 20–40 | Lubricants (C20–C60), C1–C2 gases | 869 |
| Ru/Nb2O5 | PET, PS, PC | 200–320 | 3–5 | 12–16 | 1–2 | Aromatic hydrocarbons | 864 |
| 5Ru/C | PE, LDPE | 200–225 | 20 | 16 | 25 | Liquid alkanes (C3–C13), light gases (C1–C6) | 865 |
| 5Ru/C | PP | 250 | 35 | 8–24 | 14 | Liquid alkanes (C5–C32), light gases (C1–C5) | 866 |
| Ru/FAU | LDPE, PP | 300–350 | 50 | 3 | 50 | Methane, light paraffins (C2–C11) | 867 |
| Ru/WO3/ZrO2 | LDPE | 250 | 30 | 2 | 40 | Lubricants, waxes, diesel (C4–C35) | 868 |
| Ru/C | HDPE | 220 | 30 | 1 | 2 | Lubricants, liquid fuels (C6–C38) | 870 |
| Pt/WO3/ZrO2 + zeolite | LDPE | 250 | 30 | 2 | 10 | Liquid fuels (C5–C22) | 871 |
| Pt/SrTiO3 | PE | 300 | 12 | 96 | 5 | Lubricants, waxes (Mw 200–1000 Da) | 872 |
| SiO2/Pt/SiO2 | HDPE | 300 | 14 | 24 | 88 | Fuels, lubricants (C8–C32) | 873 |
| Pt/C | PP | 300 | 15 | 24 | 10 | Lubricants (C5–C45) | 874 |
Pt-Based catalysts are another class of materials utilized in the hydrogenolysis of plastics.871–874 It has been reported that PE can be converted into lubricants and waxes (Mw 200–1000 Da) over platinum supported on strontium titanate or mesoporous shell/Pt/silica at 250–300 °C in the presence of H2 (10–17 bar).872,873 Moreover, platinum can be supported on carbon which catalyzes the conversion of PP into liquid hydrocarbons (C5–C45),874 or on WO3/ZrO2 and mixed with zeolites which catalyzes the production of liquid fuels and short-chain hydrocarbons from PE, PP or PS.871 The synergistic effects between platinum and supports (carbon/metal oxides) or co-catalysts (zeolites) are vital in these processes. Additionally, the hydrogenolysis of plastics can also be carried out over Al2O3 or SiO2 supported transition metals (Cr, Ni, Mo, Co, or Fe),875 micro–mesoporous zeolites,876 and other catalysts877 which are not discussed here.
10.2 Functionalization and reactive extrusion
Another strategy to valorize plastics is the addition of other chemical groups (e.g., aromatic ring, halogen, carboxylate, carbon–carbon unsaturated bond etc.) to these polymers via chemical methods (e.g., oxidation, halogenation, esterification, metathesis)878,879 as shown in Fig. 41. This approach is typically performed by reactive extrusion in an extruder. The products include polymers with new properties,880 liquid fuels,881 waxes,882 and platform molecules.883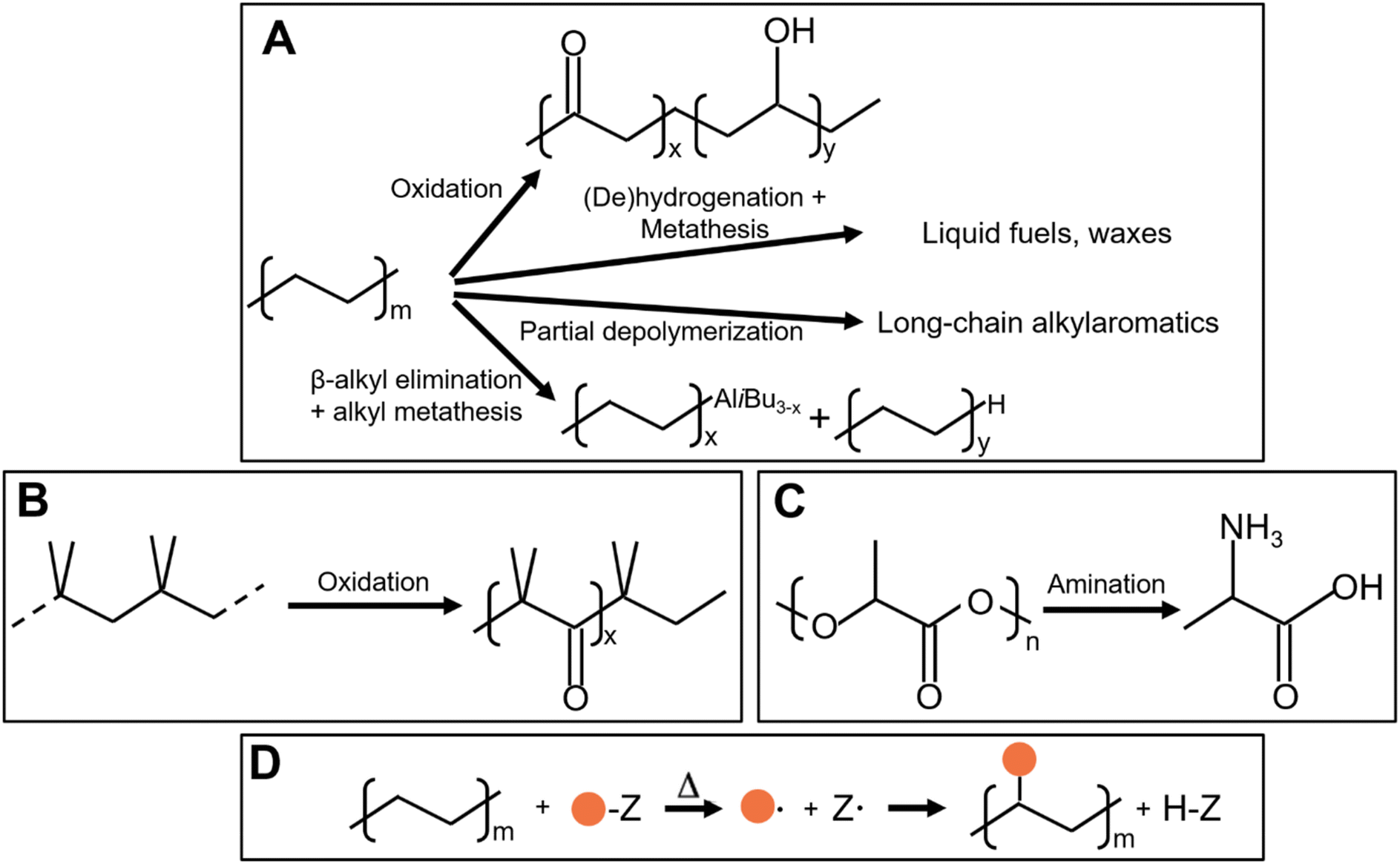 | ||
| Fig. 41 Strategies in functionalization of plastics: (A) functionalization of polyolefins; (B) oxidation of polyisobutene; (C) PLA amination; (D) reactive extrusion where orange circle represents a function group. | ||
The C–H bonds in PE (HDPE, LDPE, LLDPE) can be oxidized to ketone or hydroxyl groups, which generates functionalized polymers with unique physical properties (e.g., strong adhesion, ability to be painted with common paint etc.).880 A high-valent ruthenium-oxo catalyst (polyfluorinated ruthenium porphyrin) can be added with 2,6-dichloropyridine N-oxide to form esters from PE at 120 °C. The same complex can catalyze the oxidation of polyisobutene to oxo-polyisobutene (Fig. 41B) where the catalysts, reaction conditions, and oxidants are vital in selectively oxidizing the methylene positions and avoiding chain cleavage.884 Reactions besides oxidation can be utilized to functionalize the plastics. Kanbur et al. reported that the conversion of polyolefins (PE, PP) to shorter aliphatic alkylaluminium species can be achieved via β-alkyl elimination and heterobimetallic alkyl metathesis using organozirconium catalysts and aluminum reagents at 200 °C.885 The obtained shorter aliphatic alkylaluminium species can be further converted into alkyl alcohols, acids, halides, and hydrocarbons with a quenching process. Cross-alkane metathesis can convert PE into liquid fuels and waxes (short-chain linear alkanes) at 175 °C.881 This process consists of three steps: (1) the dehydrogenation of PE and cofed light alkane; (2) the metathesis of olefins; (3) the hydrogenation of the obtained olefins. An iridium complex can catalyze steps 1 and 3 while Re2O7/γ-Al2O3 catalyzes the olefin metathesis. The catalysts are compatible with various polymer additives and product distribution can be manipulated by the catalyst structure and reaction time.
Functionalized polymers can also be produced by dehydrogenation, sulfonylation, amination, and vitrimerization. Zinc sulfide is active in PP dehydrogenation886 while an iridium complex can catalyze the dehydration of PE and poly(1-hexane) in the p-xylene and norbornene mixture.887 For sulfonylation, it has been reported that PS can be sulfonated by chlorosulfonic acid, fuming/concentrated sulfuric acid, sulfur trioxide, and acetyl sulfate.888–891 Tian et al. reported that Ru/TiO2 can catalyze the amination of ether groups in PLA in ammonia solution at 140 °C (Fig. 41C).883 Alanine can be produced by this process with high selectivity in the absence of hydrogen. In most of the cases, other chemicals (e.g., light alkanes, water, halogens) need to be cofed for the functionalization of the waste plastics. However, the cofed species is not necessary if the carbon and hydrogen in the polymers can be utilized. One approach is to partially depolymerize the plastics. Long-chain alkylaromatics (ca. C30) can be produced from PE via controlled depolymerization on Pt/γ-Al2O3 without solvent or H2 at 280 °C.882 This process couples exothermic hydrogenolysis and endothermic aromatization which enables simultaneous C–C cleavage, ring closure, and dehydrogenation under mild conditions (280 °C). Vitrimerization is a process converting polymers to vitrimers, which behave like silica,892 are covalent networks and can change their topology by thermally activated bond-exchange reactions. Vitrimers can be produced from both thermoplastics (e.g., PBT893) and thermoset plastics (e.g. polyurethane894). The key step in vitrimerization is introducing covalent adaptable networks via addition of catalysts or cross-linkers. Although catalyst is not necessary for vitrimerization, the properties of a catalyst-containing vitrimer can be efficiently tuned.893,895
Reactive extrusion is a commercially practiced approach to install functionality of plastics.896 One example is the radical-mediated addition of maleic anhydride to PE and PP.897 One key step in reactive extrusion is the production of a pair of radicals which can be achieved through heating or photolysis of the reagent. One of the radicals cleaves a C–H bond in plastics and a chain transfer step can functionalize plastics (Fig. 41D). Thiocarbonyl amide reagent has been utilized to functionalize branched polyolefins.898 The authors demonstrated that this metal-free C–H functionalization of isotactic PP can be performed on a decagram scale forming polymers with improved adhesion to polar substrates. Recently, another study demonstrated that an ideal reagent (e.g., O-alkenylhydroxamate) enables slow chain transfer kinetics which provides various functionality to the backbone of plastics by other radical sources.899
The functionalization of waste polymers is being commercialized (e.g., Novoloop900). The company can recycle waste LDPE, HDPE, and LLDPE. Those PE wastes are shredded, cleaned, and fed into a patented, accelerated thermal oxidative decomposition (ATOD) reactor (Fig. 42).900,901 The treated PE is then heated to a temperature range from 60 to 200 °C, and oxidized into dicarboxylic acid monomers by a group of oxidation agents including oxygen, nitrous oxide, nitric oxide, and aqueous nitric acid.902 The oxidized monomers are used to produce thermoplastic polyurethane (TPU).
 | ||
| Fig. 42 The scheme of the ATOD reactor. (Extracted from the US patent US20220119616A1).902 | ||
Plastic alloys are a type of functionalize plastic where different plastic materials are functionalized to create a single plastic phase.903,904 The properties of the plastic alloy can be tuned by adding other materials (e.g., waste fibers) which enables applications of plastic alloys in automotive, electrical & electronics, construction, home appliances, and healthcare equipment.905–908 The advantages of plastic alloying are short reaction times, no need for solvents, and low operating/capital costs compared with other plastic technologies like gasification.909
Plastic alloying can introduce long chain branching of the polymer backbone to improve the properties (e.g. melt strength) of plastics. This approach has been used for functionalization of PP,909 PE,910 and polyhydroxybutyrate (PHB).906 Long chain branching can be added using peroxides, which can result in increases in melt elasticity.908 Long chain branching can alter physical properties such as the crystallinity and transition temperatures.911 The thermophysical properties including crystallinity, melting behavior and tensile properties of HDPE were shown to be influenced by di-tert butyl cumyl peroxide induced cross-linking.910 Dicumyl peroxide (DCP)-induced cross-linking in PE improves the melt strength and is dependent on peroxide concentration.912 The lifetime of the peroxide formed from DCP is relatively close to the residence time of extrusion. The generally accepted process of peroxide induced cross-linking of polymers follows three key steps:913
(1) The primary radical formation from the thermal decomposition of peroxide,
RO – OR → 2RO˙ (Primary radicals)
(2) H abstraction from polymer backbone by free radicals of peroxide generated in step (1),
RO˙ + P (polymer) → P˙ (polymer radical) + ROH
(3) the bimolecular radical recombination of polymer radicals from step (2) to generate C–C cross-links.
2P˙ → P–P (crosslinking)
There is limited information on the chemical position(s) associated with cross-linked molecular chains to form a gel network structure. Long chain branching increases the plastic molar mass, polydispersity, melt strength and strain hardening in PP.909 One key step in plastic alloying is C–H activation which can be achieved by different routes.2
Plastic alloying has been explained by inter-linking and interfacial adhesion between different polymers. Wigotsky pointed out the importance of plastic alloying to the plastic industry because of the improved physical properties of the alloys.904 The compatibilizing agents in plastics act as polymeric surfactants which lowers surface tension and promotes interfacial adhesion between polymer phases in plastic alloys.914 In addition to this, PP functionalization (i.e., carboxylation) generates compatibilizers which can be used to produce plastic alloys.915 Sakai reported a processing technology which can improve compatibility of plastic and produce plastic alloys using twin-screw extruders.916 Asay et al. reported that PVC can be utilized to produce plastic alloys because of the varied degree of miscibility between PVC and a wide range of polymers.917,918 Composite materials with improved properties can be obtained via addition of other materials. Kimura showed significant improvement in tensile strength by adding fillers to polyolefins.919 Amor et al. studied the improvements of properties of PLA with biomass, demonstrating synergetic effect of small addition of PLA on the thermo-mechanical properties.920 The above studies portray the importance of plastic alloying for the production of plastic and composite material products. However, these studies were all conducted with pure polymers by mixing two or three components. Aumnate et al. were the first to recognize that plastic waste could be also alloyed.903 In their fundamental study, they simulated plastic wastes by mixing pure components of LDPE and HDPE with PP and showed the effects of the branching of PEs on mechanical properties and crystallization behavior.
Another plastic alloying approach is the “grafting onto” method which grafts the PHB polymer onto cellulose fibers through plastic alloying processing with the use of a small amount of DCP.906,908 LDPE has also been shown to graft onto cellulose fibers, retaining the stiffness of cellulose and the flexibility of the polymer matrix.921 When the peroxide is exposed to heat during extrusion, it will decompose into strong free radicals which tend to abstract H's from the polymer and cellulose molecular chains and initiate the grafting between the two phases in composites.
10.3 Microwave-assisted conversion
Microwave radiation can be applied during catalysis to heat the reaction system, shorten reaction times, and enhance the kinetics of some chemical reactions.922 In conventional thermal heating processes, the whole reaction system (catalysts, reactor, diluent, feedstock etc.) are heated while microwave radiation enables the electromagnetic energy to be absorbed evenly and efficiently at the microwave-adsorbing points (e.g., catalyst surface).922–924 Furthermore, other researchers propose that the microwaves likely have intrinsic catalytic effects or assist in lowering the activation energy of the reactions.925Microwave-assisted depolymerization and catalytic upgrading can be separated by the reactor system.491,926 In microwave-assisted pyrolysis, LDPE is depolymerized into pyrolysis vapor and the microwave absorbent (e.g., SiC) enables even heating of the feedstock which promotes the formation of light olefins.491 The products are then catalyzed by MgO to produce more valuable chemicals (i.e., aromatics, C12+ hydrocarbons) in a subsequent reactor. This process can be modified by changing the microwave absorbent to NiO and the catalyst in the separated reactor to HY zeolite which enhances the production of olefins in the pyrolysis zone and produces gasoline fuel with high yield as well as appropriate carbon number.926 Microwave-assisted conversion can also be performed with catalysts in the liquid phase.927,928 LDPE powder can be completely degraded in 1 h of microwave irradiation at 180 °C in the presence of dilute nitric acid solution (0.1 g mL−1).927 In this process, water is used to absorb microwave energy and nitric acid catalyzes the oxidation of LDPE towards dicarboxylic acids (e.g., malonic acid, succinic acid, glutaric acid etc.). Bäckström et al.928 pointed out that HDPE can be converted to dicarboxylic acids with a similar approach and the obtained products can then be utilized to synthesize plasticizers. Addition of the plasticizer to PLA improves the physical properties of the polymers. Much faster degradation can be achieved via microwave-assisted conversion combined with appropriate catalysts. Jie and coworkers923 reported that pulverized plastics (HDPE, LDPE, PP, PS) can be deconstructed into hydrogen and high-value carbon materials (e.g., carbon nanotubes) in 30–90 s. FeAlOx plays a significant role in this process by both efficiently absorbing microwave energy, which initiates the physical heating process, and by catalyzing the depolymerization of the plastics.
Additionally, continuous processing and cofeeding of biomass can be applied to microwave-assisted conversion of plastics. Zhou et al.929 reported a continuous microwave-assisted pyrolysis system which can convert HDPE and PP to gasoline-range hydrocarbons rich in aromatic hydrocarbons. The system consists of microwave heating with a SiC mixing-ball-bed and a secondary catalyst bed with ZSM-5 and can process up to 10 kg of plastics an hour with 89.6% energy efficiency. Considering the oxygen deficiency of polyolefins, it can be used as a hydrogen source to be co-processed with hydrogen-deficient biomass via microwave heating, thus improving the quality of the bio-oil.930,931 Zeolites and metal oxides as well as the combination of these two materials have been reported to catalyze the production of hydrocarbons from a mixture of LDPE and lignin, where microwave-assisted heating is vital in achieving even and efficient internal heating.
10.4 Biocatalysis
This section will focus on the biocatalysis of other plastics as the application of biocatalysis for PET depolymerization has been discussed in Section 9. The biological breakdown of plastics by enzymes refers to an attack on water immiscible plastic polymers by appropriate microflora. Plastics are degraded by microbial flora's enzymatic activity, which results in polymer chain fragmentation into monomers.932 Degradation is caused by the activity of enzymes secreted by microorganisms, which use hydrocarbons in the polymer backbone as their major carbon source.932 Enzymatic plastic degradation proceeds in two steps.933 The first step is the decomposition of polymers into smaller molecular compounds by attacking main chains of the polymer with abiotic and/or biotic reactions. The second step is the assimilation of the decomposed polymer (monomer, dimer, or oligomers) by means of microbials and its mineralization which creates CO2, H2O, and CH4.The degradation of HDPE by exposure to white rot fungus, Bjerkandera adusta TBB-03, in a lignocellulose substrate treatment has been demonstrated.934 TBB-03 is able to effectively improve laccase activity (211.4 U L−1) in the presence of ash wood chips due to a mediator resulting from lignin decomposition. The mediator is oxidized by laccase and may play a role as a strong oxidant by producing radical intermediates, which results in the significant morphological change of HDPE.
Skariyachan and coworkers reported that thermophilic consortia of Aneurinibacillus sp. and Brevibacillus sp. showed degradation potential for LDPE, HDPE and PP films and pellets.935 The percentage weight reduction for LDPE, HDPE and PP strips in the consortia of isolates at 50 °C in 140 days was calculated to be 58.21%, 46.6% and 56.3%, respectively. The report on the enzymatic degradation associated with PVC was discussed by Khatoon et al.936Phanerocheate chrysosporium is able to degrade PVC film with 31% weight loss at 25 °C in the 4th week of the incubation period with the fungal strain. Furthermore, Sumathi et al. isolated a fungal species, Cochilobolus sp., for production of laccase, from plastic dumped soils and then determined the ability of the fungi to degrade low molecular weight PVC under lab conditions where the synthetic polymer served as the only carbon source for the Cochliobolus sp.937 They observed significant differences from the FTIR, GC-MS, and SEM results in between control and Cochilobolus sp. treated PVC.
11. Conclusions and outlook
Compared to other engineered materials (i.e., glass, metal, paper etc.), plastics are lightweight and can be easily manufactured into products for a variety of applications. Generation of plastic waste and the ratio of waste plastics in MSW have increased due to the growth of plastic production. Recycling of plastics is still far behind where it needs to be for a circular plastics economy, but improved technology is continuously being developed to efficiently increase the lifetime of plastics.11.1 Collecting and sorting of waste plastics
The first step in any plastic recycling involves collecting and separating the plastics. There is an opportunity to divert more material from landfills through an upgrade in the recycling infrastructure. This can be achieved by an increase in the number of facilities, such as MRFs, or by improving process efficiency along the recycling process, such as improved sorting by adding more optical sorters. However, this will result in additional capital costs for MRFs. Education and outreach regarding recycling is also critical, as consumers need to learn the environmental, social, and economic benefits of recycling.The operation of a MRF presents multiple challenges. One is the difficulty in finding personnel. Another challenge is the lack of predictability of the inbound stream, as each week, the received material can fluctuate according to holidays. For example, in the period of the year close to the Thanksgiving and Christmas holidays, there is an increase in food containers (from dinner parties) and fibers (ads from the shopping season). In terms of the materials collected, some MRFs view the presence of full-body shrink sleeve labels on PET bottles and aluminum cans as a challenge, as it affects the recyclability of two significant sources of income (PET and aluminum). Other challenges include the presence of films and flexible packaging, shredded paper, and packaging with mixed types of material (e.g., plastic bottles with metal closures).938
Increased contamination of the waste material stream is also an issue, especially with material collected from areas not used to having recycling programs, which often have less awareness of the waste sorting procedures. For example, lithium batteries are a growing concern due to the danger of starting fires, with 68 incidents reported in MRFs between 2013 and 2020.939,940 The challenges associated with the presence of contaminants in the bales could result in lower yields per bale, showing an opportunity to improve bale quality. Such measures would include an increase in sortation efficiency by adding more optical sorters, expansion of recycling education to reduce contamination in inbound streams, or even construction of new MRFs.
In developing countries, the waste pickers also need support. The main needs of waste pickers include improving their working conditions, learning to manage their family needs, receiving education and training, preventing violence and guidance to reduce teenage pregnancies, alcoholism and drug addiction, and training to “produce something” from the waste that gives them more income.941 Some actions that would lead to fair living wage waste picking jobs include: (a) develop strategies for precise censuses of waste picker communities and to have dynamic updates of this information; (b) integrate waste pickers in the recycling value chain; (c) harmonize recycling strategies with frontier technologies and train the waste pickers; (d) develop innovative business models for MSW management with a social perspective; (e) update and enforce the legal and regulatory framework of recycling value chains in compliance with human rights of waste pickers; (f) integrate and establish social programs for waste pickers, from public, private, local, and international organizations.
11.2 Mechanical recycling of waste plastics
Mechanical recycling works well for a very homogeneous plastic stream like HDPE milk bottles. However, contamination and other plastics decrease the quality of the plastics produced during mechanical recycling. Some plastics cannot undergo mechanical recycling or do not flow upon heating after cure (thermosets) as they cannot tolerate thermal treatment. Multi-layered plastic products (i.e., juice bottles) cannot be mechanically recycled. These multi-layered packaging materials are more challenging to recycle as each plastic in a different layer has a different melting temperature, and different plastics are often immiscible which makes them practically non-recyclable.942 Colored plastic products, for example PET containers with carbon black in them, are not detected by the sensors during the sorting stage of recycling and complicate the recycling process.943 Films produce a low quality product during mechanical recycling. To improve the quality of the products from mechanical recycling, more advanced separating techniques can be used to obtain feedstock with high a degree of homogeneity. In addition, additives can be added to the extruder to improve the quality of the resin.11.3 Pyrolysis, liquefaction, and gasification of waste plastics
Pyrolysis, liquefaction and gasification have the advantage of being able to process mixtures of plastics and more easily dealing with contamination than mechanical recycling. Several plastic pyrolysis approaches are being commercialized. Catalytic pyrolysis is used to increase product selectivity.944Despite continuing advancement of the technology, commercialization of plastics pyrolysis faces significant challenges which can be addressed by further research. Better understanding of the complex reaction mechanisms controlling depolymerization of plastics could improve selectivity to desired produce molecules. Novel approaches to process intensification will be important to overcome the combination of relatively modest chemical reaction rates and heat and mass transfer limitations that characterize many pyrolysis processes. Using process intensification to achieve economies of scale, determining optimal reactor and process conditions, and securing consistent feedstock streams will increase the profitability of pyrolysis plants. In addition to this, the reactors used in pyrolysis need to be well designed to enable efficient mass and heat transfer. A better understanding of approaches to convert the plastic pyrolysis oils into chemicals, plastics and fuels is needed as steam cracking of the oil has several challenges.
Liquefaction is another technology currently being commercialized. The major challenges of liquefaction are high costs related to the equipment and operation conditions. Supercritical HTL also increases reactor corrosion. Enabling a high feedstock mass loading in the solvent without significantly affecting liquefaction efficiency is also critical, as it is less efficient to heat a large amount of solvent and convert only a small amount of plastic waste. According to Licella, feedstock loading must be sufficiently high (i.e., 70%) for their Cat-HTR process to be economical. Developing an improved conversion strategy to produce higher quality products with minimal required upgrading is also essential. While lab studies indicate that feedstock compositions and reaction conditions strongly impact liquefaction product yield and quality, fewer studies discuss cross-interactions among different plastics. There is also a lack of information about the fate of contaminants and pollutants during liquefaction. HTL of polymers also promotes oxidative reactions, which become more noticeable under supercritical conditions. The effect of oxygenated compounds on the direct use of the crude oil or during subsequent upgrading should be addressed in future studies.
Gasification of MSW shares similar challenges and opportunities as other fossil-based gasification technologies, primarily associated with the high capital cost of gasification. Converting synthetic gas to heat and power produces energy that competes with the declining costs of solar and wind power generation which are now less than $0.04 per kW per h.945 Pursuing higher-value chemicals such as ethylene which is valued at more than $1000 per metric ton946 is possible but requires several catalytic steps (methanol synthesis and methanol to olefins). The varying composition of waste plastic streams presents an additional operational and management challenge for recycling facilities. An incentive program tailored to reduce the risk of economic losses due to market price volatility could serve as a complement or alternative to environmental regulations. Waste reduction policies and incentive programs will likely spur the development of waste gasification facilities.947
11.4 Dissolution-based approaches for waste plastics
Dissolution-based plastic recycling approaches can have advantages over other recycling technologies as they produce the virgin resins. Proper solvent selection for specific types of plastic waste is the key to the feasibility of dissolution-based plastic recycling. Many solvent systems for common polymers have been disclosed in the literature and in patents by different companies. However, when considering more complicated plastic waste feedstocks, advanced thermodynamic computational tools are a means to expedite the development and design of these processes. The recycled materials obtained via dissolution/precipitation methods could be economically competitive with virgin resins, showing the capability of these approaches to reduce plastic waste. These techniques can also be applied to complex multicomponent input streams (e.g., multilayer films) which are challenging for current mechanical recycling technologies.Several challenges remain in the design of dissolution-based recycling techniques that merit future study. The use of large amounts of solvent in these processes poses potential challenges associated with toxicity and energy consumption.631,948,949 To address these concerns, methods for solvent selection based on polymer solubility can be combined with technoeconomic and life cycle analysis techniques to assess tradeoffs in solvent selection.38,47,634 Another challenge is the removal of potential contaminants (e.g., residual inks or retained solvent) from the recovered polymers.588,950 The kinetics of dissolution also could hinder dissolution-based recycling for large amounts of plastic waste.609,651 These challenges could be addressed through additional processing steps for contaminant removal or mechanical shredding prior to dissolution; such steps will require further investigation. Finally, there are opportunities to explore the integration of dissolution-based techniques with liquid-phase chemical recycling techniques.669,951 For example, selective dissolution could be used to separate target polymers from mixed plastic waste prior to their chemical depolymerization, thereby mimicking pretreatment methods utilized in biomass conversion. In this context, solvents could be selected to promote both polymer solubility and desired reaction outcomes.
11.5 Chemical recycling approaches for polyesters and other plastics
New chemical recycling processes centered around catalysis are emerging. PET can be depolymerized via many different routes generating chemicals to produce rPET or other commodities (Fig. 28). For the hydrogenolysis of waste plastics, the product distribution can be tuned by adjusting the residence time, catalyst to polymer mass ratio, temperature, and H2 pressure etc. Detailed reaction mechanisms for the hydrogenolysis of plastics should be elucidated to provide structure–function relationships at the molecular/atomic level which will provide insights in the rational design of new catalysts with improved efficiency or cheaper metal centers. The products from hydrogenolysis are mainly paraffins and light gases. Converting these back into plastics would involve dehydrogenation followed by steam cracking or another type of processing. The cost of H2 and safety issues related to its use are major concerns in the industrialization of such processes. Continuous flow of feedstocks with uniform properties might be challenging due to the solid state of waste plastics. It would be ideal to design techniques which enable continuous feedstock flow with high polymer/catalyst ratio and short reaction times. Furthermore, noble metal centers are likely to be poisoned by the impurities in the feedstock, which also needs to be considered in the development of catalytic materials and processes for waste plastics hydrogenolysis.In the functionalization of plastics, product selectivity and atom efficiency are two challenges since side reactions (e.g., undesired chain cleavage, over-functionalized etc.) easily occur under reaction conditions and an excess of reactant is often required to provide the functional groups. The impurities in waste plastics likely lead to contaminated products or poison the catalysts, which should also be considered in developing these techniques. The removal of impurities in feedstocks needs significant effort in the sortation and pretreatment of waste plastics. Instead, virgin plastic resin can be utilized to manufacture some of the functionalized polymers. Besides, some of the processes need long reaction times (i.e., up to 4 days), which needs to be addressed in scaling up the techniques. Plastic alloying has several advantages compared to other recycling technologies. The key advantages include: (1) little sorting is needed prior to forming the plastic alloys; (2) every type of plastic (#1–#7) can be recycled via this approach; (3) less manpower is needed to operate a plastic alloy facility compared to a MRF and the secondary recycling facility. Compared to pyrolysis of waste plastics, the advantages are: (1) higher ratio of the mixed plastic waste stream can be recycled; (2) fewer numbers of steps are required which results in lower capital/operating costs; and (3) higher value materials than a plastic oil can be produced. More work on this technology is needed to understand if it can be applied to real plastic waste feedstocks, understand the relationships between the catalysts/additives and identify the properties of the plastic alloys, as well as the desirable properties of the plastic alloys.
The removal of impurities in the feedstock is also one of the challenges in microwave-assisted conversion. The impurities that can absorb microwave energy likely lead to varied local temperature, which then results in wide product distribution. Accurate parameters (i.e., microwave power, radiation intensity, magnetic permeability etc.) need to be standardized for the development of this technique. The capital cost and energy efficiency should also be considered in scaling up microwave-assisted techniques for plastic recycling. As for the biocatalytic processes, it is important to standardize and improve the efficiency of the isolation procedure for microbial species which contain plastic degrading enzymes so that pure breeding colonies of these microorganisms can be quickly screened. Enzyme production from pure colonies can be boosted by standardizing the initial step, which enhances the overall performance of biocatalytic plastic degradation.
Overall, enhancing plastic recycling requires focused efforts, particularly in chemical recycling technologies. Efficient approaches to process waste plastics need to be developed to decrease plastic waste and pollution.
Conflicts of interest
The authors declare conflict of interest.Acknowledgements
This material is based upon work supported by the U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy, Bioenergy Technologies Office under Award Number DE-EE0009285. Professor G. W. Huber has an ownership interest in Anellotech, which is commercializing the PlasTCat technology described in this review paper. This paper was also supported in part by the Institute for the Advancement of Food and Nutrition Sciences (IAFNS) through a grant from the Food Packaging Safety and Sustainability Committee. IAFNS is a nonprofit science organization that pools funding from industry collaborators and advances sciencethrough the in-kind and financial contributions from public and private sector participants. We also thank Rosalind J. McKennon for her help with requiring permission for materials and Xin Zou for her help with improving figures in this paper.References
- R. C. Thompson, S. H. Swan, C. J. Moore and F. S. Vom Saal, Our Plastic Age, The Royal Society Publishing, 2009, vol. 364, pp. 1973–1976 Search PubMed.
- R. Geyer, J. R. Jenna, and K. L. Law, Production, Use, and Fate of all Plastics ever Made, Sci. Adv., 2017, 3(7), e1700782 CrossRef PubMed.
- PlasticEurope Plastics – the Facts 2015 an Analysis of European Plastics Production, Demand and Waste Data, https://plasticseurope.org/wp-content/uploads/2021/10/2015-Plastics-the-facts.pdf (accessed Dec 06).
- A. J. Ragauskas, G. W. Huber, J. Wang, A. Guss, H. M. O'Neill, C. S. K. Lin, Y. Wang, F. R. Wurm and X. Meng, New Technologies are Needed to Improve the Recycling and Upcycling of Waste Plastics, Wiley Online Library, 2021, vol. 14, pp. 3982–3984 Search PubMed.
- Foundation, E. M. Plastics and the Circular Economy. https://archive.ellenmacarthurfoundation.org/explore/plastics-and-the-circular-economy (accessed Feb 02).
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Plastic waste inputs from land into the ocean, Science, 2015, 347(6223), 768–771 CrossRef CAS PubMed.
- National Academies of Sciences, E.; Medicine, Reckoning with the U.S. Role in Global Ocean Plastic Waste. 2021.
- S. Allen, D. Allen, F. Baladima, V. Phoenix, J. Thomas, G. Le Roux and J. Sonke, Evidence of Free Tropospheric and Long-Range Transport of Microplastic at Pic Du Midi Observatory, Nat. Commun., 2021, 12(1), 1–10 CrossRef PubMed.
- United States Environmental Protection Agency Trash-Free Waters Plastic Pollution. https://www.epa.gov/trash-free-waters/plastic-pollution (accessed Dec 06).
- C. Benson, Waste containment: Strategies and performance, Aust. Geomech., 2001, 36(4), 1–25 Search PubMed.
- K. L. Law, N. Starr, T. R. Siegler, J. R. Jambeck, N. J. Mallos and G. H. Leonard, The United States’ contribution of plastic waste to land and ocean, Sci. Adv., 2020, 6(44), eabd0288 CrossRef PubMed.
- HS Markit Analysis Indicates Need for Multibillion-Dollar Capital Spending to Achieve a “Circular Plastics Economy” by 2050. https://news.ihsmarkit.com/prviewer/release_only/slug/bizwire-2021-10-11-ihs-markit-analysis-indicates-need-for-multibillion-dollar-capital-spending-to-achieve-a-circular-plastics-economy-by-2050 (accessed Feb 02).
- S. Ilias and A. Bhan, Mechanism of the Catalytic Conversion of Methanol to Hydrocarbons, ACS Catal., 2013, 3(1), 18–31 CrossRef CAS.
- S. R. Nicholson, N. A. Rorrer, A. C. Carpenter and G. T. Beckham, Manufacturing energy and greenhouse gas emissions associated with plastics consumption, Joule, 2021, 5(3), 673–686 CrossRef CAS.
- S. D. A. Sharuddin, F. Abnisa, W. M. A. W. Daud and M. K. Aroua, A Review on Pyrolysis of Plastic Wastes, Energy Convers. Manage., 2016, 115, 308–326 CrossRef.
- S. Ügdüler, K. M. Van Geem, M. Roosen, E. I. Delbeke and S. De Meester, Challenges and Opportunities of Solvent-Based Additive Extraction Methods for Plastic Recycling, Waste Manage., 2020, 104, 148–182 CrossRef PubMed.
- V. Sinha, M. R. Patel and J. V. Patel, Pet Waste Management by Chemical Recycling: A Review, J. Polym. Environ., 2010, 18(1), 8–25 CrossRef CAS.
- A. Singh, N. A. Rorrer, S. R. Nicholson, E. Erickson, J. S. DesVeaux, A. F. Avelino, P. Lamers, A. Bhatt, Y. Zhang and G. Avery, Techno-Economic, Life-Cycle, and Socioeconomic Impact Analysis of Enzymatic Recycling of Poly (Ethylene Terephthalate), Joule, 2021, 5(9), 2479–2503 CrossRef CAS.
- A. J. Martín, C. Mondelli, S. D. Jaydev and J. Pérez-Ramírez, Catalytic Processing of Plastic Waste on the Rise, Chem, 2021, 7(6), 1487–1533 Search PubMed.
- L. O. Mark, M. C. Cendejas and I. Hermans, Cover Feature: The Use of Heterogeneous Catalysis in the Chemical Valorization of Plastic Waste (ChemSusChem 22/2020), ChemSusChem, 2020, 13(22), 5773–5773 CrossRef CAS.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Chemical and Biological Catalysis for Plastics Recycling and Upcycling, Nat. Catal., 2021, 4(7), 539–556 CrossRef CAS.
- J.-P. Lange, Managing Plastic Waste– Sorting, Recycling, Disposal, and Product Redesign, ACS Sustainable Chem. Eng., 2021, 9(47), 15722–15738 CrossRef CAS.
- T. A. Osswald and J. P. Hernández-Ortiz, Polymer processing, Modeling and Simulation, Hanser, Munich, 2006, pp. 1–651 Search PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Production, Use, and Fate of All Plastics Ever Made, Sci. Adv., 2017, 3(7), e1700782 CrossRef PubMed.
- A.-V. Ruzette and L. Leibler, Block copolymers in tomorrow's plastics, Nat. Mater., 2005, 4(1), 19–31 CrossRef CAS PubMed.
- T. Ren, M. K. Patel and K. Blok, Steam cracking and methane to olefins: Energy use, CO2 emissions and production costs, Energy, 2008, 33(5), 817–833 CAS.
- S. S. Chadwick, Ullmann's encyclopedia of industrial chemistry. Reference Services Review 1988 Search PubMed.
- IHS_Chemical, PEP Review-Ethylene Process Summary, 2015.
- M. Yang and F. You, Comparative techno-economic and environmental analysis of ethylene and propylene manufacturing from wet shale gas and naphtha, Ind. Eng. Chem. Res., 2017, 56(14), 4038–4051 CrossRef CAS.
- E. Baur, T. A. Osswald and N. Rudolph, Plastics Handbook, Hanser Publications, Munich, Germany, 2019 Search PubMed.
- A. Naranjo, Plastics testing and characterization: Industrial applications, Hanser Verlag, 2008 Search PubMed.
- T. A. Osswald and G. Menges, Materials science of polymers for engineers, Carl Hanser Verlag GmbH Co KG, 2012 Search PubMed.
- F. Fleischhaker, A. P. Haehnel, A. M. Misske, M. Blanchot, S. Haremza and C. Barner–Kowollik, Glass–Transition–, Melting–, and Decomposition Temperatures of Tailored Polyacrylates and Polymethacrylates: General Trends and Structure–Property Relationships, Macromol. Chem. Phys., 2014, 215(12), 1192–1200 CrossRef CAS.
- IAPD Thermoplastics Rectangle. https://www.iapd.org/ItemDetail?iProductCode=ITR (accessed 07 Feb).
- F. Perugini, M. L. Mastellone and U. Arena, A life cycle assessment of mechanical and feedstock recycling options for management of plastic packaging wastes, Environ. Prog., 2005, 24(2), 137–154 CrossRef CAS.
- L. Shen, E. Worrell and M. K. Patel, Open-loop recycling: A LCA case study of PET bottle-to-fibre recycling, Resour., Conserv. Recycl., 2010, 55(1), 34–52 CrossRef.
- S. Toniolo, A. Mazzi, M. Niero, F. Zuliani and A. Scipioni, Comparative LCA to evaluate how much recycling is environmentally favourable for food packaging, Resour., Conserv. Recycl., 2013, 77, 61–68 CrossRef.
- A. Schwarz, T. Ligthart, D. G. Bizarro, P. De Wild, B. Vreugdenhil and T. Van Harmelen, Plastic recycling in a circular economy; determining environmental performance through an LCA matrix model approach, Waste Manage., 2021, 121, 331–342 CrossRef CAS PubMed.
- U. Arena, M. L. Mastellone and F. Perugini, Life cycle assessment of a plastic packaging recycling system, Int. J. Life Cycle Assess., 2003, 8(2), 92–98 CrossRef.
- D. Garraín, P. Martínez, R. Vidal and M. J. Bellés, In LCA of thermoplastics recycling, Proc. 3rd International Conference of Life Cycle Assessment. Zurich, Austria. https://www.lcm2007.org/paper/168.pdf accessed July, 2007; p 2008.
- L. Rigamonti, M. Grosso and M. C. Sunseri, Influence of assumptions about selection and recycling efficiencies on the LCA of integrated waste management systems, Int. J. Life Cycle Assess., 2009, 14(5), 411–419 CrossRef CAS.
- P. N. Pressley, J. W. Levis, A. Damgaard, M. A. Barlaz and J. F. Decarolis, Analysis of material recovery facilities for use in life-cycle assessment, Waste Manage., 2015, 35, 307–317 CrossRef PubMed.
- H. H. Khoo, LCA of plastic waste recovery into recycled materials, energy and fuels in Singapore, Resour., Conserv. Recycl., 2019, 145, 67–77 CrossRef.
- A. Demetrious and E. Crossin, Life cycle assessment of paper and plastic packaging waste in landfill, incineration, and gasification-pyrolysis, J. Mater. Cycles Waste Manage., 2019, 21(4), 850–860 CrossRef CAS.
- S. Al-Salem, S. Evangelisti and P. Lettieri, Life cycle assessment of alternative technologies for municipal solid waste and plastic solid waste management in the Greater London area, Chem. Eng. J., 2014, 244, 391–402 CrossRef CAS.
- M. F. Cosate de Andrade, P. Souza, O. Cavalett and A. R. Morales, Life cycle assessment of poly (lactic acid)(PLA): Comparison between chemical recycling, mechanical recycling and composting, J. Polym. Environ., 2016, 24(4), 372–384 CrossRef CAS.
- D. Maga, M. Hiebel and N. Thonemann, Life cycle assessment of recycling options for polylactic acid, Resour., Conserv. Recycl., 2019, 149, 86–96 CrossRef.
- N. Rudolph, R. Kiesel and C. Aumnate, Understanding plastics recycling: Economic, ecological, and technical aspects of plastic waste handling, Carl Hanser Verlag GmbH Co KG, 2020 Search PubMed.
- H. Jeswani, C. Krüger, M. Russ, M. Horlacher, F. Antony, S. Hann and A. Azapagic, Life cycle environmental impacts of chemical recycling via pyrolysis of mixed plastic waste in comparison with mechanical recycling and energy recovery, Sci. Total Environ., 2021, 769, 144483 CrossRef CAS PubMed.
- S. Kaza, L. Yao, P. Bhada-Tata and F. Van Woerden, What a waste 2.0: a global snapshot of solid waste management to 2050, World Bank Publications, 2018 Search PubMed.
- U.S.EPA Plastics: Material-Specific Data. https://www.epa.gov/facts-and-figures-about-materials-waste-and-recycling/plastics-material-specific-data (accessed Feb 02).
- Parliament, E. Plastic waste and recycling in the EU: facts and figures. https://www.europarl.Europa.eu/news/en/headlines/society/20181212STO21610/plastic-waste-and-recycling-in-the-eu-facts-and-figures (accessed Feb 1).
- D. C. Wilson, L. Rodic, P. Modak, R. Soos, A. Carpintero, K. Velis, M. Iyer and O. Simonett, Global waste management outlook, UNEP, 2015 Search PubMed.
- Agency, U. S. E. P. Non-Hazardous Materials and Waste Management Hierarchy. https://www.epa.gov/smm/sustainable-materials-management-non-hazardous-materials-and-waste-management-hierarchy (accessed Dec 11).
- C. A. Bareither, C. H. Benson, M. A. Barlaz, T. B. Edil and T. M. Tolaymat, Performance of North American bioreactor landfills. I: Leachate hydrology and waste settlement, J. Environ. Eng., 2010, 136(8), 824–838 CrossRef CAS.
- M. A. Barlaz, C. A. Bareither, A. Hossain, J. Saquing, I. Mezzari, C. H. Benson, T. M. Tolaymat and R. Yazdani, Performance of North American bioreactor landfills. II: Chemical and biological characteristics, J. Environ. Eng., 2010, 136(8), 839–853 CrossRef CAS.
- Agency, U. E. P. Basic information about landfill gas. https://www.epa.gov/lmop/basic-information-about-landfill-gas (accessed Dec 11).
- R. R. Bora, R. Wang and F. You, Waste polypropylene plastic recycling toward climate change mitigation and circular economy: Energy, environmental, and technoeconomic perspectives, ACS Sustainable Chem. Eng., 2020, 8(43), 16350–16363 CrossRef CAS.
- P. Kjeldsen, M. A. Barlaz, A. P. Rooker, A. Baun, A. Ledin and T. H. Christensen, Present and long-term composition of MSW landfill leachate: a review, Crit. Rev. Environ. Sci. Technol., 2002, 32(4), 297–336 CrossRef CAS.
- T. F. Stocker, D. Qin, G.-K. Plattner, M. Tignor, S. K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex and P. M. Midgley, The physical science basis. Contribution of working group I to the fifth assessment report of the intergovernmental panel on climate change, Comput. Geom., 2013, 18(2), 95–123 Search PubMed.
- D. Mutz, D. Hengevoss, C. Hugi and T. Gross, Waste-to-energy options in municipal solid waste management a guide for decision makers in developing and emerging countries, Deutsche Gesellschaft für Internationale Zusammenarbeit (GIZ) GmbH, 2017 Search PubMed.
- D. Tonini, V. Martinez-Sanchez and T. F. Astrup, Material resources, energy, and nutrient recovery from waste: are waste refineries the solution for the future?, Environ. Sci. Technol., 2013, 47(15), 8962–8969 CAS.
- G. Finnveden, A. Björklund, M. C. Reich, O. Eriksson and A. Sörbom, Flexible and robust strategies for waste management in Sweden, Waste Manage., 2007, 27(8), S1–S8 CrossRef PubMed.
- T. Fruergaard and T. Astrup, Optimal utilization of waste-to-energy in an LCA perspective, Waste Manage., 2011, 31(3), 572–582 CrossRef CAS PubMed.
- L. Rigamonti, M. Grosso and L. Biganzoli, Environmental Assessment of Refuse–Derived Fuel Co–Combustion in a Coal–Fired Power Plant, J. Ind. Ecol., 2012, 16(5), 748–760 CrossRef CAS.
- A. Villar, J. J. Arribas and J. Parrondo, Waste-to-energy technologies in continuous process industries, Clean Technol. Environ. Policy, 2012, 14(1), 29–39 CrossRef.
- T. F. Astrup, D. Tonini, R. Turconi and A. Boldrin, Life cycle assessment of thermal Waste-to-Energy technologies: Review and recommendations, Waste Manage., 2015, 37, 104–115 CrossRef CAS PubMed.
- M. U. S. Jaganmohan, Waste Energy - Statistics & Facts. https://www.statista.com/topics/3231/waste-energy-in-the-us/#dossierKeyfigures (accessed Dec 08).
- W. E. Council, World energy resources: Waste to Energy, World Energy Council London, 2016 Search PubMed.
- (UNEP), U. N. E. P, Waste-to-Energy: Considerations for Informed Decision-Making, 2019 Search PubMed.
- (EIA), U. S. E. I. A. U. S, Waste-to-energy (Municipal Solid Waste. https://www.eia.gov/energyexplained/biomass/waste-to-energy.php (accessed Dec 7).
- (EPA), U. S. E. P. A. U. S, Energy Recovery from the Combustion of Municipal Solid Waste (MSW). https://www.epa.gov/smm/energy-recovery-combustion-municipal-solid-waste-msw (accessed Dec 9).
- (OECD)., O. f. E. C. a. D, Municipal Waste. https://www.oecd-ilibrary.org/environment/data/oecd-environment-statistics/municipal-waste_data-00601-en (accessed Dec 07).
- EPA, U. National Overview: Facts and Figures on Materials, Wastes and Recycling. https://www.epa.gov/facts-and-figures-about-materials-waste-and-recycling/national-overview-facts-and-figures-materials (accessed Dec 06).
- (DOE), U. S. D. o. E. U. S, Waste-to-Energy from Municipal Solid Wastes, 2019.
- (WHO), W. H. O. Dioxins and their effects on human health. https://www.who.int/news-room/fact-sheets/detail/dioxins-and-their-effects-on-human-health. (accessed Feb 02).
- A. Mukherjee, B. Debnath and S. K. Ghosh, A review on technologies of removal of dioxins and furans from incinerator flue gas, Proc. Environ. Sci., 2016, 35, 528–540 CrossRef CAS.
- Z. Hao, M. Sun, J. J. Ducoste, C. H. Benson, S. Luettich, M. J. Castaldi and M. A. Barlaz, Heat generation and accumulation in municipal solid waste landfills, Environ. Sci. Technol., 2017, 51(21), 12434–12442 CrossRef CAS.
- U.S.EPA, Material Recovery Facilities for Municipal Solid Waste, United States Environmental Protection Agency, 1991 Search PubMed.
- J. Pichtel, Waste Management Practices: Municipal, Hazardous, and Industrial, CRC Press, Boca Raton, FL, 2014, vol. 2 Search PubMed.
- M. L. Mastellone, R. Cremiato, L. Zaccariello and R. Lotito, Evaluation of performance indicators applied to a material recovery facility fed by mixed packaging waste, Waste Manage., 2017, 64, 3–11 CrossRef.
- H. Damgacioglu, M. Hornilla, O. Bafail and N. Celik, Recovering value from single stream material recovery facilities – An outbound contamination analysis in Florida, Waste Manage., 2020, 102, 804–814 CrossRef CAS PubMed.
- D. J. Tonjes, O. Aphale, L. Clark and K. L. Thyberg, Conversion from dual stream to single stream recycling results in nuanced effects on revenues and waste stream amounts and composition, Resour., Conserv. Recycl., 2018, 138, 151–159 CrossRef.
- G. C. Fitzgerald, J. S. Krones and N. J. Themelis, Greenhouse gas impact of dual stream and single stream collection and separation of recyclables, Resour., Conserv. Recycl., 2012, 69, 50–56 CrossRef.
- C. Lakhan, A Comparison of Single and Multi-Stream Recycling Systems in Ontario, Canada, Resources, 2015, 4(2), 384–397 CrossRef.
- ISRI, ISRI Scrap Specifications Circular 2021, Institute of Scrap Recycling Industries, Washigton, DC, 2021 Search PubMed.
- MRFF Flexible Packaging Recycling in Material Recovery Facilities Pliot. https://www.materialsrecoveryforthefuture.com/wp-content/uploads/MRFF-Pilot-Report-2020-Final.pdf (accessed 17 Dec 2021).
- GBB Supplemental Report: The Evolution of Mixed Waste Processing Facilities - Technology and Equipment Guide, Gershman, Brickner & Bratton, Inc., The American Chemistry Council, Fairfax, VA, 2016, p. 32 Search PubMed.
- T. H. Christensen and B. Bilitewski, Mechanical Treatment: Material Recovery Facilities, in Solid Waste Technology & Management, ed. T. H. Christensen, John Wiley & Sons, Ltd, Chichester, 2010, pp. 349–362 Search PubMed.
- B. Bilitewski, Mechanical Treatment: Unit Processes, in Solid Waste Technology & Management, ed. T. H. Christensen, John Wiley & Sons, Ltd, Chichester, 2010, vol. 1, pp. 319–348 Search PubMed.
- J. Beigbeder, D. Perrin, J.-F. Mascaro and J.-M. Lopez-Cuesta, Study of the physico-chemical properties of recycled polymers from waste electrical and electronic equipment (WEEE) sorted by high resolution near infrared devices, Resour., Conserv. Recycl., 2013, 78, 105–114 CrossRef.
- B. Eule, Processing of Co-mingled Recyclate Material at UK Material Recycling Facilities (MRF), RWTH Aachen, Aachen, 2013 Search PubMed.
- K. Barker, Optical sorting advances are driving the MRF of the future. https://www.recyclingproductnews.com/article/35838/optical-sorting-advances-are-driving-the-mrf-of-the-future (accessed 09 Nov 2021).
- Y. Zheng, J. Bai, J. Xu, X. Li and Y. Zhang, A discrimination model in waste plastics sorting using NIR hyperspectral imaging system, Waste Manage., 2018, 72, 87–98 CrossRef CAS PubMed.
- G. Bonifazi and S. Serranti, in Quality control by HyperSpectral Imaging (HSI) in solid waste recycling: logics, algorithms and procedures, IS&T/SPIE Electronic Imaging, San Francisco, CA, SPIE: San Francisco, CA, 2014 Search PubMed.
- M. De Biasio, T. Arnold, G. McGunnigle, R. Leitner, D. Balthasar and V. Rehrmann, In Detecting and discriminating PE and PP polymers for plastics recycling using NIR imaging spectroscopy, SPIE Defense, Security, and Sensing, Orlando, FL, SPIE, Orlando, FL, 2010 Search PubMed.
- S. Zinchik, S. Jiang, S. Friis, F. Long, L. Høgstedt, V. M. Zavala and E. Bar-Ziv, Accurate Characterization of Mixed Plastic Waste Using Machine Learning and Fast Infrared Spectroscopy, ACS Sustainable Chem. Eng., 2021, 9(42), 14143–14151 CrossRef CAS.
- J. Xia, Y. Huang, Q. Li, Y. Xiong and S. Min, Convolutional neural network with near-infrared spectroscopy for plastic discrimination, Environ. Chem. Lett., 2021, 19(5), 3547–3555 CrossRef CAS.
- S. Zhu, H. Chen, M. Wang, X. Guo, Y. Lei and G. Jin, Plastic solid waste identification system based on near infrared spectroscopy in combination with support vector machine, Adv. Ind. Eng. Polym. Res., 2019, 2(2), 77–81 Search PubMed.
- W. A. Worrell and P. A. Vesling, Solid Waste Engineering, Cengage Learning, Stamford, CT, 2012 Search PubMed.
- H. Leverenz, G. Tchobanoglous and D. B. Spencer, Recycling, in Handbook of Solid Waste Management, ed. G. Tchobanoglous and F. Kreith, McGraw-Hill, New York, NY, 2002 Search PubMed.
- K. Maile, Baler basics. https://www.recyclingtoday.com/article/baler-basics/(accessed 09 Nov 2021).
- APR Model Bale Specifications. https://plasticsrecycling.org/model-bale-specifications (accessed 09 Nov 2021).
- Machinex Machinex Expands its Baler Range with the II ram Baler Series. https://www.machinexrecycling.com/news/machinex-expands-baler-range-ii-ram-baler-series/(accessed 11 Nov 2021).
- RecyclingMarkets RecyclingMarkets.net. https://www.recyclingmarkets.net (accessed Feb 02).
- U.S.EPA, Advancing Sustainable Materials Management: 2018 Tables and Figures, United States Environmental Protection Agency, 2020 Search PubMed.
- 2020 U.S. Post-Consumer Plastic Recycling Survey. https://www.lastbeachcleanup.org/usplasticrecyclingsurvey (accessed 20 Oct 2021).
- J. Hocevar, Circular Claims Fall Flat: Comprehensive U.S. Survey of Plastics Recyclability, Greenpeace, 2020 Search PubMed.
- D. Toto, The largest 75 MRFs in North America. https://www.wastetodaymagazine.com/article/largest-north-american-material-recovery-facilities/(accessed 11 Nov 2021).
- WRAP Materials Facility Reporting Portal Q4 2019 – Commentary; Waste and Resources Action Programme, 2020.
- M. Ali and P. Courtenay, Evaluating the progress of the UK's Material Recycling Facilities: A mini review, Waste Manage. Res., 2014, 32(12), 1149–1157 CrossRef PubMed.
- COREPLA, Rapporto di Sostenibilita 2020; Consorzio Nazionale per la raccolta, il riciclo e il recupero degli imballaggi in plastica, Milan, Italy, 2020 Search PubMed.
- MISE, Studio di fattibilità nell'ambito dell'Economia Circolare mirato al riciclo delle Plastiche Miste, Ministero dello sviluppo economico, MISE (Italy), Roma, Italy, 2019 Search PubMed.
- Destatis Waste treatment plants 2019. https://www.destatis.de/EN/Themes/Society-Environment/Environment/Waste-Management/Tables/waste-treatment-plants-2019.html (accessed 11 Nov 2021).
- ADEME, Déchets chiffres-clés Édition 2020; Agence de la transition ecologique, Angers, France, 2020 Search PubMed.
- S. M. Dias, Waste pickers and cities, Environ. Urban, 2016, 28(2), 375–390 CrossRef.
- F. Gutierrez-Galicia, A. L. Coria-Paez, R. Tejeida-Padilla and E. F. Galicia-Haro, A System for the Inclusion of the Informal Recycling Sector (IRS) in Mexico City's Solid Waste Management, Sustainability, 2021, 13(22), 12490 CrossRef.
- M. Medina, Scavenger cooperatives in Asia and Latin America, Resour., Conserv. Recycl., 2000, 31(1), 51–69 CrossRef.
- C. V. Santos and J. A. G. García, El papel de los pepenadores de materiales reciclables en la gestión de residuos sólidos: los casos de Brasil y México, Revista Legislativa de Estudios Sociales y de Opinión Pública, 2019, 12(24), 87–114 Search PubMed.
- D. I. Nogueda, Life cycle assessment of PET bottle recycling: a case study for Mexico, 2013 Search PubMed.
- M. Medina, Social Inclusion in Mexico's PET Plastic Recycling, 2013.
- Análisis de costos de producción y márgenes del pet virgen y reciclado, 2011.
- Compra venta de chatarra y reciclados por kilogramos, 2022.
- P. Navarrete-Hernández and N. Navarrete-Hernández, Unleashing waste-pickers’ potential: supporting recycling cooperatives in Santiago de Chile, World Dev., 2018, 101, 293–310 CrossRef.
- O. Buenrostro and G. Bocco, Solid waste management in municipalities in Mexico: goals and perspectives, Resour., Conserv. Recycl., 2003, 39(3), 251–263 CrossRef.
- V. Montiel-Corona and J. A. Guevara-García, Impulsando a México a una Cultura de Reciclaje, in México Contemporáneo: aspectos económicos, políticos y sociales, ed. U. Veracruzana, Universidad Veracruzana, 2018 Search PubMed.
- C. Silva-Chavez, D. Caro and M. Thomsen, Environmental Assessment of Alternatives for Biowaste Treatment in Mexico City, Front. Energy Res., 2019, 7, 30 CrossRef.
- N. M. Jimenez-Martinez and R. Garcia-Barrios, The Zero Waste University Program in Mexico: A Model for Grassroots Innovations in Sustainability, Sustainability, 2020, 12(22), 9444 CrossRef.
- V. Montiel-Corona and J. Guevara-García, Impulsando a México a una Cultura de Reciclaje, in México Contemporáneo: aspectos, 2018 Search PubMed.
- C. S. Chávez, D. Caro and M. Thomsen, Environmental assessment of alternatives for biowaste treatment in Mexico City, Front. Energy Res., 2019, 7, 30 CrossRef.
- A. Flores and O. Fairuz, Policy mechanisms to reduce single-use plastic waste. Review of available options and their applicability in Mexico, World Resources Institute Mexico, 2020, pp. 50 Search PubMed.
- Netherlands Enterprise Agency, Market opportunities on Circular Economy in Mexico, 2019 Search PubMed.
- Gobierno de México, LA LEY GENERAL DE ECONOMÍA CIRCULAR. Camara de Senadores, Ed. Gobierno de México: Diario Oficial de la Federación, 2021.
- G. A. o. W. Pickers, Global Alliance of Waste Pickers. https://globalrec.org/life-and-voices/(accessed Aug 28).
- Guidance for Industry: Use of Recycled Plastics in Food Packaging (Chemistry Considerations). U.S. Food and Drug (FDA), 2021.
- N. Singh, D. Hui, R. Singh, I. P. S. Ahuja, L. Feo and F. Fraternali, Recycling of plastic solid waste: A state of art review and future applications, Composites, Part B, 2017, 115, 409–422 CrossRef CAS.
- Mechanical Recycling; European Bioplastics (EUBP), 2020.
- M. D. Jagtap, S. S. Khatavkar and T. Z. Quazi, Methods for Waste Plastic Recycling. International Journal on Recent Technologies in Mechanical and Electrical Engineering (IJRMEE) 2 (ISSN: 2349-7947), 120–122.
- FDA, U., Use of Recycled Plastics in Food Packaging: Chemistry Considerations. Food and Drug Administration, Washington, DC, 2006 Search PubMed.
- N. Malik, P. Kumar, S. Shrivastava and S. B. Ghosh, An overview on PET waste recycling for application in packaging, Int. J. Plast. Technol., 2017, 21(1), 1–24 CrossRef CAS.
- C. Prakash, S. Singh, G. Krolczyk and B. Pabla, Advances in materials science and engineering, Lect. Notes Mech. Eng., 2020, 10, 978–981 Search PubMed.
- L. Fetters, D. Lohse, D. Richter, T. Witten and A. Zirkel, Connection between polymer molecular weight, density, chain dimensions, and melt viscoelastic properties, Macromolecules, 1994, 27(17), 4639–4647 CrossRef CAS.
- R. H. Colby, L. J. Fetters and W. W. Graessley, The melt viscosity-molecular weight relationship for linear polymers, Macromolecules, 1987, 20(9), 2226–2237 CrossRef CAS.
- R. W. Nunes, J. R. Martin and J. F. Johnson, Influence of molecular weight and molecular weight distribution on mechanical properties of polymers, Polym. Eng. Sci., 1982, 22(4), 205–228 CrossRef CAS.
- F. P. La Mantia, Recycling of PVC and mixed plastic waste, ChemTec Publishing, 1996 Search PubMed.
- M. Paci and F. La Mantia, Influence of small amounts of polyvinylchloride on the recycling of polyethyleneterephthalate, Polym. Degrad. Stab., 1999, 63(1), 11–14 CrossRef CAS.
- F. La Mantia, D. Curto and R. Scaffaro, Recycling of dry and wet polyamide 6, J. Appl. Polym. Sci., 2002, 86(8), 1899–1903 CrossRef CAS.
- P. Kumar, R. K. Yadav, R. Kumar and S. Maurya, Recyling of waste plastic using extrusion process.
- K. P. Rajan, A. Gopanna and S. P. Thomas, A project based learning (PBL) Approach involving pet recycling in chemical engineering education, Recycling, 2019, 4(1), 10 CrossRef.
- B. Jabłońska, P. Kiełbasa, M. Korenko and T. Dróżdż, Physical and chemical properties of waste from PET bottles washing as a component of solid fuels, Energies, 2019, 12(11), 2197 CrossRef.
- D. L. Gall, J. Ralph, T. J. Donohue and D. R. Noguera, A group of sequence-related sphingomonad enzymes catalyzes cleavage of β-aryl ether linkages in lignin β-guaiacyl and β-syringyl ether dimers, Environ. Sci. Technol., 2014, 48(20), 12454–12463 CrossRef CAS PubMed.
- B. Brooks, K. Hays and L. Milner, Plastics recycling, in PET and Europe lead the way, 2019 Search PubMed.
- S. Cruz and M. Zanin, PET recycling: Evaluation of the solid state polymerization process, J. Appl. Polym. Sci., 2006, 99(5), 2117–2123 CrossRef CAS.
- F. Awaja and D. Pavel, Recycling of PET, Eur. Polym. J., 2005, 41(7), 1453–1477 CrossRef CAS.
- Z. O. Schyns and M. P. Shaver, Mechanical recycling of packaging plastics: A review, Macromol. Rapid Commun., 2021, 42(3), 2000415 CrossRef CAS PubMed.
- A. Brems, J. Baeyens, C. Vandecasteele and R. Dewil, Polymeric cracking of waste polyethylene terephthalate to chemicals and energy, J. Air Waste Manage. Assoc., 2011, 61(7), 721–731 CrossRef CAS.
- M. Paci and F. P. La Mantia, Influence of small amounts of polyvinylchloride on the recycling of polyethyleneterephthalate, Polym. Degrad. Stab., 1999, 63(1), 11–14 CrossRef CAS.
- G. W. Curtzwiler, E. B. Williams, A. L. Maples, N. W. Davis, T. L. Bahns, J. Eliseo De León and K. L. Vorst, Ultraviolet protection of recycled polyethylene terephthalate, J. Appl. Polym. Sci., 2017, 134(32), 45181 CrossRef.
- T. Kim and S. Jabarin, Solid–state polymerization of poly (ethylene terephthalate). II. Modeling study of the reaction kinetics and properties, J. Appl. Polym. Sci., 2003, 89(1), 213–227 CrossRef CAS.
- M. Asensio, P. Esfandiari, K. Núñez, J. F. Silva, A. Marques, J. C. Merino and J. M. Pastor, Processing of pre-impregnated thermoplastic towpreg reinforced by continuous glass fibre and recycled PET by pultrusion, Composites, Part B, 2020, 200, 108365 CrossRef CAS.
- Unilever makes PET plastic breakthrough. https://www.businessgrowthhub.com/green-technologies-and-services/green-intelligence/resource-library/unilever-makes-pet-plastic-breakthrough.
- D. Toto, Indorama Ventures highlights sustainability achievements. https://www.recyclingtoday.com/article/indorama-sustainablity-report-2020-pet-recycling/.
- Anonymous, Guidance for industry: Use of recycled plastics in food packaging: Chemistry considerations;U.S. Dept. of Health and Human Services, FDA, Center for Food Safety and Applied Nutrition, College Park, 2021 Search PubMed.
- C. Marschik, B. Löw-Baselli and J. Miethlinger, in Modeling devolatilization in single-and multi-screw extruders, AIP Conference Proceedings, AIP Publishing LLC, 2017, p. 080006 Search PubMed.
- S. Pachner, M. Aigner and J. Miethlinger, A heuristic method for modeling the initial pressure drop in melt filtration using woven screens in polymer recycling, Polym. Eng. Sci., 2019, 59(6), 1105–1113 CrossRef CAS.
- M. Mousavi, Territorial Environmental Modeling of Cement Concrete Demolition Waste (CCDW) Management with a Life Cycle Approach, Loire Bretagne University, 2018 Search PubMed.
- F. Tao, S. Dag, L.-W. Wang, Z. Liu, D. R. Butcher, H. Bluhm, M. Salmeron and G. A. Somorjai, Break-up of stepped platinum catalyst surfaces by high CO coverage, Science, 2010, 327(5967), 850–853 CrossRef CAS PubMed.
- rPET market to garner US $12.5 billion by 2026. recycling magazine 2019.
- J. A. Brown, M. Alic and M. H. Gold, Manganese peroxidase gene transcription in Phanerochaete chrysosporium: Activation by manganese, J. Bacteriol., 1991, 173(13), 4101–4106 CrossRef CAS PubMed.
- . 2020 PET RECYCLING REPORT. https://napcor.com/reports-resources/.
- Y. Wang, R. Song and D. Shen, Note: Enthalpy relaxation studies in amorphous poly (ethylene terephthalate) by Modulated DSC, J. Macromol. Sci., Part B: Phys., 1998, 37(5), 709–716 CrossRef.
- G. W. Curtzwiler, M. Schweitzer, Y. Li, S. Jiang and K. L. Vorst, Mixed post-consumer recycled polyolefins as a property tuning material for virgin polypropylene, J. Cleaner Prod., 2019, 239, 117978 CrossRef CAS.
- G. W. Curtzwiler, E. B. Williams, E. Hurban, J. Greene and K. L. Vorst, Certification markers for empirical quantification of post-consumer recycled content in extruded polyethylene film, Polym. Test., 2018, 65, 103–110 CrossRef CAS.
- S. Luzuriaga, J. Kovářová and I. Fortelný, Degradation of pre-aged polymers exposed to simulated recycling: properties and thermal stability, Polym. Degrad. Stab., 2006, 91(6), 1226–1232 CrossRef CAS.
- J. M. Soto, G. Blázquez, M. Calero, L. Quesada, V. Godoy and M. Á. Martín-Lara, A real case study of mechanical recycling as an alternative for managing of polyethylene plastic film presented in mixed municipal solid waste, J. Cleaner Prod., 2018, 203, 777–787 CrossRef CAS.
- Amut Friction Washer Cleans Contaminated PET Flakes. https://www.recyclingtoday.com/article/amut-friction-recycled-pet-washing/.
- Oh!, E. https://www.ernestny.com/eco-oh.php (accessed Feb 02).
- L. A. Pinheiro, M. A. Chinelatto and S. V. Canevarolo, The role of chain scission and chain branching in high density polyethylene during thermo-mechanical degradation, Polym. Degrad. Stab., 2004, 86(3), 445–453 CrossRef CAS.
- P. Oblak, J. Gonzalez-Gutierrez, B. Zupančič, A. Aulova and I. Emri, Processability and mechanical properties of extensively recycled high density polyethylene, Polym. Degrad. Stab., 2015, 114, 133–145 CrossRef CAS.
- K. Bhunia, S. S. Sablani, J. Tang and B. Rasco, Migration of chemical compounds from packaging polymers during microwave, conventional heat treatment, and storage, Compr. Rev. Food Sci. Food Saf., 2013, 12(5), 523–545 CrossRef CAS PubMed.
- Diversion of waste from landfill in Europe. https://www.eea.europa.eu/ims/diversion-of-waste-from-landfill.
- Anonymous Advancing Sustainable Materials Management: 2018 Fact Sheet. https://www.epa.gov/sites/production/files/2021-01/documents/<?pdb_no 2018?>2018<?pdb END?>_ff_fact_sheet_dec_2020_fnl_508.pdf (accessed 28 April).
- S. Camarero, O. Garcıa, T. Vidal, J. Colom, J. C. del Rıo, A. Gutiérrez, J. M. Gras, R. Monje, M. J. Martınez and Á.T Martınez, Efficient bleaching of non-wood high-quality paper pulp using laccase-mediator system, Enzyme Microb. Technol., 2004, 35(2–3), 113–120 CrossRef CAS.
- Z. Wen, Y. Xie, M. Chen and C. D. Dinga, China's plastic import ban increases prospects of environmental impact mitigation of plastic waste trade flow worldwide, Nat. Commun., 2021, 12(1), 1–9 CrossRef PubMed.
- F. G. Calvo-Flores and J. A. Dobado, Lignin as renewable raw material, ChemSusChem, 2010, 3(11), 1227–1235 CrossRef CAS PubMed.
- Plastics – the Facts 2019, 2019.
- Recycled Plastics in Food Packaging, 2020.
- T. D. Bugg, M. Ahmad, E. M. Hardiman and R. Rahmanpour, Pathways for degradation of lignin in bacteria and fungi, Nat. Prod. Rep., 2011, 28(12), 1883–1896 RSC.
- Food Packaging Regulation in the US; foodpackagingforum, 2013.
- FDA, Recycled Plastics in Food Packaging, FDA, 2020 Search PubMed.
- K. A. Fenlon, A. C. Johnson, C. R. Tyler and E. M. Hill, Gas–liquid chromatography–tandem mass spectrometry methodology for the quantitation of estrogenic contaminants in bile of fish exposed to wastewater treatment works effluents and from wild populations, J. Chromatogr., A, 2010, 1217(1), 112–118 CrossRef CAS PubMed.
- Anonymous Submissions on Post-Consumer Recycled (PCR) Plastics for Food-Contact Articles. https://www.cfsanappsexternal.fda.gov/scripts/fdcc/?set=RecycledPlastics&sort=Date_of_NOL&order=ASC&type=basic&search= (accessed 11 January).
- M. Lorber, A. Schecter, O. Paepke, W. Shropshire, K. Christensen and L. Birnbaum, Exposure assessment of adult intake of bisphenol A (BPA) with emphasis on canned food dietary exposures, Environ. Int., 2015, 77, 55–62 CrossRef CAS.
- V. S. Cecon, P. F. Da Silva, G. W. Curtzwiler and K. L. Vorst, The challenges in recycling post-consumer polyolefins for food contact applications: A review, Resour., Conserv. Recycl., 2021, 167, 105422 CrossRef CAS.
- M. G. Davidson, R. A. Furlong and M. C. McManus, Developments in the life cycle assessment of chemical recycling of plastic waste e A review, J. Cleaner Prod., 2021, 293, 126163 CrossRef CAS.
- R. K. Singh and B. Ruj, Time and temperature depended fuel gas generation from pyrolysis of real world municipal plastic waste, Fuel, 2016, 174, 164–171 CrossRef CAS.
- H. J. Rubel, U. Jung, C. Follette, A. Meyer zum Felder, S. Appathurai and M. Benedi Diaz, A Circular Solution to Plastic Waste, Boston Consulting Group, 2019 Search PubMed.
- H. Jeswani, C. Kruger, M. Russ, M. Horlacher, F. Antony, S. Hann and A. Azapagic, Life cycle environmental impacts of chemical recycling via pyrolysis of mixed plastic waste in comparison with mechanical recycling and energy recovery, Sci. Total Environ., 2021, 769, 15 CrossRef PubMed.
- W. Kaminsky and I. J. N. Zorriqueta, Catalytical and thermal pyrolysis of polyolefins, J. Anal. Appl. Pyrolysis, 2007, 79(1-2 SPEC. ISS.), 368–374 CrossRef CAS.
- D. Zhao, X. Wang, J. B. Miller and G. W. Huber, The Chemistry and Kinetics of Polyethylene Pyrolysis: A Process to Produce Fuels and Chemicals, ChemSusChem, 2020, 13(7), 1764–1774 CrossRef CAS.
- Y. Xue, P. Johnston and X. Bai, Effect of catalyst contact mode and gas atmosphere during catalytic pyrolysis of waste plastics, Energy Convers. Manage., 2017, 142, 441–451 CrossRef CAS.
- Y. Xue, S. Zhou, R. C. Brown, A. Kelkar and X. Bai, Fast pyrolysis of biomass and waste plastic in a fluidized bed reactor, Fuel, 2015, 156, 40–46 CrossRef CAS.
- M. L. Mastellone, F. Perugini, M. Ponte and U. Arena, Fluidized bed pyrolysis of a recycled polyethylene, Polym. Degrad. Stab., 2002, 76(3), 479–487 CrossRef CAS.
- T. Kuroki, T. Sawaguchi, S. Niikuni and T. Ikemura, Mechanism for Long-Chain Branching in the Thermal Degradation of Linear High-Density Polyethylene, Macromolecules, 1982, 15(6), 1460–1464 CrossRef CAS.
- L. Cheng, J. Gu, Y. Wang, J. Zhang, H. Yuan and Y. Chen, Polyethylene high-pressure pyrolysis: Better product distribution and process mechanism analysis, Chem. Eng. J., 2020, 385(September 2019), 123866–123866 CrossRef CAS.
- C. Berrueco, E. J. Mastral, E. Esperanza and J. Ceamanos, Production of waxes and tars from the continuous pyrolysis of high density polyethylene. Influence of operation variables, Energy Fuels, 2002, 16(5), 1148–1153 CrossRef CAS.
- S. H. Jung, M. H. Cho, B. S. Kang and J. S. Kim, Pyrolysis of a fraction of waste polypropylene and polyethylene for the recovery of BTX aromatics using a fluidized bed reactor, Fuel Process. Technol., 2010, 91(3), 277–284 CrossRef CAS.
- P. J. Donaj, W. Kaminsky, F. Buzeto and W. Yang, Pyrolysis of polyolefins for increasing the yield of monomers’ recovery, Waste Manage., 2012, 32(5), 840–846 CrossRef CAS.
- A. Aboulkas, K. El harfi and A. El Bouadili, Thermal degradation behaviors of polyethylene and polypropylene. Part I: Pyrolysis kinetics and mechanisms, Energy Convers. Manage., 2010, 51(7), 1363–1369 CrossRef CAS.
- L. A. Wall and S. Straus, Pyrolysis of polyolefins, J. Polym. Sci., 1960, 44(144), 313–323 CrossRef CAS.
- J. D. Peterson, S. Vyazovkin and C. A. Wight, Kinetics of the Thermal and Thermo-Oxidative Degradation of Polystyrene, Polyethylene and Poly(propylene), Macromol. Chem. Phys., 2001, 202(6), 775–784 CrossRef CAS.
- R. W. J. Westerhout, J. Waanders, J. A. M. Kuipers and W. P. M. van Swaaij, Kinetics of the Low-Temperature Pyrolysis of Polyethene, Polypropene, and Polystyrene Modeling, Experimental Determination, and Comparison with Literature Models and Data, Ind. Eng. Chem. Res., 1997, 36(6), 1955–1964 CrossRef CAS.
- A. Marcilla, M. I. Beltrán and R. Navarro, Evolution of products during the degradation of polyethylene in a batch reactor, J. Anal. Appl. Pyrolysis, 2009, 86(1), 14–21 CrossRef CAS.
- E. Kiran and J. K. Gillham, Pyrolysis–molecular weight chromatography: A new on–line system for analysis of polymers. II. Thermal decomposition of polyolefins: Polyethylene, polypropylene, polyisobutylene, J. Appl. Polym. Sci., 1976, 20(8), 2045–2068 CrossRef CAS.
- H. Bockhorn, A. Hornung, U. Hornung, P. Jakobströer and M. Kraus, Dehydrochlorination of plastic mixtures, J. Anal. Appl. Pyrolysis, 1999, 49(1), 97–106 CrossRef CAS.
- R. P. Lattimer and W. J. Kroenke, Mechanisms of formation of volatile aromatic pyrolyzates from poly(vinyl chloride), J. Appl. Polym. Sci., 1982, 27(4), 1355–1366 CrossRef CAS.
- Y. Tsuchiya and K. Sumi, Thermal decomposition products of polypropylene, J. Polym. Sci., Part A: Polym. Chem., 1969, 7(7), 1599–1607 CrossRef CAS.
- R. P. Lattimer, Pyrolysis field ionization mass spectrometry of hydrocarbon polymers, J. Anal. Appl. Pyrolysis, 1997, 39(2), 115–127 CrossRef CAS.
- Y. Matsuzawa, M. Ayabe, J. Nishino, N. Kubota and M. Motegi, Evaluation of char fuel ratio in municipal pyrolysis waste, Fuel, 2004, 83(11–12), 1675–1687 CrossRef CAS.
- J. Yu, L. Sun, C. Ma, Y. Qiao and H. Yao, Thermal degradation of PVC: A review, Waste Manage., 2016, 48, 300–314 CrossRef CAS PubMed.
- V. V. Krongauz, Y. P. Lee and A. Bourassa, Kinetics of thermal degradation of poly(vinyl chloride): Thermogravimetry and spectroscopy, J. Therm. Anal. Calorim., 2011, 106(1), 139–149 CrossRef CAS.
- L. Dean, Z. Dafei and Z. Deren, Mechanism and kinetics of thermo-dehydrochlorination of poly(vinyl chloride), Polym. Degrad. Stab., 1988, 22(1), 31–41 CrossRef.
- T. Y. Xie, A. E. Hamielec, M. Rogestedt and T. Hjertberg, Experimental investigation of vinyl chloride polymerization at high conversion: polymer microstructure and thermal stability and their relationship to polymerization conditions, Polymer, 1994, 35(7), 1526–1534 CrossRef CAS.
- W. H. Starnes, Structural and mechanistic aspects of the thermal degradation of poly(vinyl chloride), Prog. Polym. Sci., 2002, 27(10), 2133–2170 CrossRef CAS.
- W. H. Starnes, Overview and assessment of recent research on the structural defects in poly(vinyl chloride), Polym. Degrad. Stab., 2012, 97(9), 1815–1821 CrossRef CAS.
- B. B. Troitskii, A. S. Yakhnov, M. A. Novikova, T. G. Ganyukhina and V. N. Denisova, Effect of tacticity on thermal degradation of poly(vinyl chloride), Eur. Polym. J., 1997, 33(4), 505–511 CrossRef CAS.
- W. H. Starnes and X. Ge, Mechanism of Autocatalysis in the Thermal Dehydrochlorination of Poly(vinyl chloride), Macromolecules, 2004, 37(2), 352–359 CrossRef CAS.
- V. M. Yanborisov and S. S. Borisevich, Quantum-chemical modeling of the mechanism of autocatalytic dehydrochlorination of PVC, Theor. Exp. Chem., 2005, 41(6), 352–358 CrossRef CAS.
- T. Hjertberg and E. Sörvik, Thermal degradation of PVC. In Degradation and Stabilisation of PVC, Springer, 1984, pp. 21–79 Search PubMed.
- I. C. McNeill, L. Memetea and W. J. Cole, A study of the products of PVC thermal degradation, Polym. Degrad. Stab., 1995, 49(1), 181–191 CrossRef CAS.
- L. Ye, T. Li and L. Hong, Understanding enhanced char formation in the thermal decomposition of PVC resin: Role of intermolecular chlorine loss, Mater. Today Commun., 2021, 26, 102186 CrossRef CAS.
- J. Huang, X. Li, G. Zeng, X. Cheng, H. Tong and D. Wang, Thermal decomposition mechanisms of poly(vinyl chloride): A computational study, Waste Manage., 2018, 76, 483–496 CrossRef CAS.
- A. Lopez-Urionabarrenechea, I. De Marco, B. M. Caballero, M. F. Laresgoiti and A. Adrados, Catalytic stepwise pyrolysis of packaging plastic waste, J. Anal. Appl. Pyrolysis, 2012, 96, 54–62 CrossRef CAS.
- A. Guyot, Recent developments in the thermal degradation of polystyrene—A review, Polym. Degrad. Stab., 1986, 15(3), 219–235 CrossRef CAS.
- L. A. Wall, S. Straus, R. E. Florin and L. J. Fetters, Pyrolysis of anionic and thermally prepared polystyrenes, J. Res. Natl. Bur. Stand., Sect. A, 1973, 77(1), 157–157 CrossRef CAS.
- G. G. Cameron and G. P. Kerr, Thermal degradation of polystyrene-II the role of abnormalities, Eur. Polym. J., 1970, 6(2), 423–433 CrossRef CAS.
- L. Costa, G. Camino, A. Guyot, M. Bert and A. Chiotis, The rôle of chain ends in the thermal degradation of anionic polystyrene, Polym. Degrad. Stab., 1982, 4(4), 245–260 CrossRef CAS.
- J. Huang, X. Li, H. Meng, H. Tong, X. Cai and J. Liu, Studies on pyrolysis mechanisms of syndiotactic polystyrene using DFT method, Chem. Phys. Lett., 2020, 747(January), 137334–137334 CrossRef CAS.
- A. Rahimi and J. M. García, Chemical recycling of waste plastics for new materials production, Nat. Rev. Chem., 2017, 1(6), 0046 CrossRef.
- L. H. Buxbaum, The Degradation of Poly(ethylene terephthalate), Angew. Chem., Int. Ed. Engl., 1968, 7(3), 182–190 CrossRef CAS.
- G. Montaudo, C. Puglisi and F. Samperi, Primary thermal degradation mechanisms of PET and PBT, Polym. Degrad. Stab., 1993, 42(1), 13–28 CrossRef CAS.
- J. Huang, H. Meng, X. Luo, X. Mu, W. Xu, L. Jin and B. Lai, Insights into the thermal degradation mechanisms of polyethylene terephthalate dimer using DFT method, Chemosphere, 2022, 291, 133112 CrossRef CAS PubMed.
- B. J. Holland and J. N. Hay, The thermal degradation of poly(vinyl acetate) measured by thermal analysis–Fourier transform infrared spectroscopy, Polymer, 2002, 43(8), 2207–2211 CrossRef CAS.
- H. H. G. Jellinek, Thermal Degradation of Polystyrene and Polyethylene. Part III, Journal of Polymer Science, 1949, 4, 13–36 CrossRef CAS.
- G. Lopez, M. Artetxe, M. Amutio, J. Bilbao and M. Olazar, Thermochemical routes for the valorization of waste polyolefinic plastics to produce fuels and chemicals. A review, Renewable Sustainable Energy Rev., 2017, 73, 346–368 CrossRef CAS.
- S. M. Al-Salem, A. Antelava, A. Constantinou, G. Manos and A. Dutta, A review on thermal and catalytic pyrolysis of plastic solid waste (PSW), J. Environ. Manage., 2017, 197, 177–198 CrossRef CAS PubMed.
- E. A. W. Williams and T. Paul, The Pyrolysis of Individual Plastics and a Plastic Mixture in a Fixed Bed Reactor, J. Chem. Technol. Biotechnol., 1997, 70, 9–20 CrossRef CAS.
- U. R. Gracida-Alvarez, M. K. Mitchell, J. C. Sacramento-Rivero and D. R. Shonnard, Effect of Temperature and Vapor Residence Time on the Micropyrolysis Products of Waste High Density Polyethylene, Ind. Eng. Chem. Res., 2018, 57(6), 1912–1923 CrossRef CAS.
- J. M. Russell, U. R. Gracida-Alvarez, O. Winjobi and D. R. Shonnard, Update to “Effect of Temperature and Vapor Residence Time on the Micropyrolysis Products of Waste High Density Polyethylene”, Ind. Eng. Chem. Res., 2020, 59(22), 10716–10719 CrossRef CAS.
- D. G. Kulas, A. Zolghadr and D. Shonnard, Micropyrolysis of Polyethylene and Polypropylene Prior to Bioconversion: The Effect of Reactor Temperature and Vapor Residence Time on Product Distribution, ACS Sustainable Chem. Eng., 2021, 9(43), 14443–14450 CrossRef.
- G. W. Meindersma, Extraction of aromatics from naphtha with ionic liquids : from solvent development to pilot RDC evaluation, 2005 Search PubMed.
- D. Zhao, X. Wang, J. B. Miller and G. W. Huber, The Chemistry and Kinetics of Polyethylene Pyrolysis: A Process to Produce Fuels and Chemicals, ChemSusChem, 2020, 13, 1764–1774 CrossRef CAS PubMed.
- I. Velghe, R. Carleer, J. Yperman and S. Schreurs, Study of the pyrolysis of municipal solid waste for the production of valuable products, J. Anal. Appl. Pyrolysis, 2011, 92(2), 366–375 CrossRef CAS.
- J. A. Conesa, R. Font, A. Marcilla and A. N. Garcia, Pyrolysis of Polyethylene in a Fluidized Bed Reactor, Energy Fuels, 1994, 8(6), 1238–1246 CrossRef CAS.
- S. D. Anuar Sharuddin, F. Abnisa, W. M. A. Wan Daud and M. K. Aroua, A review on pyrolysis of plastic wastes, Energy Convers. Manage., 2016, 115, 308–326 CrossRef CAS.
- F. J. Mastral, E. Esperanza, C. Berrueco, M. Juste and J. Ceamanos, Fluidized bed thermal degradation products of HDPE in an inert atmosphere and in air-nitrogen mixtures, J. Anal. Appl. Pyrolysis, 2003, 70(1), 1–17 CrossRef CAS.
- D. Zhao, X. Wang, J. B. Miller and G. W. Huber, The Chemistry and Kinetics of Polyethylene Pyrolysis: A Process to Produce Fuels and Chemicals, ChemSusChem, 2020, 13(7), 1764–1774 CrossRef CAS PubMed.
- A. López, I. De Marco, B. M. Caballero, M. F. Laresgoiti and A. Adrados, Influence of time and temperature on pyrolysis of plastic wastes in a semi-batch reactor, Chem. Eng. J., 2011, 173(1), 62–71 CrossRef.
- S. Orozco, J. Alvarez, G. Lopez, M. Artetxe, J. Bilbao and M. Olazar, Pyrolysis of plastic wastes in a fountain confined conical spouted bed reactor: Determination of stable operating conditions, Energy Convers. Manage., 2021, 229, 113768 CrossRef CAS.
- M. Sogancioglu, G. Ahmetli and E. Yel, A Comparative Study on Waste Plastics Pyrolysis Liquid Products Quantity and Energy Recovery Potential, Energy Procedia, 2017, 118, 221–226 CrossRef CAS.
- L. Soják, R. Kubinec, H. Jurdáková, E. Hájeková and M. Bajus, High resolution gas chromatographic-mass spectrometric analysis of polyethylene and polypropylene thermal cracking products, J. Anal. Appl. Pyrolysis, 2007, 78(2), 387–399 CrossRef.
- A. Demirbas, Pyrolysis of municipal plastic wastes for recovery of gasoline-range hydrocarbons, Journal of Analytical Applied Pyrolysis, 2004, 79, 97–102 CrossRef.
- K. H. Lee, Pyrolysis of municipal plastic wastes separated by difference of specific gravity, J. Anal. Appl. Pyrolysis, 2007, 79, 362–367 CrossRef CAS.
- O. Dogu, M. Pelucchi, R. Van de Vijver, P. H. Van Steenberge, D. R. D'hooge, A. Cuoci, M. Mehl, A. Frassoldati, T. Faravelli and K. M. Van Geem, The chemistry of chemical recycling of solid plastic waste via pyrolysis and gasification: State-of-the-art, challenges, and future directions, Prog. Energy Combust. Sci., 2021, 84, 100901 CrossRef.
- R. Miandad, M. A. Barakat, A. S. Aburiazaiza, M. Rehan, I. M. I. Ismail and A. S. Nizami, Effect of plastic waste types on pyrolysis liquid oil, Int. Biodeterior. Biodegrad., 2017, 119, 239–252 CrossRef CAS.
- I. Maafa, Pyrolysis of Polystyrene Waste: A Review, Polymers, 2021, 13(2), 225 CrossRef CAS PubMed.
- Y. S. Kim, G. C. Hwang, S. Y. Bae, S. C. Yi, S. K. Moon and H. Kumazawa, Pyrolysis of polystyrene in a batch-type stirred vessel, Korean J. Chem. Eng., 1999, 16(2), 161–165 CrossRef CAS.
- J. A. Onwudili, N. Insura, and P. T. Williams, Composition of products from the pyrolysis of polyethylene and polystyrene in a closed batch reactor: Effects of temperature and residence time, J. Anal. Appl. Pyrolysis, 2009, 86, 293–303 CrossRef CAS.
- Y. Liu, J. Qian and J. Wang, Pyrolysis of polystyrene waste in a fluidized-bed reactor to obtain styrene monomer and gasoline fraction, Fuel Process. Technol., 2000, 63(1), 45–55 CrossRef CAS.
- Y. Mo, L. Zhao, Z. Wang, C.-L. Chen, G.-Y. A. Tan and J.-Y. Wang, Enhanced styrene recovery from waste polystyrene pyrolysis using response surface methodology coupled with Box–Behnken design, Waste Manage., 2014, 34(4), 763–769 CrossRef CAS PubMed.
- H.-W. B. Wong and J. Linda, Tertiary Resource Recovery from Waste Polymers vis Pyrolysis: Neat and Binary Mixture Reactions of Polypropylene and Polystyrene, Ind. Eng. Chem. Res., 2001, 40, 4716–4723 CrossRef CAS.
- C. Bradley, What's Happening in Advanced Recycling Now. https://www.plasticstoday.com/advanced-recycling/whats-happening-advanced-recycling-now (accessed January 15).
- A. B. J. Brems, C. Vandecasteele and R. Dewil, Polymeric Cracking of Waste Polyethylene Terephthalate to Chemicals and Energy, J. Air Waste Manage. Assoc., 2011, 61(7), 721–731 CrossRef CAS.
- P. Das and P. Tiwari, Thermal degradation study of waste polyethylene terephthalate (PET) under inert and oxidative environments, Thermochim. Acta, 2019, 679, 178340 CrossRef.
- M. S. Qureshi, A. Oasmaa, H. Pihkola, I. Deviatkin, A. Tenhunen, J. Mannila, H. Minkkinen, M. Pohjakallio and J. Laine-Ylijoki, Pyrolysis of plastic waste: Opportunities and challenges, J. Anal. Appl. Pyrolysis, 2020, 152, 104804 CrossRef CAS.
- H. Jia, H. Ben, Y. Luo and R. Wang, Catalytic Fast Pyrolysis of Poly (Ethylene Terephthalate) (PET) with Zeolite and Nickel Chloride, Polymers, 2020, 12(3), 705 CrossRef CAS.
- P. T. Williams and E. Slaney, Analysis of products from the pyrolysis and liquefaction of single plastics and waste plastic mixtures, Resour., Conserv. Recycl., 2007, 51(4), 754–769 CrossRef.
- H. Almohamadi, M. Alamoudi, U. Ahmed, R. Shamsuddin and K. Smith, Producing hydrocarbon fuel from the plastic waste: Techno-economic analysis, Korean J. Chem. Eng., 2021, 38(11), 2208–2216 CrossRef.
- A. H. Tullo, Plastic has a problem; is chemical recycling the solution?https://cen.acs.org/environment/recycling/Plastic-problem-chemical-recycling-solution/97/i39 (accessed January 15).
- T. Thiounn and R. C. Smith, Advances and approaches for chemical recycling of plastic waste, Journal of Polymer Science, 2020, 58(10), 1347–1364 CrossRef CAS.
- N. George and T. Kurian, Recent Developments in the Chemical Recycling of Postconsumer Poly(ethylene terephthalate) Waste, Ind. Eng. Chem. Res., 2014, 53(37), 14185–14198 CrossRef CAS.
- H. S. Wu, Strategic Possibility Routes of Recycled PET, Polymers, 2021, 13(9), 1475 CrossRef PubMed.
- W. Kaminsky, Chemical recycling of plastics by fluidized bed pyrolysis, Fuel Communications, 2021, 8, 100023 CrossRef.
- N. Miskolczi, L. Bartha and A. Angyal, Pyrolysis of Polyvinyl Chloride (PVC)-Containing Mixed Plastic Wastes for Recovery of Hydrocarbons, Energy Fuels, 2009, 23(5), 2743–2749 CrossRef CAS.
- A. Buekens, Introduction to Feedstock Recycling of Plastics, in Feedstock Recycling and Pyrolysis of Waste Plastics: Converting Waste Plastics into Diesel and Other Fuels, John Wiley & Sons, Ltd, 2006 Search PubMed.
- A. López, I. De Marco, B. M. Caballero, M. F. Laresgoiti and A. Adrados, Dechlorination of fuels in pyrolysis of PVC containing plastic wastes, Fuel Process. Technol., 2011, 92(2), 253–260 CrossRef.
- J. Scheirs and W. Kaminsky, Feedstock Recycling and Pyrolysis of Waste Plastics: Converting Waste Plastics into Diesel and Other Fuels, John Wiley & Sons, Ltd, 2006 Search PubMed.
- W. Kaminsky and H. Sinn, Pyrolysis of plastic waste and scrap tires using a fluidized-bed process, in Thermal Conversion of Solid Wastes and Biomass, ed. A. C. Society, 1980, vol. 130, pp. 423–439 Search PubMed.
- H. R. Sinn, W. Kaminsky and J. R. Janning, Processing of Plastic Waste and Scrap Tires into Chemical Raw Materials, Especially by Pyrolysis, Angew. Chem., Int. Ed. Engl., 1976, 15(11), 660–672 CrossRef.
- W. Chen, S. Shi, J. Zhang, M. Chen and X. Zhou, Co-pyrolysis of waste newspaper with high-density polyethylene: Synergistic effect and oil characterization, Energy Convers. Manage., 2016, 112, 41–48 CrossRef CAS.
- D. V. Suriapparao, A. Yerrayya, G. Nagababu, R. K. Guduru and T. H. Kumar, Recovery of renewable aromatic and aliphatic hydrocarbon resources from microwave pyrolysis/co-pyrolysis of agro-residues and plastics wastes, Bioresour. Technol., 2020, 318, 124277 CrossRef CAS PubMed.
- B. B. Uzoejinwa, X. He, S. Wang, A. E. F. Abomohra, Y. Hu and Q. Wang, Co-pyrolysis of biomass and waste plastics as a thermochemical conversion technology for high-grade biofuel production: Recent progress and future directions elsewhere worldwide, Energy Convers. Manage., 2018, 163, 468–492 CrossRef CAS.
- Z. B. Wang, G. Kiran, T. Lei and A. K. Gupta, Co-pyrolysis of waste plastic and solid biomass for synergistic production of biofuels and chemicals - A review, Prog. Energy Combust. Sci., 2021, 84, 100899 CrossRef.
- W. Ścierski, Migration of Sulfur and Nitrogen in the Pyrolysis Products of Waste and Contaminated Plastics, Appl. Sci., 2021, 11(10), 4374 CrossRef.
- N. Sakthipriya, Plastic waste management: A road map to achieve circular economy and recent innovations in pyrolysis, Sci. Total Environ., 2022, 809, 151160 CrossRef PubMed.
- L. Mingxing, W. Mei, L. Zelong and Z. Qundan, Qualitative and Quantitative Analysis of Silicon Species in Pyrolysis Oil From Waste Plastic, Acta Pet. Sin., 2021, 37(4), 909 Search PubMed.
- T. Bhaskar, T. Matsui, J. Kaneko, M. A. Uddin, A. Muto and Y. Sakata, Novel calcium based sorbent (Ca-C) for the dehalogenation (Br, Cl) process during halogenated mixed plastic (PP/PE/PS/PVC and HIPS-Br) pyrolysis, Green Chem., 2002, 4(4), 372–375 RSC.
- T. A. U. M. Bhaskar, J. Kaneko, T. Matsui, A. Muto and Y. Sakata, Pyrolysis of Polypropylene/Polyethylene/Polystyrene and Polyvinylchloride Mixed Plastics using CaCO3, Prog. Rubber, Plast. Recycl. Technol., 2004, 20(2), 163–170 CrossRef CAS.
- B. Dou, J. Gao, S. W. Baek and X. Sha, High-Temperature HCl Removal with Sorbents in a Fixed-Bed Reactor, Energy Fuels, 2003, 17(4), 874–878 CrossRef CAS.
- Y. Sakata, M. A. Uddin, K. Koizumi and K. Murata, Thermal degradation of polyethylene mixed with poly(vinyl chloride) and poly(ethyleneterephthalate), Polym. Degrad. Stab., 1996, 53(1), 111–117 CrossRef CAS.
- M. A. Uddin, Y. Sakata, Y. Shiraga, A. Muto and K. Murata, Dechlorination of Chlorine Compounds in Poly(vinyl chloride) Mixed Plastics Derived Oil by Solid Sorbents, Ind. Eng. Chem. Res., 1999, 38(4), 1406–1410 CrossRef CAS.
- M. Fukushima, B. Wu, H. Ibe, K. Wakai, E. Sugiyama, H. Abe, K. Kitagawa, S. Tsuruga, K. Shimura and E. Ono, Study on dechlorination technology for municipal waste plastics containing polyvinyl chloride and polyethylene terephthalate, J. Mater. Cycles Waste Manage., 2010, 12, 108–122 CrossRef CAS.
- A. Soler, J. Conesa and N. Ortuño, Application of Subcritical Water to Dechlorinate Polyvinyl Chloride Electric Wires, Energies, 2018, 11(10), 2612 CrossRef.
- A. Lopez-Urionabarrenechea, I. De Marco, B. M. Caballero, M. F. Laresgoiti and A. Adrados, Upgrading of chlorinated oils coming from pyrolysis of plastic waste, Fuel Process. Technol., 2015, 137, 229–239 CrossRef CAS.
- M. Kusenberg, A. Eschenbacher, M. R. Djokic, A. Zayoud, K. Ragaert, S. De Meester and K. M. Van Geem, Opportunities and challenges for the application of post-consumer plastic waste pyrolysis oils as steam cracker feedstocks: To decontaminate or not to decontaminate?, Waste Manage., 2022, 138, 83–115 CrossRef CAS PubMed.
- J. Brock, V. Valerie and G. John, The recycling myth - Big oil's solution for plastic waste littered with failure. https://www.reuters.com/investigates/special-report/environment-plastic-oil-recycling/(accessed 14 January).
- A. B. Koc and Y. Ali, Catalytic and thermal oxidative pyrolysis of LDPE in a continuous reactor system, J. Anal. Appl. Pyrolysis, 2007, 78(1), 7–13 CrossRef CAS.
- R. C. Brown, Process Intensification through Directly Coupled Autothermal Operation of Chemical Reactors, Joule, 2020, 4(11), 2268–2289 CrossRef CAS.
- J. P. Polin, C. A. Peterson, L. E. Whitmer, R. G. Smith and R. C. Brown, Process intensification of biomass fast pyrolysis through autothermal operation of a fluidized bed reactor, Appl. Energy, 2019, 249, 276–285 CrossRef CAS.
- M. Y. Wey, C. S. Lo, S. Y. Wu and Y. T. Lee, Operating parameters of autothermal pyrolysis of plastic waste in a fluidized bed, Waste Manage. Res., 1998, 16(1), 72–81 CrossRef CAS.
- M. Gholizadeh, C. Li, S. Zhang, Y. Wang, S. Niu, Y. Li and X. Hu, Progress of the development of reactors for pyrolysis of municipal waste, Sustainable Energy Fuels, 2020, 4(12), 5885–5915 RSC.
- E. Butler, G. Devlin and K. Mcdonnell, Waste Polyolefins to Liquid Fuels via Pyrolysis: Review of Commercial State-of-the-Art and Recent Laboratory Research, Waste Biomass Valorization, 2011, 2(3), 227–255 CrossRef CAS.
- D. Chen, L. Yin, H. Wang and P. He, Pyrolysis technologies for municipal solid waste: A review, Waste Manage., 2014, 34(12), 2466–2486 CrossRef CAS PubMed.
- S. H. Gebre, M. G. Sendeku and M. Bahri, Recent Trends in the Pyrolysis of Non–Degradable Waste Plastics, ChemistryOpen, 2021, 10(12), 1202–1226 CrossRef CAS PubMed.
- D. Kunii and O. Levenspiel, in Fluidization Engineering, Butterworth-Heinemann, 2nd edn, 1991 Search PubMed.
- W. Kaminsky, The Hamburg Fluidized-bed Pyrolysis Process to Recycle Polymer Wastes and Tires, in Feedstock Recycling and Pyrolysis of Waste Plastics: Converting Waste Plastics into Diesel and Other Fuels, ed. Schiers and J. K. Walter, John Wiley & Sons, Ltd, 2006, pp. 475–491 Search PubMed.
- M. Predel and W. Kaminsky, Pyrolysis of mixed polyolefins in a fluidised-bed reactor and on a pyro-GC/MS to yield aliphatic waxes, Polym. Degrad. Stab., 2000, 70(3), 373–385 CrossRef CAS.
- W. Kaminsky and J. Franck, Monomer recovery by pyrolysis of poly(methyl methacrylate) (PMMA), J. Anal. Appl. Pyrolysis, 1991, 19, 311–318 CrossRef CAS.
- S.-H. Jung, M.-H. Cho, B.-S. Kang and J.-S. Kim, Pyrolysis of a fraction of waste polypropylene and polyethylene for the recovery of BTX aromatics using a fluidized bed reactor, Fuel Process. Technol., 2010, 91(3), 277–284 CrossRef CAS.
- F. J. Mastral, E. G. P. Esperanza and M. Juste, Pyrolysis of high-density polyethylene in a fluidised bed reactor. Influence of the temperature and residence time, J. Anal. Appl. Pyrolysis, 2002, 63(1), 1–15 CrossRef CAS.
- K. B. Park, Y. S. Jeong, B. Guzelciftci and J. S. Kim, Characteristics of a new type continuous two-stage pyrolysis of waste polyethylene, Energy, 2018, 166, 343–351 CrossRef.
- T. Yoshioka, G. Grause, C. Eger, W. Kaminsky and A. Okuwaki, Pyrolysis of poly(ethylene terephthalate) in a fluidised bed plant, Polym. Degrad. Stab., 2004, 86(3), 499–504 CrossRef CAS.
- U. M. Arena and M. Laura, Fluidized Bed Pyrolysis of Plastic Wastes, in Feedstock Recycling and Pyrolysis of Waste Plastics: Converting Waste Plastics into Diesel and Other Fuels, ed. W. K. John Scheirs, John Wiley & Sons, Ltd, 2006, pp. 435–474 Search PubMed.
- J. F. B. C. Mastral and J. Ceamanos, Modelling of the pyrolysis of high density polyethylene: Product distribution in a fluidized bed reactor, J. Anal. Appl. Pyrolysis, 2007, 313–322 CrossRef CAS.
- A. M. W. Cunliffe and T. Paul, Composition of oils derived from the batch pyrolysis of tyres, J. Anal. Appl. Pyrolysis, 1998, 44(2), 131–152 CrossRef CAS.
- M. Arabiourrutia, G. Elordi, M. Olazar and J. Bilbao, Pyrolysis of Polyolefins in a Conical Spouted Bed Reactor: A Way to Obtain Valuable Products, in Pyrolysis, InTech, 2017 Search PubMed.
- M. Artetxe, G. Lopez, M. Amutio, I. Barbarias, A. Arregi, R. Aguado, J. Bilbao and M. Olazar, Styrene recovery from polystyrene by flash pyrolysis in a conical spouted bed reactor, Waste Manage., 2015, 45, 126–133 CrossRef CAS PubMed.
- R. Aguado, M. Olazar, B. Gaisán, R. Prieto and J. Bilbao, Kinetic Study of Polyolefin Pyrolysis in a Conical Spouted Bed Reactor, Ind. Eng. Chem. Res., 2002, 41(18), 4559–4566 CrossRef CAS.
- G. Elordi, M. Olazar, G. Lopez, M. Artetxe and J. Bilbao, Product Yields and Compositions in the Continuous Pyrolysis of High-Density Polyethylene in a Conical Spouted Bed Reactor, Ind. Eng. Chem. Res., 2011, 50(11), 6650–6659 CrossRef CAS.
- B. V. M. Csukas, N. Miskolczi, S. Balogh, A. Angyal and L. Bartha, Simplified dynamic simulation model of plastic waste pyrolysis in laboratory and pilot scale tubular reactor, Fuel Process. Technol., 2013, 106, 186–200 CrossRef CAS.
- B. Fekhar, V. Zsinka and N. Miskolczi, Value added hydrocarbons obtained by pyrolysis of contaminated waste plastics in horizontal tubular reactor: In situ upgrading of the products by chlorine capture, J. Cleaner Prod., 2019, 241, 118166 CrossRef CAS.
- W. J. Hall, N. M. M. Mitan, T. Bhaskar, A. Muto, Y. Sakata and P. T. Williams, The co-pyrolysis of flame retarded high impact polystyrene and polyolefins, J. Anal. Appl. Pyrolysis, 2007, 80, 406–415 CrossRef CAS.
- E. Hartulistiyoso, F. A. Sigiro and M. Yulianto, Temperature Distribution of the Plastics Pyrolysis Process to Produce Fuel at 450 °C, Proc. Environ. Sci., 2015, 28, 234–241 CrossRef CAS.
- W. K. Buah, A. M. Cunliffe and P. T. Williams, Characterization of Products from the Pyrolysis of Municipal Solid Waste, Process Saf. Environ. Prot., 2007, 85(5), 450–457 CrossRef CAS.
- S. Luo, B. Xiao, Z. Hu and S. Liu, Effect of particle size on pyrolysis of single-component municipal solid waste in fixed bed reactor, Int. J. Hydrogen Energy, 2010, 35(1), 93–97 CrossRef CAS.
- F. Campuzano, R. C. Brown and J. D. Martínez, Auger reactors for pyrolysis of biomass and wastes, Renewable Sustainable Energy Rev., 2019, 102, 372–409 CrossRef CAS.
- S. B. H. Gephart and C. John, Process and Apparatus for Producing Hydrocarbon Fuel from Waste Plastic, 2015 Search PubMed.
- S. M. Al-Salem, Y. Yang, J. Wang and G. A. Leeke, Pyro-Oil and Wax Recovery from Reclaimed Plastic Waste in a Continuous Auger Pyrolysis Reactor, Energies, 2020, 13(8), 2040 CrossRef CAS.
- J. Schabel, R. A. Schwarz, C. W. Grispin, M. A. Gencer and J. D. Hensel, Process and apparatus for producing petroleum products, US Pat., 10711202, 2017 Search PubMed.
- J. Xu, J. Yu, W. He, J. Huang, J. Xu and G. Li, Recovery of carbon black from waste tire in continuous commercial rotary kiln pyrolysis reactor, Sci. Total Environ., 2021, 772, 145507 CrossRef CAS.
- S.-Q. Li, Q. Yao, Y. Chi, J.-H. Yan and K.-F. Cen, Pilot-Scale Pyrolysis of Scrap Tires in a Continuous Rotary Kiln Reactor, Ind. Eng. Chem. Res., 2004, 43(17), 5133–5145 CrossRef CAS.
- A. Prakash and R. R. Palkar, Co-processing of plastic waste in a cement kiln: a better option, Environ. Sci. Pollut. Res., 2021, 1–11 CAS.
- Y. J. Zhang, C. Guozhao, W. Chuanshuai, W. Yinxiang and L. Weijian, Aimin, Liquid oils produced from pyrolysis of plastic wastes with heat carrier in rotary kiln, Fuel Process. Technol., 2020, 206 Search PubMed.
- F. C. S. Fantozzi, P. Bartocci and U. Desideri, Rotary Kiln Slow Pyrolysis for Syngas and Char Production from Biomass and Waste-Part I: Working Envelope of the Reactor, J. Eng. Gas Turbines Power, 2007, 129, 901–907 CrossRef CAS.
- A. Olufemi and S. Olagboye, Thermal conversion of waste plastics into fuel oil, Int. J. Petrochem. Sci. Eng., 2017, 2(8), 252–257 Search PubMed.
- S.-S. Kim and S. Kim, Pyrolysis characteristics of polystyrene and polypropylene in a stirred batch reactor, Chem. Eng. J., 2004, 98(1–2), 53–60 CrossRef CAS.
- H. Smuda, The method of obtaining liquid fuels from polyolefine wastes, 1990 Search PubMed.
- D. McNamara, C. L. M. Murray and C. Dublin, Conversion of Waste Plastics Materal to Fuel, 2012 Search PubMed.
- A. Brems, J. B. C. Vandecasteele and R. Dewil, Polymeric Cracking of Waste Polyethylene Terephthalate to Chemicals and Energy, J. Air Waste Manage. Assoc., 2011, 61(7), 721–731 CrossRef CAS PubMed.
- R. C. Brown, Process Intensification through Directly Coupled Autothermal Operation of Chemical Reactors, Joule, 2020, 4, 2268–2289 CrossRef CAS.
- M. Blazso, Composition of Liquid Fuels Derived from the Pyrolysis of Plastics, in Feedstock Recycling and Pyrolysis of Waste Plastics: Converting Waste Plastics into Diesel and Other Fuels, 2006, pp. 315–344 Search PubMed.
- TotalEnergies Doubles its Recycled Plastic Production Capacity in France. https://totalenergies.com/media/news/press-releases/totalenergies-doubles-its-recycled-plastic-production-capacity-france.
- Shell and Pryme Sign Strategic Plastic Waste to Chemicals Cooperation Agreement. https://www.shell.com/business-customers/chemicals/media-releases/2021-media-releases/shell-and-pryme-sign-cooperation-agreement.html.
- A. H. Tullo, ExxonMobil plunges into plastics recycling. https://cen.acs.org/environment/recycling/ExxonMobil-plunges-plastics-recycling/99/i38.
- M. Smalley, SABIC, BP collaborate at chemical complex in Germany. https://www.recyclingtoday.com/article/sabic-bp-collaborate-chemical-complex-germany/.
- A. Fivga and I. Dimitriou, Pyrolysis of plastic waste for production of heavy fuel substitute: A techno-economic assessment, Energy, 2018, 149, 865–874 CrossRef CAS.
- M. Larrain, S. Van Passel, G. Thomassen, U. Kresovic, N. Alderweireldt, E. Moerman and P. Billen, Economic performance of pyrolysis of mixed plastic waste: Open-loop versus closed-loop recycling, J. Cleaner Prod., 2020, 270, 122442 CrossRef CAS.
- J. Sahu, K. Mahalik, H. K. Nam, T. Y. Ling, T. S. Woon, M. S. bin Abdul Rahman, Y. Mohanty, N. Jayakumar and S. Jamuar, Feasibility study for catalytic cracking of waste plastic to produce fuel oil with reference to M alaysia and simulation using ASPEN Plus, Environ. Prog. Sustainable Energy, 2014, 33(1), 298–307 CrossRef CAS.
- U. R. Gracida-Alvarez, O. Winjobi, J. C. Sacramento-Rivero and D. R. Shonnard, System Analyses of High-Value Chemicals and Fuels from a Waste High-Density Polyethylene Refinery. Part 1: Conceptual Design and Techno-Economic Assessment, ACS Sustainable Chem. Eng., 2019, 7(22), 18254–18266 CrossRef CAS.
- M. A. A. J. Ghodrat, D. Hagare, R. Yang and B. Samali, Economic feasibility of energy recovery from waste plastic using pyrolysis technology: an Australian perspective, Int. J. Environ. Sci. Technol., 2019, 16, 3721–3734 CrossRef CAS.
- A. Tukker, H. G. L. Simons and S. Wiegerasma, Chemical Recycling of Plastics Waste (PVC and other resins), TNO Institute, 1999 Search PubMed.
- Y. Sakata, T. Bhaskar, M. Uddin, A. Muto and T. Matsui, Development of a catalytic dehalogenation (Cl, Br) process for municipal waste plastic-derived oil, J. Mater. Cycles Waste Manage., 2003, 113–124 CrossRef CAS.
- Energy, P. Plastic Energy Achieves RSB Certification For Recycling Plant In Sevilla, Spain. https://plasticenergy.com/rsb_certification_sevilla/.
- Sabic bp and sabic embark on new cooperation for products from advanced plastics recycling. https://www.sabic.com/en/news/26431-bp-and-sabic-embark-on-new-cooperation.
- Shell Plastic waste. https://www.shell.com/sustainability/environment/plastic-waste.html.
- Energy, P. Plastic Energy Announces Final Investment Decision And Start Of Construction Works For Advanced Recycling Plant In France. https://plasticenergy.com/plastic-energy-announces_fid_and_start_of_construction_works_for_advanced_recycling_plant_in_france/.
- Quantafuel expands into the UK to help solve plastic waste problem. https://www.quantafuel.com/media#/pressreleases/quantafuel-expands-into-the-uk-to-help-solve-plastic-waste-problem-3130820.
- Third Quarter 2021 Consolidated financial statements Quantafuel ASA, Quantafuel ASA, 2021 Search PubMed.
- J. M. Lee and E. W. C. Lim, Heat Transfer in a Pulsating Turbulent Fluidized Bed, Appl. Therm. Eng., 2020, 174, 115321 CrossRef.
- Dow and Fuenix enter into a partnership for the production of 100% circular plastic. https://corporate.dow.com/en-us/news/press-releases/dow-and-fuenix-enter-into-a-partnership-for-the-production-of-10.html.
- Dow Terneuzen. https://nl.dow.com/en-us/locations/terneuzen.html (accessed 2/22).
- Sandoval, D. Dow, Fuenix Ecogy Group partner on plastics recycling project. https://www.recyclingtoday.com/article/dow-to-produce-new-plastics-from-pyrolysis-oil-netherlands/(accessed 2/22).
- Ashley Plastics Renewal Facility. https://www.brightmark.com/plastics-renewal/projects/ashley-indiana.
- ORDC Freepoint Commodities to Receive $50,000 Grant. https://rail.ohio.gov/wps/portal/gov/ordc/about-ordc/news-and-events/ordc-approves-grant-for-freepoint-commodities.
- Z. Xu, S. S. Kolapkar, S. Zinchik, E. Bar-Ziv and A. G. McDonald, Comprehensive kinetic study of thermal degradation of polyvinylchloride (PVC), Polym. Degrad. Stab., 2020, 176, 109148 CrossRef CAS.
- Energy, P. Plastic Recycling: Plastic Energy, Freepoint Eco-Systems And TotalEnergies Partner On Advanced Recycling Project In The U.S. https://plasticenergy.com/plastic-energy-freepoint-eco-systems-totalenergies-advanced-recycling-project-in-the-us/.
- Shell Investment in shell energy and chemicals park singapore to bring circular chemicals to asia pacific customers. https://www.shell.com.sg/media/2021-media-releases/investment-in-shell-energy-and-chemicals-park-singapore-to-bring-circular-chemicals-to-asia-pacific-customers.html.
- Energy, P. Plastic Energy Joins Forces with WWF to Combat Plastic Pollution in Indonesia. https://plasticenergy.com/press-release-plastic-energy-join-forces-with-wwf/.
- T. M. Hundertmark, M. Mirjam, J. S. Chris, W. Theo, Christof How plastics waste recycling could transform the chemical industry. https://www.mckinsey.com/industries/chemicals/our-insights/how-plastics-waste-recycling-could-transform-the-chemical-industry (accessed January 14).
- Pryme Q3 2021 Presentation, in Pryme converts plastic waste into valuable products on an industrial scale, 2021 Search PubMed.
- H. Jiang, E. Celeste and B. Stephanis, How waste solutions company Brightmark is converting plastic waste into fuel. https://www.businessinsider.com/brightmark-plastic-waste-fuel-pollution-2020-10 (accessed 2/22).
- J. Aguado, D. P. Serrano and J. M. Escola, Fuels From Waste Plastics by Thermal and Catalytic Processes: A Review, Ind. Eng. Chem. Res., 2008, 47, 7982–7992 CrossRef CAS.
- R. Miandad, M. A. Barakat, A. S. Aburiazaiza, M. Rehan and A. S. Nizami, Catalytic pyrolysis of plastic waste: A review, Process Saf. Environ. Prot., 2016, 102, 822–838 CrossRef CAS.
- S. M. Al-Salem, A. Antelava, A. Constantinou, G. Manos and A. Dutta, A review on thermal and catalytic pyrolysis of plastic solid waste (PSW), J. Environ. Manage., 2017, 197, 177–198 CrossRef CAS PubMed.
- H. Ohkita, R. Nishiyama, Y. Tochihara, T. Mizushima, N. Kakuta, Y. Morioka, A. Ueno, Y. Namiki, S. Tanifuji, H. Katoh, H. Sunazuka, R. Nakayama and T. Kuroyanagi, Acid Properties of Silica-Alumina Catalysts and Catalytic Degradation of Polyethylene, Ind. Eng. Chem. Res., 1993, 32, 3112–3116 CrossRef CAS.
- G. Audisio, F. Bertini, P. L. Beltrame and P. Carniti, Catalytic Degradation of Polymers: Part III - Degradation of Polystyrene, Polym. Degrad. Stab., 1990, 29, 191–200 CrossRef CAS.
- A. Maity, S. Chaudhari, J. J. Titman and V. Polshettiwar, Catalytic nanosponges of acidic aluminosilicates for plastic degradation and CO2 to fuel conversion, Nat. Commun., 2020, 11(1), 1–12 CrossRef PubMed.
- R. Ochoa, H. Van Woert, W. H. Lee, R. Subramanian, E. Kugler and P. C. Eklund, Catalytic Degradation of Medium Density Polyethylene Over Silica - Alumina Supports, Fuel Process. Technol., 1996, 49, 119–136 CrossRef CAS.
- J. Aguado, D. P. Serrano, J. M. Escola, E. Garagorri and J. A. Fernández, Catalytic conversion of polyolefins into fuels over zeolite beta, Polym. Degrad. Stab., 2000, 69, 11–16 CrossRef CAS.
- M. A. Uddin, Y. Sakata, A. Muto, Y. Shiraga, K. Koizumi, Y. Kanada and K. Murata, Catalytic Degradation of Polyethylene and Polypropylene into Liquid Hydrocarbons with Mesoporous Silica, Microporous Mesoporous Mater., 1998, 21, 557–564 CrossRef.
- Y. Sakata, M. A. Uddin and A. Muto, Degradation of Polyethylene and Polypropylene into Fuel Oil by Using Solid Acid and Non-Acid Catalysts, J. Anal. Appl. Pyrolysis, 1999, 51, 135–155 CrossRef CAS.
- D. P. Serrano, J. Aguado and J. M. Escola, Catalytic Conversion of Polystyrene over HMCM-41, HZSM-5 and Amorphous SiO2-Al2O3: Comparison with Thermal Cracking, Appl. Catal., B, 2000, 25, 181–189 CrossRef CAS.
- E. Y. Hwang, J. R. Kim, J. K. Choi, H. C. Woo and D. W. Park, Performance of Acid Treated Natural Zeolites in Catalytic Degradation of Polypropylene, J. Anal. Appl. Pyrolysis, 2002, 62, 351–364 CrossRef CAS.
- B. Roozbehani, M. Motevassel, M. Mirdrikvand, S. I. Moqadam and A. Kharaghani, Gasoline Production from a Polymeric Urban Disposal Mixture using Silica–Alumina Catalyst, Clean Technol. Environ. Policy, 2017, 19, 123–136 CrossRef CAS.
- Y. Sakata, M. A. Uddin, A. Muto, Y. Kanada, K. Koizumi and K. Murata, Catalytic degradation of Polyethylene into Fuel Oil Over Mesoporous Silica (KFS-16) Catalyst, J. Anal. Appl. Pyrolysis, 1997, 43, 15–25 CrossRef CAS.
- N. Miskolczi, C. Wu and P. T. Williams, Fuels by Waste Plastics using Activated Carbon, MCM-41, HZSM-5 and Their Mixture, MATEC Web Conf., 2016, 49, 05001 CrossRef.
- D. Munir, F. Piepenbreier and M. R. Usman, Hydrocracking of a Plastic Mixture over Various Micro-Mesoporous Composite Zeolites, Powder Technol., 2017, 316, 542–550 CrossRef CAS.
- C. Ma, J. Yu, B. Wang, Z. Song, J. Xiang, S. Hu, S. Su and L. Sun, Catalytic Pyrolysis of Flame Retarded High Impact Polystyrene over Various Solid Acid Catalysts, Fuel Process. Technol., 2017, 155, 32–41 CrossRef CAS.
- Q. Huang, C. Liu, R. Wei and J. Wang, Experimental Study of Polyethylene Pyrolysis and Combustion over HZSM-5, HUSY, and MCM-41, J. Hazard. Mater., 2017, 333, 10–22 CrossRef CAS PubMed.
- S. C. Kim, B. S. Kang, B. S. Kim, Y. M. Kim, J. K. Joen and Y. K. Park, Catalytic Pyrolysis of Municipal Plastic Film Wastes over Nanoporous Al-MCM-41, J. Nanosci. Nanotechnol., 2018, 18, 1078–1082 CrossRef CAS PubMed.
- L. O. Mark, M. C. Cendejas and I. Hermans, The Use of Heterogeneous Catalysis in the Chemical Valorization of Plastic Waste (ChemSusChem 22/2020), ChemSusChem, 2020, 13, 5773–5773 CrossRef CAS.
- D. P. Serrano, J. Aguado, J. M. Escola, E. Garagorri, J. M. Rodríguez, L. Morselli, G. Palazzi and R. Orsi, Feedstock Recycling of Agriculture Plastic Film Wastes by Catalytic Cracking, Appl. Catal., B, 2004, 49, 257–265 CrossRef CAS.
- H. Juwono, L. Fauziah, I. Q. Uyun, R. Alfian, Suprapto, Y. L. Ni'Mah and I. Ulfin, Catalytic Convertion of Al-MCM-41-Ceramic on Hidrocarbon (C8 - C12) Liquid Fuel Synthesis from Polypropylene Plastic Waste, AIP Conf. Proc., 2018, 2049 Search PubMed.
- P. Verma, Y. Kuwahara, K. Mori, R. Raja and H. Yamashita, Functionalized mesoporous SBA-15 silica: recent trends and catalytic applications, Nanoscale, 2020, 12, 11333–11363 RSC.
- K. Li, S. Lee, G. Yuan, J. Lei, S. Lin, P. Weerachanchai, Y. Yang and J.-Y. Wang, Investigation into the Catalytic Activity of Microporous and Mesoporous Catalysts in the Pyrolysis of Waste Polyethylene and Polypropylene Mixture, Energies, 2016, 9, 431 CrossRef.
- Z. Zhang, K. Gora-Marek, J. S. Watson, J. Tian, M. R. Ryder, K. A. Tarach, L. López-Pérez, J. Martínez-Triguero and I. Melián-Cabrera, Recovering Waste Plastics using Shape-Selective Nano-Scale Reactors as Catalysts, Nat. Sustain., 2019, 2, 39–42 CrossRef.
- A. R. Songip, T. Masuda, H. Kuwahara and K. Hashimoto, Production of High-Quality Gasoline by Catalytic Cracking over Rare-Earth Metal Exchanged Y-Type Zeolites of Heavy Oil from Waste Plastics, Energy Fuels, 1994, 8, 136–140 CrossRef CAS.
- G. Manos, A. Garforth and J. Dwyer, Catalytic Degradation of High-Density Polyethylene on an Ultrastable-Y Zeolite. Nature of Initial Polymer Reactions, Pattern of Formation of Gas and Liquid Products, and Temperature Effects, Ind. Eng. Chem. Res., 2000, 39, 1203–1208 CrossRef CAS.
- G. Manos, I. Y. Yusof, N. Papayannakos and N. H. Gangas, Catalytic cracking of polyethylene over clay catalysts. Comparison with an ultrastable Y zeolite, Ind. Eng. Chem. Res., 2001, 40, 2220–2225 CrossRef CAS.
- I. C. Neves, G. Botelho, A. V. Machado and P. Rebelo, The effect of acidity behaviour of Y zeolites on the catalytic degradation of polyethylene, Eur. Polym. J., 2006, 42, 1541–1547 CrossRef CAS.
- M. Davoodpour, R. Tafreshi, A. A. Khodadadi and Y. Mortazavi, Two-Stage Cracking Catalyst of Amorphous Silica-Alumina on Y Zeolite for Enhanced Product Selectivity and Suppressed Coking, Korean J. Chem. Eng., 2017, 34, 681–691 CrossRef CAS.
- A. A. Ajibola, J. A. Omoleye and V. E. Efeovbokhan, Catalytic Cracking of Polyethylene Plastic Waste using Synthesised Zeolite Y from Nigerian Kaolin Deposit, Appl. Petrochem. Res., 2018, 8, 211–217 CrossRef CAS.
- N. Miskolczi, T. Juzsakova and J. Sója, Preparation and Application of Metal Loaded ZSM-5 and Y-Zeolite Catalysts for Thermo-Catalytic Pyrolysis of Real End of Life Vehicle Plastics Waste, J. Energy Inst., 2019, 92, 118–127 CrossRef CAS.
- B. P. S. Santos, D. D. Almeida, M. d. F. V. Marques and C. A. Henriques, Degradation of Polypropylene and Polyethylene Wastes Over HZSM-5 and USY Zeolites, Catal. Lett., 2019, 149, 798–812 CrossRef CAS.
- W. U. Eze, I. C. Madufor, G. N. Onyeagoro, H. C. Obasi and M. I. Ugbaja, Study on the Effect of Kankara Zeolite-Y-Based Catalyst on the Chemical Properties of LIquid Fuel from Mixed Waste Plastics (MWPs) Pyrolysis, Polym. Bull., 2020, 03116 Search PubMed.
- A. Marcilla, A. Gómez, J. A. Reyes-Labarta, A. Giner and F. Hernández, Kinetic study of polypropylene pyrolysis using ZSM-5 and an equilibrium fluid catalytic cracking catalyst, J. Anal. Appl. Pyrolysis, 2003, 68–69, 467–480 CrossRef CAS.
- Q. Zhou, L. Zheng, Y. Z. Wang, G. M. Zhao and B. Wang, Catalytic degradation of low-density polyethylene and polypropylene using modified ZSM-5 zeolites, Polym. Degrad. Stab., 2004, 84, 493–497 CrossRef CAS.
- J. G. A. P. Filho, E. C. Graciliano, A. O. S. Silva, M. J. B. Souza and A. S. Araujo, Thermo gravimetric kinetics of polypropylene degradation on ZSM-12 and ZSM-5 catalysts, Catal. Today, 2005, 107–108, 507–512 CrossRef.
- S. Boxiong, W. Chunfei, G. Binbin and W. Rui, Liangcai, Pyrolysis of waste tyres with zeolite USY and ZSM-5 catalysts, Appl. Catal., B, 2007, 73, 150–157 CrossRef.
- A. López, I. de Marco, B. M. Caballero, A. Adrados and M. F. Laresgoiti, Deactivation and regeneration of ZSM-5 zeolite in catalytic pyrolysis of plastic wastes, Waste Manage., 2011, 31, 1852–1858 CrossRef PubMed.
- A. López, I. de Marco, B. M. Caballero, M. F. Laresgoiti, A. Adrados and A. Aranzabal, Catalytic pyrolysis of plastic wastes with two different types of catalysts: ZSM-5 zeolite and Red Mud, Appl. Catal., B, 2011, 104, 211–219 CrossRef.
- J. Y. Lee, S. M. Park, S. K. Saha, S. J. Cho and G. Seo, Liquid-phase degradation of polyethylene (PE) over MFI zeolites with mesopores: Effects of the structure of PE and the characteristics of mesopores, Appl. Catal., B, 2011, 108–109, 61–71 CrossRef CAS.
- A. Coelho, L. Costa, M. M. Marques, I. M. Fonseca, M. A. Lemos and F. Lemos, The effect of ZSM-5 zeolite acidity on the catalytic degradation of high-density polyethylene using simultaneous DSC/TG analysis, Appl. Catal., A, 2012, 413–414, 183–191 CrossRef CAS.
- N. Carmo, D. Afonso, E. Santos, I. Fonseca, F. Lemos and M. A. Lemos, Coprocessing of Waste Plastic and Hydrocarbons over MFI (HZSM-5), Int. J. Chem. Kinet., 2016, 48, 329–336 CrossRef CAS.
- Y. Zhao, Z. Ye, H. Zhang, Y. Zhang and Y. Tang, Facile Fabrication and Morphology Regulation of Crossed MFI Zeolite with Improved Performance on LDPE Cracking, Ind. Eng. Chem. Res., 2019, 58, 13174–13181 CrossRef CAS.
- A. Marcilla, A. Gómez-Siurana and F. Valdés, Catalytic Cracking of Low-Density Polyethylene over H-Beta and HZSM-5 Zeolites: Influence of the External Surface. Kinetic Model, Polym. Degrad. Stab., 2007, 92, 197–204 CrossRef CAS.
- M. S. Renzini, L. C. Lerici, U. Sedran and L. B. Pierella, Stability of ZSM-11 and BETA zeolites during the catalytic cracking of low-density polyethylene, J. Anal. Appl. Pyrolysis, 2011, 92, 450–455 CrossRef CAS.
- K. Wanchai and A. Chaisuwan, Catalytic Cracking of Polypropylene Waste over Zeolite Beta, J. Chem. Mater. Res., 2013, 3, 2225–2956 Search PubMed.
- H. W. Lee, S. J. Choi, J. K. Jeon, S. H. Park, S. C. Jung and Y. K. Park, Catalytic conversion of waste particle board and polypropylene over H-beta and HY zeolites, Renewable Energy, 2015, 79, 9–13 CrossRef CAS.
- D. Munir, H. Amer, R. Aslam, M. Bououdina and M. R. Usman, Composite Zeolite Beta Catalysts for Catalytic Hydrocracking of Plastic Waste to Liquid Fuels, Mater, Renewable Sustainable Energy Rev., 2020, 9, 9 Search PubMed.
- K. Pyra, K. A. Tarach, E. Janiszewska, D. Majda and K. Góra-Marek, Evaluation of the Textural Parameters of Zeolite Beta in LDPE Catalytic Degradation: Thermogravimetric Analysis Coupled with FTIR Operando Studies, Molecules, 2020, 25(4), 926 CrossRef CAS PubMed.
- S. Y. Lee, J. H. Yoon, J. R. Kim and D. W. Park, Catalytic Degradation of Polystyrene over Natural Clinoptilolite Zeolite, Polym. Degrad. Stab., 2001, 74, 297–305 CrossRef CAS.
- S. Salamah, D. Azhari and A. Mitanto, Pyrolysis of Ethylene Vynyl Acetate Polymer by Natural Zeolite Catalyst as an Alternative Fuel, Adv. Eng. Res., 2019, 189, 54–59 Search PubMed.
- E. Erawati, Hamid and R. D. Permatasari, Pyrolysis of Polypropylene Waste with Natural Zeolite as Catalyst, AIP Conf. Proc., 2019, 2114, 050019 CrossRef.
- S. Jin, K. Cui, H. Guan, M. Yang, L. Liu and C. Lan, Preparation of mesoporous MCM-41 from natural sepiolite and its catalytic activity of cracking waste polystyrene plastics, Appl. Clay Sci., 2012, 56, 1–6 CrossRef CAS.
- W. Sriningsih, M. G. Saerodji, W. Trisunaryanti, R. Armunanto and I. I. Falah, Fuel Production from LDPE Plastic Waste over Natural Zeolite Supported Ni, Ni-Mo, Co and Co-Mo Metals, Procedia Environ. Sci., 2014, 20, 215–224 CrossRef CAS.
- I. Borovanska, M. L. Cerrada, P. Zipper and S. Djoumaliisky, Recycled Polyolefin Blends: Effect of Modified Natural Zeolite on their Properties and Morphology, Polym.-Plast. Technol. Eng., 2016, 55, 486–497 CrossRef CAS.
- L. G. Mssina, P. R. Bonelli and A. L. Cukierman, In situ Catalytic Pyrolysis of Peanut Shells Using Modified Natural Zeolite, Fuel Process. Technol., 2017, 159, 160–167 CrossRef.
- S. Salamah and A. Aktawan, Product Distribution of Pyrolysis of Polystyrene Foam Waste using Catalyst of Natural Zeolite and Nickel/Silica, IOP Conf. Series: Earth and Envir. Sci., 2018, 175 Search PubMed.
- P. Kusumo, A. Romli, M. I. Aulia and E. M. Yanuar, Fuel Oil from Municipal Plastic Waste through Pyrolysis with and without Natural Zeolite as Catalysts, E3S Web Conf., 2018, 73, 01021 CrossRef.
- E. Erawati, H. Hamid and A. A. Ilma, Pyrolysis Process of Mixed Polypropylene (PP) and High-Density Polyethylene (HDPE) Waste with Natural Zeolite as Catalyst, Molekul, 2018, 13, 106 CrossRef CAS.
- I. N. Meshkova, T. M. Ushakova, V. G. Grinev, V. A. Nikashina and L. A. Novokshonova, Use of Synthetic and Natural Zeolites for Fabricating Immobilized Olefin Polymerization Catalysts and Polyolefin-Based Composite Materials, Pet. Chem., 2019, 59, 831–837 CrossRef CAS.
- W. Ding, J. Liang and L. L. Anderson, Hydrocracking and Hydroisomerization of High-Density Polyethylene and Waste Plastic over Zeolite and Silica - Alumina-Supported Ni and Ni-Mo Sulfides, Energy Fuels, 1997, 11, 1219–1223 CrossRef CAS.
- T. Isoda, S. Maemoto, K. Kusakabe and S. Morooka, Hydrocracking of pyrenes over a nickel-supported Y-zeolite catalyst and an assessment of the reaction mechanism based on MD calculations, Energy Fuels, 1999, 13, 617–623 CrossRef CAS.
- A. Eschenbacher, F. Goodarzi, R. J. Varghese, K. Enemark-Rasmussen, S. Kegnæs, M. S. Abbas-Abadi and K. M. Van Geem, Boron-Modified Mesoporous ZSM-5 for the Conversion of Pyrolysis Vapors from LDPE and Mixed Polyolefins: Maximizing the C2-C4Olefin Yield with Minimal Carbon Footprint, ACS Sustainable Chem. Eng., 2021, 9, 14618–14630 CrossRef.
- J. M. Escola, J. Aguado, D. P. Serrano, A. García, A. Peral, L. Briones, R. Calvo and E. Fernandez, Catalytic Hydroreforming of the Polyethylene Thermal Cracking Oil over Ni Supported Hierarchical Zeolites and Mesostructured Aluminosilicates, Appl. Catal., B, 2011, 106, 405–415 CrossRef CAS.
- E. G. Fuentes-Ordóñez, J. A. Salbidegoitia, J. L. Ayastuy, M. A. Gutiérrez-Ortiz, M. P. González-Marcos and J. R. González-Velasco, High external surface Pt/zeolite catalysts for improving polystyrene hydrocracking, Catal. Today, 2014, 227, 163–170 CrossRef.
- E. G. Fuentes-Ordóñez, J. A. Salbidegoitia, M. P. González-Marcos, J. L. Ayastuy, M. A. Gutiérrez-Ortiz and J. R. González-Velasco, Pt/ITQ-6 zeolite as a bifunctional catalyst for hydrocracking of waste plastics containing polystyrene, J. Mater. Cycles Waste Manage., 2015, 17, 465–475 CrossRef.
- D. P. Serrano, J. M. Escola, L. Briones, S. Medina and A. Martínez, Hydroreforming of the oils from LDPE thermal cracking over Ni-Ru and Ru supported over hierarchical Beta zeolite, Fuel, 2015, 144, 287–294 CrossRef CAS.
- D. P. Serrano, J. M. Escola, L. Briones and M. Arroyo, Hydroprocessing of the LDPE Thermal Cracking Oil into Transportation Fuels over Pd Supported on Hierarchical ZSM-5 Catalyst, Fuel, 2017, 206, 190–198 CrossRef CAS.
- A. Hijazi, A. A. H. Al-Muhtaseb, S. Aouad, M. N. Ahmad and J. Zeaiter, Pyrolysis of Waste Rubber Tires with Palladium Doped Zeolite, J. Environ. Chem. Eng., 2019, 7, 103451 CrossRef CAS.
- K. Pyra, K. A. Tarach, A. Srebowata, I. Melián-Cabrera and K. Góra-Marek, Pd-Modified Beta Zeolite for Modulated Hydro-Cracking of Low-Density Polyethylene into a Paraffinic-Rich Hydrocarbon Fuel, Appl. Catal., B, 2020, 119070 CrossRef CAS.
- G. Manos, I. Y. Yusof, N. H. Gangas and N. Papayannakos, Tertiary Recycling of Polyethylene to Hydrocarbon Fuel by Catalytic Cracking Over Aluminum Pillared Clays, Energy Fuels, 2002, 16, 485–489 CrossRef CAS.
- A. De Stefanis, P. Cafarelli, F. Gallese, E. Borsella, A. Nana and G. Perez, Catalytic Pyrolysis of Polyethylene: A Comparison Between Pillared and Restructured Clays, J. Anal. Appl. Pyrolysis, 2013, 104, 479–484 CrossRef CAS.
- K. Li, J. Lei, G. Yuan, P. Weerachanchai, J. Y. Wang, J. Zhao and Y. Yang, Fe-, Ti-, Zr- and Al-Pillared Clays for Efficient Catalytic Pyrolysis of Mixed Plastics, Chem. Eng. J., 2017, 317, 800–809 CrossRef CAS.
- V. Patil, S. Adhikari and P. Cross, Co-pyrolysis of lignin and plastics using red clay as catalyst in a micro-pyrolyzer, Bioresour. Technol., 2018, 270, 311–319 CrossRef CAS PubMed.
- I. Vollmer, M. J. Jenks, R. Mayorga González, F. Meirer and B. M. Weckhuysen, Plastic Waste Conversion over a Refinery Waste Catalyst, Angew. Chem., 2021, 133, 16237–16244 CrossRef.
- S. C. Cardona and A. Corma, Tertiary Recycling of Polypropylene by Catalytic Cracking in a Semibatch Stirred Reactor. Use of Spent Equilibrium FCC Commercial Catalyst, Appl. Catal., B, 2000, 25, 151–162 CrossRef CAS.
- K. H. Lee, N. S. Noh, D. H. Shin and Y. Seo, Comparison of Plastic Types for Catalytic Degradation of Waste Plastics into Liquid Product with Spent FCC Catalyst, Polym. Degrad. Stab., 2002, 78, 539–544 CrossRef CAS.
- Y. H. Lin and M. H. Yang, Catalytic pyrolysis of polyolefin waste into valuable hydrocarbons over reused catalyst from refinery FCC units, Appl. Catal., A, 2007, 328, 132–139 CrossRef CAS.
- E. Rodríguez, R. Palos, A. Gutiérrez, F. J. Vela, J. M. Arandes and J. Bilbao, Effect of the FCC Equilibrium Catalyst Properties and of the Cracking Temperature on the Production of Fuel from HDPE Pyrolysis Waxes, Energy Fuels, 2019, 33, 5191–5199 CrossRef.
- E. Rodríguez, G. Elordi, J. Valecillos, S. Izaddoust, J. Bilbao, J. M. Arandes and P. Castaño, Coke Deposition and Product Distribution in the Co-Cracking of Waste Polyolefin Derived Streams and Vacuum Gas Oil Under FCC Unit Conditions, Fuel Process. Technol., 2019, 192, 130–139 CrossRef.
- K. Praveen Kumar and S. Srinivas, Catalytic Co-pyrolysis of Biomass and Plastics (Polypropylene and Polystyrene) Using Spent FCC Catalyst, Energy Fuels, 2020, 34, 460–473 CrossRef CAS.
- D. K. Ratnasari, M. A. Nahil and P. T. Williams, Catalytic Pyrolysis of Waste Plastics using Staged Catalysis for Production of Gasoline Range Hydrocarbon Oils, J. Anal. Appl. Pyrolysis, 2017, 124, 631–637 CrossRef CAS.
- D. Munir and M. R. Usman, Catalytic Hydropyrolysis of a Model Municipal Waste Plastic Mixture over Composite USY/SBA-16 Catalysts, J. Anal. Appl. Pyrolysis, 2018, 135, 44–53 CrossRef CAS.
- K. Ding, S. Liu, Y. Huang, S. Liu, N. Zhou, P. Peng, Y. Wang, P. Chen and R. Ruan, Catalytic Microwave-Assisted Pyrolysis of Plastic Waste over NiO and HY for Gasoline-Range Hydrocarbons Production, Energy Convers. Manage., 2019, 196, 1316–1325 CrossRef CAS.
- M. J. B. de Souza, T. H. A. Silva, T. R. S. Ribeiro, A. O. S. da Silva and A. M. G. Pedrosa, Thermal and Catalytic Pyrolysis of Polyvinyl Chloride using Micro/Mesoporous ZSM-35/MCM-41 Catalysts, J. Therm. Anal. Calorim., 2020, 140, 167–175 CrossRef CAS.
- M. Pires, M. Murariu, A. M. Cardoso, L. Bonnaud and P. Dubois, Thermal Degradation of Poly(lactic acid)–Zeolite Composites Produced by Melt-Blending, Polym. Bull., 2020, 77, 2111–2137 CrossRef CAS.
- P. Sudarsanam, E. Peeters, E. V. Makshina, V. I. Parvulescu and B. F. Sels, Advances in porous and nanoscale catalysts for viable biomass conversion, Chem. Soc. Rev., 2019, 48, 2366–2421 RSC.
- B. Fekhar, V. Zsinka and N. Miskolczi, Value Added Hydrocarbons Obtained by Pyrolysis of Contaminated Waste Plastics in Horizontal Tubular Reactor: In Situ Upgrading of the Products by Chlorine Capture, J. Cleaner Prod., 2019, 241, 118166 CrossRef CAS.
- J. Pérez-Ramírez, C. H. Christensen, K. Egeblad, C. H. Christensen and J. C. Groen, Hierarchical zeolites: Enhanced utilisation of microporous crystals in catalysis by advances in materials design, Chem. Soc. Rev., 2008, 37, 2530–2542 RSC.
- R. Zou, Y. Wang, L. Jiang, Z. Yu, Y. Zhao, Q. Wu, L. Dai, L. Ke, Y. Liu and R. Ruan, Microwave-assisted co-pyrolysis of lignin and waste oil catalyzed by hierarchical ZSM-5/MCM-41 catalyst to produce aromatic hydrocarbons, Bioresour. Technol., 2019, 289, 121609 CrossRef PubMed.
- Y. M. Kim, J. Jeong, S. Ryu, H. W. Lee, J. S. Jung, M. Z. Siddiqui, S. C. Jung, J. K. Jeon, J. Jae and Y. K. Park, Catalytic pyrolysis of wood polymer composites over hierarchical mesoporous zeolites, Energy Convers. Manage., 2019, 195, 727–737 CrossRef CAS.
- B. J. B. Silva, L. V. Sousa, L. R. A. Sarmento, S. L. Alencar, P. H. L. Quintela and A. O. S. Silva, Kinetic Study of Thermocatalytic Degradation of UHMWPE over Microporous and Hierarchical ZSM-23 Zeolite, Appl. Catal., B, 2020, 267, 118699 CrossRef.
- Z. Li, Z. Zhong, B. Zhang, W. Wang, G. V. S. Seufitelli and F. L. P. Resende, Catalytic fast co-pyrolysis of waste greenhouse plastic films and rice husk using hierarchical micro-mesoporous composite molecular sieve, Waste Manage., 2020, 102, 561–568 CrossRef CAS PubMed.
- J. Aguado, D. P. Serrano, J. M. Escola and L. Briones, Deactivation and regeneration of a Ni supported hierarchical Beta zeolite catalyst used in the hydroreforming of the oil produced by LDPE thermal cracking, Fuel, 2013, 109, 679–686 CrossRef CAS.
- W. Namchot and S. Jitkarnka, Catalytic pyrolysis of waste tire using HY/MCM-41 core-shell composite, J. Anal. Appl. Pyrolysis, 2016, 121, 297–306 CrossRef CAS.
- V. P. S. Caldeira, A. Peral, M. Linares, A. S. Araujo, R. A. Garcia-Muñoz and D. P. Serrano, Properties of Hierarchical Beta Zeolites Prepared from Protozeolitic Nanounits for the Catalytic Cracking of High Density Polyethylene, Appl. Catal., A, 2017, 531, 187–196 CrossRef CAS.
- K. A. Tarach, K. Pyra, S. Siles, I. Melián-Cabrera and K. Góra-Marek, Operando Study Reveals the Superior Cracking Activity and Stability of Hierarchical ZSM-5 Catalyst for the Cracking of Low-Density Polyethylene, ChemSusChem, 2019, 12, 633–638 CrossRef CAS PubMed.
- S. L. Wu, J. H. Kuo and M. Y. Wey, Thermal degradation of waste plastics in a two-stage pyrolysis-catalysis reactor over core-shell type catalyst, J. Anal. Appl. Pyrolysis, 2019, 142, 104641 CrossRef CAS.
- Y. Zhang, D. Duan, H. Lei, E. Villota and R. Ruan, Jet Fuel Production from Waste Plastics via Catalytic Pyrolysis with Activated Carbons, Appl. Energy, 2019, 251, 113337 CrossRef CAS.
- C. Wu and P. T. Williams, A Novel Ni-Mg-Al-CaO Catalyst with the Dual Functions of Catalysis and CO2 Sorption for H2 Production from the Pyrolysis-Gasification of Polypropylene, Fuel, 2010, 89, 1435–1441 CrossRef CAS.
- M. Busto, C. R. Vera and J. M. Grau, Optimal process conditions for the isomerization-cracking of long-chain n-paraffins to high octane isomerizate gasoline over Pt/SO42–ZrO2 catalysts, Fuel Process. Technol., 2011, 92, 1675–1684 CrossRef CAS.
- M. N. Siddiqui, E. V. Antonakou, H. H. Redhwi and D. S. Achilias, Kinetic analysis of thermal and catalytic degradation of polymers found in waste electric and electronic equipment, Thermochim. Acta, 2019, 675, 69–76 CrossRef CAS.
- M. N. Almustapha, M. Farooq, M. L. Mohammed, M. Farhan, M. Imran and J. M. Andresen, Modification of Acidic and Textural Properties of a Sulphated Zirconia Catalyst for Efficient Conversion of High-Density Polyethylene into Liquid Fuel, Environ. Sci. Pollut. Res., 2020, 27, 55–65 CrossRef CAS PubMed.
- C. A. Veses, R. Date, R. Date, A. Date and N. Mv, From Laboratory Scale to Pilot Plant: Evaluation of the Catalytic Co-Pyrolysis of Grape Seeds and Polystyrene Wastes with CaO, Catal. Today, 2020, 379, 87–95 CrossRef.
- A. Patel, V. Brahmkhatri and N. Singh, Biodiesel Production by Esterification of Free Fatty Acid over Sulfated Zirconia, Renewable Energy, 2013, 51, 227–233 CrossRef CAS.
- E. V. Antonakou, K. G. Kalogiannis, S. D. Stefanidis, S. A. Karakoulia, K. S. Triantafyllidis, A. A. Lappas and D. S. Achilias, Catalytic and thermal pyrolysis of polycarbonate in a fixed-bed reactor: The effect of catalysts on products yields and composition, Polym. Degrad. Stab., 2014, 110, 482–491 CrossRef CAS.
- S. Kumagai, I. Hasegawa, G. Grause, T. Kameda and T. Yoshioka, Thermal Decomposition of Individual and Mixed Plastics in the Presence of CaO or Ca(OH)2, J. Anal. Appl. Pyrolysis, 2015, 113, 584–590 CrossRef CAS.
- M. S. Khan, M. Sohail and N. S. Khattak, Conversion of Mixed Low-Density Polyethylene Wastes into Liquid Fuel by Novel CaO/SiO2 Catalyst, J. Polym. Environ., 2016, 24, 255–263 CrossRef CAS.
- L. Fan, Y. Zhang, S. Liu, N. Zhou, P. Chen, Y. Liu, Y. Wang, P. Peng, Y. Cheng and M. Addy, Ex-Situ Catalytic Upgrading of Vapors from Microwave-Assisted Pyrolysis of Low-Density Polyethylene with MgO, Energy Convers. Manage., 2017, 149, 432–441 CrossRef CAS.
- M. Utami, K. Wijaya and W. Trisunaryanti, Pt-Promoted Sulfated Zirconia as Catalyst for Hydrocracking of LDPE Plastic Waste into Liquid Fuels, Mater. Chem. Phys., 2018, 213, 548–555 CrossRef CAS.
- M. Nadjib, T. H. A. Santosa, A. Zuniardi and R. Shihabudin, Properties of co-pyrolysed palm kernel shell and plastic grocery bag with CaO as catalyst, IOP Conference Series: Earth and Environmental Science, 2018, 209 Search PubMed.
- L. S. Diaz-Silvarrey, A. McMahon and A. N. Phan, Benzoic Acid Recovery via Waste Poly(ethylene terephthalate) (PET) Catalytic Pyrolysis using Sulphated Zirconia Catalyst, J. Anal. Appl. Pyrolysis, 2018, 134, 621–631 CrossRef CAS.
- Y. Uemichi, Y. Makino and T. Kanazuka, Degradation of Polyethylene to Aromatic Hydrocarbons Over Metal-Supported Activated Carbon Catalysts, J. Anal. Appl. Pyrolysis, 1989, 14, 331–344 CrossRef CAS.
- N. Miskolczi, C. Wu and P. T. Williams, Pyrolysis of Waste Plastics Using Catalysts: Activated Carbon, MCM-41 and HZSM-5, Int. J. Chem. Eng. Appl., 2017, 8, 67–74 CAS.
- K. Sun, Q. Huang, X. Meng, Y. Chi and J. Yan, Catalytic Pyrolysis of Waste Polyethylene into Aromatics by H3PO4-Activated Carbon, Energy Fuels, 2018, 32, 9772–9781 CrossRef CAS.
- S. Kim, C. Park and J. Lee, Reduction of Polycyclic Compounds and Biphenyls Generated by Pyrolysis of Industrial Plastic Waste by Using Supported Metal Catalysts: A Case Study of Polyethylene Terephthalate Treatment, J. Hazard. Mater., 2020, 392, 122464 CrossRef CAS PubMed.
- B. Kunwar, B. R. Moser, S. R. Chandrasekaran, N. Rajagopalan and B. K. Sharma, Catalytic and Thermal Depolymerization of Low Value Post-Consumer High Density Polyethylene Plastic, Energy, 2016, 111, 884–892 CrossRef CAS.
- M. V. Singh, Waste and Virgin High-Density Poly(ethylene) into Renewable Hydrocarbons Fuel by Pyrolysis-Catalytic Cracking with a CoCO3 Catalyst, J. Anal. Appl. Pyrolysis, 2018, 134, 150–161 CrossRef CAS.
- M. V. Singh, S. Kumar and M. Sarker, Waste HD-PE plastic, Deformation into Liquid Hydrocarbon Fuel using Pyrolysis-Catalytic Cracking with a CuCO3 Catalyst, Sustainable Energy Fuels, 2018, 2, 1057–1068 RSC.
- M. V. Singh, Deformation of Virgin HD-PE, PP and Waste PP Plastics into Green Fuel via a Pyrolysis-Catalytic using a NiCO3 Catalyst, Indian Chem. Eng., 2019, 61, 254–268 CrossRef CAS.
- A. Ahamed, L. Liang, W. P. Chan, P. C. K. Tan, N. T. X. Yip, J. Bobacka, A. Veksha, K. Yin and G. Lisak, In situ catalytic reforming of plastic pyrolysis vapors using MSW incineration ashes, Environ. Pollut., 2021, 276, 116681 CrossRef CAS PubMed.
- H. Persson, I. Duman, S. Wang, L. J. Pettersson and W. Yang, Catalytic pyrolysis over transition metal-modified zeolites: A comparative study between catalyst activity and deactivation, J. Anal. Appl. Pyrolysis, 2019, 138, 54–61 CrossRef CAS.
- F. J. Mastral, E. Esperanza, P. García and M. Juste, Pyrolysis of high-density polyethylene in a fluidised bed reactor. Influence of the temperature and residence time, J. Anal. Appl. Pyrolysis, 2002, 63, 1–15 CrossRef CAS.
- Y. Xue, P. Johnston and X. Bai, Effect of catalyst contact mode and gas atmosphere during catalytic pyrolysis of waste plastics, Energy Convers. Manage., 2017, 142, 441–451 CrossRef CAS.
- D. Kunii and O. Levenspiel, Fluidization engineering, Butterworth-Heinemann, Boston, London, 1991 Search PubMed.
- Anellotech New Anellotech technology tackles plastics pollution by recycling plastic waste into chemicals. https://anellotech.com/press/new-anellotech-technology-tackles-plastics-pollution-recycling-plastic-waste-chemicals#:~:text=Pearl%20River%2C%20NY%20%E2%80%93%20December%206,to%20make%20new%2C%20virgin%20plastics. (accessed Feb 02).
- D. Sudolsky, Commercializing Renewable Aromatics for Biofuels, Biobased Chemicals and Plastics Chemical Recycling, Ind. Biotechnol., 2019, 15, 330–333 CrossRef.
- Braskem advances in research into chemical recycling of plastics. https://www.braskem.com.br/news-detail/braskem-advances-in-research-into-chemical-recycling-of-plastics (accessed Feb 02).
- Goldsberry, C. Braskem America, Encina Partner to Develop Circular Polypropylene Feedstock. https://www.plasticstoday.com/advanced-recycling/braskem-america-encina-partner-develop-circular-polypropylene-feedstock (accessed Feb 02).
- CALIENDO, H. Encina Turning Plastic Waste Into Renewable Base Chemicals and Fuels. https://www.ptonline.com/news/encina-turning-plastic-waste-into-renewable-base-chemicals-and-fuels#:~:text=Encina%20is%20a%20company%20emerging,fuels%20for%20the%20circular%20economy.&text=Encina%20has%20developed%20a%20process,plastic%20waste%20through%20catalytic%20pyrolysis. (accessed Feb 02).
- M. Luo and C. W. Curtis, Effect of reaction parameters and catalyst type on waste plastics liquefaction and coprocessing with coal, Fuel Process. Technol., 1996, 49, 177–196 CrossRef CAS.
- M. Čolnik, P. Kotnik, Ž. Knez and M. Škerget, Hydrothermal decomposition of polyethylene waste to hydrocarbons rich oil, J. Supercrit. Fluids, 2020, 169, 105136 CrossRef.
- T. Hirota and F. N. Fagan, Liquefaction of Plastics, Makromol. Chem., Macromol. Symp., 1992, 57, 161–173 CrossRef CAS.
- M. M. Taghiei, Z. Feng, F. E. Huggins and G. P. Huffman, Coliquefaction of Waste Plastics with Coal, Energy Fuels, 1994, 8, 1228–1232 CrossRef CAS.
- W. Zmierczak, X. Xiao and J. Shabtai, Depolymerization-liquefaction of plastics and rubbers. 2. Polystyrenes and styrene-butadiene copolymers, Fuel Process. Technol., 1996, 49, 31–48 CrossRef CAS.
- M. V. S. Murty, P. Rangarajan, E. A. Grulke and D. Bhattacharyya, Thermal degradation/hydrogenation of commodity plastics and characterization of their liquefaction products, Fuel Process. Technol., 1996, 49, 75–90 CrossRef CAS.
- Z. Feng, J. Rockwell, D. Bailey and G. Huffman, Direct liquefaction of waste plastics and coliquefaction of coal-plastic mixtures, Fuel Process. Technol., 1996, 49, 17–30 CrossRef CAS.
- J. Shabtai, X. Xiao and W. Zmierczak, Depolymerization-liquefaction of plastics and rubbers. 1. Polyethylene, polypropylene, and polybutadiene, Energy Fuels, 1997, 11, 76–87 CrossRef CAS.
- P. K. Ramdoss and A. R. Tarrer, High-temperature liquefaction of waste plastics, Fuel, 1998, 77, 293–299 CrossRef CAS.
- N. Shah, J. Rockwell and G. P. Huffman, Conversion of waste plastic to oil: Direct liquefaction versus pyrolysis and hydroprocessing, Energy Fuels, 1999, 13, 832–838 CrossRef CAS.
- P. T. Williams and E. Slaney, Analysis of products from the pyrolysis and liquefaction of single plastics and waste plastic mixtures, Resour., Conserv. Recycl., 2007, 51, 754–769 CrossRef.
- T. Moriya and H. Enomoto, Characteristics of polyethylene cracking in supercritical water compared to thermal cracking, Polym. Degrad. Stab., 1999, 65, 373–386 CrossRef CAS.
- G. C. Hwang, K. H. Kim, S. Y. Bae, S. C. Yi and H. Kumazawa, Degradation of High Density Polyethylene, Polypropylene and Their Mixtures in Supercritical Acetone, Korean J. Chem. Eng., 2001, 18, 396–401 CrossRef CAS.
- G. C. Hwang, J. H. Choi, S. Y. Bae and H. Kumazawa, Degradation of Polystyrene in Supercritical n-Hexane, Korean J. Chem. Eng., 2001, 18, 854–861 CrossRef CAS.
- H. Y. Shin and S. Y. Bae, Thermal decomposition of polystyrene in supercritical methanol, J. Appl. Polym. Sci., 2008, 108, 3467–3472 CrossRef CAS.
- M. S. Seshasayee and P. E. Savage, Oil from plastic via hydrothermal liquefaction: Production and characterization, Appl. Energy, 2020, 278, 115673 CrossRef CAS.
- S. H. Ng, H. Seoud, M. Stanciulescu and Y. Sugimoto, Conversion of Polyethylene to Transportation Fuels through Pyrolysis and Catalytic Cracking, Energy Fuels, 1995, 9, 735–742 CrossRef CAS.
- Z. Fang and J. A. Koziński, A study of rubber liquefaction in supercritical water using DAC-stereomicroscopy and FT-IR spectrometry, Fuel, 2002, 81, 935–945 CrossRef CAS.
- X. Su, Y. Zhao, R. Zhang and J. Bi, Investigation on degradation of polyethylene to oils in supercritical water, Fuel Process. Technol., 2004, 85, 1249–1258 CrossRef CAS.
- Y. Takeshita, K. Kato, K. Takahashi, Y. Sato and S. Nishi, Basic study on treatment of waste polyvinyl chloride plastics by hydrothermal decomposition in subcritical and supercritical regions, J. Supercrit. Fluids, 2004, 31, 185–193 CrossRef CAS.
- H. f. Zhang, X. l. Su, D. k. Sun, R. Zhang and J. c. Bi, Investigation on degradation of polyethylene to oil in a continuous supercritical water reactor, J. Fuel Chem. Technol., 2007, 35, 487–491 CrossRef CAS.
- M. Sugano, T. Shimizu, A. Komatsu, Y. Kakuta and K. Hirano, Liquefaction and dechlorination of hydrothermally treated waste mixture containing plastics with glass powder, Environ. Sci. Technol., 2011, 45, 2493–2497 CrossRef CAS PubMed.
- T. H. Pedersen and F. Conti, Improving the circular economy via hydrothermal processing of high-density waste plastics, Waste Manage., 2017, 68, 24–31 CrossRef.
- B. Bai, H. Jin, C. Fan, C. Cao, W. Wei and W. Cao, Experimental investigation on liquefaction of plastic waste to oil in supercritical water, Waste Manage., 2019, 89, 247–253 CrossRef CAS PubMed.
- W. T. Chen, K. Jin and N. H. Linda Wang, Use of Supercritical Water for the Liquefaction of Polypropylene into Oil, ACS Sustainable Chem. Eng., 2019, 7, 3749–3758 CrossRef CAS.
- J. S. dos Passosa, M. Glasius and P. Biller, Screening of common synthetic polymers for depolymerization by subcritical hydrothermal liquefaction, Process Saf. Environ. Prot., 2020, 139, 371–379 CrossRef.
- H. Jin, B. Bai, W. Wei, Y. Chen, Z. Ge and J. Shi, Hydrothermal Liquefaction of Polycarbonate (PC) Plastics in Sub-/Supercritical Water and Reaction Pathway Exploration, ACS Sustainable Chem. Eng., 2020, 8, 7039–7050 CrossRef CAS.
- K. Jin, P. Vozka, G. Kilaz, W. T. Chen and N. H. L. Wang, Conversion of polyethylene waste into clean fuels and waxes via hydrothermal processing (HTP), Fuel, 2020, 273, 1–11 CrossRef.
- P. Zhao, Z. Yuan, J. Zhang, X. Song, C. Wang, Q. Guo and A. J. Ragauskas, Supercritical water co-liquefaction of LLDPE and PP into oil: Properties and synergy, Sustainable Energy Fuels, 2021, 5, 575–583 RSC.
- B. Ciuffi, L. Rosi, E. Miliotti, G. Lotti, A. M. Rizzo and D. Chiaramonti, Batch Hydrothermal liquefaction of end-of-life plastic and oil characterization, E3S Web Conf., 2021, 238, 0–4 CrossRef CAS.
- X. Pei, X. Yuan, G. Zeng, H. Huang, J. Wang, H. Li and H. Zhu, Co-liquefaction of microalgae and synthetic polymer mixture in sub- and supercritical ethanol, Fuel Process. Technol., 2012, 93, 35–44 CrossRef CAS.
- N. Ahmad, N. Ahmad, I. M. Maafa, U. Ahmed, P. Akhter, N. Shehzad, U. e. s. Amjad and M. Hussain, Thermal conversion of polystyrene plastic waste to liquid fuel via ethanolysis, Fuel, 2020, 279, 118498 CrossRef CAS.
- H. A. Baloch, S. Nizamuddin, M. T. H. Siddiqui, N. M. Mubarak, S. Mazari, G. J. Griffin and M. P. Srinivasan, Co-liquefaction of synthetic polyethylene and polyethylene bags with sugarcane bagasse under supercritical conditions: A comparative study, Renewable Energy, 2020, 162, 2397–2407 CrossRef CAS.
- H. A. Baloch, M. T. H. Siddiqui, S. Nizamuddin, N. M. Mubarak, M. Khalid, M. P. Srinivasan and G. J. Griffin, Solvothermal co-liquefaction of sugarcane bagasse and polyethylene under sub-supercritical conditions: Optimization of process parameters, Process Saf. Environ. Prot., 2020, 137, 300–311 CrossRef CAS.
- Z. Dai, B. Hatano, J. I. Kadokawa and H. Tagaya, Effect of diaminotoluene on the decomposition of polyurethane foam waste in superheated water, Polym. Degrad. Stab., 2002, 76, 179–184 CrossRef CAS.
- M. Goto, M. Sasaki and T. Hirose, Reactions of polymers in supercritical fluids for chemical recycling of waste plastics, J. Mater. Sci., 2006, 41, 1509–1515 CrossRef CAS.
- H. Kwak, H. Y. Shin, S. Y. Bae and H. Kumazawa, Characteristics and kinetics of degradation of polystyrene in supercritical water, J. Appl. Polym. Sci., 2006, 101, 695–700 CrossRef CAS.
- M. Watanabe, H. Hirakoso, S. Sawamoto, T. Adschiri and K. Arai, Polyethylene conversion in supercritical water, J. Supercrit. Fluids, 1998, 13, 247–252 CrossRef CAS.
- M. Sugano, A. Komatsu, M. Yamamoto, M. Kumagai, T. Shimizu, K. Hirano and K. Mashimo, Liquefaction process for a hydrothermally treated waste mixture containing plastics, J. Mater. Cycles Waste Manage., 2009, 11, 27–31 CrossRef CAS.
- A. Tukker, H. De Groot, L. Simons and S. Wiegersma, Chemical Recycling of Plastics Waste (PVC and other resins), Delft, Netherlands, 1999 Search PubMed.
- A. Okuwaki, T. Yoshioka, M. Asai, H. Tachibana, K. Wakai and K. Tada, The Liquefaction of Plastic Containers and Packaging in Japan, in Feedstock Recycling and Pyrolysis of Waste Plastics: Converting Waste Plastics into Diesel and Other Fuels, ed. J. Scheirs and W. Kaminsky, John Wiley & Sons, 2006, pp. 665–708 Search PubMed.
- T. E. Loop, J. D. Flynn, G. Allan, S. C. Van Swearingen and K. O. Gaw, Biomass and waste plastics depolymerization machine and methods via supercritical water, US Pat., XTRUDX Technologies Inc, 8980143, 2015 Search PubMed.
- Y. Ishihara, K. Saido and T. Kuroki, Economic evaluation of small liquefaction plants for waste plastics, Sekiyu Gakkaishi, 2003, 46, 77–83 CrossRef CAS.
- A. Okuwaki, Feedstock recycling of plastics in Japan, Polym. Degrad. Stab., 2004, 85, 981–988 CrossRef CAS.
- Plastic Waste Management Institute, An Introduction to Plastic Recycling, Tokyo, Japan, 2019, pp. 21–22 Search PubMed.
- Licella Holdings Cat-HTR™: A Bridge to a lower carbon future, Sydney, Australia, 2019 Search PubMed.
- Mura Technology Limited Hydro-PRS Technology. https://muratechnology.com/technology/(accessed 2022-01-18).
- Licella Holdings Plastic Waste – Advanced Recycling Feasibility Study, Geelong-Altona Industrial Corridor, Sydney, Australia, 2021; pp. 26–38 Search PubMed.
- Licella Holdings Plastics Cat-HTR™. https://www.licella.com/solutions/plastic/(accessed 2022-01-18).
- Aduro Clean Technologies Inc. Valuable 21st century resources from plastic waste and other low-value feedstocks. https://adurocleantech.com/about-us/(accessed 2022-01-18).
- Aduro Clean Technologies Inc. Hydrochemolytic Plastic Upcycling. https://adurocleantech.com/hpu/(accessed 2022-01-18).
- S. Jarungthammachote and A. Dutta, Thermodynamic equilibrium model and second law analysis of a downdraft waste gasifier, Energy, 2007, 32(9), 1660–1669 CrossRef CAS.
- U. Arena, Process and technological aspects of municipal solid waste gasification. A review, Waste Manage., 2012, 32(4), 625–639 CrossRef CAS PubMed.
- U. Arena, M. L. Mastellone and F. Perugini, The environmental performance of alternative solid waste management options: a life cycle assessment study, Chem. Eng. J., 2003, 96(1–3), 207–222 CrossRef CAS.
- C. H. Bartholomew and R. J. Farrauto, Fundamentals of industrial catalytic processes, John Wiley & Sons, 2011 Search PubMed.
- J. Dong, Y. Tang, A. Nzihou, Y. Chi, E. Weiss-Hortala and M. Ni, Life cycle assessment of pyrolysis, gasification and incineration waste-to-energy technologies: Theoretical analysis and case study of commercial plants, Sci. Total Environ., 2018, 626, 744–753 CrossRef CAS PubMed.
- Y.-C. Seo, M. T. Alam and W.-S. Yang, in Gasification of Municipal Solid Waste, ed. Y. Yun, InTech, 2018, pp. 115–136 Search PubMed.
- F. N. a. Sidek, Review on effects of gasifying agents, temperature and equivalence ratio in biomass gasification process, IOP Conference Series: Materials Science and Engineering, 2020, 863(1), p. 012028 Search PubMed.
- P. Xu, Y. Jin and Y. Cheng, Thermodynamic Analysis of the Gasification of Municipal Solid Waste, Engineering, 2017, 3(3), 416–422 CrossRef.
- L. Zhao, H. Wang, S. Qing and H. Liu, Characteristics of gaseous product from municipal solid waste gasification with hot blast furnace slag, J. Nat. Gas Chem., 2010, 19(4), 403–408 CrossRef CAS.
- H. A. Arafat and K. Jijakli, Modeling and comparative assessment of municipal solid waste gasification for energy production, Waste Manage., 2013, 33(8), 1704–1713 CrossRef CAS PubMed.
- S. Chen, A. Meng, Y. Long, H. Zhou, Q. Li and Y. Zhang, TGA pyrolysis and gasification of combustible municipal solid waste, J. Energy Inst., 2015, 88(3), 332–343 CrossRef CAS.
- M. Cortazar, J. Alvarez, G. Lopez, M. Amutio, L. Santamaria, J. Bilbao and M. Olazar, Role of temperature on gasification performance and tar composition in a fountain enhanced conical spouted bed reactor, Energy Convers. Manage., 2018, 171, 1589–1597 CrossRef CAS.
- R. C. Brown, Thermochemical processing of biomass: conversion into fuels, chemicals and power, John Wiley & Sons, 2019 Search PubMed.
- G. W. Huber, S. Iborra and A. Corma, Synthesis of transportation fuels from biomass: chemistry, catalysts, and engineering, Chem. Rev., 2006, 106(9), 4044–4098 CrossRef CAS PubMed.
- A. Bolt, I. Dincer and M. Agelin-Chaab, A critical review of synthetic natural gas production techniques and technologies, J. Nat. Gas Sci. Eng., 2020, 103670 CrossRef CAS.
- M. R. Gogate, Methanol-to-olefins process technology: current status and future prospects, Pet. Sci. Technol., 2019, 37(5), 559–565 CrossRef CAS.
- C. Higman, State of the gasification industry: Worldwide gasification and syngas databases 2016 update. Available at https://www.gasification-syngas.org/uploads/downloads/2016-presentations/2016-Wed-Higman.pdf. Accessed June 2016, 8, 2017.
- Edmonton to be Site of World's First Industrial Scale Municipal Waste-to-Ethanol Facility. https://www.theautochannel.com/news/2008/06/26/091250.html (accessed Feb 02).
- Enerkem project pipeline worldwide. https://enerkem.com/company/facilities-projects/(accessed Feb 02).
- R. Voss, R. P. Lee, L. Seidl, F. Keller and M. Frhling, Global warming potential and economic performance of gasification-based chemical recycling and incineration pathways for residual municipal solid waste treatment in Germany, Waste Manage., 2021, 134, 206–219 CrossRef CAS PubMed.
- F. Di Gregorio and L. Zaccariello, Fluidized bed gasification of a packaging derived fuel: energetic, environmental and economic performances comparison for waste-to-energy plants, Energy, 2012, 42(1), 331–341 CrossRef CAS.
- J. Daniel-Gromke, N. Rensberg, V. Denysenko, M. Trommler, T. Reinholz, K. Vller, M. Beil and W. Beyrich, Anlagenbestand Biogas und Biomethan–Biogaserzeugung und-nutzung in Deutschland, DBFZ Rep., 2017,(30), 7 Search PubMed.
- S. Billiet and S. R. Trenor, 100th Anniversary of Macromolecular Science Viewpoint: Needs for Plastics Packaging Circularity, ACS Macro Lett., 2020, 9(9), 1376–1390 CrossRef CAS PubMed.
- J. G. Poulakis and C. D. Papaspyrides, The dissolution/reprecipitation technique applied on high density polyethylene: I. Model recycling experiments, Adv. Polym. Technol., 1995, 14, 237–242 CrossRef CAS.
- D. Triebert, H. Hanel, M. Bundt and K. Wohnig, Solvent-Based Recycling, in Circular Economy of Polymers: Topics in Recycling Technologies, ACS Publications, 2021, pp. 33–59 Search PubMed.
- J. Brandrup, E. H. Immergut, E. A. Grulke, A. Abe and D. R. Bloch, Polymer handbook, Wiley, New York, 1999, vol. 89 Search PubMed.
- M. Rubinstein and R. H. Colby, Polymer physics, Oxford university press, 2003 Search PubMed.
- K. Sändig, Process for producing particularly stable condensation products, US2061635, 1936.
- D. W. Young and W. J. Sparks, Preparation of purified polyesters, US2491350, 1949.
- R. B. Seymour and G. A. Stahl, Separation of waste plastics: An experiment in solvent fractionation J, Chem. Educ., 1976, 53(10), 653 CrossRef CAS.
- J. Teti, Process for polymer recovery, US3696058, 1972.
- N. C. Sidebotham, P. D. Shoemaker and C. W. Young, Polyester polymer recovery, US4064079, 1977.
- E. B. Nauman and J. C. Lynch, Polymer recycling by selective dissolution, US5198471, 1993.
- E. B. Nauman and J. C. Lynch, Polymer recycling by selective dissolution, US5278282, 1994.
- J. G. Poulakis and C. D. Papaspyrides, Recycling of polypropylene by the dissolution/reprecipitation technique: I. A model study, Resour., Conserv. Recycl., 1997, 20(1), 31–41 CrossRef.
- E. M. Kampouris, C. D. Papaspyrides and C. N. Lekakou, A model process for scrap polystyrene foam by means of solvent systems, Conserv. Recycl., 1987, 10(4), 315–319 CrossRef CAS.
- E. M. Kampouris, C. D. Papaspyrides and C. N. Lekakou, A Model Process for the Solvent Recycling of Polystyrene, Polym. Eng. Sci., 1988, 28(8), 534–537 CrossRef CAS.
- E. M. Kampouris, D. C. Diakoulaki and C. D. Papaspyrides, Solvent Recycling of Rigid Poly(Viny1 Chloride) Bottles, J. Vinyl Technol., 1986, 8(2), 79–82 CrossRef CAS.
- C. D. Papaspyrides, J. G. Poulakis and P. C. Varelides, A model recycling process for low density polyethylene, Resour., Conserv. Recycl., 1994, 12(3–4), 177–184 CrossRef.
- G. Pappa, C. Boukouvalas, C. Giannaris, N. Ntaras, V. Zografos, K. Magoulas, A. Lygeros and D. Tassios, The selective dissolution/precipitation technique for polymer recycling: a pilot unit application, Resour., Conserv. Recycl., 2001, 34(1), 33–44 CrossRef.
- J. G. Poulakis; and C. D. Papaspyrides, Dissolution/Reprecipitation: A Model Process for PET Bottle Recycling, J. Appl. Polym. Sci., 2001, 81, 91–95 CrossRef.
- Accelerating circular supply chains for plastics: A landscape of transformational technologies that stop plastic waste, keep materials in pay and grow markets. https://www.closedlooppartners.com/research/advancing-circular-systems-for-plastics/.
- B. K. C. Sharma, Sriraam Beneficial Use of Plastic Wastes. https://hdl.handle.net/2142/113454 (accessed Feb 02).
- S. R. Chandrasekaran, S. Avasarala, D. Murali, N. Rajagopalan and B. K. Sharma, Materials and energy recovery from e-waste plastics, ACS Sustainable Chem. Eng., 2018, 6(4), 4594–4602 CrossRef CAS.
- I. Georgiopoulou, G. D. Pappa, S. N. Vouyiouka and K. Magoulas, Recycling of post-consumer multilayer Tetra Pak® packaging with the Selective Dissolution-Precipitation process, Resour., Conserv. Recycl., 2021, 165, 105268 CrossRef CAS.
- K. L. Sánchez-Rivera, P. Zhou, M. S. Kim, L. D. González Chávez, S. Grey, K. Nelson, S. C. Wang, I. Hermans, V. M. Zavala and R. C. Van Lehn, Reducing Antisolvent Use in the STRAP Process by Enabling a Temperature–Controlled Polymer Dissolution and Precipitation for the Recycling of Multilayer Plastic Films, ChemSusChem, 2021, 14(19), 4317–4329 CrossRef.
- T. W. Walker, N. Frelka, Z. Shen, A. K. Chew, J. Banick, S. Grey, M. S. Kim, J. A. Dumesic, R. C. Van Lehn and G. W. Huber, Recycling of multilayer plastic packaging materials by solvent-targeted recovery and precipitation, Sci. Adv., 2020, 6(47), eaba7599 CrossRef PubMed.
- D. S. Achilias, A. Giannoulis and G. Z. Papageorgiou, Recycling of polymers from plastic packaging materials using the dissolution–reprecipitation technique, Polym. Bull., 2009, 63(3), 449–465 CrossRef CAS.
- R. Côté, Processes for recycling polystyrene waste, US20200165407A1, 2019.
- A. J. Hadi, G. F. Najmuldeen and K. B. Yusoh, Dissolution/reprecipitation technique for waste polyolefin recycling using new pure and blend organic solvents, J. Polym. Eng., 2013, 33(5), 471–481 CAS.
- A. J. Hadi, G. F. Najmuldeen and I. Ahmed, Polyolefins Waste Materials Reconditioning Using Dissolution/Reprecipitation Method, APCBEE Proc., 2012, 3, 281–286 CrossRef CAS.
- J. M. Layman, D. I. Collias, H. Schonemann and K. Williams, Method for purifying reclaimed polymers, US10961366, 2021.
- K. F. Drain, W. R. Murphy and M. S. Otterburn, A solvent technique for the recycling of polypropylene—degradation on recycling, Conserv. Recycl., 1983, 6(3), 123–137 CrossRef CAS.
- W. Chen, Y. Yang, X. Lan, B. Zhang, X. Zhang and T. Mu, Biomass-derived γ-valerolactone: efficient dissolution and accelerated alkaline hydrolysis of polyethylene terephthalate, Green Chem., 2021, 23(11), 4065–4073 RSC.
- E. Siggel and J. Klein, Dissolution and purification of polyethylene terephthalate, US2762788, 1956.
- G. S. Weeden Jr., N. H. Soepriatna and N. H. Wang, Method for efficient recovery of high-purity polycarbonates from electronic waste, Environ. Sci. Technol., 2015, 49(4), 2425–2433 CrossRef PubMed.
- C. Kartalis, J. Poulakis, C. Tsenoglou and C. Papaspyrides, Pure component recovery from polyamide 6/6 6 mixtures by selective dissolution and reprecipitation, J. Appl. Polym. Sci., 2002, 86(8), 1924–1930 CrossRef CAS.
- S. Ugduler, K. M. Van Geem, M. Roosen, E. I. P. Delbeke and S. De Meester, Challenges and opportunities of solvent-based additive extraction methods for plastic recycling, Waste Manage, 2020, 104, 148–182 CrossRef CAS PubMed.
- K. Kaiser, M. Schmid and M. Schlummer, Recycling of Polymer-Based Multilayer Packaging: A Review, Recycling, 2018, 3(1), 1 CrossRef.
- S. Ugduler, T. De Somer, K. M. Van Geem, M. Roosen, A. Kulawig, R. Leineweber and S. De Meester, Towards a Better Understanding of Delamination of Multilayer Flexible Packaging Films by Carboxylic Acids, ChemSusChem, 2021, 14(19), 4198–4213 CrossRef PubMed.
- O. Horodytska, F. J. Valdés and A. Fullana, Plastic flexible films waste management–A state of art review, Waste Manage., 2018, 77, 413–425 CrossRef CAS PubMed.
- T. Bauer, H.-G. Krebber, C. Nover and R. Saffert, Process for the recovery of a polymer from a polymer coating on a support. November 3, 2005 2005.
- K. Wohnig, Sustainable Plastic Innovation: Closing the Loop, GPCA PlastiCon, 2018 Search PubMed.
- M. Niaounakis, Recycling of Flexible Plastic Packaging, William Andrew, 2020 Search PubMed.
- R. Coker, APK deploys downstream melt processing system. https://www.brazilianplastics.com/edicao00/02_December2019.html#:~:text=December%204%2C%202019%3A%20APK%<?pdb_no 20AG?>20AG<?pdb END?>,properties%20close%<?pdb_no 20to?>20to<?pdb END?>%20virgin%20resin. (accessed March 17, 2022).
- K. Wohnig, M. Kaina, M. Fleig and H. Hanel, Solvent and method for dissolving a plastic from a solid within a suspension, DE102016015198A1, 2018.
- K. Wohnig, M. Kaina, M. Fleig and J. Jeschke, Gas-tight centrifuge for separating solids from a polymer solution, and method for separating solids from a polymer solution, DE102016015197A1, 2018.
- C. P. Ashcroft, P. J. Dunn, J. D. Hayler and A. S. Wells, Survey of solvent usage in papers published in organic process research & development 1997–2012, Org. Process Res. Dev., 2015, 19(7), 740–747 CrossRef CAS.
- A. Mäurer, M. Schlummer, F. Knappich, T. Fell, A. Berrang and R. Leizinger, Method for Recycling Polyolefin Containing Waste, EP3305839 A1, 2018.
- A. Maeurer, M. Schlummer and O. Beck, Methods for recycling plastic materials and use thereof, US8138232B2, 2006.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Tools and techniques for solvent selection: green solvent selection guides, Sustainable Chem. Processes, 2016, 4(1), 1–24 Search PubMed.
- Plasteurope VINYLOOP: Closure of operation in Italy/Phthalates issue under REACH brings down European PVC recycling project. https://www.plasteurope.com/news/Closure_of_operation_in_Italy_Phthalates_issue_under_REACH_brings_do_t240095/(accessed Feb 1).
- Caliendo, H., Unilever Introduces Process to Recycle Multilayer Sachets. https://www.ptonline.com/blog/post/unilever-introduces-process-to-recycle-multilayer-sachets- (accessed March 6, 2022).
- Plastic Recycling Plants. https://www.creacycle.de/en/creasolv-plants.html (accessed February 15, 2022).
- J. M. Layman, M. Gunnerson, H. Schonemann and K. Williams, Method for purifying contaminated polypropylene, 2017 Search PubMed.
- PureCycle Technologies transforms end-of-life carpet into its UPRP resin. https://www.recyclingtoday.com/article/purecycle-trials-carpet-recycling-using-pandg-technology/(accessed March 21, 2022).
- J. Layman, M. Gunnerson, E. Bond, A. Neltner and J. Hosmer, Articles of reclaimed polypropylene compositions, WO2017/003802Al, 2017.
- J. M. Layman, D. I. Collias, H. Schonemann and K. Williams, Method for separating and purifying polymers from reclaimed product, US11008433, 2021.
- J. M. Layman, D. I. Collias, H. Schonemann and K. Williams, Method for purifying reclaimed polyethylene, US10941269, 2021.
- Polystyrene recycling https://www.polystyvert.com/en/technology/.
- A. Walker, Process for extracting polyester from an article, WO2014/045062Al, 2013.
- Worn Again Technologies https://wornagain.co.uk/.
- P. Patel, Shay Sethi talks about the future of fabric recycling. Chemical & Engineering News, March 15, 2021 Search PubMed.
- Ambercycling. https://ambercycle.com/technology (accessed 15 Febrary, 2022).
- T. A. Cooper, in Developments in End of Life Technologies for Flexible Packaging: Multilayer Films, Barrier Structures and Pouches, Society of Plastics Engineers. Flex Pack Con., Myrtle Beach, NC, October 8 2019, ARGO Group International Myrtle Beach, NC, 2019 Search PubMed.
- Plant & Service. https://www.saperatec.de/en/plant-service.html (accessed March 26, 2022).
- D. R. Bloch, Solvents and non solvents for polymers, The Wiley Database of Polymer Properties, 2003 Search PubMed.
- Y.-B. Zhao, X.-D. Lv and H.-G. Ni, Solvent-based separation and recycling of waste plastics: A review, Chemosphere, 2018, 209, 707–720 CrossRef CAS PubMed.
- J. K. Kim, S. Thomas and P. Saha, Multicomponent Polymeric Materials, Springer, 2016, vol. 223 Search PubMed.
- B. A. Miller-Chou and J. L. Koenig, A review of polymer dissolution, Prog. Polym. Sci., 2003, 28(8), 1223–1270 CrossRef CAS.
- C. M. Hansen, The three dimensional solubility parameter, Danish Technical, Copenhagen, 1967, vol. 14 Search PubMed.
- R. W. Taft, J.-L. M. Abboud, M. J. Kamlet and M. H. Abraham, Linear solvation energy relations, J. Solution Chem., 1985, 14(3), 153–186 CrossRef CAS.
- V. Gutmann, Empirical parameters for donor and acceptor properties of solvents, Electrochim. Acta, 1976, 21(9), 661–670 CrossRef CAS.
- C. G. Swain, M. S. Swain, A. L. Powell and S. Alunni, Solvent effects on chemical reactivity. Evaluation of anion-and cation-solvation components, J. Am. Chem. Soc., 1983, 105(3), 502–513 CrossRef CAS.
- C. M. Hansen, Hansen solubility parameters: a user's handbook, CRC press, 2007 Search PubMed.
- S. Abbott and C. M. Hansen, Hansen solubility parameters in practice, Hansen-Solubility, 2008 Search PubMed.
- A. Chandrasekaran, C. Kim, S. Venkatram and R. Ramprasad, A deep learning solvent-selection paradigm powered by a massive solvent/nonsolvent database for polymers, Macromolecules, 2020, 53(12), 4764–4769 CrossRef CAS.
- P. Zhou, K. L. Sánchez–Rivera, G. W. Huber and R. C. Van Lehn, Computational Approach for Rapidly Predicting Temperature–Dependent Polymer Solubilities Using Molecular–Scale Models, ChemSusChem, 2021, 14(19), 4307–4316 CrossRef CAS.
- A.-S. Bescond and A. Pujari, PET Polymer, IHS Markit, 2020 Search PubMed.
- A. J. Martín, C. Mondelli, S. D. Jaydev and J. Pérez-Ramírez, Catalytic processing of plastic waste on the rise, Chem, 2021, 7(6), 1487–1533 Search PubMed.
- Recycling of polyethylene terephthalate bottles, ed. S. Thomas, A. V. Rane, K. Kanny, V. K. Abitha and M. G. Thomas, William Andrew, Oxford, 2018 Search PubMed.
- S. M. Al-Salem, P. Lettieri and J. Baeyens, Recycling and recovery routes of plastic solid waste (PSW): A review, Waste Manage., 2009, 29(10), 2625–2643 CrossRef CAS PubMed.
- R. D. Allen and M. I. James, Chemical Recycling of PET, in Circular Economy of Polymers: Topics in Recycling Technologies, American Chemical Society, 2021, vol. 1391, pp. 61–80 Search PubMed.
- G. W. Coates and Y. D. Y. L. Getzler, Chemical recycling to monomer for an ideal, circular polymer economy, Nat. Rev. Mater., 2020, 5(7), 501–516 CrossRef CAS.
- J. Payne and M. D. Jones, The Chemical Recycling of Polyesters for a Circular Plastics Economy: Challenges and Emerging Opportunities, ChemSusChem, 2021, 14(19), 4041–4070 CrossRef CAS PubMed.
- K. Ragaert, L. Delva and K. Van Geem, Mechanical and chemical recycling of solid plastic waste, Waste Manage., 2017, 69, 24–58 CrossRef CAS PubMed.
- A. B. Raheem, Z. Z. Noor, A. Hassan, M. K. Abd Hamid, S. A. Samsudin and A. H. Sabeen, Current developments in chemical recycling of post-consumer polyethylene terephthalate wastes for new materials production: A review, J. Cleaner Prod., 2019, 225, 1052–1064 CrossRef CAS.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Beyond Mechanical Recycling: Giving New Life to Plastic Waste, Angew. Chem., Int. Ed., 2020, 59(36), 15402–15423 CrossRef CAS PubMed.
- D. Paszun and T. Spychaj, Chemical Recycling of Poly(ethylene terephthalate), Ind. Eng. Chem. Res., 1997, 36(4), 1373–1383 CrossRef CAS.
- S. R. Nicholson, J. E. Rorrer, A. Singh,M. O. Konev, N. A. Rorrer, A. C. Carpenter, A. J. Jacobsen, Y. Román-Leshkov and G. T. Beckham, The critical role of process analysis in chemical recycling and upcycling of waste plastics, Annu. Rev. Chem. Biomol. Eng., 2022, 13 Search PubMed , https://assets.amazon.science/5a/a7/aa47ad51471ba07a68b8cfc69240/the-critical-role-of-process-analysis-in-chemical-recycling-and-upcycling-of-waste-plastics.pdf.
- B. Geyer, G. Lorenz and A. Kandelbauer, Recycling of poly(ethylene terephthalate) – A review focusing on chemical methods, eXPRESS Polym. Lett., 2016, 10, 559–586 CrossRef CAS.
- S. A. Jabarin and E. A. Lofgren, Thermal stability of polyethylene terephthalate, Polym. Eng. Sci., 1984, 24(13), 1056–1063 CrossRef.
- W. Partenheimer, Transformation of Polymers to Useful Chemical Oxidation, US6,958,373B2, 2005.
- W. Partenheimer, Valuable oxygenates by aerobic oxidation of polymers using metal/bromide homogeneous catalysts, Catal. Today, 2003, 81(2), 117–135 CrossRef CAS.
- W. Partenheimer, Methodology and scope of metal/bromide autoxidation of hydrocarbons, Catal. Today, 1995, 23(2), 69–158 CrossRef CAS.
- S. Ügdüler, K. M. Van Geem, R. Denolf, M. Roosen, N. Mys, K. Ragaert and S. De Meester, Towards closed-loop recycling of multilayer and coloured PET plastic waste by alkaline hydrolysis, Green Chem., 2020, 22(16), 5376–5394 RSC.
- M. Han, 5 - Depolymerization of PET Bottle via Methanolysis and Hydrolysis, in Recycling of Polyethylene Terephthalate Bottles, ed. S. Thomas, A. Rane, K. V. K. A. Kanny and M. G. Thomas, William Andrew Publishing, 2019, pp. 85–108 Search PubMed.
- G. P. Karayannidis and D. S. Achilias, Chemical Recycling of Poly(ethylene terephthalate), Macromol. Mater. Eng., 2007, 292(2), 128–146 CrossRef CAS.
- T. Yoshioka, M. Ota and A. Okuwaki, Conversion of a Used Poly(ethylene terephthalate) Bottle into Oxalic Acid and Terephthalic Acid by Oxygen Oxidation in Alkaline Solutions at Elevated Temperatures, Ind. Eng. Chem. Res., 2003, 42(4), 675–679 CrossRef CAS.
- V. A. Kosmidis, D. S. Achilias and G. P. Karayannidis, Poly(ethylene terephthalate) Recycling and Recovery of Pure Terephthalic Acid. Kinetics of a Phase Transfer Catalyzed Alkaline Hydrolysis, Macromol. Mater. Eng., 2001, 286(10), 640–647 CrossRef CAS.
- M. N. Siddiqui, D. S. Achilias, H. H. Redhwi, D. N. Bikiaris, K. A. G. Katsogiannis and G. P. Karayannidis, Hydrolytic Depolymerization of PET in a Microwave Reactor, Macromol. Mater. Eng., 2010, 295(6), 575–584 CrossRef CAS.
- H. I. Khalaf and O. A. Hasan, Effect of quaternary ammonium salt as a phase transfer catalyst for the microwave depolymerization of polyethylene terephthalate waste bottles, Chem. Eng. J., 2012, 192, 45–48 CrossRef CAS.
- L.-C. Hu, A. Oku, E. Yamada and K. Tomari, Alkali-Decomposition of Poly(ethylene terephthalate) in Mixed Media of Nonaqueous Alcohol and Ether. Study on Recycling of Poly(ethylene terephthalate), Polym. J., 1997, 29(9), 708–712 CrossRef CAS.
- H. Essaddam, Polyethylene terephthalate depolymerization, US9,550,713B1, 2017.
- C.-Y. Kao, B.-Z. Wan and W.-H. Cheng, Kinetics of Hydrolytic Depolymerization of Melt Poly(ethylene terephthalate), Ind. Eng. Chem. Res., 1998, 37(4), 1228–1234 CrossRef CAS.
- J. R. Campanelli, D. G. Cooper and M. R. Kamal, Catalyzed hydrolysis of polyethylene terephthalate melts, J. Appl. Polym. Sci., 1994, 53(8), 985–991 CrossRef CAS.
- G. Güçlü, T. Yalçınyuva, S. Özgümüş and M. Orbay, Hydrolysis of waste polyethylene terephthalate and characterization of products by differential scanning calorimetry, Thermochim. Acta, 2003, 404(1), 193–205 CrossRef.
- L. Zhang, J. Gao, J. Zou and F. Yi, Hydrolysis of poly(ethylene terephthalate) waste bottles in the presence of dual functional phase transfer catalysts, J. Appl. Polym. Sci., 2013, 130(4), 2790–2795 CrossRef CAS.
- D. Stanica-Ezeanu and D. Matei, Natural depolymerization of waste poly(ethylene terephthalate) by neutral hydrolysis in marine water, Sci. Rep., 2021, 11(1), 4431 CrossRef CAS PubMed.
- G. Brown and R. O'Brien, Method for recovering terephthalic acid and ethylene glycol from polyester materials, US3,952,053A, 1976.
- T. Yoshioka, T. Sato and A. Okuwaki, Hydrolysis of waste PET by sulfuric acid at 150 °C for a chemical recycling, J. Appl. Polym. Sci., 1994, 52(9), 1353–1355 CrossRef CAS.
- T. Yoshioka, N. Okayama and A. Okuwaki, Kinetics of Hydrolysis of PET Powder in Nitric Acid by a Modified Shrinking-Core Model, Ind. Eng. Chem. Res., 1998, 37(2), 336–340 CrossRef CAS.
- F. Liu, X. Cui, S. Yu, Z. Li and X. Ge, Hydrolysis reaction of poly(ethylene terephthalate) using ionic liquids as solvent and catalyst, J. Appl. Polym. Sci., 2009, 114(6), 3561–3565 CrossRef CAS.
- W. Yang, R. Liu, C. Li, Y. Song and C. Hu, Hydrolysis of waste polyethylene terephthalate catalyzed by easily recyclable terephthalic acid, Waste Manage., 2021, 135, 267–274 CrossRef CAS PubMed.
- H. Kurokawa, M.-a. Ohshima, K. Sugiyama and H. Miura, Methanolysis of polyethylene terephthalate (PET) in the presence of aluminium tiisopropoxide catalyst to form dimethyl terephthalate and ethylene glycol, Polym. Degrad. Stab., 2003, 79(3), 529–533 CrossRef CAS.
- S. Enthaler, Zinc-Catalyzed Depolymerization Reactions, Zinc Catal., 2015, 207–218 CAS.
- T. Sako, T. Sugeta, K. Otake, N. Nakazawa, M. Sato, K. Namiki and M. Tsugumi, Depolymerization of Polyethylene Terephthalate to Monomers with Supercritical Methanol, J. Chem. Eng. Jpn., 1997, 30(2), 342–346 CrossRef CAS.
- M. Hofmann, J. Sundermeier, C. Alberti and S. Enthaler, Zinc(II) acetate Catalyzed Depolymerization of Poly(ethylene terephthalate), ChemistrySelect, 2020, 5(32), 10010–10014 CrossRef CAS.
- D. D. Pham and J. Cho, Low-energy catalytic methanolysis of poly(ethyleneterephthalate), Green Chem., 2021, 23(1), 511–525 RSC.
- S. Tanaka, J. Sato and Y. Nakajima, Capturing ethylene glycol with dimethyl carbonate towards depolymerisation of polyethylene terephthalate at ambient temperature, Green Chem., 2021, 9412–9416 RSC.
- S. L. Fávaro, A. R. Freitas, T. A. Ganzerli, A. G. B. Pereira, A. L. Cardozo, O. Baron, E. C. Muniz, E. M. Girotto and E. Radovanovic, PET and aluminum recycling from multilayer food packaging using supercritical ethanol, J. Supercrit. Fluids, 2013, 75, 138–143 CrossRef.
- R. E. N. De Castro, G. J. Vidotti, A. F. Rubira and E. C. Muniz, Depolymerization of poly(ethylene terephthalate) wastes using ethanol and ethanol/water in supercritical conditions, J. Appl. Polym. Sci., 2006, 101(3), 2009–2016 CrossRef CAS.
- P. Lozano-Martinez, T. Torres-Zapata and N. Martin-Sanchez, Directing Depolymerization of PET with Subcritical and Supercritical Ethanol to Different Monomers through Changes in Operation Conditions, ACS Sustainable Chem. Eng., 2021, 9(29), 9846–9853 CrossRef CAS.
- C. S. Nunes, M. J. Vieira da Silva, D. Cristina da Silva, A. d. R. Freitas, F. A. Rosa, A. F. Rubira and E. C. Muniz, PET depolymerisation in supercritical ethanol catalysed by [Bmim][BF4], RSC Adv., 2014, 4(39), 20308–20316 RSC.
- J. R. Fernandes, L. P. Amaro, E. C. Muniz, S. L. Favaro and E. Radovanovic, PET depolimerization in supercritical ethanol conditions catalysed by nanoparticles of metal oxides, J. Supercrit. Fluids, 2020, 158, 104715 CrossRef.
- J. Xin, Q. Zhang, J. Huang, R. Huang, Q. Z. Jaffery, D. Yan, Q. Zhou, J. Xu and X. Lu, Progress in the catalytic glycolysis of polyethylene terephthalate, J. Environ. Manage., 2021, 296, 113267 CrossRef CAS PubMed.
- J. Y. Chen, C. F. Ou, Y. C. Hu and C. C. Lin, Depolymerization of poly(ethylene terephthalate) resin under pressure, J. Appl. Polym. Sci., 1991, 42(6), 1501–1507 CrossRef CAS.
- U. R. Vaidya and V. M. Nadkarni, Polyester polyols from glycolyzed PET waste: Effect of glycol type on kinetics of polyesterification, J. Appl. Polym. Sci., 1989, 38(6), 1179–1190 CrossRef CAS.
- S. Baliga and W. T. Wong, Depolymerization of poly(ethylene terephthalate) recycled from post-consumer soft-drink bottles, J. Polym. Sci., Part A: Polym. Chem., 1989, 27(6), 2071–2082 CrossRef CAS.
- G. Güçlü, A. Kaşgöz, S. Özbudak, S. Özgümüş and M. Orbay, Glycolysis of poly(ethylene terephthalate) wastes in xylene, J. Appl. Polym. Sci., 1998, 69(12), 2311–2319 CrossRef.
- S. H. Mansour and N. E. Ikladious, Depolymerization of poly(ethylene terephthalate) wastes using 1,4-butanediol and triethylene glycol, Polym. Test., 2002, 21(5), 497–505 CrossRef CAS.
- C.-H. Chen, Study of glycolysis of poly(ethylene terephthalate) recycled from postconsumer soft-drink bottles. III. Further investigation, J. Appl. Polym. Sci., 2003, 87(12), 2004–2010 CrossRef CAS.
- M. Ghaemy and K. Mossaddegh, Depolymerisation of poly(ethylene terephthalate) fibre wastes using ethylene glycol, Polym. Degrad. Stab., 2005, 90(3), 570–576 CrossRef CAS.
- K. Troev, G. Grancharov, R. Tsevi and I. Gitsov, A novel catalyst for the glycolysis of poly(ethylene terephthalate), J. Appl. Polym. Sci., 2003, 90(4), 1148–1152 CAS.
- P. Fang, B. Liu, J. Xu, Q. Zhou, S. Zhang, J. Ma and X. lu, High-efficiency glycolysis of poly(ethylene terephthalate) by sandwich-structure polyoxometalate catalyst with two active sites, Polym. Degrad. Stab., 2018, 156, 22–31 CrossRef CAS.
- H. Wang, Y. Liu, Z. Li, X. Zhang, S. Zhang and Y. Zhang, Glycolysis of poly(ethylene terephthalate) catalyzed by ionic liquids, Eur. Polym. J., 2009, 45(5), 1535–1544 CrossRef CAS.
- Q. Wang, X. Lu, X. Zhou, M. Zhu, H. He and X. Zhang, 1-Allyl-3-methylimidazolium halometallate ionic liquids as efficient catalysts for the glycolysis of poly(ethylene terephthalate), J. Appl. Polym. Sci., 2013, 129(6), 3574–3581 CrossRef CAS.
- M. Khoonkari, A. H. Haghighi, Y. Sefidbakht, K. Shekoohi and A. Ghaderian, Chemical Recycling of PET Wastes with Different Catalysts, Int. J. Polym. Sci., 2015, 2015, 124524 Search PubMed.
- H. Wang, Z. Li, Y. Liu, X. Zhang and S. Zhang, Degradation of poly(ethylene terephthalate) using ionic liquids, Green Chem., 2009, 11(10), 1568–1575 RSC.
- H. Wang, R. Yan, Z. Li, X. Zhang and S. Zhang, Fe-containing magnetic ionic liquid as an effective catalyst for the glycolysis of poly(ethylene terephthalate), Catal. Commun., 2010, 11(8), 763–767 CrossRef CAS.
- S. Cot, M. K. Leu, A. Kalamiotis, G. Dimitrakis, V. Sans, I. de Pedro and I. Cano, An Oxalate-Bridged Binuclear Iron(III) Ionic Liquid for the Highly Efficient Glycolysis of Polyethylene Terephthalate under Microwave Irradiation, ChemPlusChem, 2019, 84(7), 786–793 CrossRef CAS PubMed.
- Q. F. Yue, C. X. Wang, L. N. Zhang, Y. Ni and Y. X. Jin, Glycolysis of poly(ethylene terephthalate) (PET) using basic ionic liquids as catalysts, Polym. Degrad. Stab., 2011, 96(4), 399–403 CrossRef CAS.
- A. P. Abbott, E. I. Ahmed, R. C. Harris and K. S. Ryder, Evaluating water miscible deep eutectic solvents (DESs) and ionic liquids as potential lubricants, Green Chem., 2014, 16(9), 4156–4161 RSC.
- D. V. Wagle, H. Zhao and G. A. Baker, Deep Eutectic Solvents: Sustainable Media for Nanoscale and Functional Materials, Acc. Chem. Res., 2014, 47(8), 2299–2308 CrossRef CAS PubMed.
- E. Sert, E. Yılmaz and F. S. Atalay, Chemical Recycling of Polyethlylene Terephthalate by Glycolysis Using Deep Eutectic Solvents, J. Polym. Environ., 2019, 27(12), 2956–2962 CrossRef CAS.
- S. R. Shukla, V. Palekar and N. Pingale, Zeolite catalyzed glycolysis of poly(ethylene terephthalate) bottle waste, J. Appl. Polym. Sci., 2008, 110(1), 501–506 CrossRef CAS.
- A. M. Al-Sabagh, F. Z. Yehia, D. R. K. Harding, G. Eshaq and A. E. ElMetwally, Fe3O4-boosted MWCNT as an efficient sustainable catalyst for PET glycolysis, Green Chem., 2016, 18(14), 3997–4003 RSC.
- F. R. Veregue, C. T. Pereira da Silva, M. P. Moisés, J. G. Meneguin, M. R. Guilherme, P. A. Arroyo, S. L. Favaro, E. Radovanovic, E. M. Girotto and A. W. Rinaldi, Ultrasmall Cobalt Nanoparticles as a Catalyst for PET Glycolysis: A Green Protocol for Pure Hydroxyethyl Terephthalate Precipitation without Water, ACS Sustainable Chem. Eng., 2018, 6(9), 12017–12024 CrossRef CAS.
- Z. Laldinpuii, S. Lalhmangaihzuala, Z. Pachuau and K. Vanlaldinpuia, Depolymerization of poly(ethylene terephthalate) waste with biomass-waste derived recyclable heterogeneous catalyst, Waste Manage., 2021, 126, 1–10 CrossRef CAS PubMed.
- Z. Wang, Y. Wang, S. Xu, Y. Jin, Z. Tang, G. Xiao and H. Su, A pseudo-homogeneous system for PET glycolysis using a colloidal catalyst of graphite carbon nitride in ethylene glycol, Polym. Degrad. Stab., 2021, 190, 109638 CrossRef CAS.
- S. Najafi-Shoa, M. Barikani, M. Ehsani and M. Ghaffari, Cobalt-based ionic liquid grafted on graphene as a heterogeneous catalyst for poly (ethylene terephthalate) glycolysis, Polym. Degrad. Stab., 2021, 192, 109691 CrossRef CAS.
- A. M. Al-Sabagh, F. Z. Yehia, G. Eshaq and A. E. ElMetwally, Ionic Liquid-Coordinated Ferrous Acetate Complex Immobilized on Bentonite As a Novel Separable Catalyst for PET Glycolysis, Ind. Eng. Chem. Res., 2015, 54(50), 12474–12481 CrossRef CAS.
- I. Cano, C. Martin, J. A. Fernandes, R. W. Lodge, J. Dupont, F. A. Casado-Carmona, R. Lucena, S. Cardenas, V. Sans and I. de Pedro, Paramagnetic ionic liquid-coated SiO2@Fe3O4 nanoparticles—The next generation of magnetically recoverable nanocatalysts applied in the glycolysis of PET, Appl. Catal., B, 2020, 260, 118110 CrossRef CAS.
- K. Fukushima, O. Coulembier, J. M. Lecuyer, H. A. Almegren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. McNeil, P. Dubois, R. M. Waymouth, H. W. Horn, J. E. Rice and J. L. Hedrick, Organocatalytic depolymerization of poly(ethylene terephthalate), J. Polym. Sci., Part A: Polym. Chem., 2011, 49(5), 1273–1281 CrossRef CAS.
- K. Fukushima, D. J. Coady, G. O. Jones, H. A. Almegren, A. M. Alabdulrahman, F. D. Alsewailem, H. W. Horn, J. E. Rice and J. L. Hedrick, Unexpected efficiency of cyclic amidine catalysts in depolymerizing poly(ethylene terephthalate), J. Polym. Sci., Part A: Polym. Chem., 2013, 51(7), 1606–1611 CrossRef CAS.
- C. Jehanno, I. Flores, A. P. Dove, A. J. Müller, F. Ruipérez and H. Sardon, Organocatalysed depolymerisation of PET in a fully sustainable cycle using thermally stable protic ionic salt, Green Chem., 2018, 20(6), 1205–1212 RSC.
- N. D. Pingale and S. R. Shukla, Microwave assisted ecofriendly recycling of poly (ethylene terephthalate) bottle waste, Eur. Polym. J., 2008, 44(12), 4151–4156 CrossRef CAS.
- S. Chaudhary, P. Surekha, D. Kumar, C. Rajagopal and P. K. Roy, Microwave assisted glycolysis of poly(ethylene terepthalate) for preparation of polyester polyols, J. Appl. Polym. Sci., 2013, 129(5), 2779–2788 CrossRef.
- D. S. Achilias, H. H. Redhwi, M. N. Siddiqui, A. K. Nikolaidis, D. N. Bikiaris and G. P. Karayannidis, Glycolytic depolymerization of PET waste in a microwave reactor, J. Appl. Polym. Sci., 2010, 118(5), 3066–3073 CrossRef CAS.
- O. Saravari, P. Potiyaraj and S. Phunphoem, Preparation of Urethane Oils from Microwave-Assisted Glycolyzed Products of Waste PET Bottles, Energy Procedia, 2011, 9, 491–497 CrossRef CAS.
- M. Parrott, Chemical recycling of polyethylene terephthalate by microwave irradiation, US10,858,493B2, 2020.
- K. Fukushima, J. M. Lecuyer, D. S. Wei, H. W. Horn, G. O. Jones, H. A. Al-Megren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. McNeil, J. E. Rice and J. L. Hedrick, Advanced chemical recycling of poly(ethylene terephthalate) through organocatalytic aminolysis, Polym. Chem., 2013, 4(5), 1610–1616 RSC.
- H. Zahn and H. Pfeifer, Aminolysis of polyethylene terephthalate, Polymer, 1963, 4, 429–432 CrossRef CAS.
- R. K. Soni, M. Teotia and K. Dutt, Studies on synthesis and characterization of a novel acrylic aromatic amide oligomer of aminolysed endproducts generated from pet waste with hydrazine monohydrate and its photocuring with acrylate monomers, J. Appl. Polym. Sci., 2010, 118(2), 638–645 CAS.
- A. Bhatnagar, R. Padhan, A. Gupta and K. Agadi, An environment friendly method for disposal of waste plastics through conversion of polyethylene terephthalate (pet) polymers to anti-stripping compounds, WO2013/118057Al, 2013.
- R. K. Soni and S. Singh, Synthesis and characterization of terephthalamides from poly(ethylene terephthalate) waste, J. Appl. Polym. Sci., 2005, 96(5), 1515–1528 CrossRef CAS.
- S. R. Shukla and A. M. Harad, Aminolysis of polyethylene terephthalate waste, Polym. Degrad. Stab., 2006, 91(8), 1850–1854 CrossRef CAS.
- M. E. Tawfik and S. B. Eskander, Chemical recycling of poly(ethylene terephthalate) waste using ethanolamine. Sorting of the end products, Polym. Degrad. Stab., 2010, 95(2), 187–194 CrossRef CAS.
- K. Chan and A. Zinchenko, Conversion of waste bottles’ PET to a hydrogel adsorbent via PET aminolysis, J. Environ. Chem. Eng., 2021, 9(5), 106129 CrossRef CAS.
- K. Fukushima, G. O. Jones, H. W. Horn, J. E. Rice, T. Kato and J. L. Hedrick, Formation of bis-benzimidazole and bis-benzoxazole through organocatalytic depolymerization of poly(ethylene terephthalate) and its mechanism, Polym. Chem., 2020, 11(30), 4904–4913 RSC.
- N. D. Pingale and S. R. Shukla, Microwave-assisted aminolytic depolymerization of PET waste, Eur. Polym. J., 2009, 45(9), 2695–2700 CrossRef CAS.
- C. N. Hoang and Y. H. Dang, Aminolysis of poly(ethylene terephthalate) waste with ethylenediamine and characterization of α,ω-diamine products, Polym. Degrad. Stab., 2013, 98(3), 697–708 CrossRef CAS.
- D. S. Achilias, G. P. Tsintzou, A. K. Nikolaidis, D. N. Bikiaris and G. P. Karayannidis, Aminolytic depolymerization of poly(ethylene terephthalate) waste in a microwave reactor, Polym. Int., 2011, 60(3), 500–506 CrossRef CAS.
- P. Sharma, B. Lochab, D. Kumar and P. K. Roy, Sustainable Bis-benzoxazines from Cardanol and PET-Derived Terephthalamides, ACS Sustainable Chem. Eng., 2016, 4(3), 1085–1093 CrossRef CAS.
- M. T. Martello and M. A. Hillmyer, Polylactide–Poly(6-methyl-ε-caprolactone)–Polylactide Thermoplastic Elastomers, Macromolecules, 2011, 44(21), 8537–8545 CrossRef CAS.
- K. Tanemura, T. Suzuki, Y. Nishida and T. Horaguchi, Synthesis of the sulfonated condensed polynuclear aromatic (S-COPNA) resins as strong protonic acids, Tetrahedron, 2011, 67(6), 1314–1319 CrossRef CAS.
- Y. Luo, S. Zhang, Y. Ma, W. Wang and B. Tan, Microporous organic polymers synthesized by self-condensation of aromatic hydroxymethyl monomers, Polym. Chem., 2013, 4(4), 1126–1131 RSC.
- E. M. Krall, T. W. Klein, R. J. Andersen, A. J. Nett, R. W. Glasgow, D. S. Reader, B. C. Dauphinais, S. P. Mc Ilrath, A. A. Fischer, M. J. Carney, D. J. Hudson and N. J. Robertson, Controlled hydrogenative depolymerization of polyesters and polycarbonates catalyzed by ruthenium(II) PNN pincer complexes, Chem. Commun., 2014, 50(38), 4884–4887 RSC.
- J. A. Fuentes, S. M. Smith, M. T. Scharbert, I. Carpenter, D. B. Cordes, A. M. Z. Slawin and M. L. Clarke, On the Functional Group Tolerance of Ester Hydrogenation and Polyester Depolymerisation Catalysed by Ruthenium Complexes of Tridentate Aminophosphine Ligands, Chem. – Eur. J., 2015, 21(30), 10851–10860 CrossRef CAS PubMed.
- S. Westhues, J. Idel and J. Klankermayer, Molecular catalyst systems as key enablers for tailored polyesters and polycarbonate recycling concepts, Sci. Adv., 2018, 4(8), eaat9669 CrossRef CAS.
- Y. Kratish, J. Li, S. Liu, Y. Gao and T. J. Marks, Polyethylene Terephthalate Deconstruction Catalyzed by a Carbon-Supported Single-Site Molybdenum-Dioxo Complex, Angew. Chem., Int. Ed., 2020, 59(45), 19857–19861 CrossRef CAS PubMed.
- Y. Jing, Y. Wang, S. Furukawa, J. Xia, C. Sun, M. J. Hülsey, H. Wang, Y. Guo, X. Liu and N. Yan, Towards the Circular Economy: Converting Aromatic Plastic Waste Back to Arenes over a Ru/Nb2O5 Catalyst, Angew. Chem., Int. Ed., 2021, 60(10), 5527–5535 CrossRef CAS PubMed.
- P. Wu, G. Lu and C. Cai, Cobalt–molybdenum synergistic catalysis for the hydrogenolysis of terephthalate-based polyesters, Green Chem., 2021, 23(21), 8666–8672 RSC.
- R. Wei and W. Zimmermann, Biocatalysis as a green route for recycling the recalcitrant plastic polyethylene terephthalate, Microb. Biotechnol., 2017, 10(6), 1302–1307 CrossRef CAS PubMed.
- N. Wierckx, M. A. Prieto, P. Pomposiello, V. de Lorenzo, K. O'Connor and L. M. Blank, Plastic waste as a novel substrate for industrial biotechnology, Microb. Biotechnol., 2015, 8(6), 900–903 CrossRef PubMed.
- I. Taniguchi, S. Yoshida, K. Hiraga, K. Miyamoto, Y. Kimura and K. Oda, Biodegradation of PET: Current Status and Application Aspects, ACS Catal., 2019, 9(5), 4089–4105 CrossRef CAS.
- F. Kawai, T. Kawabata and M. Oda, Current State and Perspectives Related to the Polyethylene Terephthalate Hydrolases Available for Biorecycling, ACS Sustainable Chem. Eng., 2020, 8(24), 8894–8908 CrossRef CAS.
- R. Wei, T. Tiso, J. Bertling, K. O'Connor, L. M. Blank and U. T. Bornscheuer, Possibilities and limitations of biotechnological plastic degradation and recycling, Nat. Catal., 2020, 3(11), 867–871 CrossRef CAS.
- A. Carniel, V. d. A. Waldow and A. M. d. Castro, A comprehensive and critical review on key elements to implement enzymatic PET depolymerization for recycling purposes, Biotechnol. Adv., 2021, 52, 107811 CrossRef CAS PubMed.
- R.-J. Müller, H. Schrader, J. Profe, K. Dresler and W.-D. Deckwer, Enzymatic Degradation of Poly(ethylene terephthalate): Rapid Hydrolyse using a Hydrolase from T. fusca, Macromol. Rapid Commun., 2005, 26(17), 1400–1405 CrossRef.
- X. Hu, U. Thumarat, X. Zhang, M. Tang and F. Kawai, Diversity of polyester-degrading bacteria in compost and molecular analysis of a thermoactive esterase from Thermobifida alba AHK119, Appl. Microbiol. Biotechnol., 2010, 87(2), 771–779 CrossRef CAS PubMed.
- D. Ribitsch, E. H. Acero, K. Greimel, A. Dellacher, S. Zitzenbacher, A. Marold, R. D. Rodriguez, G. Steinkellner, K. Gruber, H. Schwab and G. M. Guebitz, A New Esterase from Thermobifida halotolerans Hydrolyses Polyethylene Terephthalate (PET) and Polylactic Acid (PLA), Polymers, 2012, 4(1), 617–629 CrossRef.
- D. Ribitsch, E. H. Acero, K. Greimel, I. Eiteljoerg, E. Trotscha, G. Freddi, H. Schwab and G. M. Guebitz, Characterization of a new cutinase from Thermobifida alba for PET-surface hydrolysis, Biocatal. Biotransform., 2012, 30(1), 2–9 CrossRef CAS.
- C. Roth, R. Wei, T. Oeser, J. Then, C. Föllner, W. Zimmermann and N. Sträter, Structural and functional studies on a thermostable polyethylene terephthalate degrading hydrolase from Thermobifida fusca, Appl. Microbiol. Biotechnol., 2014, 98(18), 7815–7823 CrossRef CAS.
- Å.M Ronkvist, W. Xie, W. Lu and R. A. Gross, Cutinase-Catalyzed Hydrolysis of Poly(ethylene terephthalate), Macromolecules, 2009, 42(14), 5128–5138 CrossRef.
- C. Silva, S. Da, N. Silva, T. Matamá, R. Araújo, M. Martins, S. Chen, J. Chen, J. Wu, M. Casal and A. Cavaco-Paulo, Engineered Thermobifida fusca cutinase with increased activity on polyester substrates, Biotechnol. J., 2011, 6(10), 1230–1239 CrossRef CAS PubMed.
- E. Herrero Acero, D. Ribitsch, A. Dellacher, S. Zitzenbacher, A. Marold, G. Steinkellner, K. Gruber, H. Schwab and G. M. Guebitz, Surface engineering of a cutinase from Thermobifida cellulosilytica for improved polyester hydrolysis, Biotechnol. Bioeng., 2013, 110(10), 2581–2590 CrossRef CAS PubMed.
- J. Then, R. Wei, T. Oeser, M. Barth, M. R. Belisário-Ferrari, J. Schmidt and W. Zimmermann, Ca2+ and Mg2+ binding site engineering increases the degradation of polyethylene terephthalate films by polyester hydrolases from Thermobifida fusca, Biotechnol. J., 2015, 10(4), 592–598 CrossRef CAS PubMed.
- M. Furukawa, N. Kawakami, A. Tomizawa and K. Miyamoto, Efficient Degradation of Poly(ethylene terephthalate) with Thermobifida fusca Cutinase Exhibiting Improved Catalytic Activity Generated using Mutagenesis and Additive-based Approaches, Sci. Rep., 2019, 9(1), 16038 CrossRef PubMed.
- C. M. Payne, B. C. Knott, H. B. Mayes, H. Hansson, M. E. Himmel, M. Sandgren, J. Ståhlberg and G. T. Beckham, Fungal Cellulases., Chem. Rev., 2015, 115(3), 1308–1448 CrossRef CAS PubMed.
- S. Sulaiman, S. Yamato, E. Kanaya, J. Kim, Y. Koga, K. Tanako and S. Kanaya, Isolation of a novel cutinase homolog with polyethylene terephthalate-degrading activity from leaf-branch compost by using a metagenomic approach, Appl. Environ. Microbiol., 2012, 78(5), 1556–1562 CrossRef CAS.
- V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M. L. Desrousseaux, H. Texier, S. Gavalda, M. Cot, E. Guémard, M. Dalibey, J. Nomme, G. Cioci, S. Barbe, M. Chateau, I. André, S. Duquesne and A. Marty, An engineered PET depolymerase to break down and recycle plastic bottles, Nature, 2020, 580(7802), 216–219 CrossRef CAS PubMed.
- A. N. Shirke, C. White, J. A. Englaender, A. Zwarycz, G. L. Butterfoss, R. J. Linhardt and R. A. Gross, Stabilizing Leaf and Branch Compost Cutinase (LCC) with Glycosylation: Mechanism and Effect on PET Hydrolysis, Biochemistry, 2018, 57(7), 1190–1200 CrossRef CAS PubMed.
- C. Sonnendecker, J. Oeser, P. K. Richter, P. Hille, Z. Zhao, C. Fischer, H. Lippold, P. Blázquez-Sánchez, F. Engelberger, C. A. Ramírez-Sarmiento, T. Oeser, Y. Lihanova, R. Frank, H.-G. Jahnke, S. Billig, B. Abel, N. Sträter, J. Matysik and W. Zimmermann, Low Carbon Footprint Recycling of Post-Consumer PET Plastic with a Metagenomic Polyester Hydrolase, ChemSusChem, 2021, 15(9), e202101062 Search PubMed.
- D. Danso, J. Chow and W. Streit, Plastics: Environmental and biotechnological perspectives on microbial degradation, Appl. Environ. Microbiol., 2019, 85(19), e01095–e01019 CrossRef CAS PubMed.
- D. Danso, C. Schmeisser, J. Chow, W. Zimmermann, R. Wei, C. Leggewie, X. Li, T. Hazen and W. Streit, New insights into the function and global distribution of polyethylene terephthalate (PET)-degrading bacteria and enzymes in marine and terrestrial metagenomes, Appl. Environ. Microbiol., 2018, 84(8), e02773–e02717 CrossRef CAS PubMed.
- S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara, K. Miyamoto, Y. Kimura and K. Oda, A bacterium that degrades and assimilates poly(ethylene terephthalate), Science, 2016, 351(6278), 1196–1199 CrossRef CAS PubMed.
- X. Han, W. Liu, J.-W. Huang, J. Ma, Y. Zheng, T.-P. Ko, L. Xu, Y.-S. Cheng, C.-C. Chen and R.-T. Guo, Structural insight into catalytic mechanism of PET hydrolase, Nat. Commun., 2017, 8(1), 2106 CrossRef PubMed.
- H. P. Austin, M. D. Allen, B. S. Donohoe, N. A. Rorrer, F. L. Kearns, R. L. Silveira, B. C. Pollard, G. Dominick, R. Duman, K. El Omari, V. Mykhaylyk, A. Wagner, W. E. Michener, A. Amore, M. S. Skaf, M. F. Crowley, A. W. Thorne, C. W. Johnson, H. L. Woodcock, J. E. McGeehan and G. T. Beckham, Characterization and engineering of a plastic-degrading aromatic polyesterase, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(19), E4350 CrossRef CAS PubMed.
- T. Fecker, P. Galaz-Davison, F. Engelberger, Y. Narui, M. Sotomayor, L. P. Parra and C. A. Ramírez-Sarmiento, Active Site Flexibility as a Hallmark for Efficient PET Degradation by I. sakaiensis PETase, Biophys. J., 2018, 114(6), 1302–1312 CrossRef CAS PubMed.
- S. Joo, I. J. Cho, H. Seo, H. F. Son, H.-Y. Sagong, T. J. Shin, S. Y. Choi, S. Y. Lee and K.-J. Kim, Structural insight into molecular mechanism of poly(ethylene terephthalate) degradation, Nat. Commun., 2018, 9(1), 382 CrossRef PubMed.
- G. J. Palm, L. Reisky, D. Böttcher, H. Müller, E. A. P. Michels, M. C. Walczak, L. Berndt, M. S. Weiss, U. T. Bornscheuer and G. Weber, Structure of the plastic-degrading Ideonella sakaiensis MHETase bound to a substrate, Nat. Commun., 2019, 10(1), 1717 CrossRef PubMed.
- H. F. Son, I. J. Cho, S. Joo, H. Seo, H.-Y. Sagong, S. Y. Choi, S. Y. Lee and K.-J. Kim, Rational Protein Engineering of Thermo-Stable PETase from Ideonella sakaiensis for Highly Efficient PET Degradation, ACS Catal., 2019, 9(4), 3519–3526 CrossRef CAS.
- B. C. Knott, E. Erickson, M. D. Allen, J. E. Gado, R. Graham, F. L. Kearns, I. Pardo, E. Topuzlu, J. J. Anderson, H. P. Austin, G. Dominick, C. W. Johnson, N. A. Rorrer, C. J. Szostkiewicz, V. Copié, C. M. Payne, H. L. Woodcock, B. S. Donohoe, G. T. Beckham and J. E. McGeehan, Characterization and engineering of a two-enzyme system for plastics depolymerization, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(41), 25476 CrossRef CAS PubMed.
- Y. Cui, Y. Chen, X. Liu, S. Dong, Y. e. Tian, Y. Qiao, R. Mitra, J. Han, C. Li, X. Han, W. Liu, Q. Chen, W. Wei, X. Wang, W. Du, S. Tang, H. Xiang, H. Liu, Y. Liang, K. N. Houk and B. Wu, Computational Redesign of a PETase for Plastic Biodegradation under Ambient Condition by the GRAPE Strategy, ACS Catal., 2021, 11(3), 1340–1350 CrossRef CAS.
- H. Lu, D. J. Diaz, N. J. Czarnecki, C. Zhu, W. Kim, R. Shroff, D. J. Acosta, B. Alexander, H. Cole, Y. J. Zhang, N. Lynd, A. D. Ellington and H. S. Alper, Deep learning redesign of PETase for practical PET degrading applications, bioRxiv, 2021 DOI:10.1101/2021.10.10.463845.
- A. Singh, N. A. Rorrer, S. R. Nicholson, E. Erickson, J. S. DesVeaux, A. F. T. Avelino, P. Lamers, A. Bhatt, Y. Zhang, G. Avery, L. Tao, A. R. Pickford, A. C. Carpenter, J. E. McGeehan and G. T. Beckham, Techno-economic, life-cycle, and socioeconomic impact analysis of enzymatic recycling of poly(ethylene terephthalate), Joule, 2021, 5(9), 2479–2503 CrossRef CAS.
- E. Erickson, T. J. Shakespeare, F. Bratti, B. L. Buss, R. Graham, M. A. Hawkins, G. König, W. E. Michener, J. Miscall, K. J. Ramirez, N. A. Rorrer, M. Zahn, A. R. Pickford, J. E. McGeehan and G. T. Beckham, Comparative Performance of PETase as a Function of Reaction Conditions, Substrate Properties, and Product Accumulation, ChemSusChem, 2021, e202101932 Search PubMed.
- R. Wei, D. Breite, C. Song, D. Gräsing, T. Ploss, P. Hille, R. Schwerdtfeger, J. Matysik, A. Schulze and W. Zimmermann, Biocatalytic Degradation Efficiency of Postconsumer Polyethylene Terephthalate Packaging Determined by Their Polymer Microstructures, Adv. Sci., 2019, 6(14), 1900491 CrossRef PubMed.
- M. C. Delpech, F. M. B. Coutinho and M. E. S. Habibe, Bisphenol A-based polycarbonates: characterization of commercial samples, Polym. Test., 2002, 21(2), 155–161 CrossRef CAS.
- E. V. Antonakou and D. S. Achilias, Recent Advances in Polycarbonate Recycling: A Review of Degradation Methods and Their Mechanisms, Waste Biomass Valorization, 2013, 4(1), 9–21 CrossRef CAS.
- J. G. Kim, Chemical recycling of poly(bisphenol A carbonate), Polym. Chem., 2020, 11(30), 4830–4849 RSC.
- R. Geyer, J. R. Jambeck and K. L. Law, Production, use, and fate of all plastics ever made, Sci. Adv., 2017, 3(7), e1700782 CrossRef PubMed.
- M. Blazsó, Thermal decomposition of polymers modified by catalytic effects of copper and iron chlorides, J. Anal. Appl. Pyrolysis, 1999, 51(1), 73–88 CrossRef.
- S.-J. Chiu, S.-H. Chen and C.-T. Tsai, Effect of metal chlorides on thermal degradation of (waste) polycarbonate, Waste Manage., 2006, 26(3), 252–259 CrossRef CAS PubMed.
- B. Subramaniam and M. A. McHugh, Reactions in supercritical fluids - a review, Ind. Eng. Chem. Process Des. Dev., 1986, 25(1), 1–12 CrossRef CAS.
- M. Watanabe, Y. Matsuo, T. Matsushita, H. Inomata, T. Miyake and K. Hironaka, Chemical recycling of polycarbonate in high pressure high temperature steam at 573 K, Polym. Degrad. Stab., 2009, 94(12), 2157–2162 CrossRef CAS.
- H. Weingärtner and E. U. Franck, Supercritical Water as a Solvent, Angew. Chem., Int. Ed., 2005, 44(18), 2672–2692 CrossRef.
- T. Yoshioka, K. Sugawara, T. Mizoguchi and A. Okuwaki, Chemical Recycling of Polycarbonate to Raw Materials by Thermal Decomposition with Calcium Hydroxide/Steam, Chem. Lett., 2005, 34(3), 282–283 CrossRef CAS.
- G. Grause, K. Sugawara, T. Mizoguchi and T. Yoshioka, Pyrolytic hydrolysis of polycarbonate in the presence of earth-alkali oxides and hydroxides, Polym. Degrad. Stab., 2009, 94(7), 1119–1124 CrossRef CAS.
- G. Grause, R. Kärrbrant, T. Kameda and T. Yoshioka, Steam Hydrolysis of Poly(bisphenol A carbonate) in a Fluidized Bed Reactor, Ind. Eng. Chem. Res., 2014, 53(11), 4215–4223 CrossRef CAS.
- G. O. Jones, A. Yuen, R. J. Wojtecki, J. L. Hedrick and J. M. García, Computational and experimental investigations of one-step conversion of poly(carbonate)s into value-added poly(aryl ether sulfone)s, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(28), 7722 CrossRef CAS PubMed.
- Y. Huang, S. Liu and Z. Pan, Effects of plastic additives on depolymerization of polycarbonate in sub-critical water, Polym. Degrad. Stab., 2011, 96(8), 1405–1410 CrossRef CAS.
- G. Grause, N. Tsukada, W. J. Hall, T. Kameda, P. T. Williams and T. Yoshioka, High-value products from the catalytic hydrolysis of polycarbonate waste, Polym. J., 2010, 42(6), 438–442 CrossRef CAS.
- E. Quaranta, Rare Earth metal triflates M(O3SCF3)3 (M=Sc, Yb, La) as Lewis acid catalysts of depolymerization of poly-(bisphenol A carbonate) via hydrolytic cleavage of carbonate moiety: Catalytic activity of La(O3SCF3)3, Appl. Catal., B, 2017, 206, 233–241 CrossRef CAS.
- X. Song, F. Liu, L. Li, X. Yang, S. Yu and X. Ge, Hydrolysis of polycarbonate catalyzed by ionic liquid [Bmim][Ac], J. Hazard. Mater., 2013, 244–245, 204–208 CrossRef CAS PubMed.
- L. Li, F. Liu, Z. Li, X. Song and S. Yu, Hydrolysis of polycarbonate using ionic liquid [Bmim][Cl] as solvent and catalyst, Fibers Polym., 2013, 14(3), 365–368 CrossRef CAS.
- M. Liu, J. Guo, Y. Gu, J. Gao and F. Liu, Degradation of waste polycarbonate via hydrolytic strategy to recover monomer (bisphenol A) catalyzed by DBU-based ionic liquids under metal- and solvent-free conditions, Polym. Degrad. Stab., 2018, 157, 9–14 CrossRef CAS.
- G. P. Tsintzou and D. S. Achilias, Chemical Recycling of Polycarbonate Based Wastes Using Alkaline Hydrolysis Under Microwave Irradiation, Waste Biomass Valorization, 2013, 4(1), 3–7 CrossRef CAS.
- L. Rosi, M. Bartoli, A. Undri, M. Frediani and P. Frediani, Synthesis of dianols or BPA through catalytic hydrolyisis/glycolysis of waste polycarbonates using a microwave heating, J. Mol. Catal. A: Chem., 2015, 408, 278–286 CrossRef CAS.
- L.-C. Hu, A. Oku and E. Yamada, Alkali-catalyzed methanolysis of polycarbonate. A study on recycling of bisphenol A and dimethyl carbonate, Polymer, 1998, 39(16), 3841–3845 CrossRef CAS.
- R. Piñero, J. García and M. J. Cocero, Chemical recycling of polycarbonate in a semi-continuous lab-plant. A green route with methanol and methanol–water mixtures, Green Chem., 2005, 7(5), 380–387 RSC.
- L. Chen, Y. Wu, Y. Ni, K. Huang and Z. Zhu, Depolymerization of polycarbonate in critical region of methanol, Acta Sci. Circumstantiae, 2004, 24(1), 60–64 Search PubMed.
- H. Jie, H. Ke, Z. Qing, C. Lei, W. Yongqiang and Z. Zibin, Study on depolymerization of polycarbonate in supercritical ethanol, Polym. Degrad. Stab., 2006, 91(10), 2307–2314 CrossRef CAS.
- D. Kim, B.-k. Kim, Y. Cho, M. Han and B.-S. Kim, Kinetics of Polycarbonate Glycolysis in Ethylene Glycol, Ind. Eng. Chem. Res., 2009, 48(2), 685–691 CrossRef CAS.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Guanidine and Amidine Organocatalysts for Ring-Opening Polymerization of Cyclic Esters, Macromolecules, 2006, 39(25), 8574–8583 CrossRef CAS.
- E. Quaranta, D. Sgherza and G. Tartaro, Depolymerization of poly(bisphenol A carbonate) under mild conditions by solvent-free alcoholysis catalyzed by 1,8-diazabicyclo[5.4.0]undec-7-ene as a recyclable organocatalyst: a route to chemical recycling of waste polycarbonate, Green Chem., 2017, 19(22), 5422–5434 RSC.
- E. Quaranta, C. C. Minischetti and G. Tartaro, Chemical Recycling of Poly(bisphenol A carbonate) by Glycolysis under 1,8-Diazabicyclo[5.4.0]undec-7-ene Catalysis, ACS Omega, 2018, 3(7), 7261–7268 CrossRef CAS PubMed.
- C. Alberti and S. Enthaler, Depolymerization of End-of-Life Poly(bisphenol A carbonate) via Alkali-Metal-Halide-Catalyzed Methanolysis, Asian J. Org. Chem., 2020, 9(3), 359–363 CrossRef CAS.
- W. Huang, H. Wang, W. Hu, D. Yang, S. Yu, F. Liu and X. Song, Degradation of polycarbonate to produce bisphenol A catalyzed by imidazolium-based DESs under metal-and solvent-free conditions, RSC Adv., 2021, 11(3), 1595–1604 RSC.
- F. Liu, Y. Xiao, X. Sun, G. Qin, X. Song and Y. Liu, Synergistic catalysis over hollow CeO2-CaO-ZrO2 nanostructure for polycarbonate methanolysis with methanol, Chem. Eng. J., 2019, 369, 205–214 CrossRef CAS.
- M. Liu, J. Guo, Y. Gu, J. Gao, F. Liu and S. Yu, Pushing the Limits in Alcoholysis of Waste Polycarbonate with DBU-Based Ionic Liquids under Metal- and Solvent-Free Conditions, ACS Sustainable Chem. Eng., 2018, 6(10), 13114–13121 CrossRef CAS.
- F. Liu, J. Guo, P. Zhao, M. Jia, M. Liu and J. Gao, Novel succinimide-based ionic liquids as efficient and sustainable media for methanolysis of polycarbonate to recover bisphenol A (BPA) under mild conditions, Polym. Degrad. Stab., 2019, 169, 108996 CrossRef.
- F. Liu, Z. Li, S. Yu, X. Cui and X. Ge, Environmentally benign methanolysis of polycarbonate to recover bisphenol A and dimethyl carbonate in ionic liquids, J. Hazard. Mater., 2010, 174(1), 872–875 CrossRef CAS PubMed.
- S. Hata, H. Goto, E. Yamada and A. Oku, Chemical conversion of poly(carbonate) to 1,3-dimethyl-2-imidazolidinone (DMI) and bisphenol A: a practical approach to the chemical recycling of plastic wastes, Polymer, 2002, 43(7), 2109–2116 CrossRef CAS.
- S. Nakano and T. Kato, A new process for producing polyamide from polyester. J. Polym. Sci., Part A: Polym. Chem., 1999, 37(10), 1413–1423 CrossRef CAS.
- F. Iannone, M. Casiello, A. Monopoli, P. Cotugno, M. C. Sportelli, R. A. Picca, N. Cioffi, M. M. Dell'Anna and A. Nacci, Ionic liquids/ZnO nanoparticles as recyclable catalyst for polycarbonate depolymerization, J. Mol. Catal. A: Chem., 2017, 426, 107–116 CrossRef CAS.
- K. Hatakeyama, T. Kojima and T. Funazukuri, Chemical recycling of polycarbonate in dilute aqueous ammonia solution under hydrothermal conditions, J. Mater. Cycles Waste Manage., 2014, 16(1), 124–130 CrossRef CAS.
- R. Arai, K. Zenda, K. Hatakeyama, K. Yui and T. Funazukuri, Reaction kinetics of hydrothermal depolymerization of poly(ethylene naphthalate), poly(ethylene terephthalate), and polycarbonate with aqueous ammonia solution, Chem. Eng. Sci., 2010, 65(1), 36–41 CrossRef CAS.
- H. J. Radusch, Poly(Butylene Terephthalate), Handbook of Thermoplastic Polyesters, 2002, pp. 389–419 Search PubMed.
- F. G. Terrade, J. van Krieken, B. J. V. Verkuijl and E. Bouwman, Catalytic Cracking of Lactide and Poly(Lactic Acid) to Acrylic Acid at Low Temperatures, ChemSusChem, 2017, 10(9), 1904–1908 CrossRef CAS PubMed.
- I. A. Shuklov, H. Jiao, J. Schulze, W. Tietz, K. Kühlein and A. Börner, Studies on the epimerization of diastereomeric lactides, Tetrahedron Lett., 2011, 52(9), 1027–1030 CrossRef CAS.
- Technology: Simple Sustainable Recycling. https://www.depoly.ch (accessed 01/08/22).
- S. L. Anderson, C. P. Ireland, B. Smit and K. Stylianou, Degradation of plastic materials into terephthalic acid (tpa), ethylene glycol and/or other monomers that form the plastic materials, WO2020/173961A1, 2020.
- F. G. Barla, T. Showalter, H. S. Su, J. Jones and I. Bobe, Methods for recycling cotton and polyester fibers from waste textiles, US10,501,599B2, 2020.
- . Threading Together the Future of Circular Fashion. https://circ.earth/(accessed Feb 02, 2022).
- Moving from Linear to Circular. https://gr3n-recycling.com/technology.html (accessed 01/08/22).
- Making Cutting Edge Science an Everyday Reality. https://www.loopindustries.com/en/about (accessed 01/08/22).
- J. Paben, Eastman offers details on $250M depolymerization plant. https://resource-recycling.com/plastics/2021/02/03/eastman-offers-details-on-250m-depolymerization-plant/(accessed 01/08/22).
- A. Naujokas and K. Ryan, Recovery process for ethylene glycol and dimethylterephthalate, US5,051,528A, 1991.
- BRING Technology. https://www.jeplan.co.jp/en/technology/(accessed 01/08/22).
- T. Hooghoudt, V. Pilippi and M. V. Artigas, Polymer Degradation, US2018/0037710A1, 2019.
- Ioniqa takes first 10 kiloton PET upcycling factory into operation. https://ioniqa.com/ioniqa-takes-first-10-kiloton-pet-upcycling-factory-into-operation/(accessed 01/08/22).
- R. D. Allen, K. M. Bajjuri, J. L. Hedrick, G. Breyta and C. E. Larson, Methods and materials for depolymerizing polyesters, US9,255,194B2, 2015.
- Technip Energies, IBM and Under Armour form joint venture to advance the possibilities of plastics recycling technology. https://www.technipenergies.com/media/news/technip-energies-ibm-under-armour-form-joint-venture-advance-possibilities-plastics-recycling (accessed 01/08/22).
- Enzymatic Recycling: Removing the Constraints of Current Processes. https://www.carbios.com/en/enzymatic-recycling/(accessed 01/08/22).
- Our Partners. https://www.carbios.com/en/partnering/(accessed 01/08/22).
- F. Akoueson, C. Chbib, S. Monchy, I. Paul-Pont, P. Doyen, A. Dehaut and G. Duflos, Identification and quantification of plastic additives using pyrolysis-GC/MS: A review, Sci. Total Environ., 2021, 773, 145073 CrossRef CAS.
- Technip announces successful launch of Carbios industrial demonstration plant. https://www.hydrocarbonprocessing.com/news/2021/11/technip-announces-successful-launch-of-carbios-industrial-demonstration-plant (accessed 01/19/2022).
- Y. Nakaji, M. Tamura, S. Miyaoka, S. Kumagai, M. Tanji, Y. Nakagawa, T. Yoshioka and K. Tomishige, Low-Temperature Catalytic Upgrading of Waste Polyolefinic Plastics into Liquid Fuels and Waxes, Appl. Catal., B, 2021, 285, 119805 CrossRef CAS.
- Y. Jing, Y. Wang, S. Furukawa, J. Xia, C. Sun, M. J. Hülsey, H. Wang, Y. Guo, X. Liu and N. Yan, Towards the Circular Economy: Converting Aromatic Plastic Waste Back to Arenes over a Ru/Nb2O5 Catalyst, Angew. Chem., 2021, 133(10), 5587–5595 CrossRef.
- J. E. Rorrer, G. T. Beckham and Y. Román-Leshkov, Conversion of Polyolefin Waste to Liquid Alkanes with Ru-Based Catalysts under Mild Conditions, JACS Au, 2020, 1(1), 8–12 CrossRef PubMed.
- J. E. Rorrer, C. Troyano-Valls, G. T. Beckham and Y. Román-Leshkov, Hydrogenolysis of Polypropylene and Mixed Polyolefin Plastic Waste over Ru/C to Produce Liquid Alkanes, ACS Sustainable Chem. Eng., 2021, 9(35), 11661–11666 CrossRef CAS.
- W.-T. Lee, F. D. Bobbink, A. P. van Muyden, K.-H. Lin, C. Corminboeuf, R. R. Zamani and P. J. Dyson, Catalytic Hydrocracking of Synthetic Polymers into Grid-Compatible Gas Streams, Cell Rep. Phys. Sci., 2021, 2(2), 100332 CrossRef CAS.
- C. Wang, T. Xie, P. A. Kots, B. C. Vance, K. Yu, P. Kumar, J. Fu, S. Liu, G. Tsilomelekis and E. A. Stach, Polyethylene Hydrogenolysis at Mild Conditions over Ruthenium on Tungstated Zirconia, JACS Au, 2021, 1(9), 1422–1434 CrossRef CAS.
- P. A. Kots, S. Liu, B. C. Vance, C. Wang, J. D. Sheehan and D. G. Vlachos, Polypropylene Plastic Waste Conversion to Lubricants over Ru/TiO2 Catalysts, ACS Catal., 2021, 11, 8104–8115 CrossRef CAS.
- C. Jia, S. Xie, W. Zhang, N. N. Intan, J. Sampath, J. Pfaendtner and H. Lin, Deconstruction of high-density polyethylene into liquid hydrocarbon fuels and lubricants by hydrogenolysis over Ru catalyst, Chem. Catal., 2021, 437–455 CrossRef CAS.
- S. Liu, P. A. Kots, B. C. Vance, A. Danielson and D. G. Vlachos, Plastic waste to fuels by hydrocracking at mild conditions, Sci. Adv., 2021, 7(17), eabf8283 CrossRef CAS PubMed.
- G. Celik, R. M. Kennedy, R. A. Hackler, M. Ferrandon, A. Tennakoon, S. Patnaik, A. M. LaPointe, S. C. Ammal, A. Heyden and F. A. Perras, Upcycling Single-Use Polyethylene into High-Quality Liquid Products, ACS Cent. Sci., 2019, 5(11), 1795–1803 CrossRef CAS PubMed.
- A. Tennakoon, X. Wu, A. L. Paterson, S. Patnaik, Y. Pei, A. M. LaPointe, S. C. Ammal, R. A. Hackler, A. Heyden and I. I. Slowing, Catalytic Upcycling of High-Density Polyethylene via a Processive Mechanism, Nat. Catal., 2020, 3(11), 893–901 CrossRef CAS.
- S. D. Jaydev, A. J. Martin and J. Pérez-Ramírez, Direct Conversion of Polypropylene into Liquid Hydrocrabons on Carbon–Supported Platinum Catalysts, ChemSusChem, 2021, 5179–5185 CrossRef CAS PubMed.
- J. Kijeński and T. Kaczorek, Catalytic degradation of polystyrene-Rapid Communication, Polimery, 2005, 50(1), 60–63 CrossRef.
- D. Munir, F. Piepenbreier and M. R. Usman, Hydrocracking of a plastic mixture over various micro-mesoporous composite zeolites, Powder Technol., 2017, 316, 542–550 CrossRef CAS.
- D. Munir, M. F. Irfan and M. R. Usman, Hydrocracking of virgin and waste plastics: A detailed review, Renewable Sustainable Energy Rev., 2018, 90, 490–515 CrossRef CAS.
- C. W. Yeung, J. Y. Teo, X. J. Loh and J. Y. Lim, Polyolefins and Polystyrene as Chemical Resources for a Sustainable Future: Challenges, Advances, and Prospects, ACS Mater. Lett., 2021, 3, 1660–1676 CrossRef CAS.
- J. B. Williamson, S. E. Lewis, R. R. Johnson III, I. M. Manning and F. A. Leibfarth, C–H Functionalization of Commodity Polymers, Angew. Chem., Int. Ed., 2019, 58(26), 8654–8668 CrossRef CAS PubMed.
- L. Chen, K. G. Malollari, A. Uliana, D. Sanchez, P. B. Messersmith and J. F. Hartwig, Selective, Catalytic Oxidations of C–H Bonds in Polyethylenes Produce Functional Materials with Enhanced Adhesion, Chem, 2021, 7(1), 137–145 CAS.
- X. Jia, C. Qin, T. Friedberger, Z. Guan and Z. Huang, Efficient and Selective Degradation of Polyethylenes into Liquid Fuels and Waxes under Mild Conditions, Sci. Adv., 2016, 2(6), e1501591 CrossRef PubMed.
- F. Zhang, M. Zeng, R. D. Yappert, J. Sun, Y.-H. Lee, A. M. LaPointe, B. Peters, M. M. Abu-Omar and S. L. Scott, Polyethylene Upcycling to Long-Chain Alkylaromatics by Tandem Hydrogenolysis/Aromatization, Science, 2020, 370(6515), 437–441 CrossRef CAS PubMed.
- S. Tian, Y. Jiao, Z. Gao, Y. Xu, L. Fu, H. Fu, W. Zhou, C. Hu, G. Liu and M. Wang, Catalytic Amination of Polylactic Acid to Alanine, J. Am. Chem. Soc., 2021, 16358–16363 CrossRef CAS PubMed.
- L. Chen, K. G. Malollari, A. Uliana and J. F. Hartwig, Ruthenium-Catalyzed, Chemoselective and Regioselective Oxidation of Polyisobutene, J. Am. Chem. Soc., 2021, 143(12), 4531–4535 CrossRef CAS PubMed.
- U. Kanbur, G. Zang, A. L. Paterson, P. Chatterjee, R. A. Hackler, M. Delferro, I. I. Slowing, F. A. Perras, P. Sun and A. D. Sadow, Catalytic Carbon-Carbon Bond Cleavage and Carbon-Element Bond Formation Give New Life for Polyolefins as Biodegradable Surfactants, Chem, 2021, 7(5), 1347–1362 CAS.
- J. De Andrés, A. Aguilar and J. Domenech, Kinetic Study of the Catalytic Effect of Zinc(II) Ion on the Dehydrogenation of Atactic Polypropylene by Sulfur, J. Macromol. Sci., Chem., 1990, 27(2), 213–223 CrossRef.
- A. Ray, K. Zhu, Y. V. Kissin, A. E. Cherian, G. W. Coates and A. S. Goldman, Dehydrogenation of Aliphatic Polyolefins Catalyzed by Pincer-Ligated Iridium Complexes, Chem. Commun., 2005,(27), 3388–3390 RSC.
- D. Brown and R. Lowry, Molecular Weight Standards from Sulfonation of Polystyrene, J. Polym. Sci., Polym. Chem. Ed., 1979, 17(4), 1039–1046 CrossRef CAS.
- J. E. Coughlin, A. Reisch, M. Z. Markarian and J. B. Schlenoff, Sulfonation of Polystyrene: Toward the “Ideal” Polyelectrolyte, J. Polym. Sci., Part A: Polym. Chem., 2013, 51(11), 2416–2424 CrossRef CAS.
- F. Kučera and J. Jančář, Preliminary Study of Sulfonation of Polystyrene by Homogeneous and Heterogeneous Reaction, Chem. Pap., 1996, 50(4), 224–227 Search PubMed.
- F. Kučera and J. Jančář, Homogeneous and Heterogeneous Sulfonation of Polymers: a Review, Polym. Eng. Sci., 1998, 38(5), 783–792 CrossRef.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Silica-Like Malleable Materials from Permanent Organic Networks, Science, 2011, 334(6058), 965–968 CrossRef CAS PubMed.
- Y. Zhou, R. Groote, J. G. Goossens, R. P. Sijbesma and J. P. Heuts, Tuning PBT Vitrimer Properties by Controlling the Dynamics of the Adaptable Network, Polym. Chem., 2019, 10(1), 136–144 RSC.
- L. Yue, V. S. Bonab, D. Yuan, A. Patel, V. Karimkhani and I. Manas-Zloczower, Vitrimerization: A Novel Concept to Reprocess and Recycle Thermoset Waste via Dynamic Chemistry, Global Challenges, 2019, 3(7), 1800076 CrossRef PubMed.
- W. Denissen, J. M. Winne and F. E. Du Prez, Vitrimers: Permanent Organic Networks with Glass-Like Fluidity, Chem. Sci., 2016, 7(1), 30–38 RSC.
- C. Hopmann, M. Adamy and A. Cohnen, Introduction to reactive extrusion, Reactive Extrusion: Principles and Applications, 2017, pp. 1–10 Search PubMed.
- D. Shi, J. Yang, Z. Yao, Y. Wang, H. Huang, W. Jing, J. Yin and G. Costa, Functionalization of isotactic polypropylene with maleic anhydride by reactive extrusion: mechanism of melt grafting, Polymer, 2001, 42(13), 5549–5557 CrossRef CAS.
- J. B. Williamson, C. G. Na, R. R. Johnson III, W. F. Daniel, E. J. Alexanian and F. A. Leibfarth, Chemo-and regioselective functionalization of isotactic polypropylene: A mechanistic and structure–property study, J. Am. Chem. Soc., 2019, 141(32), 12815–12823 CrossRef CAS.
- T. J. Fazekas, J. W. Alty, E. K. Neidhart, A. S. Miller, F. A. Leibfarth and E. J. Alexanian, Diversification of aliphatic C–H bonds in small molecules and polyolefins through radical chain transfer, Science, 2022, 375(6580), 545–550 CrossRef CAS.
- M. Butcher, Novoloop says it's worked out how to upcycle plastic waste, raises $11M Series A. https://techcrunch.com/2022/02/14/novoloop-says-its-worked-out-how-to-upcycle-plastic-waste-raises-11m-series-a/?guccounter=1&guce_referrer=aHR0cHM6Ly93d3cubm92b2xvb3AuY29tLw&guce_referrer_sig=AQAAAE528UVdX0V9achO5yivPlUNGIzR1f8PLRKsCcthW3CsLPIoejxo919fF-v3V1VOzHfddFhDZTejDx-7tT3B77CJJysbfJTz0R-hEz6Ri0wVOiLkEYHLfRKK4BX1cV-E8uQyaMk91b0EYbwOmzpH7GiDocq-P0j-NO3CZ49bX4xH.
- Novoloop upcycling. https://www.novoloop.com/technology (accessed 2/22/2022).
- J. Y. Yao, Y. W. Wang, T. Muppaneni, R. Shrestha, J. L. Roy and G. D. Figuly, Methods for the decomposition of contaminated plastic, Waste, 2020, 1–57 Search PubMed.
- C. Aumnate, N. Rudolph and M. Sarmadi, Recycling of polypropylene/polyethylene blends: Effect of chain structure on the crystallization behaviors, Polymers, 2019, 11(9), 1456 CrossRef CAS PubMed.
- V. Wigotsky, Alloys and blends home in on demanding design specs, Plast. Eng., 1988, 44(11), 25–32 Search PubMed.
- L. Wei, A. McDonald, C. Freitag and J. Morrell, Effects of wood fiber esterification on properties, weatherability and biodurability of wood plastic composites, Polym. Degrad. Stab., 2013, 98, 1348–1361 CrossRef CAS.
- L. Wei, A. McDonald and N. Stark, Grafting of Bacterial Polyhydroxybutyrate (PHB) onto Cellulose via In Situ Reactive Extrusion with Dicumyl Peroxide, Biomacromolecules, 2015, 16, 1040–1049 CrossRef CAS PubMed.
- L. Wei and A. McDonald, Peroxide induced cross-linking by reactive melt processing of two biopolyesters: Poly(3-hydroxybutyrate) and poly(-lactic acid) to improve their melting processability, J. Appl. Polym. Sci., 2015, 132, 41724 Search PubMed.
- L. Wei, N. Stark and A. McDonald, Interfacial improvements in biocomposites based on poly(3-hydroxybutyrate) and poly(3-hydroxybutyrate-co-3-hydroxyvalerate) bioplastics reinforced and grafted with α-cellulose fiber, Green Chem., 2015, 17, 4800–4814 RSC.
- S. Stanic, T. Koch, K. Schmid, S. Knaus and V.-M. Archodoulaki, Upcycling of polypropylene with various concentrations of peroxydicarbonate and dilauroyl peroxide and two processing steps, J. Appl. Polym. Sci., 2021, e50659 CrossRef.
- H. Khonakdar, J. Morshedian, U. Wagenknecht and S. Jafari, An investigation of chemical crosslinking effect on properties of high-density polyethylene, Polymer, 2003, 44, 4301–4309 CrossRef CAS.
- R. Rasal, A. Janorkar and D. Hirt, Poly(lactic acid) modifications, Prog. Polym. Sci., 2010, 35, 338–356 CrossRef CAS.
- H. Zhou, Z. Wang, G. Xu, X. Wang, B. Wen and S. Jin, Preparation of Crosslinked High density Polyethylene Foam Using Supercritical CO2 as Blowing Agent, Cell. Polym., 2017, 36(4), 167–182 CrossRef CAS.
- M. Takamura, T. Nakamura, S. Kawaguchi, T. Takahashi and K. Koyama, Molecular characterization and crystallization behavior of peroxide-induced slightly crosslinked poly(L-lactide) during extrusion, Polym. J., 2010, 42, 600–608 CrossRef CAS.
- N. Gaylord, Compatibilizing agents: Structure and function in polyblends, J. Macromol. Sci., Chem., 1989, 26, 1211–1229 Search PubMed.
- S. C. Manning and R. B. Moore, Carboxylation of polypropylene by reactive extrusion with functionalized peroxides for use as a compatibilizer in polypropylene/polyamide–6, 6 blends, J. Vinyl Addit. Technol., 1997, 3(2), 184–189 CrossRef CAS.
- T. Sakai, Report on the state of the art: Reactive processing using twin-screw extruders, Adv. Polym. Technol., 1991, 11, 99–108 CrossRef CAS.
- R. Asay, M. Hein and D. Wharry, Blends and alloys of vinyl—technology and applications, J. Vinyl Addit. Technol., 1993, 15, 76–81 CrossRef CAS.
- M. Hein, Elastomeric alloys of PVC, J. Vinyl Addit. Technol., 1994, 16, 208–212 CrossRef CAS.
- Y. Kimura, Co-rotating Twin Screw Extruder Compounding Technologies, Plast. Eng., 2014, 70, 32–36 CrossRef.
- A. Amor, N. Okhay, A. Guinault, G. Miquelard-Garnier, C. Sollogoub and M. Gervais, Combined compatibilization and plasticization effect of low molecular weight poly(Lactic acid) in poly(lactic acid)/poly(3-hydroxybutyrate-co-3-hydroxyvalerate) blends, eXPRESS Polym. Lett., 2018, 12, 114–125 CrossRef CAS.
- S. Sapieha, P. Allard and Y. Zang, Dicumyl peroxide-modified cellulose/LLDPE composites, J. Appl. Polym. Sci., 1990, 41, 2019–2048 CrossRef.
- S. Horikoshi and N. Serpone, Role of Microwaves in Heterogeneous Catalytic Systems, Catal. Sci. Technol., 2014, 4(5), 1197–1210 RSC.
- X. Jie, W. Li, D. Slocombe, Y. Gao, I. Banerjee, S. Gonzalez-Cortes, B. Yao, H. AlMegren, S. Alshihri and J. Dilworth, Microwave-Initiated Catalytic Deconstruction of Plastic Waste into Hydrogen and High-Value Carbons, Nat. Catal., 2020, 3(11), 902–912 CrossRef CAS.
- C. Ludlow-Palafox and H. A. Chase, Microwave-Induced Pyrolysis of Plastic Wastes, Ind. Eng. Chem. Res., 2001, 40(22), 4749–4756 CrossRef CAS.
- V. Palma, D. Barba, M. Cortese, M. Martino, S. Renda and E. Meloni, Microwaves and Heterogeneous Catalysis: A Review on Selected Catalytic Processes, Catalysts, 2020, 10(2), 246 CrossRef CAS.
- K. Ding, S. Liu, Y. Huang, S. Liu, N. Zhou, P. Peng, Y. Wang, P. Chen and R. Ruan, Catalytic Microwave-Aassisted Pyrolysis of Plastic Waste over NiO and HY for Gasoline-Range Hydrocarbons Production, Energy Convers. Manage., 2019, 196, 1316–1325 CrossRef CAS.
- E. Bäckström, K. Odelius and M. Hakkarainen, Trash to Treasure: Microwave-Assisted Conversion of Polyethylene to Functional Chemicals, Ind. Eng. Chem. Res., 2017, 56(50), 14814–14821 CrossRef.
- E. Bäckström, K. Odelius and M. Hakkarainen, Designed from Recycled: Turning Polyethylene Waste to Covalently Attached Polylactide Plasticizers, ACS Sustainable Chem. Eng., 2019, 7(12), 11004–11013 CrossRef.
- N. Zhou, L. Dai, Y. Lyu, H. Li, W. Deng, F. Guo, P. Chen, H. Lei and R. Ruan, Catalytic Pyrolysis of Plastic Wastes in a Continuous Microwave Assisted Pyrolysis System for Fuel Production, Chem. Eng. J., 2021, 418, 129412 CrossRef CAS.
- Q. Bu, Y. Liu, J. Liang, H. M. Morgan Jr., L. Yan, F. Xu and H. Mao, Microwave-Assisted Co-Pyrolysis of Microwave Torrefied Biomass with Waste Plastics using ZSM-5 as a Catalyst for High Quality Bio-Oil, J. Anal. Appl. Pyrolysis, 2018, 134, 536–543 CrossRef CAS.
- L. Fan, P. Chen, Y. Zhang, S. Liu, Y. Liu, Y. Wang, L. Dai and R. Ruan, Fast Microwave-Assisted Catalytic Co-Pyrolysis of Lignin and Low-Density Polyethylene with HZSM-5 and MgO for Improved Bio-Oil Yield and Quality, Bioresour. Technol., 2017, 225, 199–205 CrossRef CAS PubMed.
- N. Mohanan, Z. Montazer, P. K. Sharma and D. B. Levin, Microbial and enzymatic degradation of synthetic plastics, Front. Microbiol., 2020, 11, 2837 Search PubMed.
- M. Suzuki, Y. Tachibana and K.-i. Kasuya, Biodegradability of poly (3-hydroxyalkanoate) and poly (ε-caprolactone) via biological carbon cycles in marine environments, Polym. J., 2021, 53(1), 47–66 CrossRef CAS.
- B. R. Kang, S. B. Kim, H. A. Song and T. K. Lee, Accelerating the biodegradation of high-density polyethylene (HDPE) using Bjerkandera adusta TBB-03 and lignocellulose substrates, Microorganisms, 2019, 7(9), 304 CrossRef CAS.
- S. Skariyachan, A. A. Patil, A. Shankar, M. Manjunath, N. Bachappanavar and S. Kiran, Enhanced polymer degradation of polyethylene and polypropylene by novel thermophilic consortia of Brevibacillus sps. and Aneurinibacillus sp. screened from waste management landfills and sewage treatment plants, Polym. Degrad. Stab., 2018, 149, 52–68 CrossRef CAS.
- N. Khatoon, A. Jamal and M. I. Ali, Lignin peroxidase isoenzyme: a novel approach to biodegrade the toxic synthetic polymer waste, Environ. Technol., 2019, 40(11), 1366–1375 CrossRef CAS PubMed.
- T. Sumathi, B. Viswanath, A. Sri Lakshmi and D. SaiGopal, Production of laccase by Cochliobolus sp. isolated from plastic dumped soils and their ability to degrade low molecular weight PVC, Biochem. Res. Int., 2016, 2016 Search PubMed.
- ASTRX ASTRX Review of Material Flow at MRFs and Reprocessors; Sustainable Packaging Coalition and The Recycling Partnership, 2018 Search PubMed.
- Staub, C. MRF operator: Lithium-ion batteries are ‘ticking time bombs’. https://resource-recycling.com/recycling/2021/04/13/mrf-operator-lithium-ion-batteries-are-ticking-time-bombs/(accessed 15 Nov 2021).
- U.S.EPA, An Analysis of Lithium-ion Battery Fires in Waste Management and Recycling; United States Environmental Protection Agency, 2021 Search PubMed.
- CEDOC. Proyecto diagnóstico participativo situacional y propuesta de programa de intervención con Mujeres Pepenadoras de la Unidad Habitacional Manuel M. López, Tláhuac 2014 Search PubMed.
- N. C. Maragou, A. Makri, E. N. Lampi, N. S. Thomaidis and M. A. Koupparis, Migration of bisphenol A from polycarbonate baby bottles under real use conditions, Food Addit. Contam., Part A, 2008, 25(3), 373–383 CrossRef CAS PubMed.
- D. Chiche and J.-M. Schweitzer, Investigation of competitive COS and HCN hydrolysis reactions upon an industrial catalyst: Langmuir-Hinshelwood kinetics modeling, Appl. Catal., B, 2017, 205, 189–200 CrossRef CAS.
- J. Aguado, D. P. Serrano and J. M. Escola, Fuels from Waste Plastics by Thermal and Catalytic Processes: A Review, Ind. Eng. Chem. Res., 2008, 47(21), 7982–7992 CrossRef CAS.
- E. Asmelash, G. Prakash, R. Gorini and D. Gielen, Role of IRENA for global transition to 100% renewable energy, in Accelerating the transition to a 100% renewable energy era, Springer, 2020; pp. 51–71 Search PubMed.
- L., F. Price of ethylene worldwide from 2017 to 2021. https://www.statista.com/statistics/1170573/price-ethylene-forecast-globally/#:~:text=The%20average%20price%<?pdb_no 20of?>20of<?pdb END?>%20ethylene,comparison%20with%20the%20previous%20year. (accessed Feb 02).
- C. Mukherjee, J. Denney, E. Mbonimpa, J. Slagley and R. Bhowmik, A review on municipal solid waste-to-energy trends in the USA, Renewable Sustainable Energy Rev., 2020, 119, 109512 CrossRef CAS.
- S. Das, C. Liang and J. B. Dunn, Life Cycle Assessment of Polymers and Their Recycling, in Circular Economy of Polymers: Topics in Recycling Technologies, ACS Publications, 2021, pp. 143–170 Search PubMed.
- P. G. Jessop, Searching for green solvents, Green Chem., 2011, 13(6), 1391–1398 RSC.
- J. García-Turiel and B. Jérôme, Solvent retention in thin polymer films studied by gas chromatography, Colloid Polym. Sci., 2007, 285(14), 1617–1623 CrossRef.
- I. Vollmer, M. J. Jenks, M. C. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van Der Laan, F. Meirer, J. T. Keurentjes and B. M. Weckhuysen, Beyond mechanical recycling: Giving new life to plastic waste, Angew. Chem., Int. Ed., 2020, 59(36), 15402–15423 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc02588d |
| This journal is © The Royal Society of Chemistry 2022 |