
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltraflexible all-in-one supercapacitors with high capacitance and ultrastable cycle performance enabled by wood cellulose network†
Shaoqiu
Ke‡
ab,
Di
Xie‡
b,
Kai
Zhang
b,
Fangchao
Cheng
 *ab and
Yiqiang
Wu
*a
*ab and
Yiqiang
Wu
*a
aCollege of Material Science and Engineering, Central South University of Forestry and Technology, Changsha 410004, China. E-mail: fangchaocheng@gxu.edu.cn; wuyq0506@126.com
bGuangxi Key Laboratory of Processing for Nonferrous Metallic and Featured Materials, School of Resources, Environment and Materials, Guangxi University, Nanning 530004, China
First published on 11th January 2022
Abstract
Herein, we demonstrate a structure-enabled strategy to construct an ultrastable, high-performance, ultraflexible all-in-one supercapacitor with a one-piece wood cellulose network/polyaniline/multiwalled carbon nanotube composite as both the electrodes and the separator. A cellulose network was used as the carrier and separator, and multiwalled carbon nanotube and polyaniline acted as conducting materials. Excellent flexibility was achieved by combining the cellulose network with a polyvinyl alcohol-based electrolyte. Natural directional channels in the cellulose network and the gradual distribution of the polyaniline and carbon nanotube within the structure enabled the construction of an integrated supercapacitor with reduced contact resistance between layers and improved electronic/ionic conductivity and stability. The supercapacitors resulted in a high specific areal capacitance of 0.71 F cm−2, as well as ultrastable cycle performance, which led to a 106.68% retention of the specific capacitance after 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 loops at a current density of 3 A g−1. The current study provides a nature-inspired strategy to rationally design and construct environmentally friendly high-performance and ultrastable energy storage devices.
000 loops at a current density of 3 A g−1. The current study provides a nature-inspired strategy to rationally design and construct environmentally friendly high-performance and ultrastable energy storage devices.
1. Introduction
At present, excessive consumption of fossil fuels and consequential environmental pollution compel countries to search for the sustainable development of new energy and advanced technologies associated with energy storage.1 Supercapacitors, as one of the most important energy storage devices, can not only store energy like traditional batteries, but also have the advantages of ultra-high capacitance, high power density, rapid charging, long lifetimes, and excellent low-temperature performance and safety, and have been widely applied in electronics products, electric vehicles, military applications, and other industrial fields.2 Due to the popularity of electronic wearables, flexible supercapacitors (FSCs) are designed to operate without performance degradation under various strain conditions, such as bending, folding, and even twisting, which are particularly suitable for wearable electronics.3 In general, the performance of FSCs mainly depends on electrodes, and multifarious electrode materials have been used to construct FSCs, including graphene,4,5 carbon nanotube (CNT),6 reduced graphene oxide,4,5 conductive polymers,7 MnO2,8 and other materials.9 Among them, natural renewable materials, such as cellulosic materials, have drawn much attention as flexible electrode matrixes due to their biodegradability and environment friendliness. Cellulosic materials10 (e.g., cellulose nanofiber, bacterial cellulose, cellulose nanocrystal, wood, and paper) have been combined with conducting polymers,5 carbon nanomaterials,6 and metal oxides11 to form electrode materials for FSCs, resulting in high performance for energy storage and outstanding flexibility. However, FSCs prepared with cellulosic materials cannot take full advantage of the unique, naturally endowed structure of lignocellulosic biomass and cannot achieve high energy/power density, long cycle lives, and high mass loading simultaneously.12,13 Wood is a widely used material with unique porous hierarchical cellular structures originating from its natural growth.14 It is a promising concept to use wood as a template to produce electrode materials due to the naturally complex meshstructures of wood, which can provide a series of microreactors for other polymers or functional materials to form directional and functional composites.15,16 Recently, we prepared a highly conductive cellulose network/polyaniline composite as a promising electrode material by removing hemicellulose and lignin from wood while retaining the cellulosic skeleton structure of the wood.17 However, cellulose network-based conductive composites have poor flexibility due to the stiffness of the original wood. Therefore, further investigations are needed to realize the high energy/power density, long life cycles, and flexibility of naturally sourced, environmentally friendly cellulose network-based FSCs.In addition to electrode materials, the electrolyte is an essential component that plays a crucial role in the performance of FSCs; it usually involves the liquid, gel phase, or all-solid-state forms.18 Due to the advantages of low cost, high reliability, and high ionic conductivity (about 10−4 to 10−1 S cm−1), gel electrolytes are commonly used in FSCs.19 Unfortunately, most FSCs using gel electrolytes (e.g., the polyvinyl alcohol (PVA)–acid system) exhibit poor structural integrity and low mechanical strength due to the nature of liquid gels.20 However, PVA-based gel electrolytes can be converted into robust, self-standing hydrogels through freezing-thawing cycles.21 This may allow us to enhance the flexibility and mechanical strength of FSCs with the introduction of PVA-based gel electrolytes.
The structure design of FSCs is also of great importance in making full use of active electrode materials and facilitating ion and electron transport, which is particularly true when a thick electrode is utilized. Traditional FSCs have a multilayered structure that consists of two solid electrodes, a separator in between, and the electrolytes.22 However, the laminated configuration results in a large interface contact resistance because of the multiple interfaces among the layers, which hinders the fast charge and ion transport from the cathode to the anode.23 Furthermore, during the continuous bending or stretching cycles, the relative displacement between the different layers may lead to the electrode delamination of FSCs, which will undoubtedly reduce the performance of the device.23 In this case, decreasing the layer component number of FSCs and introducing an all-in-one structure into the FSCs may be beneficial to the stability of the FSCs and reduce the interface contact resistance, thus resulting in high performance for energy storage. As discussed above, the freezing-thawing treatment of PVA-based gel electrolytes can not only enhance the flexibility and mechanical properties of FSCs, but also allow for the assembly of all the components (electrodes, separator, and collectors) of FSCs without adhesive. Additionally, if the separator is substituted with an intermediate structure between electrodes constructed with gel electrolytes and a cellulose network, a novel all-in-one configuration of FSCs can be realized by fine-tuning the preparation process of the electrode materials, and the purpose of improving the energy storage performance and stability of FSCs would be expected to achieve by introducing the unique configuration and structure.
To verify this hypothesis, a novel type of all-in-one FSCs was developed based on a single-piece cellulose network originating from solid wood. In the structure of the FSCs, a piece of cellulose network/polyaniline/multiwalled carbon nanotube (CPM) composite acts as both the electrodes and separator, where the two surface parts containing polyaniline (PANI) and multiwalled carbon nanotube (MWCNT) act as the two electrodes, and the intermediate part of the composites between the two electrodes can be directly used as the separator. The PVA-based gel electrolytes can fix all components of the FSCs as well as act as an adhesive to fix the carbon cloth collectors. The cellulose network/polyaniline/multiwalled carbon nanotube (CPM) composites were synthesized by combining the chemical fractionation of solid wood, the immersion of MWCNT, in situ partial growth of PANI, and the coating of MWCNT onto the surface of the composites. The FSCs were assembled by the impregnation of the PVA-based gel electrolytes followed by freezing-thawing cycles. The FSCs exhibited high specific areal capacitance (up to 0.71 F cm−2), ultrahigh cyclic stability (about 106.68% retention after 50000 loops at 3 A g−1), promising ionic conductivity (up to 1.92 × 10−2 S cm−1), and stable performance retention under various folding statuses. Moreover, the fabrication strategy of the all-in-one FSCs shows wide applicability, and different cellulosic materials, such as cotton cloth and cellulose paper, can be employed to construct FSCs with this method. The design strategy of all-in-one FSCs with a cellulose network provides a new direction for the preparation of integrated-structure, high-performance, safe, and reliable naturally sourced energy storage devices and may promote the sustainable and innovative development of wearable devices.
2. Experimental
2.1 Materials
Carboxylated multiwalled carbon nanotube with diameters of 10–20 nm was obtained from Jiangsu Xianfeng Nanomaterial Technology Co., Ltd (China). Phosphoric acid (H3PO4, 98%) was purchased from Lianjiang Ailian Chemical Reagent Co., Ltd (China). Polyvinyl alcohol (≥97%) was obtained from Tianjin Damao Chemical Reagent Factory (China). Potassium hydroxide (KOH, ≥85%) was purchased from Chengdu Jinshan Chemical Reagent Co., Ltd (China). Acetic acid (CH3COOH, ≥99.5%) was purchased from Chengdu Cologne Chemical Co., Ltd (China). Anhydrous ethanol (≥99.7%) was obtained from Tianjin Fuyu Fine Chemical Co., Ltd (China). Phytic acid (PA, 70%), sodium chlorite (NaClO2, 98%), ammonium persulfate (≥98%), and aniline (≥99.5%) were provided by Shanghai Aladdin Bio-Chem Technology Co., Ltd (China). Balsa wood (Ochroma lagopus Swartz) was purchased from Zhuhai Dechi Technology Co., Ltd (China). All reagents were used as received.2.2 Fractionation, impregnation and polymerization processes
Balsa Wood samples (2 g) with dimensions of 15 × 15 × 1 mm were immersed in aqueous KOH solution (2.5 M, 50 mL) at room temperature for 12 h. Then, the mixture was refluxed for 3 h at 90 °C with magnetic stirring to remove most of the hemicellulose and part of lignin. Next, the sample was rinsed repeatedly with deionized water until the pH was equal to 7. The treated samples were put into a 150 mL conical flask, and 65 mL deionized water, 0.5 mL glacial acetic acid, and sodium chlorite (0.7 g) were successively added to the flask. The mixture was heated at 75 °C for 1 h, and more glacial acetic acid (0.5 mL) and sodium chlorite (0.7 g) were subsequently added. The above process was repeated three times to obtain white, purified wood samples. The as-prepared samples were rinsed repeatedly with deionized water until the pH equaled 7 and was stored in anhydrous ethanol to obtain a white cellulose network block.The weight percent gain (WPG) of the cellulose network sample during the in situ polymerization process was calculated with the following eqn (1):
| WPG = ((M1 − M)/M) × 100% | (1) |
Then, MWCNT solution (0.15 mg mL−1, 10 mL) was infiltrated into a cellulose network sample by vacuum filtration, and the sample was subsequently immersed in a solution containing aniline (4.58 mL), phytic acid (6.59 mL), and deionized water (20 mL) under vacuum for 0.3 h. Subsequently, the ammonium persulfate solution, which was prepared by dissolving ammonium persulfate (1.82 g) in 10 mL deionized water, was slowly dropped into the above system containing the cellulose network sample at 5 °C. The mixture was kept at 0 °C for 6 h to allow the in situ polymerization of the aniline monomer by diffusing APS into the cellulose network sample. After the in situ polymerization process, the sample was repeatedly rinsed with deionized water and anhydrous ethanol until the pH reached 7, and then dried in a freeze-dryer for 24 h. Narrow strips with widths of about 2.5 mm were cut off from four edges of the dried block to obtain the CPM composite sample with dimensions of 10 × 10 × 1 mm (L × W × H). To analyze the effect of MWCNT on the electrochemical performance of the composites, CP composites were also prepared using the same method without the infiltration of MWCNT.
2.3 Fabrication of the supercapacitor prototype based on CPM block
The PVA-based electrolyte was prepared by mixing H3PO4 (1 g), deionized water (10 mL), and polyvinyl alcohol (PVA) powder (1 g) and then heating the mixture at 85 °C with magnetic stirring until it became clear; the solution was then kept at 85 °C for another 1 h without stirring. Then the electrolyte was fully immersed into the CPM blocks by vacuum impregnation. Subsequently, MWCNT was coated on the surface of the CPM blocks and adhered to the composites with the electrolyte. Different MWCNT loadings of 0.6, 2.8, 5, and 7.2 mg cm−2 resulted in corresponding electrode samples called CPM0, CPM1, CPM2, and CPM3, respectively. Two carbon cloth collectors were adhered to the surfaces of the CPMX (i.e. CPM0, CPM1, CPM2, and CPM3) samples with the electrolyte to assemble various CPMX supercapacitors. The FSCs based on CPMX composites were obtained after three cycles of freezing-thawing treatment with 12 h of freezing at −20 °C and 3 h of thawing at room temperature in each cycle.2.4 Characterizations
| d = nλ/2sinθ | (2) |
| CrI = ((I002 − Iam)/I002) × 100% | (3) |
For the three-electrode tests, the area specific capacitance C (F cm−2) was calculated with eqn (4):
| C = (∫IdU)/(2vSU) | (4) |
The electrochemical properties of the as-prepared supercapacitors were also characterized with CV and GCD methods. CV and GCD measurements were conducted with a potential window from −0.6 V to 0.6 V. The scan rates for CV and GCD measurement were same as those for the three-electrode tests. EIS analysis was also performed with the same settings as those for the three-electrode tests. The area specific capacitance Cs (F cm−2), the gravimetric specific capacitance Cg (F g−1), and the bulk ionic conductivity σi (S cm−1) were calculated with eqn (5) and (6):
| Cs = (∫IdU)/(vSU) | (5) |
| Cg = 4 (IΔt)/(mU) | (6) |
| σi = d/(RS) | (7) |
| E = (1/2) × Cs × U2 | (8) |
| P = E/t | (9) |
3. Results and discussion
The construction of all-in-one FSCs was realized in this study with the help of the straightforward cellular structure of the cellulose network, the controllable partial deposition of PANI, and the integrated assembly with PVA-based gel electrolytes. As graphically presented in Fig. 1a, a free-standing cellulose network retaining the structure and porosity of the original wood was produced by the chemical fractionation of solid wood. The three-layer structure of CPM composites was subsequently constructed through the immersion of MWCNT and in situ partial growth of PANI followed by cutting the four edges of the composites. The electrolyte was fully injected into the CPM composites by vacuum impregnation, and then MWCNT was coated on the surface to obtain CPMX (X = 0, 1, 2, 3) composites. The CPMX-based FSCs were finally formed after introducing carbon cloth collectors and conducting three freezing-thawing cycles (Fig. 1a). The CPMX composites endowed the FSCs with an integrated all-in-one structure containing both electrodes and a separator (Fig. 1b). The two surface parts contained active materials (i.e., MWCNT and PANI) that can act as the electrodes as well as an intermediate part of the composites between the two electrodes, where there is no distribution of active materials; thus, it can be directly used as the separator. The as-prepared FSCs exhibited excellent flexibility, which allowed them to be wound around a finger and serve as a power source for wearable devices (Fig. 1c). Due to the extensive availability of wood, the CPMX-based FSCs possess scalable characteristics, and a scale-up sample of CPM composites with dimensions of 10 × 12 × 2 cm is presented in Fig. 1d. The CPM electrode materials that featured super flexibility can endure various forms of deformation such as bending, curling, twisting, and folding (Fig. 1e). Tensile tests were performed on the all-in-one FSCs to verify the adhesion strength between the current collector, MWCNT layer and CPM composites. The results showed that the all-in-one supercapacitor exhibited a promising tensile strength of 169.70 KPa (Fig. S1, ESI†). Moreover, the present preparation strategies of the CPMX electrodes as well as the corresponding FSCs showed excellent applicability on other cellulose materials commonly used in daily life, such as cotton cloth and cellulose paper. The CPM composites based on cotton cloth and cellulose paper can also produce a three-layer structure by the in situ partial growth of PANI (Fig. 1f), which can be utilized as an electrode material for the all-in-one FSCs.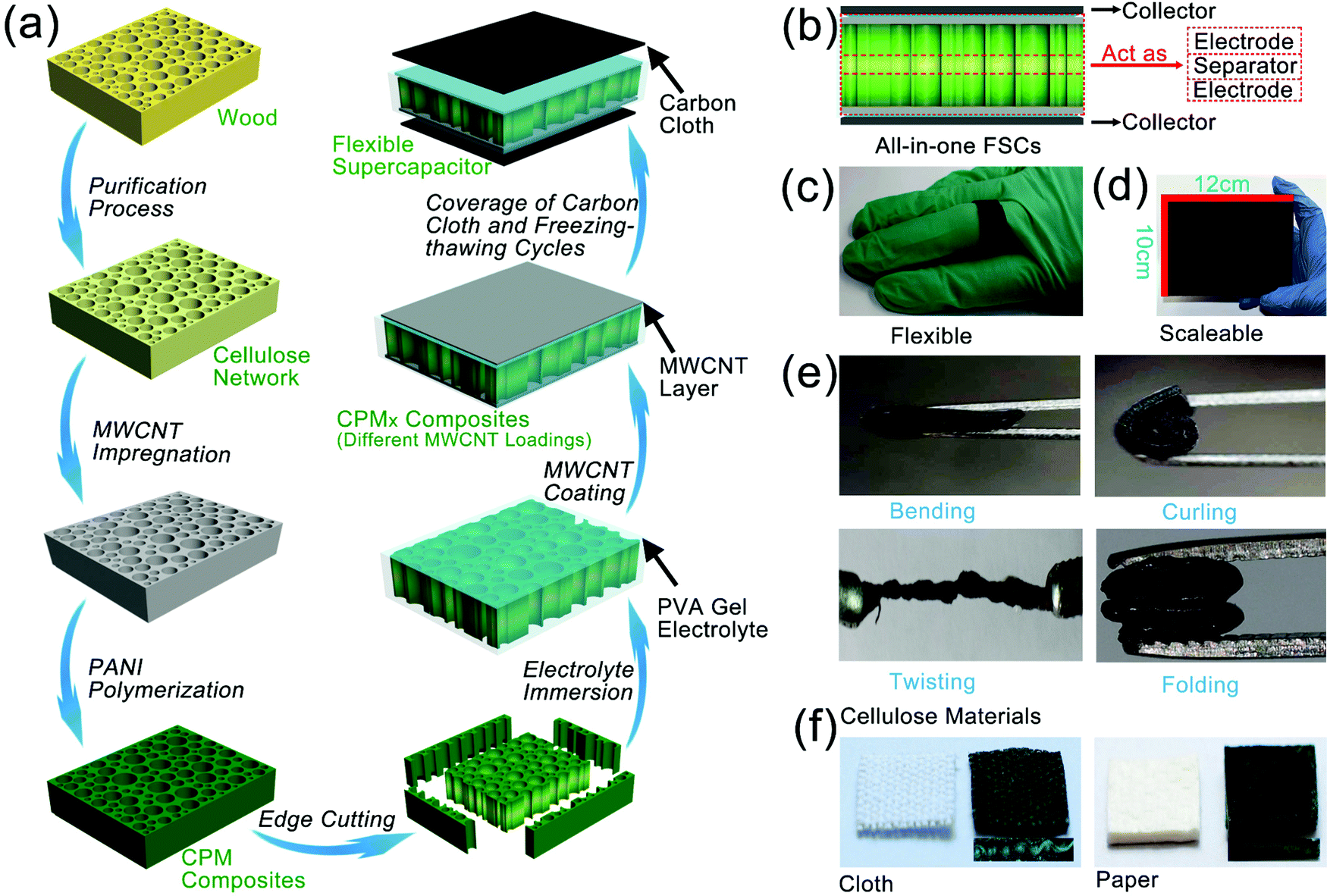 | ||
| Fig. 1 Construction procedures and characteristics of the CPMX composites and the corresponding FSCs: (a) schematic diagram of the preparation process of the all-in-one FSCs, (b) the structure of the all-in-one FSCs, (c) flexibility of the CPMX-based FSC, (d) scalable CPM composites, (e) various deformation forms of CPMX composites, and (f) CPM composites based on different cellulose materials. | ||
Fig. 2 demonstrates the morphologies and characterizations of products in various stages of the preparation process of the CPM composites. A white cellulose network block with high porosity (Fig. 2a) was firstly produced by the chemical fractionation of the solid wood with alkali and sodium chlorite, which can dissolve hemicellulose and decompose lignin into water-soluble compositions through the reactions of phenolic and alcoholic hydroxyls.24 The fractionation process can enhance the nanostructure of the cellulose network by removing most of the hemicellulose and lignin distributed between the cellulose microfibrils, which was confirmed by the disappearance of the characteristic peaks of hemicellulose and lignin in the Fourier-transform infrared spectra (Fig. S2, ESI†). Cellulose microfibril bundles were observed on the cross-section of the cellulose network (Fig. 2b). The process can also deconstruct the structures of pit pairs on the wood cell wall to produce micron-scale holes connecting the adjacent wood cells (Fig. S2, ESI†). The enhanced hierarchical porous structure makes the cellulose network an excellent matrix for further compounding and modification, in order to produce electrode materials and energy storage devices.17 It is reported that the introduction of MWCNT can reduce the ion diffusion impedance of PANI by forming a continuous phase for charge transfer, thereby improving the capacity, stability, and functional characteristics of the PANI-based electrode.25 In this study, MWCNT was firstly infiltrated into the cellulose network before the in situ polymerization of PANI to construct a cross network with PANI in the next step. SEM results showed a gradient distribution of MWCNT on the inner wall of the wood cell lumen, and the MWCNT distribution gradually decreased from the surface to the center (Fig. 2e and Fig. S3a–c, ESI†). TEM images illustrate the multiple-wall structure of MWCNT (Fig. 2c). It should be noted that a low mass loading of MWCNT (0.6 mg cm−2) was employed to avoid a possible short circuit when the composites were used as the integrated electrodes for the FSCs (Fig. S2, ESI†). The as-prepared cellulose network/MWCNT complex was then impregnated with aniline monomers, and the in situ deposition of PANI was realized through the chemical oxidative polymerization of aniline.26 Extremely dense distribution of PANI was observed on the cross-section of the CPM composites (Fig. 2f). Additionally, inside the cell lumen, PANI was found to be intensively deposited on the MWCNT surface to form a cross-network and anchor the MWCNT to the inner cell wall (Fig. 2g). Interestingly, the PANI deposition on the inner cell wall presented a gradual change from the surface to the interior that can be seen from the longitudinal section of the sample (Fig. 2a), and the mass loading and distribution of PANI in the CPM composites can be tuned by varying the reaction time and PA concentration (Fig. S2, ESI†). Thus, a three-layer structure of the CPM composites can be formed through regulating the mass loading of PANI (Fig. S2, ESI†); the corresponding weight percent gain for PANI ranged from 41.0% to 80.4%. The layered structure enabled the subsequent preparation of integrated electrode materials and the construction of all-in-one FSCs.
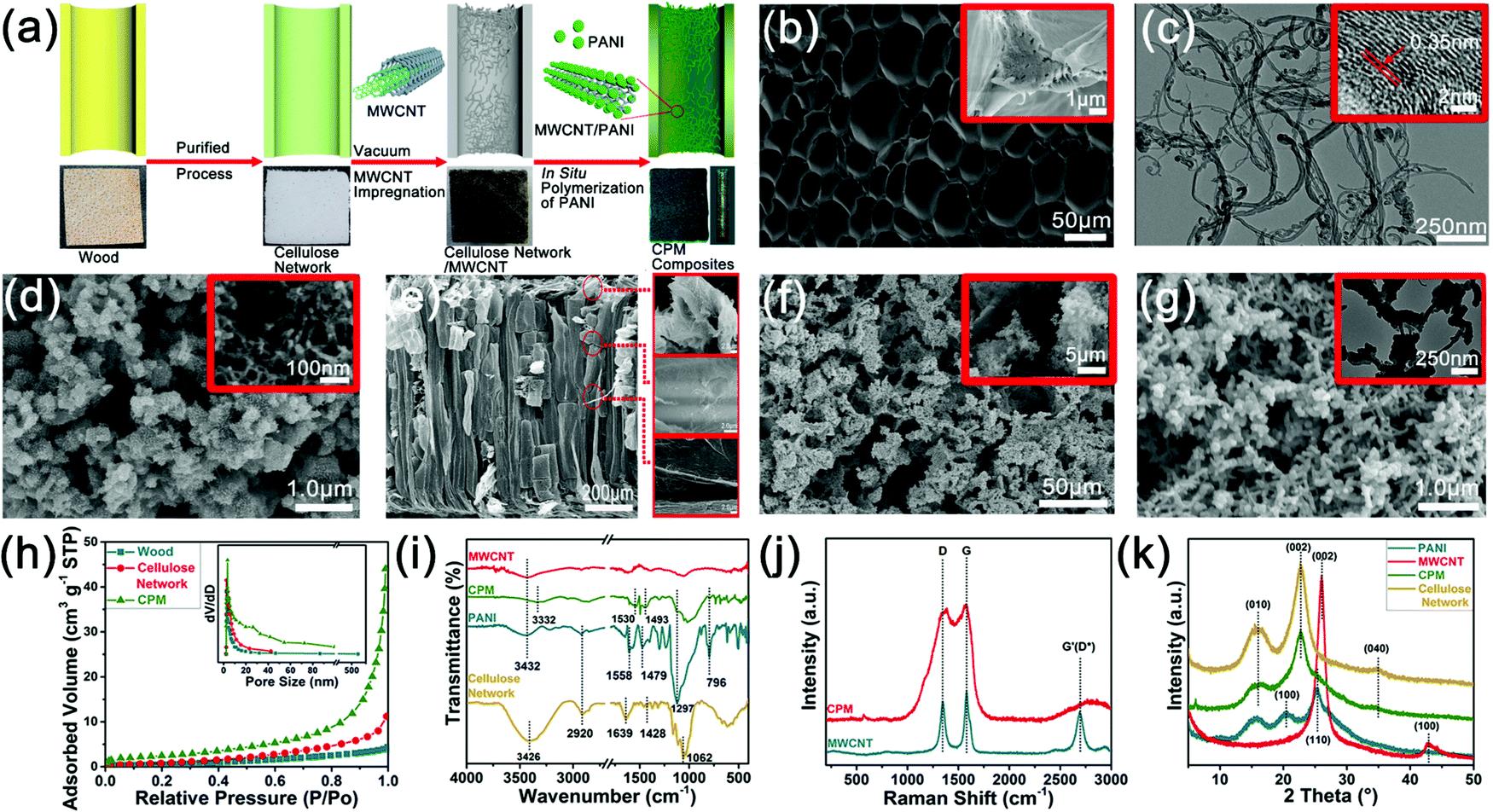 | ||
| Fig. 2 The morphologies and characterizations of various products during the preparation process of the CPM composites: (a) schematic diagrams of the wood cell lumen during the preparation process of CPM composites and the photographs of the corresponding samples, (b) SEM images for the cellulose network, (c) TEM images for MWCNT, (d) FESEM images for PANI, (e) SEM images for the longitudinal section of the cellulose network/MWCNT sample, (f) SEM images for the cross section (surface) of CPM composites, (g) SEM and TEM images for the MWCNT/PANI complex in CPM composites, (h) nitrogen adsorption–desorption curve and the pore size distribution of wood, cellulose network, and CPM composites, (i) FTIR spectra of the MWCNT, cellulose network, PANI, and CPM composites, (j) Raman spectra of MWCNT, PANI, and CPM composites, and (k) X-ray diffraction patterns of the MWCNT, cellulose network, PANI, and CPM samples. | ||
The influence of the fractionation and in situ polymerization process on the porosity of the wood matrix was investigated by comparing the BET surface area and pore volume of the wood, cellulose network, and CPM samples (Fig. 2h). The BET surface area of the cellulose network was 3.382 m2 g−1, which was slightly higher than 1.817 m2 g−1 of wood, indicating that the fractionation process can remove most of the hemicelluloses and lignin fractions of wood and enhance its porosity and microstructure.27 By comparison, the CPM composite exhibited a much higher BET surface area (8.773 m2 g−1) than that of the cellulose network and wood samples. Moreover, the comparison of pore size distribution curves (Fig. 2h) illustrated that the CPM composite possessed a much higher pore volume (0.065 cm3 g−1) relative to that of the cellulose network (0.017 cm3 g−1) and wood (0.006 cm3 g−1). Compared with cellulose network and wood, the pore size in the CPM composite showed a wider distribution within the range of micropore and mesoporous structures (Fig. 2h). These results suggest that the cellulose network had abundant nanoscale pore distribution despite its specific surface area being small. The introduction of PANI and MWCNT enriched the pore structure of the CPM composites, especially the micropores and mesopores.
The interactions of cellulose, PANI, and MWCNT in the CPM composites were analyzed by FTIR, Raman spectra, and XRD analyses. Most of the characteristic peaks for cellulose, PANI, and MWCNT were present in the FTIR spectra of the CPM composites (Fig. 2i). Compared with three respective spectra, red shift and a decreased intensity of the O–H stretching absorption peak can be observed in the spectrum of the CPM (Fig. 2i), which indicated that the incorporation of PANI and MWCNT partly weakened the native hydrogen bonding of the cellulose, and that these three components formed a new hydrogen bonding network in the CPM composites.28 Consistent with the above deduction, XRD patterns confirmed the decrease of cellulose crystallinity with the introduction of PANI and MWCNT; the relative crystallinity of the cellulose decreased from 60.23% for the cellulose network to 54.97% for the CPM composites (Fig. 2k). The decrease in crystallinity may be mainly attributed to the infiltration of aniline monomers into the cellulose microstructure and the subsequent in situ formation of PANI.28 The Raman spectra of the CPM composites revealed high similarity with those of PANI and MWCNT, indicating the successful in situ polymerization of PANI around MWCNT (Fig. 2j). The Raman spectra also confirmed that the incorporation of PANI had no obvious influence on the multiwall structure of MWCNT.
During the preparation process of the CPM composites, MWCNT was infiltrated into the cellulose network to form an ionic conductive complex with PANI, and an improvement on the electrochemical performance was expected to be achieved. To test this hypothesis, two kinds of composites were prepared based on the cellulose network and PANI for comparison: CPM composites with MWCNT and cellulose network/PANI (CP) composites without MWCNT. The effects of the incorporation of MWCNT on the electrochemical performance of the electrodes were evaluated by three-electrode tests with CV, GCD, and electrochemical impedance spectroscopy methods (Fig. 3). The cyclic voltammetry curves (Fig. 3a–c) at different scan rates confirmed the evident capacitive nature of the CP and CPM composite electrodes. Two pairs of redox peaks for PANI can be observed in the CV curves at a scanning rate of 2 mV s−1 (Fig. 3c), which should be attributed to the embedding and stripping of H+ from H2SO4 electrolytes in PANI, which was also the main source of capacitance.29,30 The cellular structures in the cellulose network can act as microreactors for the in situ preparation of PANI along the inner wall of the wood cell lumen. Considering the fact that the H+ doping degree of PANI mainly determined its conductivity,31 the ion may primarily transmit along the cell lumen in this case. Thus, the microporous structure formed by the cellulose network and PANI can provide ion channels for electrolyte penetration and ion transmission when the composites are used as electrode materials. Compared to the CP electrode, a more visible curvature of the CV curve for the CMP electrode implied its stronger pseudocapacitive feature, and the comparison of the integral areas demonstrated the much higher capacitance of the CPM electrode. The CPM electrode provided a promising areal specific capacitance of 0.27 F cm−2 at a scan rate of 2 mV s−1 in the three-electrode test, while the CP electrode resulted in an areal specific capacitance of 0.14 F cm−2 at the same scan rate; this was mainly attributed to the incorporation of MWCNT in the CPM electrode, which increases the specific surface area of the sample and causes the chain of PANI molecules linked together to have no contact with each other as well as the corresponding enhanced electron transfer and electrochemical reactivity.29 The Nyquist diagram (Fig. 3d) of the CPM electrode showed a lower interface resistance than that of the CP electrode (2.66 Ohm vs. 3.00 Ohm), indicating the higher electronic/ion transmission rate for the CPM electrode.30 The bulk ionic conductivities of the CP and CPM electrodes were also determined with the EIS method (Fig. 3d). The CPM electrode showed a slightly higher ionic conductivity of 3.76 × 10−2 S cm−1 than that of the CP electrode (3.33 × 10−2 S cm−1), which was consistent with the above interfacial resistance results. The galvanostatic charge−discharge curves for the CP and CPM electrodes at different current densities (Fig. 3e and f) exhibited approximately symmetrical triangular shapes, which reflected their promising conductivity properties. The CPM electrode exhibited much better gravimetric specific capacitances (186.58 F g−1vs. 110.52 F g−1) than that of the CP electrode. In addition, the Coulombic efficiency for the CPM electrode reached 93.9%, which confirmed its high electronic/ion transmission rate.30 The promising conductivity and specific capacitance of the as-prepared CPM composite makes it an important candidate for electrode materials. However, it is also suitable to act as the integrated electrode material for all-in-one supercapacitors when considering its outstanding advantage of possessing an integrated structure.
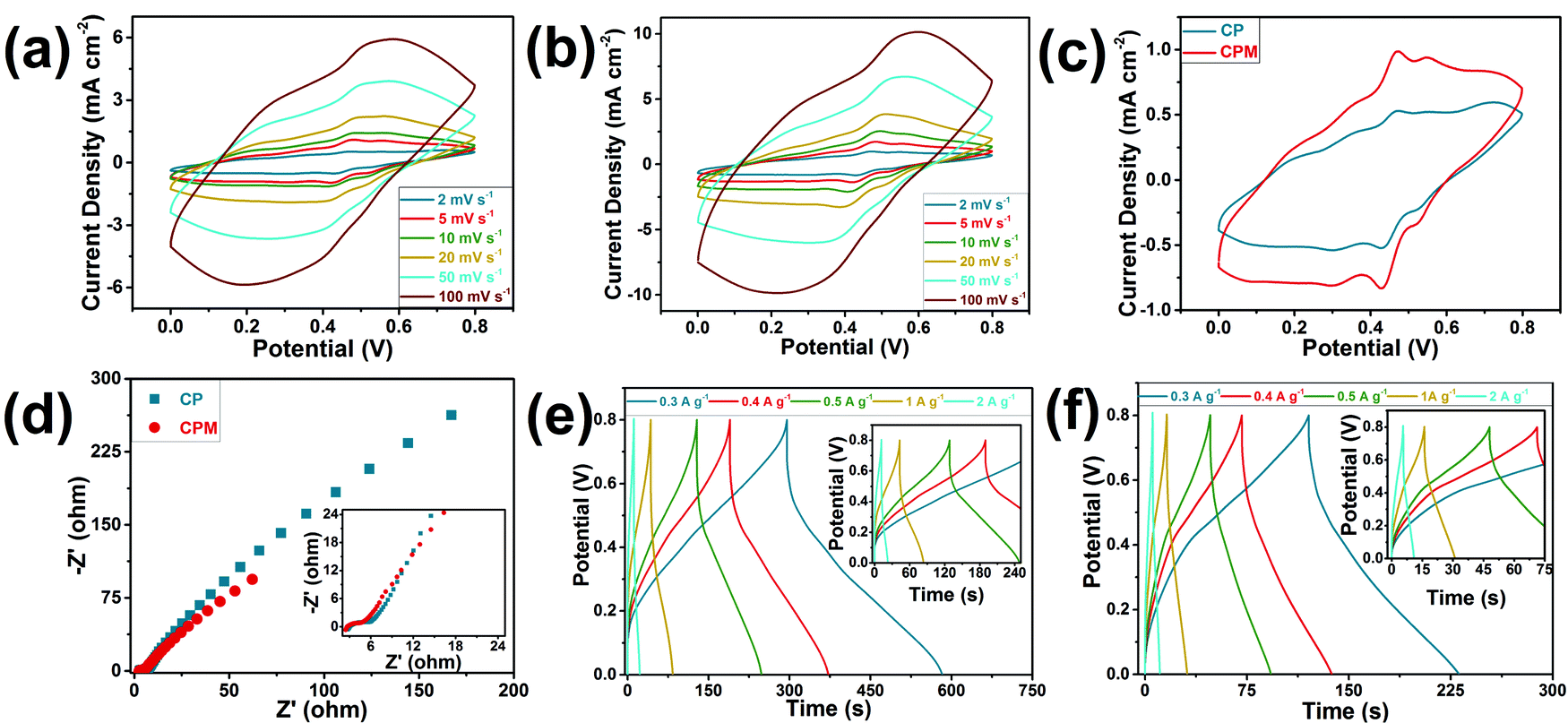 | ||
| Fig. 3 Electrochemical performance of the wood-derived electrode materials in a three-electrode test. CV curves for (a) CP and (b) CPM electrodes under various sweep rates. (c) CV curves of the CP and CPM electrodes at a sweep rate of 2 mV s−1. (d) Nyquist diagrams for CP and CPM. GCD curves for (e) CP and (f) CPM electrodes under various current densities. | ||
Given the integrated structure of the CPM composites, we proposed that an integrated all-in-one FSC device can be obtained based on a single CPM block. Corresponding to the electrode–separator–electrode structure of the traditional supercapacitor, two surface parts of the CPM block acted as the electrodes in the integrated all-in-one FSC, and the middle part acted as the separator (Fig. 1b). Additionally, the CPM composites showed uneven and porous surfaces on the cross sections (Fig. 2f). In order to obtain flat surfaces of the CPM composite electrodes and improve their electrochemical energy storage performance, different loadings of MWCNT were coated on the surfaces of the CPM composites and adhered to the surface with electrolytes to form the CPMX (X = 0, 1, 2, and 3) integrated electrode materials. The integrated FSC was then assembled by the coverage of carbon clothes on a single CPMX block as the collectors and the subsequent freezing-thawing cycles of PVA-based gel electrolytes. The freezing-thawing treatment improved the physical cross-linking degree of the PVA-based gel electrolytes,32 and the gel electrolytes were thus converted into ultraflexible solid materials, resulting in an excellent flexibility of the as-prepared FSCs (Fig. 1b).
Fig. 4 presents the electrochemical properties of the integrated all-in-one FSCs based on the CPMX composites. The CV curves of all the FSCs exhibited a rectangular shape (Fig. 4a), indicating their promising capacitive behavior. The characteristic redox peaks of PANI can also be observed on the CV diagrams of all the FSCs at a scan rate 2 mV s−1.37 The specific areal capacitance of the FSC based on CPM0 (i.e. without the coating of MWCNT) was 0.12 F cm−2 at a sweep rate of 2 mV s−1. The FSCs based on the CPMX composites with the coating of MWCNT (CPM1, CPM2, and CPM3) showed much higher specific areal capacitance of up to 0.71 F cm−2 (CPM2); the capacitance increased first and then decreased as the mass loading of MWCNT increased from 0 mg cm−2 to 7.2 mg cm−2 (Fig. 4c). During the assembly process of the FSCs, MWCNT was coated on the surface after the complete infiltration of the electrolyte. It was adhered by the gel electrolyte and fixed on the composite surfaces through the solid conversion of the electrolyte with the freezing-thawing treatment to ensure full contact between the coated MWCNT and the CPM electrode. Appropriate loading of the MWCNT can be stably stacked in the holes on the CPM surfaces, while a further increase of the MWCNT loading may lead to a loose structure of the electrode materials (Fig. S3d–f, ESI†) and thus insufficient contact in the electrode material and electrolyte.38 Consequently, the MWCNT loading of 7.2 mg cm−2 (CPM3) resulted in a lower specific areal capacitance of 0.65 F cm−2 than that for the loading of 5.0 mg cm−2 (CPM2). Additionally, the CV curves of the FSCs at various scan rates indicated their ideal capacitive behaviors (Fig. S4, ESI†). The GCD curves of the FSCs with different MWCNT loadings (CPM0,1,2,3) were also recorded under various current densities to evaluate their charge–discharge performance (Fig. S4, ESI†). The GCD curves for FSCs under the current density of 0.3 A g−1 exhibited typical symmetrical triangle-shape charge–discharge profiles, indicating the fast charge/discharge properties of the as-prepared FSCs (Fig. 4d). Similar to the CV results, the GCD curves confirmed that MWCNT loading played a crucial role on the capacitance performance of the FSCs. A maximum gravimetric specific capacitance of 384.69 F g−1 was obtained with the sample based on CPM1 composites under a current density of 0.3 A g−1. These results also demonstrated that an appropriate MWCNT loading was beneficial to the capacitance performance of the FSCs. However, the optimal MWCNT loading as determined by the GCD results was different from that determined by thee CV results, and thus the appropriate MWCNT loading can be selected according to different specific capacitance requirements (areal, gravimetric, or volumetric). The capacitance performance of FSCs showed a certain degree of stability under various current densities. For example, the gravimetric specific capacitance for CPM1 remained at 240.56 F g−1 under a current density of 2 A g−1 (Fig. 4e). Notably, GCD curves under various current densities showed no obvious IR drop, and the Coulombic efficiency remained at approximately 100% during the charge–discharge cycles (Fig. S4, ESI†). A comparative study confirmed that the electrodes prepared by soaking the electrolyte and subsequently applying MWCNT and PANI showed much lower area and weight specific capacitances than that of the above-mentioned sample (Fig. S5, ESI†). Therefore, it is necessary to coat the wooden substrate with MWCNT and PANI before electrolyte immersion.
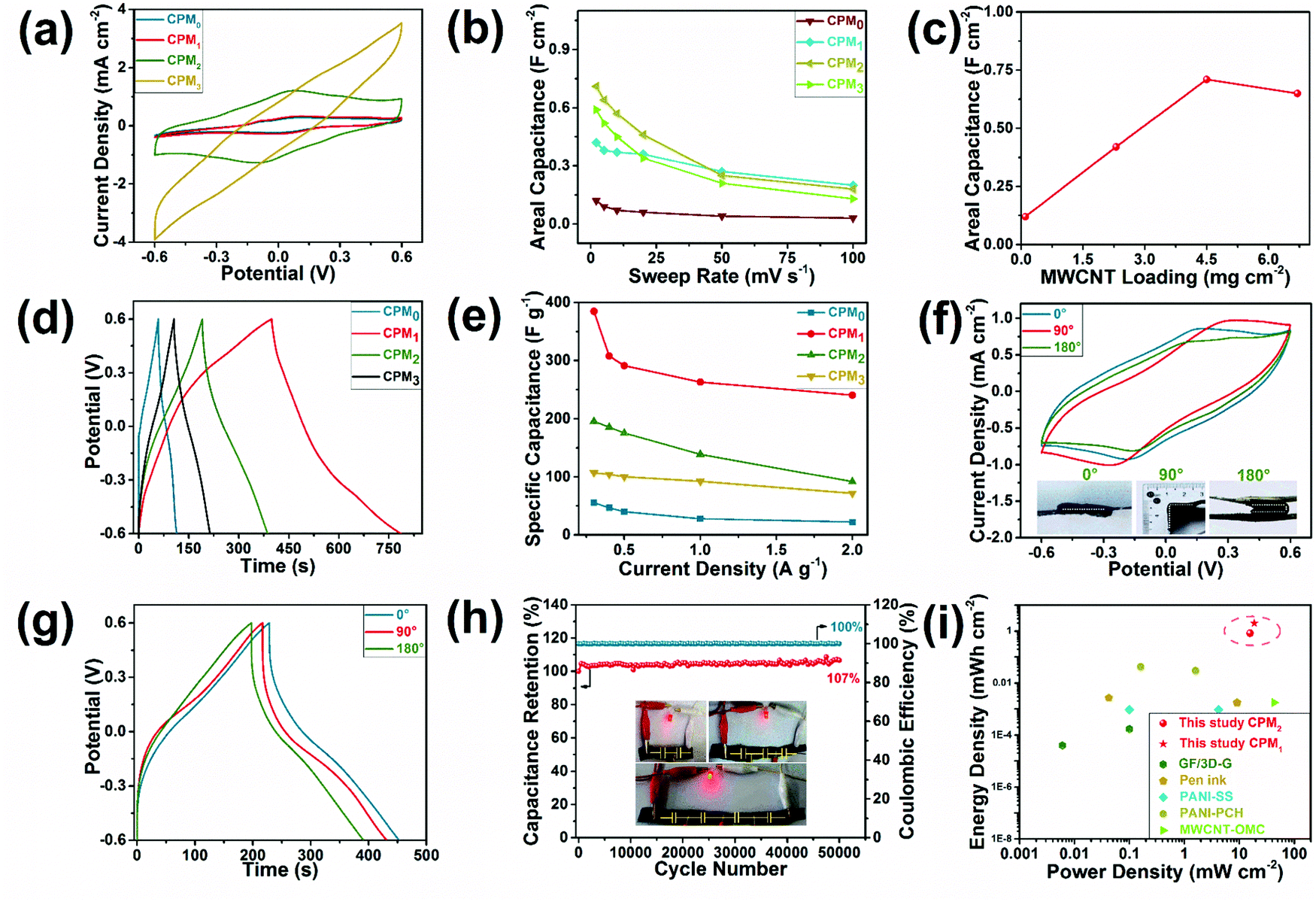 | ||
| Fig. 4 Electrochemical performance of the integrated FSCs based on single CPM blocks with MWCNT of various weights: (a) CV curves of the CPMX-based FSCs at 2 mV s−1, (b) areal specific capacitance at different sweep rates, (c) specific areal capacitance of the FSCs with various mass loadings of MWCNT, (d) GCD curves of the FSCs at 0.3 A g−1, (e) gravimetric specific capacitance under different current densities, (f) CV curves for FSCs folded at different angles at 2 mV s−1, (g) GCD curves for FSCs bent in different angles under 0.3 A g−1, (h) capacitance retention and Coulombic efficiency of the CPM2-based FSC in the cyclic charge and discharge test under 3 A g−1 (inlet: LED lighting with the FSCs), and (i) comparison of Ragone plots of the present and reported supercapacitor devices (GF/3D-G,6 Pen ink,33 PANI-SS,34 PANI-PCH,35 and MWCNT-OMC36). | ||
Fig. 4f and g illustrates the capacitance performance of the FSCs device under various folding states. The CV curves of the FSCs with different folding angles showed limited changes at a scanning rate of 2 mV s−1. The specific areal capacitance of the FSCs based on CPM1 composites were 0.42, 0.39, and 0.34 F cm−2 for the folding angles of 0°, 90°, and 180°, respectively. The CV curves show a rectangular shape and notable redox peaks, and the capacitance retention was up to 80.95% for the folding angle of 180°. The GCD curves of the FSCs in various folding states under a current density of 0.3 A g−1 exhibited similar triangular shapes, indicating that the FSCs showed fast charging and discharging performance under the folding states (Fig. 4g). Excellent flexibility and high capacitance retention under folding states provide great possibilities for the application of CPMX-based FSCs in wearable devices.
The consecutive charge–discharge cycles (Fig. 4h) showed the ultrastable cycle performance of the CPM2-based FSCs, and the specific capacitance retention reached 106.68% after 50000 cycles under a current density of 3 A g−1. Moreover, even at an elevated current density (5 A g−1), the specific capacitance retention reached 96.41% (Fig. S4g, ESI†), although there was a decay of capacitance at the early cycling stage, which may result from the partial pulverization of the thin, top PANI layer and uneven CNT coating.23 The slight increase of capacitance at the final stage may result from the effect of a mesoporous structure in the MWCNT and cellulose network on the charge transmission and storage in the FSCs,39 as the dominant mesoporous distribution in CPM composites was confirmed by the BET results (Fig. 2h). In addition, the activation of the FSCs via electrochemical cycles could increase the contact area between the electrode material and electrolyte, which could also contribute to the improvement of the specific capacitance.40 In addition, the series connection circuit of several FSC devices showed realistic applications, such as lighting up a light-emitting diode (LED) light (Fig. 4h). Although the charging time was slightly extended, the charging voltage of the CPM2-based FSCs after 50000 cycles reached approximately 2.7 V at a current density of 5 A g−1, thereby lighting up a LED successfully (Fig. S4g, ESI†).
The FSC device based on CPM1 and CPM2 showed a promising energy density of 2.01/0.83 mW h cm−2 at a power density of 18.77/15.67 mW cm−2 (at a scan rate of 2 mV s−1). The comparison on Ragone plots of the present and reported FSC prototypes confirmed the extremely high energy density and power density of the as-prepared FSCs (Fig. 4i). This may be attributed to the abundant porous structure of the cellulose network, which can provide a large place for the deposition of the active substances. The effective deposition of the active substances thus greatly increased the capacitance and energy density per unit area.41 Furthermore, the integrated structure was conducive to charge transfer in the supercapacitors and contributed to the improvement of energy storage performance in the present study.23
Fig. 5 summarizes the different structures of the conventional supercapacitors and the present integrated all-in-one FSC, the proposed working mechanism of the present FSCs, and the electrochemical performance of the FSCs with various structural prototypes. Generally, two electrodes and a separator between them are needed to assemble a conventional supercapacitor, while only an integrated electrode material was necessary to construct the integrated FSCs in this study (Fig. 5a). The present strategy eliminated the complicated assembly steps of the conventional supercapacitors and reduced the contact resistance between interfaces. Additionally, the working mechanism of the integrated all-in-one FSC was proposed as a hybrid supercapacitor combined electrical double-layer capacitor with faradaic pseudocapacitor according to the literature, as shown in Fig. 5b.42 During the charging process, the external circuit resulted in the directional movement of electrons and produced a potential difference between the electrodes. In order to maintain the overall electrical neutrality, the anions and cations in the electrolyte moved to the anode and cathode, respectively, and thus a dense double electric layer was formed in the interface between the MWCNT and electrolyte.40,42 Meanwhile, a redox reaction of PANI occurred by losing or gaining electrons, and the resulting directional movement of the anions and cations in the electrolyte produced faradaic pseudocapacitance.43,44 The superposition of these two capacitances increased the charge storage capacity of the FSC device. In the discharge process, electrons flowed from the external circuit to the anode, which made the potential difference between the anode and cathode disappear, and the anions and cations on the surface of the electrode material broke away from the attraction of their respective electrodes and migrated back to the electrolyte, thus returning to the previous state of irregular distribution.42–44 Additionally, the ion transport capacity of the pure PVA gel electrolyte, PVA gel electrolyte/cellulose network, and PVA gel electrolyte/cellulose paper separator was evaluated by comparing their ionic conductivities with an EIS method in a three-electrode system. The sample of PVA gel electrolyte/cellulose network showed a much higher ionic conductivity (1.85 × 10−2 S cm−1) than the pure PVA gel electrolyte (6.33 × 10−3 S cm−1) and the PVA gel electrolyte/cellulose paper separator (3.45 × 10−3 S cm−1) (Fig. S6, ESI†). This may be due to the directional porous structure of the cellulose network. The PVA gel electrolyte possessed a relatively compact structure, and the existence of a cellulose network in the PVA gel electrolyte promoted ion transport by providing directional channels for the ion transport.41 These results confirmed the above inference on the promotion effect of the cellulose network on the ion transport in the FSCs.
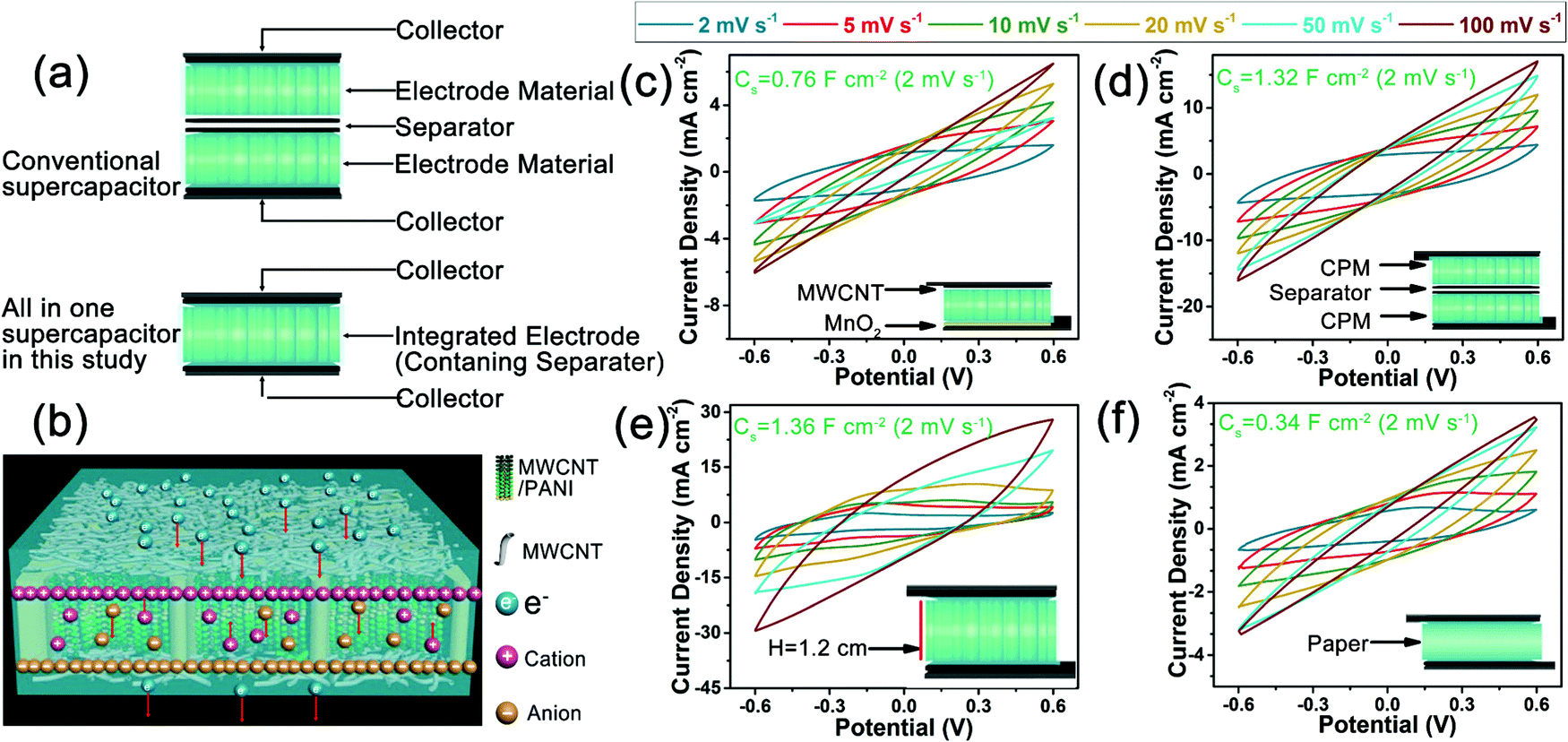 | ||
| Fig. 5 The structural features, working mechanism, and expansibility of the presented FSCs: (a) different structures of conventional supercapacitors and FSCs in this study, (b) proposed working mechanism of the FSCs, and CV curves under various scan rates for (c) asymmetric FSC, (d) conventional FSC, (e) ultra-thick FSC, and (f) paper based FSC. | ||
Various structural prototypes of FSCs were also studied to investigate the applicability of the present preparation strategy of FSCs. Manganese dioxide (MnO2), as an alternative to MWCNT, and was coated on one side of the CPM composites to assemble asymmetric FSCs. The CV curve at a scan rate of 2 mV s−1 resulted in a higher specific areal capacitance of 0.76 F cm−2 than that of the corresponding symmetric FSC based on CPM2 composites (Fig. 5c). The introduction of MnO2 and the asymmetric structure enabled the excellent electrochemical performance of the FSCs, which provides a new idea for designing novel biomass-based supercapacitors with excellent electrochemical performance.45 Moreover, we also prepared a conventional-structure FSC device with two CPM1 electrodes and a commercial cellulose separator (Fig. 5d), and, compared with that of the all-in-one CPM1 FSC, a much higher specific areal capacitance of 1.32 F cm−2 was achieved through the CV test at a scan rate of 2 mV s−1; this was mainly attributed to the much higher mass loading (up to 19.18 mg cm−2) of active substances (MWCNT and PANI) within unit area due to the utilization of two CPM electrodes. Additionally, the maximum volumetric capacitance of the FSCs based on two pieces of CPM2 was calculated based on the thickness of the whole device (2.2 mm, including the collectors), which was much less than the all-in-one FSC (3.45 F cm−3vs. 5.92 F cm−3) based on one piece of CPM2 (1.2 mm) in this study. The results indicated the integrated structure had the advantages of improving the energy storage performance of FSCs when compared with the conventional structure. An ultra-thick FSC device assembled with a 1.2 cm-thick CPM1 block also showed excellent electrochemical performance (Fig. 5e). The CV curve of the ultra-thick FSC at the scan rate of 2 mV s−1 exhibited an ultrahigh specific areal capacitance of 1.36 F cm−2. The ultrahigh mass loading of active substances for the ultra-thick FSC led to the excellent electrochemical performance, and the porous structure of the cellulose network endowed the as-prepared FSCs with an outstanding tunability on the mass loading of active substances. In addition to the wooden cellulose network, the commonly used cellulose paper was used as an electrode supporting material to prepare the all-in-one FSCs (Fig. 5f). The paper-based FSC gave a promising specific areal capacitance of 0.34 F cm−2, which was slightly lower than that of the FSC based on CPM1. This may be attributed to the relatively compact structure and the lack of directional channels in the cellulose paper. Based on the above analyses, it can be found that the present strategy for the assembly of FSCs showed great extensibility on the electrode material and supercapacitor structure.
The present results demonstrated that the CPM composite can be used as an important electrode material for high-efficiency and ultrastable energy storage applications. Additionally, it should be noted that the hierarchical porous structure of the cellulose network provided an extremely wide space for its performance improvement, which can be used as a platform for the development of green and sustainable high-performance electrode materials.
4. Conclusion
This paper presents a new type of integrated FSC with an integrated CPMX composite block acting as both the electrodes and separator. The combination of the straightforward hierarchical structure of the wood cellulose network with high-electrochemical-active PANI and MWCNT in the CPMX composites endowed the FSC with a unique structure for ion/electron transmission and an ideal electrochemical energy storage performance; the specific areal capacitance of the as-prepared FSCs based on CPM2 reached up to 0.71 F cm−2. The as-prepared FSCs also demonstrated ultrastable cycle performance, and a specific capacitance retention of 106.68% was achieved after 50000 cycles at a current density of 3 A g−1, which showed a significant advantage over reported FSCs. The present study provides a new strategy for the design and construction of high-performance and ultrastable FSCs using renewable natural resources for wearable electronics applications.
Author contributions
Shaoqiu Ke: conceptualization, methodology, writing – original draft, validation, investigation; Di Xie: conceptualization, methodology, writing – original draft, validation, investigation; Kai Zhang: methodology, investigation; Fangchao Cheng: conceptualization, writing – origin draft, writing – review & editing, supervision, funding acquisition; Yiqiang Wu: conceptualization, writing – review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the support of the Project Funded by China Postdoctoral Science Foundation (2017M620361), the Natural Science Foundation of Guangxi (2020GXNSFAA297028; 2016GXNSFCA380025), and the Scientific Research Foundation of Guangxi University (XGZ150513).References
- C. Yuan, L. Yang, L. Hou, L. Shen, X. Zhang and X. W. Lou, Energy Environ. Sci., 2012, 5, 7883–7887 RSC.
- D. Wei, S. J. Wakeham, T. W. Ng, M. J. Thwaites, H. Brown and P. Beecher, Electrochem. Commun., 2009, 11, 2285–2287 CrossRef CAS.
- L. Nyholm, G. Nyström, A. Mihranyan and M. Strømme, Adv. Mater., 2011, 23, 3751–3769 CAS.
- C. Shao, T. Xu, J. Gao, Y. Liang, Y. Zhao and L. Qu, Nanoscale, 2017, 9, 12324–12329 RSC.
- R. Liu, L. Ma, S. Huang, J. Mei, E. Li and G. Yuan, J. Phys. Chem. C, 2016, 120, 28480–28488 CrossRef CAS.
- J. Ren, W. Bai, G. Guan, Y. Zhang and H. Peng, Adv. Mater., 2013, 25, 5965–5970 CrossRef CAS PubMed.
- J. Wang, L. Dong, C. Xu, D. Ren, X. Ma and F. Kang, ACS Appl. Mater. Interfaces, 2018, 10, 10851–10859 CrossRef CAS PubMed.
- A. Sumboja, C. Y. Foo, X. Wang and P. S. Lee, Adv. Mater., 2013, 25, 2809–2815 CrossRef CAS PubMed.
- X. Lu, Y. Zeng, M. Yu, T. Zhai, C. Liang, S. Xie, M.-S. Balogun and Y. Tong, Adv. Mater., 2014, 26, 3148–3155 CrossRef CAS PubMed.
- Y. Zhang, L. Zhang, K. Cui, S. Ge, X. Cheng, M. Yan, J. Yu and H. Liu, Adv. Mater., 2018, 30, 1801588 CrossRef PubMed.
- M. M. Pérez-Madrigal, M. G. Edo and C. Alemán, Green Chem., 2016, 18, 5930–5956 RSC.
- H.-P. Cong, X.-C. Ren, P. Wang and S.-H. Yu, Energy Environ. Sci., 2013, 6, 1185–1191 RSC.
- Z. Li, Y. Mi, X. Liu, S. Liu, S. Yang and J. Wang, J. Mater. Chem., 2011, 21, 14706–14711 RSC.
- A. R. de Arellano-Lopez, J. Martinez-Fernandez, P. Gonzalez, C. Dominguez, V. Fernandez-Quero and M. Singh, Int. J. Appl. Ceram. Technol., 2004, 1, 56–67 CrossRef CAS.
- H. Song, S. Xu, Y. Li, J. Dai, A. Gong, M. Zhu, C. Zhu, C. Chen, Y. Chen, Y. Yao, B. Liu, J. Song, G. Pastel and L. Hu, Adv. Energy Mater., 2018, 8, 1701203 CrossRef.
- F. Jiang, T. Li, Y. Li, Y. Zhang, A. Gong, J. Dai, E. Hitz, W. Luo and L. Hu, Adv. Mater., 2018, 30, 1703453 CrossRef PubMed.
- S. Ke, T. Ouyang, K. Zhang, Y. Nong, Y. Mo, Q. Mo, Y. Wei and F. Cheng, Macromol. Mater. Eng., 2019, 304, 1900112 CrossRef.
- H. Gao and K. Lian, RSC Adv., 2014, 4, 33091–33113 RSC.
- N. A. Choudhury, S. Sampath and A. K. Shukla, Energy Environ. Sci., 2009, 2, 55–67 RSC.
- M. Kaempgen, C. K. Chan, J. Ma, Y. Cui and G. Gruner, Nano Lett., 2009, 9, 1872–1876 CrossRef CAS PubMed.
- K. Chen, J. Liu, X. Yang and D. Zhang, Mater. Sci. Eng., C, 2017, 78, 520–529 CrossRef CAS PubMed.
- F. Béguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef PubMed.
- K. Wang, X. Zhang, C. Li, X. Sun, Q. Meng, Y. Ma and Z. Wei, Adv. Mater., 2015, 27, 7451–7457 CrossRef CAS PubMed.
- M. Zhu, J. Song, T. Li, A. Gong, Y. Wang, J. Dai, Y. Yao, W. Luo, D. Henderson and L. Hu, Adv. Mater., 2016, 28, 5181–5187 CrossRef CAS PubMed.
- S. X. Xiong, X. K. Zhang, R. Wang, Y. Z. Lu, H. F. Li, J. Liu, S. Li, Z. Qiu, B. H. Wu, J. Chu, X. Q. Wang, R. L. Zhang, M. Gong and Z. M. Chen, J. Polym. Res., 2019, 26, 90 CrossRef.
- T.-M. Wu and Y.-W. Lin, Polymer, 2006, 47, 3576–3582 CrossRef CAS.
- D. Bondeson, A. Mathew and K. Oksman, Cellulose, 2006, 13, 171–180 CrossRef CAS.
- D. Xu, X. Xiao, J. Cai, J. Zhou and L. Zhang, J. Mater. Chem. A, 2015, 3, 16424–16429 RSC.
- T. Liu, L. Finn, M. Yu, H. Wang, T. Zhai, X. Lu, Y. Tong and Y. Li, Nano Lett., 2014, 14, 2522–2527 CrossRef CAS PubMed.
- D. Ge, L. Yang, L. Fan, C. Zhang, X. Xiao, Y. Gogotsi and S. Yang, Nano Energy, 2015, 11, 568–578 CrossRef CAS.
- J.-C. Chiang and A. G. MacDiarmid, Synth. Met., 1986, 13, 193–205 CrossRef CAS.
- H. Huang, J. Yao, L. Li, F. Zhu, Z. Liu, X. Zeng, X. Yu and Z. Huang, J. Mater. Sci., 2016, 51, 8728–8736 CrossRef CAS.
- Y. Meng, Y. Zhao, C. Hu, H. Cheng, Y. Hu, Z. Zhang, G. Shi and L. Qu, Adv. Mater., 2013, 25, 2326–2331 CrossRef CAS PubMed.
- Y. Fu, X. Cai, H. Wu, Z. Lv, S. Hou, M. Peng, X. Yu and D. Zou, Adv. Mater., 2012, 24, 5713–5718 CrossRef CAS PubMed.
- Y. Fu, H. Wu, S. Ye, X. Cai, X. Yu, S. Hou, H. Kafafy and D. Zou, Energy Environ. Sci., 2013, 6, 805–812 RSC.
- W. He, J. P. Li, J. X. Tian, H. Jing and Y. J. Li, Polym. Compos., 2018, 39, 537–543 CrossRef CAS.
- W. Zhao, Y. Li, S. Wu, D. Wang, X. Zhao, F. Xu, M. Zou, H. Zhang, X. He and A. Cao, ACS Appl. Mater. Interfaces, 2016, 8, 34027–34033 CrossRef CAS PubMed.
- W. Liu, S. Wang, Q. Wu, L. Huan, X. Zhang, C. Yao and M. Chen, Chem. Eng. Sci., 2016, 156, 178–185 CrossRef CAS.
- L. Hao, X. Li and L. Zhi, Adv. Mater., 2013, 25, 3899–3904 CrossRef CAS PubMed.
- D. Shin, Y. Ko and J. Cho, RSC Adv., 2016, 6, 21844–21853 RSC.
- Y. Wu and C. B. Cao, Sci. China: Mater., 2018, 61, 1517–1526 CAS.
- S. Goswami, G. R. Dillip, S. Nandy, A. N. Banerjee, A. Pimentel, S. W. Joo, R. Martins and E. Fortunato, Electrochim. Acta, 2019, 316, 202–218 CrossRef CAS.
- W. Chen, R. B. Rakhi and H. N. Alshareef, J. Phys. Chem. C, 2013, 117, 15009–15019 CrossRef CAS.
- L. B. Chen, Y. R. Chen, J. F. Wu, J. W. Wang, H. Bai and L. Li, J. Mater. Chem. A, 2014, 2, 10526–10531 RSC.
- N. Choudhary, C. Li, J. Moore, N. Nagaiah, L. Zhai, Y. Jung and J. Thomas, Adv. Mater., 2017, 29, 1605336 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma01046h |
| ‡ These authors made equal contributions to this paper. |
| This journal is © The Royal Society of Chemistry 2022 |