
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-stimuli responsive and intrinsically luminescent polymer metallogel through ring opening copolymerization coupled with thiol-ene click chemistry†
Chindhu
Soman
a,
Alphy
Sebastian
a,
Malay Krishna
Mahato
a,
U. V.
Varadaraju
b and
Edamana
Prasad
 *a
*a
aDepartment of Chemistry, Indian Institute of Technology of Madras, Chennai 600036, India. E-mail: pre@iitm.ac.in; Fax: +91-44-2257-4202; Tel: +91-44-2252-4232
bInternational Advanced Research Centre for Powder Metallurgy and New Materials(ARCI), Chennai 600113, India
First published on 23rd May 2022
Abstract
Luminescent polymer based metallogels have gained considerable interest due to their wide range of applications in the fields of drug delivery, tissue engineering, sensing, and optical systems. One of the challenges in the area is to tune the properties of metallogels for a given application, which is largely controlled by the choice of the functional groups in the polymer and the binding metal ions. In the present study, efforts are made to utilize ring opening copolymerization followed by functionalization by thiol-ene click chemistry to attach desirable functional groups to the polymer for preparing polymer based metallogels. A norbornene-alt-cyclohexene oxide based polymer is synthesized to develop an intrinsically luminescent metallogel using lanthanide metal–ligand complexation. The optical properties of the polymer metallogel are modulated via a dual channel process where a cluster induced intrinsic emission surpasses resonance energy transfer mechanism, leading to cool white light emission from the metallogel [CIE coordinates (0.33, 0.37);correlated color temperature 5752 K], with reversible vapochromism and irreversible chemochromism. Our studies suggest that the combination of ring opening co-polymerization and thiol-ene click chemistry is a potential design strategy for preparing polymer based metallogels with multi-stimuli responsive properties.
Introduction
In the recent past, supramolecular materials with self-healing and environmentally benign stimuli responsiveness have drawn considerable attention due to their wide applications in actuating and sensing,1–6 separation and catalysis,7–10 light-harvesting,11–15 and, biomedical engineering.16–18 Supramolecular gels, a class of supramolecular materials, are of particular interest due to their pivotal role in developing drug encapsulation, controlled guest release, smart coatings, and sensors.19 Different non-covalent interactions such as H-bonding and π–π interactions are widely used to construct supramolecular gels. Formation of gels by self-assembly of metal–ligand coordination enhances toughness and shape memory of the self-assembled system. Furthermore, metal–ligand coordination results in increased stimuli-responsiveness of the gels, owing to the dynamic and reversible nature of the bonds.20–25Introduction of metal ion into gels brings intriguing changes to redox, electronic, optical, and catalytic properties,26–28 and endow materials with versatile functionalities such as tunable emission, catalytic activity, proton conductivity, gas storage, and sensing.29–39 Recently, to gain better control over the luminescent properties for use in solid-state lighting and large panel displays, transition metal ions and lanthanides are introduced into gels comprising small organic molecules and functionalized polymers.40–49 However, compared to the transition metal, the intrinsic optical properties of lanthanides, such as long lifetime, sharp emission, intense luminescence and resistance to photo-bleaching, make lanthanides a better choice for constructing luminescent supramolecular gels.50 Chen et al. utilized “tuning stoichiometry” approach with lanthanides [Eu(III)/Tb(III)] to develop white light-emitting polymer based metallogels.31 Similarly, a white light-emitting metallogel containing lanthanide was also reported by Sutar et al. by coordinating amphiphilic, tripodal low-molecular-weight gelator (L) that contains 4,4′,4-[1,3,5-phenyl-tri(methoxy)]-tris-benzene core and 2,2′:6′,2′′-terpyridyl termini.51 In another attempt, poly (aryl ether) dendron was utilized for the formation of white light emitting gel in presence of organic chromophore and lanthanide ions [Eu(III)].52 Such studies clearly establish the presence of resonance energy transfer (RET) from organic groups to lanthanide ions as the fundamental working mechanism behind the production of white light. Hence, choice of organic ligand and lanthanide ion is critical to regulate luminescent properties of the metallogel.
The dynamic nature of ligand–metal coordination in supramolecular gels attracted special attention because it allows the materials to exhibit multi-stimuli responsive behavior to various stimuli such as heat, pH, electricity, and ions.53–58 For example, Mahapatra et al. developed a white light-emitting supramolecular gel using mixed Eu(III)/Tb(III) with 4′-p-halophenyl-2,2′:6′,2′′-terpyridine (L-X, X = F, Cl) ligands, and the resulting gel exhibited stimuli-responsive behavior towards pH as well as temperature.59 In a previous work, a polymer-based white light-emitting hydrogel containing Eu(III), and Tb(III) was reported by Zhu et al. and the resultant metallogel was responsive to multiple stimuli including pH, metal ions and temperature.60 Recently, lanthanide based white light emitting metallogel was reported from our group which responds to a wide range of pH and temperature conditions.61
In all of the above-mentioned studies, polymers were functionalized with various molecular groups such as amine, hydroxyl, and carboxyl to develop multi-stimuli responsive gels, in presence of a metal ion. The studies also reveal that the above approaches demand multiple synthetic steps for the polymer functionalization. One of the challenges that exists is the limited ways through which the functional groups can be introduced into the polymer backbone, which in turn restricts the choices of metal–ligand coordination, leading to a narrow window for tuning the stimuli-responsive properties of the metallogel. In the current work, it is hypothesized that this difficulty can be overcome by ring-opening copolymerization method followed by simple click-chemistry to introduce various functional groups in post-polymerization steps. Thus, a novel supramolecular polymer metallogel is designed and developed by self-assembly of carboxylate functionalized poly(norbornene anhydride-alt-cyclohexene oxide) containing lanthanide ions (Eu(III) and Tb(III)) and the intrinsic light emission as well as stimuli-responsive nature of the self-assembled systems have been examined. The intricacies associated with the intrinsic emission and resonance energy transfer, leading to white light emission from the metallogel, has also been investigated. In addition, the stimuli-responsive nature of the white light emitting gel was examined in presence of vapors and anions to verify vapochromism and chemochromism.
Experimental section
Materials
Unless otherwise stated, all reagents are purchased from commercial sources and used without further purification. All air-sensitive reactions were carried out under Argon atmosphere in the Mbrown glove box. Norbornene anhydride was sublimed under vacuum at 110 °C. Cyclohexene oxide was distilled over CaH2 under Argon atmosphere. Bis(triphenylphosphine)iminium chloride and thioglycolic acid were purchased from Sigma and used directly. Tetrahydrofuran (THF) was refluxed and distilled over sodium-benzophenone under argon.Characterization
1H and 13C NMR data were recorded on a Bruker 400 MHz spectrometer in DMSO-d6 and CDCl3 solvent. IR spectra were collected from a JASCO FT/IR-4100 spectrometer at room temperature. JASCO V-660 spectrophotometer was used for recording UV spectra. Fluorescence spectra were recorded on a Horiba Jobin Yuvon Fluoromax-4 fluorescence spectrofluorometer. Rheology experiments were performed on Anton-Paar (MCR 102) using a parallel plate geometry on a Peltier plate. Bruker ultrafleXtreme MALDI-TOF spectrometer was used for recording MALDI-TOF spectra. SEM images were collected with HITACHI S 4800 FE-SEM instrument. In a time correlated single-photon counting (TCSPC) setup, time-resolved luminescence studies were recorded on a Horiba Jobin Yvon FluoroCube apparatus. The light source was a 350 nm nano-LED with a 1 MHz pulse repetition rate. A scatterer was used to capture the instrument response function (IRF) (Ludox AS40 colloidal silica, Sigma-Aldrich). Fixing the emission wavelength at 620 nm captured the excited-state decay of the samples. The decay was fitted to the following tri-exponential decay (eqn (1)) using IBH software (DAS6), where I is the luminescence lifetime and Ai is the amplitude of the related decay.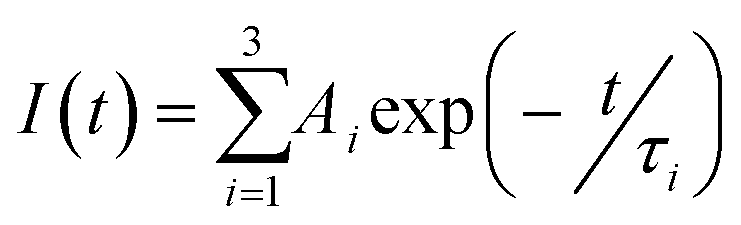 | (1) |
The following calculation was used to compute the average fluorescence lifetime.(τav)
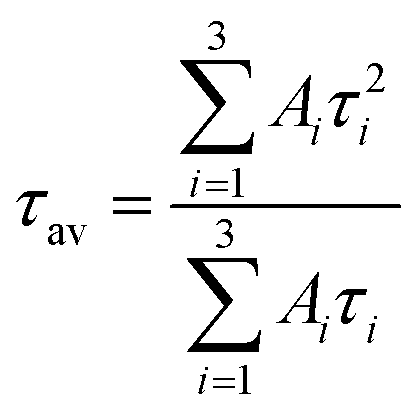 | (2) |
Procedure for the ring opening copolymerization of cyclohexene oxide (CHO) and norbornene anhydride (NA)
To avoid moisture, the polymerization was carried out under an argon atmosphere in a glove box using a procedure reported in the literature.62 Norbornene anhydride (NA) (1 g, 3.5 mmol) was charged in a 20 mL reaction tube. After that, bis(triphenylphosphine)iminium chloride ([PPN]Cl) (17 mg, 0.017 mmol), and cyclohexene oxide(CHO) (1.23 mL, 6.9 mmol) were added to the above reaction tube. The reaction tube was then sealed with Teflon and stirred at 110 °C for 12 h. The reaction tube was allowed to cool down to room temperature and excess methanol was added to the reaction mixture. After 2 hour stirring, the reaction mixture was filtered. The solid product was repeatedly washed with methanol to remove unreacted monomers, low molecular weight oligomers and excess catalyst and finally dried under vacuum which yielded the desired polyester. 1H-NMR (TMS, CDCl3, 400 MHz): δ = 6.3 (m, 2H, CH = CH); δ = 4.6 (s, 2H, CH–CH); δ = 3.2 (m, 4H, CH); δ = 1.5 (m, 8H, CH2); δ = 2.0(s, 2H, CH2).Postpolymerization reaction (thiol-ene reaction)
Based on the reported procedure for the postpolymerization,62 polyester (0.5 g, 1.9 mmol) and azobisisobutyronitrile (AIBN) (3 mg, 0.02 mmol) were taken in a schlenk tube containing 5 mL of THF under argon atmosphere. To obtain a clear homogenous solution, the above mixture was stirred for 10 minutes. Then mercaptoacetic acid (0.2 mL, 2.8 mmol) was added to the above reaction mixture and heated at 70 °C in an oil bath for 3 hours. The polymerization was terminated by adding distilled water into the reaction tube. Finally, the obtained white precipitated was washed with distilled water and dried under vacuum which yield carboxylic acid functionalized polyester. 1H-NMR (TMS, CDCl3, 400 MHz): δ = 12.8 (s, 1H, COOH); δ = 4.6 (s, 2H, CH–CH); δ = 3.8 (s, 2H, CH2); δ = 3.2 (m, 4H, CH); δ = 1.5 (m, 8H, CH2); δ = 2.0(s, 2H, CH2).Results and discussions
Synthesis and characterization of polyester and corresponding Eu(III) and Tb(III) metallogels
The polyester was synthesized by ring-opening copolymerization using bis(triphenylphosphine)iminium chloride ([PPN]Cl) as a catalyst. In a subsequent step the main chain of the polyester was functionalized with the carboxylic acid group using thiol-ene click chemistry as shown in Scheme 1 (details of synthesis are given in experimental section).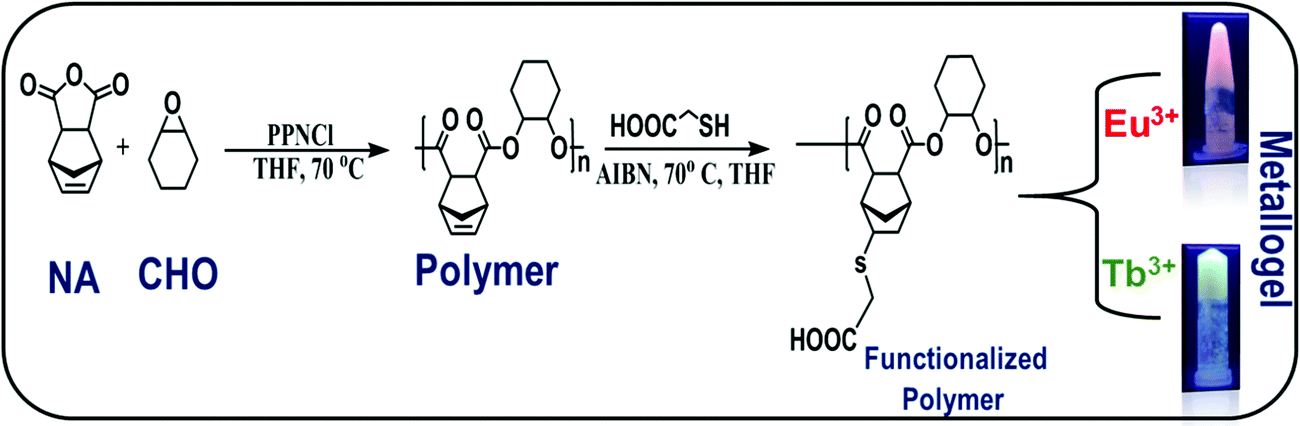 | ||
| Scheme 1 Synthetic scheme of polymer, functionalized polymer and metallogels. | ||
The polyester was characterized using 1H NMR spectroscopy, gel permeation chromatography (GPC), MALDI-TOF, and TGA analysis. The structures of the pristine and functionalized polyester were confirmed by 1H NMR as shown in Fig. S1 and S2 (ESI†), respectively. 1H NMR showed a sharp peak at 4.7 ppm, indicating that the polyester is fully alternating. The disappearance of multiplets at 6–6.5 ppm suggests the substitution of carboxyl ligand to the unsaturated norbornene group in the polymer backbone. Additionally, the complete disappearance of the unsaturated peak suggests that the functionalization of the polymer in thiol-ene reaction is more than 99%. Fig. S3 (ESI†) shows the results of the gel permeation chromatography (GPC) of the pristine and functionalized polymer. The results indicate that the number average molecular weight of the pristine polymer is 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 920 Da with dispersity index (Mw/Mn) value of 1.30. However, no peaks of the functionalized polyester were detected by GPC, presumably due to the adhesion of carboxylic functional groups of the functionalized polymer with the polystyrene gels used in GPC column. MALDI-TOF spectrum of functionalized polyester shows sharp peaks around m/z = 4817 and m/z = 8205 indicating the formation of the polymer (ESI† Fig. S4). Fig. S5 (ESI†) shows the results of thermo-gravimetric analysis (TGA) of the functionalized polymer. The weight loss in the temperature range 120–180 °C is attributed to loss of water molecules. The onset of decomposition of the polymer is ∼250 °C and the decomposition is complete at ∼420 °C.
920 Da with dispersity index (Mw/Mn) value of 1.30. However, no peaks of the functionalized polyester were detected by GPC, presumably due to the adhesion of carboxylic functional groups of the functionalized polymer with the polystyrene gels used in GPC column. MALDI-TOF spectrum of functionalized polyester shows sharp peaks around m/z = 4817 and m/z = 8205 indicating the formation of the polymer (ESI† Fig. S4). Fig. S5 (ESI†) shows the results of thermo-gravimetric analysis (TGA) of the functionalized polymer. The weight loss in the temperature range 120–180 °C is attributed to loss of water molecules. The onset of decomposition of the polymer is ∼250 °C and the decomposition is complete at ∼420 °C.
The functionalized polymer was found to be insoluble in non-polar organic solvents due to the presence of highly polar carboxyl groups in the polymer chain (ESI† Table S1). The gelation ability of the functionalized polymer was tested in different solvents by varying the concentration, temperature and sonication but, stable gels could not be obtained. However, addition of Ln(OAc)3 [Ln = Eu(III), Tb(III)] (3 mg) to the functionalized polymer in DMF solution(10 mg mL−1) followed by heating and gentle shaking resulted in the formation of a stable gel almost instantaneously (Scheme 1). This observation clearly indicates that metal–ligand coordination is one of the major driving forces to form the gel. The formation of the stable gel upon addition of the lanthanide salt was confirmed by the vial inversion test as shown in Fig. S6 (ESI†). The gel was stable for several months at room temperature. This is the first report of supramolecular metallogel formed from lanthanides and polyesters, synthesized through highly controlled ring opening copolymerization followed by facile functionalization by carboxyl group via thiol-ene click chemistry. The complexation of metal and polymer that leads to metallogels formation was confirmed by IR spectroscopy. Fig. 1 exhibits the comparison of IR spectra of the functionalized polymer with the corresponding metallogels of Eu(III) and Tb(III). The functionalized polymer showed characteristic carbonyl stretching frequency at 1674 cm−1. However, the carbonyl stretching frequency in the polymer in presence of Eu(III) and Tb(III) was found to be at 1658 cm−1. The observed decrease in carbonyl frequency (16 cm−1) clearly indicates the coordination of the carboxyl group of the polymer with the lanthanides (Eu(III) and Tb(III)).
 | ||
| Fig. 1 (a) FTIR spectra of functionalized polymer (black line), polymer and Eu(OAc)3 (red line), polymer and Tb(OAc)3 (blue line). (b) Zoomed spectra of carbonyl peak position of polymer (black line), Eu gel(red line) and Tb gel(blue line). | ||
Further, the intensity of the peak at 3500 cm−1 corresponding to hydroxyl stretching frequency of the polymer, was found to decrease with increasing concentration of the lanthanides (ESI† Fig. S7). This observation further confirms that the polymer carboxylic acid group indeed coordinated with the metal ion; the carboxylic acid group (–COOH) converts –COO− upon formation of metallogels. It is worth mentioning that other lanthanide salts such as Ln(SO4)3 and Ln(ClO4)3 could not form gel which suggests that only lanthanide acetate could deprotonate the –COOH groups to form –COO−, which is essential for metal coordination.
Further to understand the morphology of the obtained gel, scanning electron microscopy (SEM) images were recorded. The gels were dried in a desiccator, under vacuum for two hours prior taking the images. Fig. 2 shows the SEM micrographs of the metallogels containing Eu(III) and Tb(III) ions. Both the metallogels show highly crosslinked aggregated fibrous morphology. The size of the fiber was comparatively small as generally observed in the typical gels which is consistent with the earlier literature.61 The highly crosslinked aggregates clearly validates the formation of cluster upon metalation of the polymer. Additionally, same morphology of the metallogels indicates there is no phase separation and both the metal ions binds in similar pattern during the assembly formation with the polymers.
 | ||
| Fig. 2 SEM images of Eu(left) and Tb(right) gels dried on the silicon plates (scale bars, 500 nm). | ||
To understand the mechanical properties of the obtained gels, rheological studies (frequency and strain sweeps) were carried out. Fig. 3a and c show the results of the frequency dependent rheological study of Eu(III) and Tb(III) metallogels, respectively. The moduli values of both Eu gel and Tb gels were independent of applied frequency in the frequency range tested. The dominance of storage modulas over loss moduli in the tested frequency range further confirmed the viscoelastic nature of metallogels. This observation is in line with other cross-linked polymers.59,63 Also, in both cases, the higher value of G′ compared to that of G′′ indicates their solid-like character. Fig. 3b and d show the results of strain sweep rheological measurements for Eu(III) and Tb(III) containing metallogels, respectively. The results indicate that the storage modulus remains unchanged for both the cases at lower strain amplitudes, till yield strain is reached. Further increase in the strain amplitude results in decrease in G′ suggesting gel–sol conversion. The magnitude of storage moduli was found to be similar for Eu(III) gel (6.04 kPa) and Tb(III) gel (4.4 kPa). However, the yield strain of Eu(III) metallogel is 5%, whereas Tb(III) metallogel yields at 16.5%.
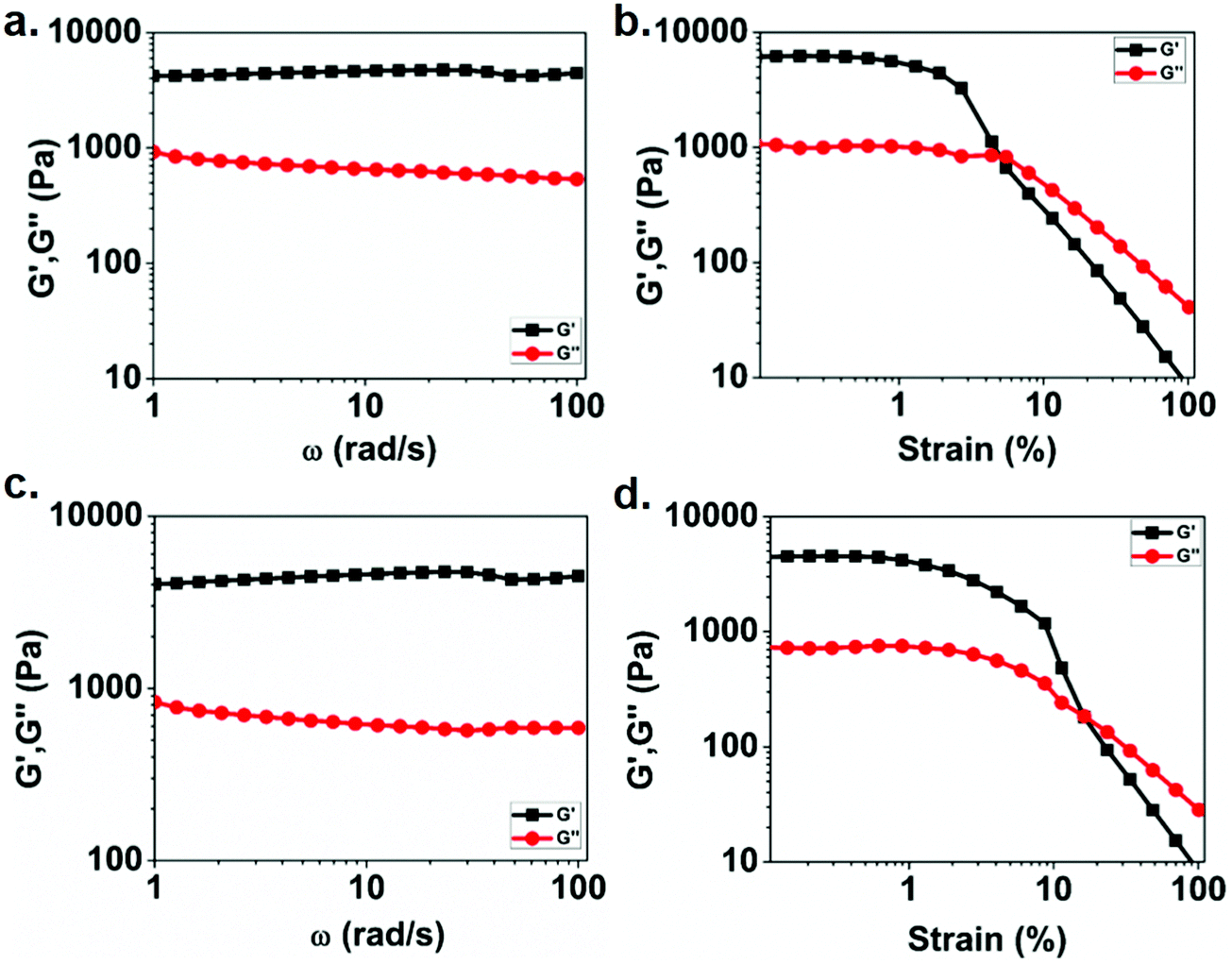 | ||
| Fig. 3 At a strain amplitude of 0.1 percent, the frequency dependence of the storage modulus G′ (■) and loss modulus G′′ (●) is shown: (a) Eu(III), and (c) Tb(III). The corresponding strain sweeps for (b) Eu(III) and (d) Tb(III) are also displayed. | ||
Photo-physical analysis of Ln(III) mediated self-assembly of functionalized polyester in solution
To understand the formation of Ln(III) mediated self-assembly of the functionalized polymer, the UV-vis. absorption spectra of the polymer in DMF were recorded with the increasing concentration of lanthanide ions. Fig. 4a and c show the absorption spectra of polymer in presence of Eu(III) and Tb(III), respectively. The absorption spectra for the functionalized polymer exhibit a broad band from 250 nm to 600 nm. However, upon addition of lanthanide ions (both Eu(III) and Tb(III)), the absorbance was found to increase in both cases. The changes in the absorption spectra can be either due to the complex formation or additive absorption by the individual component. To understand the exact reason behind changes the absorption spectra, separate absorption spectra were recorded for polymer and lanthanides with similar concentration. Individual absorption spectrum along with the additive absorption are shown in Fig. S8 (ESI†). The results indicate that polymer absorption upon addition of lanthanides does not match with the additive absorption spectrum, which confirms the complex formation between polymer and lanthanides. In addition, the appearance of turbidity upon addition of lanthanides to an initially clear solution again indicates the complex formation. Further, to quantify the binding ratio of the metal with the functionalized polymer, Jobs plots were made using the absorption spectra. Fig. 4b and d show the Jobs plot of the polymer with Eu(III) and Tb(III), respectively. The results suggest that the absorbance gradually increases up to 0.3 equiv. of Eu(III) as well as Tb(III), followed by a decrease in the absorbance upon further addition of lanthanide ions, indicating a 3:1 ligand–metal binding ratio.
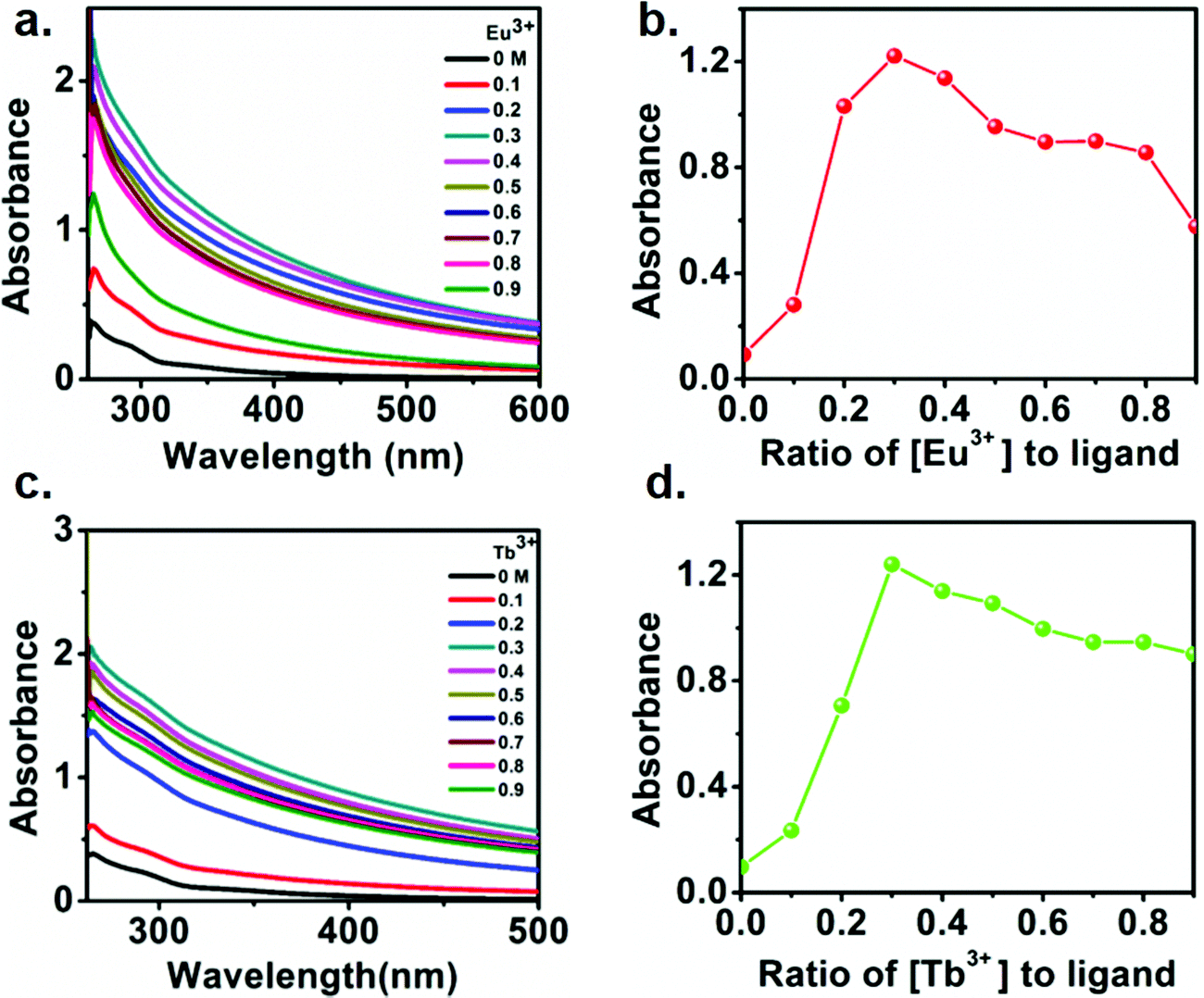 | ||
| Fig. 4 Changes in the UV-vis absorption spectra upon titrating the polymer (3.1 × 10−3 M) with 0 → 0.9 equivalence of (a) Eu(OAc)3 and (c) Tb(OAc)3 and, the corresponding Jobs plots (b) and (d), respectively (absorbance collected at 335 nm). | ||
In order to understand the electronic interaction between the polymer and lanthanides, fluorescence spectrum of the metallo-polymer was recorded by exciting the sample at 335 nm. Fig. 5a and c respectively show the luminescent spectra of the polyester solution with increasing concentration of Eu(III) and Tb(III). The results show a broad luminescence (black line) from 350 nm to 450 nm for the polymer solution. Notably, two sharp emission peak was appeared to observe along with the polymer emission band upon addition of lanthanides. The appearance of characteristics sharp emission band of lanthanides clearly indicates the ability of the polymer to act as a sensitizing antenna for lanthanides, since direct excitation of lanthanides are Laporte forbidden transition. The observed sharp emission peak at 591 and 615 nm for Eu(III) containing polyester are due to transitions from 5D0–7F1 and 5D0–7F2 state. On the other hand, The emission peaks at 488, 544, 582, 620 nm for Tb(III) are assigned to the deactivation of 5D4 of Tb(III) excited state to 7Fj (6–3) states.50 The addition of Eu(III) ion to the functionalized polymer solution resulted in an increase in the intensity of the emission band of Eu(III) up to 0.3 equivalent of Eu(III), followed by a decline in emission intensity with further addition of Eu(III) (Fig. 5b). The same behavior was observed in the case of Tb(III) ion titration with the functionalized polymer (Fig. 5c). These results confirm that the complex formation of functionalized polymer with the Ln(III) metal ion with a 3:1 ratio. It is interesting to note that the polymer exhibits blue luminescence though it does not contain any conventional fluorophore. This issue has been addressed in the literature and is described as clustering triggered emission.64–68 Considering the structure of the polymer in the present study, it is reasonable to attribute the polymer intrinsic emission to the aggregation of the oxygen terminals. To understand the mechanism of the clustered triggered emission, a single and double units of the polymer (cluster) structures were optimized using B3LYP/3-21G level density functional theory. The optimized structure of polymer (ESI† Fig. S9) indicates that a number of short intra (1.73, 2.26, 2.62, 2.59 Å) and inter (2.28, 2.29, 2.71, 2.50, 2.66 Å) molecular H-bonding are feasible in the polymer which can potentially restrict the molecular motion and rigidify the polymer. Furthermore, several intra (2.24, 2.72, 2.26, 2.26 Å) and inter (2.64, 1.52, 1.51, 2.67, 2.73 Å) molecular O⋯O electronic interactions can be present with significant shorter length than the sum of the van der Waals (vdW) radii (3.04 Å)67 which result in effective three-dimensional space conjugation of the lone pairs.
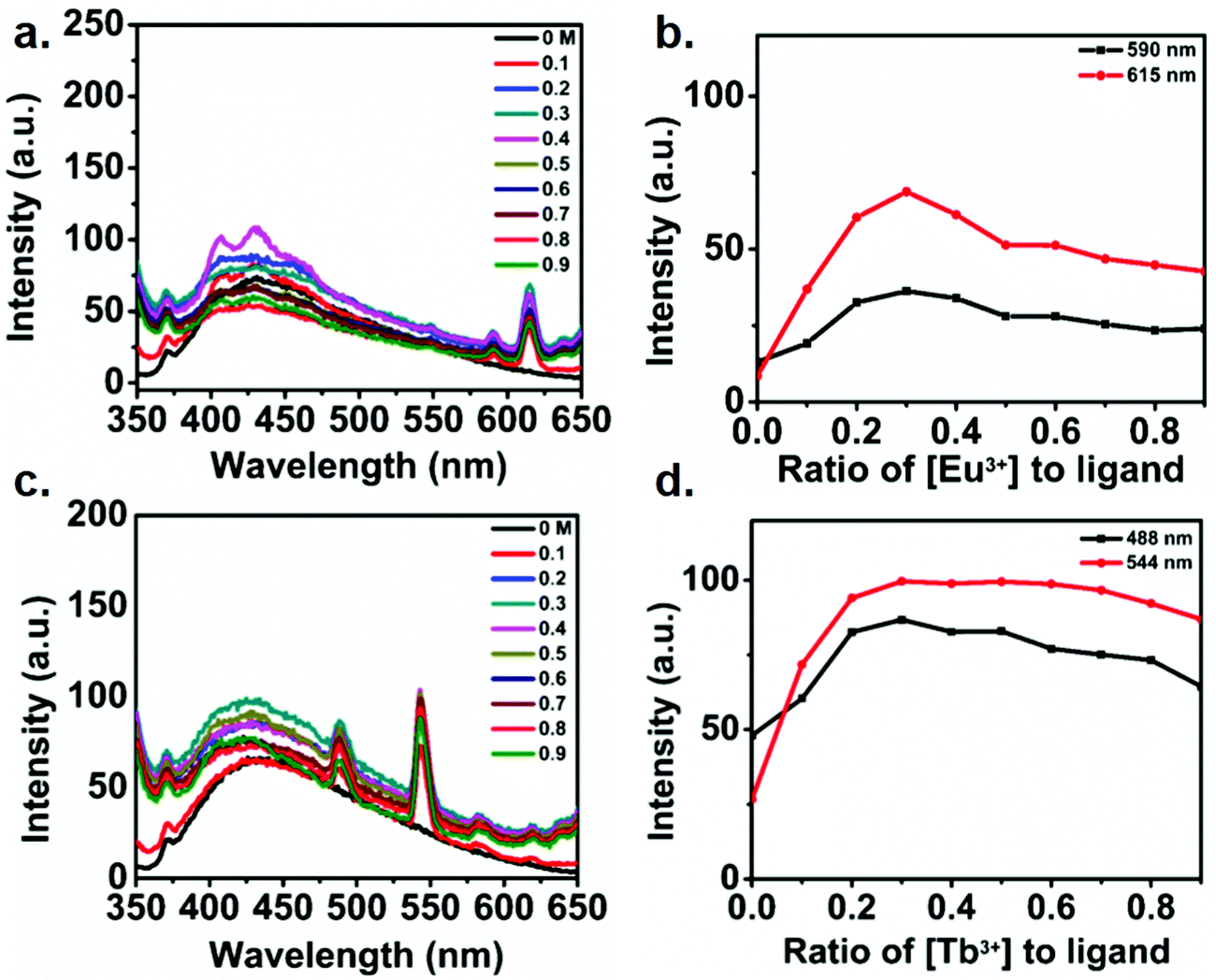 | ||
| Fig. 5 Changes in (a) the emission spectra upon titrating polymer (3.1 × 10−3 M) with Eu(OAc)3 (0 → 0.9 equiv.), (b) experimental binding isotherms (●) and their corresponding fits (−) for the increase in Eu(OAc)3 concentration, (c) changes in the emission spectra upon titrating polymer (3 × 10−3 M) with Tb(OAc)3 (0 → 0.9 equiv.), (d) experimental binding isotherms (●) and their corresponding fits (−) for the titration with Tb(OAc)3. Both titrations were carried out in DMF at 298 K and the spectra were recorded under 335 nm excitation. | ||
In order to understand the mechanism of the intrinsic emission, the electron density on the HOMO and LUMO level were calculated. Fig. 6c shows the results of electron density on the HOMO and LUMO level of single and double unit of the polymer (cluster). In the single unit of polymer, the electron density was distributed throughout all oxygen centre, suggesting significant O⋯O intramolecular electronic interaction. The electron density on the LUMO and LUMO+1 level of the cluster were found to be distributed between the neighboring unit, which clearly suggest the extended delocalization of electron in the excited state and further support the hypothesis of the clustering triggered emission in the present polymer. Further to confirm this phenomena, a control experiment was carried out by taking the emission spectra of polymer with the non-photoactive metal Zn-acetate. The results are shown in Fig. S10 (ESI†) which indicates the emission intensity of the polymer was enhanced in presence of Zn2+ which confirmed the enhancement of the polymer is due to the cluster formation.
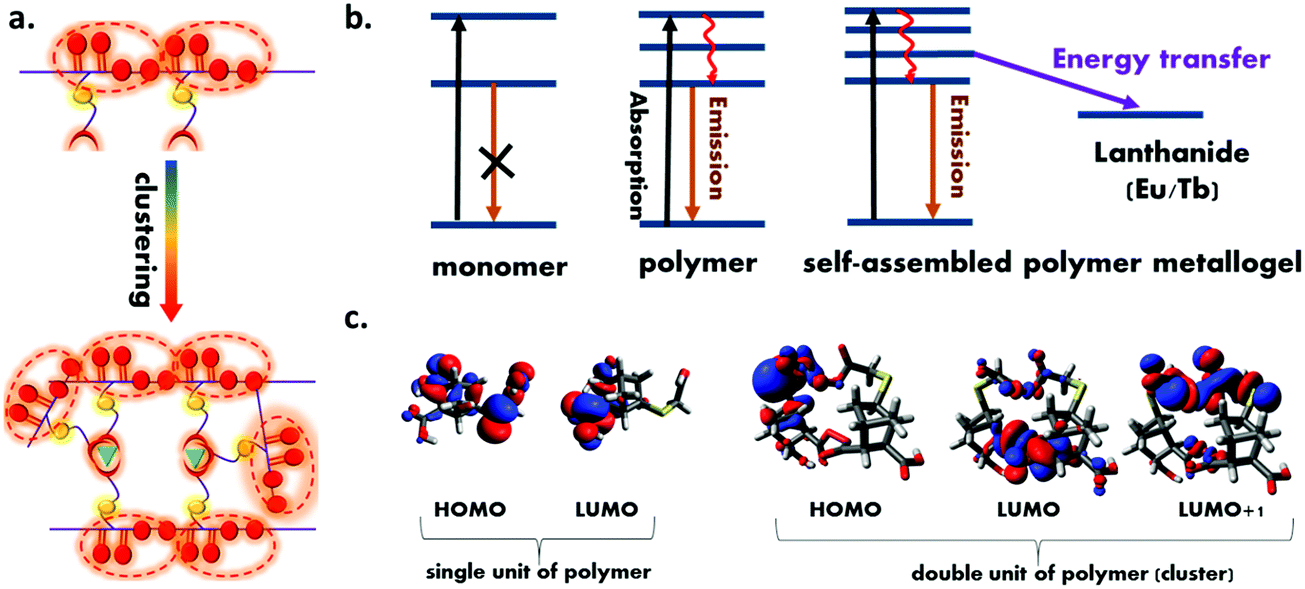 | ||
| Fig. 6 (a) Schematic illustration of the mechanism of clustering triggered emission of non-conventional luminophores. (b) Schematic representation of all the photophysical process in the present system (c) the electron density distribution on HOMO and LUMO of the single and double unit (cluster) of the polymer obtained using the B3LYP/3-21G level of density functional theory. | ||
Moreover, the results of the emission spectra indicate that upon successive addition of lanthanides, the sharp lanthanides centered emission intensity was increased due to the resonance energy transfer from the polymer to lanthanides.52 In order to provide direct evidence on resonance energy transfer, excitation spectrum of both the polymer metallogels were recorded (ESI† Fig. S11). The excitation spectrum shows a broad peak in the range 350–450 nm for both cases upon collecting the emission at lanthanide centered emission which clearly validates that energy is transferred from the polymer to the lanthanides.61 Further time resolved decay experiment and relative quantum yield (QY) calculation were performed. Table S2 (ESI†) shows the emission QY of the polymer in presence and absence of lanthanides. Fig. S12 (ESI†) shows the results of the time resolved decay spectra of the polymer and corresponding metallopolymer. The results clearly indicate the faster decay of the polymer emission in presence of both the lanthanides. This observation suggests that the polymer excited state involved additional deactivation channel by which emission lifetime decreases. The average lifetime value of all the samples were calculated (ESI,† Table S3) which shows the lifetime of the polymer changes from 3.98 ns to 3.53 ns and 3.38 ns with the Eu and Tb respectively which clearly validates the resonance energy transfer from polymer to the Eu and Tb. It is to be noted that the emission band of the polymer (donor) was found to increase as the amount of lanthanide ions was increased, which is in contrast with the normal FRET. When lanthanides are added to the polymer, the carboxylate group of the polymer binds to the lanthanides, resulting in more polymer self-assembly, which leads in more oxygen clusters and rigidification69 of the polymer metallogel (Fig. 6a). The cluster formation and resonance energy transfer occur simultaneously, however, cluster formation triggered emission may outcompete the energy transfer which could be the most probable reason behind increased emission of polymer with the increasing concentration of the lanthanides ion. In Fig. 6b, all of the process that are taking place in the present study are depicted.
All the above experimental evidence clearly suggest that the mechanism of gelation mainly involves the deprotonation of –COOH groups with the addition of Ln(III) acetates and their simultaneous coordination with Ln(III) ions to form a supramolecular network with the metal ligand binding ratio 1:3. This supramolecular network then entraps solvent molecules in its fibrous network to form a viscoelastic gel. Based on the above mechanistic investigation, the structure of the obtained gels can be schematically represented as shown in Fig. 7a.
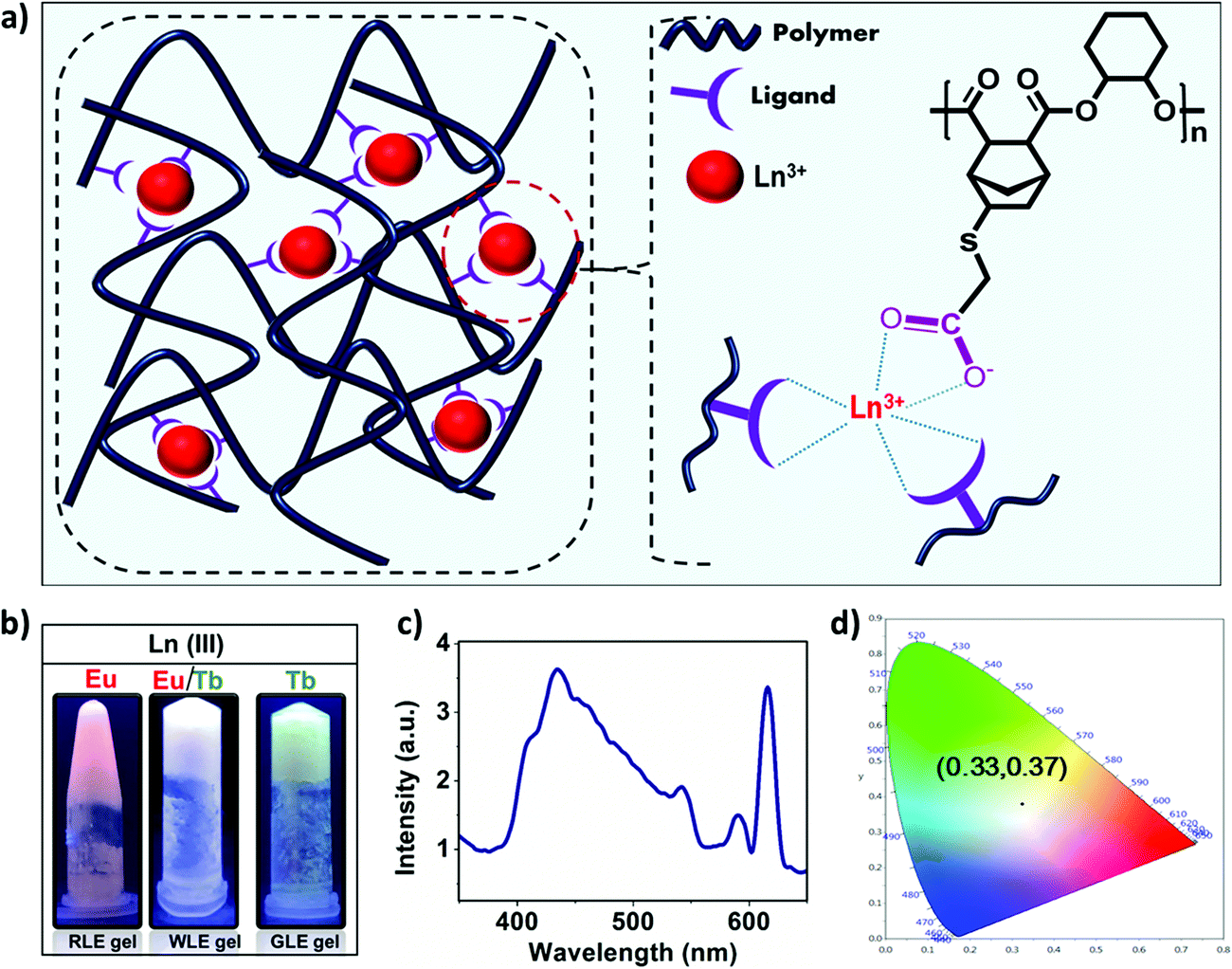 | ||
| Fig. 7 (a) Schematic representation of crosslinking of polymer chains via lanthanide coordination.(b) Photograph of the white light-emitting metallogel under UV light (λex = 365 nm, 2.7 wt% polyester in DMF) from the red light emitting (RLE) and green light emitting (GLE) metallogels. (c) The emission spectrum of WLE metallogels(λex = 335 nm, 12 mg mL−1) and (d) CIE(1931) diagram for the white light emitting gel. | ||
Having established the formation of red and green emitting metallogels with Eu(III) and Tb(III), respectively, we have tried to modulate the emission of the metallogel in presence of both the lanthanide ions. The concentration of the polymer was kept fixed and molar ratio of the Eu(III) and Tb(III) was varied to achieve white light emission. Interestingly, white light emission from the metallogel was observed at [Tb(III)] = 15 mM and [Eu(III)] = 11 mM, and [polymer] = 83 mM.
Fig. 7b shows the photograph of the as prepared white light emitting (WLEM), red light emitting (RLE) and green light emitting (GLE) metallogels. Fig. 7c and d depicts the photoluminescence spectrum of white light-emitting mixed metallogel(WLEM) and the corresponding CIE diagram which indicates the CIE coordinates are x = 0.33 and y = 0.37 respectively. The correlated color temperature (CCT) of the white light emission from metallogels was estimated using McCamy's formula. The CCT value of 5752 K indicates that the gel emits cool white light, which suggests that the obtained WLEM metallogel potentially could be used in electronic flashbulbs and fluorescent lights.
Stimuli-responsive behavior
The broad emission spectrum of the WLEM inspired us to investigate the stimuli responsive behavior of the WLEM. To explore the mechano-response stimuli behavior, the WLEM was sonicated for thirty minutes in a water bath at room temperature. No changes were observed in the metallogels upon sonication. This clearly indicates that the WLEM is exceptionally stable compared to other previously reported white light emitting metallogels.31,51 This observation also led us to conclude that the dynamic M–L bond is quite stable in the lanthanide–polymer metallogel, which prompted us to verify the response from the WLEM in a range of temperatures from 30 to 100 °C. The gel was found to be stable up to 90 °C, followed by an irreversible phase transition to solution (Fig. S13, ESI†). Further, to investigate the potential application of the WLEM as stimuli responsive material, vapochromic and chemochromic behavior of the gel have been studied. The WLEM was exposed to the vapour of the trifluoroacetic acid (TFA) for two minutes followed by exposure to triethylamine (TEA) for six hours. The results of this experiment are displayed in Fig. 8a, which indicate that upon exposure to TFA, the white light emitting metallogel immediately changes to bluish sol. However, upon exposure to TEA, the solution is turned to white light emitting gel again. The quick transition from gel to sol is due to the increment of pH of the environment in presence of the TFA, which leads to the protonation of carboxyl groups and thereby promotes breakage of the gel network. On the other hand, TEA again deprotonates the carboxylic acid group which ultimately results in the formation of gel network again. The sol–gel transition was also confirmed by the disappearance of the Eu(III) and Tb(III) emission peaks in the luminescence spectrum of WLEM in presence of TFA (Fig. 8c). The disappearance of emission peaks in presence of TFA directly indicates that the sensitization of lanthanides decreased due to the breakage of metal ligand bond (M–L) which ultimately results in the formation of sol. The Gel–Sol reversibility experiment was carried out with five hours regular time interval and it was found to be reversible up to 4 cycles. The same experiment was repeated three times and same results have been observed. Next, to investigate the chemochromism, the WLEM was exposed to tetra-n-butylammonium fluoride (TBAF). The response of WLEM to F− ion is displayed in Fig. 8b. Upon exposure to F−, the gel immediately changes to sol with slight blue colour. Contrary to the vapochromism, the chemochromism does not show the reversibility of the sol. The observed irreversibility in the case of fluoride ion is attributed to the precipitation of LnF3.31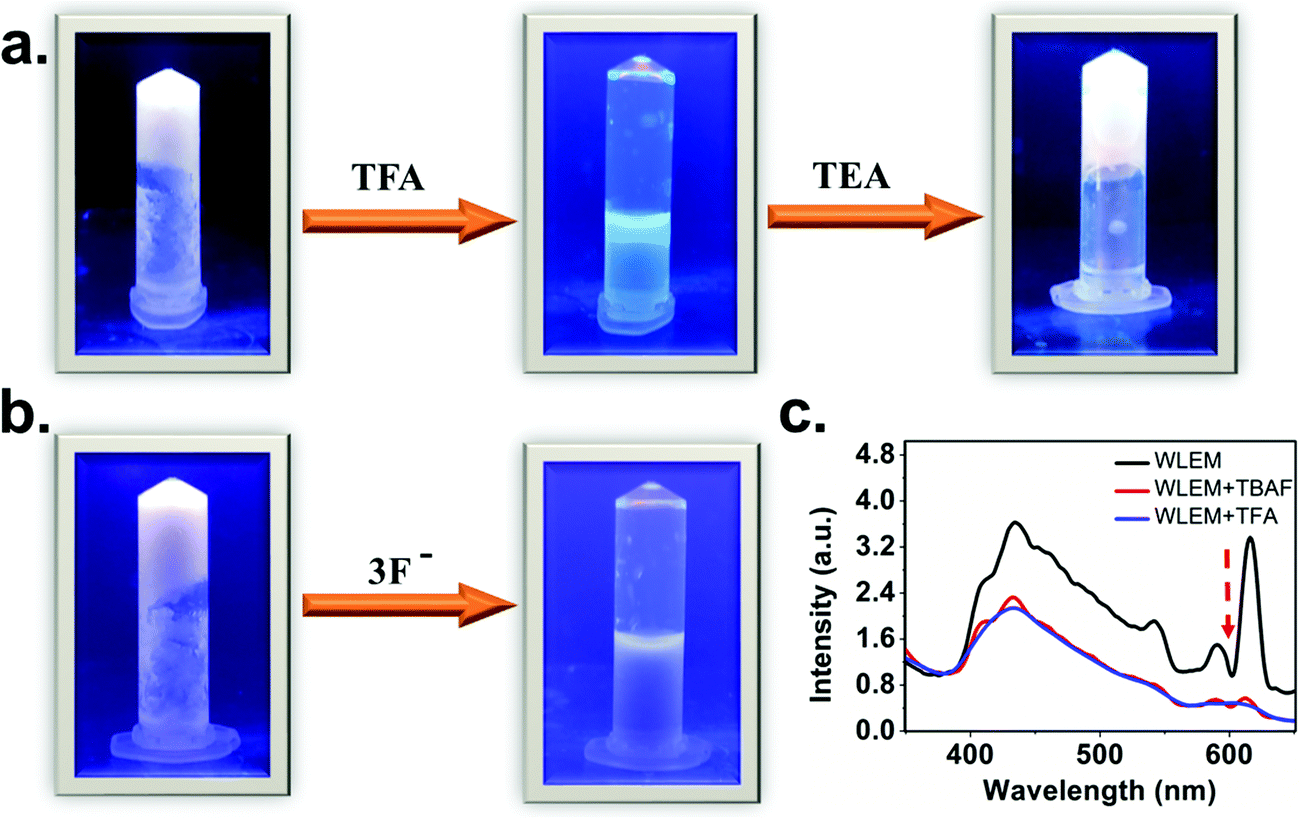 | ||
| Fig. 8 (a) Stimuli-responsive emission, phase transitions and color changes of WLE metallogel by pH triggered vapochromism. (b) Stimuli-responsive emission, phase transitions and color changes of WLE metallogels by F-induced chemochromism. All of the photographs were taken under a UV lamp (λex = 365 nm) (c) Photoluminescence spectra of WLE metallogel in presence of tertiary butyl ammonium fluoride (TBAF) and trifluoroacetic acid (TFA). | ||
Conclusion
A novel intrinsically emitting polymer metallogel, containing lanthanide ions [Eu(III) & Tb(III)] and a functionalized polymer, is reported. The complex formation between the polymer and the lanthanide ions is established with 1:3 metal to ligand ratio. The as-prepared metallogel has been characterized using FTIR, SEM and rheology measurements. Further, the emission of the metallogel is modulated by changing the relative stoichiometry of the Eu(III) and Tb(III) and a white light emitting metallogel is developed. The emission modulation was feasible due to the interplay between the intrinsic emission and resonance energy transfer in the system and reveals a facile approach to engineer the emission through the entire visible spectrum. Furthermore, by taking the advantages of dynamic M–L bond, the as prepared white light emitting metallogel (WLEM) is utilized to respond towards various stimuli such as pH and anions. The results indicate that the reversible vapochromism and irreversible chemochromism of the functional polymer-based metallogels can potentially be utilized to prepare intelligent coating materials and paints that are responsive to environmental changes.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank the Department of Chemistry, IISER Pune, for providing GPC facilities. C. S. wants to thank IIT Madras for fellowship. A. S. and M. K. M thank UGC and CSIR New Delhi India, respectively for the fellowship. The authors also thank the Department of Chemistry and SAIF, IIT Madras, for other instrumental facilities.Notes and references
- M. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960–2004 CrossRef CAS PubMed.
- G. K. Veits, K. K. Carter, S. J. Cox and A. J. McNeil, J. Am. Chem. Soc., 2016, 138, 12228–12233 CrossRef CAS PubMed.
- A. Goujon, G. Mariani, T. Lang, E. Moulin, M. Rawiso, E. Buhler and N. Giuseppone, J. Am. Chem. Soc., 2017, 139, 4923–4928 CrossRef CAS PubMed.
- H. Wang, X. Ji, Z. Li and F. Huang, Adv. Mater., 2017, 29, 1606117 CrossRef PubMed.
- X. Ji, R. T. Wu, L. Long, X. S. Ke, C. Guo, Y. J. Ghang, V. M. Lynch, F. Huang and J. L. Sessler, Adv. Mater., 2018, 30, 1–6 Search PubMed.
- S. Lim, Y. Kuang and H. A.-M. Ardoña, Front. Chem., 2021, 9, 1–21 Search PubMed.
- F. Trausel, F. Versluis, C. Maity, J. M. Poolman, M. Lovrak, J. H. Van Esch and R. Eelkema, Acc. Chem. Res., 2016, 49, 1440–1447 CrossRef CAS PubMed.
- B. O. Okesola and D. K. Smith, Chem. Soc. Rev., 2016, 45, 4226–4251 RSC.
- X. Ji, R. T. Wu, L. Long, C. Guo, N. M. Khashab, F. Huang and J. L. Sessler, J. Am. Chem. Soc., 2018, 140, 2777–2780 CrossRef CAS PubMed.
- Y. Zhang, P. P.-Y. Chan and A. E. Herr, Angew. Chem., Int. Ed., 2018, 57, 2357–2361 CrossRef CAS PubMed.
- A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109–122 RSC.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed.
- P. Duan, N. Yanai, H. Nagatomi and N. Kimizuka, J. Am. Chem. Soc., 2015, 137, 1887–1894 CrossRef CAS PubMed.
- C. Felip-León, S. Díaz-Oltra, F. Galindo and J. F. Miravet, Chem. Mater., 2016, 28, 7964–7972 CrossRef.
- T. Gorai and U. Maitra, Angew. Chem., Int. Ed., 2017, 56, 10730–10734 CrossRef CAS PubMed.
- H. J. Moon, D. Y. Ko, M. H. Park, M. K. Joo and B. Jeong, Chem. Soc. Rev., 2012, 41, 4860–4883 RSC.
- S. Saxena, C. E. Hansen and L. A. Lyon, Acc. Chem. Res., 2014, 47, 2426–2434 CrossRef CAS PubMed.
- S. Zhang, A. M. Bellinger, D. L. Glettig, R. Barman, Y. A.-L. Lee, J. Zhu, C. Cleveland, V. A. Montgomery, L. Gu, L. D. Nash, D. J. Maitland, R. Langer and G. Traverso, Nat. Mater., 2015, 14, 1065–1071 CrossRef CAS PubMed.
- S. Banerjee, R. K. Das and U. Maitra, J. Mater. Chem., 2009, 19, 6649–6687 RSC.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS PubMed.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2 Search PubMed.
- M. Zhang, D. Xu, X. Yan, J. Chen, S. Dong, B. Zheng and F. Huang, Angew. Chem., Int. Ed., 2012, 51, 7011–7015 CrossRef CAS PubMed.
- H. Chen, X. Ma, S. Wu and H. Tian, Angew. Chem., Int. Ed., 2014, 53, 14149–14152 CrossRef CAS PubMed.
- W. Zheng, L. J. Chen, G. Yang, B. Sun, X. Wang, B. Jiang, G. Q. Yin, L. Zhang, X. Li, M. Liu, G. Chen and H. B. Yang, J. Am. Chem. Soc., 2016, 138, 4927–4937 CrossRef CAS PubMed.
- W. Zheng, G. Yang, N. Shao, L. J. Chen, B. Ou, S. T. Jiang, G. Chen and H. B. Yang, J. Am. Chem. Soc., 2017, 139, 13811–13820 CrossRef CAS PubMed.
- H. Wu, J. Zheng, A. L. Kjøniksen, W. Wang, Y. Zhang and J. Ma, Adv. Mater., 2019, 31, 1–23 Search PubMed.
- W. J. Peveler, J. C. Bear, P. Southern and I. P. Parkin, Chem. Commun., 2014, 50, 14418–14420 RSC.
- Z. Xie, F. Wang and C. Y. Liu, Adv. Mater., 2012, 24, 1716–1721 CrossRef CAS PubMed.
- P. Sutar, V. M. Suresh and T. K. Maji, Chem. Commun., 2015, 51, 9876–9879 RSC.
- H. B. Aiyappa, S. Saha, P. Wadge, R. Banerjee and S. Kurungot, Chem. Sci., 2015, 6, 603–607 RSC.
- P. Chen, Q. Li, S. Grindy and N. Holten-Andersen, J. Am. Chem. Soc., 2015, 137, 11590–11593 CrossRef CAS PubMed.
- M. Martínez-Calvo, O. Kotova, M. E. Möbius, A. P. Bell, T. McCabe, J. J. Boland and T. Gunnlaugsson, J. Am. Chem. Soc., 2015, 137, 1983–1992 CrossRef PubMed.
- H. Lee, S. H. Jung, W. S. Han, J. H. Moon, S. Kang, J. Y. Lee, J. H. Jung and S. Shinkai, Chem. – Eur. J., 2011, 17, 2823–2827 CrossRef CAS.
- S. Barman, J. A. Garg, O. Blacque, K. Venkatesan and H. Berke, Chem. Commun., 2012, 48, 11127–11129 RSC.
- B. Xing, M. F. Choi and B. Xu, Chem. – Eur. J., 2002, 8, 5028–5032 CrossRef CAS PubMed.
- Y. X. Ye, W. L. Liu and B. H. Ye, Catal. Commun., 2017, 89, 100–105 CrossRef CAS.
- J. H. Lee, S. Kang, J. Y. Lee and J. H. Jung, Soft Matter, 2012, 8, 6557–6563 RSC.
- T. Feldner, M. Häring, S. Saha, J. Esquena, R. Banerjee and D. D. Díaz, Chem. Mater., 2016, 28, 3210–3217 CrossRef CAS.
- S. Sarkar, S. Dutta, P. Bairi and T. Pal, Langmuir, 2014, 30, 7833–7841 CrossRef CAS PubMed.
- S. Bhattacharyya, A. Chakraborty, K. Jayaramulu, A. Hazra and T. Kumar Maji, Chem. Commun., 2014, 50, 13567–13570 RSC.
- O. Kotova, R. Daly, C. M.-G. Dos Santos, P. E. Kruger, J. J. Boland and T. Gunnlaugsson, Inorg. Chem., 2015, 54, 7735–7741 CrossRef CAS PubMed.
- O. Kotova, R. Daly, C. M.-G. Dos Santos, M. Boese, P. E. Kruger, J. J. Boland and T. Gunnlaugsson, Angew. Chem., Int. Ed., 2012, 51, 7208–7212 CrossRef CAS PubMed.
- L. V. Meyer, F. Schönfeld and K. M. Buschbaum, Chem. Commun., 2014, 50, 8093–8108 RSC.
- D. E. Barry, D. F. Caffrey and T. Gunnlaugsson, Chem. Soc. Rev., 2016, 45, 3244–3274 RSC.
- Y. Liu, D. Tu, H. Zhu and X. Chen, Chem. Soc. Rev., 2013, 42, 6924–6958 RSC.
- Q. Y. Yang, M. Pan, S. C. Wei, K. Li, B. Bin Du and C. Y. Su, Inorg. Chem., 2015, 54, 5707–5716 CrossRef CAS PubMed.
- W. X. Feng, S. Y. Yin, M. Pan, H. P. Wang, Y. N. Fan, X. Q. Lü and C. Y. Su, J. Mater. Chem. C, 2017, 5, 1742–1750 RSC.
- S. Mohapatra, S. Adhikari, H. Riju and T. K. Maji, Inorg. Chem., 2012, 51, 4891–4893 CrossRef CAS PubMed.
- K. Nath, A. Husain and P. Dastidar, Cryst. Growth Des., 2015, 15, 4635–4645 CrossRef.
- J. C.-G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- P. Sutar and T. K. Maji, Inorg. Chem., 2017, 56, 9417–9425 CrossRef CAS PubMed.
- P. Kumar, S. Soumya and E. Prasad, ACS Appl. Mater. Interfaces, 2016, 8, 8068–8075 CrossRef CAS.
- D. W.-R. Balkenende, C. A. Monnier, G. L. Fiore and C. Weder, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- P. Howlader, B. Mondal, P. C. Purba, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2018, 140, 7952–7960 CrossRef CAS PubMed.
- X. Ji, W. Chen, L. Long, F. Huang and J. L. Sessler, Chem. Sci., 2018, 9, 7746–7752 RSC.
- E. B. Berda, L. F. Deravi, E. J. Foster, Y. Simon and M. M. Thuo, Macromolecules, 2019, 52, 6339–6341 CrossRef CAS.
- X. Zhang, L. Chen, K. H. Lim, S. Gonuguntla, K. W. Lim, D. Pranantyo, W. P. Yong, W. J.-T. Yam, Z. Low, W. J. Teo, H. P. Nien, Q. W. Loh and S. Soh, Adv. Mater., 2019, 31, 1–48 Search PubMed.
- X. Q. Wang, W. Wang, W. J. Li, L. J. Chen, R. Yao, G. Q. Yin, Y. X. Wang, Y. Zhang, J. Huang, H. Tan, Y. Yu, X. Li, L. Xu and H. B. Yang, Nat. Commun., 2018, 9, 1–11 CrossRef PubMed.
- T. Singha Mahapatra, H. Singh, A. Maity, A. Dey, S. K. Pramanik, E. Suresh and A. Das, J. Mater. Chem. C, 2018, 6, 9756–9766 RSC.
- Q. Zhu, L. Zhang, K. Van Vliet, A. Miserez and N. Holten-Andersen, ACS Appl. Mater. Interfaces, 2018, 10, 10409–10418 CrossRef CAS PubMed.
- A. Sebastian, M. K. Mahato and E. Prasad, Soft Matter, 2019, 15, 3407–3417 RSC.
- B. Han, L. Zhang, B. Liu, X. Dong, I. Kim, Z. Duan and P. Theato, Macromolecules, 2015, 48, 3431–3437 CrossRef CAS.
- J. Wang, S. Sun, B. Wu, L. Hou, P. Ding, X. Guo, M. A. Cohen Stuart and J. Wang, Macromolecules, 2019, 52, 8643–8650 CrossRef CAS.
- D. A. Tomalia, B. Klajnert-Maculewicz, K. A.-M. Johnson, H. F. Brinkman, A. Janaszewska and D. M. Hedstrand, Prog. Polym. Sci., 2019, 90, 35–117 CrossRef CAS.
- Q. Zhou, B. Cao, C. Zhu, S. Xu, Y. Gong, W. Z. Yuan and Y. Zhang, Small, 2016, 12, 6586–6592 CrossRef CAS PubMed.
- J. Wang, L. Xu, S. Zhong, Y. Yang, G. Feng, Q. Meng, Y. Gao and X. Cui, Polym. Chem., 2021, 12, 7048–7055 RSC.
- Q. Zhou, T. Yang, Z. Zhong, F. Kausar, Z. Wang, Y. Zhang and W. Z. Yuan, Chem. Sci., 2020, 11, 2926–2933 RSC.
- K. Fahmeeda, Y. Tianjia, Z. Zihao, Z. Yongming and Y. Wang Zhang, Chem. Res. Chin. Univ., 2021, 37, 177–182 CrossRef.
- N. L. Leung, N. Xie, W. Yuan, Y. Liu, Q. Wu, Q. Peng, Q. Miao, J. W. Lam and B. Z. Tang, Chem. – Eur. J., 2014, 20, 15349–15353 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00109h |
| This journal is © The Royal Society of Chemistry 2022 |