
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA promising scalable route to construct GO-based laminate membranes for antifouling ultrafiltration†
Yusen
Meng
,
Yuqing
Qiao
,
Haifeng
Zhou
,
Jingye
Li
 and
Bowu
Zhang
*
and
Bowu
Zhang
*
MOE Key Lab of Resource Chemistry, Shanghai Key Lab of Rare Earth Functional Materials, College of Chemistry and Materials Science, Shanghai Normal University, Shanghai, 200234, China
First published on 14th April 2022
Abstract
Graphene oxide (GO) laminate membranes have been deemed as promising membrane materials for a variety of applications, but they suffer from poor stability in solvents and difficulty in large-scale preparation. Herein, a scalable method was used to fabricate GO-based laminate membranes in a large area by dip-coating a GO/polyvinyl alcohol (PVA) mixed dispersion on a commercial filter paper, followed by borate crosslinking for enhancing the stability. Micromorphological investigation demonstrated that an even and continuous GO/PVA layer was constructed on a filter paper, which exhibited obviously laminated assembly architecture. Membrane testing confirmed that the water flux and molecular weight cut-off (MWCO) of the obtained GO/PVA composite membrane could be facilely tuned by changing the content of GO or PVA in the mixed dispersion. Moreover, multiple recycling tests demonstrated that about 86% of the water flux of the GO/PVA composite membrane was recovered by water cleaning even after 4 cycles, exhibiting good anti-protein-fouling performance. Additionally, the composite membrane also showed good tolerance to acidic and alkaline solutions, organic dye separation capability and excellent antibacterial property. This research affirmed that dip-coating a GO/PVA mixed dispersion is a potential way for the large-scale preparation of GO-based laminate membranes and broadening the prospects of GO-based laminate membranes in various separation fields.
Introduction
As an emerging membrane material, graphene oxides (GOs) have attracted extensive attention in recent decades, due to their unique single-atom thickness and two-dimensional structure, and incredible multifunction in antibacterial property, amphiphilicity and processibility,1–3 which have been used as fillers to prepare mixed-matrix membranes,4 or building blocks to construct laminated GO membranes.5 Particularly, the laminated GO membrane has unhindered water transport through the two-dimensional frictionless nanochannel network composed of nonoxidized regions of adjacent stacked GO sheets,6 and tunable nano-size sieves for small molecules and ions, which emerges the great prospect of using GO membranes in water purification and solute separation.7 However, in fact, the practical application of laminated GO membranes is facing challenges from the poor stability under aqueous conditions and the difficulty to batch preparation in a large area. As it is well known, the laminated GO membranes have an inherent tendency to swelling in water due to the high hydrophilicity of sheets,8 causing significant interlayer distance (d-spacing) enlargement and even structural collapse, thereby losing their separation properties,6,9 which heavily limits their practical applications,10 especially under aqueous conditions. Therefore, enhancing the mechanical and structural stability of GO-based membranes in solutions is another major technical prerequisite towards practical applications. To date, many methods including partial reduction,11,12 crosslinking,13–15 nanomaterial intercalating,16 and cation complexing17–19 have been developed to solve this stability problem. Therein, the crosslinking method is comparatively suitable for massive production due to its low cost, high efficacy and easy implementation.Till date, the most frequently used method to produce GO-based laminate membranes is the self-assembly of GO flakes by filtration of GO dispersions on a porous filter support,20 which requires large volumes of liquid, significant time, labor and energy, making its scalability a disadvantage. For laminated GO membrane construction, other solution processes such as layer-by-layer assembly,21 spin-coating,22 and drop-coating23 are in a similar dilemma to scale-up production. Gao's group24 and Kim's group25 almost at the same time reported that GO sheets capably form liquid crystals (LCs) in dispersions for their high aspect ratio and excellent dispersibility in water. With the increase in concentration, the GO dispersion usually goes through an isotropic-to-nematic phase transition (forming LCs) at around 0.23 wt% of concentration,26 which enables it to form highly oriented graphene-based fibers,27 films,28 aerogels,29etc., on a large scale via solution processes since the dispersion viscosity increases and the plasticity reinforces in the LC texture. For instance, Akbari et al. prepared high-concentration GO LCs by concentrating the dilute dispersion with superabsorbent polymer hydrogel beads, and fabricated large-area graphene-based nanofiltration membranes by casting GO LCs on a porous support coupled with hydrazine vapor reduction.30 Kim et al. fabricated a deoxygenated GO dispersion with high concentration (10 mg mL−1) similar to the large-scale preparation of continuous nanofiltration membranes by slot-die coating.31 While the GO LC is still a non-Newtonian fluid,32 highly relying on GO concentration, average aspect ratio of GO sheets, and the liquid environment, such as pH or ionic concentration,26 lack of enough colloidal stability, which leads to macroscopic discontinuous structure defects such as voids or holes,30 in the casting process affects the separation performance of products. To improve the continuity of membranes, a highly concentrated GO dispersion is the necessary prerequisite for laminated GO membrane preparation by the casting method, which complicates the production process and pushes up the cost. However, the GO suspensions with high concentrations (typically above 1 wt% of composition) would vitrify and exhibit a solid-like elastic behavior.33 This gelation of GO handicaps the uniform alignment of GO in macroscopic assemblies, thus degrading the relevant materials’ performances.34 Therefore, the formation of GO LCs at a very low GO concentration with good colloidal stability is an intriguing pathway to construct the laminated GO membrane via low-cost and high-throughput solution processes. The incorporation with polymer additives has been confirmed as an effective way for the improvement of GO LCs’ stability,35 and GO alignment in composites.36
Polyvinyl alcohol (PVA) is an inexpensive, biofriendly and stable polymer, which is compatible with GO sheets at molecular level,37 and can crosslink with GO sheets by chemical conjugation,38 thereby enhancing the mechanical strength and structural stability of the GO/PVA nanocomposite. As reported by Li et al., PVA-intercalated GO membranes were facilely achieved, which could toughly withstand ultrasonication destruction and showed stable and reliable desalination performance.39 In our experiments, we found that the addition of small amounts of PVA solution into the dilute GO dispersion could effectively increase the colloid stability and viscosity of the latter, which can uniformly spread on a porous support. Inspired by this, we here designed a simple method to fabricate GO-based laminate membranes in a large area by dip-coating a GO/PVA mixed dispersion followed by borate crosslinking. A commercial porous filter paper, made from herbs in nature, is abundant, cheap, renewable and rich in hydroxyl groups. In this study, it was used as the porous support for the GO/PVA membrane, instead of the common synthetic polymer membranes, to enhance the interfacial combination between the GO/PVA layer and the support, because the hydroxyl groups on the interface could be crosslinked by sodium borate. The rheological investigation demonstrated the GO/PVA mixed dispersion has higher viscosity than that of GO dispersion with the same content of GO sheets, and exhibited an LC-like viscoelastic behavior. Via dip-coating and borate crosslinking, it was easy to fabricate large-area and continuous GO-based laminate membranes (∅ 20 cm) with good stability in water. The developed membranes exhibited excellent antifouling and antibacterial properties, and controllable water permeability and solute rejection, which is affected by the GO and PVA concentration in the mixed LCs. Since dip-coating is a popular way of creating a thin and uniform coating onto flat or cylindrical substrates for industrial purposes,40,41 it is expectable that the dip-coating method proposed in this work would facilitate the commercialization and practical application of GO-based laminate membranes.
Experimental
Chemicals and materials
Qualitative filter paper (mainly composed of cotton fibers with a pore size of 30–50 μm), graphite powder (<20 μm), sulfuric acid (H2SO4, 96%), potassium permanganate (KMnO4), hydrogen peroxide (H2O2, 30%), potassium persulfate (K2S2O8), phosphorus pentoxide (P2O5), PVA granules (1799), ethanol, N-methyl-2-pyrrolidone (NMP), acetic acid and sodium borate were purchased from Sinopharm Chemical Reagent Co., Ltd (China). Polyethylene glycol (PEG) and polyethylene oxide (PEO) with different molecular weights, methylene blue (MB), congo red (CR), acid red 29 (AR29), bismarck brown R (BBR), bovine serum albumin (BSA) and lysozyme (LZ) were obtained from Shanghai Aladdin Biochemical Technology Co., Ltd. Unless otherwise stated, all chemicals were used as purchased without further purification, and the water was purified using a lab-scale ultra-pure water purification system (Smart S15, Hitech Instruments Co., Ltd, Shanghai, China).Synthesis of GO sheets
The GO sheets used in this study were synthesized by a modified Hummers’ method,42 with details as follows. Graphite powder (5 g), concentrated H2SO4 (300 mL), K2S2O8 (4.2 g) and P2O5 (6.2 g) were added into a 500 mL flask and mixed well. After reacting at 80 °C for 5 h, the mixture was cooled to room temperature, and diluted with 2 L of water. Subsequently, vacuum filtration was carried out using a 0.45 μm cellulose acetate membrane with vacuum assistance to separate the solid in the above mixture. Washed with abundant water, the black cake from vacuum filtration was naturally dried at room temperature for 2 days, which is called the pre-oxidized graphite. The pre-oxidized graphite powder was dispersed in 200 mL concentrated H2SO4 in the ice-water bath (0 °C), and 15 g of KMnO4 was slowly added under gentle stirring. When a uniform mixture was obtained, further 2 h stirring at 35 °C was implemented. Then, 2 L of water was added into the mixture under stirring; subsequently, the color of the diluted mixture gradually turned to bright yellow, accompanied by dropping 20 mL of 30% H2O2 solution. After 2 days of standing, the supernatant was discarded and the residual mixture was mechanically stirred for 24 h. The obtained bright yellow dispersion was put into a dialysis bag and immersed in plenty of deionized water until the pH value of the dialysate was close to neutral (pH 5–7). Finally, the dialyzed dispersion was filtrated using two layers of non-woven fabric as a filter medium to remove the black impurities and obtain the purified GO dispersion. For measuring the contention, 20 mL of GO dispersion was added in a watch glass, evaporated in an oven at 80 °C and further dried in a vacuum oven at 60 °C. The GO content was calibrated by weighing the residual GO in watch glass. The obtained GO sheets were characterized by atomic force microscopy (AFM) (Fig. S1, ESI†) for measuring their areal size (∼15 μm) and height profile (∼1.1 nm).Fabrication of GO/PVA composite membranes
Scheme 1 depicts the diagram of GO/PVA composite membranes via dip-coating and borate crosslinking, as described next. PVA granules were dissolved in water at 90 °C under vigorous stirring to form 5 wt% of PVA solution. Following this, 50 mL of the prepared GO dispersion was first subjected to ultrasonic treatment for 10 min and then mixed with a certain amount of PVA solution and water to obtain a series of GO/PVA mixtures containing different mass ratios of GO and PVA. To obtain even and continuous GO/PVA composite membranes, a modified dip-coating method was designed and performed in steps as follows: first, a piece of commercial filter paper was laid and fixed on a stainless-steel wire sieve (80 mesh) using tape, and then immersed in a GO/PVA mixture at room temperature for 10 min. After that, the filter paper was taken out from the mixture, subsequently incubated at 50 °C for 1 h, and then immersed in a 5 wt% sodium borate solution for 12 h. Finally, the composite membrane was taken out, washed with plenty of water for 15 min to remove the residual sodium borate on the membrane, and dried in an air-drying oven at 40 °C overnight. For the sake of contrast, a series of GO/PVA composite membranes were prepared from the GO/PVA mixed dispersions with different GO and PVA contents. For brevity, the borate crosslinked GO/PVA composite membranes were named according to the GO and PVA contents of mixed dispersions. For example, the composite membrane obtained from a GO/PVA dispersion containing 6 mg mL−1 GO and 1.8 mg mL−1 PVA could be denoted as G6P1.8 membrane. Correspondingly, the un-crosslinked composite membrane prepared from a GO/PVA dispersion containing 6 mg mL−1 GO and 1.8 mg mL−1 PVA was named Uc-G6P1.8 membrane. The rest may be inferred by analogy. | ||
| Scheme 1 Preparation diagram of stable GO/PVA composite membranes via dip-coating and borate crosslinking. | ||
Membrane performance test
The water performance of the composite membrane was evaluated by a customized ultrafiltration module with 12.56 cm2 of effective filtration area through the dead-end filtration mode. To obtain a stable pure water flux (PWF), plenty of water was permeated through the composite membrane under 1 bar of pressure and 360 rpm of magnetic stirring until the effluent became constant. The following formula was used to calculate the PWF of the concerned GO/PVA composite membranes: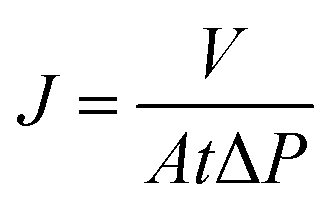 | (1) |
Meanwhile, the rejection ability of membranes was assessed by 100 ppm of aqueous solutions containing PEG or PEO with different molecular weights. The rejection ability of the membranes was measured using eqn (2):
| R = (C0 − Cp)/C0 × 100% | (2) |
Determination of molecular weight cut-off (MWCO), mean effective pore size and distribution
If ignoring the interactions of solutes to the pores of membranes, the MWCO, mean effective pore size (μp), and pore size (dp) distribution could be evaluated by neutral solute separation experiments. Here, PEG with 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 (10 kDa) and 20000 g mol−1 (20 kDa) of average molecular weight and PEO with 100000 g mol−1 (100 kDa), 300000 g mol−1 (300 kDa) and 600000 g mol−1 (600 kDa) of average molecular weight were selected as the solutes for the preparation of aqueous feed solutions. The separation experiments were performed on a dead-end ultrafiltration cell (50 mL) under 1 bar of pressure and 360 rpm of magnetic stirring by using 100 ppm of solute solution as feed. A total organic carbon tester (TOC-VCPN, Shimadzu, Japan) was used to determine the solute concentration in the feed and permeate. The Stokes diameter (ds, nm) could be related to the molecular weight (M, g mol−1) as follows:43
000 g mol−1 (10 kDa) and 20000 g mol−1 (20 kDa) of average molecular weight and PEO with 100000 g mol−1 (100 kDa), 300000 g mol−1 (300 kDa) and 600000 g mol−1 (600 kDa) of average molecular weight were selected as the solutes for the preparation of aqueous feed solutions. The separation experiments were performed on a dead-end ultrafiltration cell (50 mL) under 1 bar of pressure and 360 rpm of magnetic stirring by using 100 ppm of solute solution as feed. A total organic carbon tester (TOC-VCPN, Shimadzu, Japan) was used to determine the solute concentration in the feed and permeate. The Stokes diameter (ds, nm) could be related to the molecular weight (M, g mol−1) as follows:43
For PEG:
| ds = 33.46 × 10−12 × M0.557 | (3) |
For PEO:
| ds = 20.88 × 10−12 × M0.587 | (4) |
The normal logarithm of solute rejection (R) has a linear relationship with solute ds. Here, μp is defined as the ds at R = 50%, and the geometric standard deviation (σp) is determined as the ratio of ds at R = 84.13% over that at R = 50%, and the MWCO is the molecular weight of the solute that rejected 90% (R = 90%) by the membrane.43 The pore size distribution of membranes can be expressed as the following probability density function:43
 | (5) |
Antibacterial activity test
The antibacterial property of the composite membrane to Escherichia coli (ATCC 8739) was tested by TüV SüD Products Testing (Shanghai) Co. Ltd. The test was implemented with reference to ISO 22196:2011 (Measurement of antibacterial activity on plastics and other non-porous surfaces), with details as follows: the tested membranes were cut into 50 × 50 mm dimension and wiped with 70% ethanol in water. Then, the specimens were stacked in 250 mL wide-mouth jars with screw caps, respectively. After this, 0.2 mL of test inoculum with a bacterial density of 9.0 × 105 CFU mL−1 (colony forming units per millilitre) and 0.05% Polysorbate 80 was put onto each sample. The surface area of the cover film was 1600 mm2. After inoculation (0 h), 100 mL neutralizing solution was added to each of the jars as quickly as possible. By shaking the jars vigorously for 1 min, serial dilutions (such as 10, 102, and 103) were prepared with water and plated on nutrient agar. A same inoculation procedure was carried out on another test sample and control sample. After culturing the inoculated membranes at 37 °C for 24 h, 100 mL of neutralizing solution was added to the jars under vigorous stirring for 1 min, and serial dilutions with water were made and plated on nutrient agar. All plates were incubated at 37 °C for 48 h. From the microbial count results obtained, the antibacterial activity (R) was calculated using the following equation:| R = logUt − logAt | (6) |
| Reduction = (U0 − At)/U0 × 100% | (7) |
Fouling resistance test
Two proteins, BSA and LZ, were chosen as model organic pollutants to assess the fouling resistance of membranes to proteins in the filtration process. In the static adsorption experiment of the test membrane, the G6P1.8 membrane (12.56 cm2) was first immersed in 40 mL BSA or LZ solution (1 g L−1) for 5 hours. The BSA or LZ solution before and after static adsorption was tested by the G6P1.8 membrane, and the eluants from the G6P1.8 membranes were absorbed from the BSA or LZ solution by UV absorption spectra. The adsorption capacity of the membrane can be calculated by comparison. After evaluating the initial PWF (Jw1) of the membrane, 40 mL BSA or LZ solution (1 g L−1) was injected into a dead-end ultrafiltration cell (50 mL) and filtered under 1 bar and 360 rpm magnetic stirring. Then, the membrane was rinsed by plenty of water for 15 min without disassembly, and used to treat 40 mL protein solution again under the same conditions. After 4 repeated cycles, the membrane was unloaded from the device and washed thoroughly with water for 15 min to recover the membrane. The water permeance of the recycled membrane (Jw2) was then tested following the above-described step. Based on this, the flux recovery rate of the membrane was determined to reflect the anti-protein-fouling performance of the membranes as follows: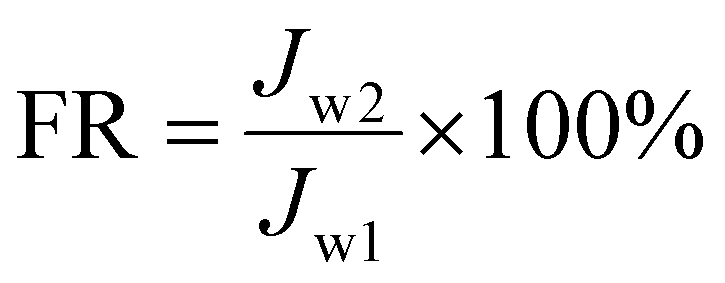 | (8) |
Dye removal test
Four organic dyes including methyl blue (799.8 Da), congo red (696.7 Da), chromotropic acid 2R (468.4 Da) and bismarck brown R (461.4 Da) were selected to characterize the ability of the GO/PVA composite membranes to remove organic dyes from water. The four organic dyes were dissolved in water to obtain a dye solution of concentration 10 mg L−1. In the dye removal test, a cross-flow ultra-/nano-filtration system was used at an applied pressure of 1 bar. The dye concentration of the filtrate was assayed by UV-Vis spectrophotometry to determinate the rejection of dyes according to eqn (2).Instruments and characterization
Fourier transform infrared (FT-IR) spectra were recorded using a Thermo Scientific Nicolet iS10 FT-IR spectrometer in the range of 4000–600 cm−1 in an attenuated total reflectance (ATR) module. Raman spectra were recorded using a Renishaw inVia plus laser Raman spectrometer at an excitation wavelength of 638 nm by scanning in the range of 500–2000 cm−1. The water contact angles were measured using an Attention Theta system (KSV Instruments Ltd., Helsinki, Finland). A 5 μL water droplet was dropped onto the surface of membranes (dried in an air oven at 60 °C overnight) in air atmosphere and the profile of the droplet was recorded and analyzed using the software from the instrument manufacturer. Every membrane was tested 3 times at different positions at least for averaging the values. The micro-morphologies of membranes were observed using a field-emission scanning electron microscope (FE-SEM, S4800, Hitachi, Japan) at an accelerating voltage of 5 kV. To access the stability of the composite membrane in aqueous solutions, the XRD patterns of membranes in both dry and wet states were recorded using an X-ray diffractometer (D/MAX2200, Rigaku Co., Japan) equipped with a copper source of monochromatic radiation (Cu Kα, λ = 0.15418 nm) operating at 30 mA and 40 kV. The tests were carried out in the reflection mode at room temperature with 2θ in the range of 5–15°, at a scanning speed of 10 deg min−1 and a step size of 0.02°. Before testing, the dry membrane was stored in a desiccator at room temperature overnight, and the wet one was immersed in deionized water for 5 h and then blotted with paper. The polarizing microscopic image was obtained using a LEICA DM750P polarizing microscope in the polarizing module. Dynamic viscosity was measured using a rheometer (MCR 102, Anton Paar GmbH, Austria) at a rotational frequency of 10 s−1 at 25 °C.Results and discussion
Fig. 1(a) shows that the GO dispersion (∼6 mg mL−1) was dripping down the dropper, while the GO/PVA (GO 6 mg mL−1, PVA 1.8 mg mL−1) mixed dispersion exhibited a continuous and slow flow, demonstrating the enhanced viscoelastic property of the GO dispersion after PVA addition, which could be attributed to the formation of hydrogen bonding between PVA chains and GO sheets, thickening the dilute GO dispersions (6 mg mL−1).44 The observation via a polarizing optical microscope exhibits that the GO sheets were disorderly dispersed in a dispersion, but became well-aligned in the presence of PVA similar to the behavior of liquid crystals (LCs) (Fig. 1(b)), which also reflected the enhanced viscoelastic property. To investigate the enhanced viscoelastic property of the GO/PVA dispersion, the dynamic viscosity of the mixed dispersions was measured using a rotational rheometer, which contained GO and PVA of different concentrations. It is shown that the dynamic viscosity of GO/PVA dispersions increased remarkably along with the GO concentration (Fig. 1(c)) and PVA content (Fig. 1(d)). Peculiarly, the GO/PVA (GO 6 mg mL−1, PVA 1.8 mg mL−1) dispersion showed a dynamic viscosity as high as 877 mPa s, rivaling that of GO dispersions with 40 mg mL−1 of GO content.30 This phenomenon implies that the GO/PVA dispersion could be capable of fabricating a GO-based laminate membrane through scalable processes such as doctor-blade casting30,45 and slot-die coating.31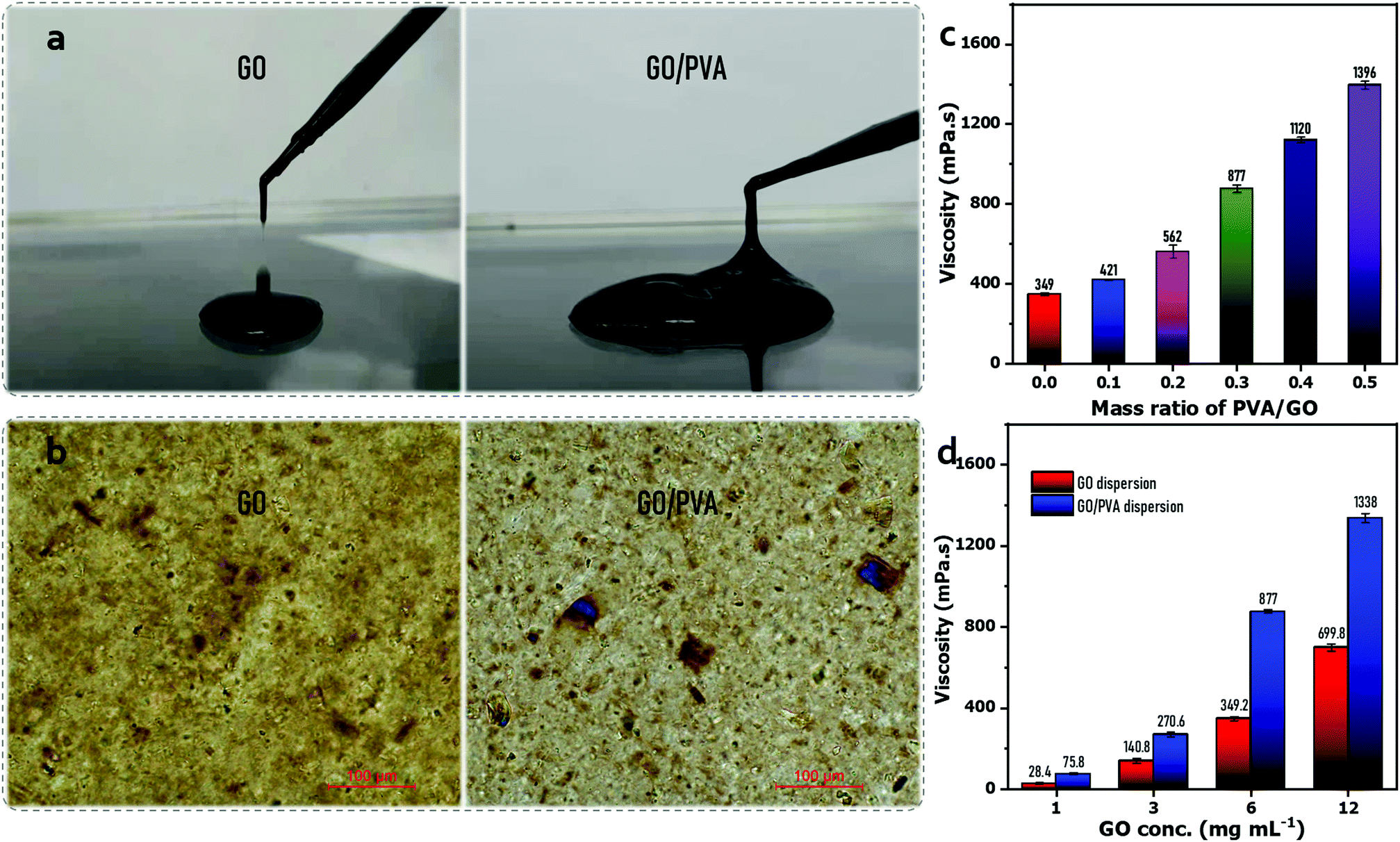 | ||
| Fig. 1 (a) Viscoelastic property of the GO (∼6 mg mL−1) dispersion and GO/PVA (GO 6 mg mL−1, PVA 1.8 mg mL−1) mixed dispersion. (b) Polarizing optical microscopic image of the GO (6 mg mL−1) dispersion and GO/PVA (GO 6 mg mL−1, PVA 1.8 mg mL−1) mixed dispersion. (c) Dynamic viscosity of the GO dispersion and GO/PVA (PVA 1.8 mg mL−1) mixed dispersion over the GO concentration. (d) Dynamic viscosity of GO/PVA dispersions (GO 6 mg mL−1) over PVA concentration. | ||
In this work, we designed a dip-coating route to fabricate a GO-based laminate membrane, as presented in Scheme 1. A large-area GO/PVA composite membrane (∅ ≥ 20 cm) was prepared easily (Fig. S2, ESI†) and the color appeared uniform and darkened along with the increase in GO content of mixed dispersions (Fig. S3, ESI†). Fig. 2(a) shows the peaks around 1649 cm−1 and 1421 cm−1, belonging to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CC of GO sheets, respectively, which disappeared after compounding with PVA, which could be attributed to the molecular level compatibility between GO sheets and PVA chains, and the high absorption intensity of the abundant hydroxyl group of PVA in IR zone. The G6P1.8 membrane exhibits a stronger and wider peak at around 3450 cm−1, ascribed to the fact that its hydroxyl groups (–OH) were much more than that of GO, while the peak of the borate crosslinked membrane was obviously weakened, affirming the decreased hydroxyl groups on PVA chains due to borate crosslinking. Additionally, a new peak near 1300 cm−1 (stretching vibration of B–O–C) appeared, indicating the successful borate crosslinking.46
O and CC of GO sheets, respectively, which disappeared after compounding with PVA, which could be attributed to the molecular level compatibility between GO sheets and PVA chains, and the high absorption intensity of the abundant hydroxyl group of PVA in IR zone. The G6P1.8 membrane exhibits a stronger and wider peak at around 3450 cm−1, ascribed to the fact that its hydroxyl groups (–OH) were much more than that of GO, while the peak of the borate crosslinked membrane was obviously weakened, affirming the decreased hydroxyl groups on PVA chains due to borate crosslinking. Additionally, a new peak near 1300 cm−1 (stretching vibration of B–O–C) appeared, indicating the successful borate crosslinking.46
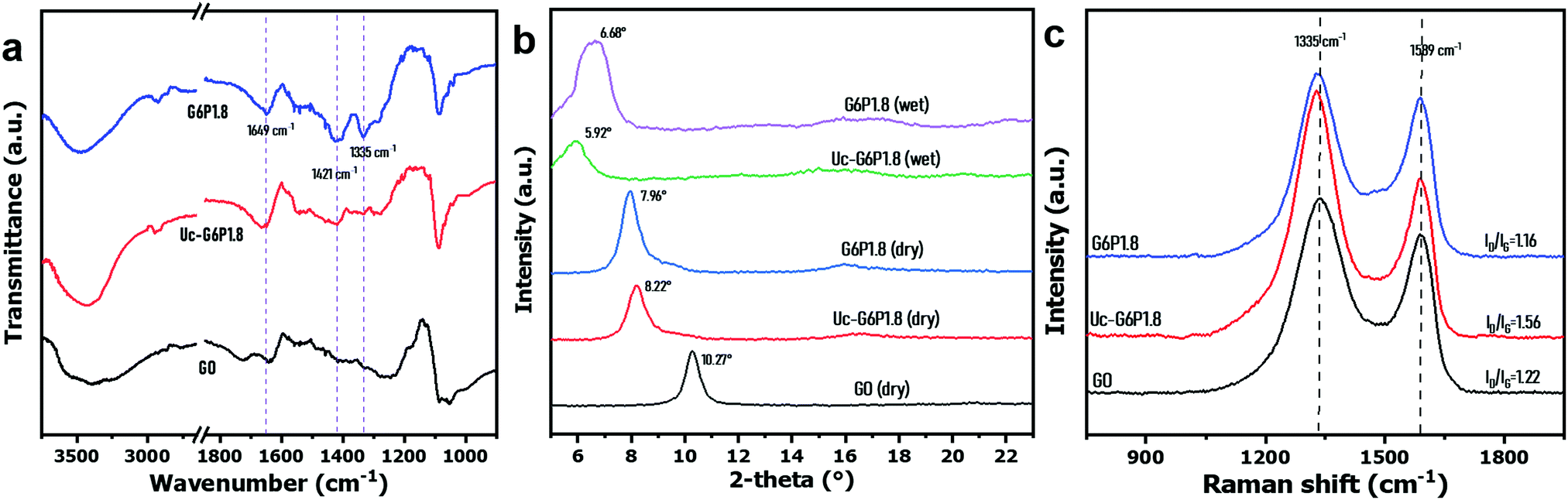 | ||
| Fig. 2 (a) FT-IR spectra, (b) X-ray diffraction pattern and (c) Raman spectra of the GO membrane and GO/PVA composite membrane before and after borate crosslinking (prepared from the GO/PVA dispersion: GO 6 mg mL−1, PVA 1.8 mg mL−1). | ||
The XRD patterns (Fig. 2(b)) reveal that the GO membrane has a diffraction peak (002) at 10.3°, but the peak shifted to 8.2° by PVA intercalation,47 and further to 8.0° by borate crosslinking. As for the GO-based laminated membrane, the undesirable swelling in water is the evil of performance deterioration and a challenge to the practical application. Herein, the XRD patterns of membranes in dry and wet states were compared to assess the swelling resistance of membranes before and after borate crosslinking. It was found that the diffraction peak (002) of Uc-G6P1.8 membrane shifted to 5.92° after water immersion for 5 h, while that of the wet G6P1.8 membrane just positioned at 6.68°, demonstrating that the borate crosslinking effectively improved the swelling resistance of the resulting GO/PVA composite membrane. Furthermore, the mechanical property in water of the composite membrane was evaluated by ultrasonication treatment. First, the composite membrane was placed in hot water (∼100 °C) for 5 min, and then treated under ultrasonication for 15 min. It was found the membrane before borate crosslinking was obviously broken, while the one after borate crosslinking remained intact. It reflects that the borate crosslinking leads to the formation of tough internal connections, and the mechanical property of the composite membrane was significantly improved (Fig. S4, ESI†).
Raman spectroscopy is frequently used to characterize the impurity, structural defects, and irregularities of GO-based materials by comparing the signal intensity of D-band to G-band (ID/IG).48 As Fig. 2(c) shows, the ID/IG values of the GO membrane, and GO/PVA composite membrane before and after borate crosslinking are 1.22, 1.56 and 1.16, respectively, indicating that the presence of PVA remarkably increases the impurities in the composite, while the borate crosslinking treatment removed redundant PVA and created robust cross-linkage between PVA and GO sheets and, therefore, resulted in lower ID/IG values of the G6P1.8 membrane than that of the GO membrane. To some extent, the borate crosslinking enhanced the regularity of the GO/PVA composite membrane.
The membrane micromorphology was observed by scanning electron microscopy (SEM). Fig. 3(a) shows that the commercial filter paper is composed of ribbon-like natural fiber bundles, with rough surface micromorphology and 20–80 μm pore size. Although the GO sheets just with about 15 μm of areal size are possible to penetrate into the pore of the filter paper in a dip-coating process, the strong hydrogen bonding would occur between PVA and the filter paper because of the plenty of hydroxyl groups on PVA and carboxyl groups on cellulose, which blocks the pores of the filter paper and is beneficial to GO sheets depositing on the surface of filter paper. However, the GO sheets also tend to form hydrogen bonds with PVA and the filter paper owing to the existence of oxygen-containing groups. Additionally, the 2D micro-structure makes it more liable to deposition on the surface of the filter paper and forms a laminated assembly layer based on the bridging and layer stacking action. The SEM image shows that there is an even and continuous skin layer formed on the surface of the filter paper after dip-coating with GO/PVA mixed dispersions (Fig. 3(b)). After borate crosslinking, the composite membrane still kept its even and continuous structure without any visible voids or holes on the membrane surface (Fig. 3(c)), demonstrating the stability of the composite membrane under aqueous conditions. Furthermore, the higher the GO concentration, the smoother the surface (Fig. S5, ESI†). Owing to the difficulty to make a brittle fracture of the filter paper in liquid nitrogen, we prepared a freestanding GO membrane and a G8P2.4 membrane to investigate the cross-sectional micromorphology of the composite membrane by dip-coating a GO dispersion (8 mg mL−1) and a GO/PVA mixed dispersion (GO 8 mg mL−1, PVA 2.4 mg mL−1) onto a commercial PVDF membrane with 0.22 μm pore size. As shown in Fig. 3(d), the G8P2.4 membrane exhibits a regular laminated stacking structure and about 2.5 μm skin thickness, proving the orderly aligned behavior of the GO/PVA mixed dispersion dip-coated onto the filter paper surface. As a comparison, the GO membrane from dip-coating shows an irregular laminated assembly with 1.8 μm thickness (Fig. S6, ESI†). This observation supports the point about the enhanced irregularity of laminar membrane by PVA addition and borate crosslinking. At the same time, it was observed that the fibers of the filter paper were uniformly covered by GO/PVA composites to form a uniform and continuous skin layer.
 | ||
| Fig. 3 Surface SEM images of (a) filter paper, and GO/PVA composite membrane (b) before and (c) after borate crosslinking. (d) Cross-sectional SEM image of the cross-linked GO/PVA composite membrane. The composite membrane was prepared from the GO/PVA mixed dispersion with 8 mg mL−1 of GO and 2.4 mg mL−1 of PVA. | ||
The surface of the filter paper was evenly coated by the GO/PVA laminate layer in success, which endows the permeation peculiarity of GO-based laminates to the composite membranes. In the dead-end filtration system (25 °C, 1 bar), the PWF of the composite membranes was significantly negatively affected by the GO and PVA concentrations of the mixed dispersions (Fig. 4(a)). This can be explained by the increase in permeation resistance through the composite membranes, as the higher GO and PVA concentrations in mixed dispersions result in a thicker GO/PVA skin layer on the surface of the filter paper. Besides this, the intercalated PVA chains in GO laminates inevitably occupy part of the channel, and thus, retard the water transmission by steric hindrance and hydrogen bonding attraction between PVA and water molecules.47 However, the capability of holding back the macromolecular organic matter of the composite membranes has been distinctly enhanced with the increase in GO and PVA concentrations. It can be observed in Fig. 4(b) that the composite membranes obtained from low GO or PVA concentration hardly reject PEG or PEO with a molecular weight ranging from 10 kDa to 600 kDa, unless the GO concentration increases to 6 mg mL−1. The molecular weight cut-off (MWCO) and mean effective pore size were calculated by the relationship curve between the rejection rate and molecular weight of PEG or PEO (Fig. 4(b)) on the basis of a well-documented method.43 The MWCO of G8P2.4, G6P1.8 and G6P1.2 membranes were 347 kDa, 415 kDa and 541 kDa, respectively, and the corresponding mean pore diameters were 14.3, 11.0 and 9.5 nm, respectively, which indicates that the obtained composite membranes can be used for the ultrafiltration (UF) process.49 In addition, the pore size distribution of the GO/PVA composite membranes narrows, and the probable pore size decreases with the increase in GO or PVA content (Fig. 4(c)), reflecting that the filter fineness of composite membranes is easily customized by controlling the GO or PVA concentration in the mixed dispersions.
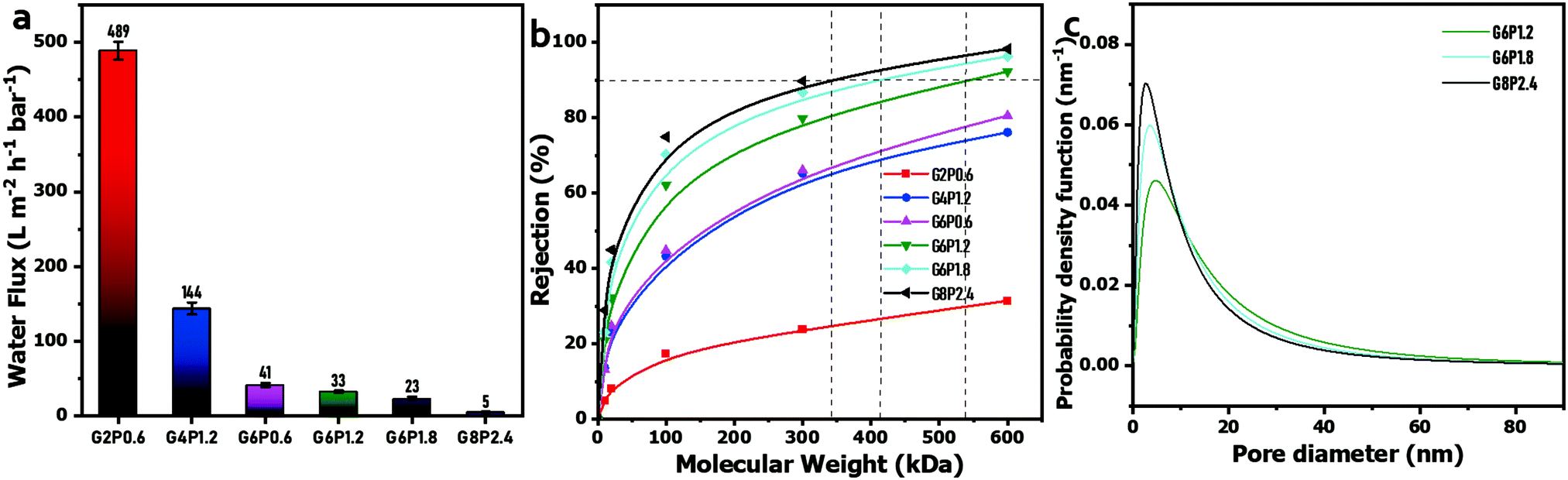 | ||
| Fig. 4 (a) PWF of GO/PVA composite membranes. (b) Plot of solute rejection versus molecular weight. (c) Pore size distribution of the GO/PVA composite membranes. | ||
Owing to its super water absorption and high porosity, the water contact angle of the commercial filter paper is hard to be measured by water dropping. After GO/PVA dip-coating and borate crosslinking, the obtained composite membranes show a water contact angle from 52° to 86°, increasing with the increase in GO and PVA contents in mixed dispersions (Fig. 5(a)). This is probably because the formation of more dense skin layers is induced by borate crosslinking, which is also reflected by the water permeation test (Fig. 4(a)). In addition, PVA-borate originally is a hydrophobic compound. With the increase in the concentration of PVA in the mixed dispersion, more PVA-borate was formed in the composite membrane, eventually, resulting in a higher water contact angle. This is also the reason that the water contact angle of composite membranes after borate crosslinking was higher than those membranes without crosslinking (Fig. S7, ESI†).
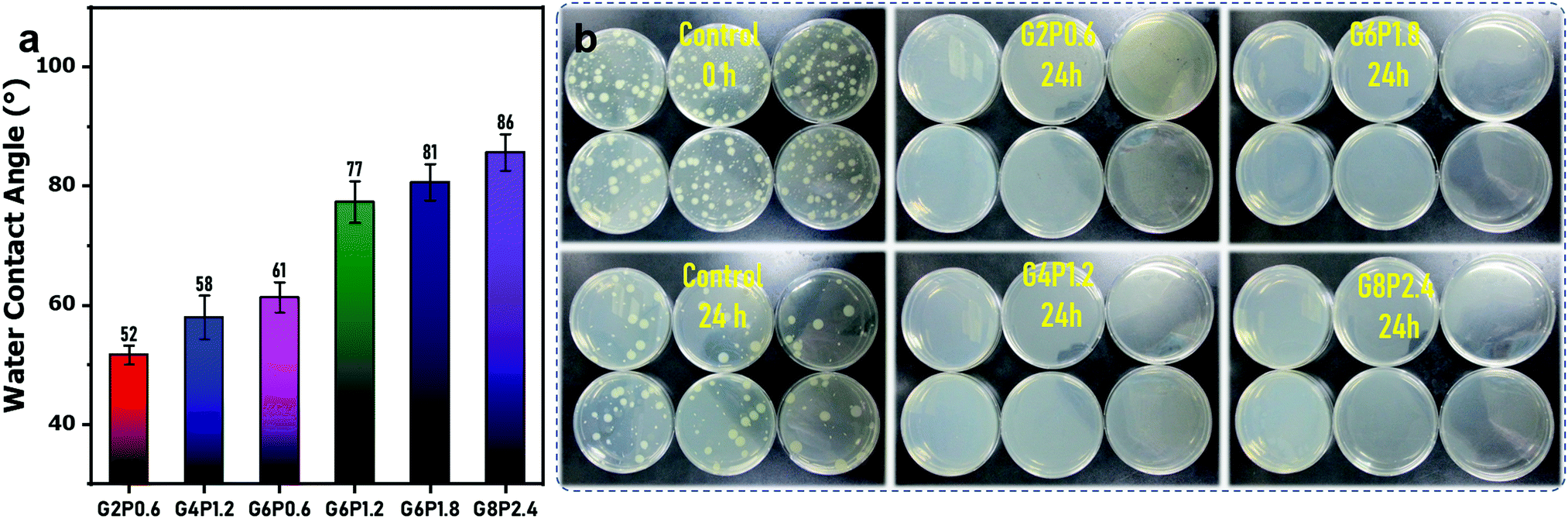 | ||
| Fig. 5 (a) Water contact angle of the composite membranes. (b) The colony of Escherichia coli cultured on the G2P0.6 membrane, G4P1.2 membrane, G6P1.8 membrane and G8P2.4 membrane. The control sample was provided by TÜV SÜD microbiological lab (Shanghai, China). | ||
It is well known that GO sheets have outstanding antibacterial properties, which is thought to be mediated by physicochemical interactions between GO and microorganisms.50,51 Three mainstream mechanisms have been proposed, namely, nanoknives from sharp edges,52 oxidative stress,50 and encapsulation or trapping of bacterial membranes by flexible GO sheets.53 As a novel antibacterial material, GO has advantages over its counterparts due to its excellent dispersibility, low cytotoxicity and good compatibility,54 which make it a promising candidate endowing antibacterial performance to various GO-embedded functional materials. Here, antibacterial testing was carried out to assess the antibacterial performance of the borate-crosslinked GO/PVA composite membranes by an international test standard (ISO 22196-2011). As the report shows, all the four GO/PVA composite membranes (G2P0.6, G4P1.2, G6P1.8 and G8P2.4) exhibited Log values of antimicrobial activity for Escherichia coli up to 6.6. That is to say, 99.9999% of the inoculated Escherichia coli were killed on the surface of those four membranes (Fig. 5(b)). As it is known, microbial proliferation and spreading on the membrane surface would result in severe membrane biofouling, further diminish the membrane performance and cause a series of operational problems such as increased trans-membrane pressure drop, decreased normalized flux and altered membrane selectivity.55 Therefore, the antibacterial membrane surface could suppress the microbial attachment and propagation, and thus, avert the bacteria-induced membrane fouling. Besides this, the antibacterial membrane could also play a role in sterilization to produce cleaner water from membrane filtration. To this point, the excellent antibacterial property of the borate-crosslinked GO/PVA composite membrane would be beneficial for its practical application in water purification or some other sterile operation.
Considering that the G6P1.8 membrane has a good balance between water flux and solute rejection, it was selected as the representative sample to evaluate the anti-protein-fouling performance and resistance to chemical washing and hot water. First, a static adsorption test was implemented by immersing the G6P1.8 membrane (12.56 cm2) in 40 mL BSA or LZ solution (1 g L−1) for 5 h. As Fig. 6(a) shows, the UV absorption around 280 nm of the BSA or LZ solution is almost interlapped before and after immersion of the G6P1.8 membrane, and the eluants from washing the immersed G6P1.8 membrane have no obvious UV adsorption. Based on the tiny difference of UV adsorption, the adsorption capacity of BSA and LZ on the G6P1.8 membrane was calculated as 33.4 μg cm−2 and 22.3 μg cm−2, respectively. Furthermore, a dynamic antifouling filtration was used to evaluate the recyclability of the G6P1.8 membrane. It is depicted that the flux of the G6P1.8 membrane decreased by about 32% when using BSA or LZ solution as feed, and about 95% PWF can be recovered just by washing with water for 15 min (Fig. 6(b) and (c)). Even after 4 cycles of fouling and washing, about 86% PWF of the G6P1.8 membrane was still recovered, whether it was contaminated by BSA or LZ, affirming the excellent antifouling performance and recyclability of the G6P1.8 membrane.
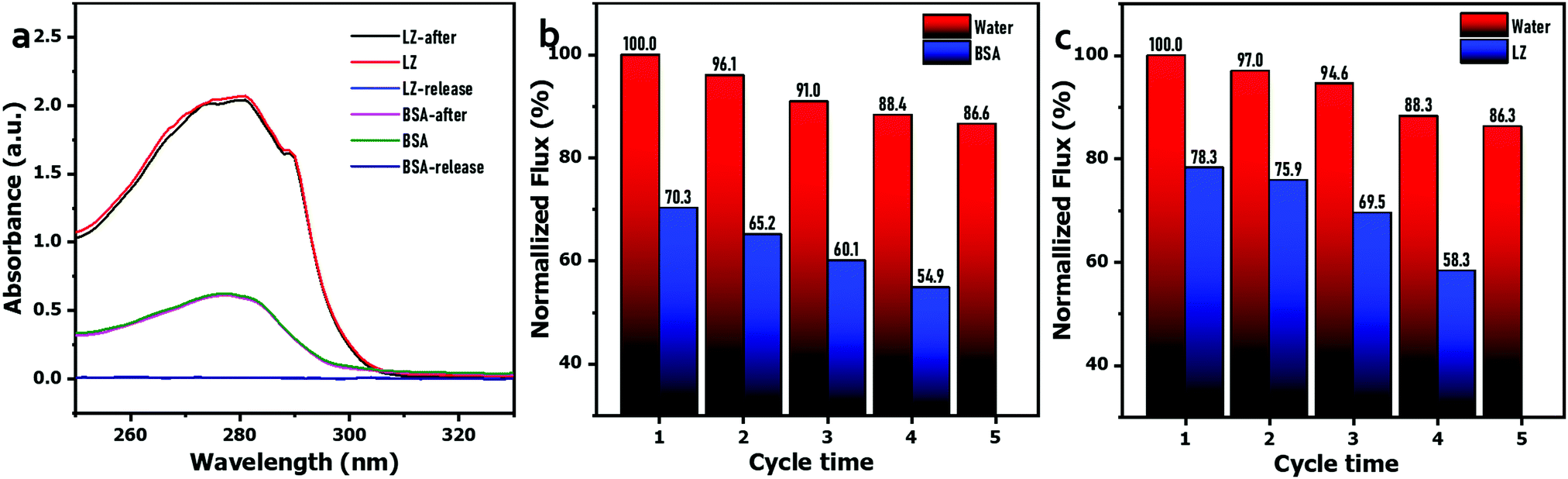 | ||
| Fig. 6 (a) UV absorption spectra of the BSA or LZ solution before and after static adsorption by the G6P1.8 membrane, and the eluants from the G6P1.8 membranes absorbed from the BSA or LZ solution. The recyclability test of the G6P1.8 membrane by fouling in (b) BSA and (c) LZ solutions. | ||
Membrane fouling is the main cause of membrane performance deteriorating in use, resulting in a lower rejection rate and a lower flux, which shorten the service life of membrane materials and drive the operation cost up. As an integral part of the operation of the membrane system, membrane cleaning is required to remove the foulants aggregated on the membrane surface and pore inside and restore the separation performance. Generally, it is easy to remove inorganic fouling by hydraulic washing or backwashing, but not including the organic fouling or biofouling, which are so stubborn that acidic or alkaline cleaning agents are often used in the cleaning process. These chemical cleaning treatments are apt to corrode the selective layer, even damage the membrane integrity, and finally lead to the scrap of membrane.56 Therefore, resistance to chemical cleaning agents is also a requirement for high-performance membranes. Herein, the G6P1.8 membranes were treated by soaking in acidic (HCl, 1 mol L−1) and alkaline (NaOH, 1 mol L−1) solutions, respectively, to evaluate the tolerance to the chemical cleaning process. Fig. 7(a) shows the water contact angle of the G6P1.8 membrane to be decreased after immersion in HCl or NaOH solutions, which is attributed to the hydrolysis of the acetate portion from the PVA under strong acidic or alkaline conditions.57 The FT-IR spectra of the G6P1.8 membrane before and after the above-mentioned chemical immersions show that the peaks near 1660 cm−1 (ascribed to acetate groups) weakened obviously after chemical immersions (Fig. 7(b)), confirming the acetate hydrolysis of the PVA under acidic or alkaline conditions,57 and the acetate absorption intensity of the NaOH-soaked G6P1.8 membrane is weaker than that of the HCl-soaked G6P1.8 membrane, indicating the higher hydrolysis degree of the former, which is affirmed by the lower water contact angle of the NaOH-soaked G6P1.8 membrane (Fig. 7(a)). Meanwhile, the peak around 1310 cm−1 (stretching vibration of B–O–C) still exists without any obvious drop, indicating no hydrolysis of the borate groups in the G6P1.8 membrane. Owing to the acetate hydrolysis, the PVA chains should be rearranged to some extent,57 which causes a slight change in the PWF and rejection ability of the G6P1.8 membrane. As it is shown in Fig. 7(c) and (d), the PWF of the HCl-soaked G6P1.8 membrane increased to 28.6 L m−2 h−1 bar−1, and the retention effect of PEO with 300 kDa of molecular weight still remained above 80%. Furthermore, the recyclability test exhibits that the flux of the G6P1.8 membrane treated by soaking in HCl or NaOH solutions still retained more than 80% of recovery rate (Fig. 7(e) and (f)) even after 4 cycles. It follows that the GO/PVA composite membrane can maintain its membrane performance whether it is cleaned with acidic or alkaline reagents, meaning its good performance in terms of chemical stability, cleanliness and tolerance.
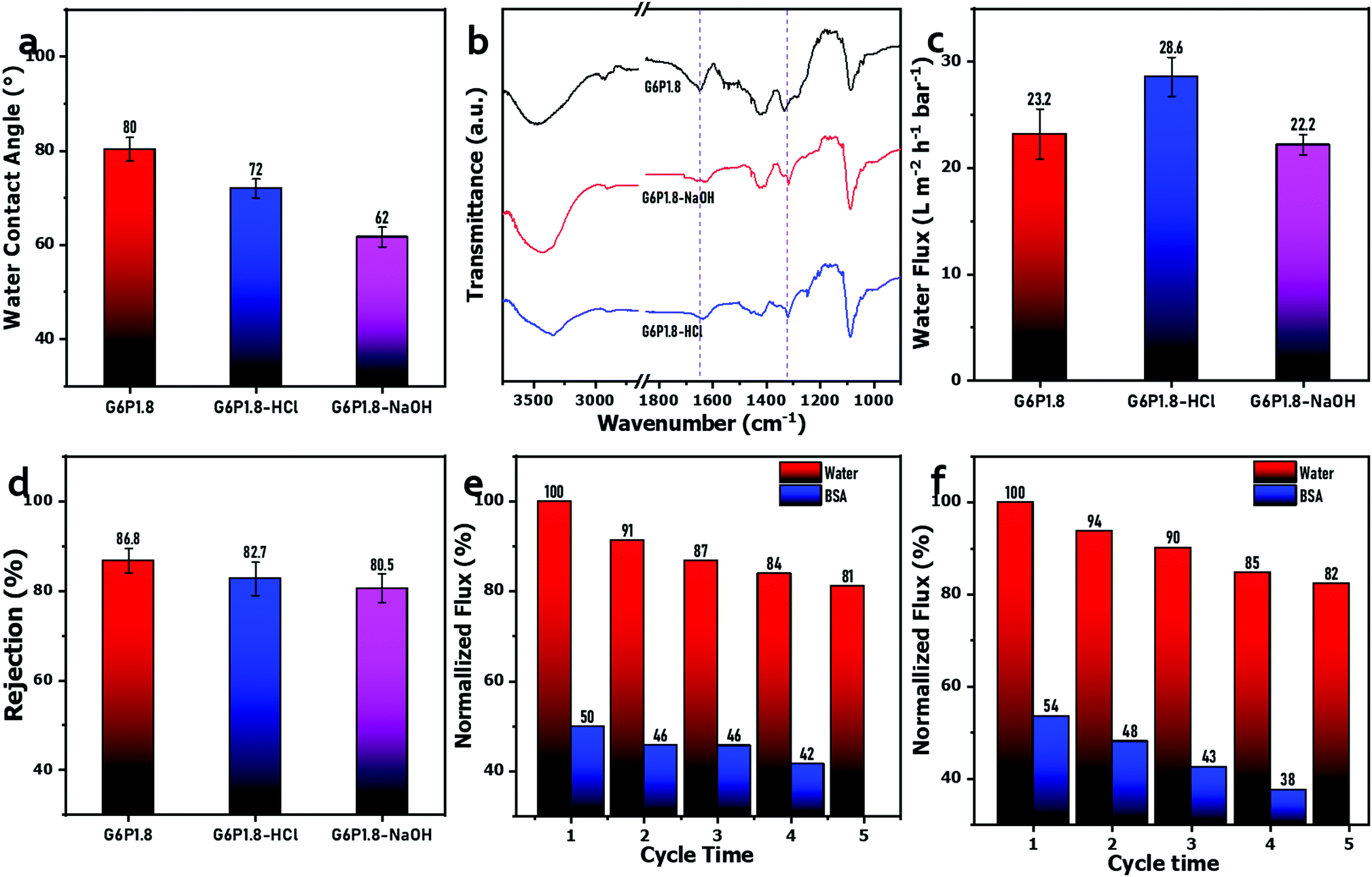 | ||
| Fig. 7 (a) Water contact angle, (b) FT-IR spectra, (c) PWF and (d) rejection rate (PEO with 300 kDa of molecular weight) of the G6P1.8 membrane before and after 24 h soaking in a HCl solution (1 mol L−1) or NaOH solution (1 mol L−1). The multiple recycling filtration test of the G6P1.8 membrane after soaking in (e) HCl and (f) NaOH solutions, using a BSA solution as the feed and water as the cleaning reagent. | ||
Additionally, the permeation of organic solvents through the G6P1.8 membrane was also tested via dead-end filtration, and is depicted in Fig. 8(a). It can be seen that the fluxes of NMP, acetic acid, and ethanol as the filtration solution are just 5.97 L m−2 h−1 bar−1, 7.88 L m−2 h−1 bar−1, and 9.55 L m−2 h−1 bar−1, respectively, which are significantly lower that of pure water (23.9 L m−2 h−1 bar−1). The flux is negatively correlated with the molecular weight of the organic solvent, mainly because the water molecule has a small volume and can easily pass through the pores of the membrane, while the organic solvent molecules have a larger size and a higher steric hindrance, making them difficult to transport through the composite membrane. This permeation difference between water and organic solvents endows the GO/PVA composite membrane with the potential for the separation of organic solvent aqueous mixtures. Furthermore, we also tested the performance of the G6P1.8 membrane for organic dye solution separation via a cross-flow filtration system. As shown in Fig. 8(b), the filtration flux of the G6P1.8 membrane for the four dye solutions was 83.4%, 76.7%, 66.8% and 64% of the PWF, corresponding to the solutions of AR29, BBR, CR and MB, respectively, indicating the good flux retention of the membrane under working conditions. In the meantime, the rejection rate of the four above-mentioned dyes was 43.8%, 47.8%, 55.7% and 67.1%, respectively (Fig. 8(c)). The trends of flux and rejection rate well conformed to the molecular weight sequence of the four dyes, revealing the size selectivity of the G6P1.8 membrane. However, it should be noted that the molecular weight of AR29 is slightly higher than that of BBR, but it has a higher flux but a lower rejection rate than those of BBR. This is because AR29 is a negatively charged molecule with two sulfonic groups, and subjected to strong electrostatic repulsion from the negatively charged GO sheets in the G6P1.8 membrane, thus resulting in low aggregation of dye molecules on the surface of the membrane. BBR is a positively charged molecule containing four amino groups, prone to enrichment on the membrane surface by electrostatic attraction, thus causing the concentration polarization of dyes, which negatively affects the separation efficiency of the membrane. According to Fig. 4(b), there is about 70% PEO with 100 kDa molecular weight rejected by the G6P1.8 membrane, which is much larger than that of MB (799.80 Da). This demonstrates that not only the size-selectivity and electrostatic interaction, but also the π–π interaction between dye molecules and GO sheets play their role in the dye separation by the GO/PVA composite membrane.
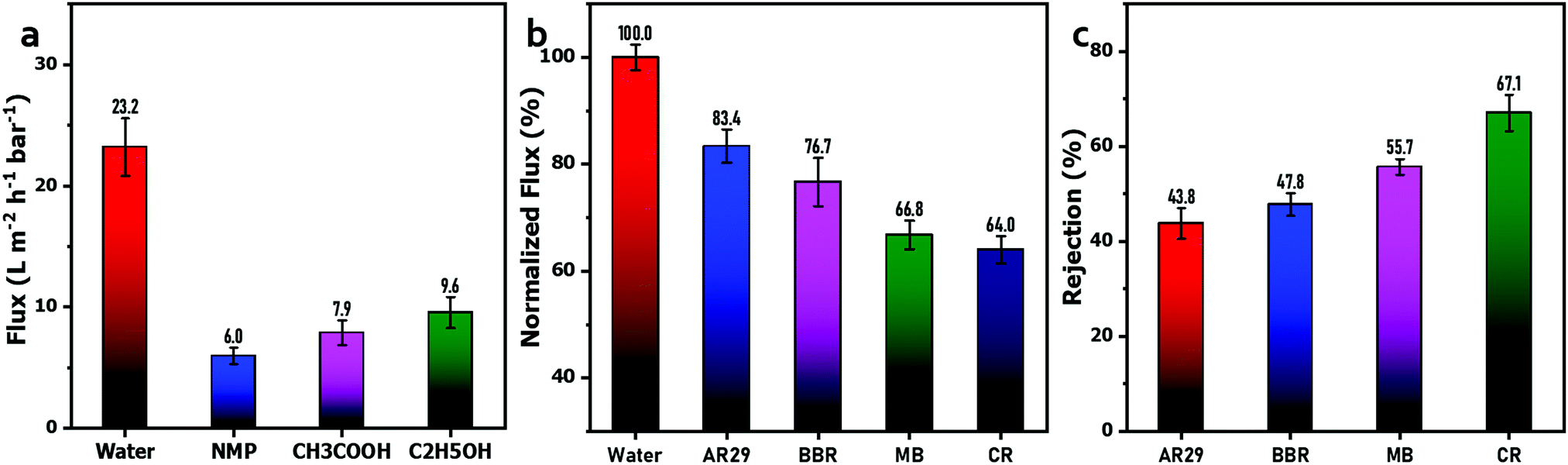 | ||
| Fig. 8 (a) Flux of C2H5OH, CH3COOH and NMP through the G6P1.8 membrane; the separation performance of the G6P1.8 membrane for organic dye solutions: (b) flux and (c) rejection retention. The concentration of bismarck brown R (BBR, 461.39 Da), acid red 29 (AR29, 468.4 Da), congo red (CR, 696.68 Da), and methyl blue (MB, 799.80 Da) was 1 g L−1 (pH ∼ 7). | ||
Conclusions
In summary, a promising scalable method based on dip-coating and borate crosslinking has been developed to construct a large-sized, continuous-structure GO-based laminate composite membrane using a GO/PVA mixed dispersion as the coating solution and a commercial filter paper as the support. The obtained composite membrane had an ultrathin skin layer with a remarkable laminate structure and a smooth surface. The apparent mean pore size and distribution, and the MWCO of the composite membrane could be effectively controlled by adjusting the GO or PVA concentration in the mixed dispersion, which made it meet the different filtration requirements, including microfiltration (MF) and ultrafiltration (UF). Furthermore, the composite membrane exhibited excellent antibacterial properties, low protein adsorption (including BSA and LZ) and good fouling resistance to protein solution filtration. It was found that the PWF recovery ratio of the representative composite membrane, G6P1.8 membrane, was up to about 86% even though it went through 4 cycles of BSA fouling and water washing, indicating its good antifouling performance and recyclability. Besides, there was just a very slight change in the membrane performance after the composite membrane was soaked in a HCl (1 mol mL−1) solution or a NaOH (1 mol mL−1) solution, which reflected that it can withstand the strong acidic and alkaline solution etching and maintain its performance in separation and fouling resistance. All in all, this research provided a convenient and promising method for the large-scale preparation of GO-based laminate membranes, which would really promote the development and application of GO-based functional membranes.Author contributions
Yusen Meng: Investigation, methodology, validation, data analysis, visualization, writing – original draft. Yuqing Qiao & Haifeng Zhou: Investigation, data analysis. Jingye Li: Funding acquisition, resources, data analysis. Bowu Zhang: Project design and administration, investigation, data analysis, conceptualization, writing – review & editing, supervision, resources, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grants 11875313 and 12075153). The authors cordially thank the supports from Shanghai Engineering Research Centre of Green Energy Chemical Engineering.Notes and references
- G. Liu, W. Jin and N. Xu, Chem. Soc. Rev., 2015, 44, 5016–5030 RSC.
- J. Zhu, X. Meng, J. Zhao, Y. Jin, N. Yang, S. Zhang, J. Sunarso and S. Liu, J. Membr. Sci., 2017, 535, 143–150 CrossRef CAS.
- H. M. Hegab and L. Zou, J. Membr. Sci., 2015, 484, 95–106 CrossRef CAS.
- S. Cong, H. Li, X. Shen, J. Wang, J. Zhu, J. Liu, Y. Zhang and B. Van der Bruggen, J. Mater. Chem. A, 2018, 6, 17854–17860 RSC.
- A. Ali, M. Aamir, K. H. Thebo and J. Akhtar, Chem. Rec., 2020, 20, 344–354 CrossRef CAS PubMed.
- R. R. Nair, H. A. Wu, P. N. Jayaram, I. V. Grigorieva and A. K. Geim, Science, 2012, 335, 442–444 CrossRef CAS PubMed.
- N. Zhang, W. Qi, L. Huang, E. Jiang, J. Bao, X. Zhang, B. An and G. He, Chin. J. Chem. Eng., 2019, 27, 1348–1360 CrossRef CAS.
- B. Rezania, N. Severin, A. V. Talyzin and J. P. Rabe, Nano Lett., 2014, 14, 3993–3998 CrossRef CAS PubMed.
- Z. Wang, F. He, J. Guo, S. Peng, X. Q. Cheng, Y. Zhang, E. Drioli, A. Figoli, Y. Li and L. Shao, Mater. Adv., 2020, 1, 554–568 RSC.
- B. Mi, Science, 2014, 343, 740–742 CrossRef CAS PubMed.
- H. Liu, H. Wang and X. Zhang, Adv. Mater., 2015, 27, 249–254 CrossRef CAS PubMed.
- R. Yi, X. Xia, R. Yang, R. Yu, F. Dai, J. Chen, W. Liu, M. Wu, J. Xu and L. Chen, Carbon, 2021, 172, 228–235 CrossRef CAS.
- Y. Gu, J. Zhao, H. Zhou, H. Jiang, J. Li, B. Zhang and H. Ma, Carbon, 2021, 183, 830–839 CrossRef CAS.
- F. Yan, C. Yu, B. Zhang, T. Zou, H. Zhao and J. Li, RSC Adv., 2017, 7, 1326–1335 RSC.
- J. Guo, H. Bao, Y. Zhang, X. Shen, J.-K. Kim, J. Ma and L. Shao, J. Membr. Sci., 2021, 619, 118791 CrossRef CAS.
- L. Chen, N. Li, Z. Wen, L. Zhang, Q. Chen, L. Chen, P. Si, J. Feng, Y. Li, J. Lou and L. Ci, Chem. Eng. J., 2018, 347, 12–18 CrossRef CAS.
- Y. Gu, B. Zhang, J. Li, M. Yu, L. Li and J. Li, J. Membr. Sci., 2020, 613, 118519 CrossRef CAS.
- L. Chen, G. Shi, J. Shen, B. Peng, B. Zhang, Y. Wang, F. Bian, J. Wang, D. Li, Z. Qian, G. Xu, G. Liu, J. Zeng, L. Zhang, Y. Yang, G. Zhou, M. Wu, W. Jin, J. Li and H. Fang, Nature, 2017, 550, 380–383 CrossRef CAS PubMed.
- K. Ching, B. Lian, G. Leslie, X. Chen and C. Zhao, Carbon, 2020, 170, 646–657 CrossRef CAS.
- J. Ye, B. Zhang, Y. Gu, M. Yu, D. Wang, J. Wu and J. Li, ACS Appl. Nano Mater., 2019, 2, 6611–6621 CrossRef CAS.
- X. Yan, W. Tao, S. Cheng, C. Ma, Y. Zhang, Y. Sun and X. Kong, Chemosphere, 2020, 256, 127118 CrossRef CAS PubMed.
- D. I. Petukhov, E. A. Chernova, O. O. Kapitanova, O. V. Boytsova, R. G. Valeev, A. P. Chumakov, O. V. Konovalov and A. A. Eliseev, J. Membr. Sci., 2019, 577, 184–194 CrossRef CAS.
- W. S. Chong, S. X. Gan, H. M. Al-Tuwirit, W. Y. Chong, C. S. Lim and H. Ahmad, Mater. Chem. Phys., 2020, 249, 122970 CrossRef CAS.
- Z. Xu and C. Gao, ACS Nano, 2011, 5, 2908–2915 CrossRef CAS PubMed.
- J. E. Kim, T. H. Han, S. H. Lee, J. Y. Kim, C. W. Ahn, J. M. Yun and S. O. Kim, Angew. Chem., Int. Ed., 2011, 50, 3043–3047 CrossRef CAS PubMed.
- R. Narayan, J. E. Kim, J. Y. Kim, K. E. Lee and S. O. Kim, Adv. Mater., 2016, 28, 3045–3068 CrossRef CAS PubMed.
- Z. Xu and C. Gao, Nat. Commun., 2011, 2, 571 CrossRef PubMed.
- Z. Liu, Z. Li, Z. Xu, Z. Xia, X. Hu, L. Kou, L. Peng, Y. Wei and C. Gao, Chem. Mater., 2014, 26, 6786–6795 CrossRef CAS.
- Z. Xu, Y. Zhang, P. Li and C. Gao, ACS Nano, 2012, 6, 7103–7113 CrossRef CAS PubMed.
- A. Akbari, P. Sheath, S. T. Martin, D. B. Shinde, M. Shaibani, P. C. Banerjee, R. Tkacz, D. Bhattacharyya and M. Majumder, Nat. Commun., 2016, 7, 10891 CrossRef CAS PubMed.
- J. H. Kim, Y. Choi, J. Kang, E. Choi, S. E. Choi, O. Kwon and D. W. Kim, J. Membr. Sci., 2020, 612, 118454 CrossRef CAS.
- J. Zhang, S. Seyedin, Z. Gu, N. Salim, X. Wang and J. M. Razal, Part. Part. Syst. Charact., 2017, 34, 1600396 CrossRef.
- Y. H. Shim, K. E. Lee, T. J. Shin, S. O. Kim and S. Y. Kim, Mater. Horiz., 2017, 4, 1157–1164 RSC.
- U. N. Maiti, J. Lim, K. E. Lee, W. J. Lee and S. O. Kim, Adv. Mater., 2014, 26, 615–619 CrossRef CAS PubMed.
- Y. H. Shim, K. E. Lee, T. J. Shin, S. O. Kim and S. Y. Kim, ACS Nano, 2018, 12, 11399–11406 CrossRef CAS PubMed.
- S. J. Mun, Y. H. Shim, G. W. Kim, S. H. Koo, H. Ahn, T. J. Shin, S. O. Kim and S. Y. Kim, Nanoscale, 2021, 13, 2720–2727 RSC.
- J. Liang, Y. Huang, L. Zhang, Y. Wang, Y. Ma, T. Guo and Y. Chen, Adv. Funct. Mater., 2009, 19, 2297–2302 CrossRef CAS.
- X. Li, T. Liu, D. Wang, Q. Li, Z. Liu, N. Li, Y. Zhang, C. Xiao and X. Feng, ACS Appl. Mater. Interfaces, 2018, 10, 21672–21680 CrossRef CAS PubMed.
- J. Sun, X. Qian, Z. Wang, F. Zeng, H. Bai and N. Li, J. Membr. Sci., 2020, 599, 117838 CrossRef CAS.
- M. Joshi and B. S. Butola, in Advances in the Dyeing and Finishing of Technical Textiles, ed. M. L. Gulrajani, Woodhead Publishing, 2013, pp. 355–411, DOI:10.1533/9780857097613.2.355.
- G. Wypych, in Handbook of Fillers, ed. G. Wypych, ChemTec Publishing, 4th edn, 2016, pp. 793–821 DOI:10.1016/B978-1-895198-91-1.50020-8.
- H. K. Dave and K. Nath, J. Water Process. Eng., 2016, 14, 124–134 CrossRef.
- P. H. H. Duong, S. Chisca, P.-Y. Hong, H. Cheng, S. P. Nunes and T.-S. Chung, ACS Appl. Mater. Interfaces, 2015, 7, 3960–3973 CrossRef CAS PubMed.
- H. Bai, C. Li, X. Wang and G. Shi, J. Phys. Chem. C, 2011, 115, 5545–5551 CrossRef CAS.
- E. Yang, H. E. Karahan, K. Goh, C. Y. Chuah, R. Wang and T.-H. Bae, Carbon, 2019, 155, 129–137 CrossRef CAS.
- N. Wang, L. Zhao, C. Zhang and L. Li, J. Appl. Polym. Sci., 2016, 133, 43246 Search PubMed.
- Y. Gu, J. Zhao, H. Jiang, J. Li and B. Zhang, Carbon, 2022, 191, 243–254 CrossRef CAS.
- P. Pachfule, D. Shinde, M. Majumder and Q. Xu, Nat. Chem., 2016, 8, 718–724 CrossRef CAS PubMed.
- A. Lee, J. W. Elam and S. B. Darling, Environ. Sci.: Water Res. Technol., 2016, 2, 17–42 RSC.
- S. Liu, T. H. Zeng, M. Hofmann, E. Burcombe, J. Wei, R. Jiang, J. Kong and Y. Chen, ACS Nano, 2011, 5, 6971–6980 CrossRef CAS PubMed.
- J. Chen, H. Peng, X. Wang, F. Shao, Z. Yuan and H. Han, Nanoscale, 2014, 6, 1879–1889 RSC.
- W. Hu, C. Peng, W. Luo, M. Lv, X. Li, D. Li, Q. Huang and C. Fan, ACS Nano, 2010, 4, 4317–4323 CrossRef CAS PubMed.
- O. Akhavan, E. Ghaderi and A. Esfandiar, J. Phys. Chem. B, 2011, 115, 6279–6288 CrossRef CAS PubMed.
- X. Zou, L. Zhang, Z. Wang and Y. Luo, J. Am. Chem. Soc., 2016, 138, 2064–2077 CrossRef CAS PubMed.
- H. J. de Vries, E. Kleibusch, G. D. A. Hermes, P. van den Brink and C. M. Plugge, Water Res., 2021, 198, 117163 CrossRef CAS PubMed.
- M. F. Rabuni, N. M. Nik Sulaiman and N. Awanis Hashim, Desalin. Water Treat., 2015, 1–12 Search PubMed.
- Y. Gu, B.-W. Zhang, Z. Guo, J.-H. Li, M. Yu, L.-F. Li and J.-Y. Li, Nucl. Sci. Tech., 2021, 32, 70 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d1ma01111a |
| This journal is © The Royal Society of Chemistry 2022 |