
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmergence of long afterglow and room temperature phosphorescence emissions from ultra-small sulfur dots†
Karthika S.
Sunil‡
a,
Kommula
Bramhaiah‡
a,
Srayee
Mandal
a,
Subhajit
Kar
a,
Neena S.
John
 b and
Santanu
Bhattacharyya
*a
b and
Santanu
Bhattacharyya
*a
aIndian Institute of Science Education and Research, Transit Campus, Govt. ITI Building (Transit Campus), Engg. School Road, Berhampur, Odisha-760010, India. E-mail: santanub@iiserbpr.ac.in
bCentre for Nano and Soft Matter Sciences (CeNS), Arkavathi Campus, Shivanapura, Bangaluru-562162, India
First published on 6th January 2022
Abstract
Sulfur dots (S-dots) are one of the most recently developed non-metallic luminescent nanomaterials and possess several advantages over traditional inorganic quantum dots (QDs). Here, we have synthesized highly luminescent ultra-small S-dots using a facile single-step mechanical grinding-assisted technique without consuming additional energy in terms of heat, time, pressure, and harsh chemical treatment. The morphological features are characterized by high-resolution transmission electron microscopy (HR-TEM), and are further correlated with elemental analysis by X-ray photoelectron spectroscopy (XPS) and Fourier transform infra-red (FTIR) spectroscopy. The results suggest the formation of highly homogenous ultra-small S-dots with specific functional groups on the surface. The photophysical properties of the as-synthesized S-dots are characterized by steady-state and time-resolved photoluminescence (PL) spectroscopy. Furthermore, a temperature-dependent PL study has been carried out and it is correlated with the structural features of S-dots. Finally, these ultra-small S-dots show efficient room-temperature phosphorescence and long-term afterglow (up to 5 s) while embedded into B2O3 and biuret matrixes. The mechanistic insight behind the phosphorescence emission, the kinetics of phosphorescence, and furthermore their correlation with elemental/structural features has been investigated in depth. Finally, a reversible transformation of room temperature phosphorescence to thermally activated delayed fluorescence has been explored by a detailed temperature-dependent study. Furthermore, we have also presented the applicability of these smart materials in anti-counterfeiting applications and security devices.
Introduction
Afterglow emission/room temperature phosphorescence is a unique optical phenomenon with a long lifetime, in which a substance releases energy in the form of photons after the removal of the excitation source.1–5 Materials exhibiting long afterglow emission can be utilized in various applications such as biological imaging, printable inks, optoelectronic displays, light-emitting diodes, lightening, anti-counterfeiting, and security systems.1,5,6 Usually, room-temperature phosphorescence (RTP) materials include rare-earth metal-containing inorganic phosphors, organometallic complexes, and pure organic materials.1,7–10 However, these traditional afterglow materials suffer from several disadvantages, which include the usage of expensive raw materials, relatively complex synthesis, and/or purification processes. Moreover, the usual fabrication techniques are commonly energy consuming and not environmentally sustainable too. More specifically, rare-earth-based afterglow materials having strong phosphorescence emission and/or thermally activated delayed fluorescence (TADF) properties need further stringent conditions.1,3,11,12 Likewise, an efficient crystalline state is a common requirement for organic-based afterglow materials. As a result, no afterglow emission can be observed in the solution state or dispersion state for organic materials due to the enhanced rotational and vibrational motions, which initiate non-radiative relaxation pathways. Moreover, the dissolved oxygen and water molecules can quench the excited triplet state through energy transfer to form singlet oxygen.11 In addition, it is noteworthy that the organic luminescent materials rarely exhibit a long afterglow emission due to the rapid quenching of triplet excitons by means of a high non-radiative deactivation rate through inter/intramolecular interactions.1,11,13–15 In this regard, inorganic quantum dots (QDs) can be efficient alternatives. However, the major constituents of most of the traditional inorganic QDs are heavy metal-based, such as CdS, CdSe, CdTe, HgTe, PbS, etc. Therefore, the major challenges are their potential toxicity and environmentally hazardous nature.5,12 As a result, excessive utilization of traditional inorganic QDs in solid-state devices can be another major concern in the very near future. Henceforth, to be very specific, it is significant to develop new generation materials that can show a long afterglow/efficient phosphorescence emission by employing a facile and cost-effective approach. Furthermore, the constituent materials should be readily available, environmentally sustainable and low-toxicity, and should have other superior properties over traditional materials.Considering this, researchers are looking for efficient alternative materials that can overcome all the difficulties. In connection with this, luminescent nanomaterials made of pure elements such as carbon dots, graphene QDs, silicon QDs, and phosphorous dots have been extensively studied for the last few years.11,16–20 Recently, S-dots have attracted great attention owing to their intricate electronic properties and promising applicability as emitting and catalytic materials.21–23 Sulfur is earth-abundant and has been used in many fields such as the production of sulfuric acid, lithium–sulfur batteries, potassium–sulfur batteries, supercapacitors, and sulfur-modified polymeric and organic transformation materials.21,22,24,25 Furthermore, it is already well known that elemental sulfur usually possesses many fascinating properties such as high alkaline metal storage ability, high molar refraction, high transparency in the IR region, pesticide properties, and antioxidizing as well as antimicrobial activities.22,25,26 However, the luminescence nature of ultrasmall S-dots has been rarely explored to date as a potential alternative to traditional inorganic QDs. Currently, very few reports are available regarding the facile synthesis of luminescent S-dots. Initially, Li's group reported the fabrication of S-dots employing a phase interfacial reaction, where CdS QDs were converted to S-dots by dissociation of CdS into Cd2+ and oxidation of S2− into bulk elemental sulfur using HNO3. These S-dots exhibited a weak blue emission with a quantum yield of 0.5%.27 Later, Shen et al. fabricated luminescent S-dots via etching bulk sulfur with the help of NaOH followed by passivation with PEG.28 Furthermore, the luminescence of S-dots was increased employing hydrogen peroxide to etch the surface of the S-dot, resulting in the creation of more surface states.28,29 A few more recent reports are also available explaining the fabrication of a 2D layered morphology from 0D S-dots employing a single-step hydrothermal approach.29 Despite effective fabrication of luminescent S-dot materials by these approaches, they suffer from numerous drawbacks such as harsh reaction conditions, complex operation, low photoluminescence quantum yield (PLQY), high power consumption, and high cost, which significantly limits the large scale production and practical applicability of S-dots. Henceforth, it is highly desirable to adopt new approaches for facile and low-cost preparation methods for S-dot production.
Considering this, we have come up with a facile, low-cost, and rapid method, i.e. the mechanical grinding assisted synthesis approach using a domestic mechanical grinder for the fabrication of ultra-small S-dots from sublimed sulfur, polyethylene glycol (PEG), and sodium hydroxide (NaOH) as initial precursors. The as-synthesized ultra-small S-dots are highly monodispersed in nature and they show excellent aqueous solubility with high photoluminescence quantum yields. Morphological and elemental characterization was performed by high-resolution transmission electron microscopy (HR-TEM), X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared (FTIR) spectroscopy. The photophysical properties of the as-synthesized S-dots were characterized by steady-state and time-resolved photoluminescence spectroscopy. Furthermore, the temperature-dependent photoluminescence properties were further correlated with the structural features of these ultrasmall dots. Finally, the as-synthesized S-dots were embedded in B2O3 and biuret matrixes to study the solid-state emitting properties. We have achieved efficient room-temperature phosphorescence (RTP) and a long-term afterglow of almost up to 5 seconds. To our knowledge, this is the first report explaining the RTP and strong afterglow for S-dots. Beyond this, we have also achieved temperature-dependent thermally activated delayed fluorescence at moderately higher temperatures and a reversible transformation between the fluorescence and phosphorescence emissions. This is further supported by the temperature-dependent kinetic study. Furthermore, we have critically investigated the role of matrix crystallinity on the room temperature phosphorescence and after-glow emission by means of a detailed structure–property correlation. Finally, we have also shown the applicability of these typical materials in anti-counterfeiting applications and security devices. In general, the current investigation will open up new prospects for further development of this new class of luminescent nanomaterials in numerous photonics, optoelectronics, and sensing devices.
Results and discussion
Recently, S-dots have been extremely attracted by the scientific community as a new class of non-metallic fluorescent nanomaterials.21–23,29 However, most of the synthetic methods include harsh chemical/mechanical treatment and energy/time-consuming processes. Therefore, it is highly recommended to explore the possible synthesis approaches with low-cost, facile, and biocompatible methods for the fabrication of S-dots. Herein, for the first time, we have used a facile, low-cost approach for the fabrication of novel luminescent S-dots employing a one-step, top-down, domestic mechanical grinding assisted approach (Fig. 1a). As such, the needed reaction time is drastically reduced to <60 min, while in general chemical treatment needs hours to obtain fluorescent sulfur dots. In the course of the mechanical grinding process, the colorless solution changes to a light yellow, orange, and finally red-colored solution. The obtained product was centrifuged to remove bigger and unreacted compounds. After centrifugation, the supernatant was collected and dialyzed followed by the vacuum drying method. Due to the hydrophilic surface functional groups and the existence of PEG as passivating/capping reagents, S-dots are well dispersed in the normal aqueous medium. The plausible reaction mechanism can be shown as follows: initially, the bulk sulfur powder reacts with NaOH and forms smaller particles. These smaller particles were passivated and stabilized by the PEG molecules. The sodium sulfides formed at the initial reaction time can react with the bulk sulfur and form sodium polysulfides with spherical particles.21,22,28,29| 3S + 6NaOH → 2Na2S + Na2SO3 + H2O |
| (x − 1)S + Na2S → Na2Sx |
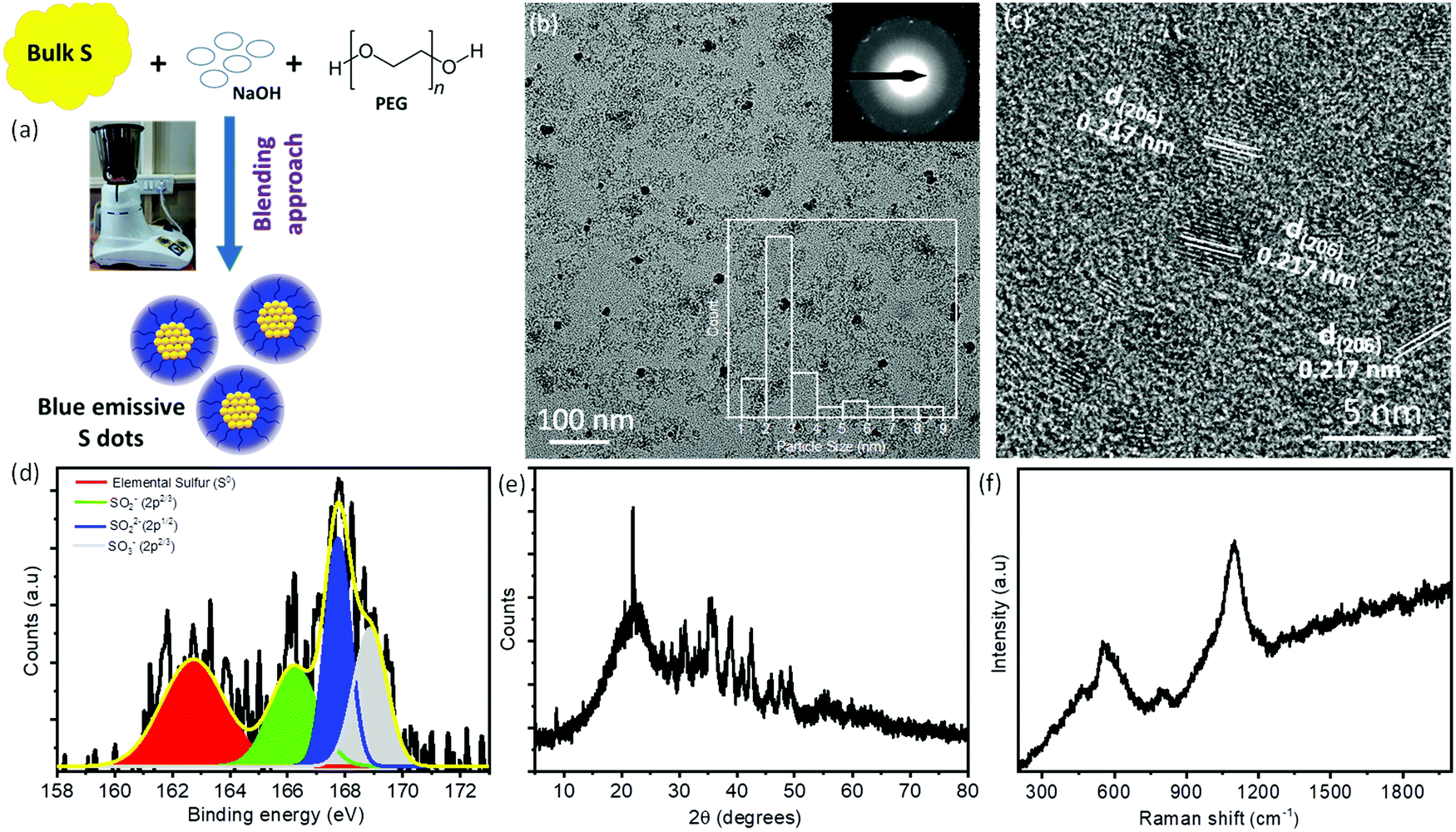 | ||
| Fig. 1 (a) Schematic representation of mechanical grinding assisted synthesis of luminescent sulfur dots. (b) TEM images of S-dots; the top inset shows the SAED pattern and the bottom inset shows the particle size histogram. (c) HRTEM images of S-dots along with lattice fringes. (d) Deconvoluted S 2p XPS spectra, (e) XRD spectra and (f) Raman spectra of S-dots. | ||
Furthermore, the chemical/elemental compositions of S-dots are confirmed by the XPS study. Fig. 1d displays the high-resolution XPS spectra of S2p and it is deconvoluted into four different peaks, which are located at 162.8 eV, 166.2 eV, 167.8 eV, and 168.9 eV, respectively. The peak located at 162.8 eV corresponds to the elemental sulfur, whereas, the peaks around 166.2 eV, 167.8 eV, and 168.9 eV correspond to the oxidized form of sulfur such as SO2− (2p2/3), SO22− (2p1/2), and SO3− (2p2/3), respectively.23,28–30,32 Furthermore, the deconvoluted spectra of O1s exhibit three distinguished peaks at around 530.9 eV, 532.6 eV, and 534.8 eV, respectively. These peaks correspond to S–O, C–O/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and adsorbed H2O molecules on S-dots, respectively (Fig. S1a, ESI†). The deconvoluted C1s spectra exhibit peaks at around 284.8 eV, 285.3 eV, and 286.4 eV, which correspond to C–C, C–O, and C–OH groups, respectively (Fig. S1b, ESI†). From the XPS results, we can conclude that the as-synthesized S-dots are mainly composed of elemental sulfur along with various sulfite, sulfonyl, and sulfonate functional groups on the surface. Therefore, the oxidized sulfur groups hanging over the surface of the S-dots make them hydrophilic in nature. All these results are in good agreement with the previous reports. To determine the crystallinity and phase of S-dots, we have performed XRD measurements and depicted the spectra in Fig. 1e. The S-dots display S8 orthorhombic (JCPDF: 83-2285) and S6 hexagonal (JCPDF: 74-1654) mixed phases.23,28,30 Furthermore, we have utilized Raman spectroscopy to observe the sulfur species over the S-dots. The measured Raman spectra (Fig. 1f) exhibit three distinct peaks at around 560, 800, and 1100 cm−1.29,31,33,34 The broad peaks ranging from 500 cm−1 to 650 cm−1 correspond to the formation of shorter chain polysulfides such as S4− and S5−. The medium intense peak at around ∼800 cm−1 can be assigned to the stretching vibrations of C–O groups, which are coming from the PEG molecules over the S-dots. In addition, the strong intense peak at around ∼1100 cm−1 belongs to the stretching vibration of the S–O bond.23,28,31,33 The Raman results suggest that the as-synthesized S-dots are composed of shorter chain polysulfides as a core along with surface S–O groups. Moreover, the S-dots are passivated or covered with PEG molecules. To determine the functional groups and interactions between the PEG and S-dots, we have performed FTIR measurements and depicted them in Fig. S2 (ESI†). The S-dots exhibit a broad peak at around 3426 cm−1 and a moderately intense peak at around 1637 cm−1, which correspond to the stretching vibrations of the adsorbed water molecules and hydroxyl groups (–OH) present over PEG or the S-dot, respectively. Furthermore, the prominent peaks at around 2941 cm−1 and 2864 cm−1, and the stronger peak at around 1450 cm−1 can be assigned to the stretching vibrations of C–H, and the deformation band of –CH2 groups of alkyl chains or SO groups, respectively.28,30,35 The bands at around 1130 cm−1 and 1007 cm−1 can be assigned to the stretching vibrations of the –C–OH and C–O–C groups of PEG molecules attached to the surface of the S-dots. In addition, the band at around 860 cm−1 corresponds to the bending vibration of –C–C–H groups. Moreover, the medium intense peaks were observed at around 670 cm−1 and 549 cm−1 corresponding to the S–O and S–S linkage stretching vibrations, respectively.22,23,28,30 Therefore, from the FTIR spectra, it is evident that PEG and S-dots along with some surface groups are observed. Furthermore, no other additional peaks were observed, which suggests that there is no chemical interaction between PEG and S-dots.
O, and adsorbed H2O molecules on S-dots, respectively (Fig. S1a, ESI†). The deconvoluted C1s spectra exhibit peaks at around 284.8 eV, 285.3 eV, and 286.4 eV, which correspond to C–C, C–O, and C–OH groups, respectively (Fig. S1b, ESI†). From the XPS results, we can conclude that the as-synthesized S-dots are mainly composed of elemental sulfur along with various sulfite, sulfonyl, and sulfonate functional groups on the surface. Therefore, the oxidized sulfur groups hanging over the surface of the S-dots make them hydrophilic in nature. All these results are in good agreement with the previous reports. To determine the crystallinity and phase of S-dots, we have performed XRD measurements and depicted the spectra in Fig. 1e. The S-dots display S8 orthorhombic (JCPDF: 83-2285) and S6 hexagonal (JCPDF: 74-1654) mixed phases.23,28,30 Furthermore, we have utilized Raman spectroscopy to observe the sulfur species over the S-dots. The measured Raman spectra (Fig. 1f) exhibit three distinct peaks at around 560, 800, and 1100 cm−1.29,31,33,34 The broad peaks ranging from 500 cm−1 to 650 cm−1 correspond to the formation of shorter chain polysulfides such as S4− and S5−. The medium intense peak at around ∼800 cm−1 can be assigned to the stretching vibrations of C–O groups, which are coming from the PEG molecules over the S-dots. In addition, the strong intense peak at around ∼1100 cm−1 belongs to the stretching vibration of the S–O bond.23,28,31,33 The Raman results suggest that the as-synthesized S-dots are composed of shorter chain polysulfides as a core along with surface S–O groups. Moreover, the S-dots are passivated or covered with PEG molecules. To determine the functional groups and interactions between the PEG and S-dots, we have performed FTIR measurements and depicted them in Fig. S2 (ESI†). The S-dots exhibit a broad peak at around 3426 cm−1 and a moderately intense peak at around 1637 cm−1, which correspond to the stretching vibrations of the adsorbed water molecules and hydroxyl groups (–OH) present over PEG or the S-dot, respectively. Furthermore, the prominent peaks at around 2941 cm−1 and 2864 cm−1, and the stronger peak at around 1450 cm−1 can be assigned to the stretching vibrations of C–H, and the deformation band of –CH2 groups of alkyl chains or SO groups, respectively.28,30,35 The bands at around 1130 cm−1 and 1007 cm−1 can be assigned to the stretching vibrations of the –C–OH and C–O–C groups of PEG molecules attached to the surface of the S-dots. In addition, the band at around 860 cm−1 corresponds to the bending vibration of –C–C–H groups. Moreover, the medium intense peaks were observed at around 670 cm−1 and 549 cm−1 corresponding to the S–O and S–S linkage stretching vibrations, respectively.22,23,28,30 Therefore, from the FTIR spectra, it is evident that PEG and S-dots along with some surface groups are observed. Furthermore, no other additional peaks were observed, which suggests that there is no chemical interaction between PEG and S-dots.
To obtain insights into the optical properties of the as-synthesized S-dots, we have recorded UV-visible absorption spectra, steady-state and time-resolved PL spectra, and temperature-dependent PL spectra (as depicted in Fig. 2). From the absorption spectra of the S-dots (Fig. 2a), one can observe that there are three distinctive absorption bands centered at around 221 nm, 305 nm, and 365 nm. The band at 221 nm corresponds to the n–σ* transition of non-bonding electrons present in the S-dots. Generally, heteroatoms such as S and O containing non-bonding electrons exhibit the n–σ* transition in the range from 150 nm to 250 nm. The bands at around 305 nm and 365 nm correspond to the S22− and S82− species formed over the surface of the S-dots.23,28,29 The PL spectra of the S-dots exhibit maxima at around 434 nm while exciting at 330 nm. For further investigations, regarding the fundamental photophysical properties of the as-synthesized S-dots, steady-state PL (Fig. 2b) spectra have been investigated in the aqueous medium upon excitation in the range from 270 nm to 500 nm. From Fig. 2b, we can clearly state that the S-dots mainly exhibit the excitation-dependent PL emission. As the excitation wavelengths vary in the range between 270 and 500 nm, the emission peak maximum gradually shifts towards the longer wavelengths (Fig. S3, ESI†). The excitation-dependent PL properties suggest the multistate emissive properties of S-dots due to various surface states/defects during the formation of S-dots, and they exhibit a stable fluorescence emission (Fig. S4, ESI†). Furthermore, these as-synthesized S-dots employing a mechanical grinding approach exhibit a stable behavior for months. The observed absorbance and PL emissions along with the PL excitation spectra of these S-dots after 3 months are comparable with the initial measurements and are displayed in Fig. S5 (ESI†). To obtain more insight into the fluorescence emission behavior, we have carried out detailed time-resolved fluorescence lifetime measurements at a 340 nm excitation wavelength, which are depicted in Fig. 2c. The decay curves follow biexponential decay kinetics. The faster component corresponds to the faster recombination of initially generated excitons, while the slower component corresponds to the surface states. The main characteristic features of the decay kinetics remain almost similar at longer excitation wavelengths, which indicates the contributions from multichromophoric centers in S-dots at the various excitation wavelengths (depicted in Table S1, ESI†).
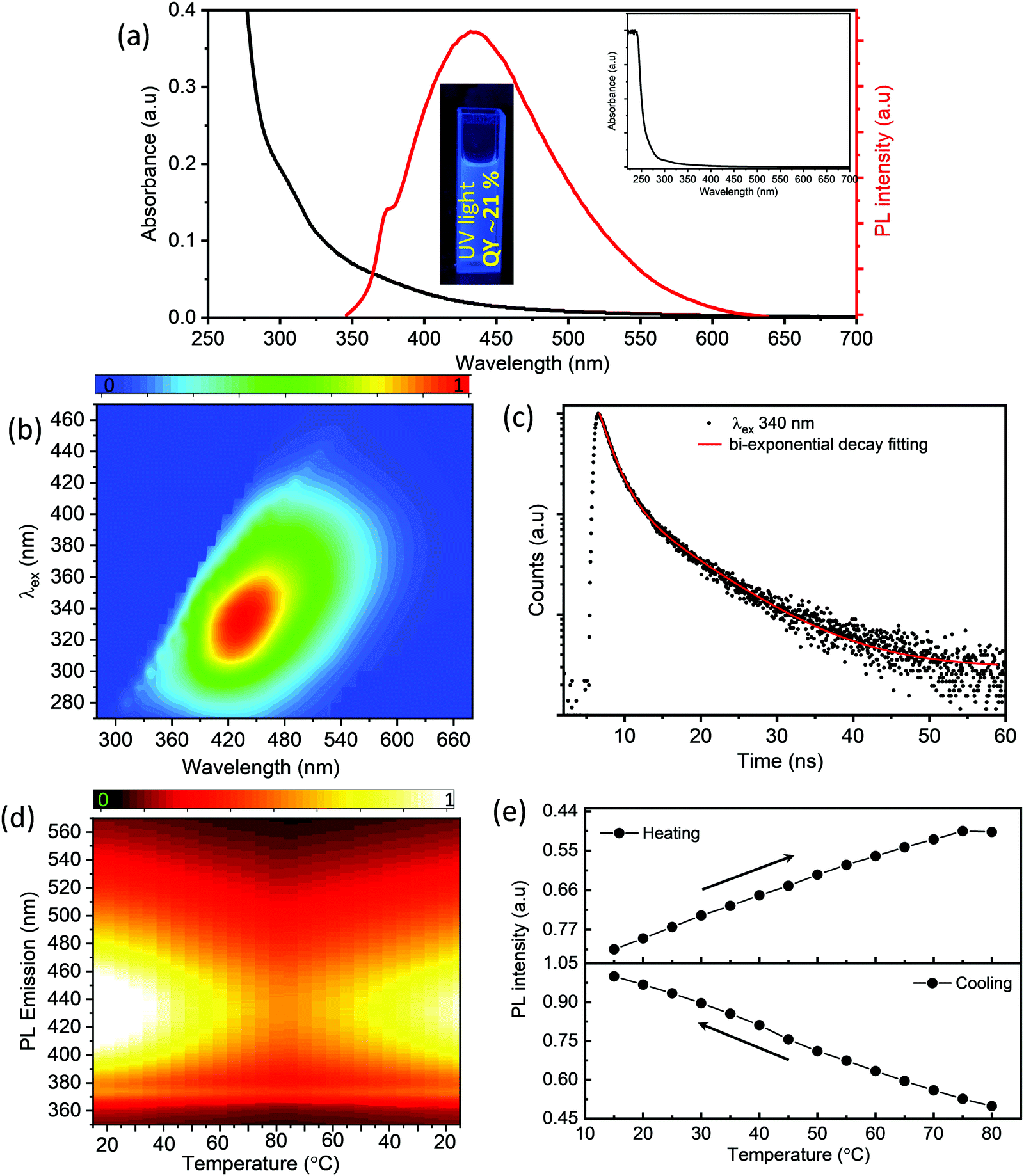 | ||
| Fig. 2 (a) UV-vis absorption and PL spectra of S-dots (the inset shows the full range absorbance spectra and photographs under UV light irradiation), (b) PL spectra of S-dots at different excitation wavelengths, (c) time-resolved lifetime measurements at 340 nm excitation wavelength, (d) thermo-responsive PL spectra of S-dots by varying the temperature (heating and cooling) and (e) plot of F/F0vs. temperature (heating and cooling process). | ||
For further clarification, we have studied the thermo-responsive PL properties of the as-synthesized S-dots (depicted in Fig. 2d). Fig. 2d shows the PL intensities of the S-dots as a function of temperature ranging from 15 °C to 80 °C (heating process), and 80 °C to 15 °C (cooling process) by changing 5 °C per step and keeping the S-dot aqueous solution for 2 min at each temperature to attain equilibrium. The results indicate that the as-synthesized S-dots could be a potential candidate as a temperature sensor. Upon excitation at 330 nm, the S-dots showed an emission wavelength of 432 nm. Besides, the emission wavelength and FWHM of the PL peak, the S-dots also exhibit an insignificant shift and broadening over the measured temperature range (see Fig. S6, ESI†). From Fig. 2d, one can observe that the emission intensity progressively decreases, as the temperature increases, and the PL intensity recovers on cooling to the initial temperature. One can observe from Fig. 2d that the temperature response range of the as-synthesized S-dots employing the blending approach shows sensitivity through a much wider range compared to the other reports.36–39 For a better understanding of the temperature over the fluorescence intensity of the S-dots, we have plotted the relative fluorescence intensity (F/F0) of the S-dots as a function of temperature and this is depicted in Fig. 2e, where F0 and F are the fluorescence intensities at the initial (15 °C) and elevated temperatures, respectively. It was observed that the fluorescence intensity of the S-dots decreases up to 51.2%, with increasing the temperature from 15 °C to 80 °C (the heating process). On the other hand, the fluorescence intensity almost recovered back to its original value, when the temperature falls back from 80 °C to 15 °C (the cooling process). This behavior might be because of the thermally activated non-radiative trapping of excited carriers. At the higher temperature, a large number of excited carriers relax through nonradiative ways, which is mainly due to the several vibrational states originating from surface functional groups.38,39 Furthermore, the reversibility and stability of the temperature-dependent fluorescence of the S-dots were measured through thermal cycling experiments (Fig. S7, ESI†). The thermal reversibility of the S-dots was assessed by measuring the recovery through multiple heating (70 °C) and cooling (10 °C) cycles, and keeping the S-dot aqueous solution for 5 min at each temperature to attain the equilibrium. The results show that the initial fluorescence intensity does not significantly change by the multiple heating and cooling cycles. The S-dot NPs have displayed on–off PL properties. It is already established that S-dots are efficient emitting materials while dispersed in a solvent medium, especially in an aqueous medium. Furthermore, it can be a potentially good candidate to develop solid-state optical materials for various optoelectronic devices.21–23 In addition to the solid-state fluorescence, room temperature long-term afterglow emission is also highly desirable, where we can harvest the triplet state for phosphorescence. Meanwhile, the major challenge is aggregation-induced quenching in the solid-state which increases the nonradiative relaxation processes, as well as enhances the triplet quenching through vibrational relaxation.3,5,10 To be very precise, the afterglow emission of S-dots can originate from the n → π* transition of the surface states/surface functional groups. To obtain the enhanced afterglow emission or room temperature phosphorescence, one should improve the typical intersystem crossing (ISC) by improving vibrational coupling and/or stabilizing the triplet states by enhancing spin–orbit coupling.10,11,40 Therefore, to harvest the triplet state of S-dots, we can suppress the vibrational and rotational motions of surface functional groups by fixing their motions either through covalent or non-covalent bonding. This process permits the protection of the excited triplet states for the generation of afterglow emissions. For this, we have embedded the luminescence S-dots in various matrixes including B2O3 and biuret, and varied the S-dot weight from 1 mg to 6 mg in the matrix. The 3 mg S-dot in the B2O3 matrix displayed a higher phosphorescence emission (Fig. S8, ESI†). The schematic illustration of the synthesis procedure for the long afterglow emissive S-dot/B2O3 and S-dot/biuret composites is shown in Fig. S9 (ESI†). The S-dots are dispersed in boric oxide (B2O3) and biuret, which are facilely obtained by one-pot heating treatment of S-dots and boric acid or urea dispersion in an aqueous solution at a higher temperature. It was computationally shown that the ISC from a singlet (S1) state to a triplet state (Tn) can be enhanced by increasing the (n, π*) transition component of the excited triplet (Tn) state that is nearby to the singlet (S1) state.41–43 Generally, carbonyl and sulfonyl groups are extensively used in organic-based afterglow materials to increase the (n, π*) character of the singlet and triplet states owing to the presence of lone pairs on the oxygen atoms.42,43 The S-dot nanomaterials synthesized through the mechanical grinding approach mainly have elemental sulfur (S0) as the core and sulfite, sulfonyl, and sulfonate functional groups on the surface (as depicted in the XPS spectra; Fig. 1e). Furthermore, the solid-state optical responses such as fluorescence and phosphorescence emissions along with the excitation spectra of S-dots embedded in the B2O3 and biuret matrixes are studied using photoluminescence measurements at different delay times (0 ms and 2000 ms). Fig. 3a and b display the solid-state phosphorescence (PL), fluorescence excitation (FL excitation), and fluorescence emission spectra (FL) of S-dot–B2O3 and S-dot–biuret composites. The excitation spectra show a peak at around 370 nm corresponding to the oxidized sulfur groups/surface functional groups. The prompt fluorescence and phosphorescence emission maxima of S-dot–B2O3 and S-dot–biuret composites are at 420 nm and 495 nm, respectively. The time-resolved phosphorescence decay curves of S-dot–B2O3 and S-dot–biuret composites are measured and depicted in Fig. 3c. The decay curves have to be fitted with the biexponential fitting equation and the average phosphorescence lifetimes for the S-dot/B2O3 and S-dot/biuret composites are 419.6 ms and 434.48 ms, respectively. It is well known that the RTP and longtime afterglow emission mostly depend on the effective protection of the excited triplet state. Upon incorporation of luminescent S-dots into highly crystalline materials through covalent or hydrogen bonds, we have observed strong RTP. However, when the crystallinity decreases, the matrix rigidity decreases. As a result, non-radiative relaxation pathways increase which eventually decrease the RTP. On the other hand, when the matrix rigidity increases, the nonradiative relaxation pathways get reduced, resulting in enhanced RTP and longtime afterglow emissions.4,44,45
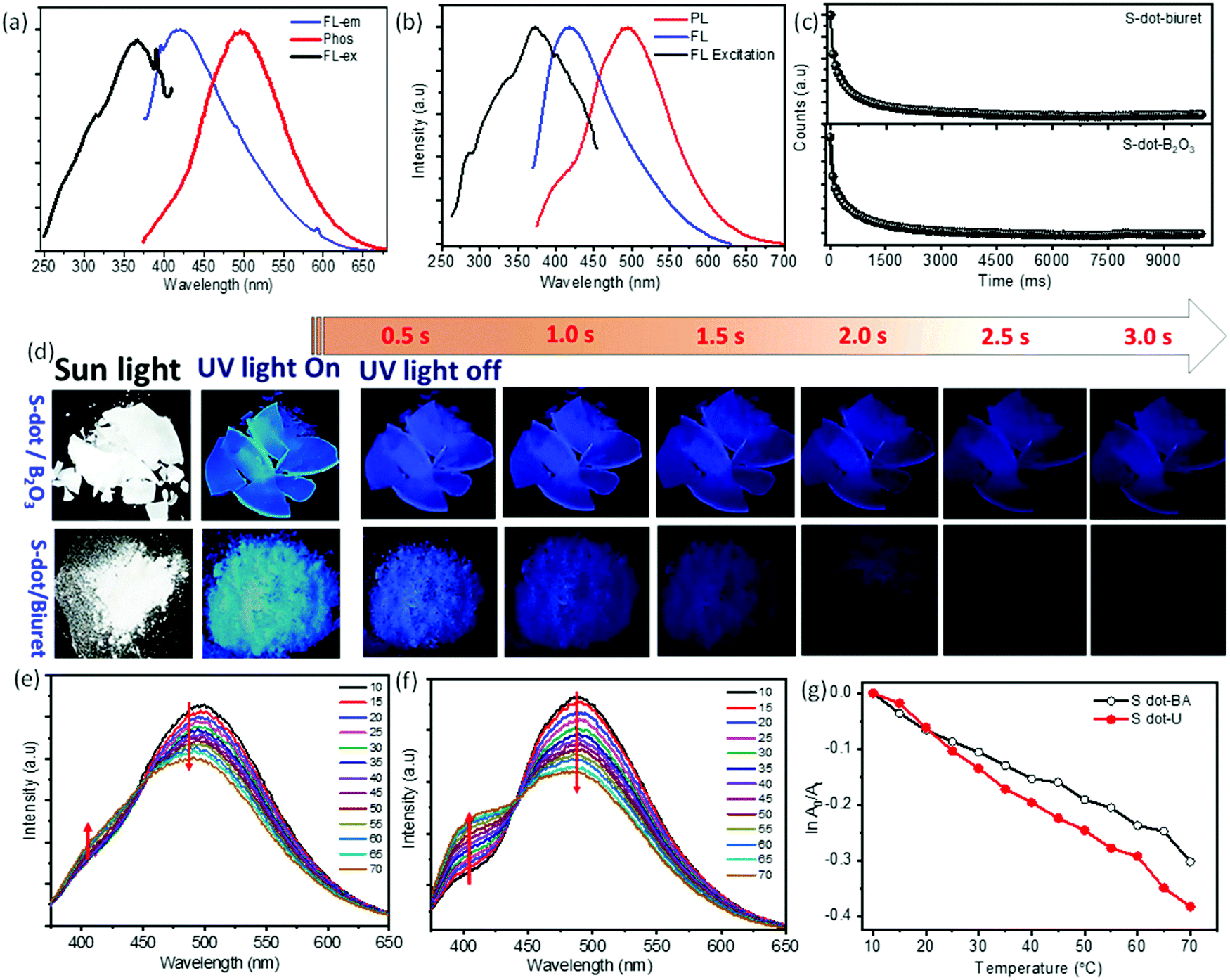 | ||
Fig. 3 (a) Solid-state fluorescence spectra (FL), and phosphorescence spectra (PL) along with fluorescence excitation spectra (FL excitation) of the S-dot/B2O3 composite. (b) Solid-state fluorescence, and phosphorescence emission spectra along with excitation spectra of S-dot/biuret composites. (c) Phosphorescence lifetime data of S-dot–B2O3 and S-dot–biuret composites. (d) Digital photographs of the S-dot/B2O3 composite and S-dot/biuret composite before and after turning off the UV light (∼365 nm ± 10 nm). (e) Thermo-responsive phosphorescence spectra of the S-dots/B2O3 composite by varying the temperature (the heating process). (f) Thermo-responsive phosphorescence spectra of the S-dots/biuret composite by varying the temperature (the heating process). (g) Plot ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) A0/Atversus temperature. A0/Atversus temperature. | ||
In the same context, to understand the effect of the pH on the phosphorescence emission while encapsulated in a solid matrix, we have synthesized both the S-dot–B2O3 and S-dot–biuret nanocomposites at various pHs, i.e. -acidic (pH = 3), neutral (pH = 7), and basic (pH = 9). For the S-dot–B2O3 nanocomposite, at the acidic pH (pH = 3), we observed a maximum crystallinity of the matrix and long afterglow emission. As we change the pH from acidic to basic, B2O3 becomes amorphous in nature (see XRD in Fig. S10a, ESI†). Whereas, in the case of the urea matrix, the matrix phase changes from biuret cyanuric acid (JCPDF No. 047-1938, see XRD spectra in Fig. S10b, ESI†) phase to mixed phases such as biuret cyanuric acid, and biuret hydrate (JCPDF No. 047-1938 and JCPDF No. 011-0719, see XRD spectra in Fig. S10b, ESI†) phase, respectively, while converting from the basic medium to the acidic medium. In the mixed phase, vibrational and rotational motions are enhanced, resulting in enhanced non-radiative relaxation pathways. As a result, the room temperature phosphorescence and afterglow emission decrease in an acidic medium, in the case of the biuret matrix (Fig. S11, ESI†).7,40,45–47
Fig. 3b displays the photographs of S-dot/B2O3 and S-dot/biuret composite powders with the excitation light (∼365 nm) on and off. It can be seen that the S-dot/B2O3 composite powder can emit strong fluorescence emission under UV light irradiation, and emit bright blue phosphorescence, which can be manifestly observed by the naked eye up to 5 s after turning off the light irradiation source, while this is 3 s for the S-dot/biuret composite powder. The corresponding luminescence videos of the S-dot/B2O3 and S-dot/biuret composite powders are shown in the ESI† (Videos S1 and S2, ESI†). Furthermore, we have investigated the effect of the temperature on the afterglow emission of the as-synthesized S-dot/B2O3 and S-dot/biuret composites by collecting afterglow emission spectra at temperatures ranging from 10 °C to 70 °C during both the heating and cooling processes, respectively (depicted in Fig. 3e and f, respectively). With increasing the temperature from 10 °C to 70 °C, the afterglow emission for the S-dot/B2O3 and S-dot/biuret composite powders corresponding to the prompt fluorescence at ∼415 nm increases, while the emission intensity corresponding to phosphorescence at ∼495 nm decreases. Usually, lower temperature favors the phosphorescence emission, while a reasonably higher temperature is beneficial for thermally activated delayed fluorescence (TADF) through reverse intersystem crossing. Therefore, the above results support the possible TADF process at higher temperatures. However, phosphorescence predominates over the TADF process. It is noteworthy that the afterglow due to phosphorescence recovers and TADF gradually decreases while cooling from 70 °C to 10 °C (Fig. S12, ESI†). The lifetimes of the phosphorescence of the S-dot–B2O3 and S-dot–biuret composites are measured during both the heating (70 °C), and cooling (10 °C) processes and are depicted in Fig. S13 (ESI†). The decay curves show that the phosphorescence decay time decreases as the temperature increases from 10 °C to 70 °C, which is due to the enhanced reverse intersystem crossing process commonly observed for RTP materials (Fig. S12, ESI†).48 The details of the lifetime values are shown in Table S2 (ESI†). Considering this tunable behavior of phosphorescence emission, it can be used as a temperature sensor by plotting lnA0/Atversus temperature (as depicted in Fig. 3g). A0 and At represent the phosphorescence intensity at the initial and elevated temperatures, respectively. The S-dot/B2O3 and S-dot/biuret composite powders display a good linearity and values as follows: lnA0/At = −0.00452T + 0.03296 (R2 = 99), and lnA0/At = −0.00624T + 0.06025 (R2 = 99), respectively. However, the S-dot–biuret composite displays better temperature-dependent phosphorescence properties. Henceforth, we have measured the cycling stability for the S-dot–biuret composite. The stability of the temperature-dependent phosphorescence of the S-dot–biuret composite is evaluated through multiple heating (70 °C), and cooling (10 °C) processes, and is shown in Fig. S14 (ESI†). The stability curves display a good response. Moreover, the phosphorescence and TADF emissions of the S-dot–biuret composite can almost revert back to its initial value. The phosphorescence intensities at different temperatures for each cycle are almost unchanged, indicating the excellent reversibility of the system. Finally, based on the above results, the S-dot/B2O3 and S-dot/biuret composite systems are stable and robust for temperature sensing.
Anticounterfeiting applications
The unique features of long afterglow emission and RTP lifetime of the as-synthesized S-dot–B2O3 and S-dot–biuret composites allow us to utilize them as a kind of smart material, which can be employed for anticounterfeiting and encryption applications. As displayed in Fig. 4, security graphics (a rose flower with the leaves) and patterns (letters) are composed of green emissive carbon dots (CDs) and S-dot–B2O3 and S-dot–biuret composites. Here, the CD powder with green fluorescence emission and not having RTP features is used as a reference.49 Under UV light (365 nm) excitation, the rose flower with the leaves and the letters such as “S, D, O, T” are visible and display different fluorescent colors. Once the UV light (365 nm) is switched off, the rose flower leaves and the letters “D and T” coated with the green emissive CD powder are invisible. Whereas in the case of the rose flower and the letters “S and O” coated with the S-dot–B2O3 and S-dot–biuret composites they display a bright blue emission with UV light (365 nm) excitation. After the UV light is switched off, the color of the rose flower and the letters “S and O” exhibited a bluish-green emission, which can be seen with the naked eye. Henceforth, this afterglow emissive nature of these as-synthesized S-dot–B2O3 and S-dot–biuret composites could be used for the application of counterfeiting and information protection systems. | ||
| Fig. 4 Digital photographs of information anticounterfeiting and encryption obtained from the long afterglow emissive S-dot/B2O3 and S-dot biuret composites before and after turning off the UV light (365 nm). | ||
Conclusion
In conclusion, we have synthesized ultra-small S-dots by using a mechanical grinding-based single-step facile approach. The morphological and elemental studies confirm the formation of tiny S-dots (∼2.5 nm) with specific functionalities on the surface. The XRD data prove the existence of S8 orthorhombic and S6 hexagonal mixed phases. The steady-state and time-resolved PL studies further describe the photophysics of these typical dots and are correlated with the morphological/structural features. The temperature-dependent PL study further supports the existence of defect states due to specific surface functionalities on the surface. Finally, these S-dots show efficient room-temperature phosphorescence and long-term afterglow inside the B2O3 and biuret matrixes. To our knowledge, this is the first report of S-dot phosphorescence and room temperature afterglow. Beyond this, at high temperature, prominent thermally activated delayed fluorescence has been observed and it is reversible with the phosphorescence emission depending on the applied temperature. Finally, we have shown the prospects of these typical materials in anti-counterfeiting applications and security devices. Overall, the present investigation will open up new possibilities regarding the facile synthesis of this unique class of luminescent materials and their several prospects in solid-state photonics, optoelectronics, and sensory devices.Experimental details
Materials
The sublimated bulk sulfur and polyethylene glycol 400 (PEG-400) were purchased from Sigma-Aldrich. Sodium hydroxide (NaOH), boric acid, and urea were purchased from LOBA chemicals. All of the chemicals were of analytical grade and were used without any further purification. Milli Q water was used throughout the experiments.Synthesis of S-dots
S-Dots were synthesized employing a mechanical grinding approach. The typical procedure is as follows: 3.36 g of sublimed sulfur and 7.2 mL of PEG-400 were dispersed in 300 mL of ultra-pure water followed by the addition of 9.96 g of NaOH. The whole solution was transferred to a domestic grinding machine. The grinder was sealed with a tight lid and ground for 60 min. After the reaction time, the product was collected and centrifuged at 10000 rpm to remove the unreacted and bulk particles. The supernatant was collected and dialyzed employing a 1 kD dialysis membrane. The solution was collected and used as a mother solution for further characterization.
Synthesis of S-dot/B2O3 and S-dot/biuret composites
Briefly, 3 g of boric acid was dissolved in 40 mL of ultrapure water followed by the addition of 3 mg (varied the S-dot weight) of S-dots in a 100 mL glass beaker. The whole solution was ultra-sonicated for 10 min. Then, the glass beaker was covered with aluminum (Al) foil to prevent the fast evaporation of water vapor. Afterward, the glass beaker was placed in a hot air oven at 180 °C for 5 h. After the reaction, the beaker was naturally cooled down to room temperature. At the end of the reaction, a colorless solid residue was observed. The colorless material was collected and utilized for further characterization. Various S-dot/B2O3 composites were prepared using the same procedure by changing the S-dot concentration (1 mg, 2 mg, 3 mg, 4 mg, and 6 mg).The S-dot/biuret composite was also prepared using the S-dots (3 mg) and urea (3 g) as the initial reactants without changing the synthesis procedure.
Characterization
The morphology and crystallinity of the as-synthesized S-dots were imaged employing a TALOS F200S G2 transmission electron microscope (TEM) with an accelerating voltage of 200 kV. For imaging, a few aliquots of diluted S-dot samples were drop casted over holey carbon-coated copper grids. Then, the grids were dried under a vacuum. Room temperature steady-state photoluminescence (PL) measurements and their thermo-responsive properties were done using a Horiba Jobin Yvon Fluorolog-3 spectrofluorometer, equipped with a 300 W xenon lamp as an excitation source and Peltier cooling setup. For these measurements, the S-dot samples were dispersed in Milli-Q water. The presence of various surface groups and elements of the S-dots was studied using X-ray photoelectron spectroscopy (XPS), on a PHI 5000 VersaProbe III with the multi-technique system. For this, the S-dot samples were drop cast over the doped Si substrate. X-ray diffractograms were obtained using a BENCHTOP powder X-ray diffractometer (XRD) (Rigaku Miniflex 600) equipped with a 600 W X-ray copper tube. The samples were coated over a glass substrate (1 cm × 1 cm). Raman spectra were recorded using a RENISHAW invia Raman microscope equipped with a 532 nm laser source. For this, the S-dot samples were drop cast over the glass substrate, and dried in a vacuum. FTIR measurements were done using a BRUKER TENSOR II instrument. For this, the S-dot powder was mixed with KBr powder and pellets were made. Time-resolved fluorescence lifetime measurements were measured using a fluorescence spectrometer LifeSpec II (EDINBURGH Instruments) with a 340 nm UV-LED (EPLED Series), 375 nm, and 450 nm LASER as excitation sources. The relative PL-QY was measured using a standard dye, quinine sulfate. In brief, quinine sulfate with a PL QY of 0.55 in 0.1 M H2SO4 (refractive index (η) = 1.33) solution was taken as a reference. Initially, the absorbance and PL emissions of quinine sulfate were measured by UV-visible absorption and photoluminescence (PL) measurements. Furthermore, the PL QY of the as-synthesized S-dots in the aqueous medium was calculated according to the following equation: QY = QYqs·(FS-dots/Fqs)·(Aqs/AS-dot)·(ηS-dot/ηqs)2. Here, A, F, and η represent the absorbance at the excitation wavelength, integrated PL emission intensity calculated from the area under the emission peak on the same wavelength scale, and the refractive index of the solvent, respectively.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
K. B. acknowledges IISER Berhampur for providing him an institute postdoctoral fellowship. S. M. and S. K. acknowledge IISER Berhampur for the institute PhD fellowship. The authors acknowledge DST-SERB (grant numbers/2019/000026) for financial support and S. B. acknowledges IISER Berhampur for the initiation grant (IG/21082018). The authors acknowledge the Central Advance Instrument Facility (CAIF) at IISER Berhampur.References
- S. Xu, R. Chen, C. Zheng and W. Huang, Adv. Mater., 2016, 28, 9920–9940 CrossRef CAS PubMed.
- K. Jiang, Y. Wang, C. Cai and H. Lin, Chem. Mater., 2017, 29, 4866–4873 CrossRef CAS.
- Y. Jiang, J. Huang, X. Zhen, Z. Zeng, J. Li, C. Xie, Q. Miao, J. Chen, P. Chen and K. Pu, Nat. Commun., 2019, 10, 2064 CrossRef PubMed.
- Y. Sun, X. Zhang, J. Zhuang, H. Zhang, C. Hu, M. Zheng, B. Lei and Y. Liu, Carbon, 2020, 165, 306–316 CrossRef CAS.
- Y. Li, M. Gecevicius and J. Qiu, Chem. Soc. Rev., 2016, 45, 2090–2136 RSC.
- L. Ai, Y. Yang, B. Wang, J. Chang, Z. Tang, B. Yang and S. Lu, Sci. Bull., 2021, 66, 839–856 CrossRef CAS.
- F. Clabau, X. Rocquefelte, S. Jobic, P. Deniard, M.-H. Whangbo, A. Garcia and T. Le Mercier, Chem. Mater., 2005, 17, 3904–3912 CrossRef CAS.
- Y. Zhao, X.-G. Yang, X.-M. Lu, C.-D. Yang, N.-N. Fan, Z.-T. Yang, L.-Y. Wang and L.-F. Ma, Inorg. Chem., 2019, 58, 6215–6221 CrossRef CAS PubMed.
- S. Yang, D. Wu, W. Gong, Q. Huang, H. Zhen, Q. Ling and Z. Lin, Chem. Sci., 2018, 9, 8975–8981 RSC.
- L. Bian, H. Shi, X. Wang, K. Ling, H. Ma, M. Li, Z. Cheng, C. Ma, S. Cai, Q. Wu, N. Gan, X. Xu, Z. An and W. Huang, J. Am. Chem. Soc., 2018, 140, 10734–10739 CrossRef CAS PubMed.
- K. Jiang, Y. Wang, Z. Li and H. Lin, Mater. Chem. Front., 2020, 4, 386–399 RSC.
- J. Xu and S. Tanabe, J. Lumin., 2019, 205, 581–620 CrossRef CAS.
- J. Liu, Z. Ma, Z. Li, Y. Liu, X. Fu, H. Jiang, Z. Ma and X. Jia, J. Mater. Chem. C, 2021, 9, 3257–3263 RSC.
- A. D. Sontakke, J.-M. Mouesca, V. Castaing, A. Ferrier, M. Salaün, I. Gautier-Luneau, V. Maurel, A. Ibanez and B. Viana, Phys. Chem. Chem. Phys., 2018, 20, 23294–23300 RSC.
- S. Reineke and M. A. Baldo, Sci. Rep., 2014, 4, 3797 CrossRef PubMed.
- M. Park, H. S. Kim, H. Yoon, J. Kim, S. Lee, S. Yoo and S. Jeon, Adv. Mater., 2020, 32, 2000936 CrossRef CAS PubMed.
- M. Martini, G. Spinolo, A. Vedda and C. Arena, Solid State Commun., 1994, 91, 751–756 CrossRef CAS.
- X. Niu, Y. Yi, L. Meng, H. Shu, Y. Pu and X. Li, J. Phys. Chem. C, 2019, 123, 25775–25780 CrossRef CAS.
- S. Kundu, K. Bramhaiah and S. Bhattacharyya, Nanoscale Adv., 2020, 2, 5130–5151 RSC.
- B. Kommula and S. Bhattacharyya, Mater. Adv., 2022, 3, 142–172 RSC.
- H. Jin, Y. Sun, Z. Sun, M. Yang and R. Gui, Coord. Chem. Rev., 2021, 438, 213913 CrossRef CAS.
- P. Gao, G. Wang and L. Zhou, ChemPhotoChem, 2020, 4, 5235–5244 CrossRef CAS.
- A. Pal, F. Arshad and M. P. Sk, Adv. Colloid Interface Sci., 2020, 285, 102274 CrossRef CAS PubMed.
- G. Nikiforidis, M. C. M. van de Sanden and M. N. Tsampas, RSC Adv., 2019, 9, 5649–5673 RSC.
- J. Wang, L. Yin, H. Jia, H. Yu, Y. He, J. Yang and C. W. Monroe, ChemSusChem, 2014, 7, 563–569 CrossRef CAS PubMed.
- Y. Hwa and E. J. Cairns, ChemElectroChem, 2020, 7, 3927–3942 CrossRef CAS.
- S. Li, D. Chen, F. Zheng, H. Zhou, S. Jiang and Y. Wu, Adv. Funct. Mater., 2014, 24, 7133–7138 CrossRef CAS.
- L. Shen, H. Wang, S. Liu, Z. Bai, S. Zhang, X. Zhang and C. Zhang, J. Am. Chem. Soc., 2018, 140, 7878–7884 CrossRef CAS PubMed.
- L. Shen, J. Wei, Z. Liu, Z. Bai, Y. Li, D. Zhang and C. Zhang, Chem. Mater., 2020, 32, 10476–10481 CrossRef CAS.
- L. Xiao, Q. Du, Y. Huang, L. Wang, S. Cheng, Z. Wang, T. N. Wong, E. K. L. Yeow and H. Sun, ACS Appl. Nano Mater., 2019, 2, 6622–6628 CrossRef CAS.
- Y. Shi, P. Zhang, D. Yang and Z. Wang, Chem. Commun., 2020, 56, 10982–10988 RSC.
- N. S. A. Manan, L. Aldous, Y. Alias, P. Murray, L. J. Yellowlees, M. C. Lagunas and C. Hardacre, J. Phys. Chem. B, 2011, 115, 13873–13879 CrossRef CAS PubMed.
- H. Wang, Z. Wang, Y. Xiong, S. V. Kershaw, T. Li, Y. Wang, Y. Zhai and A. L. Rogach, Angew. Chem., Int. Ed., 2019, 58, 7040–7044 CrossRef CAS PubMed.
- Y. Song, J. Tan, G. Wang, P. Gao, J. Lei and L. Zhou, Chem. Sci., 2020, 11, 772–777 RSC.
- Y. Shi, P. Zhang, D. Yang and Z. Wang, Chem. Commun., 2020, 56, 10982–10988 RSC.
- J.-R. Macairan, D. B. Jaunky, A. Piekny and R. Naccache, Nanoscale Adv., 2019, 1, 105–113 RSC.
- Y. Yang, W. Kong, H. Li, J. Liu, M. Yang, H. Huang, Y. Liu, Z. Wang, Z. Wang, T.-K. Sham, J. Zhong, C. Wang, Z. Liu, S.-T. Lee and Z. Kang, ACS Appl. Mater. Interfaces, 2015, 7, 27324–27330 CrossRef CAS PubMed.
- K. Bramhaiah, R. Bhuyan, S. Mandal, S. Kar, R. Prabhu, N. S. John, M. Gramlich, A. S. Urban and S. Bhattacharyya, J. Phys. Chem. C, 2021, 125, 4299–4309 CrossRef CAS.
- S. Kar, K. Bramhaiah, N. S. John and S. Bhattacharyya, Chem. – Asian J., 2021, 16, 1138–1149 CrossRef CAS PubMed.
- S. Wu, Z. Pan, R. Chen and X. Liu, Long afterglow phosphorescent materials, Springer, 2017 Search PubMed.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS PubMed.
- Z. Xu, C. Climent, C. M. Brown, D. Hean, C. J. Bardeen, D. Casanova and M. O. Wolf, Chem. Sci., 2021, 12, 188–195 RSC.
- Q. Zhang, D. Tsang, H. Kuwabara, Y. Hatae, B. Li, T. Takahashi, S. Y. Lee, T. Yasuda and C. Adachi, Adv. Mater., 2015, 27, 2096–2100 CrossRef CAS PubMed.
- M. A. Bryden and E. Zysman-Colman, Chem. Soc. Rev., 2021, 50, 7587–7680 RSC.
- J. Jia, W. Lu, Y. Gao, L. Li, C. Dong and S. Shuang, Talanta, 2021, 231, 122350 CrossRef CAS PubMed.
- Z. Li, Y. Han, F. Nie, M. Liu, H. Zhong and F. Wang, Angew. Chem., Int. Ed., 2021, 60, 8212–8219 CrossRef CAS PubMed.
- Z.-W. Li, L.-Y. Peng, X.-F. Song, W.-K. Chen, Y.-J. Gao, W.-H. Fang and G. Cui, J. Phys. Chem. Lett., 2021, 12, 5944–5950 CrossRef CAS PubMed.
- J. Liu, H. Zhang, N. Wang, Y. Yu, Y. Cui, J. Li and J. Yu, ACS Mater. Lett., 2019, 1, 58–63 CrossRef CAS.
- R. Bhuyan, K. Bramhaiah and S. Bhattacharyya, J. Colloid Interface Sci., 2022, 605, 364–372 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Deconvoluted O 1s, and C 1s XPS spectra, PL and PLE spectra of S-dots in the solution phase. Temperature-dependent stability plot, XRD spectra of S-dot/B2O3 and S-dot/biuret composites at various pH values, and plot of phosphorescence intensity versus S-dot weight in the B2O3 matrix, temperature-dependent phosphorescence, and TADF plots along with stability plots. Long afterglow emission videos of S-dot/B2O3 and S-dot/biuret composites under UV light illumination. See DOI: 10.1039/d1ma00902h |
| ‡ Equal contributions. |
| This journal is © The Royal Society of Chemistry 2022 |