
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIncreased performance of an all-organic redox flow battery model via nitration of the [4]helicenium DMQA ion electrolyte†
Jules
Moutet
,
David
Mills
,
Md Mubarak
Hossain
and
Thomas L.
Gianetti
 *
*
University of Arizona, Department of Chemistry and Biochemistry, Tucson, AZ, USA. E-mail: tgianetti@arizona.edu
First published on 10th November 2021
Abstract
Redox flow batteries (RFBs), through their scalable design and virtually unlimited capacity, are promising candidates for large-scale energy storage. While recent advances in the development of redox-active bipolar organic molecules satisfy the prerequisites for the pioneering emergence of symmetrical all-organic redox flow batteries (SORFBs), problems of low durability or low energy density remain a bottleneck for their wide-spread application. The present work reports that nitration of the [4]helicenium dimethoxyquinacridinium (DMQA+) ion core (NO2C+) results in a significantly enhanced electrochemical performance of DMQA+ as the electrolyte for SORFBs. The physical and kinetic properties of NO2C+ were evaluated by cyclic voltammetry (CV) and UV-visible spectroscopy in acetonitrile and compared to those of its precursor (HC+). The ability for electron storage of NO2C+ was investigated in three different types of static H-cell experiments. In the first experiment, NO2C+ provided an open circuit voltage (OCV) of 2.24 V resulting in demonstrated good stability, as well as high coulombic (>98%) efficiencies, over more than 200 charge/discharge cycles. In the second experiment, a charge–discharge cycling over the entire redox window of NO2C+ (OCV > 3 V) resulted in 80 cycles at a potential energy density above 12 W h L−1. During the last experiment, a bipolarization stress-test was performed in which NO2C+ demonstrated a remarkable durability of 90 cycles at 100% load with a perfect retention of capacity and coulombic efficiency. The enhanced electrochemical performance of this redox material highlights that DMQA+ ions are robust and versatile materials for the emergence of SORFBs.
Introduction
Despite the increased need for renewable energy, the intermittent availability of these energy sources prevents them from meeting the essential requirement of a constant supply.1 Incorporation of efficient storage systems, such as batteries, is a growing need for optimal integration of renewable energies into the grid.2–4 Currently, the most widespread electric energy storage systems (EESs) are mainly based on high-performance lithium-ion batteries.5,6 However, the availability of this metal is quickly being overtaken by the current demand.7,8 One promising technology to be incorporated as an EES is that of redox flow batteries (RFBs).9–13 RFBs store energy in liquid electrolytes that contain dissolved redox-active species, which are pumped from storage tanks to an electrochemical cell. The conversion between electrical and chemical energies occurs at the cell, resulting in a decoupled power and capacity. As a consequence, their capacity is virtually unlimited since it relies on the volume of the electrolyte stored in the tanks, which makes RFBs suitable for large-scale electricity storage.14 Currently, aqueous all-vanadium-based RFBs (VRFBs), which use water-soluble vanadium ions in different oxidation states as redox-active species, are considered a robust choice and are an established EES technology.10,15–17 VRFBs are also an outstanding example of a symmetrical RFB using only one element in both electrolytes, allowing the crossing of electrolytes to result only in self-discharge of the battery and not in persistent cross-contamination.18 Although other metal-based electrolytes have been explored,19–22 the use of metal-based RFBs is limited due to their high cost and toxicity.23,24 Driven by the continuous demand for low-cost and large-scale organic RFBs (ORFBs), redox-active organic materials (ROMs) have emerged as attractive and powerful systems thanks to their sustainable, synthetically tunable, and potentially inexpensive raw materials.25–32To be competitive with existing VRFBs, special attention must be paid to the ORFB energy density (Ed). This is defined as Ed = nFVcellCactive0.5 (Ed in W h L−1)26 where n is the number of electrons in the redox process, F is the Faraday constant in A h L−1, Vcell is the open circuit voltage, and Cactive is the concentration of redox-active species (equal to the equivalent electron concentration at the cathode and the anode). Evidently, with the aim of increasing the energy density of ORFBs, three parameters must be addressed:
The solubility of the ROM, which is already commonly adjustable in aqueous media,29,33,34 but is still rarely optimized for non-aqueous solvents.35,36
The open circuit voltage (OCV), which in the case of a symmetrical RFB, corresponds to a potential gap between the redox species.
The number of electrons involved and/or the number of redox processes of the ROM.
While impressive advances have been made in recent years in the field of aqueous RFBs,37–39 the overall ROM performance is inherently limited by the narrow electrochemical window of water (<1.23 V). Therefore, research for providing high energy density ORFB systems has already started to move towards non-aqueous (NA) systems based on organic solvents, with wider electrochemical stability window to develop the so called NAORFBs.40–49
We have recently demonstrated that 1,3-dipropylodimethoxyquinolinoacridinium tetrafluoroborate salt helicene (denoted HC+, see Scheme 1)41 possess remarkable performance as an electrolyte material in symmetrical ORFBs via its three stable HC˙++/HC+/HC˙ redox states. The HC+ ions showed impressive redox stability, and a high OCV of 2.12 V in a static RFB model.50 Here, we investigate the study and use of a derivative of this DMQA+ after introducing an electron withdrawing group as an electrolyte for high-voltage RFBs. Access to a nitro-derivative was easily achieved by the post-synthetic nitration of HC+ under acidic conditions in dichloromethane (Scheme 1).51 This nitro-helicenium ion [nPr-DMQANO2][BF4], denoted as NO2C+ throughout this report, is expected to fulfill the three points mentioned above to increase the energy density. Herein, we report the in-depth study of the electronic behaviors of NO2C+ as an improved ROM for NAORFBs.
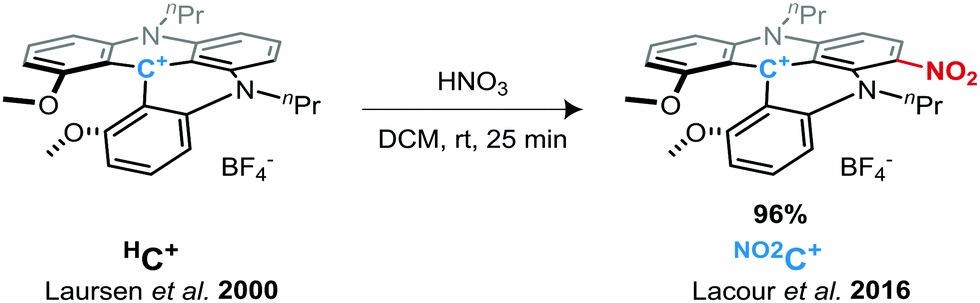 | ||
| Scheme 1 Nitration of N,N′-dialkyl-1,13-dimethoxyquinacrinidium (DMQA+) HC+ to form NO2C+. | ||
Experimental
Chemicals, reagents, and synthesis of NO2C+
All solvents were purified by a solvent purification system (SPS) or distilled over the drying agents indicated. Dried solvents and liquid reagents were transferred by oven-dried syringes or hypodermic syringes. The supporting electrolyte tetrabutylammonium hexafluorophosphate (TBAPF6) was recrystallized three times from ethanol, then dried at 80 °C under vacuum for three days prior to use. The racemic mixture of 1,13-dimethoxy-6-nitro-5,9-dipropyl-9,13b-dihydro-5H-quinolino[2,3,4-kl]acridinium tetrafluoroborate salt (NO2C+) was prepared according to Lacour et al.'s report and was purified by crystallization via slow vapor diffusion of Et2O in concentrated DCM or CH3CN solutions.51UV-vis measurements
Absorption spectra were recorded on a ThermoScientific Evolution 220 UV-Visible spectrophotometer at 25 °C in dried CH3CN at different concentrations. All recorded samples were prepared in a Vigor glovebox under an inert N2 atmosphere with H2O/O2 concentrations maintained below 1 ppm. A 3 mL gas-tight cuvette (Quartz Cuvette Self Masking Screw Cap) was used for anaerobic acquisition of all spectra.Electrochemical studies
Electrochemical analyses were conducted inside a N2-filled MBraun glovebox using a BioLogic SP-200 potentiostat/galvanostat and the EC-Lab® software (v11.33) from BioLogic Science Instruments. For convenience, all potentials are given versus the internal reference electrode AgNO3/Ag.Cyclic voltammograms (CV) were measured in a three-electrode electrochemical cell, consisting of a platinum wire counter electrode, a AgNO3/Ag reference electrode (0.01 M AgNO3 in 0.1 M TBAPF6 in CH3CN), and a platinum working electrode (0.031 cm2, CH Instrument, Inc.). The working electrode was polished prior to each recording using aluminium oxide in polishing paper and anhydrous CH3CN to remove residual particles. CVs were recorded at different scan rates (10, 25, 75, 100, 250, 400, and 500 mV s−1) in an CH3CN electrolyte containing 1 mM NO2C+ and 0.1 M TBAPF6.
Static cell experiments
Galvanostatic cycling experiments under static conditions were carried out in a custom-made glass H-cell (see Fig. S12, ESI†) with a classical three electrode system within a silver reference electrode (0.01 M AgNO3/Ag in 0.1 M TBAPF6 in CH3CN). A 2 mm fine porous glass frit (4–5.5 μm hole diameter) was used as the separator43,44 and 10 mL of NO2C+ solution (0.1 M TBAPF6) was used to study the cycling in the cell. Reticulated vitreous carbon (RVC) electrodes (100 ppi Duocel®) were cut into rods of the dimensions 0.5 cm × 0.5 cm × 4 cm and positioned about 2 cm deep in solution (active surface ∼ 33 cm2 per electrode). To rule out contamination processes, the electrodes were singly used. A constant current followed by a constant voltage galvanostatic charging (CCCV GCPL protocol) at |5| mA current was applied via RVC electrodes. During charge–discharge experiments the compartments of both cells were continuously stirred at 1000 rpm.Results and discussion
Electrochemical studies
During this work, acetonitrile was chosen as the model solvent, since it provides an exceptionally wide electrochemical window (∼6.0 V), has a positive impact on energy density,40,46 and has proved to be suitable with DMQA+.50Cyclic voltammetry experiments in acetonitrile (0.1 M TBAPF6) showed three single-electron redox events for NO2C+ (Fig. 1a, b solid black line and Fig. S1, ESI†), contrasting with the two redox events of its parent HC+ (Fig. 1b, dotted grey line). In its initial oxidation state, NO2C+ forms a pair with a BF4− anion (Fig. 1a blue frame). Differential voltammetry (DPV) experiments (Fig. 1b transparent colored plot, and Fig. S2, ESI†) were performed to accurately obtain the half-wave potential of the redox processes (E1/2) observed. The single-electron oxidation event observed at EOx1/2 = 1.35 V, corresponds to the formation of the dication radical NO2C˙++ whose two charges are then compensated by the BF4− and PF6− anions present in solution. The potential of this redox couple NO2C˙++/NO2C+ appears 310 mV higher than the one obtained for HC+ (EOx1/2 = 0.98 V) due to the electron withdrawing group NO2. Upon scanning to the anodic region of potential, the one-electron reduction of this nitro-[4]helicenium leads to the formation of the neutral radical NO2C˙ at potential ERed11/2 = −0.89 V. The NO2C˙/NO2C+ redox event occurs at a potential shifted by +250 mV compared to the event observed with HC+ (ERed1/2 = −1.14 V). Finally, a second reversible mono-electronic event in reduction occurs at ERed21/2 = −1.67 V. This latter is attributed to the redox pair NO2C˙/NO2C˙˙− corresponding to the formation of the biradical anionic state NO2C˙˙−, whose charge will be compensated by the presence of TBA+ in solution.51,52
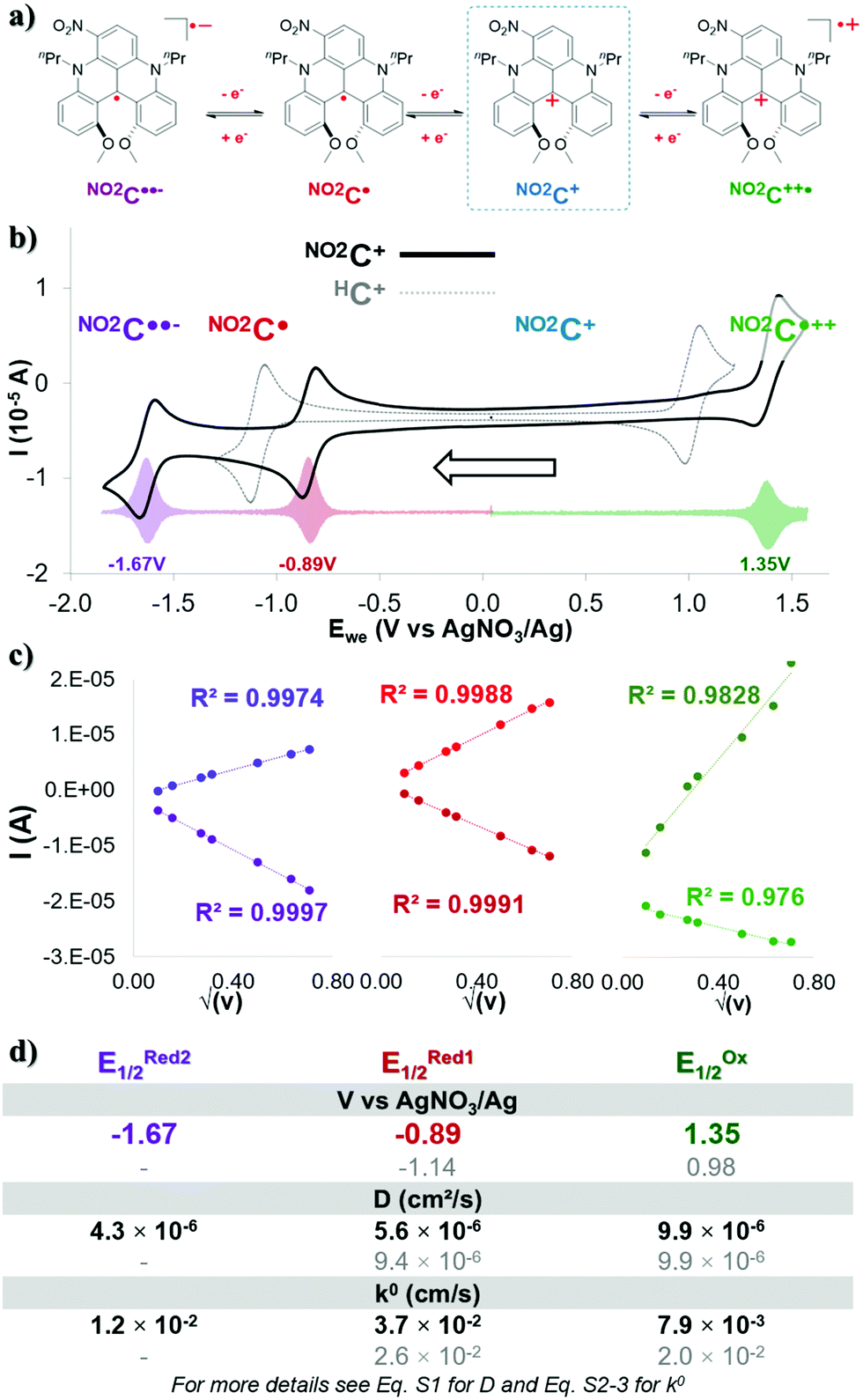 | ||
| Fig. 1 (a) Four different oxidation states of NO2C+. Counter-anions and counter-cations have been removed for clarity. (b) CV at scan rate of 100 mV s−1 for NO2C+ in black, HC+ in grey and (c) measured intensity (I) reported as a function of the square root of the scan rate (10–500 mV s−1) for the 3 electronic processes of 1.0 mM NO2C+ solution in 0.1 M TBAPF6/CH3CN. (d) D and k0 determined by studying CVs at different scan rates from 10 to 500 mV s−1 for NO2C+ in black, HC+ in grey. | ||
The reversibility of these electronic processes has been investigated through the acquisition of CV at different scan rates in a three-electrode cell (see Fig. S3, ESI†). The exhibited linear relation between measured intensity (I) and the square root of the scan rate (Fig. 1c and Fig. S4, ESI†) indicates that the electrolyte is freely diffusing, and a complete reversibility of the three electronic processes is maintained at each scan rate. The essential electrochemical kinetic parameters were extracted from these plots (Fig. 1d). The Randles–Sevcik equation application (eqn S1), gave access to an experimental diffusion coefficient (D) value of 9.9 × 10−6 cm2 s−1 for the NO2C˙++/NO2C+ pair. It is an identical value to the one exhibited by HC+ (Fig. 1d in grey D(E11/2Ox) = 9.9 × 10−6 cm2 s−1) and slightly higher than the value for a recent catholyte for ORFB application.34,36,43,47,48 On the other hand the values for the reductive processes for NO2C+/NO2C˙ (D = 5.6 × 10−6 cm2 s−1) and NO2C˙/NO2C˙˙− (D = 4.3 × 10−6 cm2 s−1) couples are approximately twice as low overall as the value for the HC+ reduction process (D(E11/2Red) = 9.4 × 10−6 cm2 s−1) and are similar to the values for recent anolytes studied.36,41,42,47,53,54
The useful work reported by Lavagnini et al. was used to determine the electron transfer rate constant (k0) using these D-values (Fig. 1d and eqn S2, S3, ESI†).55,56 The values of k0 = 3.7 × 10−2 cm s−1 for NO2C+/NO2C˙ and k0 = 1.2 × 10−2 cm s−1 for NO2C˙/NO2C˙˙− redox couples are quite similar, and their average provides a value close to that of the couple HC+/HC˙ (k0 = 2.6 × 10−2 cm s−1). However, the k0 = 7.9 × 10−3 cm s−1 value of NO2C˙++/NO2C+ is lower than that observed for HC+ (k0 = 2.0 × 10−2 cm s−1) and lower than that identified for the NO2C+ reduction phenomena which are both comparable to redox shuttles used in NAORFBs.42,43,46
We attribute the slight differences in parameters observed between each process due to the loss of symmetry of the [4]helicenium core by the addition of the –NO2 moiety. The electrokinetic parameters obtained suggest that this electrolyte is suitable for ORFB application. We could expect in a symmetrical use, an OCV of 2.24 V. The presence of a second process in reduction, accessible by a second mono-electronic transfer implies the possible access to two electron storage with NO2C˙˙− formation, with a high OCV of 3.02 V.
Saturation concentration of the different oxidation states
The saturation concentrations of NO2C+ and NO2C˙ in acetonitrile were evaluated using UV-vis spectroscopy (see Fig. S5–S8, ESI†). Despite numerous attempts to synthesize and isolate NO2C˙++ in its pure form, our efforts have been unfruitful. The NO2C˙˙− formation via an alkaline double-reduction (KC8) has been also investigated. Crystals were formed at low temperature (−35 °C) but were highly unstable upon increasing temperatures and insufficiently stable to be used for obtaining a UV-Vis calibration curve or further analysis, even with pre-cooled CH3CN solubilization. This observation is supported by the DFT calculations (Tables S5, S6 and Fig. S9–S11, ESI†), indicating a predominance of the more reactive triplet state for NO2C˙˙− (see Fig. S11, ESI†). In pure CH3CN, NO2C+sat exhibits a saturation concentration of 154 mM, whereas that for NO2C˙sat is slightly lower with a value of 110 mM. In both states, the solubility exhibited is above >0.1 M. The introduction of a nitro group modifies the electrochemical behavior of the DMQA scaffold, and also significantly increases its solubility (HC+sat = 32 mM vs.NO2C+sat = 154 mM in acetonitrile). This increased solubility will lead to a higher energy density of Ed(NO2C+) = 9.25 W h L−1 compared to that of Ed(HC+) = 1.84 W h L−1. In a single late-functionalization step, the energy density of the previous ROM was increased by a factor of 5.Galvanostatic cycling with potential limitation cycling
The electrochemical stability of the electrolyte NO2C+ was evaluated by bulk electrolysis charge/discharge cycling using a three-electrode H-cell as model for a redox flow cell setup. Although it does not allow working at high concentrations, Hansmann et al. have shown that such electrochemical cell designs are acceptable test beds for organic ROMs in RFB performance.42,44Mono-polarization cycling, OCV = 2.24 V
We first investigated the use of this NO2C+ as an electrolyte for ORFB application in a symmetrical step. Both sides of the cell were equally filled with a 1.0 mM solution of NO2C+ (0.1 M TBAPF6/CH3CN) (Fig. 2a). One cycle corresponds to a charge followed by a discharge. During the charge, in the “catholyte” compartment (the Ew side of the cell) NO2C+ is oxidized to NO2C˙++, while in the “anolyte” compartment (Ec of the cell) NO2C+ is reduced to NO2C˙.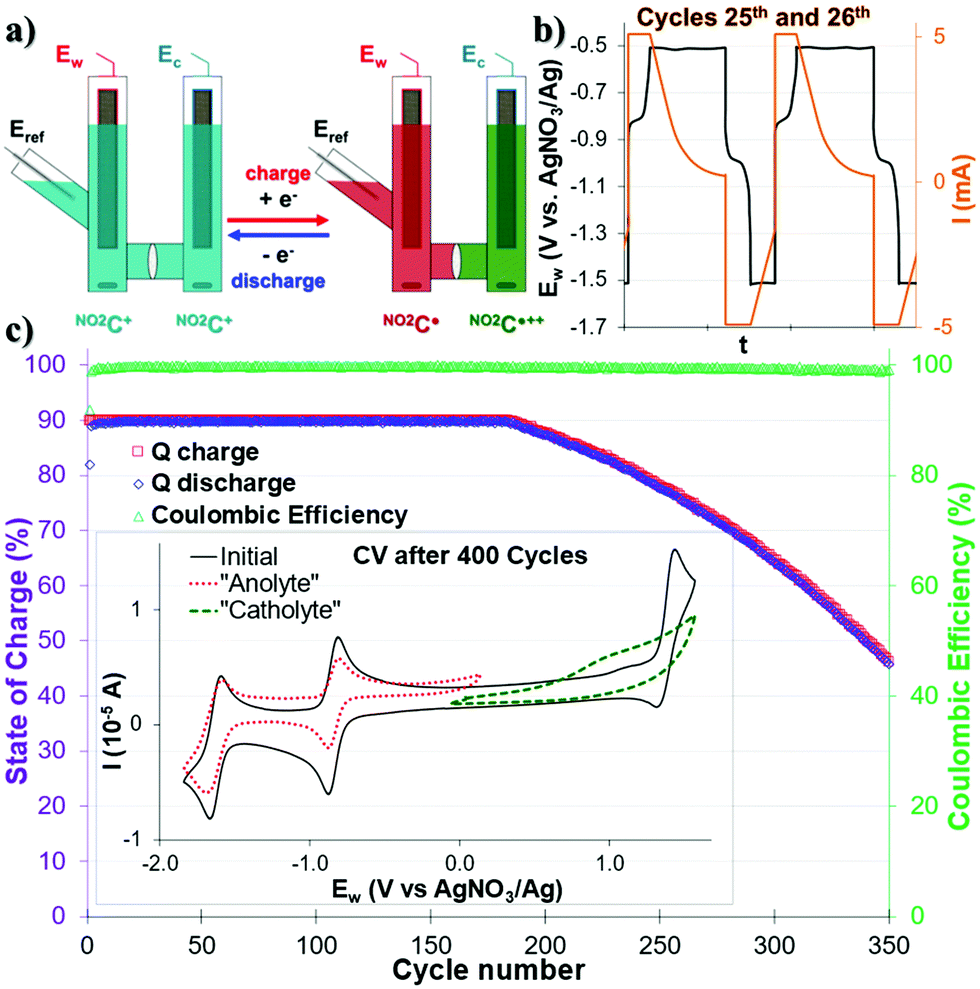 | ||
| Fig. 2 (a) Scheme showing the mono-electronic charge–discharge cycles in a symmetric H-cell. The color code is indicative of the electrolyte oxidation degree. (b) Zoom-in of Ewe during the 25th and 26th cycles of cycling and species formation in the Eref compartment. (c) Q of charge–discharge processes and Coulombic efficiency monitoring over 350 cycles of mono-electronic exchange. Inset shows CV analysis at 100 mV s−1 of the respective contents of each side of the cell after 400 cycles. | ||
In the first instance, mono-electronic transfer at 90% of theoretical capacity (90% state of charge, “SOC”) cycles (Fig. 2 and Fig. S13, S14, ESI†) were performed. A constant current followed by a constant voltage galvanostatic charging (CCCV GCPL protocol) of |5| mA was applied with potential boundaries at −1.49 V and −0.49 V vs. Eref. During the charge–discharge cycles two plateaux at ca. −0.92 V and −0.86 V in the Ew voltage curves were observed and assigned to the electronic processes of the NO2C+/NO2C˙ redox couple (Fig. 2b and Fig. S15, ESI†). Satisfactorily, the cell monitoring (Fig. 2c) shows that the Coulombic efficiency (CE, green triangle) remains constant and close to 100% throughout the experiment. Furthermore, the capacity Q (red and blue squares) exhibited a plateau, with an initial capacity (90% SOC or Qtheoretical) retention over ∼200 cycles. The decrease in the capacity started at the 186th cycle, and the system then lost 0.32% capacity retention per cycle, reaching a value of Q = 50% Qtheoretical at the 340th cycle. The cycling was stopped at the 400th cycle and the final capacity reached was Qfinal = 33% Qtheoretical. Differences in solution mobility and speed of electronic exchange led to faster system fatigue and electrolyte deterioration.
The CV analysis of each compartment after 400 monopolarization cycles of the H-cell revealed that the catholytic compartment is strongly degraded with a broadened and poorly defined profile, highlighting a deterioration of the oxidation process of NO2C+ into NO2C˙++ (Fig. 2c). In the anolytic compartment, we noted a beginning of fatigue after 400 cycles (see Fig. S15, ESI†) with the ERed11/2 process of NO2C+ to NO2C˙ decreased slightly in intensity and resolution. Thus, although the introduction of a NO2 group seems to have a negative effect on the robustness of our electrolyte, this comes along with a dramatic increase in the solubility of this ROM. In a symmetrical cell with mono-polarization it allowed increasing the gap between the two redox events in oxidation and reduction, resulting in a theoretical Ed = 9.25 W h L−1, much higher than the 1.84 W h L−1 of its parent ion HC+50 that can be delivered for almost 200 cycles. It has also been noted that, the monopolarization cycles seemed to not affect the second electronic transfer ERed21/2 at −1.67 V which led us to expect a resilient behaviour during 2-electron reduction storage experiments.
Double reduction cycling, OCV = 3.02 V
The use of multi-electron storage in order to increase the energy density of a system is currently being studied for ARFBs,35,45,57,58 but remains underexplored for NARFBs.34,36Since NO2C+ is able to store two electrons, via two one-electron processes in the anodic potential (first the NO2C˙ state, then the NO2C˙˙− state) we studied the viability of a two-electron storage ORFB through a H-cell. In this configuration, the cell is loaded asymmetrically with the same ROM on both sides: on the catholyte side the NO2C+ concentration is twice as high as that on the anolyte side (ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, for the same 0.1 M TBAPF6 supporting salt) (Fig. 3a). In this way, the following battery will be able to store twice as much current, accessing a high voltage of 3.02 V, without cross-contamination risk and increasing the amount of ROM needed by only half. If we consider CmaxNO2C+ = 154 mM with OCV of 3.02 V, a maximum value of Ed = 12.47 W h L−1 could be calculated for this double reduction cycling test, i.e. 6.8 times more energy stored than in the unsubstituted helicenium HC+.50
:1, for the same 0.1 M TBAPF6 supporting salt) (Fig. 3a). In this way, the following battery will be able to store twice as much current, accessing a high voltage of 3.02 V, without cross-contamination risk and increasing the amount of ROM needed by only half. If we consider CmaxNO2C+ = 154 mM with OCV of 3.02 V, a maximum value of Ed = 12.47 W h L−1 could be calculated for this double reduction cycling test, i.e. 6.8 times more energy stored than in the unsubstituted helicenium HC+.50
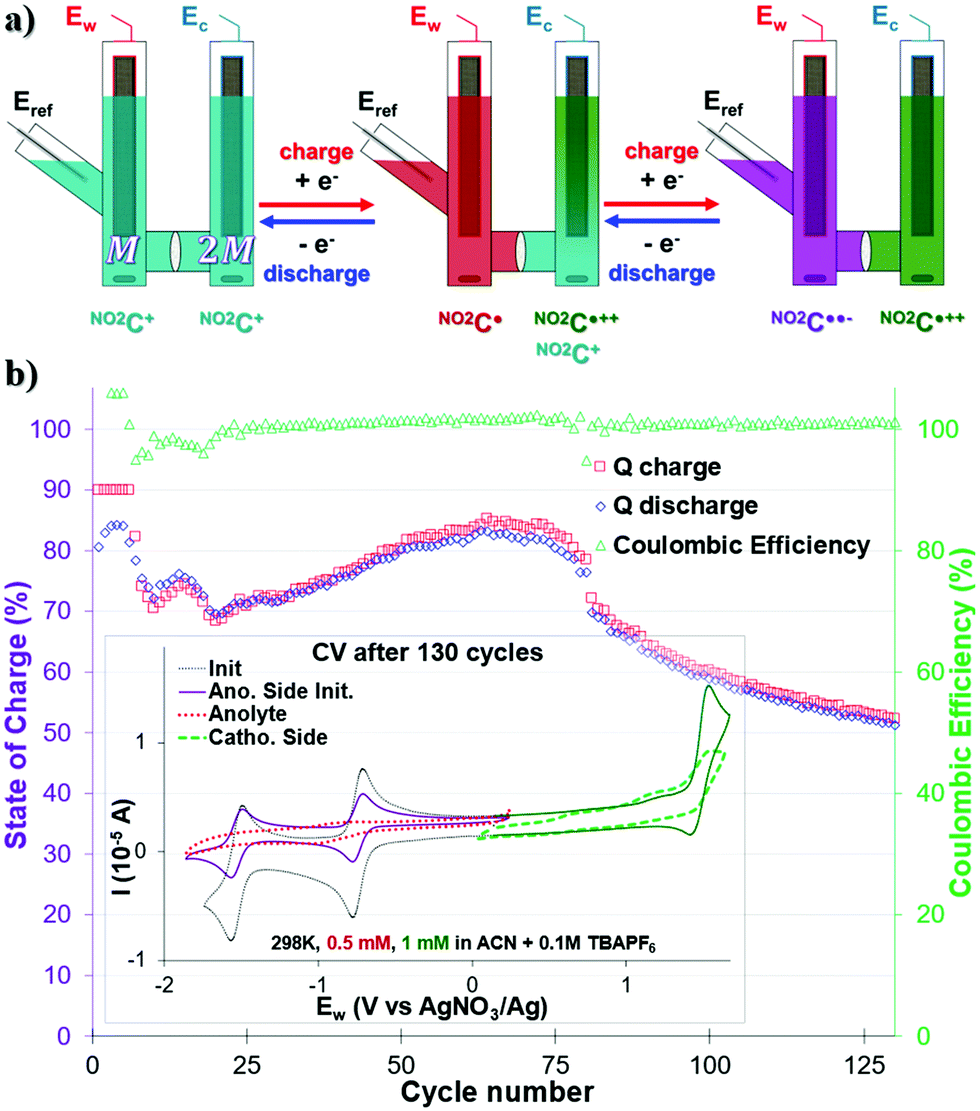 | ||
| Fig. 3 (a) Scheme showing the bi-electronic reduction charge–discharge cycles in a 1:2 NO2C+ concentration H-cell. The color code is indicative of the electrolyte oxidation degree. (b) Q of charge–discharge processes and Coulombic efficiency monitoring over 130 cycles of bi-electronic exchange. Inset shows CV analysis at 100 mV s−1 of the respective contents of each side of the cell after 130 cycles. | ||
Before any test, the anolyte side was checked via CV to verify the reversibility of the two processes ERed11/2 and ERed21/2 in the initial state. The concentration of the anolyte compartment – half of that of the catholyte compartment – is distinguishable by a current intensity twice as low as that of the catholyte solution used (Fig. 3b inset purple trace).
A CCCV GCPL protocol of |5| mA with potential boundaries at −2.07 V and −0.49 V vs. Eref limited at 90% SOC was then applied and monitored over 125 cycles (Fig. 3b). It was noted that despite an excellent CE close to 100%, the capacity of our system fluctuated a lot over the course of the experiment. Thus, as early as the 6th cycle, it was noted that the capacity of our system decreased from 90% to 70% in cycle 10. We observe a “rebound” between cycles 10 and 20, leading to the capacity of our system being <70% Qtheoretical. The capacity of our system slightly increases then to reach a value of 84% in cycle 74, and then quickly drops. It then loses 0.75% per capacity cycle, reaching a value of 53% in the 125th cycle. This type of behavior also observed by Binnemans' group for a double reduction of their ROM, has not yet been elucidated.36 Unfortunately, no clearly defined plateau in the Ew voltage curves was observed during the charge–discharge cycles (see Fig. S17, ESI†). The intensity of the current used and the low ROM concentration of the monitored compartment are probably to blame (0.5 mM at |5| mA).
The CV analysis of each compartment after 130 bi-reduction cycles of the H-cell (Fig. 3b inset and Fig. S17, ESI†) provided further information. As noted for the monopolarization experiment, the catholytic compartment consisting of the NO2C+ into NO2C˙++ process degraded and exhibited a broadened profile (Fig. 3b inset green trace). The anodic compartment, which was stable during the monopolarization experiment, shows a serious deterioration of the electro-active material after 130 cycles (Fig. 3b inset red trace). The electronic processes ERed11/2 and ERed21/2 are almost indistinguishable, and the electrochemical profile is severely altered, more than the cathodic compartment.
In view of these results, one can conclude that the low stability of NO2C˙˙−, previously noted and supported by DFT calculations (see Fig. S9–S11, ESI†), is one of the weak points of the system. Furthermore, the appearance of a “bounce” in the capacity between cycle 10 and 20 could be linked to diffusion effects. Indeed, our H-cell model is a so-called “static” model, and in the configuration where NO2C+ is present in a 1:2 ratio, diffusion effects at the interface, which is here a porous frit and not a selective membrane, may have a major role. Nevertheless, while the robustness of the system needs to be improved, the simple addition of an NO2 group onto the DMQA core allowed access to a second process in reduction, and a very high voltage battery of 3.02 V.
Bipolarization cycling, “stress-test”
The symmetrical properties of NO2C+, where the system can be charged independently by transferring electrons into one or the other side of the cell, allow us to carry out bi-electronic cycling tests by polarizing the battery in the opposite direction. Given the poor results obtained in the process of ERed21/2, we did not consider it necessary to carry out this experiment with NO2C˙˙− species as anolyte. This protocol consists of a highly stressful two-electron charge and discharge process on each side of the cell at 100% SOC. This means that the species NO2C˙++ and NO2C˙ will be formed successively in each compartment over time (Fig. 4a). These two-electron cycling processes, labeled “Stress Test” are likely to shorten the life of the battery and are not meant to describe the energy storage ability of our electrolyte under these conditions.44 The experiment will however provide information about the sturdiness of this electrolyte.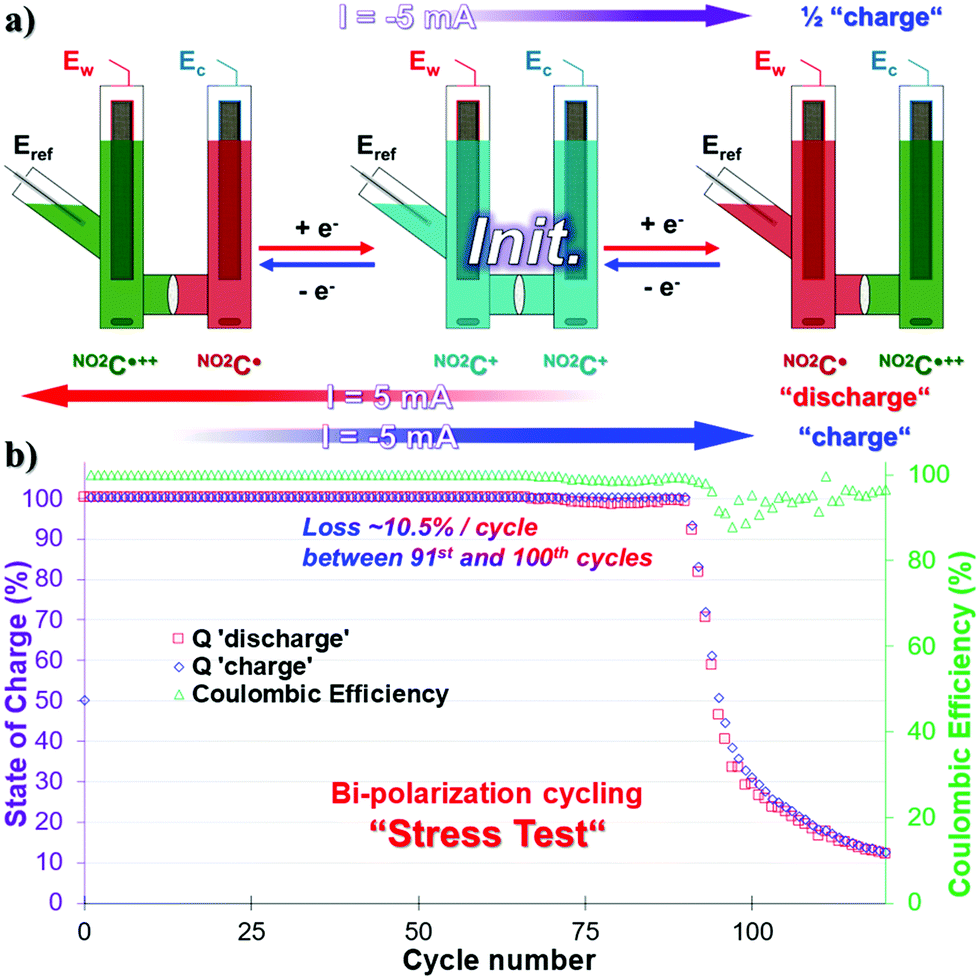 | ||
| Fig. 4 (a) Scheme showing the bi-electronic charge–discharge cycles in a symmetric H-cell as stress test. The color code is indicative of the electrolyte oxidation degree. (b) Q of charge–discharge processes and Coulombic efficiency monitoring over 120 cycles of bi-electronic exchange. | ||
A CCCV GCPL sequence with a |5| mA current, setting potential boundaries at 1.55 V and −1.49 V vs. Eref, was therefore started from the initial state of this symmetrical battery (Fig. 4a). In the first step, the working electrode side of the cell was “charged” with electrons with a cutoff potential of −1.49 V for full reduction of NO2C+ to NO2C˙ (Fig. 4a, purple arrow). Then a positive 5 mA current was applied at 1.55 V (Fig. 4a, red arrow) and two electrons were sequentially exchanged, taking NO2C˙ to NO2C˙++. Finally, the current was reversed to −1.49 V to “charge” the electrons in the working electrode side of the cell back to NO2C˙ (Fig. 4a, blue arrow). The continuous alternation of red and blue steps corresponds to a cycle. Unfortunately, over the course of the experiment, monitoring the value of Ew voltage curves is not representative of the conversion processes of NO2C˙ to NO2C+ and NO2C+ to NO2C˙++ (see Fig. S20 and S21, ESI†).
As shown by the green triangles, the CE was constant and close to 100% until the 90th cycle, with a quasi-constant full capacity Q = 100% SOC for two-electron cycling. Then a drastic drop in Q with an irrecoverable capacity loss of 70% capacity between the 91st and 100th cycles was observed, and the system then degraded more slowly to reach a value of 12.5% SOC in the 120th cycle (Fig. 4b). This was associated with a slight decrease in CE starting at the 91st cycle which varies between 90% and 98% until the end of the experiment. This electrolyte fatigue has been correlated to the degradation of the species during the oxidation phenomenon in NO2C˙++ and is probably the source of weakness of our battery.
However, during the stress test the NO2C+ demonstrated a higher robustness than its parent HC+. Indeed, 90 cycles could be reached without degradation of the capacity, i.e. +12.5% compared to HC+.50 We can directly correlate this robustness improvement to the increase in the solubility of NO2C+ and more specifically to the increase in solubility of the radical dication NO2C˙++ compared to HC˙++ which allows a longer cyclability over time under extremely stressful conditions.
Conclusions
The need to evolve RFBs tends towards the use of organic bipolar redox-active molecules to promote a more robust and reliable design.49 This is an elegant solution to the challenge of cross-contamination and membrane costs. In the case of NARFB, the choice of the solvent will allow a very large potential window and increase the accessible energy density. The question of ROM solubility remains a critical point. During this study, we set out to improve these different parameters. Thus, by a quasi-quantitative late nitration of our previous ROM, we were able to:boost the solubility of our C+ electrolyte by 4.8 times allowing a longer lifetime during the stress test,
expand the open circuit voltage (OCV) between EOx1/2 and ERed11/2 by 120 mV (monopolarization),
increase the number of reversible electronic events to three, with a second event, allowing an OCV EOx1/2 − ERed21/2 of 3.02 V (double reduction cycling).
Thus, despite the moderate stability of NO2C++˙ and NO2C−˙˙, this simple modification of the DMQA+ core allowed us to multiply the initial energy density by 5 times in the case of monoelectronic cycling. Moreover, the double anode reduction cycling demonstrated an Ed value of 12.5 W h L−1 that is closer to the one offered by current aqueous VRFBs (15–25 W h L−1).59
The simplicity of the structural modification and the results obtained underline the value that the DMQA core presents as a ROM library for symmetrical ORFBs.
Author contributions
T. L. G. and J. M. wrote the manuscript, planned the experiments, and discussed their conclusions. J. M. conducted all experiments; D. M. performed the DFT calculations; M. M. H. synthesized NO2C+.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the University of Arizona, the Research Corporation for Science Advancement, Cottrell Scholarship 2021, and the Salt River Project for financially supporting this work.References
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- S. P. S. Badwal, S. S. Giddey, C. Munnings, A. I. Bhatt and A. F. Hollenkamp, Front. Chem., 2014, 2, 1–28 CAS.
- T. M. Gür, Energy Environ. Sci., 2018, 11, 2696–2767 RSC.
- J. B. Goodenough, M. S. Whittingham and A. Yoshino, Rep. - Swed. Acad. Eng. Sci. Finl., 2019, 1 Search PubMed.
- A. Masias, J. Marcicki and W. A. Paxton, ACS Energy Lett., 2021, 621–630 CrossRef CAS.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- M. Winter, B. Barnett and K. Xu, Chem. Rev., 2018, 118, 11433–11456 CrossRef CAS PubMed.
- J. Rugolo and M. J. Aziz, Energy Environ. Sci., 2012, 5, 7151–7160 RSC.
- G. L. Soloveichik, Chem. Rev., 2015, 115, 11533–11558 CrossRef CAS PubMed.
- M. Park, J. Ryu, W. Wang and J. Cho, Nat. Rev. Mater., 2017, 2, 16080 CrossRef CAS.
- L. F. Arenas, C. Ponce de León and F. C. Walsh, Curr. Opin. Electrochem., 2019, 16, 117–126 CrossRef CAS.
- D. G. Kwabi, Y. Ji and M. J. Aziz, Chem. Rev., 2020, 120, 6467–6489 CrossRef CAS PubMed.
- P. Alotto, M. Guarnieri and F. Moro, Renewable Sustainable Energy Rev., 2014, 29, 325–335 CrossRef CAS.
- W. Lu, X. Li and H. Zhang, Phys. Chem. Chem. Phys., 2017, 20, 23–35 RSC.
- H. R. Jiang, W. Shyy, M. C. Wu, R. H. Zhang and T. S. Zhao, Appl. Energy, 2019, 233–234, 105–113 CAS.
- H. R. Jiang, J. Sun, L. Wei, M. C. Wu, W. Shyy and T. S. Zhao, Energy Storage Mater., 2020, 24, 529–540 CrossRef.
- R. A. Potash, J. R. McKone, S. Conte and H. D. Abruña, J. Electrochem. Soc., 2016, 163, A338–A344 CrossRef CAS.
- W. Wang, S. Kim, B. Chen, Z. Nie, J. Zhang, G.-G. Xia, L. Li and Z. Yang, Energy Environ. Sci., 2011, 4, 4068–4073 RSC.
- P. J. Cabrera, X. Yang, J. A. Suttil, K. L. Hawthorne, R. E. M. Brooner, M. S. Sanford and L. T. Thompson, J. Phys. Chem. C, 2015, 119, 15882–15889 CrossRef CAS.
- Y. A. Gandomi, D. S. Aaron, J. R. Houser, M. C. Daugherty, J. T. Clement, A. M. Pezeshki, T. Y. Ertugrul, D. P. Moseley and M. M. Mench, J. Electrochem. Soc., 2018, 165, A970–A1010 CrossRef CAS.
- M. Park, E. S. Beh, E. M. Fell, Y. Jing, E. F. Kerr, D. Porcellinis, M. Goulet, J. Ryu, A. A. Wong, R. G. Gordon, J. Cho and M. J. Aziz, Adv. Energy Mater., 2019, 9, 1900694 CrossRef.
- DOE Office of ARPR-E. ARPA-E, GRIDS Program Overview, https://arpa-e.energy.gov/sites/default/files/documents/files/GRIDS_ProgramOverview.pdf Search PubMed.
- U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES Public Health Service Agency for Toxic Substances and Disease Registry, ATSDR's Toxicological Profiles, CRC Press, 2012.
- T. B. Schon, B. T. McAllister, P.-F. Li and D. S. Seferos, Chem. Soc. Rev., 2016, 45, 6345–6404 RSC.
- J. Winsberg, T. Hagemann, T. Janoschka, M. D. Hager and U. S. Schubert, Angew. Chem., Int. Ed., 2017, 56, 686–711 CrossRef CAS PubMed.
- X. Wei, W. Pan, W. Duan, A. Hollas, Z. Yang, B. Li, Z. Nie, J. Liu, D. Reed, W. Wang and V. Sprenkle, ACS Energy Lett., 2017, 2, 2187–2204 CrossRef CAS.
- T. Janoschka, C. Friebe, M. D. Hager, N. Martin and U. S. Schubert, ChemistryOpen, 2017, 6, 216–220 CrossRef CAS PubMed.
- Y. Ding, C. Zhang, L. Zhang, Y. Zhou and G. Yu, Chem. Soc. Rev., 2018, 47, 69–103 RSC.
- R. F. Service, Science, 2018, 362, 508–509 CrossRef CAS PubMed.
- J. Luo, B. Hu, M. Hu, Y. Zhao and T. L. Liu, ACS Energy Lett., 2019, 4, 2220–2240 CrossRef CAS.
- R. P. Fornari and P. de Silva, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 1–16 Search PubMed.
- S. Jin, Y. Jing, D. G. Kwabi, Y. Ji, L. Tong, D. De Porcellinis, M.-A. Goulet, D. A. Pollack, R. G. Gordon and M. J. Aziz, ACS Energy Lett., 2019, 4, 1342–1348 CrossRef CAS.
- J. Chai, A. Lashgari, Z. Cao, C. K. Williams, X. Wang, J. Dong and J. Jimmy Jiang, ACS Appl. Mater. Interfaces, 2020, 12, 15262–15270 CrossRef CAS PubMed.
- H. Chen, Z. Niu, J. Ye, C. Zhang, X. Zhang and Y. Zhao, ACS Appl. Energy Mater., 2021, 4, 855–861 CrossRef CAS.
- P. Geysens, Y. Li, I. Vankelecom, J. Fransaer and K. Binnemans, ACS Sustainable Chem. Eng., 2020, 8, 3832–3843 CrossRef CAS.
- K. Lin, R. Gómez-Bombarelli, E. S. Beh, L. Tong, Q. Chen, A. Valle, A. Aspuru-Guzik, M. J. Aziz and R. G. Gordon, Nat. Energy, 2016, 1, 16102 CrossRef CAS.
- P. Leung, T. Martin, M. Liras, A. M. Berenguer, R. Marcilla, A. Shah, L. An, M. A. Anderson and J. Palma, Appl. Energy, 2017, 197, 318–326 CrossRef CAS.
- B. Hu, J. Luo, M. Hu, B. Yuan and T. L. Liu, Angew. Chem., Int. Ed., 2019, 58, 16629–16636 CrossRef CAS PubMed.
- K. Gong, Q. Fang, S. Gu, S. F. Y. Li and Y. Yan, Energy Environ. Sci., 2015, 8, 3515–3530 RSC.
- X. Wei, W. Duan, J. Huang, L. Zhang, B. Li, D. Reed, W. Xu, V. Sprenkle and W. Wang, ACS Energy Lett., 2016, 1, 705–711 CrossRef CAS.
- K. H. Hendriks, S. G. Robinson, M. N. Braten, C. S. Sevov, B. A. Helms, M. S. Sigman, S. D. Minteer and M. S. Sanford, ACS Cent. Sci., 2018, 4, 189–196 CrossRef CAS PubMed.
- Y. Yan, S. G. Robinson, M. S. Sigman and M. S. Sanford, J. Am. Chem. Soc., 2019, 141, 15301–15306 CrossRef CAS PubMed.
- P. W. Antoni, T. Bruckhoff and M. M. Hansmann, J. Am. Chem. Soc., 2019, 141, 9701–9711 CrossRef CAS PubMed.
- L. Tong, Y. Jing, R. G. Gordon and M. J. Aziz, ACS Appl. Energy Mater., 2019, 2, 4016–4021 CrossRef CAS.
- A. Korshunov, M. J. Milner, M. Grünebaum, A. Studer, M. Winter and I. Cekic-Laskovic, J. Mater. Chem. A, 2020, 8, 22280–22291 RSC.
- X. Xing, Q. Liu, W. Xu, W. Liang, J. Liu, B. Wang and J. P. Lemmon, ACS Appl. Energy Mater., 2019, 2, 2364–2369 CrossRef CAS.
- Y. Yan, T. P. Vaid and M. S. Sanford, J. Am. Chem. Soc., 2020, 142, 17564–17571 CrossRef CAS PubMed.
- M. Li, J. Case and S. D. Minteer, ChemElectroChem, 2021, 8, 1215–1232 CrossRef CAS.
- J. Moutet, J. M. Veleta and T. L. Gianetti, ACS Appl. Energy Mater., 2021, 4, 9–14 CrossRef CAS.
- I. H. Delgado, S. Pascal, A. Wallabregue, R. Duwald, C. Besnard, L. Guénée, C. Nançoz, E. Vauthey, R. C. Tovar, J. L. Lunkley, G. Muller and J. Lacour, Chem. Sci., 2016, 7, 4685–4693 RSC.
- A. C. Shaikh, J. Moutet, J. M. Veleta, M. M. Hossain, J. Bloch, A. V. Astashkin and T. L. Gianetti, Chem. Sci., 2020, 11, 11060–11067 RSC.
- B. Huskinson, M. P. Marshak, C. Suh, S. Er, M. R. Gerhardt, C. J. Galvin, X. Chen, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Nature, 2014, 505, 195–198 CrossRef CAS PubMed.
- X. Yu and A. Manthiram, Sustainable Energy Fuels, 2018, 2, 1452–1457 RSC.
- R. S. Nicholson, Anal. Chem., 1965, 37, 1351–1355 CrossRef CAS.
- I. Lavagnini, R. Antiochia and F. Magno, Electroanalysis, 2004, 16, 505–506 CrossRef CAS.
- C. DeBruler, B. Hu, J. Moss, X. Liu, J. Luo, Y. Sun and T. L. Liu, Chemistry, 2017, 3, 961–978 CrossRef CAS.
- J. Luo, B. Hu, C. Debruler and T. L. Liu, Angew. Chem., Int. Ed., 2018, 57, 231–235 CrossRef CAS PubMed.
- A. Clemente and R. Costa-Castelló, Energies, 2020, 13, 4514 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00914a |
| This journal is © The Royal Society of Chemistry 2022 |