
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of acrylonitrile side chain-appended π-conjugated polymers by a Suzuki cross-coupling polycondensation and a Knoevenagel condensation, and their optical properties†
Haruka
Yamauchi‡
a,
Mahiro
Nakabayashi‡
a,
Maiko
Kawada
a,
Yuto
Hino
a,
Syunya
Inayama
a,
Hiroki
Tanikubo
a and
Shotaro
Hayashi
 *ab
*ab
aSchool of Environmental Science and Engineering, Kochi University of Technology, 185 Tosayamada Miyanokuchi, Kami, Kochi, 782-8502, Japan. E-mail: hayashi.shotaro@kochi-tech.ac.jp
bResearch Center for Molecular Design, Kochi University of Technology, Japan
First published on 7th March 2022
Abstract
Acrylonitrile is a highly valuable unit for the design of donor–acceptor systems and luminescent π-conjugated molecular materials. Acrylonitrile-based polymers are fundamental structures in π-conjugated polymers. In contrast to the frequently reported introduction of acrylonitrile units into the main chain, we report herein their introduction into the side chain. In the present study, we carried out two syntheses: (1) a post-polymerization modification reaction in which polymers with acrylonitrile units in their side chains were produced via a Knoevenagel condensation between a π-conjugated polymer containing aldehyde groups and a cyano group-containing molecule; and (2) a one-pot process comprising a simultaneous Suzuki cross-coupling polycondensation and a Knoevenagel condensation. We obtained a high-molecular-weight π-conjugated polymer with an acrylonitrile side chain that we used to fabricate a free-standing film. The π-conjugated polymer with alkoxyphenyl acrylonitrile in its side chains exhibited charge transfer interaction based on its donor–acceptor system. It also demonstrated favourable fluorescence properties as a solid or in solution.
Introduction
π-Conjugated molecules and polymers have attracted considerable attention owing to their potential usefulness in organic devices.1 π-Conjugated molecular scaffolds that can transform into higher analogues while retaining their original skeletons are useful for designing functional molecules with enhanced and/or additional properties.2 Scaffolds with favorable reaction sites for π-expansion are noteworthy because they allow the stepwise and selective incorporation of desired functionalities.Acrylonitrile is a functional unit that has attracted attention in the field of π-conjugated molecules.3 Molecules containing the acrylonitrile unit have potential as n-type semiconductors owing to the strong electron-withdrawing effect of the cyano group. In addition, many derivatives of such molecules exhibit aggregation-induced emission (AIE), aggregation-induced enhanced emission (AIEE), and mechanofluorochromism (MFC), and can be used to produce efficient solid-state fluorescent materials.4 Therefore, such molecular structures have attracted considerable attention as solid materials for crystal engineering.5 Acrylonitrile units have also been widely used in polymers.6 Many fluorescent polymers with such units in their main chains have been reported, and linear π-conjugated polymers are being investigated for use in various devices. Network polymers also luminesce efficiently owing to the introduction of the acrylonitrile unit, and may be useful for producing solid-state sensors for the detection of chemical species.6 However, to the best of our knowledge there have been no reports on the synthesis of π-conjugated polymers with arylacrylonitrile side chains. Designing a new polymer structure is very interesting. Previously, we have synthesized various fluorescent substances comprising acrylonitrile-containing π-conjugated molecules using aldehyde groups to form conjugated scaffolds.7 Aldehyde groups can be used to introduce acrylonitrile units via Knoevenagel condensation. Knoevenagel condensation enables the ready expansion of a π system. It also facilitates the introduction of electron acceptors and the realization of solid fluorescence.
Both the synthesis of π-conjugated polymers by the polymerization of specific monomers, and the post-polymerization modification of π-conjugated polymers with reactive substituents have advantages and disadvantages.8 However, the selective post-polymerization modification or substitution of polymers has been very helpful in the construction of various functional materials such as graft, block, pendant, and network polymers.8 The thermal, optical, and other properties of polymers can be improved by efficient post-polymerization modification of the polymer chain. π-Conjugated polymers containing acrylonitrile groups in their main chains have been reported.6 Therefore, we considered the introduction of arylacrylonitrile groups into the side chains by post-polymerization modification. Herein, we report: (1) a post-polymerization modification reaction in which polymers with acrylonitrile units in their side chains were produced via a Knoevenagel condensation between a π-conjugated polymer containing aldehyde groups and a cyano group-containing molecule; and (2) a one-pot process comprising a simultaneous Suzuki cross-coupling polycondensation and a Knoevenagel condensation. We also investigated the optical properties of the obtained π-conjugated polymers.
Results and discussion
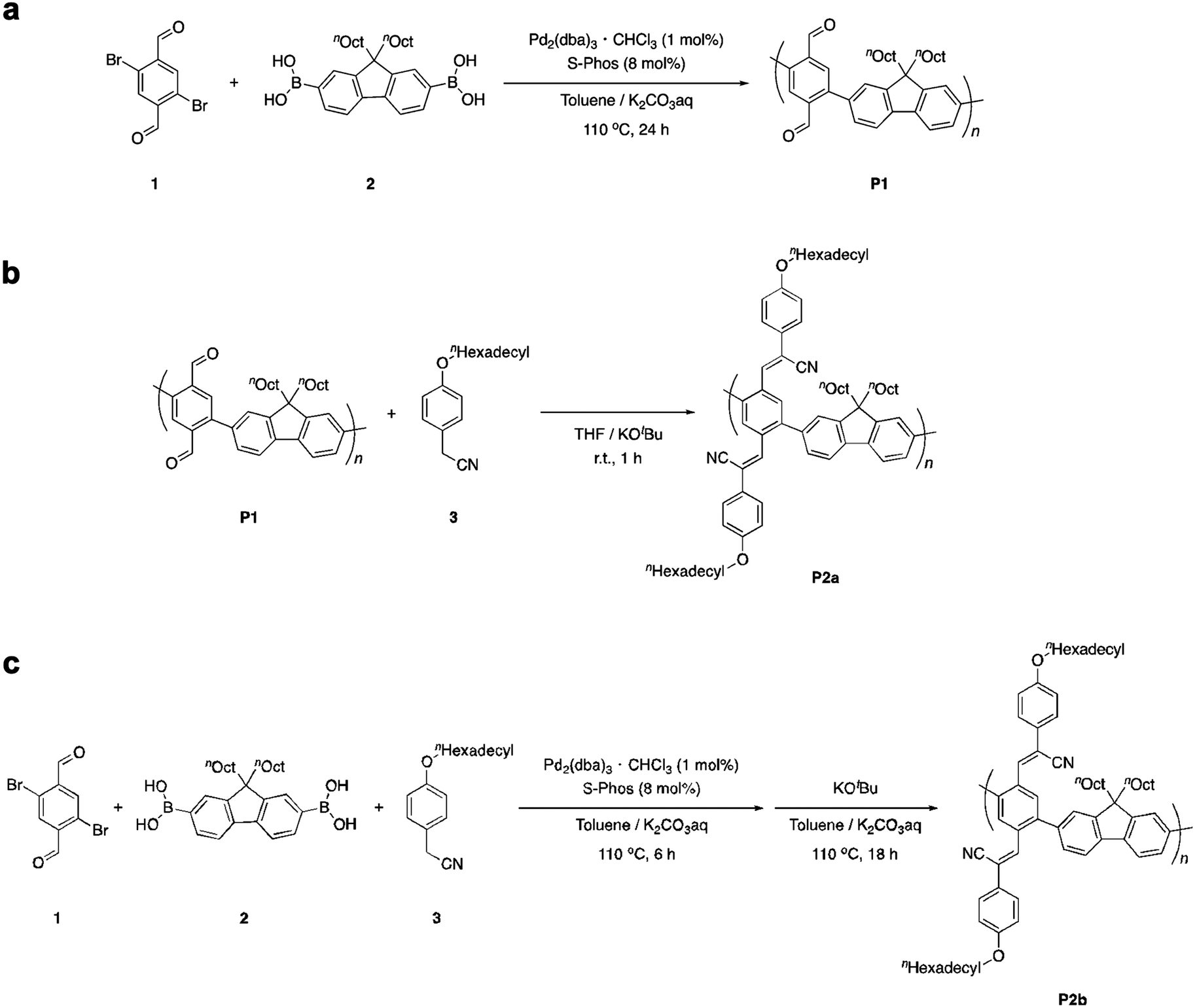
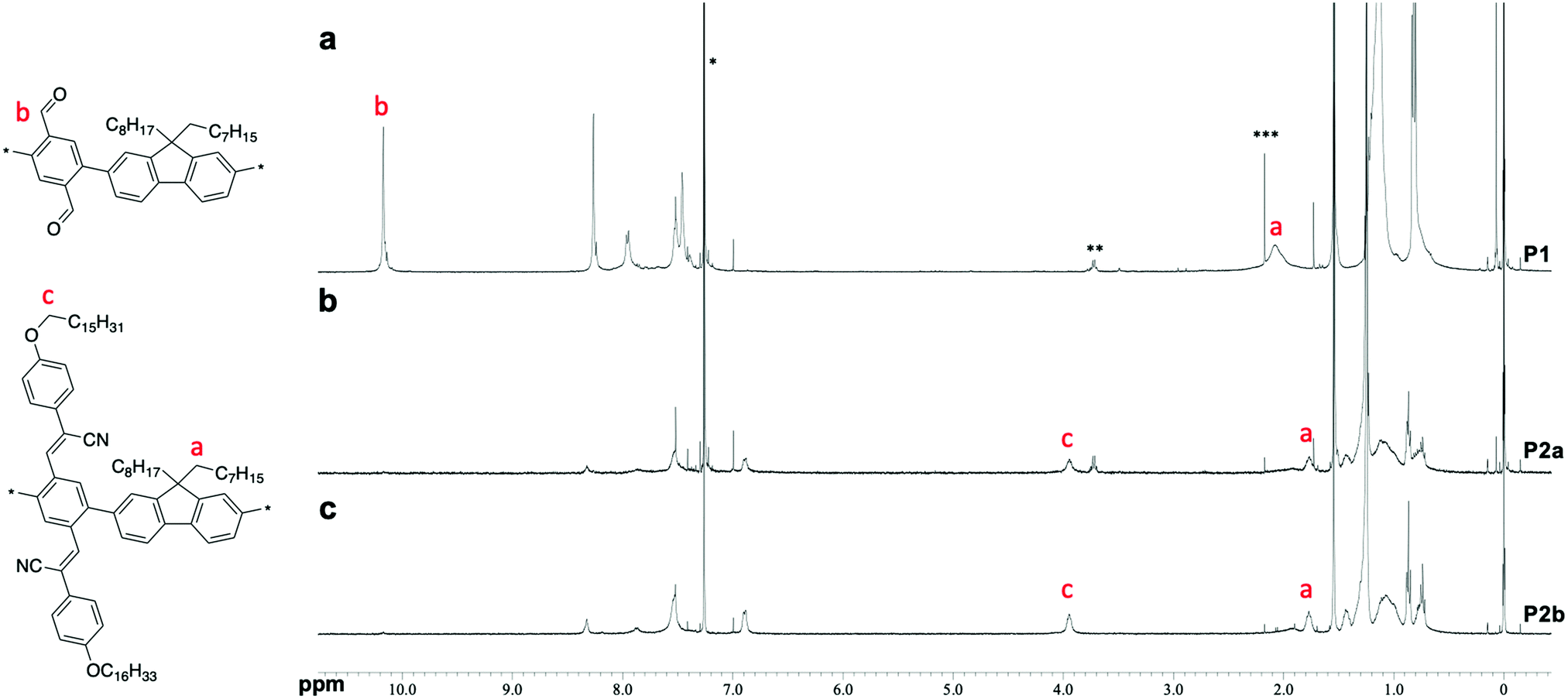
Poly(phenylene-2,5-dicarbaldehyde-alt-9,9-dioctylfluorene) (hereafter referred to as P1) was prepared by the Suzuki–Miyaura coupling polymerization of 9,9-dioctylfluorene-2,7-diboronic acid ester and 1,4-dibromobenzene-2,5-dicarbaldehyde using a Pd2(dba)3 chloroform adduct with S-Phos in a toluene/2 M potassium carbonate solution at 100 °C for 24 h (Scheme 1a). After purification by reprecipitation in methanol, P1 was obtained as a white powder. The structure of the polymer was determined by proton nuclear magnetic resonance (1H NMR) analysis (Fig. 1a). The unit ratio of phenylene-2,5-dicarbaldehyde to 9,9-dioctylfluorene can be determined from the integral ratio of an aldehyde proton of phenylene (b: 10.17 ppm) and an α-methylene proton of 9,9-dioctylfluorene (a: 2.08 ppm). As a result, the unit ratio of phenylene and fluorene was calculated to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. An acrylonitrile-appended polymer was prepared via the Knoevenagel condensation of P1 with 4-methoxybenzeneacetonitrile in the presence of t-BuOK in tetrahydrofuran (THF) at room temperature for 1 h. However, the polymeric product was insoluble. To obtain a soluble product, the polymer was synthesized by condensing P1 with 4-hexadodecyloxybenzeneacetonitrile (Scheme 1b). After removal of the excess unreacted compound (4-dodecyloxybenzeneacetonitrile), a modified P2 (hereafter referred to as P2a) was obtained as a yellow powder. Comparing the 1H NMR spectra obtained before and after the Knoevenagel condensation of P1, the signal attributable to the aldehyde groups (b: 10.17 ppm) almost disappeared during the reaction, indicating successful post-polymerization modification (Fig. 1a and b, respectively). However, a signal attributable to the α-methylene protons of the dodecyloxy groups (c: 3.95 ppm) appeared following the substitution with 4-dodecyloxyphenylacrylonitrile. Furthermore, the signal attributable to the α-methylene protons of 9,9-dioctylfluorene shifted to a higher magnetic field (a: 1.92 ppm). This downfield shift is induced by the reducing ring-current shielding effect of fluorene rings.9 The formation of acrylonitrile caused a twist in the main chain, resulting in a shift in the signal to a higher magnetic field. The relative intensity of the peak attributable to the methylene protons was in good agreement with the theoretical value when the α-methylene protons of the octyl groups in the 9,9-dioctylfluorene units were used as a standard. This also indicated that almost all the aldehyde groups had been converted to acetonitrile groups (>99%). In addition, Fourier-transform infrared spectroscopy (FTIR) analysis of P2 revealed that the characteristic peak attributable to aldehyde groups at 1690 cm−1 disappeared and a peak attributable to nitrile groups appeared at 2212 cm−1 (Fig. S1, ESI†). Gel permeation chromatography (GPC) analysis was conducted to determine the relative molecular weights and polydispersity indices of the products in comparison with those of a polystyrene standard. The molecular weights of P1 [Mn: 35000, Mw: 177000, Mw/Mn: 5.04] and P2a [Mn: 29000, Mw: 67000, Mw/Mn: 2.34] differed slightly before and after polymer modification (Fig. 2). This suggests that the GPC (hydrodynamic volume) molecular weight decreased owing to the large twist in the main chain, although the post-polymerization modification increased the molecular weight substantially.
:1. An acrylonitrile-appended polymer was prepared via the Knoevenagel condensation of P1 with 4-methoxybenzeneacetonitrile in the presence of t-BuOK in tetrahydrofuran (THF) at room temperature for 1 h. However, the polymeric product was insoluble. To obtain a soluble product, the polymer was synthesized by condensing P1 with 4-hexadodecyloxybenzeneacetonitrile (Scheme 1b). After removal of the excess unreacted compound (4-dodecyloxybenzeneacetonitrile), a modified P2 (hereafter referred to as P2a) was obtained as a yellow powder. Comparing the 1H NMR spectra obtained before and after the Knoevenagel condensation of P1, the signal attributable to the aldehyde groups (b: 10.17 ppm) almost disappeared during the reaction, indicating successful post-polymerization modification (Fig. 1a and b, respectively). However, a signal attributable to the α-methylene protons of the dodecyloxy groups (c: 3.95 ppm) appeared following the substitution with 4-dodecyloxyphenylacrylonitrile. Furthermore, the signal attributable to the α-methylene protons of 9,9-dioctylfluorene shifted to a higher magnetic field (a: 1.92 ppm). This downfield shift is induced by the reducing ring-current shielding effect of fluorene rings.9 The formation of acrylonitrile caused a twist in the main chain, resulting in a shift in the signal to a higher magnetic field. The relative intensity of the peak attributable to the methylene protons was in good agreement with the theoretical value when the α-methylene protons of the octyl groups in the 9,9-dioctylfluorene units were used as a standard. This also indicated that almost all the aldehyde groups had been converted to acetonitrile groups (>99%). In addition, Fourier-transform infrared spectroscopy (FTIR) analysis of P2 revealed that the characteristic peak attributable to aldehyde groups at 1690 cm−1 disappeared and a peak attributable to nitrile groups appeared at 2212 cm−1 (Fig. S1, ESI†). Gel permeation chromatography (GPC) analysis was conducted to determine the relative molecular weights and polydispersity indices of the products in comparison with those of a polystyrene standard. The molecular weights of P1 [Mn: 35000, Mw: 177000, Mw/Mn: 5.04] and P2a [Mn: 29000, Mw: 67000, Mw/Mn: 2.34] differed slightly before and after polymer modification (Fig. 2). This suggests that the GPC (hydrodynamic volume) molecular weight decreased owing to the large twist in the main chain, although the post-polymerization modification increased the molecular weight substantially.
 | ||
| Scheme 1 Synthetic strategy for π-conjugated polymers having an acrylonitrile side chain. (a and b) Post-polymerization process: synthesis of the polymers with an acrylonitrile unit in the side chain by Knoevenagel condensation on π-conjugated polymers with aldehyde. (c) One-pot process 2: synthesis of the polymer by step-wise Suzuki cross-coupling polycondensation and Knoevenagel condensation. | ||
 | ||
| Fig. 1 1H NMR spectra of the polymers in CDCl3 at room temperature. (a) P1. (b) P2a. (c) P2b. * chloroform. ** ethanol. *** acetone. | ||
 | ||
| Fig. 2 GPC trace of the polymers. | ||
Because both the Suzuki cross-coupling polycondensation and the Knoevenagel condensation require the presence of a base, we attempted to synthesize P2 using a one-pot reaction system. We speculated that the Knoevenagel condensation would proceed regardless of the type of base, and anticipated that even an aqueous potassium carbonate solution would suffice. Therefore, we conducted a one-pot synthesis involving the Suzuki cross-coupling polycondensation and the Knoevenagel condensation of a three-component system comprising 9,9-dioctylfluorene-2,7-diboronic acid ester, 1,4-dibromobenzene-2,5-dicarbaldehyde, and 4-dodecyloxybenzeneacetonitrile using a Pd2(dba)3 chloroform adduct with S-Phos in a toluene/2 M potassium carbonate solution (K2CO3 aq.), followed by the addition of t-BuOK (Scheme 1c). Since Knoevenagel condensation did not progress sufficiently with only K2CO3 aq., the addition of t-BuOK after 6 h was performed for a sufficient reaction. We obtained a powder after purification by reprecipitation in methanol. There was no aldehyde signal (b: 10.17 ppm) in the 1H NMR spectrum of the polymer. Therefore, we concluded that the reaction had not proceeded sufficiently. This indicates that a potassium carbonate solution is inadequate for the Knoevenagel condensation. Thus, to increase the reactivity of the Knoevenagel condensation in this one-pot process, we added 4-dodecyloxybenzeneacetonitrile together with the strong and bulky base t-BuOK (Scheme 1c). The 1H NMR spectrum of P2b agreed well with that of P2a (Fig. 1c). There was a faint aldehyde signal but the conversion efficiency was obviously high. Moreover, the molecular weight of P2b [Mn: 40000, Mw: 136000, Mw/Mn: 4.47] was convincingly high (Fig. 2). This suggests that the solubility of the polymers and catalytic reactivity were enhanced by performing Knoevenagel condensation in the reaction system. Therefore, we demonstrated that it was possible to obtain the target polymer by the one-pot process. Film-forming, optical and electrochemical properties of P2 were displayed by P2b due to similar data of P2a and P2b in all results.
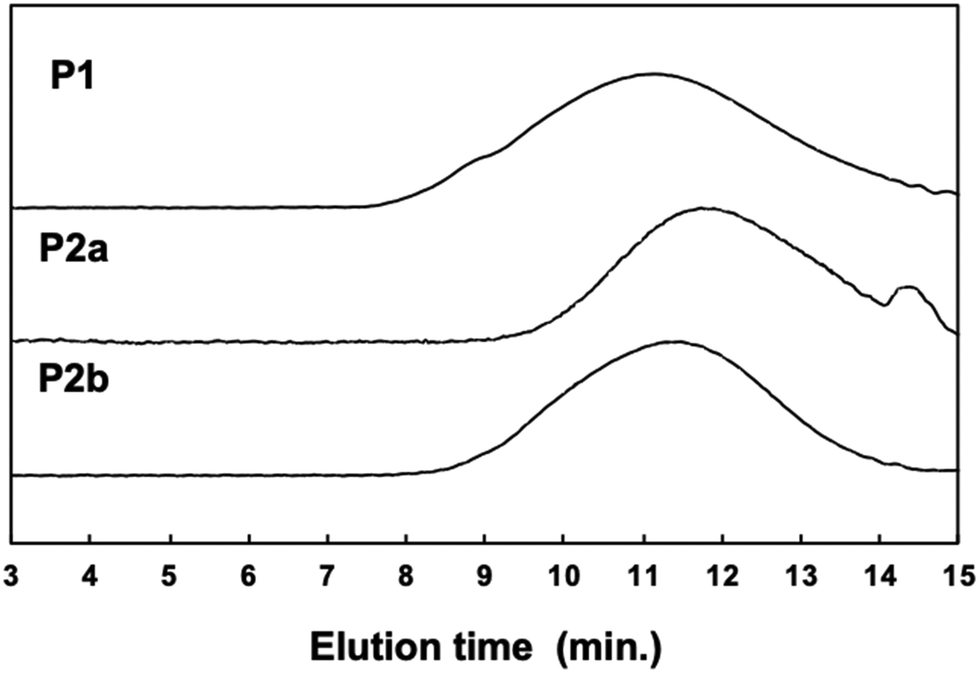
The film-forming behaviour and toughness of a polymer depend on the length of its segments and on its crystallinity. We prepared large free-standing films by casting a chloroform solution of P2 (Fig. 3a and b). Both P1 and P2 films are a similar yellow color, but the P2 film shows higher luminescence than the P1 film. The films were tough and suitable for bending and rolling (Fig. 3c).
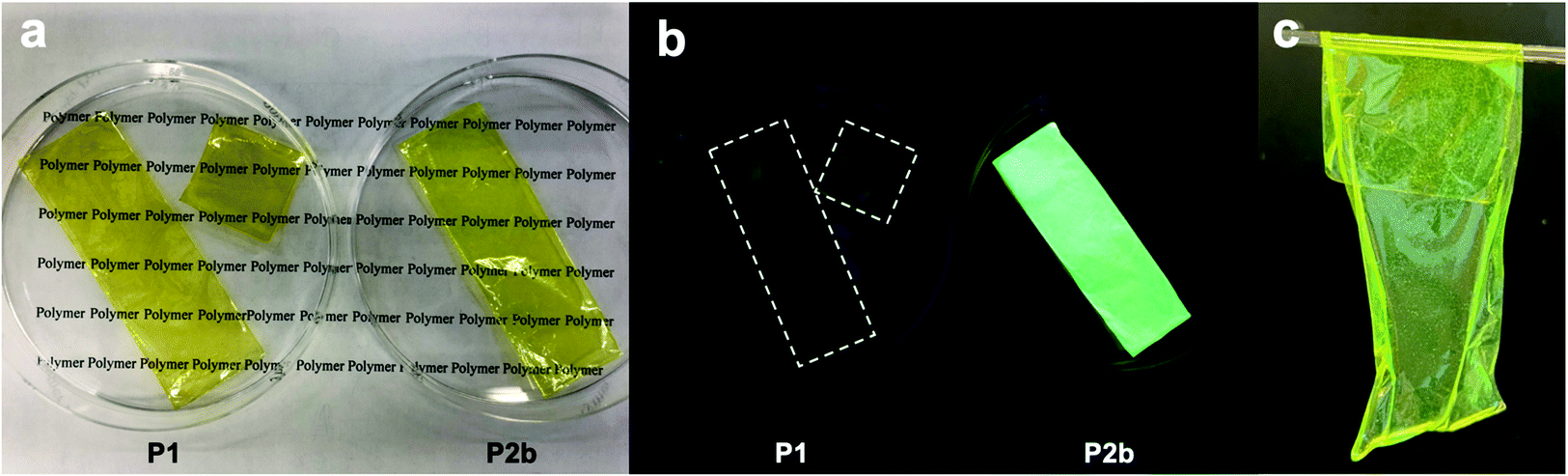 | ||
| Fig. 3 Free-standing films. (a) P1 and P2 under ambient light. (b) P1 and P2 under UV light. (c) Flexible feature of the P2 film. | ||
Thermal analyses of the polymer revealed information about its crystallinity and thermal stability. Differential scanning calorimetry (DSC) analyses of P1 and P2 revealed no glass transition or melting peaks (Fig. S2, ESI†). Thermogravimetric (TG) analyses showed that the 5% decomposition (Td) temperatures of P1 and P2 were 402 and 394 °C, respectively (Fig. S3, ESI†).
The UV-vis absorption spectra of P1 and P2 in chloroform solution and in film form are shown in Fig. 4a. The π–π* transition bands of the polymers occurred at 331 and 350 nm, respectively. The π–π* transition band of P2 appeared in a lower energy region than that of P1. However, lower energy bands appeared at 395 nm in the spectra of both P1 and P2. The geometries of the copolymer repeat units (motifs) of P1 and P2 were optimized using density functional theory (DFT) with the B3LYP function and the 6-311G basis set (Fig. 5). Solvent-free default convergence criteria were used to calculate the energy-minimized structures. The minimized geometries confirmed that both polymers had twisted (less planar) structures between phenylene and fluorene. The optimized geometries of P1 and P2 suggest delocalization of the highest occupied molecular orbital (HOMO). However, the lowest unoccupied molecular orbital (LUMO) contours of the P1 and P2 motifs indicate localization on the phenylene. These findings were substantiated by the spectra of P1 and P2 in dichloromethane, which revealed a low energy absorption onset and a broad absorption band for P2, and a relatively lower energy absorption onset for P1 (Fig. 4a). Here, we simulated the absorption bands of both P1 and P2 by TD-DFT calculation (B3LYP/6-311G). The simulated spectrum of P2 was slightly blue-shifted from that of P1 (Fig. S4, ESI†). There were slight red shifts between the solution and film spectra of the π-conjugated polymers P1 and P2 (Fig. 4b). In general, the aggregation of π-conjugated polymers tends to produce a lower energy absorption band owing to π–π interactions between the polymer segments. However, it has been suggested that these polymers are amorphous films in which low energy absorption bands are not obvious owing to the formation of crystal structures between the polymers.
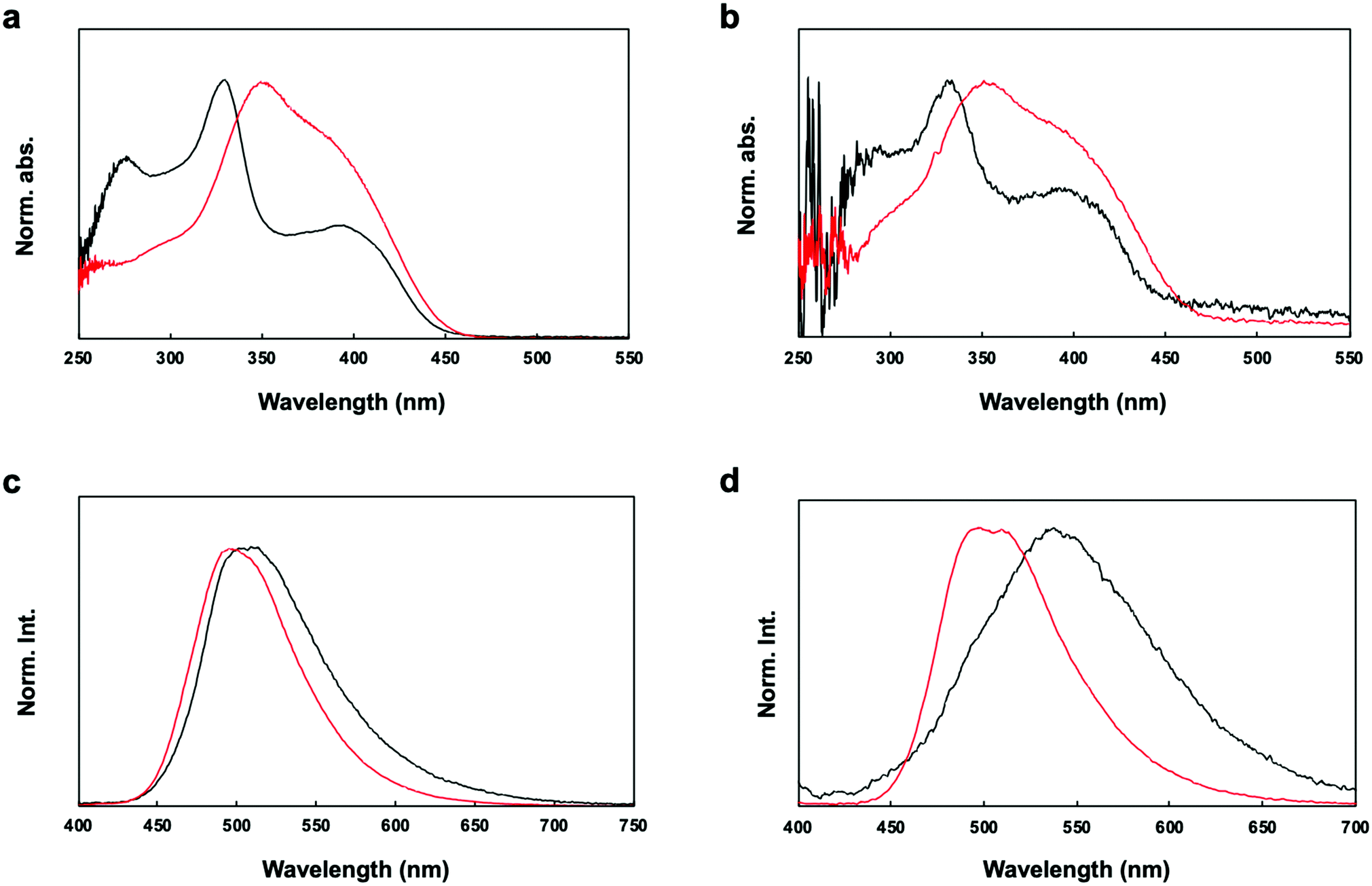 | ||
| Fig. 4 Optical properties of the polymer. Black: P1. Red: P2. (a) UV-vis absorption spectra of the polymers in chloroform. (b) UV-vis absorption spectra of the polymers in film. (c) Fluorescence spectra of the polymers in chloroform. (d) Fluorescence spectra of the polymers in film. | ||
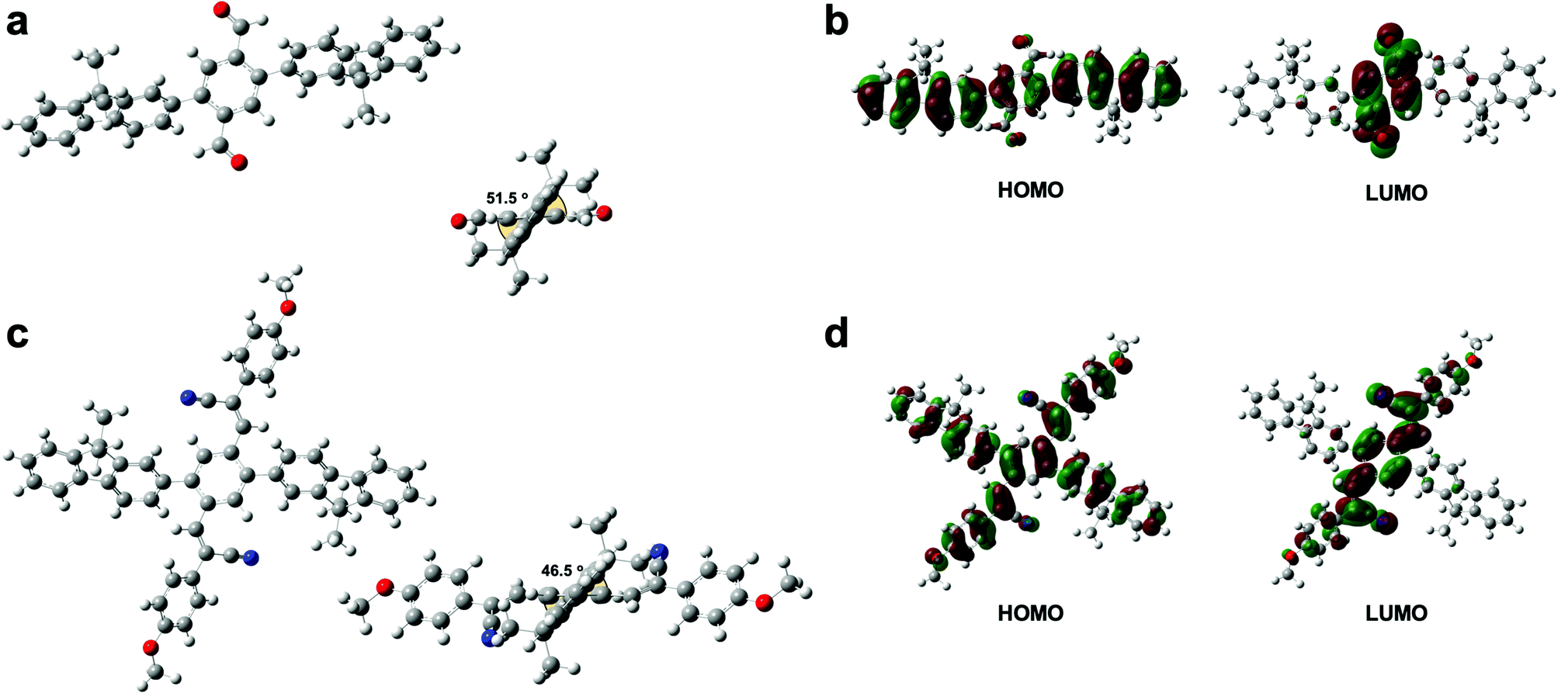 | ||
| Fig. 5 Calculated results by DFT B3LYP/6-311G(d) with the Gaussian 03 program. (a) Optimal structure of the model motif of P1. (b) Electron-cloud distributions of the HOMO and LUMO model motif of P1 (isovalue 0.02). (c) Optimal structure of the model motif of P2. (d) Electron-cloud distributions of the HOMO and LUMO model motif of P2 (isovalue 0.02). | ||
The fluorescence spectra of P1 and P2 in chloroform revealed bands at 596 and 496 nm, respectively (Fig. 4c). However, the fluorescence spectra of P1 and P2 in film form showed red-shifted bands compared with those in solution (596 and 496 nm, respectively) (Fig. 4d). It is worth noting that the quantum efficiency values of P2 in dichloromethane (24%) and film form (19%) were higher than the quantum efficiency values of P1 in dichloromethane (5%) and film form (2%). Therefore, the efficiency was enhanced by the addition of the acrylonitrile moiety. P1 with an electron-accepting aldehyde side chain is quenched by energy transfer from the excited phenylene-fluorene main chain, but P2 is considered to have shown relatively high quantum efficiency because it is dominated by the properties of the acrylonitrile side chain.
We conducted cyclic voltammetry analyses to determine the electrochemical properties of the π-conjugated polymers. A three-electrode system comprising a glassy carbon working electrode covered with the polymers, a platinum counter electrode, and a silver reference electrode was used in an electrolytic solution of 0.1 M Bu4NBF4/CH3CN (Fig. S5, ESI†). The onset oxidation potential of P2 (Eonset,ox = 1.21 V) was slightly lower than that of P1 (Eonset,ox = 1.66 V). The onset reduction potential of P2 (Eonset,red = −0.75 V) was much higher than that of P1 (Eonset,red = −1.51 V). This can be explained by the strong electron-withdrawing effect of the acrylonitrile unit.
Conclusion
In the present study, we demonstrated the facile synthesis of a fluorescent π-conjugated polymer with an alkoxyphenylacrylonitrile side chain via: (1) a step-wise process and (2) a one-pot process comprising a Suzuki cross-coupling polycondensation and a Knoevenagel post-polymerization modification. π-Conjugated polymers with aldehyde groups are useful because they allow structural transformation, particularly into π-extended analogues. The modification of the acrylonitrile unit gave charge transfer and highly fluorescent properties compared with the π-conjugated prepolymer. Thus, new polymer structural design of the acrylonitrile-side chain is a candidate for flexible luminescent devices. The polymer obtained herein could be useful for further transformation into, for example, macromolecules with acrylonitrile skeletons or solid-state fluorescence-active materials with tuneable emission properties. Further studies on polymer synthesis are currently in progress.Experimental
Chemicals
2,5-Dibromobenzene-1,4-dicarbaldehyde (TCI), 9,9-dioctylfluorene-2,7-diboronic acid (Aldrich), Pd2(dba)3·CHCl3 (Aldrich), and S-Phos (Kanto) were used as received. All solvents were used as received. Synthesis of 4-methoxybenzeneacetonitrile was performed according to a previous report.7bMeasurements
Liquid-state 1H and 13C NMR spectra were recorded on a JEOL EX-400 spectrometer. UV-vis absorption spectra were obtained on an Ocean Optics USB4000-XR1 fiber spectrometer with a DH2000-BAL tungsten halogen light source. Photoluminescence spectra were obtained on an Ocean Optics frame fiber spectrometer with a 365 nm LSM series LED light source (Ocean Photonics). Absolute quantum yield was obtained by a frame fiber spectrometer with integrated sphere (Ocean Photonics). Excitation was performed by the 365 nm LSM series LED light source (Ocean Photonics). Polymer films were prepared as follows: The solution of the polymer in chloroform was dropped on a glass substrate, and then dried under air. DSC analysis was performed by a Shimadzu DSC-60 plus, which measured during heating from room temperature to 0–250 °C at a heating rate of 20 °C min−1 in nitrogen. TG analysis was performed by a Hitachi STA7200RV, which measured during heating from room temperature to 600 °C at a heating rate of 10 °C min−1 in nitrogen. SEC analyses were performed using a Tosho HLC-8420GPC EcoSEC Elite (TSKgel SuperMultipore HZ-M column) with THF as the eluent after calibration using polystyrene standards. IR spectra were obtained on a JASCO FTIR-4100 spectrometer. Cyclic voltammetry measurements were performed by an ALS 400C. A three-electrode system equipped with a glassy carbon working electrode, platinum counter electrode and Ag/AgCl as a reference electrode was used in an electrolytic solution of acetonitrile containing 0.1 M tetraethylammonium tetrafluoroborate. The CV chart was displayed with ferrocene standard.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. H. acknowledges JST FOREST Program (no. JPMJFR211W) and a KAKENHI (Grant-in-Aid for Scientific Research B: no. 18H02052, Grant-in-Aid for Scientific Research on Innovative Areas ‘π-figuration’: no. 17H05171 and ‘coordination asymmetry’: no. 19H04604) of the Japan Society for the Promotion of Science (JSPS). This work was performed under the Cooperative Research Program of “Network Joint Research Center for Materials and Devices”.Notes and references
- (a) S. Santhosh Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef PubMed; (b) A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644–656 CrossRef CAS PubMed; (c) G. M. Newbloom, K. M. Weigandt and D. C. Pozzo, Macromolecules, 2012, 45, 3452–3462 CrossRef CAS; (d) S. Hayashi and T. Koizumi, Angew. Chem., Int. Ed., 2016, 55, 2701–2704 CrossRef CAS PubMed; (e) S. Hayashi and T. Koizumi, Chem. – Eur. J., 2018, 24, 8507–8512 CrossRef CAS PubMed; (f) S. Hayashi, S. Yamamoto, D. Takeuchi, Y. Ie and K. Takagi, Angew. Chem., Int. Ed., 2019, 57, 17002–17008 CrossRef PubMed; (g) S. Hayashi, Polym. J., 2019, 51, 813–823 CrossRef CAS; (h) S. Hayashi, Symmetry, 2020, 2022 CrossRef CAS; (i) S. Hayashi, T. Koizumi and N. Kamiya, ChemPlusChem, 2019, 84, 247–251 CrossRef CAS PubMed; (j) S. Hayashi and T. Koizumi, CrystEngComm, 2019, 21, 5990–5994 RSC; (k) S. Hayashi, F. Ishiwari, T. Fukushima, S. Mikage, M. Tashiro, Y. Imamura and M. Katouda, Angew. Chem., Int. Ed., 2020, 59, 16195–16201 CrossRef CAS PubMed; (l) J. Lin, Z. Yu, W. Zhu, G. Xing, Z. Lin, S. Yang, L. Xie, C. Niu and W. Huang, Polym. Chem., 2013, 4, 477–483 RSC; (m) N. Adachi, Y. Tatewaki, H. Shirai and M. Kimura, Polym. J., 2009, 41, 132–137 CrossRef CAS; (n) S. Hayashi, A. Takigami and T. Koizumi, Chem. – Asian J., 2018, 13, 2014–2018 CrossRef CAS PubMed; (o) S. Hayashi, Mater. Adv., 2020, 1, 632–638 RSC; (p) Y. Hino and S. Hayashi, Chem. – Eur. J., 2021, 27, 17595–17600 CrossRef CAS PubMed.
- (a) M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS PubMed; (b) A. S. Goldmann, M. Glassner, A. J. Inglis and C. P. Barner-Kowollik, Macromol. Rapid Commun., 2013, 34, 810–849 CrossRef CAS PubMed; (c) E. Blasco, M. B. Sims, A. S. Goldmann, B. S. Sumerlin and C. Barner-Kowollik, Macromolecules, 2017, 50, 5215–5252 CrossRef CAS; (d) K. A. Günay, P. Theato and H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1–28 CrossRef.
- (a) H.-J. Kim, D. R. Whang, J. Gierschner, C. H. Lee and S. Y. Park, Angew. Chem., Int. Ed., 2015, 54, 4330 CrossRef CAS PubMed; (b) B.-K. An, J. Gierschner and S. Y. Park, Acc. Chem. Res., 2012, 45, 544 CrossRef CAS PubMed; (c) B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS PubMed; (d) H.-C. Yeh, W.-C. Wu, Y.-S. Wen, D.-C. Dai, J.-K. Wang and C.-T. Chen, J. Org. Chem., 2004, 69, 6455–6462 CrossRef CAS PubMed; (e) Y. Zhang, G. Zhuang, M. Ouyang, B. Hu, Q. Song, J. Sun, C. Zhang, C. Gu, Y. Xu and Y. Ma, Dyes Pigm., 2013, 98, 486–492 CrossRef CAS; (f) S. Shin, S. H. Gihm, C. R. Park, S. Kim and S. Y. Park, Chem. Mater., 2013, 25, 3288–3295 CrossRef CAS.
- (a) H. Wang, E. Zhao, J. W. Y. Lam and B. Z. Tang, Mater. Today, 2015, 18, 365 CrossRef CAS; (b) R. Zhan, Y. Pan, P. N. Manghnani and B. Liu, Macromol. Biosci., 2017, 17, 1600433 CrossRef PubMed; (c) R. Hu, Y. Kang and B. Z. Tang, Polym. J., 2016, 48, 359 CrossRef CAS; (d) R. Hu, N. L. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494 RSC; (e) Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liua and J. Xu, Chem. Soc. Rev., 2012, 41, 3878 RSC.
- S. Zhao, H. Yamagishi, O. Oki, Y. Ihara, N. Ichiji, A. Kubo, S. Hayashi and Y. Yamamoto, Adv. Opt. Mater., 2021, 2101808 Search PubMed.
- S. Hayashi, S. Yamamoto, K. Nishi, A. Asano and T. Koizumi, Polym. J., 2019, 51, 1055–1061 CrossRef CAS.
- (a) S. Hayashi, R. Hirai, S. Yamamoto and T. Koizumi, Chem. Lett., 2018, 47, 1003–1005 CrossRef CAS; (b) S. Hayashi, M. Sakamoto, F. Ishiwari, T. Fukushima, S. Yamamoto and T. Koizumi, Tetrahedron, 2019, 75, 1079–1084 CrossRef CAS; (c) S. Hayashi, Mol. Syst. Des. Eng., 2021, 6, 503–507 RSC.
- (a) J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS; (b) S. Hayashi, S. Inagi and T. Fuchigami, Macromolecules, 2009, 42, 3755–3760 CrossRef CAS; (c) S. Hayashi, S. Inagi and T. Fuchigami, Synth. Met., 2009, 159, 1792–1795 CrossRef CAS; (d) S. Hayashi, A. Asano and T. Koizumi, Polym. Chem., 2011, 2, 2764–2766 RSC; (e) S. Hayashi, Y. Togawa, Y. Kojima and T. Koizumi, Polym. Chem., 2016, 7, 5671–5686 RSC; (f) Y. Matumura, H. Nishiyama, S. Inagi and I. Tomita, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 2519–2525 CrossRef; (g) F. Zheng, S.-E. Tan, Y. Yanamoto, N. Shida, H. Nishiyama, S. Inagi and I. Tomita, NPG Asia Mater., 2020, 12, 41 CrossRef CAS.
- M. Taguchi, N. Suzuki and M. Fujiki, Polym. J., 2013, 45, 1047–1057 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ma00113f |
| ‡ Authors with equal contribution. |
| This journal is © The Royal Society of Chemistry 2022 |