
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMesoporous zinc platinate and platinum nanotubes: insights into the formation mechanism and their catalytic activity†
Shwetha
Shetty
 a,
Ankita
Mathur
b,
Neha
Sakhuja
c,
Dipanwita
Chatterjee
a,
Subhajit
Kundu
a,
Aditi
Halder
d,
Navakanta
Bhat
c and
N.
Ravishankar
*a
a,
Ankita
Mathur
b,
Neha
Sakhuja
c,
Dipanwita
Chatterjee
a,
Subhajit
Kundu
a,
Aditi
Halder
d,
Navakanta
Bhat
c and
N.
Ravishankar
*a
aMaterials Research Centre, Indian Institute of Science, 560012, Bangalore, India. E-mail: nravi@iisc.ac.in
bSchool of Engineering, IIT-Mandi, Kamand-175005, Himachal Pradesh, India
cCentre for Nanoscience and Engineering, Indian Institute of Science, 560012, Bangalore, India
dSchool of Basic Sciences, IIT-Mandi, Kamand-175005, Himachal Pradesh, India
First published on 3rd August 2022
Abstract
Mesoporous nanostructures with their unique morphology, high accessible surface area, and numerous active sites have a distinct microstructural advantage over ultrafine nanoparticles. To incorporate hierarchical porosity, sacrificial template-based approaches are beneficial as they have the advantage of efficient rendering of morphology and simultaneous removal of the template material. It is also crucial to study the behavior of the template material and its reactivity with the deposited material under the reaction conditions. Herein, we have proposed an ultrafast microwave technique to synthesize mesoporous nanotubes of a novel phase zinc platinate (Zn2PtO4) using ZnO as a sacrificial template. We have observed that under the ultrafast kinetics offered by the microwave, aided with reaction temperature, the core ZnO reacts with the adhering hydrolyzed Pt precursor complex to form a mesoporous zinc platinate shell. By designing control experiments, we have attributed the mechanism of formation to a modified sacrificial template-assisted hydrolysis (STAH) process. The controlled, simple reduction step of Zn2PtO4 nanotubes using NaBH4 results in the formation of mesoporous Pt nanotubes. The mesoporous nature of Zn2PtO4 nanotubes and enhanced surface area (54 m2 g−1) aids in highly sensitive (down to 300 ppb) and selective sulphur dioxide (SO2) detection at room temperature. The mesoporous Pt nanotubes show exceptional catalytic activity towards the methanol oxidation reaction (MOR) and oxygen reduction reaction (ORR) which is attributed to their unique morphology. This synthesis strategy offers a unique approach to designing mesoporous structures of other zinc-based mixed metal oxides and conveniently converting them to their active metal counterparts.
1. Introduction
Nanotubes with porous shells and hollow cores are attractive as they exhibit superior electrical and thermal conductivities along with multiple interfaces (inner, outer, and pore walls) which are readily accessible for reactive molecules and guest species. Additional advantages such as low density, high surface area, and less usage of material make them find extensive use in diverse fields such as catalysis, sensing, energy storage, Surface-Enhanced Raman Spectroscopy (SERS), on-the-spot drug delivery, and enzyme encapsulation.1–4 Engineering bimodal types of pores of sizes <2 nm (microporous) or 2–50 nm (mesoporous) having long hollow channels >50 nm (macroporous) hierarchically can give rise to novel functionality for materials.5–8 Mesoporous materials have pore sizes adequate to host and immobilize large molecules like enzymes or DNA and are hence attractive in adsorption (or separation) and bio-sensing.9–12 The presence of innumerable diffusion channels in these materials, which connect the surface to the bulk, enables extended reaction time leading to an enhanced response to chemical and electrical signals for the analyte molecules.13Thermodynamically, introducing porosity in a crystalline solid is not feasible due to its inherent nature towards the formation of a close-packed structure.14 In our earlier work, we have shown that reaction-limited aggregation can be a strategy to make mesoporous metal nanostructures.15 The strategy is to limit the close packing order to nanoscopic dimensions and incorporate void spaces between them. Most of the synthesis methods involve pore directing agents in the form of short or long-chain polymers to create mesoporous structures of required pore dimensions.16 Dealloying is another commonly used technique where the selective dissolution of a chemically active metal from an alloy creates a self-supporting bicontinuous pore-ligament structure of the noble metal.6,17–19 However, the dictating factors for porosity are the degree of alloying between the constituent metals and the surface diffusivity of the noble metal constituent, which determine the size of the pores and ligaments thereby limiting the versatility of this technique.20,21 Literature shows that template-assisted methods are the best to form porous structures of a variety of materials.5,22–25 Different methodologies are adopted for metal nucleation within the controlled and confined spaces provided by the hard or soft templates to form porous structures of a variety of materials with precise control over porosity and morphology.5,26 The disadvantage is that the second step of template removal may be detrimental to the integrity of the porous structure and require harsher conditions.27 A successful pore replication also depends on favorable interaction between the precursor material and the template, which requires that the surface properties of hard templates be modified.28 Sacrificial templates and galvanic replacement strategies are handy in this regard as the whole process of introducing porosity and template removal happens simultaneously making the process simple, reproducible, and suitable for large-scale production.29 Easily etchable metals such as silver, tellurium, nickel, and metal oxide supports such as zinc oxide or copper oxide are commonly used to synthesize porous noble metal nanostructures with guided morphologies.30–39 However, the challenge here is that the reaction kinetics control the efficient pore formation and many times the template dissolution is time-consuming due to anisotropic etching.40 Therefore, developing an ultrafast low-cost one-step scalable process for the synthesis of surfactant-free mesoporous nanostructures is promising.
Pt and Pt-based nanomaterials are widely used as commercial electrode materials in polymer electrolyte membrane fuel cells (PEMFCs) as it offers excellent hydrogen and alcohol oxidation capabilities along with superior selectivity towards the oxygen reduction reaction.24 Pt alloyed with suitable oxophilic elements (such as Ru, Ni, Fe, Sn, etc.) shows better catalytic activity towards methanol oxidation due to the bifunctional mechanism, ligand, and geometric strain effects.41–44 Introducing porosity in a catalyst imparts high surface area and contiguous ligaments that provide electron pathway channels together improving mass transport and interfacial reaction processes beneficial in catalysis and sensing.28 In a PEMFC assembly, mesoporous Pt offers excellent mass-specific activity.45 Mesoporous Pt is also attractive for biomedical applications due to the feasibility of imaging, photothermal effect, and drug loading/delivery capability.46,47 Platinum oxide has excellent physicochemical properties and is a versatile catalyst due to the presence of oxygenated Pt, which evokes the bifunctional mechanism in effectively oxidizing the adsorbed CO species.48–50 Oxidized Pt is also shown to be an excellent hydrogen sensor.51 The hybrid PtO-ZnO shows enhanced catalytic and sensing properties as compared to ZnO due to the modification of the band structure.52,53 There has been a consistent effort to synthesize mesoporous Pt nanostructures with tailored architecture over the years.54,55 However, the reports on bulk scale synthesis of mesoporous Pt/Pt oxide-based nanostructures are limited. Novel compositions of mixed Pt oxide and ZnO can bring in a cost-effective strategy for materials development and for realizing novel functionalities and applications.
Sensor materials, which belong to the type of chemiresistors that are semiconductor metal oxides like ZnO, SnO2, WO3, and V2O5, have been used for the detection of several toxic gases like NH3, SO2, H2S, and NO2.56 SO2 is one of the noxious pollutants released from the petrochemical industry, power plants, metal smelting, food industry, and processing of wine and requires precise detection and monitoring.57 These toxic emissions from industrial and anthropogenic sources are responsible for global warming, ozone depletion, and health hazards.58 Though chemiresistors have attractive attributes like high sensitivity, thermal stability, and high shelf-life, they suffer from a drawback of high operating temperature. This is one of the important concerns with respect to power consumption, increase in circuit complexity, and cost due to the incorporation of micro heaters beneath the sensors. Moreover, the high-temperature operation may cause structure or morphology transformations in the sensing film over a period.59 The need of the hour is to develop new materials that can detect SO2 with high sensitivity and selectivity at room temperature.
Herein, we have proposed a simple, one-step, surfactant-free, microwave-induced aqueous phase scalable synthesis method to produce mesoporous zinc platinate (Zn2PtO4) nanotubes using high aspect ratio ZnO nanorods as sacrificial templates and controlled conversion of this novel phase into mesoporous Pt nanotubes. ZnO is a versatile template as it promotes the hydrolysis of metal precursors by self-dissolution resulting in the formation of hollow/porous structures on top of that ZnO can be synthesized in a variety of forms in 0D, 1D, and 3D, which helps in engineering the morphology of these porous structures.60,61 Additionally, the microwave technique offers an ultrafast rapid synthesis route with efficient energy utilization, a cost-effective and industrially viable option for the bulk synthesis of porous structures.62 The literature shows that extreme conditions of high oxygen partial pressure, high temperature, and prolonged reaction time are required for the synthesis of Zn2PtO4.63,64 In our case, the weak reducing ability of water along with superior reaction kinetics provided by the microwave reaction results in the adsorption-induced mixing of [PtCl6]− with dielectrically heated ZnO to form the zinc platinate phase. Based on control experiments we propose a modified sacrificial template accelerated hydrolysis (STAH) process wherein the template also reacts and participates in the formation of the shell material.60,65–67 We have carried out detailed electron microscopy studies to understand the mechanism of formation of the phase and morphology. The absence of any size controlling organic ligands gives these porous nanotubes free surfaces suitable for catalytic applications. The high surface area, the oxidation state of Pt, and the hierarchical porosity with one-dimensional morphology of Zn2PtO4 motivated us to test the gas sensing capabilities of this material and we have shown that indeed Zn2PtO4 shows high sensitivity, selectivity, and reproducibility towards SO2 sensing at room temperature. The mesoporous Pt nanotubes show excellent electrochemical methanol oxidation capabilities, and the ORR activity is comparable to that of the commercial platinum catalyst.
2. Experimental
2.1 Synthesis of ZnO nanorods
ZnO nanorods are synthesized by the modified Pacholski method.68,69 Zinc acetate (14.75 g, 0.76 M) is dissolved in methanol at 65 °C under vigorous stirring and a solution of potassium hydroxide (7.4 g, 4 M) in methanol is slowly added. The mixture is aged for 2 hours at 60 °C to evaporate about half the volume of the solvent. This concentrated solution is transferred to a 50 mL Teflon vessel and then sealed in a stainless-steel autoclave. This is heated to 120 °C for 8 hours. The white precipitate obtained is washed well with methanol and dried in an oven.2.2 Synthesis of porous nanotubes
10 mg of the as-prepared ZnO nanotube powder is dispersed in Milli-Q water and sonicated for 2 min. To this, 12 mM hexachloroplatinic acid is added and stirred well for it to dissolve. The entire mixture is transferred to a microwave transparent vessel provided with the Discover-CEM S-class microwave oven. Microwave heating is carried out under closed vessel conditions with high stirring. The reaction temperature is set at 200 °C for 5 min. The microwave power is set at 150 W, which ensures a ramping time of 3 min. After the system is cooled, the vessel is taken out. The obtained yellow powder is separated from the solvent by centrifugation at 2000 rpm for 2 min. The pH of the supernatant is checked using a glass pH electrode which is previously calibrated using standard pH 4 and pH 7 solutions. The obtained yellow powder is washed well with water until the pH is neutral. The final wash is done with acetone and the powder is collected by centrifugation and vacuum dried. This sample is labeled as pZPO-NTs. It is then re-dispersed in 50 mL of Milli-Q water by magnetic stirring. To this, sodium borohydride solution (10 mg mL−1) is added dropwise. The solution is stirred for another 30 min and centrifuged to separate the black powder, which is washed thoroughly with water and acetone, and dried in the oven. This sample is labeled as pPt-NTs (for detailed experimental methods and characterization techniques refer to the ESI†).3. Results and discussion
ZnO is a versatile template to synthesize novel material nanostructures with hollow interiors due to the ease of etching of ZnO in an acidic medium or reducing atmosphere.67,70–76 In our work, porous platinum nanotubes are synthesized by the microwave-induced reaction between chloroplatinic acid and ZnO nanorods in an aqueous medium. The dual role of ZnO nanorods as sacrificial templates and as morphology-directing agents guides the formation of porous nanotubes during the microwave reaction. To begin with, ZnO nanorods of the required aspect ratio are synthesized by the hydrothermal method. The length of the nanorods can be tuned by changing the incubation time during the hydrothermal synthesis.77 X-Ray diffraction (XRD) and scanning electron microscopy (SEM) studies show that high-aspect-ratio phase-pure (hexagonal wurtzite, P63mc, ICDD-00-036-1451) nanorods are formed (Fig. S1, ESI†). The nanorods have a hexagonal prismatic morphology with diameters ranging from 20–90 nm and lengths of 0.5–2 μm.The methodology for the synthesis of porous nanotubes using ZnO nanorods as sacrificial templates is shown in Fig. 1. ZnO nanorods, dispersed in an aqueous solution of platinum precursor (H2PtCl6), are subjected to microwave heating at 200 °C. The yellow product obtained is stable under ambient conditions and designated as pZPO-NTs. X-ray diffraction (XRD) studies (Fig. 2a) show the presence of 3 broad peaks with maxima at 18.4°, 34.8°, and 59.4°, respectively. The particle size is ∼1.7 nm as calculated from the Scherrer formula considering the Gaussian width of the first peak. Broadening due to sub 2 nm particle size along with the absence of peaks corresponding to ZnO or Pt phases confirms the single phase, and the peaks can be indexed to the inverse spinel face-centered cubic Zn2PtO4 phase (ICDD 00-023-1499).64 The STEM-HAADF image shows that the core ZnO is completely etched resulting in the formation of hollow porous tubes irrespective of the diameter or the length of the nanorods (Fig. 2b). Also, the newly formed shell is nearly uniform in thickness, in the range of 10–15 nm, and it is made up of tiny particles of size ∼1–2 nm as confirmed from the bright field transmission electron microscopy (BF-TEM) of this sample (Fig. 1e). STEM-EDS elemental mapping (Fig. 2c) and the line scan from a single nanotube (inset in Fig. 2b) show the presence of all 3 elements Zn, Pt and O. X-ray photoelectron spectroscopy (XPS) studies of the as-synthesized pZPO-NTs are shown in Fig. 2d. Pt4f core level spectra from this sample show that platinum is in a higher oxidation state of +2 and +4. The binding energy values 72.6 eV, 74.6 eV, and 75.7 eV can be assigned to Pt(OH)2, PtO2, and PtCl4, respectively, based on the standard NIST database.51,78,79 The 72.6 eV doublet is also assigned to the PtO phase.80 All Pt species are constrained to have a doublet separation of 3.3 eV and the FWHM is kept the same for 4f 7/2 and 4f 5/2 peaks of each pair. Even after the reaction, PtCl4 may still be present in the precursor state adhered to the sample surface. The presence of PtCl4 is always found in H2PtCl6 solution-impregnated solid oxide supports.79 The Zn 2p signal mainly comes from Zn2+ species at a binding energy value of 1022.0 eV. A small shoulder peak at the higher binding energy side at 1023.4 eV is from ZnCl2 species adhered to the surface of the porous pZPO-NTs.81 From XPS data, a quantitative measurement of surface composition can also be done after background subtraction of the peak intensities and normalizing with respect to the relative sensitivity factor.82 The relative sensitivity factor values for the elements Zn 2p, Pt 4f, and O 1s are 5.589, 5.575, and 0.71, respectively, as mentioned in the Kratos instrument database. The composition ratio measured from the normalized area under the peak for Zn 2p and Pt 4f is 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for the Zn2PtO4 phase. The diffuse reflectance spectra for ZnO nanorods and pZPO-NTs are shown in Fig. S2a (ESI†). Pure ZnO is white in color and does not absorb in the visible range and has absorption maxima in the IR range; the absorption edge in the zinc platinate phase is red-shifted as compared to pure ZnO and seen at around 500 nm. The multiple absorption bands present in zinc platinate may be due to the presence of oxygen vacancies.83,84 Since the particle size in the zinc platinate phase is between 1–2 nm, which exists in an inverse spinel structure with a large lattice parameter of ∼8.5 Å, it is understandable that the structure may be non-stoichiometric and suffers from severe surface stresses. In spinel or inverse spinel materials, the presence of 2 or more different transition metal cations in different coordination environments (octahedral, tetrahedral, or mixed) can cause multiple LMCT (ligand to metal charge transfer) and IVCT (Intra valence charge transfer) transitions giving rise to multiple absorption bands in the visible and near IR region of the wavelength spectrum.85,86 Tauc plots (Fig. S2b and c, ESI†) show a direct band gap of 3.2 eV for ZnO nanorods indicating the highly crystalline stoichiometric nature of the sample. pZPO-NTs show an indirect band gap of 1.8 eV. Due to the lack of data on the optical properties of the zinc platinate phase, reference to the Materials Project (MP, Lawrence Berkeley National Laboratory, Berkeley, CA) database is made which lists Zn2PtO4 as one of the metastable phases, and the calculated band gap value using LDA and GGA with a 50% underestimation is 0.8 eV.87
:1 for the Zn2PtO4 phase. The diffuse reflectance spectra for ZnO nanorods and pZPO-NTs are shown in Fig. S2a (ESI†). Pure ZnO is white in color and does not absorb in the visible range and has absorption maxima in the IR range; the absorption edge in the zinc platinate phase is red-shifted as compared to pure ZnO and seen at around 500 nm. The multiple absorption bands present in zinc platinate may be due to the presence of oxygen vacancies.83,84 Since the particle size in the zinc platinate phase is between 1–2 nm, which exists in an inverse spinel structure with a large lattice parameter of ∼8.5 Å, it is understandable that the structure may be non-stoichiometric and suffers from severe surface stresses. In spinel or inverse spinel materials, the presence of 2 or more different transition metal cations in different coordination environments (octahedral, tetrahedral, or mixed) can cause multiple LMCT (ligand to metal charge transfer) and IVCT (Intra valence charge transfer) transitions giving rise to multiple absorption bands in the visible and near IR region of the wavelength spectrum.85,86 Tauc plots (Fig. S2b and c, ESI†) show a direct band gap of 3.2 eV for ZnO nanorods indicating the highly crystalline stoichiometric nature of the sample. pZPO-NTs show an indirect band gap of 1.8 eV. Due to the lack of data on the optical properties of the zinc platinate phase, reference to the Materials Project (MP, Lawrence Berkeley National Laboratory, Berkeley, CA) database is made which lists Zn2PtO4 as one of the metastable phases, and the calculated band gap value using LDA and GGA with a 50% underestimation is 0.8 eV.87
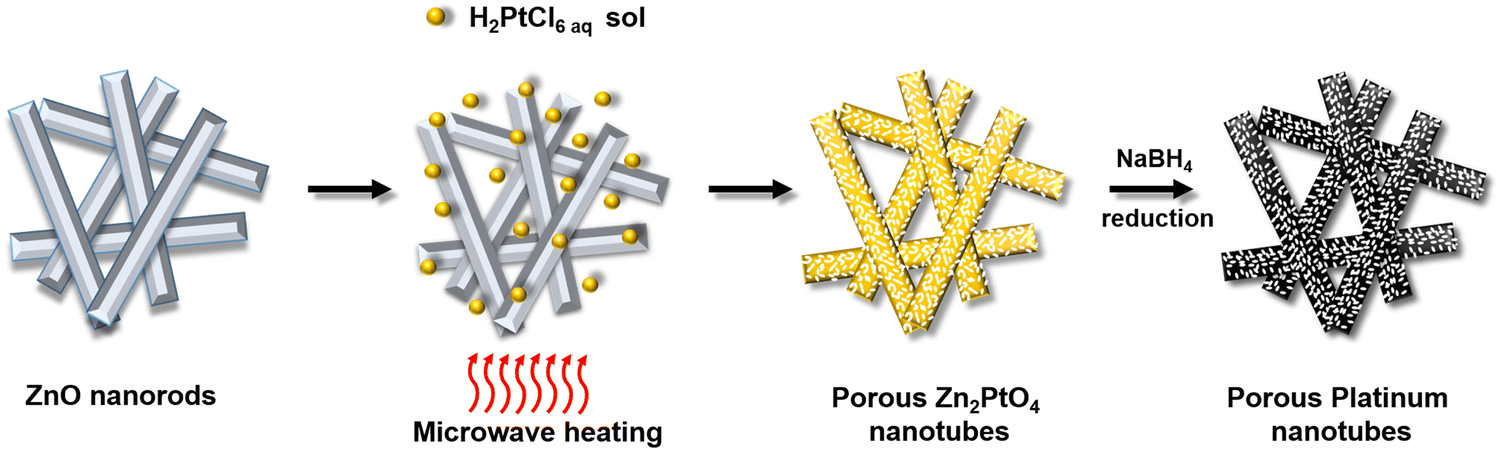 | ||
| Fig. 1 Schematic of the formation of porous zinc platinate nanotubes and their conversion to porous platinum nanotubes. | ||
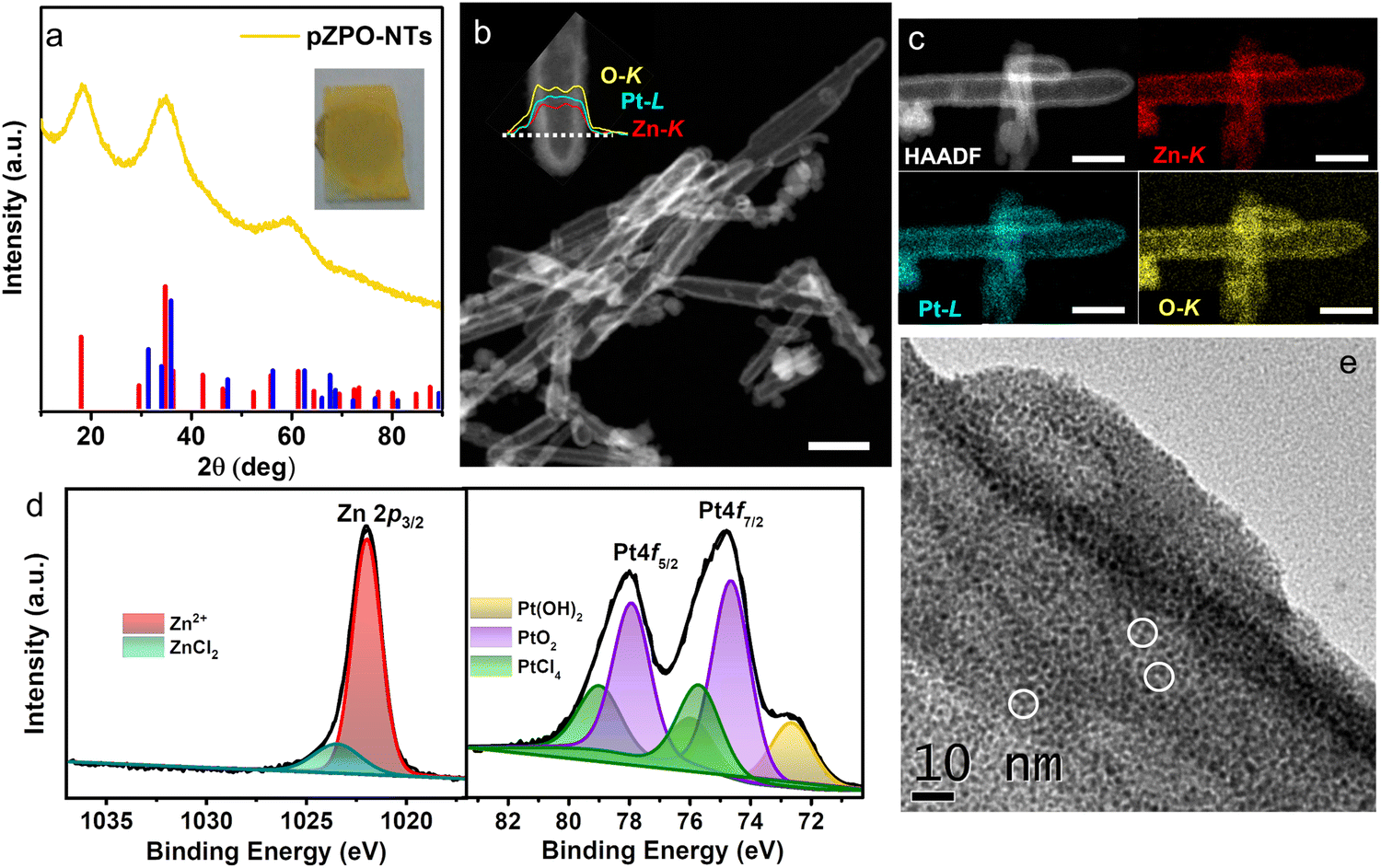 | ||
| Fig. 2 (a) XRD pattern from as-synthesized porous zinc platinate nanotubes (pZPO-NTs) which can be matched to ICDD file 00-023-1499 for the standard zinc platinate phase (red line). The standard XRD pattern for the ZnO phase (ICDD-00-036-145) is shown in blue. The inset shows a photograph of the as prepared pZPO-NTs film on a glass substrate. (b) HAADF-STEM images of pZPO-NTs confirming the hollow interior and porous nature of the nanotubes. The inset shows the STEM-EDS line scan taken along the diameter of a nanotube for the Zn-K, Pt-L, and O-K lines. (c) STEM-EDS elemental mapping for Zn-K, Pt-L and O-K, respectively, taken from a single nanotube showing the distribution of the elements. (d) Core level elemental XPS scans from Zn 2p 3/2 and Pt 4f from the pZPO-NTs sample. Zn exists in +2 oxidation state and Pt exists in multiple oxidation states of +2 and +4. Traces of ZnCl2 are found on the surface of the nanotubes. The precursor PtCl4 signal is also seen in a trace amount on the surface of the sample. (e) High magnification BF-TEM image of a single pZPO-NT showing that the shell is composed of fine crystallites of size between 1–2 nm with a well-developed pore network. Pores are highlighted by white circles. The scale bar in (b) is 200 nm and (c) is 70 nm. | ||
Zn2PtO4 is a rare phase and only a couple of papers exist in the literature which show the synthesis methodology. Muller and Roy have shown that by annealing a mixture of Zn (OH)2 and platinum black at 900 °C under an oxygen partial pressure of 200 atm for a prolonged duration (several days), light yellow-green colored Zn2PtO4 could be synthesized.64 Kjelin and Palmqvist have synthesized nanoparticles of Zn2PtO4 by mixing PtCl4 with ZnO in an Ar glovebox and annealing in a sealed quartz ampule under an N2 atmosphere in a tube furnace.63 Interestingly, a few research groups have also observed the formation of the zinc platinate phase at the interface between Pt and ZnO during non-equilibrium synthesis processes such as combustion synthesis, thin film growth, or aerosol condensation.88–90 Microwave synthesis is known to offer ultrafast reaction kinetics due to the rapid and uniform heating of the dielectric ZnO support and the reaction medium.91 The pressure generated inside the microwave reaction vessel is ∼250 psi (∼15 atm) at the reaction temperature of 200 °C, which also aids the reaction kinetics.
To understand the mechanism of the formation of the zinc platinate phase, a set of control experiments is performed. To start with, the microwave reduction of the H2PtCl6 precursor is performed maintaining the exact conditions for the formation of pZPO-NTs in the absence of ZnO nanorods. No product is formed and the precursor solution remains clear but the color changes to deeper yellow (Fig. S3a, ESI†). The absence of any product formation shows that homogeneous nucleation of metallic Pt or PtO2 is not possible under these reaction conditions. UV-Visible absorption spectroscopy of the solution before and after this control reaction displays peaks at 208 nm and 262 nm, which are assigned to ligand to metal charge transfer (LMCT) transitions of ligand σ and π orbitals of PtCl62− ions.92 After the MW reaction, the solution turns deeper yellow with a decrease in pH from 3 to ∼1 and the corresponding absorption spectra show lower intensity for both the peaks and a slightly elevated intensity in the 235 nm region suggesting the hydrolysis of H2PtCl6 into chloro-aquo complexes of the type H2[PtClnOH6−n] due to the exchange of Cl− with water. The released Cl− ions into the solvent increase the acidity of the medium.93,94 In the presence of ZnO nanorods, a clear supernatant and a yellow product are obtained after the microwave reaction (Fig. S3b, ESI†). The UV-Vis absorption spectra for the supernatant show no traces of [PtCl6]2− ions suggesting a complete removal of the platinum precursor. The pH of the supernatant is ∼5 at this stage, suggesting that the dissolution of ZnO nanorods increases the basicity of the medium. Even a prolonged microwave reaction period of 1 hour yields only the zinc platinate phase and not the Pt phase. The nanotubes are porous as earlier, but the shells are slightly thicker than that formed from the 5 min reaction. The shell thickness is in the range of 17–23 nm. The BF-TEM images for the porous pZPO-NTs nanotubes are shown in Fig. S4a and b (ESI†). The formation of the phase is confirmed by XRD (Fig. S4c, ESI†). This suggests that under microwave synthesis conditions, only zinc platinate is the stable phase formed and not Pt.
To check the effect of pH on the synthesis, the dried powder of pZPO-NTs is then re-dispersed in an aqueous medium and subjected to microwave heating at 200 °C for an hour. BF-TEM images show porous Pt nanotubes with larger Pt crystallites of size ∼5 nm dispersed all along the nanotubes (Fig. S4d and e, ESI†) and the corresponding XRD pattern shows crystalline Pt phase (Fig. S4f, ESI†). We believe that the zinc platinate phase is stable under acidic conditions of pH < 7. At neutral pH, it decomposes to Pt. The rate of hydrolysis of precursor salts and the dissolution of ZnO depends on temperature. To see the effect of temperature on the reaction mechanism, the microwave reaction is carried out at 100 °C. BF-TEM images of the white product obtained show that the nanorods are still intact but have a rough surface due to the partial etching in the acidic medium (Fig. S5, ESI†). The absence of porous nanotubes indicates that the reaction kinetics are not conducive at low temperatures.
Based on the above observations, a possible mechanism pathway for the formation of pZPO-NTs is proposed in accordance with the sacrificial template accelerated hydrolysis (STAH) mechanism proposed earlier.60 This mechanism is also invoked to explain the formation of porous VO2 nanotubes65 or porous PdO nanotubes.67 But in all these structures, the template does not participate in the formation of the final product. In our case, we believe that the weak reducibility of water prevents the reduction of the platinum precursor, thereby maintaining its +4 valence state. Unlike VO2 and PdO, platinum oxide formation is not feasible under these reaction conditions. Alternatively, the metastable zinc platinate formation is favorable due to the thermal and kinetic effects of the microwave synthesis, which help in the faster diffusion of Zn2+ ions. The reaction pathway for the formation of porous zinc platinate nanotubes is shown in the ESI† (Scheme S1). The dissolution of ZnO accelerates the hydrolysis of Pt complex ions, which in turn promotes the formation of zinc platinate in the proximity of the ZnO surface, preserving the morphology of the nanorods, aided by the superior kinetics provided by microwave reaction. The reaction proceeds if the pH of the medium is acidic enough to dissolve the ZnO. With the increase in the concentration of Zn2+ ions in solution due to the dissolution of ZnCl2, the pH of the medium increases, which arrests the STAH mechanism.65
The TEM studies of pZPO-NTs reveal that the zinc platinate phase undergoes reduction upon exposure to the electron beam and this process is very rapid and occurs within a few minutes upon exposure. Under a reduced dose of the incident electron beam, this transformation is captured as shown in Fig. S6 (ESI†). During this reduction, the particle size coarsens to ∼3 nm and the tube shrinks in diameter by ∼10 nm. Earlier reports have shown that zinc platinate nanoparticles are stable under the electron beam.63,95 The instability observed in our case may be due to the surface stresses generated in extremely small crystallite sizes of 1–2 nm, which may accommodate at maximum 2-unit cells of the inverse spinel zinc platinate (lattice parameter ∼8.5 Å). Following this observation, a controlled reduction step is designed using sodium borohydride as the reducing agent at room temperature. An aqueous solution of sodium borohydride (10 mg mL−1) is added dropwise to a dilute dispersion of pZPO-NTs in water. The color of the solution changes gradually from yellow to black over the duration of 20 minutes resulting in the formation of porous platinum tubes. The SEM image from this sample shows smooth porous shells with hollow cores (Fig. 3a). XRD data for the freshly prepared porous Pt shows only sharp FCC peaks, which can be indexed to (111), (200), (220), and (311) of a fully formed Pt phase (Fig. 3d). The peaks due to zinc platinate or ZnO are not observed. BF-TEM and STEM-HAADF studies show that the porosity is uniform with the formation of tiny ∼3 nm crystallites of Pt (Fig. 3b and c). The STEM-EDS elemental maps (Fig. 3d) and line scan (inset of Fig. 3c) from a single nanotube show the presence of Pt, Zn and O. XPS studies of the porous Pt nanotubes show a pure metallic Pt phase (Fig. S7, ESI†). The binding energy values are 71 eV and 74.3 eV, respectively, for the Pt4f doublet, which matches with the zero oxidation state of Pt.96 We also see a substantial amount of Zn 2p signal in this sample indicating that zinc in the form of Zn2+is still present at 1020.9 eV. The shift of nearly 1 eV in the binding energy of Zn2+ as compared to that of the zinc platinate phase is observed, which may be due to the change in the coordination environment.97 A small peak at the higher binding energy at 1022.9 eV is assigned to Zn(OH)2.98 Composition analysis from XPS shows the relative atomic ratio of Pt:Zn as 1:0.7. (Relative atomic ratios for Pt and Zn are listed in Table S1, ESI†).
 | ||
| Fig. 3 (a) SEM micrograph of the as-synthesized porous Pt nanotubes (pPt-NTs). (b) Low magnification BF-TEM and (c) HAADF-STEM images respectively of pPt-NTs confirming the hollow interior and porous nature of the nanotubes. The inset shows the STEM-EDS line scan taken along the diameter of a nanotube for Zn-K, Pt-L, and O-K lines. (d) XRD pattern from as-synthesized pPt-NTs, which can be matched to ICDD file 00-001-1190 for the standard Pt phase (black line). The standard Zn2PtO4 phase (ICDD 00-023-1499) and standard ZnO phase (ICDD-00-036-1451) are shown in red and blue, respectively. (e) STEM-EDS elemental mapping for Zn-K, Pt-L and O-K, respectively, taken from a single nanotube showing the distribution of the elements. Scale bar in (c) is 200 nm and in (e) is 200 nm. | ||
The highly porous nature of pZPO-NTs and pPt-NTs suggests enhanced specific surface area; estimation of the surface area is useful in predicting the catalytic efficiency. The specific surface area and porosity are evaluated from nitrogen sorption isotherms at 77 K (Fig. S8a, ESI†). The samples show type Iva adsorption isotherms. The capillary condensation and desorption hysteresis at higher P/P0 values (0.4–1) confirm the presence of mesopores.99 The asymptotic rise in the adsorption curve at P/P0 > 0.9 indicates the presence of macropores (size >50 nm), which is evident from the hollow nature of the nanotubes.100 The Barrett–Joyner–Halenda (BJH) pore size distribution shows a maximum pore diameter of 5.2 nm for pZPO-NTs (Fig. S8b, ESI†) and for pPt-NTs, the value is 6 nm (Fig. S8c, ESI†). Along with this, both the materials show a macropore size of ∼44 nm displaying the hierarchically porous nature. The Brunauer–Emmett–Teller (BET) surface area measured from the multipoint analysis is 54 m2 g−1 for pZPO-NTs and 46 m2 g−1 for pPt-NTs. Thus, both the samples display superior 1D hollow mesoporous structures of hierarchical porosity along with high surface area, which is beneficial in catalysis and sensing applications.
The electrocatalytic activity of porous nanotubes is evaluated by studying the methanol oxidation reaction (MOR) in an alkaline medium using cyclic voltammetry (CV). The MOR curves for pZPO-NTs are shown in Fig. 4a. The peak current for MOR increases with successive scans and peak potential shifts to higher values. The variation of MOR peak current and MOR peak potential with the number of CV cycles is shown in Fig. 4b for 700 cycles. For the first 100 cyclic scans, the peak MOR potential is 0.8 V vs. RHE but it increases linearly with the number of cycles and reaches a value of 1.01 V vs. RHE at the end of 200 cycles. For the next 460 cycles, the rate of increase in potential is not drastic and the potential value settles at 1.06 V vs. RHE at the end of a total of 700 cycles. The MOR peak current also shows similar behavior. The current increases in a steep manner for the initial 200 cycles and then remains constant for the rest of the cycles.
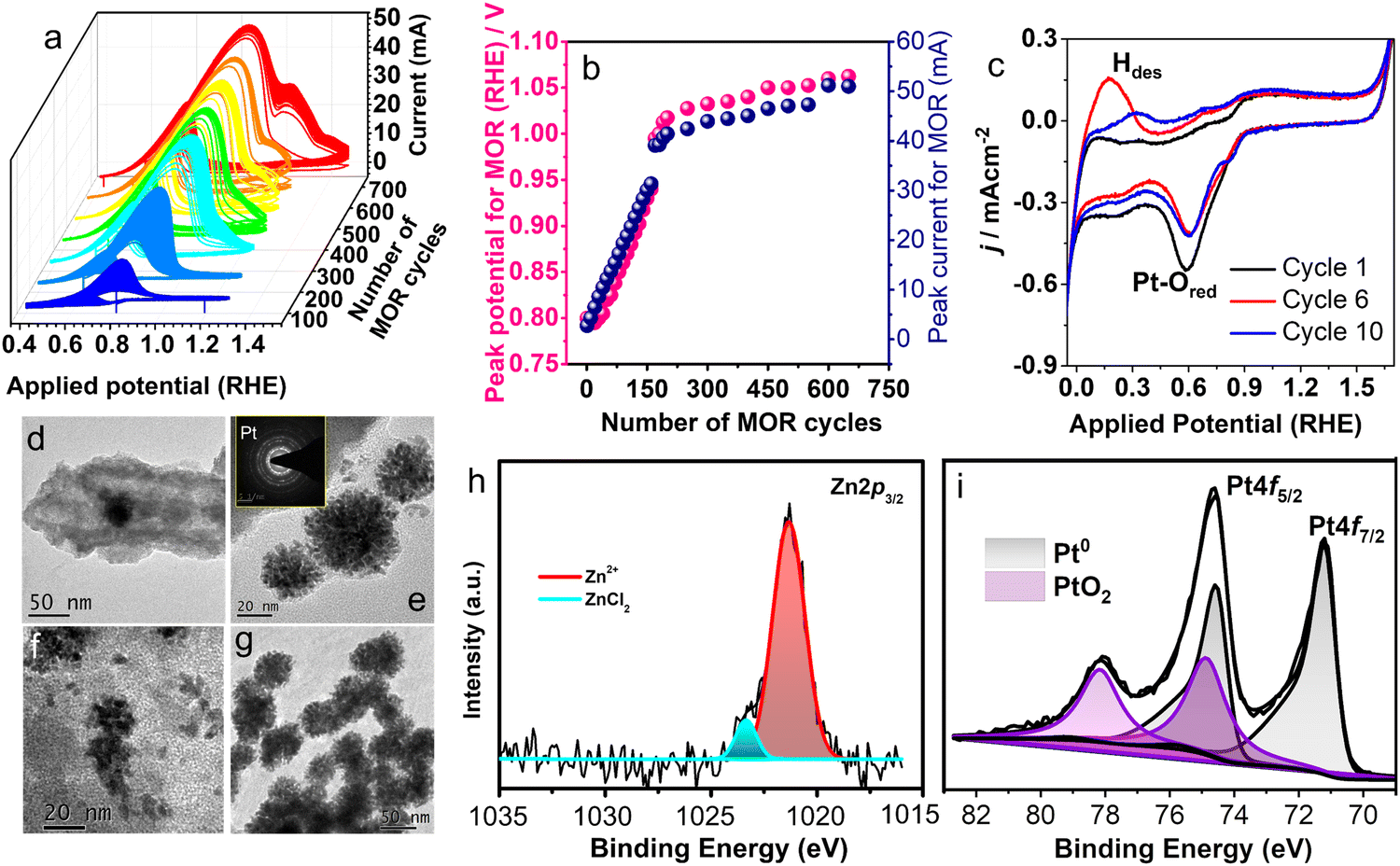 | ||
| Fig. 4 (a) Electrocatalytic MOR activity with pZPO-NTs carried out at a scan rate of 40 mV S−1 in electrolyte solution of 0.5 M KOH +1 M CH3OH shows the variation in the catalytic behaviour with successive scanning up to 700 cycles. (b) MOR peak potential and peak current variation with number of cycles. (c) Variation in the hydrogen desorption peak shape with a successive number of cycles in 0.5 M KOH solution at a scan rate of 40 mV S−1. (d–f) Formation of dendritic nanoparticles of Pt in pZPO-NTs after performing 50 cycles of MOR. (g) Morphology of the pZPO-NTs catalyst taken post electrocatalysis studies after 700 cycles of MOR. (h) and (i) XPS analysis performed post electrocatalysis of pZPO-NTs showing the Pt0 state as a prominent phase along with PtO2 phase. | ||
The BF-TEM images of the sample electrode after electrocatalytic studies show the formation of dendritic nanoparticles of Pt after 50 cycles of MOR (Fig. 4d–f). After 700 cycles, dendritic nanoparticle clusters of almost uniform size are observed (Fig. 4g). XPS studies show an intense metallic Pt 4f doublet at binding energy values 71.2 eV and 74.6 eV, respectively, and significant amounts of PtO2 at 74.9 eV and 78 eV, respectively (Fig. 4i). The composition ratio of Pt:Zn from XPS is 1:0.2 (Table S1 in the ESI†), which suggests the dissolution of the ZnO phase during the MOR. The potential cycles between −0.1 V to 1.7 V (RHE) in argon gas purged 0.5 M KOH (Fig. 4c) solution show that there is a substantial Pt–O reduction current around 0.6 V during the reverse scan, though the Pt–O formation current is not substantial in the forward scan (between 0.4 V and 1.6 V) indicating that more Pt–O is getting reduced than is formed. Also, the shape of the hydrogen desorption peak in the potential region between 0 V to 0.4 V is found to change and the position of the peak shifts to higher potential values with successive cycles. This may suggest that initially single atoms or clusters of Pt form, which have weak binding energy with hydrogen and eventually as the clusters grow the hydrogen binds strongly to these clusters thereby making the desorption difficult. It has been observed during metal electrodeposition that the nucleation and growth of the deposited metal result in dendritic structures.101,102 The computationally predicted Pourbaix diagram, which predicts the electrochemical stability of metastable phases, shows that Zn2PtO4 transforms into Pt and HZnO2− ions under the potential ranges and pH conditions used.87
Preliminary cyclic voltammetry experiments showed that Zn can leach out systematically from the porous nanotubes in the presence of high alkalinity, thereby enhancing the current. An ex situ dissolution procedure for the ZnO phase is performed by dispersing the as-synthesized porous Pt nanotubes in a dilute HCl/H2O solution of pH 3 for an hour. This sample is labeled as pPt-NTs-E. The porous morphology of the nanotubes is intact after the acid treatment as well as the nano Pt crystallite size. However, we have observed that some of the nanotubes are opening into sheets (Fig. S9 and inset of Fig. S9b, ESI†). This is due to the thinning of the Pt shells during the etching of ZnO, similar to observations we have made in PdO nanotubes, confirming that the pH of the medium has a significant effect on the integrity of the tube morphology.67 The MOR activity for pPt-NTs-E and pPt-NTs is compared with that of the commercial Pt/C sample (Fig. 5). Both the samples show good methanol oxidation activity compared to Pt/C with a similar onset MOR potential of ∼0.5 V (RHE) (Fig. 5a). The acid-treated sample shows a 75% increase in mass activity (35 mA mg−1) and a 30% increase in specific activity (0.5 mA cm−2) compared to pPt-NTs (Fig. 5a and d). The MOR peak potential is observed at 1.01 V and 1.02 V for the treated and as-prepared nanotubes, respectively, which are ∼170 mV less than that for commercial Pt/C (1.17 V) demonstrating the efficiency of porous nanotubes in catalyzing the methanol oxidation at lower overpotentials (Fig. 5f). Chronoamperometric studies (Fig. 5c) show that the samples are stable and no significant changes in the oxidation current are observed in the measured period of 4000 seconds. The ratio of oxidation currents in the forward and backward direction of the MOR curve, If/Ib (which gives an indication of efficient oxidation of unwanted CO species) >1.2 for the porous samples compared to commercial Pt/C (If/Ib = 1.0). Removal of Zn in acid-treated porous Pt nanotubes slightly reduces the If/Ib (Fig. 5f). Electrochemical Impedance Spectroscopy (EIS) studies were performed at the onset potential in alkaline conditions (0.1 M KOH) (Fig. S13, ESI†). The solution resistance (Rs) for pPt-NTs was found to be comparable to commercial Pt/C, indicating that pPt-NTs also possess equivalent surface charge. The charge-transfer resistance (Rct) of both the materials, as depicted from the semi-circle, is also comparable suggesting that both the samples have comparable electrical conductivities. However, the removal of ZnO from porous Pt nanotubes has significant benefits in enhancing the current. This may be due to the removal of the resistive paths provided by the highly insulating amorphous ZnO in between the current-carrying Pt nanoparticles.103
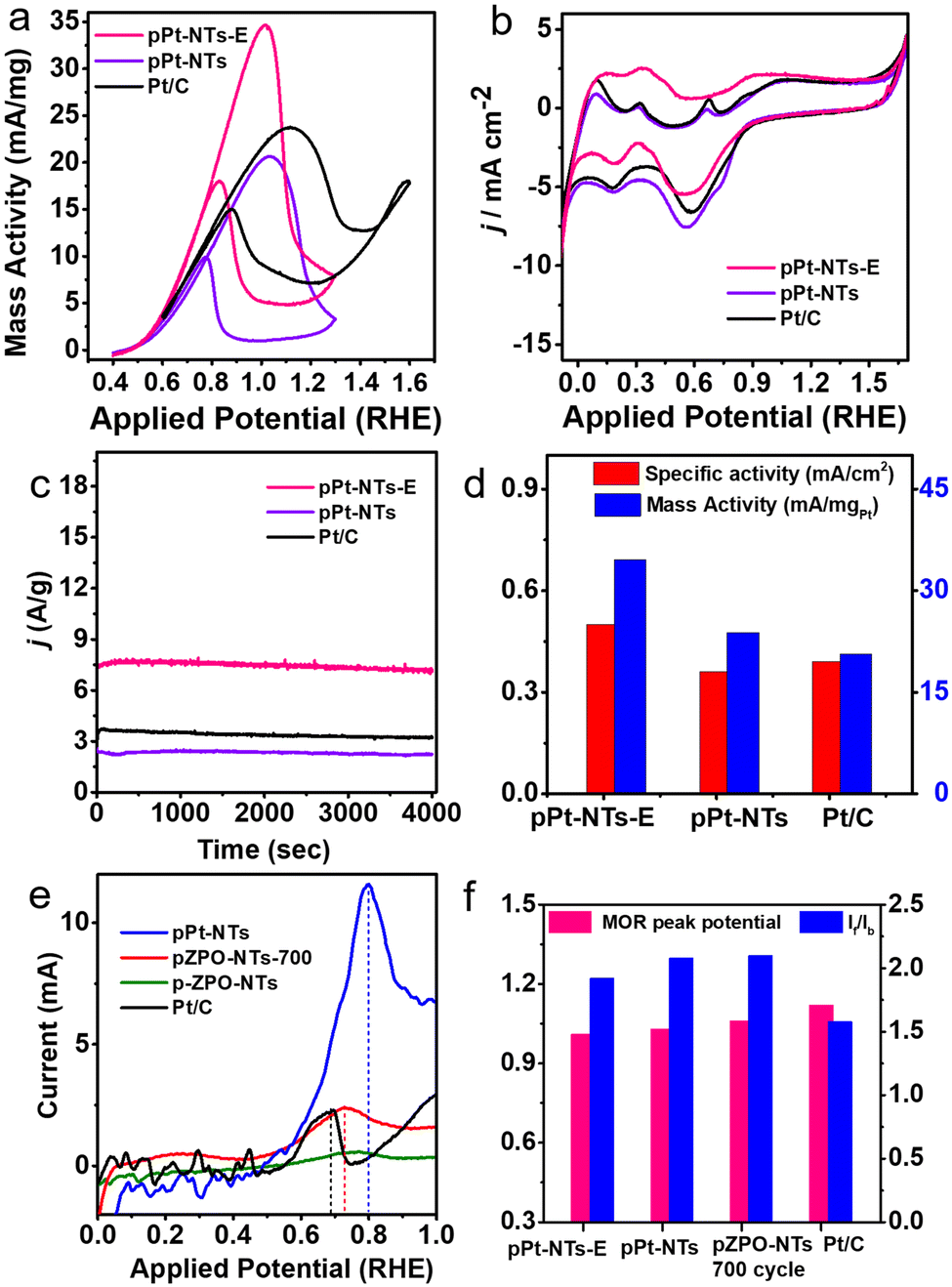 | ||
| Fig. 5 CV scans for pPt-NTs-E, pPt-NTs and Pt/C (a) for the mass activity towards MOR in 0.5 M KOH + 1 M CH3OH solution (b) for ECSA measurements in Ar purged 0.5 M KOH solution. (c) Chronoamperometry studies at 1.0 V (vs. RHE) in Ar purged 0.5 M KOH solution. (d) Specific activity and mass activity comparison. (e) First half of the CV cycle of CO-stripping studies performed on freshly prepared pPt-NTs, pZPO-NTs after 700 cycles of MOR, freshly prepared pZPO-NTs and commercial Pt/C at a scan rate of 10 mV S−1. (f) Comparison of MOR peak potential and If/Ib ratio. The porous samples show better catalytic activity with less overpotentials for the MOR with higher If/Ib values compared to commercial Pt/C. | ||
To understand the surface properties, CO-stripping voltammetry is performed on fresh pZPO-NTs, pZPO-NTs after 700 cycles of MOR, fresh pPt-NTs, and commercial Pt/C. The electrolyte (0.5 M KOH) is saturated in CO by continuously bubbling the gas through the solution for 15 min at a potential where the hydrogen desorption from the anode is favored (0.1 V vs. RHE). This is to ensure that enough active sites are available for CO to adsorb to form a monolayer on the catalyst surface. The potential sweep is started from this potential at a scan rate of 10 mV S−1. Only the first half of the full CV cycle is displayed in Fig. 5e. The CO-stripping potential for pPt-NTs, pZPO-NTs-700 (after 700 cycles of MOR), pZPO-NTs, and commercial Pt/C with respect to RHE is 0.8 V, 0.73 V, 0.75 V, and 0.7 V, respectively. The porous nanotubes show slightly higher overpotentials for CO stripping albeit their activities are much better than commercial Pt/C (Fig. 5d) due to the available active sites.
We believe that the high surface area and hierarchically porous nature are responsible for improving the MOR activity. TEM studies of porous Pt nanotubes after electrocatalysis show that connected networks of highly crystalline Pt ligaments form maintaining the hollow tubular core (Fig. S10, ESI†). The combined structure of crystalline ligaments and hollow channels is beneficial in enhancing the efficiency of methanol oxidation. There is still a trace amount of Zinc present in the +2 oxidation state at BE value 1021.1 eV as revealed by the XPS studies (Fig. S10c and d, ESI†). The Pt 4f doublet shows purely metallic Pt0 species at BE 71.2 eV and 74.6 eV, respectively. The surface of the sample also has PtO2 at BE values of 74.8 eV and 78 eV, respectively. The composition ratio of Pt:Zn in this sample is 1:0.1. As the oxidized species of an element is more hydrophilic in nature, it helps in the removal of CO by the beneficial Langmuir–Hinshelwood bi-functional mechanism when the hydroxyl group bonded to the higher oxidation state Pt can effectively oxidize the CO attached to the neighboring metallic Pt.104 The ORR performance (details are in ESI†) of pPt-NTs is investigated in oxygen saturated 0.1 M KOH medium at different rotation rates of the glassy carbon electrode. The ORR activity of pPt-NTs is comparable to commercial Pt/C (Fig. S11 and S12, ESI†). The pPt-NTs follow a direct pathway of electron reduction of O2 to H2O through a 4-electron transfer process without involving the formation of reaction intermediates. These results are in accordance with the earlier observations, thus confirming the enhanced electron and mass transfer facilitated by mesoporous nature.105
The sensing studies are performed on pZPO-NTs in an interdigitated electrode configuration (see ESI,† for more details). Initially, the sensor is exposed to different concentrations of SO2 at room temperature and the recorded dynamic response shows n-type behavior in the presence of SO2 (Fig. 6a). It can precisely detect in the range of 0.3 ppm to 3 ppm with response magnitudes varying from 8.9% to 85.27%, respectively, and demonstrates a sensitivity of 31%/ppm towards SO2 at room temperature with complete recovery on the release of SO2 gas (Fig. 6b). The response in this range is found to be linear with a correlation coefficient of 0.98. The response and recovery are swift within 10 s to 0.3 ppm SO2 (Fig. 6c). However, it took 128 s to recover when 3 ppm SO2 was released, which may be attributed to the highly porous nature of these nanotubes. The gas diffuses through the pores and hence takes longer to desorb. Nevertheless, it could be detected rapidly and reached a near plateau within 30 s for all the concentrations. The selectivity of the sensor is examined by purging different oxidizing and reducing gases such as H2, NH3, acetone, NO2, H2S, and ethanol along with SO2. The sensor exhibits specific detection of SO2 at room temperature (Fig. 6d). To investigate the reproducibility and reliability of the sensor, it is subjected to five consecutive cycles of 1 ppm SO2 (Fig. S16a, ESI†). Although the outcome is repeatable, a baseline drift, common in chemiresistive gas sensors, is observed, which is possibly due to the variations in ambient humidity levels since the sensor is operated at room temperature. The memory effect is determined experimentally by carrying out the concentration sweep from low to high and vice-versa, thereby computing the hysteresis error (Fig. S16b and c, ESI†). The hysteresis error is ±3.55, which is indicative of an insignificant memory effect on the sensor. To investigate the inter-device variations, four different devices are tested for all concentrations of SO2 and the variations are well within the tolerance limit of the sensor as seen in Fig. 6b. The sensor performance was compared with the existing works on the detection of SO2 in the literature and is summarised in Table S3 (ESI†). Among all, pZPO-NTs displayed swift and highly sensitive detection at room temperature. They could resolve the signal down to 0.3 ppm, which seems to be one of the lowest limits of detection (LOD) claimed among the reported ones at room temperature. The underlying mechanism behind the sensing can be explained by the well-established space charge theory in metal oxides57 and is discussed in the ESI.† Furthermore, the exemplary room temperature sensing can be ascribed to the mesoporous nature of these nanotubes, which adds to the enhancement of the surface area (54 m2 g−1) and hence the sensitivity.
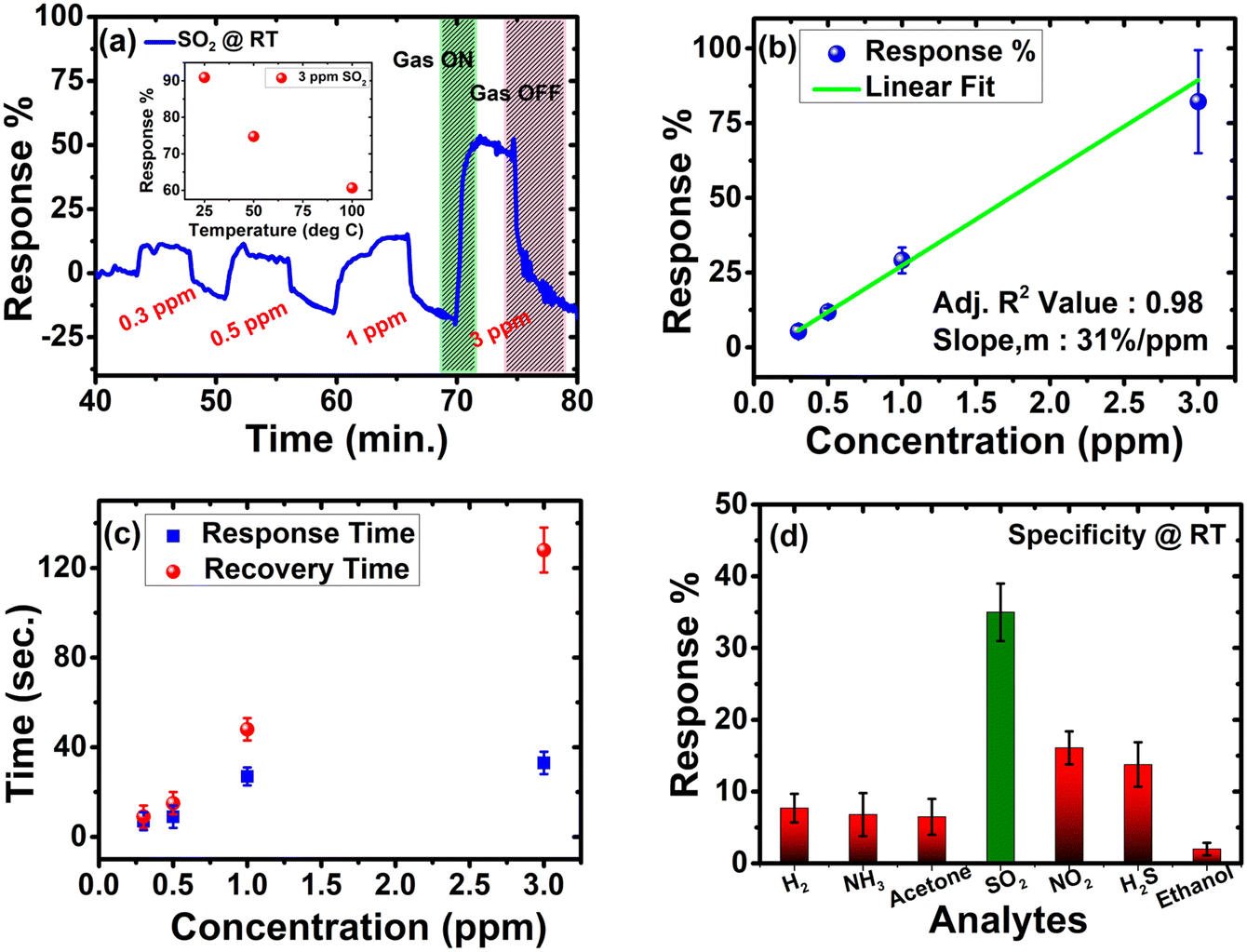 | ||
| Fig. 6 (a) Dynamic response plot for different concentrations of SO2 at room temperature; the inset shows the temperature dependent behaviour of SO2 towards pZPO-NTs, (b) linear fitting of the response vs concentration plot, (c) response time and recovery time for different H2S concentrations and (d) specificity towards different gases at 1 ppm at room temperature. | ||
4 Conclusions
In summary, our study shows the template-based synthesis of mesoporous zinc platinate and platinum nanotubes using ZnO nanorods as sacrificial templates. Unlike the other similar synthesis of mesoporous materials, in our case ZnO actively participates in the product formation and aided by the superior kinetics of microwave reaction, we can synthesize mesoporous zinc platinate nanotubes. Zinc platinate nanotubes form only under the superior reaction kinetics provided by the microwave reaction conditions. By suitably designing control experiments, we have proposed a reaction pathway that follows sacrificial template-assisted hydrolysis (STAH). A simple chemical reduction using NaBH4 can reduce the zinc platinate nanotubes into platinum nanotubes maintaining the morphology and porosity. Through N2 adsorption–desorption isotherms, we have confirmed the formation of a hollow mesoporous 1D morphology of the nanotubes with exceptionally high specific surface area with a pore diameter of ∼5 nm and 6 nm for zinc platinate and platinum nanotubes, respectively. Electrocatalytic methanol oxidation studies are performed on these porous nanotubes, and they are found to have excellent activity as compared to commercial Pt catalysts. The ORR ability of porous Pt nanotubes is comparable to commercial Pt catalysts and the mechanism follows direct 4 electron transfer with the least amount of intermediate H2O2 formation. Furthermore, we could observe highly sensitive (31%/ppm) detection of mesoporous zinc platinate nanotubes towards SO2 at room temperature with swift recovery at room temperature without an external stimulus. This could be an ideal candidate for monitoring SO2 emissions in industry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Advanced Facilities for Microscopy and Microanalysis (AFMM), Micro and Nano Characterization Facility (MNCF) and Materials Research Centre (MRC) at IISc for providing the materials characterization facilities. SS and NR acknowledge the funding provided by Nanomission, SERB and DST, Government of India. N. S. and N. B. thank the facilities provided by the Centre for Nanoscience and Engineering (CeNSE) and National Nano Fabrication Facility (NNFC) at IISc. N. S. and N. B. acknowledge the financial support from MHRD, MeitY and DST, Government of India. A. M. and A. H. acknowledge the Advanced Material Research Centre (AMRC) at IIT Mandi for providing the facilities. Also, MHRD and DST, Government of India for funding. We thank Prof. Ramananda Chakrabarti, CEaS, IISc for providing the ICP-MS facility.References
- W. Li, J. Liu and D. Zhao, Nat. Rev. Mater., 2016, 1, 16023 CrossRef CAS.
- C. Li, Q. Li, Y. V. Kaneti, D. Hou, Y. Yamauchi and Y. Mai, Chem. Soc. Rev., 2020, 49, 4681–4736 RSC.
- S. S. Park and C. S. Ha, Adv. Funct. Mater., 2018, 28, 1–29 Search PubMed.
- J. Wang, Q. Ma, Y. Wang, Z. Li, Z. Li and Q. Yuan, Chem. Soc. Rev., 2018, 47, 8766–8803 RSC.
- B. R. Thompson, T. S. Horozov, S. D. Stoyanov and V. N. Paunov, J. Mater. Chem. A, 2019, 7, 8030–8049 RSC.
- Z. Qi and J. Weissmüller, ACS Nano, 2013, 7, 5948–5954 CrossRef CAS PubMed.
- K. Miszta, J. De Graaf, G. Bertoni, D. Dorfs, R. Brescia, S. Marras, L. Ceseracciu, R. Cingolani, R. Van Roij, M. Dijkstra and L. Manna, Nat. Mater., 2011, 10, 872–876 CrossRef CAS PubMed.
- T. X. T. Sayle, K. Kgatwane, P. E. Ngoepe and D. C. Sayle, J. Mater. Chem. A, 2016, 4, 6456–6464 RSC.
- O. V. Shulga, K. Jefferson, A. R. Khan, V. T. D’Souza, J. Liu, A. V. Demchenko and K. J. Stine, Chem. Mater., 2007, 19, 3902–3911 CrossRef CAS PubMed.
- H. Qiu, L. Lu, X. Huang, Z. Zhang and Y. Qu, Bioresour. Technol., 2010, 101, 9415–9420 CrossRef CAS PubMed.
- J. H. Han, H. Boo, S. Park and T. D. Chung, Electrochim. Acta, 2006, 52, 1788–1791 CrossRef CAS.
- P. Daggumati, Z. Matharu and E. Seker, Anal. Chem., 2015, 87, 8149–8156 CrossRef CAS PubMed.
- R. Raccis, A. Nikoubashman, M. Retsch, U. Jonas, K. Koynov, H. J. Butt, C. N. Likos and G. Fytas, ACS Nano, 2011, 5, 4607–4616 CrossRef CAS PubMed.
- B. Szczęśniak, J. Choma and M. Jaroniec, Chem. Commun., 2020, 56, 7836–7848 RSC.
- B. Viswanath, S. Patra, N. Munichandraiah and N. Ravishankar, Langmuir, 2009, 25, 3115–3121 CrossRef CAS PubMed.
- Y. Li, B. P. Bastakoti, V. Malgras, C. Li, J. Tang, J. H. Kim and Y. Yamauchi, Angew. Chem., 2015, 127, 11225–11229 CrossRef.
- C. Mahr, P. Kundu, A. Lackmann, D. Zanaga, K. Thiel, M. Schowalter, M. Schwan, S. Bals, A. Wittstock and A. Rosenauer, J. Catal., 2017, 352, 52–58 CrossRef CAS.
- Z. Lu, C. Li, J. Han, F. Zhang, P. Liu, H. Wang, Z. Wang, C. Cheng, L. Chen, A. Hirata, T. Fujita, J. Erlebacher and M. Chen, Nat. Commun., 2018, 9, 1–7 CrossRef CAS.
- L. Wang and Y. Yamauchi, J. Am. Chem. Soc., 2013, 135, 16762–16765 CrossRef CAS PubMed.
- A. Kloke, F. Von Stetten, R. Zengerle and S. Kerzenmacher, Adv. Mater., 2011, 23, 4976–5008 CrossRef CAS PubMed.
- S. Rudi, L. Gan, C. Cui, M. Gliech and P. Strasser, J. Electrochem. Soc., 2015, 162, F403–F409 CrossRef CAS.
- V. Malgras, H. Ataee-Esfahani, H. Wang, B. Jiang, C. Li, K. C. W. Wu, J. H. Kim and Y. Yamauchi, Adv. Mater., 2016, 28, 993–1010 CrossRef CAS PubMed.
- S. Yang and X. Luo, Nanoscale, 2014, 6, 4438–4457 RSC.
- A. Kloke, F. Von Stetten, R. Zengerle and S. Kerzenmacher, Adv. Mater., 2011, 23, 4976–5008 CrossRef CAS PubMed.
- J. Zhang and C. M. Li, Chem. Soc. Rev., 2012, 41, 7016–7031 RSC.
- M. Pérez-Page, E. Yu, J. Li, M. Rahman, D. M. Dryden, R. Vidu and P. Stroeve, Adv. Colloid Interface Sci., 2016, 234, 51–79 CrossRef PubMed.
- P. K. Chen, N. C. Lai, C. H. Ho, Y. W. Hu, J. F. Lee and C. M. Yang, Chem. Mater., 2013, 25, 4269–4277 CrossRef CAS.
- L. Zu, W. Zhang, L. Qu, L. Liu, W. Li, A. Yu and D. Zhao, Adv. Energy Mater., 2020, 10, 1–14 Search PubMed.
- X. Xia, Y. Wang, A. Ruditskiy and Y. Xia, Adv. Mater., 2013, 25, 6313–6333 CrossRef CAS PubMed.
- J. Chen, J. M. McLellan, A. Siekkinen, Y. Xiong, Z. Y. Li and Y. Xia, J. Am. Chem. Soc., 2006, 128, 14776–14777 CrossRef CAS PubMed.
- E. Zhang, F. Ma, J. Liu, J. Sun, W. Chen, H. Rong, X. Zhu, J. Liu, M. Xu, Z. Zhuang, S. Chen, Z. Wen and J. Zhang, Nanoscale, 2018, 10, 21703–21711 RSC.
- K. Cai, Z. Lv, K. Chen, L. Huang, J. Wang, F. Shao, Y. Wang and H. Han, Chem. Commun., 2013, 49, 6024 RSC.
- L. Hai-Wei, L. Shuo, G. Jun-Yan, W. Shang-Bing, W. Lei and Y. Shu-Hong, Adv. Mater., 2009, 21, 1850–1854 CrossRef.
- K. D. Gilroy, P. Farzinpour, A. Sundar, R. A. Hughes and S. Neretina, Chem. Mater., 2014, 26, 3340–3347 CrossRef CAS.
- Y. Sun, H. Yang, X. Yu, H. Meng and X. Xu, RSC Adv., 2015, 5, 70387–70394 RSC.
- Y. W. Lee, M. A. Lim, S. W. Kang, I. Park and S. W. Han, Chem. Commun., 2011, 47, 6299 RSC.
- B. S. Choi, Y. W. Lee, S. W. Kang, J. W. Hong, J. Kim, I. Park and S. W. Han, ACS Nano, 2012, 6, 5659–5667 CrossRef CAS PubMed.
- Z. Wang, D. Luan, C. M. Li, F. Su, S. Madhavi, F. Y. C. Boey and X. W. Lou, J. Am. Chem. Soc., 2010, 132, 16271–16277 CrossRef CAS PubMed.
- F. Hong, S. Sun, H. You, S. Yang, J. Fang, S. Guo, Z. Yang, B. Ding and X. Song, Crystal Growth Design, 2011, 11, 3694–3697 CrossRef CAS.
- X. Xia, Y. Wang, A. Ruditskiy and Y. Xia, Adv. Mater., 2013, 25, 6313–6333 CrossRef CAS PubMed.
- C. Cui, L. Gan, M. Heggen, S. Rudi and P. Strasser, Nat. Mater., 2013, 12, 765–771 CrossRef CAS PubMed.
- B. D. McNicol and R. T. Short, J. Electroanal. Chem., 1977, 81, 249–260 CrossRef CAS.
- W. Sugimoto, K. Aoyama, T. Kawaguchi, Y. Murakami and Y. Takasu, J. Electroanal. Chem., 2005, 576, 215–221 CrossRef CAS.
- P. J. Kulesza, I. S. Pieta, I. A. Rutkowska, A. Wadas, D. Marks, K. Klak, L. Stobinski and J. A. Cox, Electrochim. Acta, 2013, 110, 474–483 CrossRef CAS PubMed.
- M. T. Y. Paul and B. D. Gates, Sci. Rep., 2019, 9, 1–9 CrossRef CAS PubMed.
- B. Fu, M. Dang, J. Tao, Y. Li and Y. Tang, J. Colloid Interface Sci., 2020, 570, 197–204 CrossRef CAS PubMed.
- P. Díez, E. Lucena-Sánchez, A. Escudero, A. Llopis-Lorente, R. Villalonga and R. Martínez-Máñez, ACS Nano, 2021, 15, 4467–4480 CrossRef PubMed.
- A. Rednyk, V. Johánek, I. Khalakhan, M. Dubau, M. Vorokhta and V. Matolín, Int. J. Hydrogen Energy, 2016, 41, 265–275 CrossRef CAS.
- N. R. Manwar, A. A. Chilkalwar, K. K. Nanda, Y. S. Chaudhary, J. Subrt, S. S. Rayalu and N. K. Labhsetwar, ACS Sustainable Chem. Eng., 2016, 4, 2323–2332 CrossRef CAS.
- N. Bingwa, M. J. Ndolomingo, J. H. Noh, N. Antonels, E. Carleschi, B. P. Doyle, M. Haumann and R. Meijboom, Mol. Catal., 2020, 492, 110978 CrossRef CAS.
- P. K. Basu, S. Kallatt, E. A. Anumol and N. Bhat, J. Appl. Phys., 2015, 117, 224501 CrossRef.
- R. M. Mohamed, A. A. Ismail, M. W. Kadi, A. S. Alresheedi and I. A. Mkhalid, ACS Omega, 2021, 6, 6438–6447 CrossRef CAS PubMed.
- L. Zheng, J. Xie, X. Liu, C. Yang, W. Zheng and J. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 46267–46276 CrossRef CAS PubMed.
- G. S. Attard, P. N. Bartlett, N. R. B. Coleman, J. M. Elliott, J. R. Owen and J. H. Wang, Science (1979), 1997, 278, 838–840 CAS.
- Y. Li, Y. Liu, J. Li, D. Xiong, X. Chen, M. Liu, Z. Zhong, V. Malgras and Y. Bando, Mater. Horiz., 2020, 7, 489–494 RSC.
- H. Ji, W. Zeng and Y. Li, Nanoscale, 2019, 11, 22664–22684 RSC.
- K. Wetchakun, T. Samerjai, N. Tamaekong, C. Liewhiran, C. Siriwong, V. Kruefu, A. Wisitsoraat, A. Tuantranont and S. Phanichphant, Sens. Actuators, B, 2011, 160, 580–591 CrossRef CAS.
- C. Mora, D. Spirandelli, E. C. Franklin, J. Lynham, M. B. Kantar, W. Miles, C. Z. Smith, K. Freel, J. Moy, L. V. Louis, E. W. Barba, K. Bettinger, A. G. Frazier, J. F. Colburn IX, N. Hanasaki, E. Hawkins, Y. Hirabayashi, W. Knorr, C. M. Little, K. Emanuel, J. Sheffield, J. A. Patz and C. L. Hunter, Nat. Climate Change, 2018, 8, 1062–1071 CrossRef CAS.
- Y. Liu, J. Parisi, X. Sun and Y. Lei, J. Mater. Chem. A, 2014, 2, 9919–9943 RSC.
- J. Liu, Y. Li, H. Fan, Z. Zhu, J. Jiang, R. Ding, Y. Hu and X. Huang, Chem. Mater., 2010, 22, 212–217 CrossRef CAS.
- A. Kolodziejczak-Radzimska and T. Jesionowski, Materials, 2014, 7, 2833–2881 CrossRef CAS PubMed.
- R. Jiang, Y. Da, X. Han, Y. Chen, Y. Deng and W. Hu, Cell Rep. Phys. Sci., 2021, 2, 100302 CrossRef CAS.
- P. Kjellin and A. E. C. Palmqvist, J. Mater. Sci., 2008, 43, 7250–7253 CrossRef CAS.
- O. Muller and R. Roy, Mater. Res. Bull., 1969, 4, 39–43 CrossRef CAS.
- X. Li and R. E. Schaak, Chem. Mater., 2019, 31, 2088–2096 CrossRef CAS.
- Y. W. Lee, M. A. Lim, S. W. Kang, I. Park and S. W. Han, Chem. Commun., 2011, 47, 6299 RSC.
- S. Kundu and N. Ravishankar, Nanoscale, 2016, 8, 1462–1469 RSC.
- C. Pacholski, A. Kornowski and H. Weller, Angew. Chem., Int. Ed., 2002, 41, 1188–1191 CrossRef CAS PubMed.
- A. Leelavathi, G. Madras and N. Ravishankar, J. Am. Chem. Soc., 2014, 136, 14445–14455 CrossRef CAS PubMed.
- H. J. Fan, M. Knez, R. Scholz, K. Nielsch, E. Pippel, D. Hesse, M. Zacharias and U. Gösele, Nat. Mater., 2006, 5, 627–631 CrossRef CAS PubMed.
- R. Rémiás, Á. Kukovecz, M. Darányi, G. Kozma, S. Varga, Z. Kónya and I. Kiricsi, Eur. J. Inorg. Chem., 2009, 3622–3627 CrossRef.
- J. Paiz, J. Fitch, E. Peterson, T. Hough, W. Barnard and A. Datye, Cryst. Res. Technol., 2014, 49, 699–707 CrossRef CAS.
- I. Hölken, G. Neubüser, V. Postica, L. Bumke, O. Lupan, M. Baum, Y. K. Mishra, L. Kienle and R. Adelung, ACS Appl. Mater. Interfaces, 2016, 8, 20491–20498 CrossRef PubMed.
- X. Li and R. E. Schaak, Chem. Mater., 2019, 31, 2088–2096 CrossRef CAS.
- Y. V. Kaneti, N. L. Wulan Septiani, I. Saptiama, X. Jiang, B. Yuliarto, M. J. A. Shiddiky, N. Fukumitsu, Y. M. Kang, D. Golberg and Y. Yamauchi, J. Mater. Chem. A, 2019, 7, 3415–3425 RSC.
- S. Tripathi, R. Bose, A. Roy, S. Nair and N. Ravishankar, ACS Appl. Mater. Interfaces, 2015, 7, 26430–26436 CrossRef CAS PubMed.
- A. Leelavathi, G. Madras and N. Ravishankar, Phys. Chem. Chem. Phys., 2013, 15, 10795 RSC.
- J. E. Drawdy, G. B. Hoflund, S. D. Gardner, E. Yngvadottir and D. R. Schryer, Surf. Interface Anal., 1990, 16, 369–374 CrossRef CAS.
- S. D. Jackson, J. Willis, G. D. McLellan, G. Webb, M. B. T. Keegan, R. B. Moyes, S. Simpson, P. B. Wells and R. Whyman, J. Catal., 1993, 139, 191–206 CrossRef CAS.
- G. M. Bancroft, I. Adams, C. D. Bennewitz, J. D. Brown and W. D. Westwood, Anal. Chem., 1975, 47, 586–588 CrossRef CAS.
- Y. Ji, F. Zhang, F. Yu, J. Zhang and J. Zhang, Catalysts, 2018, 8, 99 CrossRef.
- C. D. Wagner, L. E. Davis, M. V. Zeller, J. A. Taylor, R. H. Raymond and L. H. Gale, Surf. Interface Anal., 1981, 3, 211–225 CrossRef CAS.
- J. Wang, Z. Wang, B. Huang, Y. Ma, Y. Liu, X. Qin, X. Zhang and Y. Dai, ACS Appl. Mater. Interfaces, 2012, 4, 4024–4030 CrossRef CAS PubMed.
- S. Shetty, S. K. Sinha, R. Ahmad, A. K. Singh, G. Van Tendeloo and N. Ravishankar, Chem. Mater., 2017, 29, 9887–9891 CrossRef CAS.
- G. B. Andreozzi, V. D’Ippolito, H. Skogby, U. Hålenius and F. Bosi, Phys. Chem. Miner., 2019, 46, 343–360 CrossRef CAS.
- L. I. Granone, A. C. Ulpe, L. Robben, S. Klimke, M. Jahns, F. Renz, T. M. Gesing, T. Bredow, R. Dillert and D. W. Bahnemann, Phys. Chem. Chem. Phys., 2018, 20, 28267–28278 RSC.
- Materials Project, mp-35647, 2014 DOI:10.17188/1207022.
- X. Xing, T. Chen, R. Zhao, Z. Wang and Y. Wang, Sens. Actuators, B, 2018, 254, 227–238 CrossRef CAS.
- P. Singh, A. Kumar, Deepak and D. Kaur, J. Cryst. Grow., 2007, 306, 303–310 CrossRef CAS.
- V. M. Djinovic, L. T. Mancic, G. A. Bogdanovic, P. J. Vulic, G. del Rosario, T. J. Sabo and O. B. Milosevic, J. Mater. Res., 2005, 20, 102–113 CrossRef CAS.
- E. A. Anumol, P. Kundu, P. A. Deshpande, G. Madras and N. Ravishankar, ACS Nano, 2011, 5, 8049–8061 CrossRef CAS PubMed.
- D. L. Swihart and W. R. Mason, Inorg. Chem., 1970, 9, 1749–1757 CrossRef CAS.
- C. M. Davidson and R. F. Jameson, Trans. Faraday Soc., 1965, 61, 2462 RSC.
- I. V. Znakovskaya, Y. A. Sosedova, E. M. Glebov, V. P. Grivin and V. F. Plyusnin, Photochem. Photobiol. Sci., 2005, 4, 897 CrossRef CAS PubMed.
- V. M. Djinovic, L. T. Mancic, G. A. Bogdanovic, P. J. Vulic, G. del Rosario, T. J. Sabo and O. B. Milosevic, J. Mater. Res., 2005, 20, 102–113 CrossRef CAS.
- S. D. Cameron and D. J. Dwyer, Surf. Sci., 1986, 176, L857–L862 CrossRef CAS.
- M. W. Gaultois and A. P. Grosvenor, J. Phys. Chem. C, 2010, 114, 19822–19829 CrossRef CAS.
- G. Deroubaix and P. Marcus, Surf. Interface Anal., 1992, 18, 39–46 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- F. J. Sotomayor, K. A. Cychosz and M. Thommes, Acc. Mater. Surf. Res, 2018, 3, 34–50 Search PubMed.
- D. P. Barkey, J. Electrochem. Soc., 1989, 136, 2207 CrossRef CAS.
- K. Manthiram, Y. Surendranath and A. P. Alivisatos, J. Am. Chem. Soc., 2014, 136, 7237–7240 CrossRef CAS PubMed.
- Y. Huang, Z. Shen, Y. Wu, X. Wang, S. Zhang, X. Shi and H. Zeng, RSC Adv., 2016, 6, 17867–17872 RSC.
- A. Rednyk, A. Ostroverkh and V. Johánek, Int. J. Hydrogen Energy, 2017, 42, 29254–29261 CrossRef CAS.
- C. Li, T. Sato and Y. Yamauchi, Angew. Chem., 2013, 125, 8208–8211 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00090c |
| This journal is © The Royal Society of Chemistry 2022 |