
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Inverse photochromism in viologen–tetraarylborate ion-pair complexes: optical write/microwave erase switching in polymer matrices†
Willy G.
Santos
 *ab,
Darya S.
Budkina
a,
Pedro F. G. M.
da Costa
b,
Daniel
R. Cardoso
b,
Alexander N.
Tarnovsky
a and
Malcolm D. E.
Forbes
*a
*ab,
Darya S.
Budkina
a,
Pedro F. G. M.
da Costa
b,
Daniel
R. Cardoso
b,
Alexander N.
Tarnovsky
a and
Malcolm D. E.
Forbes
*a
aCenter for Photochemical Sciences, Department of Chemistry, Bowling Green State University, Bowling Green, OH 43403, USA
bChemical Institute of São Carlos, University of São Paulo - USP, CP 780, 13560-970 São Carlos, SP, Brazil. E-mail: willy_glen@yahoo.com.br
First published on 31st January 2022
Abstract
With the aim to construct a new type of photoswitchable photochromic material modulated by specific radiation in the microwave region, the spin dynamics of radical pairs (RPs) from ion-pair complexes between viologen and tetraarylborate compounds have been investigated in the presence of microwave (μw) radiation, using steady-state electron paramagnetic resonance (SSEPR) to follow the radical pair (RP) dynamics. This strategy is realized by excitation of the charge transfer (CT) absorption band of the ion-pair complex in the solid phase (powders and dispersed in polymer matrices) at 410 nm, which leads to electron transfer from borate to viologen, producing RPs. In the singlet excited state or Partially Separated Charge (PSC) state, an electron transfer process occurs between the ions, and the subsequent (purple) viologen radical is observed as a Fully Charge Separated (FCS) state. In solid state SSEPR experiments, μw radiation deactivates the FSC state by inducing back electron transfer, which subsequently increases the population of a Partially Separated Charge (PSC) state, recovering the initial color of the ion-pair complex. State-of-the-art photophysical and photochemical studies show that deactivation of the FSC state can take place using μw radiation on the RPs in a switchable, reversible fashion. The results have potential impact for a number of applications including photo-writing and photo-erasing processes and spintronics. Examples of laser writing using a polymer matrix to lock the relative positions of the radicals, and then erasing the color using microwaves, are presented and discussed.
Introduction
Photochromism of viologen type molecules is the reversible transformation of the viologen species between two forms, non-radical (V2+) and radical (V+˙).1,2 The radical form has different UV-vis absorption spectra from non-radical species that the absorbance is only observed in the ultraviolet region. In this sense, the electronic conversion of non-radical to the radical species is possible by UV-absorption of electromagnetic radiation.3–6 The thermodynamically stable non-radical form is transformed by irradiation into the less stable radical form, having a different absorption band around 600 nm for the methyl viologen compound, which can be reverted thermally to the first form.4It is well known that less common photochromic compounds have inverse photochromism (blue → yellow) such as observed in thermal reactions of viologen compounds.4 Yet an understanding of how thermal effects may induce back electron transfer is still somewhat lacking in the literature.
Stable ion-pair systems, such as viologen-borate based compounds developed in our laboratories over the past several years, can exhibit interesting photoinduced color changes, where the most stable form is pale yellow or orange and acquires coloration when irradiated with blue light (400–450 nm).7 This property often derives from a photocatalyzed electron transfer reaction from the borate anion to the viologen dication. Heretofore, this photochromic property of viologen has been found mainly in solution and liquid crystals, but few cases are observed in the solid state, in particular in crystalline solids.4–6,8,9
In viologen–borate ion pair systems such as shown in Scheme 1, charge separation involves a redox reaction between a photoexcited electron donor (D*) and an electron acceptor (A), resulting in the production of the ion pair (D* + A → D+ + A−).7 In fact, for such systems, the efficiency of the photoinduced electron-transfer processes is dependent on the rate of the thermal back-electron-transfer reaction (D+ + A− → D* + A). A significant challenge in theoretical and experimental in this field is control of the rate of forward and back electron transfers in the excited states. This requires extensive knowledge of ground and excited state structural properties and their relationship to intrinsic electronic properties.2,5,6
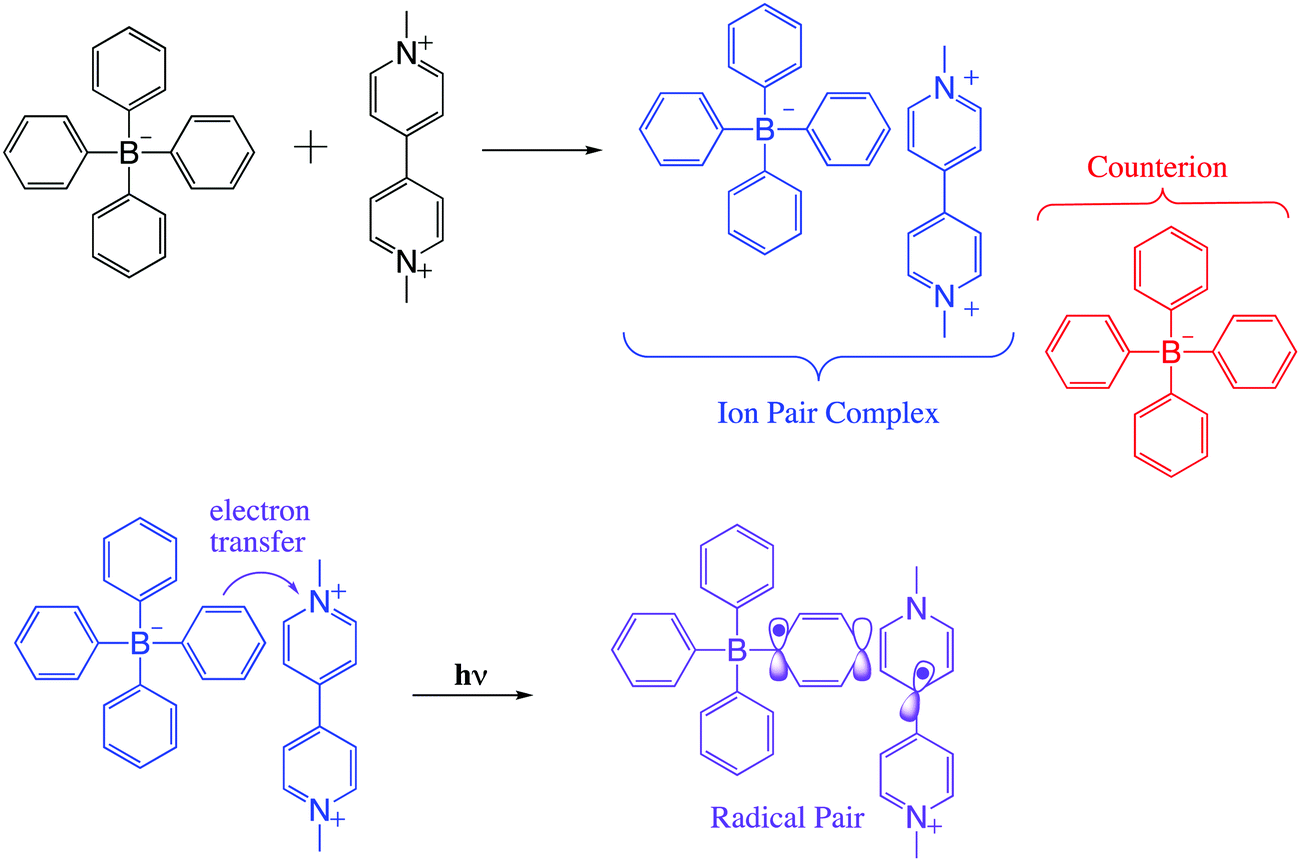 | ||
| Scheme 1 General molecular structure of the methyl viologen tetraarylborate ion pair complex, and the radical pair formed between them after light excitation. | ||
Controlled manipulation of charge and spin angular momentum in molecular and supramolecular systems is at the forefront of many emerging technologies, including electrochromic displays, molecular electronics, liquid crystal displays, memory switching, artificial photosynthesis, and solar-energy conversion.2,11–14 Ion-pair complexes based on viologen structure are of significant interest in this regard, as they can exhibit charge transfer (CT) character in their excited singlet states as one of their decay paths.7
Spin selectivity in such processes can be exploited using electromagnetic radiation as an external trigger, which can lead to precise control of the motion of charge and spin on the molecular and nanometer scales. Considering this idea, we present here results on a methylviologen–tetraarylborate ion pair system (Scheme 1) that show promise for such control using microwave induced state switching. This photoinduced electron transfer pathway can lead to diverse optical functionality such as photochromism, as previously observed by us.7
Background
Photoinduced electron transfer (ET) reactions in host–guest systems involving viologen species are characterized by the presence of viologen radical cations (V+˙), after a single ET event from the donor to the viologen.15–17 These reactions come about because the V2+/V+˙ redox couple has a low and reversible standard redox potential for an organic species: E°(V2+,V+˙) = −0.689 V vs. the normal hydrogen electrode (NHE).2,13,18 In liquid solution, donor–acceptor systems involving the viologen cation radical often lead to long-lived radical species. Usually, the ability to control the lifetimes of the radicals in such systems is exercised by manipulating the electronic coupling between the electron donor and acceptor structures, and using high temperatures (>320 K) to decrease the lifetime of the ensuing radicals.Compared with metal-based electron donors, the excited states of tetraarylborate moieties provide unique advantages for photo-induced reduction reactions. For example, with light-driven electron donation, oxidation of arylborate anions occurs because of elevated values of oxidation (1.0–1.3 V, vs. NHE) for the borates in the ground state.7,19–21 Using viologen and tetraarylborate as a donor–acceptor system, after visible light absorption of the complex, the arylborate anion becomes an efficient electron donor as predicted by the well-known Rehm–Weller equation (eqn (1)).
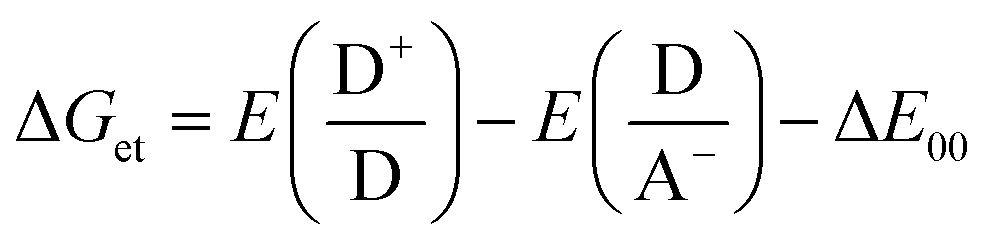 | (1) |
The Gibbs free energy change for the electron transfer reaction from the singlet excited state of these complexes is expected to be around −0.9 eV.7,20 A partially charge separated state (PCS) is commonly observed in CT complexes.2,11,22 For a fully charge transferred state (FCS), one-electron transfer accompanied by a change in valence leads to structures classified as radical pairs (RPs, bottom right of Scheme 1).2,22 Photochromic compounds that undergo ET such as viologen–tetraarylborate ion-pair complexes, are examples of systems that generally (and preferentially) produce the FCS state (RPs) after light excitation (eqn (2) and (3)).
 | (2) |
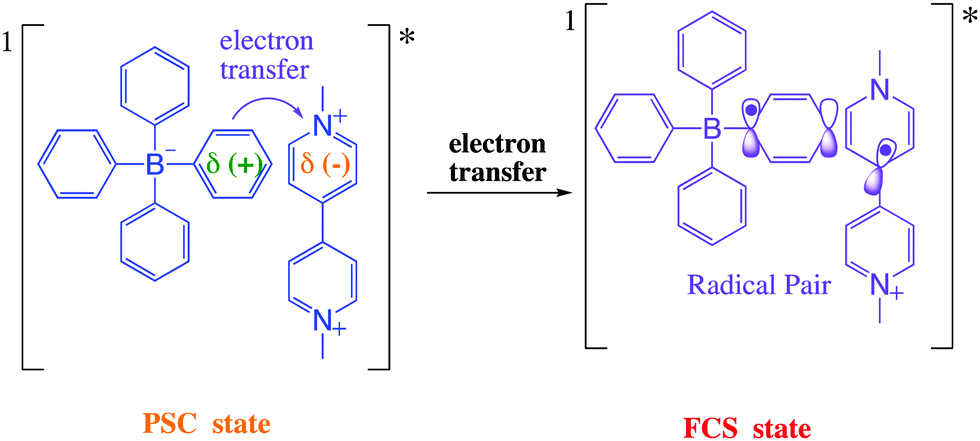 | (3) |
After excitation in the charge transfer band of the complex, the singlet excited state is populated and the formation and/or deactivation (e.g. fluorescence process) of the PSC state may be followed by ultrafast transient absorption experiments.2,7,22–24 ET to the FCS is facile when the PSC singlet excited state has enough energy to form a singlet radical pair state (1FCS*) that may evolve to a triplet radical pair state (3FCS*) by intersystem crossing.2,7,22–24 These mechanistic steps are shown in eqn (4)–(6).
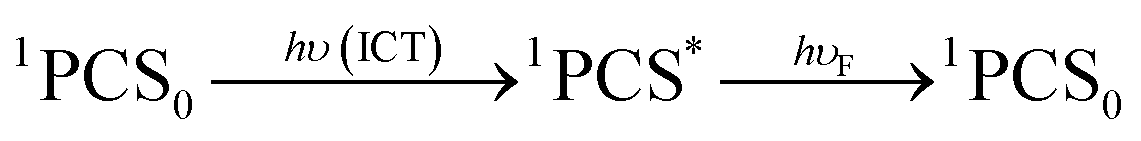 | (4) |
| 1PCS* → 1FCS* | (5) |
 | (6) |
In solution, in particular in polar solvents such as acetonitrile (ACN), dissociation of the radicals occurs, and the solvated viologen radical may be identified by EPR (eqn (7)–(8)).7
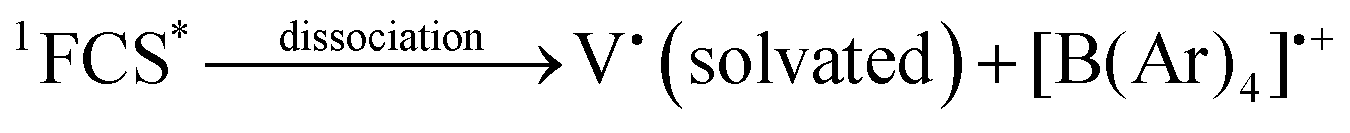 | (7) |
 | (8) |
The evolution of the PCS state to the FSC state proceeds as a result of interplay between local magnetic fields at each radical site, and the electron spin exchange interaction.2,23–25 Of special relevance to the ion-pair complex systems used in this work are the non-covalent interactions involving aromatic groups, such as π–π stacking, cation–π and anion–π interactions, which play important roles in the physiochemical properties of radicals.7,20,25–28
In this paper, we will mostly focus on the photoinduced phenomena using visible and microwave irradiation to promote the photochemical changes. To be more specific, we focus on electron donor–acceptor systems based on an ion pair complex of a viologen dication and tetraarylborate anion in which photoinduced electron transfer from the tetraarylborate (donor) to the viologen (acceptor) produces a radical pair (RP) characterized as the FCS state. Into the FSC state, the lifetime of the radical pair is modulated by microwave radiation, which indicates a new methodology based on microwave irradiation to modulate the optical switching effects of radical pairs.
Experimental
Materials and methods
Ion-pair complexes (viologen–tetraarylborate) were obtained by mixing viologen dichloride (1 × 10−3 mol L−1) and potassium tetraarylborate (2 × 10−3 mol L−1) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 methanol:H2O solution. After stirring for 2 hours at 298 K, a coloured compound precipitates from the solution. The solution was filtered, the solid was washed with methanol (150 mL) and dried in desiccator at low pressure. Following recrystallization, the resulting complex was used in all experiments.
:1 methanol:H2O solution. After stirring for 2 hours at 298 K, a coloured compound precipitates from the solution. The solution was filtered, the solid was washed with methanol (150 mL) and dried in desiccator at low pressure. Following recrystallization, the resulting complex was used in all experiments.
Thin films of the copolymer poly(ethylene vinyl acetate – PEVA) were prepared with ion-pair complexes incorporated in the polymer matrix. The sequence of film preparation was as follows: 3.30 mg of the ion-pair complex was dissolved in THF and a 60 mg amount of the polymer was added to the same solution. After mixing, the resulting solution was transferred to a Teflon plate and kept at room temperature for 24 hours. After this time period, the polymer solution was dried at 313 K for 12 hours and dried again at 313 K for 12 hours at low pressure.
Spectroscopic measurements
 | (9) |
:50 into two beams. The first beam sent to a TOPAS-C optical parametric amplifier to produces 480, and 350 nm pulses, which were used for sample excitation. The second beam was attenuated, sent through a computer-controlled optical stage to adjust the time delay with respect to the excitation pulse, and then focused onto a 2 mm CaF2 window to produce a white-light continuum (wlc) probe spanning the 345–690 nm range. The wlc probe beam was focused to a 75 μm diameter spot at the sample and overlapped nearly collinearly (angle, 1°) with the excitation beam focused to a 150 μm diameter spot. A fraction of the wlc probe beam was split off before the sample to be utilized as a reference for the correction of the shot-to-shot pulse-intensity fluctuations. The probe (after the sample) and reference beams were dispersed by a spectrograph and recorded using a dual CCD detector synchronized to the 1 kHz repetition rate. The difference between the decadic logarithms of a probe-to-reference intensity ratio measured at a specific position of the optical stage for excitation on and off represents the change in the sample absorbance (ΔA) at the corresponding delay time. The solutions were circulated through a flow cell with a 2 mm path length. The zero-time delay is obtained from the non-resonant electronic response from neat solvents measured at the same experimental conditions. The typical excitation energy was 3.1 μJ pulse−1 and the linearity of ΔA signals with excitation energy confirmed that single-photon excitation is responsible for the measured data. The polarization of the excitation beam was set at the magic angle (54.7°) with respect to the probe beam to eliminate solute rotational signals. All experiments were performed at 21 °C.
DFT calculations
Time dependent – density functional theory (DFT) calculations were performed with the Gaussian 09 (G09) program package, employing the functional B3LYP. The basis set 6-31+(d) was used to optimize the gas phase singlet or doublet geometries.Results and discussion
UV-VIS and fluorescence measurements
The specific chemical structures of four different ion-pair complexes are depicted in Scheme 2. The complexation reaction of two novel complexes (1b:2b and 1b:2a) follows the same chemical precedent reported in our previous work for the 1a:2a and 1b:2a complexes.7 | ||
| Scheme 2 Molecular structures of several ion pair complexes formed between viologen and tetraarylborate. | ||
In contrast with X-ray diffraction observed for compounds 1a:2a and 1b:2a, the absence of a diffraction signal for the 1b:2b and 1b:2a complexes made it impossible to determine the molecular structure of the respective crystals.
The new compounds 1b:2b and 1b:2c exhibit interesting photochromic behaviour (Fig. 1) similar to other two compounds (1a:2a and 1b:2a) previously reported by Santos and co-workers.7 Their colour clearly changes upon irradiation at 410 nm at room temperature in the presence of air. Fig. 1 and Fig. S1 (ESI†) illustrate photochromic changes for all compounds investigated in this work.
 | ||
| Fig. 1 (a) Four charge-transfer complexes with different coloration in saturated solution and solid phase (powder). (b) Photochromic changes of the 1b:2b (top side) and 1b:2c (bottom side) complexes during and after irradiation at 410 nm; T = 298 K. | ||
The solid product formed after complexation of the precursor salts (sodium tetraphenylborate and methylviologen dichloride) shows an intense visible coloration. The colour of the complexes arises via interaction between viologen (1a or 1b) and tetraarylborate (2a, 2b or 2c) ions, when these two ions come within a certain distance (∼3 Å).7
UV-vis absorption spectra in the solid phase (powder) and in THF:ACN solution (5:1) for complexes 1b:2b and 1b:2c are displayed in Fig. 2, along with kinetic decay profiles and solid state spectra for comparison. The ion-pair complexes formed exhibit new optical absorption bands attributed to a charge transfer (CT) absorption band. Fig. 2a and b show the fluorescence spectra of the 1b:2b and 1b:2c complexes in tetrahydrofuran solution, respectively. Analysis of the stimulated emission process reveal three-time components for all complexes. See Table S1 in ESI† for the percent amplitude and decay time constant values.
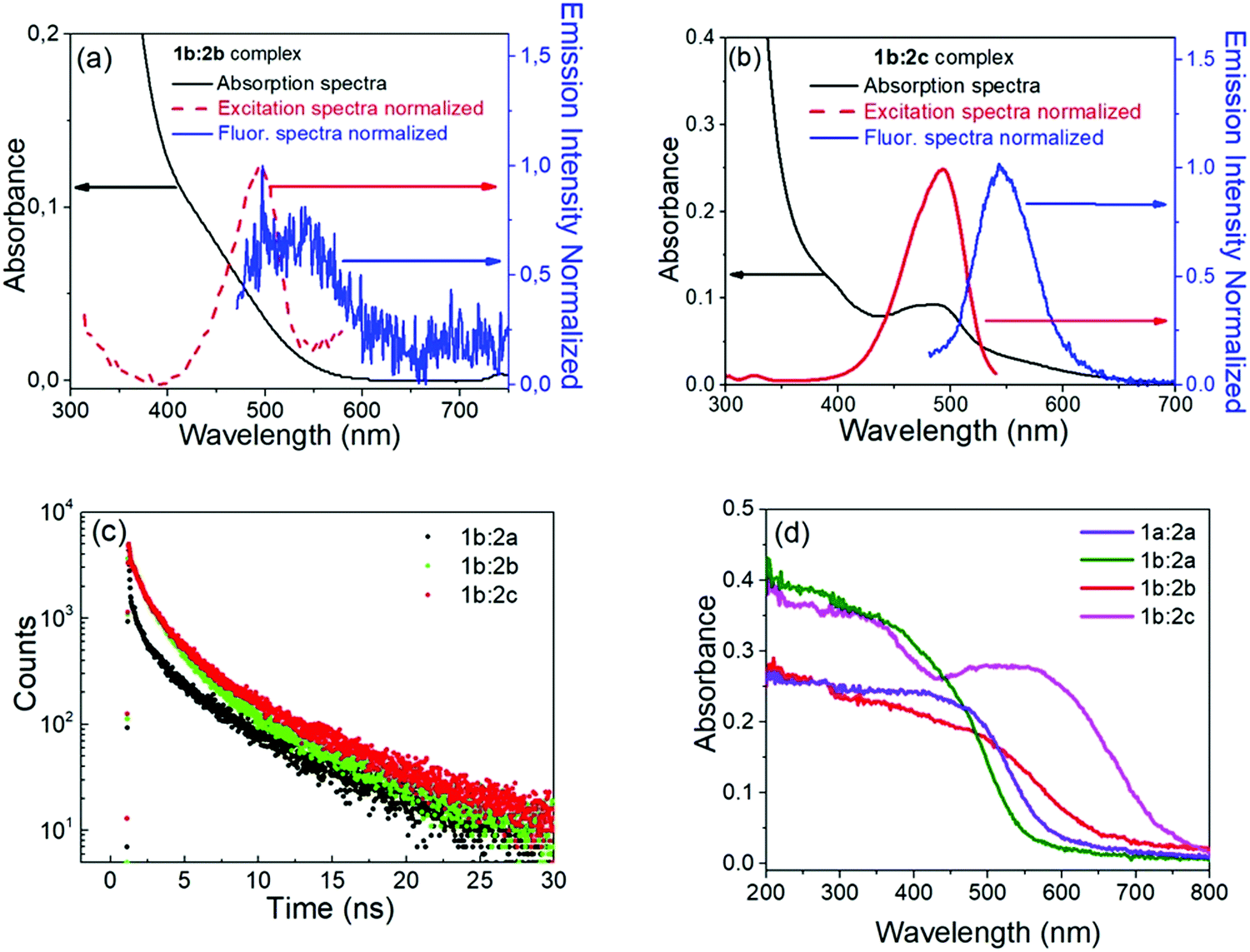 | ||
| Fig. 2 (a) and (b) Absorption and emission spectra of 1b:2b and 1b:2c complexes in THF solution, respectively. Black lines represent the absorption spectra; red dashed lines represent the fluorescence excitation spectra (obs = 600 nm); blue lines represent fluorescence excitation spectra (λobs = 600 nm); blue lines represent the fluorescence emission spectra (λexc = 410 nm), (c) Fluorescence emission decay profiles, (d) UV-vis absorption in the solid phase (powder). | ||
Analysing the percentage amplitude (Bi%) for 1b:2b and 1b:2c complexes, the first-component B1 weight decreases with concomitant increase of other two components, indicating the presence of two lower energy states for these long-lived species. The second long-lived component (B2%, τ2) was previously attributed to the β-conformer of the viologen structure with partially charge transfer character (1PCT-character). For the third component, which is attributed to the fully charge transfer state (FCS), the percentage amplitude in the decay curve increases when the para-substituent is replaced by weakelectron-withdrawing groups like chloro (Cl) and phenyl (Ph) substituents. Also, these groups enhance π–π stacking interactions between both ions, which in turn favours full charge transfer from the borate anion to the viologen cation.
The decay lifetime of the third component (τ3) decreases when H (1b:2a) is replaced by Cl (1b:2b) or Ph (1b:2c) substituents. The time evolution may be understood by considering delocalization of the electron charge transferred over two groups of the π-system. This has the effect of enlarging the overlap integral between the wavefunctions localized on the borate and viologen radicals. Therefore, the non-radiative decay rate constant between the states involved is expected to increase, in accordance with the Fermi–Wentzel golden rule.
In addition to the π–π interaction of both ions in the excited state, MESP (molecular electrostatic potential) has also been used to support the concept of the more efficient acceptor character of the viologen ion in the LUMO level. Fig. S2 (ESI†) shows the MESP surface for the methylviologen (1a) and benzylviologen (1b) structures.
The HOMO and LUMO of the 1a ion show MESP surfaces with electropositive potential in the pyridinium groups (Fig. S2, ESI†). However, for 1b ion, the LUMO shows high values of the electropositive potential in the phenyl-group, which is expected to improve the electron-orbital coupling process through π–π interactions between the phenyl groups of the 1b and 2a ions. For the 1a:2a complex, the π–π interaction is found between the phenyl group and the pyridinium group of the 1a and 2a ions, respectively.
Raman measurements
Substantial alterations of the pyridinium groups of both the 1a and 2a dication structures are predicted upon formation of the complexes between them and the tetraarylborate anion. This opens the possibility for the utilization of Raman spectroscopy for the analysis of the complex structure in the solid phase. To target specifically Raman-active modes associated with charge-transfer excitation, the resonantly-enhanced Raman scattering technique was employed to investigate the real contribution of the Ph-substituent into the viologen structure (1b ion), in which the excitation wavelength was tuned to 780 nm to have spectral overlap with the CT band of the complex. Fig. 3 shows Raman spectra of the precursor salts (e.g., potassium tetra(chlorophenyl)borate and benzylviologen bromide) and their complex form with tetraphenylborate (2a). For the complex in the solid phase (powder), the absorption of the continuous 780 nm laser irradiation leads to a build-up of moderate concentration of the biradical species and the viologen radical, which may be identified through their specific Raman features. Comparison of results from DFT vibrational frequency computations with the experimental Raman spectra greatly facilitates the spectral assignments. For the pyridinium group of the precursor 1a salt, the band centred at 1554 cm−1 (1552 cm−1 for 1b salt) is assigned to a1 symmetry and the band at 1659 cm−1 and 1306 cm−1 (1651 and 1300 cm−1 for the 1b salt) to b2 symmetry.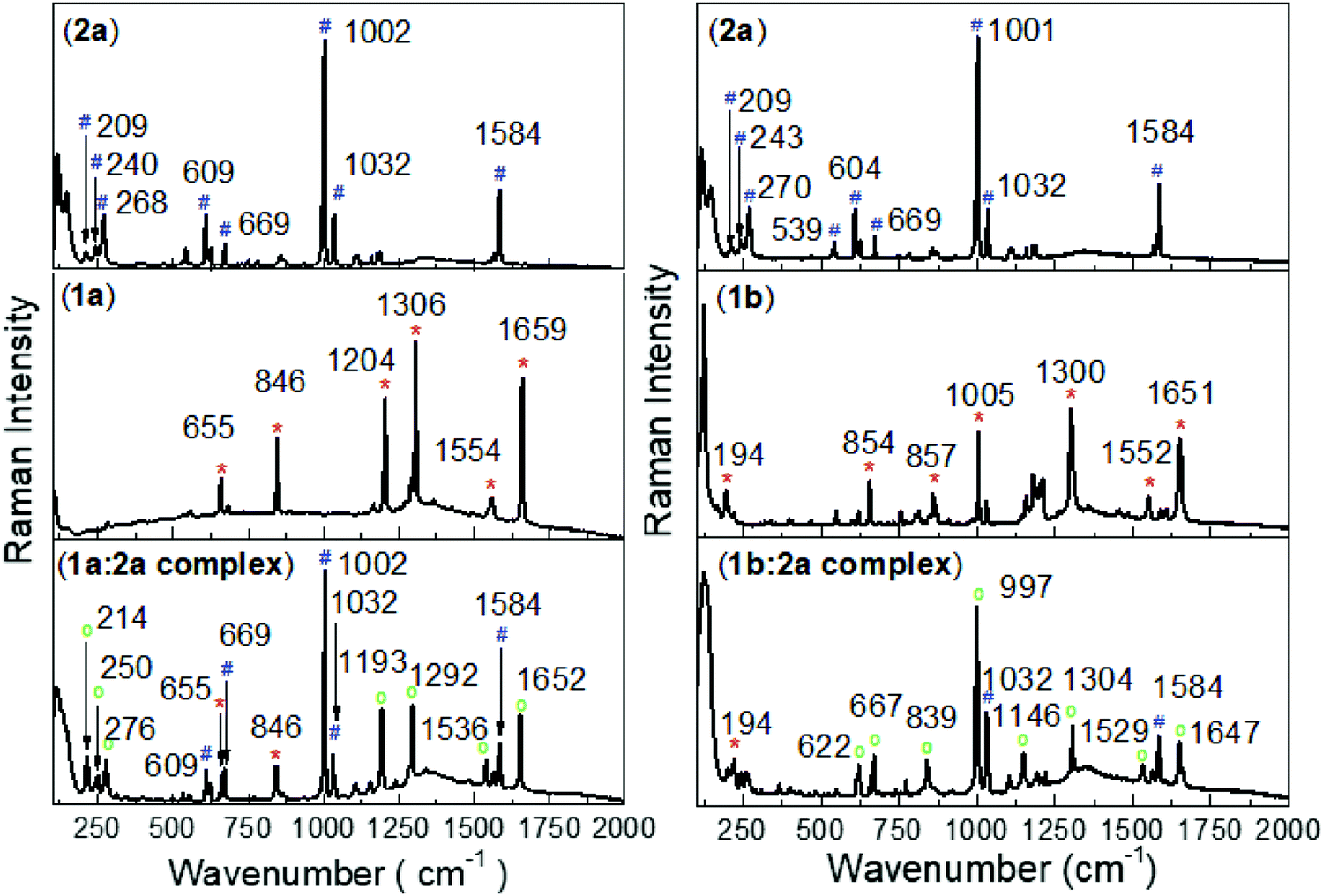 | ||
Fig. 3 Raman spectra of the precursor salts and their complex forms. (2a) potassium tetraphenylborate; (1a) methyl viologen dichloride; (1b) benzylviologen dichloride. λlaser = 780 nm. Characteristic Raman features of (#) potassium tetraarylborate, (*) viologen dichloride and ( ) the ion pair complex. ) the ion pair complex. | ||
During 780 nm irradiation, the complex form becomes excited and the presence of the viologen radical is confirmed by changes in the signals from the a1 and b2 symmetries. The Raman spectra of the complex shows an absence of the band in the precursor salts. The rise of new bands at 1652, 1536 and 1292 cm−1 (1647, 1529 and 1304 cm−1 for the 1b:2a complex) are attributed to the growth of the viologen radical species.
In the Raman spectra of the 1b ion (precursor salt), b2 symmetry is also observed at 1005 cm−1 and assigned to the Ph substituent. In the complex form, the b2 symmetry of the phenyl group of the viologen structure is shifted to 997 cm−1, indicating strong evidence for a lowest vibronic species formed via π–π interaction between the phenyl-groups of the 1b and 2a ions. As expected, the absence of the Ph substituent in the 1a structure is followed by the absence of the b2 transition (∼997 cm−1) in the Raman-spectra of the 1a:2a complex.
Ultrafast transient absorption studies
In order to unravel the relevant dynamics, one must use techniques that can resolve these ultrafast processes. In particular, the UV-vis absorption ultrafast spectroscopy uses a laser pulses with a time duration on the order of femto second. The pump pulse excites the ion pair complex and provides initiation of the photochemical process (e.g. charge transfer and/or electron transfer processes). In this sense, to better characterize the photochemical processes of the new complexes (1b:2b and 1b:2c) as well as the borate precursor salts at short times, we performed femtosecond transient absorption (fs-TA) experiments (Fig. 4) in solution.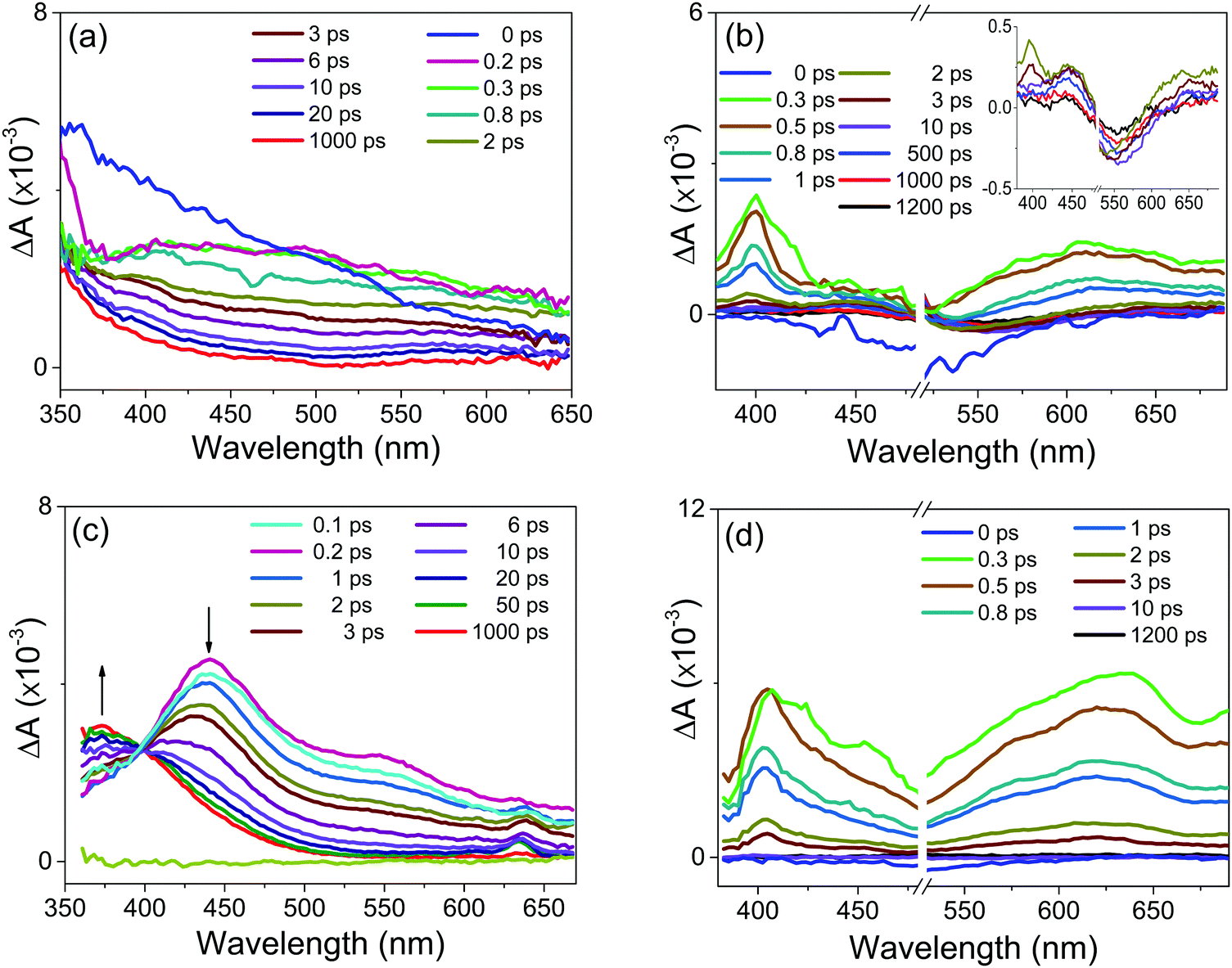 | ||
| Fig. 4 Transient absorption spectra in 1-propanol at 298 K for: (a) potassium tetra(p-chlorophenyl)borate or 2b anion; (b) 1b:2b complex; (c) potassium tetra(biphenyl)borate or 2c anion; (d) 1b:2c complex. Graph inset: TA-spectrum acquired after the delay time of 2 ps; the vertical scale is absorption in mOD units. | ||
Following excitation with a sub-50 fs 260 nm pulse, the potassium salts of 2b (Fig. 4a) or 2c (Fig. 4c) undergo sub-100 fs relaxation from the Franck–Condon states into a locally excited state (around 370 nm) or a low-lying excited state, seen as the build-up of the transient absorption band in the visible range between 400 nm and 650 nm.
The visible transient absorption spectra around 400–650 nm are attributed to absorption from the twisted intramolecular charge transfer (TICT) state as previously proposed by us for the sodium tetraphenylborate salt (2a) in polar solvents (i.e., acetonitrile, methanol, and ethanol).19 The results observed for the sodium 2a, 2b or 2c samples show that the TICT absorption band decreases with a concomitant increase of the triplet–triplet absorption band around 350–450 nm. The latter has a long decay lifetime (>1.2 ns).
For the 1b:2b and 1b:2c complexes, the fs-TA experiments were carried out utilizing excitation at 500 nm. The transient absorption spectra starting from 1 ps are similar for both complexes (Fig. 4b and d). The strongest transient absorption signals around 400 nm and 600 nm are assigned to the PCS state. Such charge separations were also detected in the ultrafast transient absorption spectra previously described for 1a:2a and 1b:2a complexes.7
After 20 ps, the transient absorption curve in Fig. 4b shows a negative signal decaying with a time constant of about 400 ps. This negative signal is assigned to the stimulated emission process because its decay is in the same time range as the one the fluorescence decay lifetime observed at 550 nm. In parallel to the negative signal evolution, a concomitant rise of positive transient absorption around 450 nm takes place, which can be attributed to the triplet–triplet absorption of tetraarylborate anion, such as previously reported for complex 1a:2a.7 This spectral and temporal behaviour suggests the existence of an efficient ISC process due to the heavy atom effect (e.g., the Cl atom) or the promotion by the conjugated Ph–Ph fragment (e.g., 1a:2c and 1b:2c complexes) present in the tetraarylborate structure. Following the fast and efficient ISC process, the population in the triplet state increases, as observed.
For the 1b:2c complex, the stimulated emission signal around 550 nm is of small amplitude (Fig. 2b), which can be attributed to the presence of a competing fast non-radiative process. This non-radiative pathway will compete with the intersystem crossing pathway, so that triplet–triplet absorption for the 1b:2c complex is expected to be of a lesser amplitude. In agreement with this suggestion, the transient absorption spectrum of this complex after 10 ps does not show any significant triplet–triplet absorption.
Steady state electron paramagnetic resonance (SSEPR)
The SSEPR technique was used to identify and follow the paramagnetic photoproducts of the ion-pair complex after different irradiation times (410 nm), thus elucidating the kinetics of photochemical reactions involved in the viologen radical generation. In this investigation, the use of different intensities of microwave of irradiation was also applied to study the effect of intensity on the viologen radical deactivation process. As will be demonstrated below, this implies an ability to switch the magnetic properties of the radical on and off using microwaves.Complexes 1b:2b and 1b:2c are SSEPR silent before irradiation. After irradiation at 410 nm, they exhibit SSEPR signals around g = 2.000, which is similar to that observed from the viologen radical (MV+˙) in the literature.20 The photochromism mentioned above is simultaneously observed, and is attributed to reversible interconversion between the MV+˙ and MV2+. See Fig. 5 for SSEPR signals of all complexes at different times of irradiation.
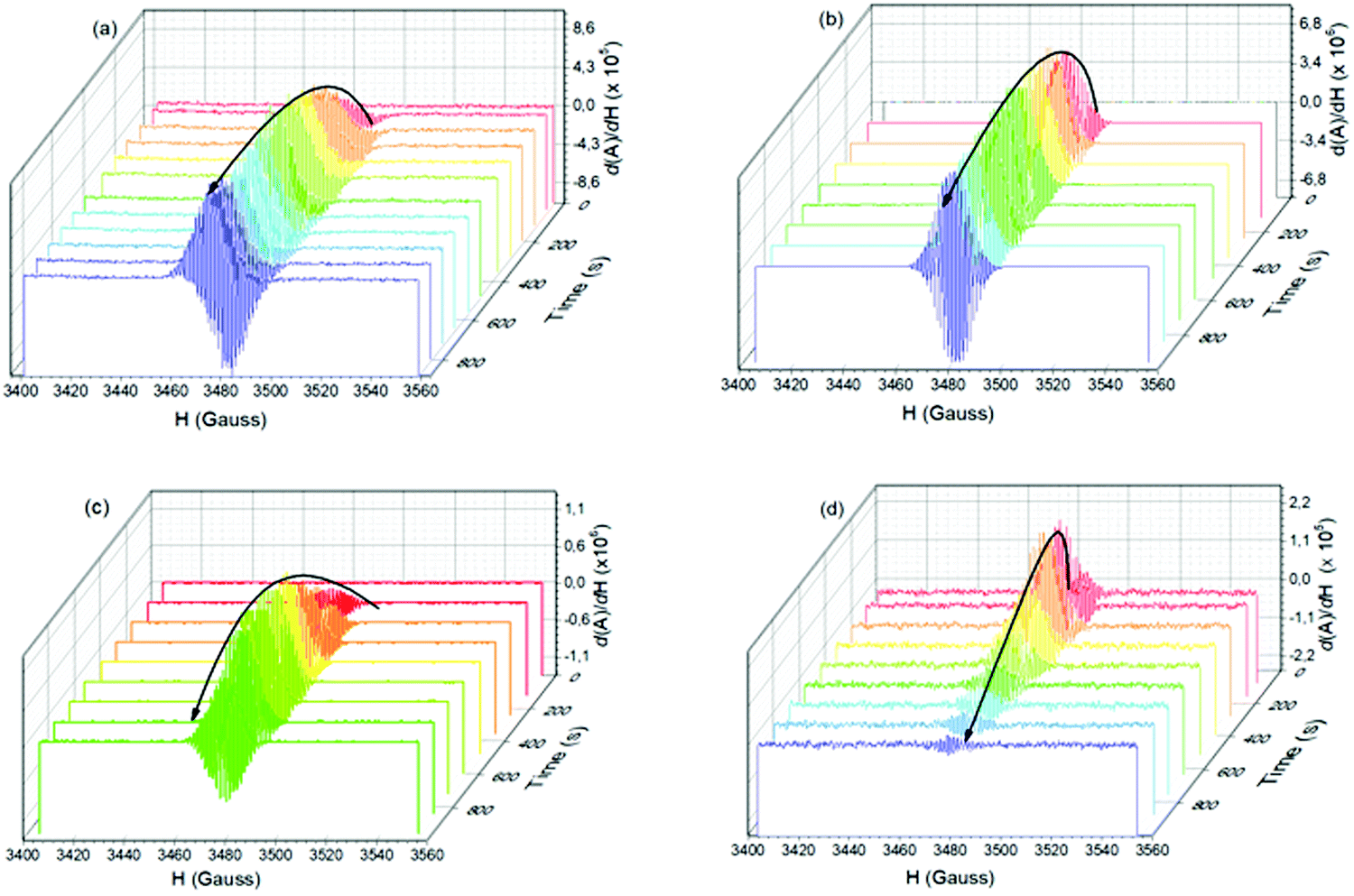 | ||
| Fig. 5 3D SSEPR spectra recorded in time-scan mode at different time irradiation (λirr = 410 nm), and for different complex system in THF:ACN solution (5:1): (a) 1a:2a, (b) 1b:2a, (c) 1b:2b, (d) 1b:2c complexes. | ||
Under continuous irradiation at 410 nm, the 3D spectra of all complexes show different dynamics for the viologen radical signal. The lifetime of the viologen radical can be explained by the nature of the π–π stacking process between viologen and the different borate structures into complex form, which may have some influence on the back electron transfer (BET) process as depicted in eqn (10). The presence of the dimer radical was ruled out due to the absence of the dimer absorption band at 562 nm in the UV-vis spectra.1
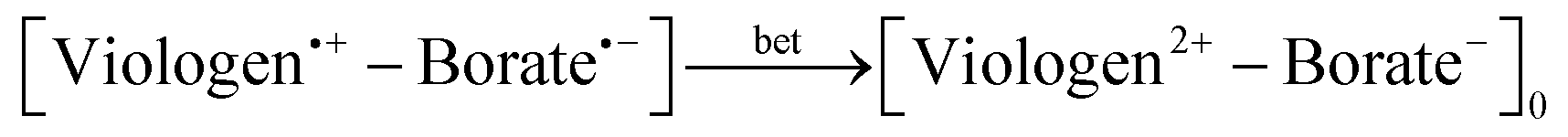 | (10) |
The hyperfine coupling constants for all complexes are summarized in Table S2 (ESI†). See ESI† for the experimental and simulated SSEPR spectra (Fig. S3, ESI†).
Fig. 6 shows SSEPR spectra of the 1b:2b and 1b:2c complexes in the presence of the spin-trap 3,3,5,5-tetramethyl-1-pyrroline N-oxide (TMPO) in THF:ACN (5:1 v/v) solution. At the beginning of the irradiation experiments at 410 nm, the radical adduct detected is attributed to the phenyl radical, as consequence of one-electron oxidation and subsequently phenyl release from the tetraphenylborate structure. This same phenyl radical adduct was previously characterized by Santos and co-workers for 1a:2a and 1b:2a compounds.7
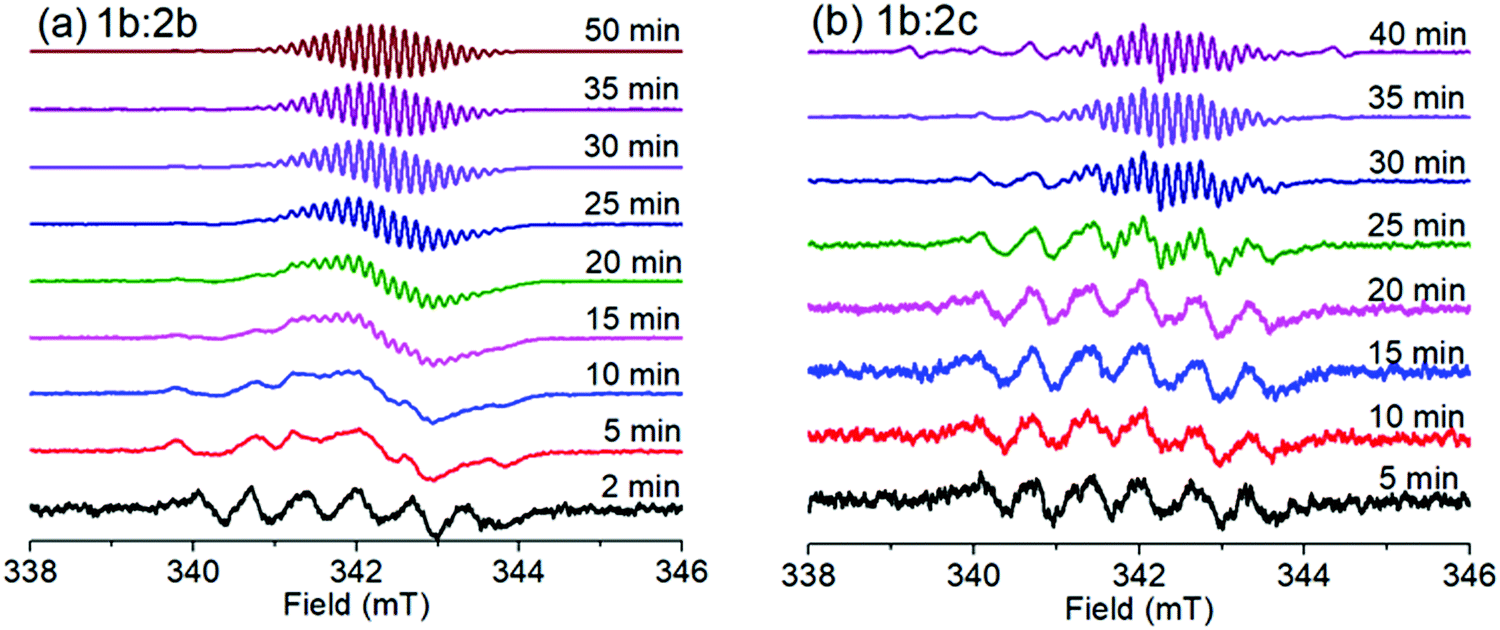 | ||
| Fig. 6 Time evolution of the maximum EPR signal of (a) 1b:2b complex and (b) 1b:2c complex recorded at different times of irradiation in THF:ACN (5:1 v/v) solvent, [complex] = 10−3 mol L−1; λirr = 410 nm. | ||
In the presence of spin traps, the viologen radical clearly is also observed in the spectra after 25 minutes of irradiation, indicating that the viologen radical may be released from the complex after a few minutes of irradiation. The lifetime of this SSEPR signal is quite long (τ > 40 min), which has been reported by other authors for non-complexed viologen radicals.29–32 In the absence of spin traps, the complexed viologen radical is detected within the first few minutes of irradiation, with a lifetime of a few minutes (see Fig. 5).
For the 1a:2c ion pair complex, the initial radical observed at 5 min of irradiation is also observed at 40 min. The initial radical observed is attributed to the adduct radical between biphenyl radical (released from tetra(biphenyl)borate anion) and 3,3,5,5-tetramethyl-1-pyrroline N-oxide (TMPO), producing the Biphenyl-TMPO radical adduct. The chemical stability of this adduct radical certainly is increased by resonance effect into the aromatic structure. In this sense, the phenyl-TMPO radical species is expected to have a short lifetime when compared to biphenyl-TMPO radical species because the biphenyl radical has an adjacent aromatic ring. The long lifetime decay of free viologen radical (>60 min) (non-complexed form) was not influenced by others free radicals such as biphenyl-TMPO radical species or phenyl-TMPO radical.
After irradiation of the complex in solution, the original complexed structure is lost, producing triphenyl borane and other products.7 However, in the solid phase, after forward electron transfer to the viologen acceptor, recombination from the radical pair MV+˙/Borate˙ leads to partial recovery of the initial ground state of the complex with a mono-exponential decay of the radical-signal. Scheme 3 summarizes our proposed mechanism for these processes.
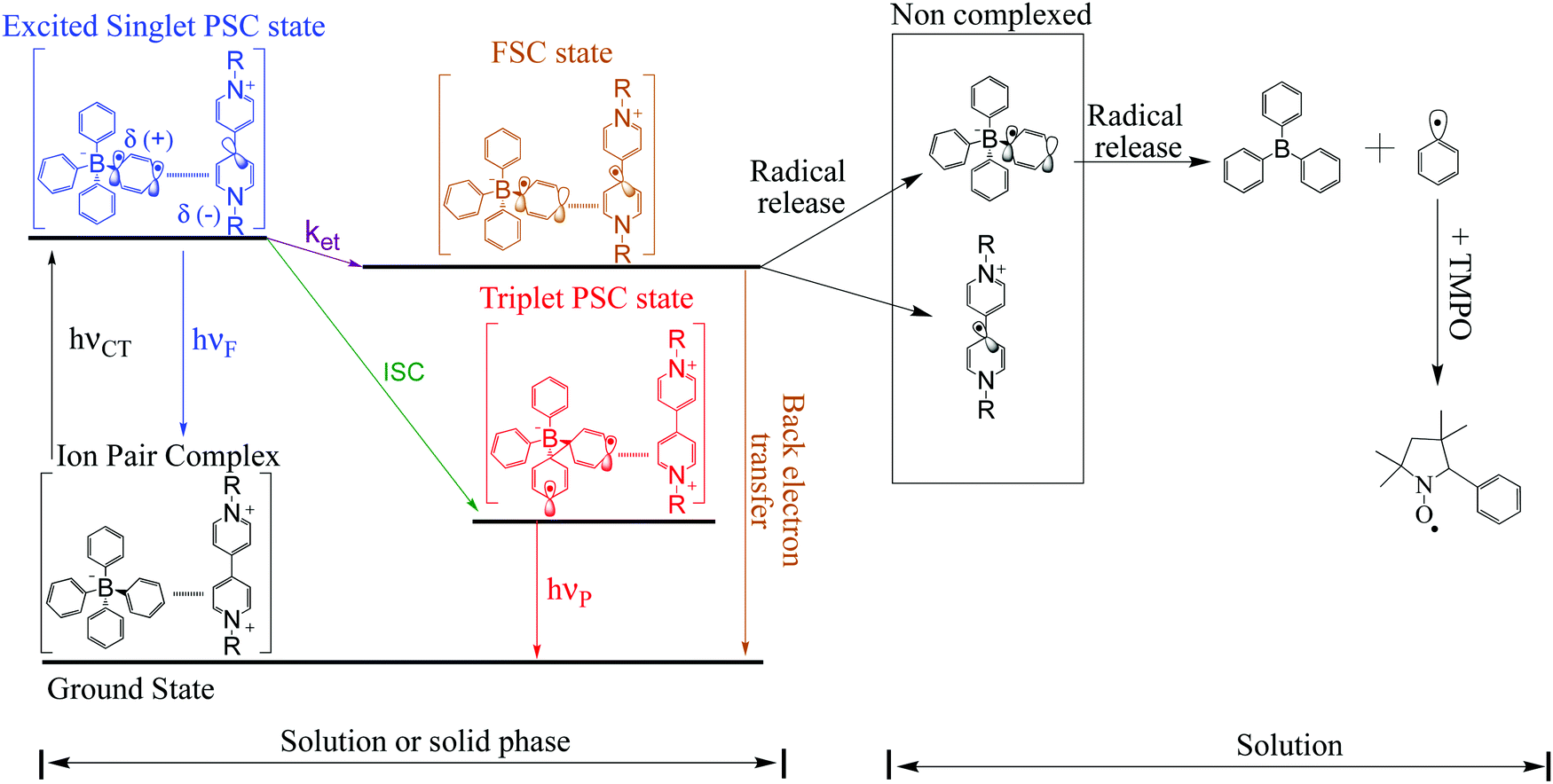 | ||
| Scheme 3 Schematic diagram for FSC-state and PCS-state deactivation. | ||
Viologens are intrinsically polar in terms of charge distribution and when subjected to oscillating microwave radiation they rapidly vibrate within the field. So the applied microwave (μw) field and may results in dielectric change and/or result in a heating process.
From the literature it is well known that the lifetimes of viologen radicals decrease when the temperature is increased. We postulate that a new pathway for modulation of the lifetime of viologen radicals may be possible when microwave radiation is applied to these complexes. As a consequence of this radical deactivation, a “photo-erasing” process is plausible, especially when it is noted that viologen radical loss is followed by a decrease of its absorption band in the visible range (λabs max = 600 nm).
To investigate the effects of resonant μw intensity on the viologen/borate complex system, the incident light at 410 nm was switched on and off at different intensities of μw excitation, using a fixed magnetic field at 346 mT (g ≈ 2.005), which corresponds to the maximum intensity for the viologen radical resonance. Fig. 7 shows the 2D time evolution spectra of the viologen radical in the complex form.
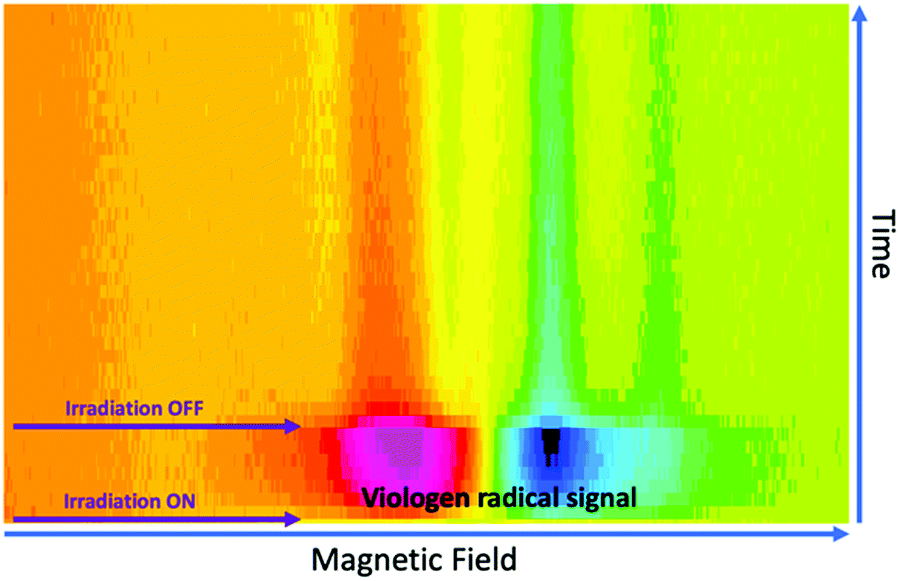 | ||
| Fig. 7 Time evolution of the SSEPR signal from the viologen radical moiety in the 1a:2a complex. The SSEPR signals were recorded with a line width of 1 Gauss and 5 mW of microwave power. | ||
In free solution, when the microwave power intensity is increased, no effect is observed on the lifetime of the viologen radical. However, in the solid state, the lifetime of the 1a radical decreases when the microwave power intensity is increased, as shown in Fig. 8.
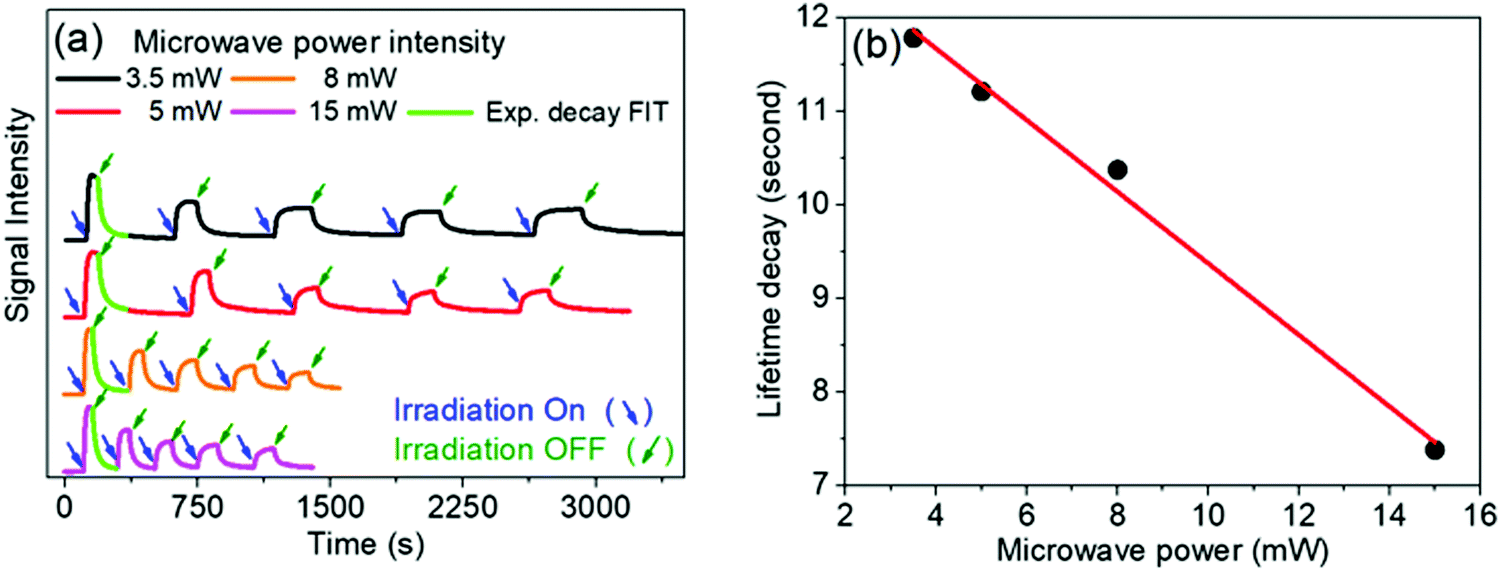 | ||
| Fig. 8 (a) Time evolution of the EPR signal at 346 mT recorded at different microwave power intensity. Sample: 1a:2a complex in solid phase (powder). (b) Direct correlation between microwave power and lifetime decay. | ||
For the viologen radical deactivation process, external μw radiation may induce relaxation transitions, promoting additional mixing between different states (PSC and FSC). Additional mixing can affect the RP spin state dynamics in the complex. Indeed, the mechanism for the back-ET process will be efficient if both unpaired electrons in the FSC-complex system encounter oppositely polarized spins, and thus transfer their magnetization to regenerate the singlet ground state, as shown in Scheme 4.
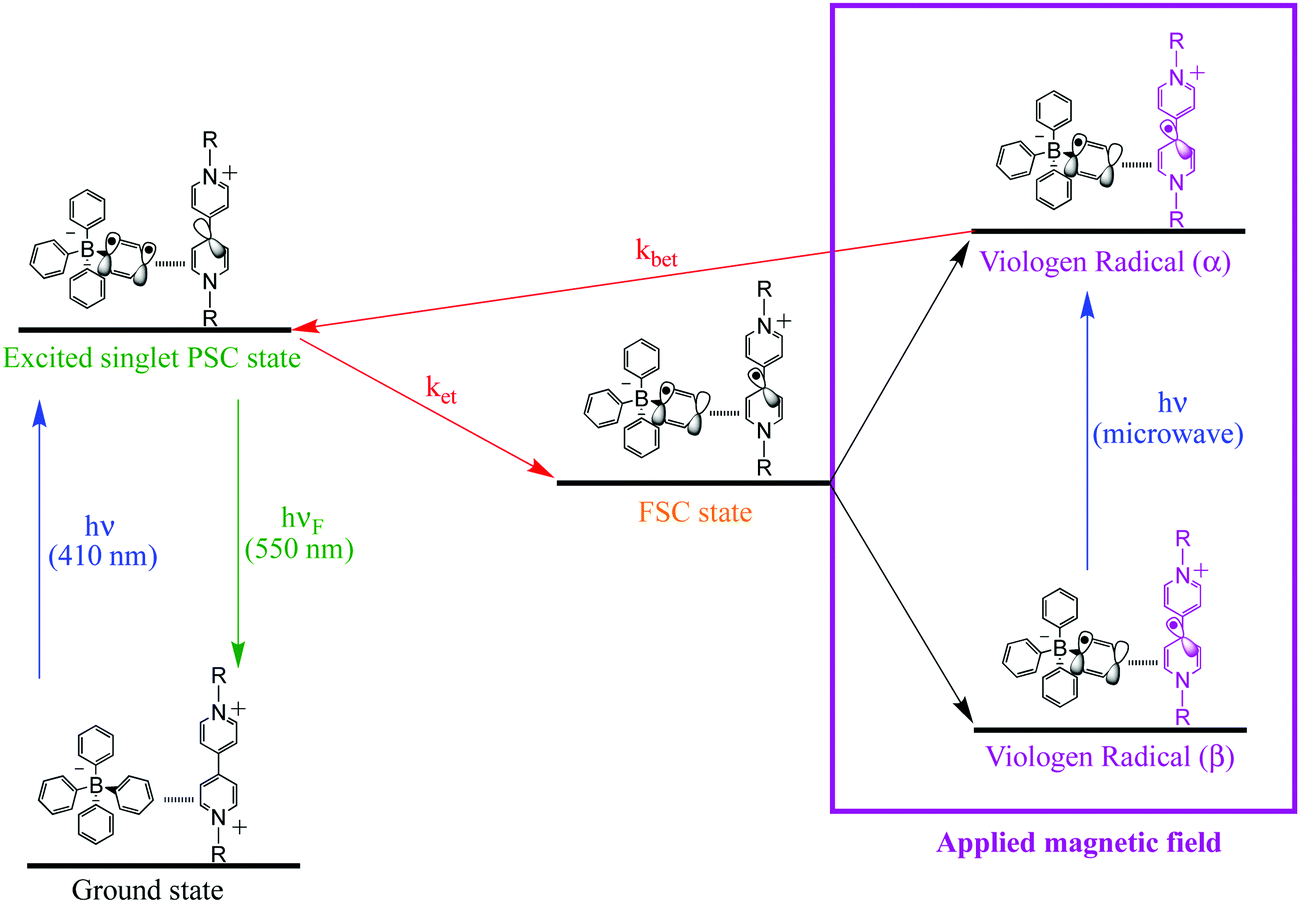 | ||
| Scheme 4 Schematic diagram for PSC-state deactivation, in the presence of magnetic field. MW = microwave radiation used to increase the yield of the viologen (α) radical. | ||
The electron spin–spin exchange interaction is exponentially distance dependent and directly reflects the donor–acceptor electronic coupling, specially into host–guest supramolecular systems where π–π stacking interaction may be observed.20,28,32–36 In fact, the presence of microwave radiation increases the yield of the viologen (α) species. This in turn increases the recombination of radicals to form excited PSC-complex species, which may be deactivated by radiative (fluorescence) and non-radiative processes (thermal deactivation) to return to the ground state. For the other complexes in powder media, time evolutions of their SSEPR spectra are shown in Fig. 9.
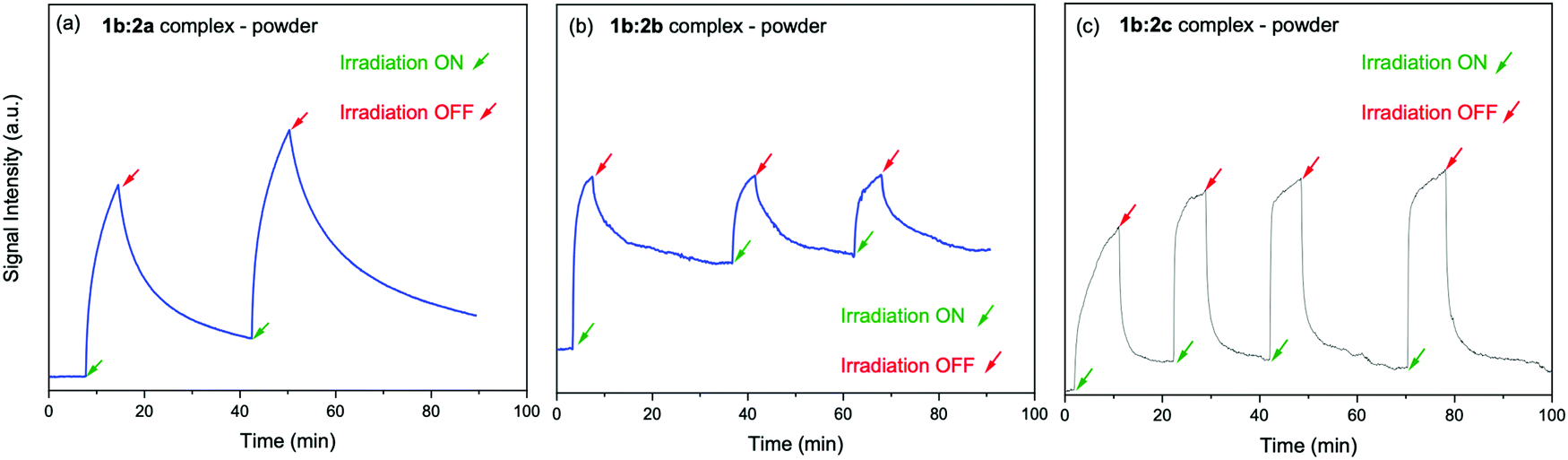 | ||
| Fig. 9 Time evolution of the EPR signal at 346 mT recorded in the presence and absence of light irradiation at 410 nm. Powder samples: (a) 1b:2a, (b) 1b:2b, (c) 1b:2c complexes. | ||
Dynamics of viologen radical species in a polymer matrix
After reduction of the viologen dication (V2+), a coloured viologen cation radical (V+˙) species is generally observed within the UV-photolytic systems under anaerobic conditions.1,2 However, the produced Viologen radical cation radical (V+˙) is highly sensitive to air oxidation, recovering the initial viologen dication after electron transfer to oxygen molecules. To avoid the photobleaching process of V+˙, this species can be embedded in a polymer matrix and the resulting radicals are stabilized by the surrounding solid matrix via thermal reverse electron transfer and/or air oxidation.5,10The 1a:2a complex was prepared in a polymer matrix (polyethylene vinyl acetate, poly(EVA)) to exemplify the use of this type of complex as an active material for optoelectronic devices. In the ESI,† a video is presented to show the use of the 1a:2a complex-polymer in a direct laser writing technique. Fig. 10 shows the ion pair complex incorporated into the polymer matrix, as well as screen shots of the writing and erasure processes.
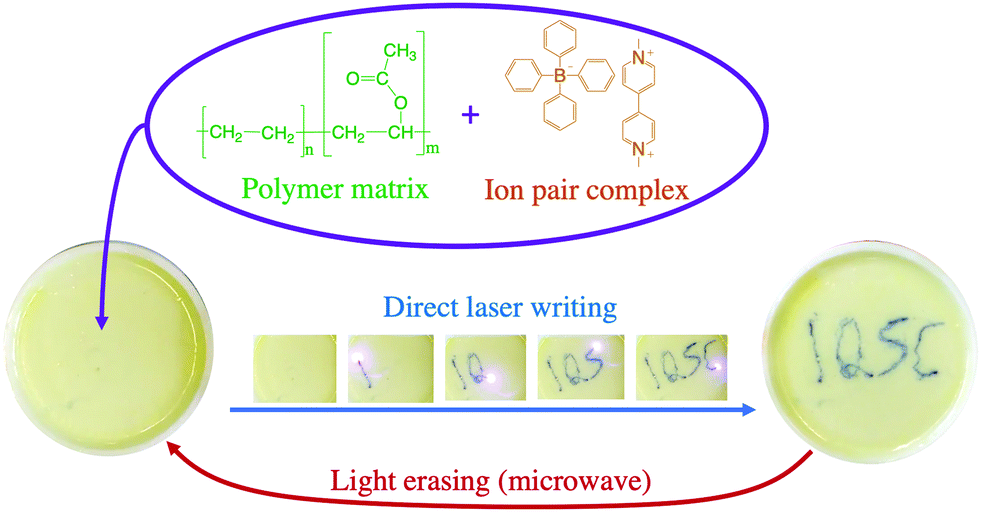 | ||
| Fig. 10 Example of laser writing and microwave induced erasing process using ion pair complex 1a:2a embedded in a poly(EVA) matrix. | ||
All ion pair compounds show an interesting photochromic transformation from yellow/orange to black upon visible irradiation. Color reversion is accomplished by irradiation with μw irradiation, as shown in Fig. 11. Indeed, when the intensity of the μw radiation is increased, the lifetime of the viologen radical concomitantly decreases.
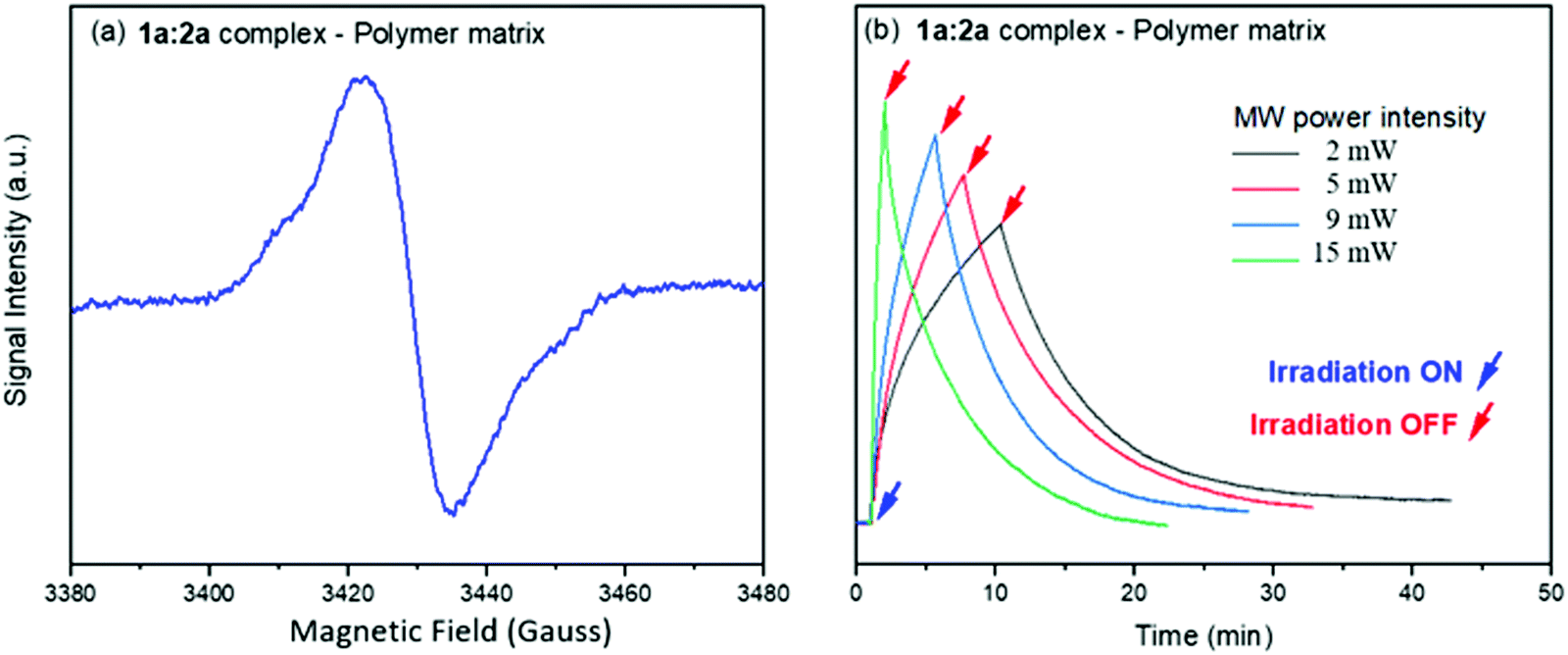 | ||
| Fig. 11 (a) SSEPR spectrum of the 1a:2a complex in thin film of PEVA. (b) Time profiles of the SSEPR signal of the viologen radical at different μw powers (in mW) in the polymer matrix, in the presence and absence of light irradiation at 410 nm. | ||
Conclusions
The excited state transient absorption events and stimulated emission of viologen–borate complexes of all ion pair complexes based on this viologen–tetraarylborate framework have been investigated in detail via excitation of the charge-transfer band around 400–700 nm. UV-vis absorbance and stimulated emission results indicate that the para-substituted tetraphenylborate may significantly alter the photophysical characteristics of the ion-pair complexes (e.g. fluorescence lifetimes and the absorption region of the complex). Using ultrafast transient absorption (TA), the formation of a charge-separated state was observed upon excitation with a femtosecond (fs) laser at 400 nm.After excitation, the FSC state is generated and observed as a radical pair. The viologen radical in the radical pair was investigated in the presence of magnetic fields and μw radiation, using the SSEPR technique. The photochromic behaviour reveals that the lifetimes of the viologen radicals may be modulated from hours to seconds when the intensity of the μw irradiation is increased.
It is logical to extend the application of these ion-pair complexes as photochromic materials in solid phase, for example in a direct laser writing process. The use of μw radiation is a plausible way to decrease the lifetime of these and other chromophoric radicals for application in photo-erasing processes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge support from FAPESP (2012/19823-4,2015/13756-1, and 17/01189-0). WGS thanks the Sisfóton Laboratory Project – CNPq and Sidney J. L. Ribeiro's group for the new research DTI-A fellowship and financial support (382034/2021-0). DRC thanks the National Council for Scientific and Technological Development (CNPq) for a research productivity fellowship. MDEF and ANT acknowledge continued strong support from the US National Science Foundation (CHE-1464817 and CHE-1900541 for MDEF, CHE-1626420 and DMR-1006761 for ANT).References
- K. Nchimi-Nono, P. Dalvand, K. Wadhwa, S. Nuryyeva, S. Alneyadi, T. Prakasam, A. C. Fahrenbach, J. C. Olsen, Z. Asfari, C. Platas-Iglesias, M. Elhabiri and A. Trabolsi, Chem. – Eur. J., 2014, 20, 7334–7344 CrossRef CAS PubMed.
- L. Liu, Q. Liu, R. Li, M. S. Wang and G. C. Guo, J. Am. Chem. Soc., 2021, 143, 2232–2238 CrossRef CAS PubMed.
- R. Pardo, M. Zayat and D. Levy, Chem. Soc. Rev., 2011, 40, 672–687 RSC.
- G. Xu, G. C. Guo, M. S. Wang, Z. J. Zhang, W. T. Chen and J. S. Huang, Angew. Chem., Int. Ed., 2007, 46, 3249–3251 CrossRef CAS PubMed.
- M. Nanasawa, Y. Matsukawa, J. J. Jin and Y. Haramoto, J. Photochem. Photobiol., A, 1997, 109, 35–38 CrossRef CAS.
- K. Madasamy, D. Velayutham, V. Suryanarayanan, M. Kathiresan and K. C. Ho, J. Mater. Chem. C, 2019, 7, 4622–4637 RSC.
- W. G. Santos, D. S. Budkina, V. M. Deflon, A. N. Tarnovsky, D. R. Cardoso and M. D. E. Forbes, J. Am. Chem. Soc., 2017, 139, 7681–7684 CrossRef CAS PubMed.
- K. B. Yoon and J. K. Kochi, Society, 1988, 6586–6588 CAS.
- L. K. Cadman, M. F. Mahon and A. D. Burrows, Faraday Discuss., 2021, 225, 414–430 RSC.
- L. A. Vermeulen, J. L. Snover, L. S. Sapochak and M. E. Thompson, J. Am. Chem. Soc., 1993, 11767–11774 CrossRef CAS.
- M. C. Sekhar, S. Paul, A. De and A. Samanta, ChemistrySelect, 2018, 3, 2675–2682 CrossRef CAS.
- T. G. Zhan, T. Y. Zhou, F. Lin, L. Zhang, C. Zhou, Q. Y. Qi, Z. T. Li and X. Zhao, Org. Chem. Front., 2016, 3, 1635–1645 RSC.
- J. Chen, K. Wu, B. Rudshteyn, Y. Jia, W. Ding, Z. Xie, V. S. Batista and T. Lian, J. Am. Chem. Soc., 2016, 138, 884–892 CrossRef CAS PubMed.
- H.-H. Li, P. Wang, X.-H. Chao, C.-C. Lin, A.-W. Gong and Z.-R. Chen, J. Cluster Sci., 2015, 26, 851–862 CrossRef CAS.
- T. Miura and M. R. Wasielewski, J. Am. Chem. Soc., 2011, 133, 2844–2847 CrossRef CAS PubMed.
- N. E. Horwitz, B. T. Phelan, J. N. Nelson, C. M. Mauck, M. D. Krzyaniak and M. R. Wasielewski, J. Phys. Chem. A, 2017, 121, 4455–4463 CrossRef CAS PubMed.
- N. E. Horwitz, B. T. Phelan, J. N. Nelson, M. D. Krzyaniak and M. R. Wasielewski, J. Phys. Chem. A, 2016, 120, 2841–2853 CrossRef CAS PubMed.
- S. M. Kim, J. H. Jang, K. K. Kim, H. K. Park, J. J. Bae, W. J. Yu and I. H. Lee, J. Am. Chem. Soc., 2009, 1–3 Search PubMed.
- W. G. Santos, J. Pina, H. D. Burrows, M. Forbes and D. R. Cardoso, Photochem. Photobiol. Sci., 2016, 15, 1124–1137 CrossRef CAS PubMed.
- W. G. Santos, D. S. Budkina, S. H. Santagneli, A. N. Tarnovsky, J. Zukerman-Schpector and S. J. L. Ribeiro, J. Phys. Chem. A, 2019, 123, 7374–7383 CrossRef CAS PubMed.
- W. G. Santos, F. Mattiucci and S. J. L. Ribeiro, Macromolecules, 2018, 51, 7905–7913 CrossRef CAS.
- N. L. Bill, M. Ishida, Y. Kawashima, K. Ohkubo, Y. M. Sung, V. M. Lynch, J. M. Lim, D. Kim, J. L. Sessler and S. Fukuzumi, Chem. Sci., 2014, 5, 3888–3896 RSC.
- F. Roth, M. Borgwardt, L. Wenthaus, J. Mahl, S. Palutke, G. Brenner, G. Mercurio, S. Molodtsov, W. Wurth, O. Gessner and W. Eberhardt, Nat. Commun., 2021, 12, 1–7 Search PubMed.
- Y. Hou, X. Zhang, K. Chen, D. Liu, Z. Wang, Q. Liu, J. Zhao and A. Barbon, J. Mater. Chem. C, 2019, 7, 12048–12074 RSC.
- T. Miura, J. Phys. Chem. B, 2013, 117, 6443–6454 CrossRef CAS PubMed.
- D. Fazzi, M. Barbatti and W. Thiel, J. Am. Chem. Soc., 2016, 138, 4502–4511 CrossRef CAS PubMed.
- K. Fukuda and M. Nakano, J. Phys. Chem. A, 2014, 118, 3463–3471 CrossRef CAS PubMed.
- H.-W. Schmidt, ChemPhysChem, 2013, 14, 1818–1829 CrossRef PubMed.
- R. Dabestani, K. J. Reszka and M. E. Sigman, J. Photochem. Photobiol., A, 1998, 117, 223–233 CrossRef CAS.
- P. C. Lee, K. Schmidt, S. Gordon and D. Meisel, Chem. Phys. Lett., 1981, 80, 242–247 CrossRef CAS.
- P. M. S. Monk, R. D. Fairweather, M. D. Ingram and J. A. Duffy, J. Chem. Soc., Perkin Trans. 2, 1992, 2039–2041 RSC.
- A. M. Rakhi and K. R. Gopidas, Chem. Phys. Lett., 2015, 618, 192–197 CrossRef CAS.
- E. N. Ushakov, V. A. Nadtochenko, S. P. Gromov, A. I. Vedernikov, N. A. Lobova, M. V. Alfimov, F. E. Gostev, A. N. Petrukhin and O. M. Sarkisov, Chem. Phys., 2004, 298, 251–261 CrossRef CAS.
- H. A. Meylemans and N. H. Damrauer, Inorg. Chem., 2009, 48, 11161–11175 CrossRef CAS PubMed.
- H. Yonemura and M. D. E. Forbes, Photochem. Photobiol., 2015, 91, 672–677 CrossRef CAS PubMed.
- E. H. Witlicki, S. S. Andersen, S. W. Hansen, J. O. Jeppesen, E. W. Wong, L. Jensen and A. H. Flood, J. Am. Chem. Soc., 2010, 132, 6099–6107 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods used for each spectroscopic measurements, fluorescence lifetimes and relative amplitudes, hyperfine coupling constants for EPR spectral simulations, UV-VIS spectra of the photolyzed ion–pair complexes in the liquid phase, DFT results, video showing the direct laser writing and microwave erasing processes for four polymer/complex systems. See DOI: 10.1039/d1ma01030a |
| This journal is © The Royal Society of Chemistry 2022 |