
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAutophagy targeting nanoparticles in rheumatoid arthritis and osteoarthritis
Akshay
Vyawahare
,
Anas
Ahmad
 ,
Kanika
,
Aneesh
Ali
,
Puspita
Saha
,
Vemana
Gowd
and
Rehan
Khan
*
,
Kanika
,
Aneesh
Ali
,
Puspita
Saha
,
Vemana
Gowd
and
Rehan
Khan
*
Department of Chemical Biology, Institute of Nano Science and Technology, Sector 81, Knowledge City, Sahibzada Ajit Singh Nagar, Mohali, Punjab 140306, India. E-mail: rehankhan@inst.ac.in; Tel: +91-172-2210075
First published on 13th April 2022
Abstract
Nanotechnology has been proven to be effective several times in the treatment of various diseases. Nanomedicine is a growing field of research for the development of novel drug delivery systems. Targeting autophagy with the help of nanotechnology based drug delivery might be beneficial over conventional ways. Autophagy is a degenerative process involving multiple steps to recycle essential components of cells, that goes into the senescence phase. It has a prominent role in pathogenesis as well as the homeostasis of cells involved in rheumatoid arthritis (RA) and osteoarthritis (OA). The health of chondrocytes is maintained by autophagy and dysregulation of autophagy is a major contributing factor in the death of chondrocytes. The autoimmune response to chondrocytes and synovium is a major trigger that ultimately leads to apoptosis of cells and then autophagy. Chondrocytes are stimulated by IL-17, IL-1 and TNF-α to secrete cartilage degrading metalloproteinases. Unhealthy conditions of cartilage reduce the number of chondrocytes and layers of articular cartilage, resulting in severe pain due to the movement of joints involving friction. Autophagy plays a major role in osteoclastogenesis in RA. The death of chondrocytes is a major hallmark of OA progression that involves bone degradation due to the complete removal of the cartilage layer. There are reports entailing that the inhibition of autophagy might be protective and can reduce bone erosion and osteoclast formation. In this review, we discussed the role of autophagy in OA and RA including signaling molecules, current therapies and nanotechnological advancements in treating these diseases.
1. Introduction
Autophagy is the biological process by which a cell starts to die automatically and its essential components are recycled to balance the source of energy to meet environmental stress or nutrient stress.1 The term ‘autophagy’, is derived from Greek words, which mean ‘eating of self’, and was first used by Christian de Duve.2 In housekeeping, it also has a role in extracting misfolded or aggregated proteins, removing damaged cell organelles, endoplasmic reticulum, peroxisomes, and evading pathogens. It is employed as a survival mechanism, although it destroys unwanted cells, recycles essential components and is utilized for the growth of other cells.1,3 Macroautophagy, microautophagy, and chaperone-induced autophagy (CMA) are three forms of autophagy. The non-selective mechanisms of autophagy, such as macroautophagy, involve the formation of a double membrane compartment called a phagophore for cargo degradation, whereas microautophagy employs direct destruction of cargo. A chaperone protein in chaperone-mediated autophagy, e.g. Hsc-70, plays a crucial role in the translocation of targeted proteins across the lysosomal membrane, which is recognized by the lysosomal membrane protein 2A (LAMP-2A) membrane receptor and further established or degraded accordingly.4,5 The induction of autophagy is triggered by various stimuli, including starvation, infection, physical and chemical stressors, and metabolic perturbations. The detailed mechanism of autophagy is explained in Fig. 2. It should be noted that irregularities in autophagy may lead to various disorders; thus it should be homeostatically maintained.6 Autophagy plays a role in various inflammatory diseases and in this review, we will delineate its significance in its role in osteoarthritis (OA) and rheumatoid arthritis (RA).The significant involvement of autophagy in RA and OA paves the way for targeting the disease through nanotechnology based drug delivery due to its unconventional ease of access to molecules involved in the progression of autophagy. Nanotechnology based drug delivery has significant advantages over the traditional methodologies.7 It basically comes with the repurposing of the existing drug molecules coming with several disadvantages such as low solubility, unwanted side effects and poor biodistribution. The beauty of nanotechnology based drug carriers is that they overcome the disadvantages of the drug and ultimately make it stupendous while exerting its therapeutic efficacy. The robustness of efficacy potentially depends on the carrier system.8
Osteoarthritis is a joint disease that mainly involves the destruction of cartilage and several other surrounding tissues. It comes with a socioeconomic burden as well as a mental and physical burden to the affected individual. It also includes remodeling of subarticular bone, weakening of periarticular muscles, synovial inflammation, ligamentous laxity, and osteophyte formation (Fig. 1). These changes occur due to an imbalance between repair and damage of joint tissue.9 Chondrocytes are able to rescue the articular cartilage matrix involved in structural changes or damages. However, the ability of chondrocytes is limited and gets lowered with age because of irregular responses to anabolic stimuli and cell death. Hence, it is necessary to keep chondrocytes intact and healthy, which is a vital component for maintaining the cartilage matrix intact and damage-free.10 The articular cartilage is always maintained in low oxygen surroundings.11 It is reported that there exists an oxygen gradient in the cartilage.12 The hypoxia-inducible factor (HIF) plays a major role under these hypoxic conditions, HIF-1α and HIF-2α have a crucial role to play in the survival, development, and differentiation of chondrocytes under vascular hypoxia. Higher expression of HIF-1α in chondrocytes can be a major adaptation to survive in the hypoxic conditions of cartilage. HIF-2α is also highly expressed in osteoarthritis compared to normal mice cartilage.11,12 Increased plate chondrocytes cause an increase in HIF-1α mediated glycolytic activity, which elevates the levels of AMP, resulting in AMP kinase activation, which downregulates the mTOR pathway and finally results in the activation of the autophagy.13 Osteoarthritis induced by surgery in PPARγ knock-out mice showed increased cartilage destruction and chondrocyte apoptosis. The overabundance of inflammatory markers abides by overexpression of mTOR and suppression of key autophagy markers.14 This is how autophagy is related to osteoarthritis progression; there are various other mechanisms by which autophagy can play a major role in the progression of osteoarthritis, which will be explained further in this review.
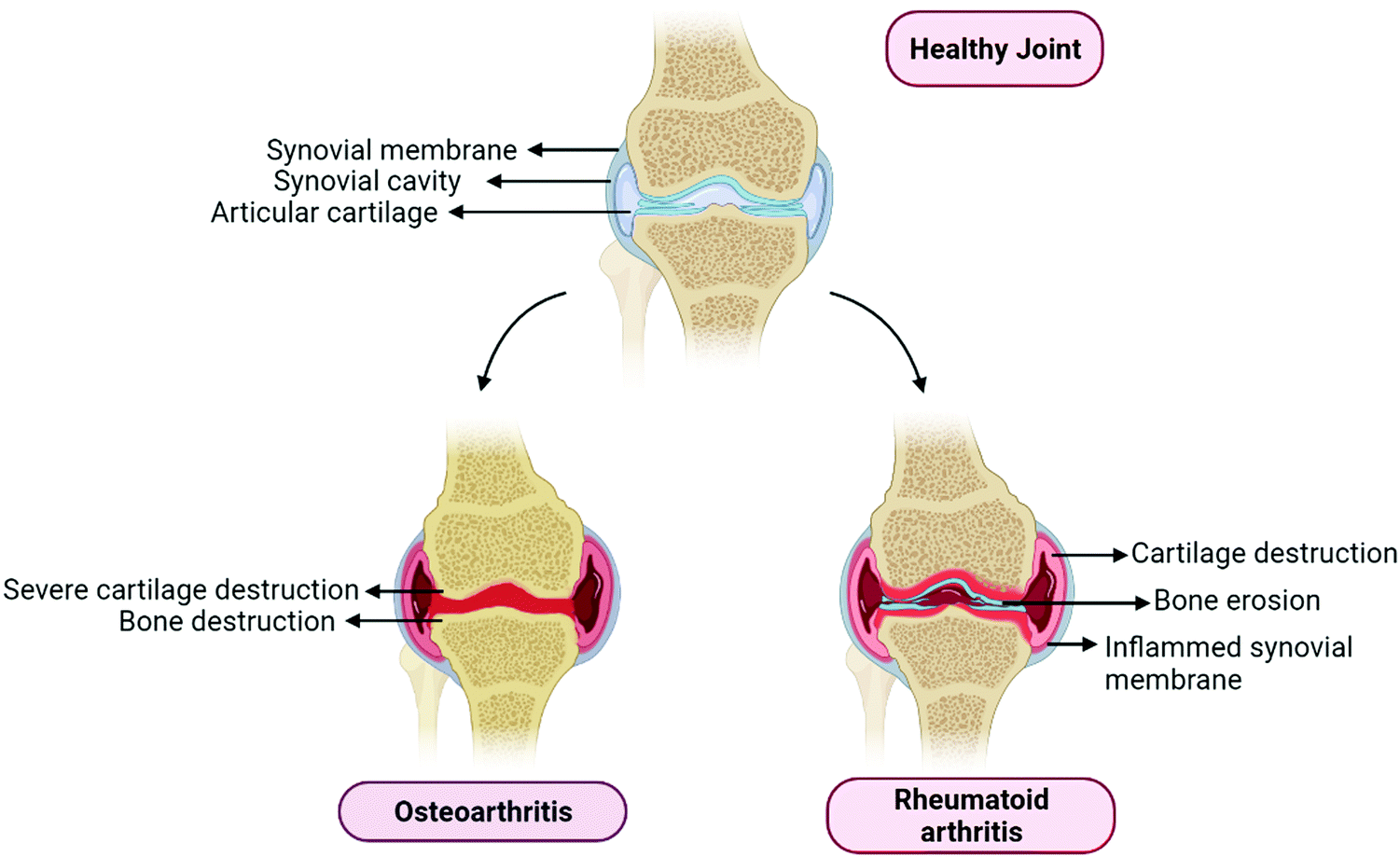 | ||
| Fig. 1 Representation of the basic differences between osteoarthritis and rheumatoid arthritis. OA Joints represents the complete removal of the articular cartilage involving severe bone destruction. RA involves the bone erosion, articular cartilage reduction, synovial inflammation and synovial fluid reduction. | ||
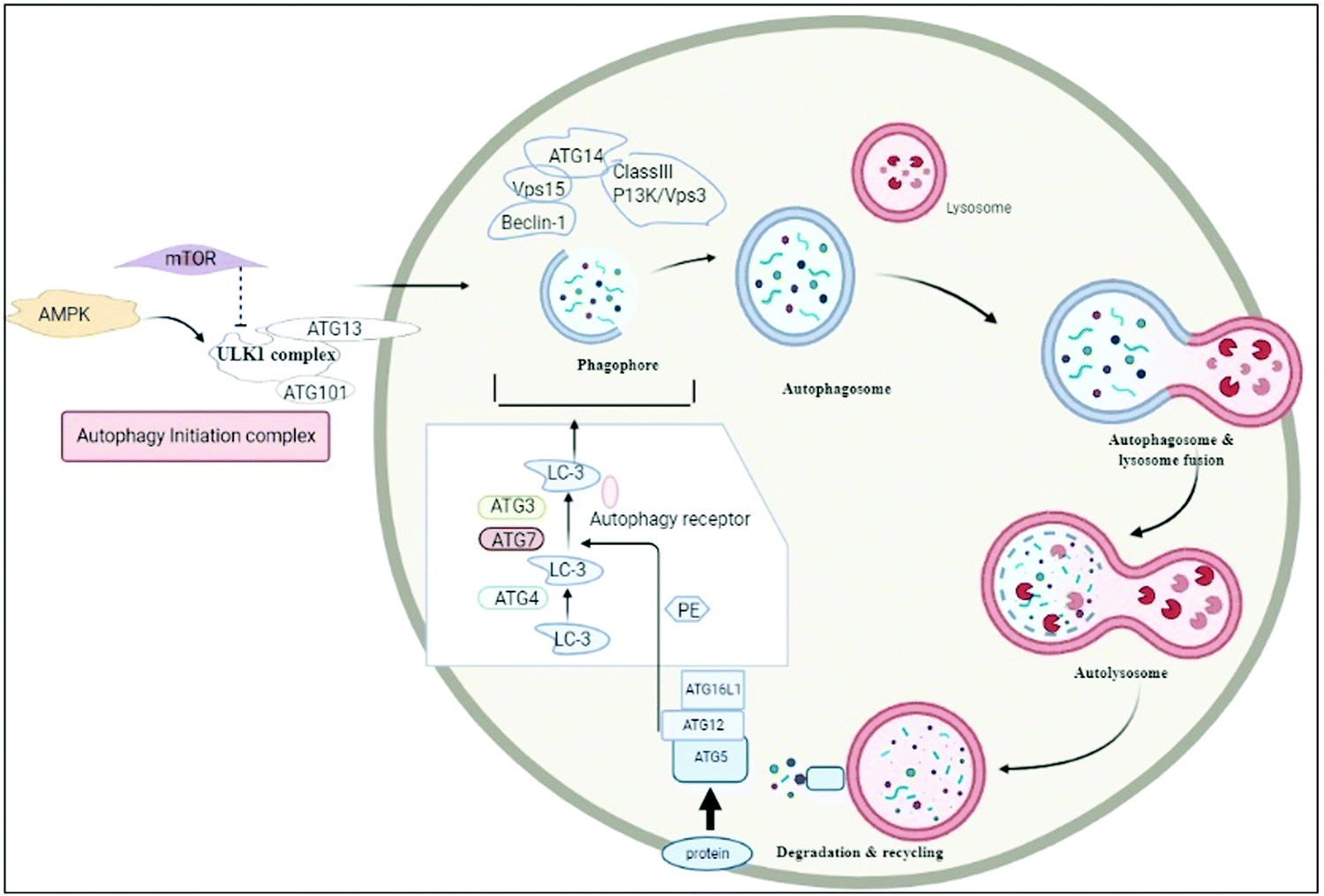 | ||
| Fig. 2 Process of autophagy and signalling molecules involved in the progression of the autophagy. Autophagy is initiated by AMPK, whereas it is suppressed by mTOR. AMPK stimulates the autophagy initiation complex. This further recruits phagophores, which nucleates and engulfs cell organelles; further this elongates into autophagosome, and this process is governed by LC-3. Lysosome gets fused with autophagosome results in the formation of the autophagolysosome, which is further subjected to degradation by lysosomal enzymes. | ||
Rheumatoid arthritis (RA) is a chronic autoimmune disease that mainly affects the joints and bones of individuals. It primarily affects females, as compared with males and is more prominently observed in the elderly population. It mainly targets the edges of synovial joints; symptoms include arthralgia, redness, swelling, and akinesia (Fig. 1).15 Till now there is no cure for the disease, however, the therapeutic goal is to lower the disease activity state.16 Rheumatoid arthritis is divided into two types depending on the type of the presence or absence of anti-citrullinated protein antibodies (ACPAs). ACPA positive and ACPA negative are two types of rheumatoid arthritis. ACPAs are found to be in 67% of patients with rheumatoid arthritis, which can be used as a marker for the diagnosis of early and non-differentiated arthritis, which is an indication of the progression of RA.17 RA has been characterized by the presence of synovitis or swelling of the synovial membrane, which mainly occurs in symmetrical small joints. Swelling of joints is an outer characteristic of inflammation to the synovial membrane, which further activates the immune response; hence it is called autoimmune disease as explained in Fig. 1. Various types of cells and cytokines play a crucial part in the progression of RA. Leukocytes and various pro-inflammatory mediators invade the synovial membrane, triggering cascades of inflammatory events that initiate fibroblast-like synoviocyte (FLS) interactions with innate immune responses including mast cells, monocytes, macrophages and dendritic cells, and adaptive immune response cells, including B cells and T cells. This triggers an autoimmune response that eventually causes bone degradation and cartilage damage, and increases the number of osteoclasts and synoviocytes.13,16
2. Role of autophagy in osteoarthritis and rheumatoid arthritis
Autophagy plays a dual role in the progression of OA and RA. With the perspective of elucidating the protective and destructive roles of autophagy in OA and RA, we have endeavored to bring together the reports from various studies on these aspects.2.1. Osteoarthritis
Osteoarthritis is a major age-related degenerative disease typified by articular cartilage impairment, osteophyte formation and stiffening of joints. OA is common in elderly people severely affects joints and related sub-structural components of joints such as bone synovium, meniscus, ligament, capsule, and muscles.10 The implication of OA mainly occurs by degeneration of articular cartilage, which plays a fundamental role in articulation by providing a smooth and lubricated surface, and facilitating movement between joints without friction and accelerating the transfer of pressure with a low frictional coefficient.18 In addition, OA is also characterized by very little turnover of the cells, such as chondrocytes as they play a vital role in the fate of extracellular matrix and their synthesis, thus providing an adequate environment to maintain the integrity of articular cartilage.19,20 Autophagy has also been involved in the maintenance of cellular homeostasis. Furthermore, it also serves as a regulating factor and plays a crucial role in the maturation and aging process of chondrocytes.21 The intense effect of autophagy was found in the rat OA model in the superficial layer of bones and intermediate zones of articular cartilage.22 During OA, the death of chondrocytes and associated extracellular matrix loss are the main causing factors behind articular degeneration; however, the mechanism of death is not known clearly so far. This series of findings suggested the possible role of both apoptotic and non-apoptotic signals. Furthermore, evidence also suggests that autophagy plays a vital role in cell survival, whereas the suppression of autophagy-related genes could mediate cell death, indicating the protective role of autophagy via the promotion of cell survival.23,24 In OA, there has been an elevated expression of autophagic LC-3 puncta, whereas it was reported normal in the case of healthy chondrocytes, aging may reduce the autophagy activity, which might protect the young cartilage.25 Monosodium urate crystals have been reported to significantly reduce the chondrocyte viability along with an increase in LC-3II level, which is the primary sign of autophagy activation. Articular degeneration is mainly caused by the death of chondrocytes by the process of apoptosis.26 Carames and Blanco reported that downregulated or loss of expression of the ULK 1, LC-3 and Beclin-1 leads to stimulating the process of apoptosis and lastly the cell death of chondrocytes) and simultaneously there is an upregulation of the poly(ADP ribose) polymerase (PARP) p85 expression along with the degradation of articular cartilage.25 It is also reported that a high level of ROS can damage mitochondria, which may induce apoptosis, and autophagy has been reported to be closely associated with apoptosis, there have been reports indicating a decreased autophagy in OA. Thus, it is assumed that the induction of autophagy as a therapeutic target in OA, would significantly protect chondrocytes from excessive apoptosis27 Chondroptosis is a term coined to describe apoptosis in chondrocytes, however, studies have demonstrated that chondrocytes do not follow the standard apoptosis pathway. Increased autophagic vacuoles, ER, Golgi, primary lysosomes, extensive blebbing, and infiltration of the cellular material into the perilacunar matrix and lacunae are the prominent features of chondroptosis.28 Mammalian target of rapamycin (mTOR) is the primary repressor of autophagy along with cell survival mechanism. However, the detailed mechanism has yet to be addressed (Fig. 3). In a recent study, scientists created mTOR knockout cartilage to better understand the role of mTOR in the progression of OA. The authors demonstrated that mTOR ablation resulted in a significant increase in autophagy signaling and thus protected mice from OA.29 The novel approach of IL-1β worsened the alteration of the OA-like genes expression and apoptotic signal,30 whereas it has been reported that OA-like gene expression changes devoid of the IL-1β, which is not affected by the inhibition and activation of autophagy. Studies reported that apoptosis and autophagy share common features such as autophagy playing a role in the protection of young cartilage by inducing the death of osteoarthritic chondrocytes and protecting cells under stress conditions, however, this process might lead to apoptosis and cell death. The protein named Bcl-2, which was originally known as beclin-1 act as the primary molecule involved in autophagosome formation and also necessary for binding to the BH-3 receptor domain of Bcl-2 or Bcl-XL.31 The activation of autophagy has been initiated by disturbing the complex named beclin 1/Bcl-2 by Bcl-2 as further it also affects the BH-3 domain of beclin-1.32,33 Autophagic response was found to be highly adaptive and mainly meant to avoid cell death in OA, as various reports have put forward that upregulation of apoptosis in the absence of autophagy depicting the relation with chondrocyte substitution, which is directly connected to dysregulated calcification of cartilage.34,35 The above stated information clearly depicts that autophagy might play a dual role in cartilage homeostasis (Fig. 4) that it may be cytoprotective as well as it can be harmful by helping in disease progression.33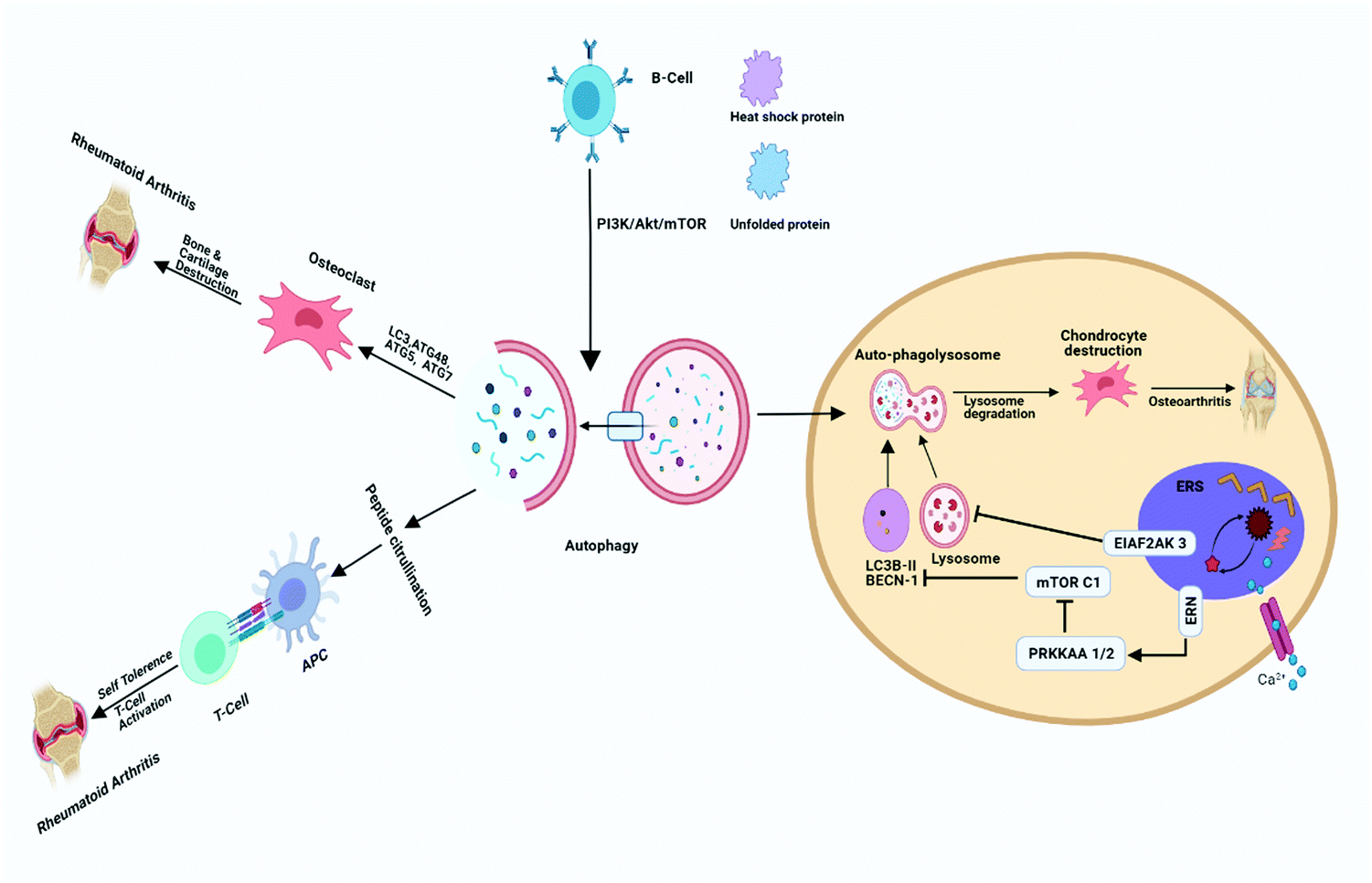 | ||
| Fig. 3 Role of autophagy in osteoarthritis and rheumatoid arthritis. Autophagy gets activated via stimulation through B cells, in the presence of LC3, ATG48, ATG5, ATG7 and activates the osteoclast involved in the bone degradation ultimately resulting in RA. Endoplasmic reticulum stress in chondrocytes activates or open Ca2+ channel and stimulates ERN and subsequently stimulates p-PRKAA1/2 and stimulates mTORC1 and results in the inhibition of autophagy. EIAF2AK3 stimulated by endoplasmic reticulum stress inhibits lysosomal degradation. Inhibition of autophagy initiation and lysosomal degradation might be helpful to prevent the worsening of OA. | ||
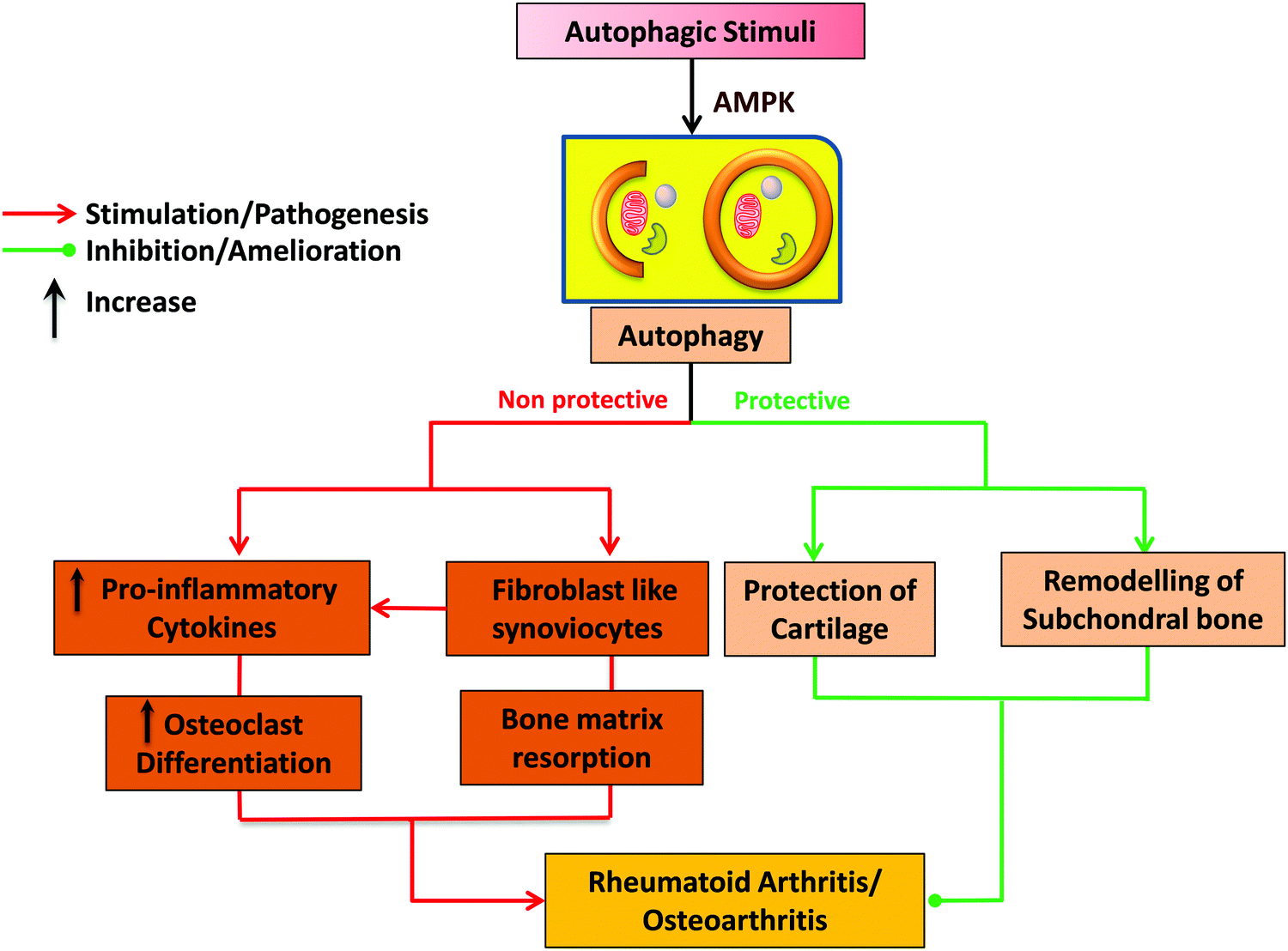 | ||
| Fig. 4 The dual role of autophagy in pathophysiology of RA and OA. The dual role includes the protective and destructive nature of autophagy for maintaining the homeostasis. | ||
2.2. Rheumatoid arthritis
A growing body of evidence demonstrates that autophagy dysregulation pathways play a vital role in the pathogenesis of autoimmune diseases in general and particularly in the case of rheumatoid arthritis (RA). Indeed, consequences of autophagy dysregulation are closely associated with the generation of citrullinated short peptides along with resistance to the apoptosis mechanism in the pathogenesis of RA disease.36–38 This close relationship between autophagy and RA has provided a very interesting platform to research autophagy involvement in autoimmune RA. In the pathophysiology of RA, immune cell activation takes place against self-antigens, which leads to the production of autoantibodies as pathognomonic biomarkers of this disorder. The activation and involvement of various immune cells such as B and T cells, synovial fibroblasts, macrophages, osteocytes, chondrocytes and thus the consequent release of different inflammatory mediators often lead to dysregulated autophagous mechanisms.10,20,39 Researchers have also acknowledged autophagy's contribution to present cytoplasmic antigens along with MHC class II moieties, which play a significant part in the acquired immune response mainlining of self-tolerance. Emerging data also suggest that autophagy plays a reasonable role in the central tolerance mechanism in RA. Furthermore, autophagy defects, as well as resultant loss of self-tolerance, also provide a cause of multiple indicators of autoimmunity reported in rheumatoid arthritis.40Researchers have also pointed towards the participation of a dysregulated autophagous mechanism in presenting self-antigen to immature T cells in the thymus and colocalization of LC3-II with lysosomes where MHC-peptide complexation occurs. In an experimental animal model of arthritis, autophagy inhibition is associated with a reduction in bone erosion as well as reduced osteoclast numbers, thus suggesting a pivotal role of autophagy in bone tissue degradation in arthritis (Fig. 3).40,41 This evidence significantly establishes that downregulation of autophagy may provide useful outcomes in bone resorption during RA and the role of autophagy both in innate and adaptive immune regulation can provide attractive therapeutic approaches in RA by autophagy modulation.42 Kato et al. investigated the role of autophagy in the regulation of stress-induced cell death in RA by treating synovial fibroblasts (RASF) with endoplasmic reticulum (ER) stress inducer thapsigargin (TG). They observed that TG administration instigates autophagy, which induces caspase-3 dependent cell death, thereby establishing the protective role of autophagy in apoptosis by inhibition of proteasome formation. Finally, it was concluded that autophagy plays a dual role in the regulation of cell death pathways under severe ER stress conditions (Fig. 4).43
Aberrant cellular homeostasis in RA results in hyperactive CD4+ T cells that resist undergoing the process of the apoptotic pathway and consequently leading to the pathogenicity of these immune cells in the autoimmune process. A better understanding of these processes concerned with these aberrant homeostatic pathways can significantly contribute to the in-depth realization of the disease process. In this regard, it was investigated how dysregulation of autophagy in CD4+ cells of patients with RA causes the disturbance of T cell homeostasis. It is suggested that the magnitude of autophagy is considerably enhanced in CD4+ T cells of RA patients and this also corroborates with the results obtained from the activated CD4+ T cells in vitro. Furthermore, inhibition of autophagy appreciably reverses the apoptosis resistance observed in CD4+ T cells of patients with RA. These results led to some novel insights into the pathophysiology of RA and emphasize the significance of autophagy as a hopeful therapeutic target.44
Another mechanism correlating autophagy with RA is the involvement of the HLA class II association, where CD4 T cells are vital regulators of the pathological immune response. These CD4 cells in RA are less efficient in glucose metabolism and ATP generation and are susceptible to apoptosis due to an insufficient glycolytic enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3). This deficiency also directs these cells towards autophagy to procure energy from it as an alternative source. This glycolytic enzyme also plays a role in autophagy regulation and its metabolic reprogramming and deficiency also render these cells in RA energy deficient and autophagy deprived senescent stages.35 The further relative theory between autophagy in RA postulates aberrant apoptosis leading to monumental hyperplasia and thus reduced apoptosis and its relation to autophagy was analysed in the synovial tissue of patients with RA. Apoptosis in synovial tissue was detected by TUNEL assay while autophagy was assessed by immunoblotting of markers such as Beclin-1 and LC3. Reduced TUNEL-positive cells in synovial tissue from RA showed enhanced expression of beclin-1 and LC3, thereby exhibiting an inverse relationship between apoptosis and autophagy in RA and it was concluded that defective apoptosis in RA synovial tissue may be the result of enhanced autophagy.45
A recent report by Fernandez-Rodriguez et al. revealed that cell death via autophagy and limited angiogenic response are implicated in the pathogenesis of RA and that the pharmacological agent resveratrol reduces synovial hyperplasia, chemokine release and activation and oxidative damage in an animal model of immune induced arthritis. Investigation of autophagic flux by resveratrol and its relation with inflammatory mediators such as C-reactive proteins and interleukins revealed that the effects of resveratrol on the autophagic proteins LC3 and Beclin-1 were correlated with enhanced autophagosome formation and accumulation of p62, which is associated with autophagic degradation dysfunction. Expression of the autophagic marker LC3 was significantly higher in the arthritis group than in the control. The authors concluded that resveratrol has the capability of engendering the non-canonical Beclin1 independent autophagy pathway, which is intertwined with inflammation in acute arthritis.46 It is already been known that dysregulated autophagy is one of the prominent reasons for autoimmune diseases including RA. Further, the involvement of autophagy in RA and its relation with arsenic trioxide, which is used for the treatment of RA, was established. It is observed that arsenic trioxide ameliorates RA symptoms by effectively boosting autophagic reflux through sufficient downregulation of the p62 marker accompanied by low inflammation and eventually reducing catabolic proteins.47
Janus tyrosine kinases play a vital role in exacerbating the pathophysiology of RA by regulating multiple cytokines as well other immune functions. Orally administrable JAK inhibitor tofacitinib inhibits jak 1, jak 2 and jak 3, and has demonstrated a great efficacy against RA. The role of tofacitinib was investigated in the context of autophagy in RA via its involvement in the degradation of intracellular components. Also, enhanced autophagy is correlated with dysregulated apoptosis of RA fibroblast-like synoviocytes. It was stated that tofacitinib reduced the expression of autophagy marker LC3-II in vitro and this was confirmed by analysing the reduction in autophagic vacuoles by specific fluorescent dyes. A new mechanism of action of tofacitinib in the context of autophagy regulation was established, which is helpful for the in-depth understanding of the correlation between autophagy and rheumatoid arthritis.48
3. Targeting autophagy in RA and OA
3.1. Nanoparticulate drug delivery
Drug-mediated targeted treatment for a particular disease can be achieved in two ways, viz. active targeting and passive targeting. Active targeting is achieved by conjugating or tagging a ligand, molecule, peptide, or protein on a carrier encapsulating a drug molecule. These conjugated ligands are chosen on the basis of the requirement to specifically target the site where the expression of this ligand–receptor could be found and overexpressed. In this way, the ligands will guide the carrier system toward the site due to the ligand's strong affinity towards the site where the receptors are overexpressed. Active targeting avoids the non-specific binding to the healthy sites so as to avoid its unwanted side effects. Active targeting fulfills the requirement of an ideal drug delivery system. Whereas, passive targeting is somewhat different from active targeting; they are designed in such a way to deliver the drug to the site of infection or inflammation.49 Disease conditions such as RA and OA can increase angiogenesis. The development of vascular network disease conditions is not proper and they are leaky in nature. Therefore, taking the advantage of the nature of the vessels, we can specifically target the site, avoiding the healthy tissues. Passive targeting of macrophages via a nanoparticulate drug delivery system is one of the strategic approaches to treating the disease.50 Macrophages are abundantly present on the site of inflammation; hence, nanoparticles might be phagocytosed by the macrophages themselves to exert their effect on the site.51Autophagy plays a role in the regulation of osteoclast differentiation and bone resorption in TNF-α dependent process, which implies that the modulation of autophagy can suppress structural damage in RA. Targeting the intricate cellular signalling pathways and reaching the cryptic sites of tissues by using the conventional delivery systems appear to be a daunting task. Nanotechnological advancements meet those needs as the favourable design of nanotherapeutics enables them to penetrate deep into the tissues and release the cargo, where they can exert their therapeutic effect.52 Low bioavailability, low permeability, low biodistribution and low cellular uptake pose formidable barriers in conventional drug treatments. However, these challenges can be overcome by the application of nanomedicine and its associated drug delivery.52,53 It has various advantages over conventional drug delivery systems, such as low drug resistance, low toxicity, high selectivity, high specificity and high bioavailability.54 The use of nanoparticulate drug delivery systems like liposomes,55 polymeric nanoparticles, and micelles have been reported to have various advantages, however, many researchers have also tried the use of metallic nanoparticles such as gold, silver and iron oxide NPs along with autophagy modulator drugs in various diseases.56,57
Neng-Yu Lin and Jörg Distler studied the role of pro-inflammatory cytokines such as IL-1 and IL-6 and TLR ligands, which are able to stimulate autophagy in several cell types. Hence, knowing the role of these signalling pathways in RA, they hypothesized that the autophagy stimulation could significantly ameliorate the pathogenesis of RA. In light of this, the authors concluded that the modulation of autophagy may prevent osteoclast differentiation, and activationwhich further inhibit bone erosion.58–60 Therefore, targeting autophagy in arthritis treatment is a promising new strategy and researchers are more focused on drugs that are involved in autophagy modulation with the help of nanotechnology in order to improve the efficient and targeted drug delivery and thus overcome several side effects.57
In recent times more research is focused on targeting molecular signalling pathways, which could provide a promising therapeutic output. The prominent role of autophagy is found not only in innate and adaptive immune regulation but also in immune system cell homeostasis. However, it will not be surprising to know that autophagy inhibition results in a therapeutic improvement in RA progression.56 Targeting autophagy or related pathways required a thorough understanding of how autophagy works. Several signalling molecules are crucial for autophagy progression.61 Genetic screening of yeast reported that there are 37 Atg genes. However, most Atg genes and proteins are evolutionarily conserved in humans.62 One study discussed the role of rapamycin, which inhibits mTOR and also led to a concomitant reduction of progression of systematic lupus erythematosus (SLE). Chloroquine, a well-known drug used to treat malaria, has also been reported to inhibit antigen presentation to T cells. Also, the differentiation of osteoclast precursors further formed into mature osteoclasts in vitro and in vivo in which autophagy is involved. Tumor necrosis factor (TNF) was found to stimulate autophagy in various cell types, which are firmly related to the pathogenesis of OA. However, there is no study that reports the anti-TNF effect of drugs on autophagy.43,63
3.2. Nanotechnology-based drug delivery systems to target autophagy
Folic acid receptors are highly expressed on macrophages and are rarely expressed in normal cells. Folic acid conjugates are exclusively used to target macrophages that have a high expression of FA receptors. In a study conducted by Cao et al., the authors prepared folic acid conjugated PLGA NPs, and encapsulated them with dexamethasone phosphate, which is an anti-rheumatoid drug. They came to the conclusion that there is an increase in the cellular uptake of dexamethasone phosphate in macrophages and also decreased production or synthesis of pro-inflammatory cytokines, such as IL-1 and TNF-α and along with nitric oxide (NO) inside the activated macrophages, whereas lower uptake was found in normal cells.64 Blocking the signalling of PI3K by a chemical compound 3-methyladenine (3-MA), which inhibits autophagy at an early stage; the formation of autophagosome is one of the crucial experiments used to analyse autophagy inhibition, imposing detrimental effects by promoting the progression of RA.63 A clinical study conducted by Li Zhu et al. to determine the involvement of autophagy in RA showed that autophagy is highly activated in the synovial tissue of patients with active RA; they also reported that this level was higher as compared with that in patients with OA.65Methotrexate (MTX) is an inhibitor of dihydrofolate reductase (DHFR), an enzyme involved in the synthesis of folic acid. MTX inhibits the production of pro-inflammatory cytokines, such as IL-1b, IL-6, IL-15, IL-18 and TNF-α, which play a major role in the induction of apoptosis. The autophagy modulator rapamycin is developed in the formulation as an immunoliposome targeting anti-E-selectin, in vitro results were promising for activating endothelial cells, whereas they also found that there is suppression of migration of endothelial cells, inflammatory cytokine expressions such as TNF-α for the treatment of inflammatory disorders such as RA and OA.66 In a study, the combined use of drugs such as 3-MA and chloroquine led to the reduced expression of PI3K and apoptosis induction via PI3K/AKT signalling, which ultimately resulted in amelioration of the inflammatory response and further inhibition of proliferation of RA-FLS (fibroblast-like synoviocyte) due to apoptotic death.67 Scientists have recently demonstrated that engineered bioactive silica-based nanoparticles for the stimulation of differentiation and mineralization of cells involved in the development of bone. These nanoparticles are having inherent biological activity and induce autophagy via the processing of LC3β-I to LC3β-II, the major factor triggering the formation of the autophagosome that is highly reliant on the ERK1/2.68 Another example of the NP-induced autophagy PAMAM G3NPs via mTOR resulting in the accumulation of LC-3 observed on the A549 cell knockdown TSC-2 gene.69 PEGylated solid lipid nanoparticles have shown efficacy in the treatment of RA, it has been reported that PEG-SLN enhances the incorporation of TNFα siRNA effectively. The systemic administration of this resulted in the restoration of paw thickness and degradation of bone in RA. Peptide drug delivery is one of the innovative approaches to target autoimmune diseases, peptide-like RGD, VIP, and HAP-1 showed good efficiency for drug delivery.70 Cyclodextrin is a biocompatible polymer recently being used for drug delivery purposes; methylprednisolone is a glucocorticoid that was used as a drug for the treatment of RA. The results showed reduced severity of arthritis and joint inflammation after administration of the NP.71 The overall summary of the nanotechnology-based drug delivery system is explained in Table 1.
| Type of nanocarrier | Loaded drug/agents | Mechanism | Target | Ref. |
|---|---|---|---|---|
| Chitosan NP | IL-1ra cDNA | Competitive inhibition of IL-1ra by antagonism | Folate receptor | 72 |
| Nanogold | Nanogold | Suppression of NF-κB via COX-2 inhibition | VEGF | 73 |
| Core/shell NP | Diclofenac | COX-2 inhibition | Magnetic field | 74 |
| Liposomes | Prednisolone | Anti-inflammatory | EPR | 75 |
| PLGA/PLA copolymers | β-Methasone | Binds glucocorticoid receptor and change gene expression | EPR/macrophages | 51 |
| Lipid microspheres | Indomethacin | Inhibit prostaglandins production | EPR/macrophages | 76 |
| Nanocapsules | Indomethacin | Inhibit prostaglandins production | EPR | 51 |
| PAMAM dendrimers | Flurbiprofen | COX inhibition | EPR | 77 |
| Chitosan NP | siRNA against TNF | Inhibit the TNF production | Macrophages | 78 |
| PLGA NP | Collagen | Lymphocytes | 79 | |
| Iron oxide nanoparticles | Dexamethasone | Binds glucocorticoid receptor and change gene expression | Synovial fibroblasts | 80 |
| Cyclodextrin polymers | Methylprednisolone | Anti-inflammatory | EPR/Macrophages | 71 |
| PLGA-cRGD NP | Binds with integrin receptor | STAT1 siRNA | 81 | |
| PEGylated SLN | Nucleic acid | DNA replication | TNF-α siRNA | 70 |
| MTX-HSA NP | Secreted protein acidic and rich in cysteine (SPARC) | Inhibit DHFR | MTX | 70 |
4. Signalling molecules involved in autophagy in RA and OA
Autophagy is a complex process through which cells decay their own components and can be positively or negatively governed by various factors starting from the initiation and maturation of autophagosomes. Autophagy can be positively regulated by beclin-1, AMPK and ULK-1, while it is negatively regulated by mTOR and PI3K/AKT.82 In this context, we will discuss the signalling molecules involved in autophagy in RA and OA and their significance as novel therapeutic targets as mentioned in Table 2.| Gene | Cell location | Function | Relation with diseases | Mol mass (kDA) | Ref. |
|---|---|---|---|---|---|
| ATG1 (Ulk1) | Cytoplasm | Initiation of the autophagy | Necessary for the longevity induced by AMPK and rapamycin | 10 | 83 |
| ATG11 | Preautophagosomal structure | Autophagosome vacuole fusion | Required for longevity induced by rapamycin | 135 | 83 |
| bec-1 (Beclin1) | Allosteric regulator of VPS-34 | Required for longevity induced by mTOR inhibition | 52 | 84 | |
| ATG7 | Cytosol | E1-like enzyme for the Atg5–Atg12 complex and the Atg8 conjugation systems | Required for longevity induced by dietary restriction, rapamycin and spermidine | 71 | 85 |
| ATG5 | Cytoplasm, autophagic vacuole | Conjugated by Atg12 | Required for longevity induced by dietary restriction by methionine restriction | 33 | 86 |
| ATG8 | Cytoplasm | Phagophore elongation and cargo recruitment | Required for longevity induced by dietary restriction by methionine restriction | 18 | 86 |
| ATG15 | Vacuolar lumen | Putative lipase required for intravacuolar disintegration of autophagic bodies | Required for longevity induced by dietary restriction | 58 | 84 |
| vps-34 | Kinase that produces PI3P to enable recruitment of machinery that forms autophagosomes | Required for longevity induced by mTOR inhibition, dietary restriction, germline ablation, | 100 | 87 | |
| ATG13 | Extrinsic to membrane | Regulatory subunit of Atg1 signalling complex | Required for vesicle formation during autophagy and involved in Atg9p, Atg23p, and Atg27p cycling | 83 | 88 |
| mTOR | Mitochondria | Associated with PI3K/Akt | Suppression of the autophagy | 289 | 89 |
| AMPK | Stimulated by metabolic stresses, such as glucose deprivation, hypoxia, ischemia, heat shock, or oxidative stress | AMPK directly inhibit mTOR also involved in the autophagy induction | 63 | 37 |
4.1. Citrulline
Citrullination is the chemical process of conversion of arginine to citrulline by the enzyme peptidylarginine deiminase (PAD). Citrullination plays a prime role in RA pathogenesis; autoantibodies associated with citrullinated peptides mainly lead to a poor prognosis.56 Inflammatory arthritis comes with an early sign of increased osteoclastogenesis and particularly of ACPA-Positive RA. Researchers have demonstrated that dendritic cells (DC's) can be metamorphosed into osteoclast (OC) precursors via escalation of protein citrullination and binding of ACPAs to the cell surface, which enhances the release of IL-8 by cells. This trans-differentiation of the DC's into OC's can be a novel mechanism that is involved in bone destruction in ACPA-positive RA that can be controlled by blocking of citrullination or IL-8 association with its receptor.90 However, since we began to know about the involvement of enzyme PAD in autophagy and the presence of stimuli with nutrient scarcity stimulates the presentation of citrullinated peptides in B cells, hence researchers suggested that citrullination can be a biomarker for autophagy.914.2. PI3K/AKT/mTOR signalling pathway
Autophagy is negatively regulated by PI3K/AKT/mTOR signalling cascade in arthritis or in overall cases. Inhibition of the PI3K/AKT/mTOR signalling pathway was found to escalate autophagy in articular chondrocytes and reduce the inflammatory response in osteoarthritic rats. Cytokines such as IL-1β are highly active in chondrocyte metabolism, where IL-1β is known to be involved in the inhibition of hyaline cartilage type II collagen synthesis, which subsequently results in chondrocyte degeneration and thus inhibition of chondrocyte proliferation.92 Beclin-1 is a member of the ATG family and plays a crucial role in autophagosome formation. They form multiprotein complexes with intracellular components such as mitochondrial, endoplasmic reticulum and nuclear membranes, followed by the initiation of autophagosome formation.93 It was also found that there was concomitant downregulation of Beclin-1 with AKT/mTOR upregulation and consequently, there was a suppression of autophagy along with downregulation of Atg 5 and Atg 7. The role of inflammation is imperative in cell fate specification of rat chondrocytes, cell cycle and suppression of autophagy.94 Therefore, PI3K/AKT/mTOR is regarded as a promising molecular target and thus promoting autophagy.4.3. ULK-1 (UNC-51-like kinase 1) complex
ULK-1 complex positively regulates autophagy, and is an initiator of autophagy along with ATG13 (autophagy-related protein 13), FIP200 (focal adhesion kinase family interacting protein of 200 kDa), and ATG101 and is mainly involved in autophagosome formation. ULK-1 phosphorylates along with ATG13 and FIP200 promote early autophagosome formation. This complex regulates selective autophagy and contributes to various disease pathogenesis such as RA and OA. mTORC1 negatively regulates the ULK-1 complex. ULK-1 is a potential target for the development of drugs or molecules targeting autophagy.93,95 One of the highly selective inhibitors of ULK and ULK2 is SBI-0206965 (2-((5-bromo-2-((3,4,5-trimethoxyphenyl)amino)pyrimidin-4-yl)oxy)-N-methylbenzamide). Furthermore, it also inhibits Beclin and vps34. It is reported that SBI-0206965 induces apoptosis by inhibiting autophagy.96 Another molecule such as MRT68921 N-(3-[[5-cyclopropyl-2[[3-(4-morpholinylmethyl)phenyl]amino]-4-pyrimidinyl]amino]propyl]cyclobutane carboxamide dihydrochloride) is reported to inhibit ULK1. It decreases autophagosome maturation by inhibiting the conversion of LC3-1 to LC3-II.954.4. AMP-activated protein kinase (AMPK)
The deprivation of glucose, nutrients and oxygen results in cellular stress, ultimately causing an alteration in the AMP/ATP ratio. The phosphorylation of AMP to AMPK results in the activation of autophagy initiation complexes such as ULK-1, which further phosphorylated and recruit other complexes, which result in autophagosome formation.93 Hence, it can be said that AMPK is an upstream inducer of autophagy. Metformin is a drug that stimulates AMPK; scientists have studied metformin against the KRN (K/BXN) arthritis mice model. They concluded that metformin stimulates the macrophage AMPK activity and this results in the suppression of mTORC1 activity, which ultimately amplified autophagic flux and inhibited the production of inflammatory cytokines via STAT1 suppression.97 Metformin and simvastatin are drugs found to activate the AMPK, whereas simvastatin is an indirect activator of MAPK.67,97 Several reports have reported their role as autophagy modulators in various cancers and showed a significant effect. Metformin exhibits its anticancer effect by impacting complex I of the electron transport chain, mTORC1, and AMPK.98 Very few studies have been conducted to test the therapeutic effect of metformin in the treatment of RA and OA by targeting autophagy. Simvastatin has also been reported to have a role in the treatment of RA.994.5. Light chain 3 (LC3)
Microtubule-associated protein 1A/1B (MAP1A/B) light chain 3 (LC3) is a protein ubiquitously distributed in the cellular system including arthritis. It is originally found in three forms (LC1, LC2, LC3) firmly associated with MAP1A and MAP1B.100 Cytosolic forms are LC3-I (conjugated to phosphatidylethanolamine) to form LC3 phosphatidylethanolamine conjugate (LC3-II) hence LC3-II is considered an autophagic marker.101 After the formation of autophagosomes, they get fused with lysosomes to form autolysosomes. At the same time, intra-autophagosomal components are degraded by hydrolases, while LC3-II gets degraded in the autolysosomal lumen. LC3-II can be used as a marker of starvation-induced autophagy.954.6. Tumor necrosis factor and interleukin
Autophagy is induced by proinflammatory cytokines such as TNF-α, IL-1, IL-17, and IL-16, whereas it is blocked by IL-13, IL-33, IL-4 and IL-10; simultaneously, autophagy regulates the synthesis and release of these cytokines.102,103 These simultaneous processes have drawn the attention of researchers and have now become the topics of a major research area.Osteoclasts are the primary cells involved in bone destruction in RA that leads to articular damage and systemic osteoporosis. Osteoclastogenesis is the process by which osteoclasts are differentiated from their precursors. This process highly depends on the presence of M-CSF (macrophage colony-stimulating factor) and RANKL (receptor activator of NF-κB ligand).60,104–106 Along with these processes, TNF-α, which is a pro-inflammatory cytokine, has a role in osteoclast differentiation and is found to escalate the osteoclast differentiation. So, it can be said that in mice, overexpressing TNF showed increased osteoclastogenesis.58,107 Interleukins have various roles in modulating and controlling autophagy, interleukins such as IL-1β are said to play a role in the pathogenesis of acute pancreatitis. IL-1β dysregulates autophagic flux and activates trypsinogen, which plays a role in the pathogenesis.107 Researchers have interpreted that modulation of IL-1β can be beneficial in the case of arthritis. IL-17 was found to cause mitochondrial dysfunction in FLS RA, which is found to increase the autophagosome formation, and enhanced FLS survival.37,108 Various TNF inhibitors have been tried to enhance the autophagy process. Sirolimus is well known as rapamycin, it is very hydrophobic and exhibits very poor bioavailability, and initially, it was used as an antifungal agent, and then because of its TNF blocking property, used as an immunosuppressive agent. Everolimus is a derivative of sirolimus, which has more solubility and bioavailability compared to sirolimus.108
5. Future perspectives
Nowadays, research has been more focused on targeting the root cause of diseases, which includes disturbances in signalling pathways or molecules involved in various processes. However, traditional drug targets were more related to targeting enzymes or molecules, which are critical for the progression of diseases. No doubt, both of these approaches had a significant impact on the therapeutic escalation of disease, but somehow current research is found to be more involved in overcoming challenges, which are curbing the bioavailability of drugs and ultimately reducing side effects. Current research is the ultimate fate of traditional research because modern techniques and genomic understanding take us far away from the traditional ways of treating diseases. In the modern era, we have ample techniques, which give an accurate diagnosis of disease. RA and OA are those diseases that are not detected at early stages; they are predominantly diagnosed at the symptomatic stage. These are elder age diseases and at this stage, cure of the diseases becomes difficult. The aim of disease management is symptomatic treatment as there is severe pain in both cases of RA and OA.For the development of effective treatment, one should have a thorough knowledge of disease pathophysiology. We have summarized disease pathophysiology, the role of autophagy in the pathogenesis of RA and OA, various signalling molecules with a vital role in autophagy progression along with current therapeutic options available for treatment. We also elaborated on the recent development of formulations that target autophagy in RA and OA. However, unfulfilled requirements of treatment and patients' unresponsiveness to currently available treatments make it necessary to develop better treatment options. Nanotechnology, nowadays, is most followed in every sphere of life. In the near future, we will see nano-formulations that are approved for the treatment of RA and OA. It is not surprising that research is mostly focused on the repurposing of drugs rather than creating new ones. In coming times, we need to clearly define the mechanism underlying drug targeting autophagy and possible side effects, along with drug interaction. Novel molecules targeting signaling pathways in autophagy are further explored in cancer-like diseases. Nowadays, they should be explored and need to be tested for RA and OA.
6. Conclusion
Incidences of RA and OA increase with aging and are related to modern living lifestyles. In recent times more research is focused on autophagy and its relation with OA and RA pathogenesis; however, its detailed mechanism is still unknown. Autophagy plays a dual role in chondrocyte homeostasis and hence modulation of autophagy can be a major target for the treatment of osteoarthritis and rheumatoid arthritis. OA and RA are currently treated with existing NSAIDs, opioids and other anti-inflammatory drugs, which only treat symptoms; however, so far, no effective cure has been found for these diseases. Knowing the prominent role of autophagy in RA and OA, scientists make it easier to overcome barriers to treating the disease by taking the advantage of nanotechnology. Various nanocarriers were formulated to target autophagy effectively and have proved to be significant in treating OA and RA. The involvement of autophagy in disease progression can offer novel ways such as targeting signalling molecules, which not only relieve severe symptoms of disease but can effectively provide treatment for OA and RA.Abbreviations
| ACPAs | Anti-citrullinated protein antibodies |
| ATG | Autophagy-related protein |
| CMA | Chaperone mediated autophagy |
| DHFR | Dihydrofolate reductase |
| DMOADs | Disease-modifying osteoarthritic drugs |
| ER | Endoplasmic reticulum |
| FIP200 | Focal adhesion kinase family interacting protein of 200 kDa |
| FLS | Fibroblast like synoviocyte |
| HCQ | Hydroxychloroquine |
| HIF | Hypoxia inducing factor |
| HLA | Human leukocyte antigen |
| LAMP-2A | Lysosomal-associated membrane protein 2A |
| LEF | Leflunomide |
| M-CSF | Macrophage colony-stimulating factor |
| MAP1A/B | Microtubule-associated protein 1A/1B |
| MHC | Major histocompatibility complex |
| MTX | Methotrexate |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| PARP | Poly(ADP ribose) polymerase |
| PPARγ | Peroxisome proliferator-activated gamma |
| RANKL | Receptor activator of NF-κB ligand |
| SSZ | Sulfasalazine |
| STAT | Signal transducer and activator of transcription |
| TNF | Tumor necrosis factor |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labelling |
| ULK-1 | (unc-51-like kinase 1) complex |
| mTOR | Mammalian target of rapamycin |
| 3 MA | 3 Methyl adenine |
Author contributions
AV contributed from introduction to the role of autophagy in RA. AA contributed to Current therapeutic options in OA and RA. KB contributed to Targeting autophagy in RA and OA. AA Contributed to signalling molecules involved in RA and OA. MMA contributed to the Future perspective and conclusion. RK formulated the concepts and reviewed the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work is supported by Department of Science and Technology (DST), SERB with grant no. CRG/2019/004018. AV is thankful to Institute of Nano Science and Technology, Mohali for providing Senior Research fellowship. AA is thankful to Institute of Nano Science and Technology, Mohali for providing a Senior Research Fellowship. K is thankful to SERB (CRG/2019/004018), for providing a Junior Research Fellowship. A. Ali is thankful to CSIR for providing junior research fellowship.References
- D. Glick, S. Barth and K. F. Macleod, Autophagy: Cellular and molecular mechanisms, J. Pathol., 2010, 221(1), 3–12, DOI:10.1002/path.2697.
- R. L. Deter and C. De Duve, Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes, J. Cell Biol., 1967, 33(2), 437–449, DOI:10.1083/jcb.33.2.437.
- G. Mariño and C. López-Otín, Autophagy: Molecular mechanisms, physiological functions and relevance in human pathology, Cell. Mol. Life Sci., 2004, 61(12), 1439–1454, DOI:10.1007/s00018-004-4012-4.
- A. R. Issa, J. Sun, C. Petitgas, A. Mesquita, A. Dulac, M. Robin, B. Mollereau, A. Jenny, B. Chérif-Zahar and S. Birman, The lysosomal membrane protein LAMP2A promotes autophagic flux and prevents SNCA-induced parkinson disease-like symptoms in the drosophila brain, Autophagy, 2018, 14(11), 1898–1910, DOI:10.1080/15548627.2018.1491489.
- P. Saftig, W. Beertsen and E. L. Eskelinen, LAMP-2: A control step fot phagosome and autophagosome maturation, Autophagy, 2008, 4(4), 510–512, DOI:10.4161/auto.5724.
- S. Tavakol, M. Ashrafizadeh, S. Deng, M. Azarian, A. Abdoli, M. Motavaf, D. Poormoghadam, H. Khanbabaei, E. G. Afshar, A. Mandegary, A. Pardakhty, C. T. Yap, R. Mohammadinejad and A. P. Kumar, Autophagy modulators: Mechanistic aspects and drug delivery systems, Biomolecules, 2019, 9(10), 530, DOI:10.3390/biom9100530.
- W. H. De Jong and J. B. Paul, Drug delivery and nanoparticles: Applications and hazards, Int. J. Nanomed., 2008, 3(2), 133–149 CrossRef CAS PubMed.
- O. C. Farokhzad and R. Langer, Impact of nanotechnology on drug delivery, ACS Nano, 2009, 3(1), 16–20, DOI:10.1021/nn900002m.
- A. Litwic, M. H. Edwards, E. M. Dennison and C. Cooper, Epidemiology and burden of osteoarthritis, Br. Med. Bull., 2013, 105(1), 185–199, DOI:10.1093/bmb/lds038.
- Y. S. Li, F. J. Zhang, C. Zeng, W. Luo, W. F. Xiao, S. G. Gao and G. H. Lei, Autophagy in osteoarthritis, Jt. Bone Spine, 2016, 83(2), 143–148, DOI:10.1016/j.jbspin.2015.06.009.
- P. I. Milner, T. P. A. Fairfax, J. A. Browning, R. J. Wilkins and J. S. Gibson, The effect of O2 tension on PH homeostasis in equine articular chondrocytes, Arthritis Rheum., 2006, 54(11), 3523–3532, DOI:10.1002/art.22209.
- J. Malda, C. A. van Blitterswijk, M. van Geffen, D. E. Martens, J. Tramper and J. Riesle, Low oxygen tension stimulates the redifferentiation of dedifferentiate adult human nasal chondrocytes, Osteoarthr. Cartil., 2004, 12(4), 306–313, DOI:10.1016/j.joca.2003.12.001.
- V. Srinivas, J. Bohensky and I. M. Shapiro, Autophagy: A new phase in the maturation of growth plate chondrocytes is regulated by HIF, MTOR and AMP kinase, Cells Tissues Organs, 2008, 189(1–4), 88–92, DOI:10.1159/000151428.
- F. Dell’Accio and J. Sherwood, PPARγ/MTOR signalling: Striking the right balance in cartilage homeostasis, Ann. Rheum. Dis., 2015, 74(3), 477–479, DOI:10.1136/annrheumdis-2014-206884.
- Q. Guo, Y. Wang, D. Xu, J. Nossent, N. J. Pavlos and J. Xu, Rheumatoid arthritis: Pathological mechanisms and modern pharmacologic therapies, Bone Res., 2018, 6(1), 1–14, DOI:10.1038/s41413-018-0016-9.
- F. Ometto, C. Botsios, B. Raffeiner, P. Sfriso, L. Bernardi, S. Todesco, A. Doria and L. Punzi, Methods used to assess remission and low disease activity in rheumatoid arthritis, Autoimmun. Rev., 2010, 9(3), 161–164, DOI:10.1016/j.autrev.2009.07.001.
- K. Nishimura, D. Sugiyama, Y. Kogata, G. Tsuji, T. Nakazawa, S. Kawano, K. Saigo, A. Morinobu, M. Koshiba, K. M. Kuntz, I. Kamae and S. Kumagai, Meta-analysis: Diagnostic accuracy of anti-cyclic citrullinated peptide antibody and rheumatoid factor for rheumatoid arthritis, Ann. Intern. Med., 2007, 146(11), 797–808, DOI:10.7326/0003-4819-146-11-200706050-00008.
- A. J. Sophia Fox, A. Bedi and S. A. Rodeo, The basic science of articular cartilage: Structure, composition, and function, Sports Health, 2009, 1(6), 461–468, DOI:10.1177/1941738109350438.
- R. F. Loeser, Aging and osteoarthritis: The role of chondrocyte senescence and aging changes in the cartilage matrix, Osteoarthr. Cartil., 2009, 17(8), 971–979, DOI:10.1016/j.joca.2009.03.002.
- W. J. Jeong, J. Bu, L. J. Kubiatowicz, S. S. Chen, Y. S. Kim and S. Hong, Peptide–nanoparticle conjugates: A next generation of diagnostic and therapeutic platforms?, Nano Convergence, 2018, 5(1), 1–18, DOI:10.1186/s40580-018-0170-1.
- C. Vinatier, E. Domínguez, J. Guicheux and B. Caramés, Role of the inflammation-autophagy-senescence integrative network in osteoarthritis, Front. Physiol., 2018, 9(Jun), 1–25, DOI:10.3389/fphys.2018.00706.
- C. Arias, N. Saavedra, K. Leal, B. Vásquez, D. S. P. Abdalla and L. A. Salazar, Histological evaluation and gene expression profiling of autophagy-related genes for cartilage of young and senescent rats, Int. J. Mol. Sci., 2020, 21(22), 1–15, DOI:10.3390/ijms21228607.
- T. Hara, K. Nakamura, M. Matsui, A. Yamamoto, Y. Nakahara, R. Suzuki-Migishima, M. Yokoyama, K. Mishima, I. Saito, H. Okano and N. Mizushima, Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice, Nature, 2006, 441(7095), 885–889, DOI:10.1038/nature04724.
- M. Komatsu, S. Waguri, T. Chiba, S. Murata, J. I. Iwata, I. Tanida, T. Ueno, M. Koike, Y. Uchiyama, E. Kominami and K. Tanaka, Loss of autophagy in the central nervous system causes neurodegeneration in mice, Nature, 2006, 441(7095), 880–884, DOI:10.1038/nature04723.
- B. Caramés, N. Taniguchi, S. Otsuki, F. J. Blanco and M. Lotz, Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis, Arthritis Rheum., 2010, 62(3), 791–801, DOI:10.1002/art.27305.
- H. S. Hwang, C. M. Yang, S. J. Park and H. A. Kim, Monosodium urate crystal-induced chondrocyte death via autophagic process, Int. J. Mol. Sci., 2015, 16(12), 29265–29277, DOI:10.3390/ijms161226164.
- C. Fleury, B. Mignotte and J. L. Vayssière, Mitochondrial Reactive Oxygen Species in Cell Death Signaling, Biochimie, 2002, 84(2–3), 131–141, DOI:10.1016/S0300-9084(02)01369-X.
- T. Aigner, H. A. Kim and H. I. Roach, Apoptosis in osteoarthritis, Rheum. Dis. Clin. North Am., 2004, 30(3), 639–653, DOI:10.1016/j.rdc.2004.04.002.
- Y. Zhang, F. Vasheghani, Y. H. Li, M. Blati, K. Simeone, H. Fahmi, B. Lussier, P. Roughley, D. Lagares, J. P. Pelletier, J. Martel-Pelletier and M. Kapoor, Cartilage-specific deletion of mtor upregulates autophagy and protects mice from osteoarthritis, Ann. Rheum. Dis., 2015, 74(7), 1432–1440, DOI:10.1136/annrheumdis-2013-204599.
- H. Sasaki, K. Takayama, T. Matsushita, K. Ishida, S. Kubo, T. Matsumoto, N. Fujita, S. Oka, M. Kurosaka and R. Kuroda, Autophagy modulates osteoarthritis-related gene expression in human chondrocytes, Arthritis Rheum., 2012, 64(6), 1920–1928, DOI:10.1002/art.34323.
- M. Almonte-Becerril, F. Navarro-Garcia, A. Gonzalez-Robles, M. A. Vega-Lopez, C. Lavalle and J. B. Kouri, Cell death of chondrocytes is a combination between apoptosis and autophagy during the pathogenesis of osteoarthritis within an experimental model, Apoptosis, 2010, 15(5), 631–638, DOI:10.1007/s10495-010-0458-z.
- S. Sinha and B. Levine, The autophagy effector beclin 1: A novel BH3-only protein, Oncogene, 2008, 27(1), S137–S148, DOI:10.1038/onc.2009.51.
- J. Chang, W. Wang, H. Zhang, Y. Hu, M. Wang and Z. Yin, The dual role of autophagy in chondrocyte responses in the pathogenesis of articular cartilage degeneration in osteoarthritis, Int. J. Mol. Med., 2013, 32(6), 1311–1318, DOI:10.3892/ijmm.2013.1520.
- Y. Y. Gu, J. Chen, Z. L. Meng, W. Y. Ge, Y. Y. Bian, S. W. Cheng, C. K. Xing, J. L. Yao, J. Fu and L. Peng, Research progress on osteoarthritis treatment mechanisms, Biomed. Pharmacother., 2017, 93, 1246–1252, DOI:10.1016/j.biopha.2017.07.034.
- Z. Yang, H. Fujii, S. V. Mohan, J. J. Goronzy and C. M. Weyand, Phosphofructokinase deficiency impairs atp generation, autophagy, and redox balance in rheumatoid arthritis T cells, J. Exp. Med., 2013, 210(10), 2119–2134, DOI:10.1084/jem.20130252.
- H. Yang, Y. Wen, M. Zhang, Q. Liu, H. Zhang, J. Zhang, L. Lu, T. Ye, X. Bai, G. Xiao and M. Wang, MTORC1 coordinates the autophagy and apoptosis signaling in articular chondrocytes in osteoarthritic temporomandibular joint, Autophagy, 2020, 16(2), 271–288, DOI:10.1080/15548627.2019.1606647.
- J. Kim, M. Kundu, B. Viollet and K.-L. Guan, AMPK and MTOR regulate autophagy through direct phosphorylation of Ulk1, Nat. Cell Biol., 2011, 13(2), 132–141, DOI:10.1038/ncb2152.
- E. K. Kim, J. E. Kwon, S. Y. Lee, E. J. Lee, D. S. Kim, S. J. Moon, J. Lee, S. K. Kwok, S. H. Park and M. L. Cho, IL-17-mediated mitochondrial dysfunction impairs apoptosis in rheumatoid arthritis synovial fibroblasts through activation of autophagy, Cell Death Dis., 2017, 8(1), 1–10, DOI:10.1038/cddis.2016.490.
- M. Kasai, I. Tanida, T. Ueno, E. Kominami, S. Seki, T. Ikeda and T. Mizuochi, Autophagic compartments gain access to the MHC class II compartments in thymic epithelium, J. Immunol., 2009, 183(11), 7278–7285, DOI:10.4049/jimmunol.0804087.
- V. L. Crotzer and J. S. Blum, Autophagy and its role in MHC-mediated antigen presentation, J. Immunol., 2009, 182(6), 3335–3341, DOI:10.4049/jimmunol.0803458.
- M. Aichinger, C. Wu, J. Nedjic and L. Klein, Macroautophagy substrates are loaded onto MHC class II of medullary thymic epithelial cells for central tolerance, J. Exp. Med., 2013, 210(2), 287–300, DOI:10.1084/jem.20122149.
- Y. Matsuzawa-Ishimoto, S. Hwang and K. Cadwell, Autophagy and inflammation, Annu. Rev. Immunol., 2018, 36, 73–101, DOI:10.1146/annurev-immunol-042617-053253.
- M. Kato, C. Ospelt, R. E. Gay, S. Gay and K. Klein, Dual role of autophagy in stress-induced cell death in rheumatoid arthritis synovial fibroblasts, Arthritis Rheumatol., 2014, 66(1), 40–48, DOI:10.1002/art.38190.
- J. van Loosdregt, M. Rossetti, R. Spreafico, M. Moshref, M. Olmer, G. W. Williams, P. Kumar, D. Copeland, K. Pischel, M. Lotz and S. Albani, Increased autophagy in CD4+ T cells of rheumatoid arthritis patients results in T-cell hyperactivation and apoptosis resistance, Eur. J. Immunol., 2016, 46(12), 2862–2870, DOI:10.1002/eji.201646375.
- K. Xu, P. Xu, J. F. Yao, Y. G. Zhang, W. K. Hou and S. M. Lu, Reduced apoptosis correlates with enhanced autophagy in synovial tissues of rheumatoid arthritis, Inflamm. Res., 2013, 62(2), 229–237, DOI:10.1007/s00011-012-0572-1.
- J. A. Fernández-Rodríguez, M. Almonte-Becerril, O. Ramil-Gómez, S. Viñas-Diz, L. Hermida-Carballo, Á. Vela-Anero, Á. Concha, M. Camacho-Encina, F. J. Blanco and M. J. Lopez-Armada, AB0121 resveratrol-enhanced autophagic flux reduces severity of experimental rheumatoid arthritis, Ann. Rheum. Dis., 2018, 77(Suppl. 2), 1254, DOI:10.1136/annrheumdis-2018-eular.4468.
- W. Wang, C. Li, Z. Zhang and Y. Zhang, Arsenic trioxide in synergy with vitamin D rescues the defective VDR-PPAR- γ functional module of autophagy in rheumatoid arthritis, PPAR Res., 2019, 2019, 1–11, DOI:10.1155/2019/6403504.
- M. Vomero, M. Caliste, C. Barbati, T. Colasanti, F. Spinelli, F. Ceccarelli, C. Perricone, A. Finucci, M. Speziali, A. I. Celia, M. Bombardieri, F. Conti, G. Valesini and C. Alessandri, Ab0145 the inhibition of Jak pathway was associated with reduction of autophagy in synoviocytes from rheumatoid arthritis patients, Ann. Rheum. Dis., 2019, 78(Suppl. 2), 1530–1531, DOI:10.1136/annrheumdis-2019-eular.5445.
- M. F. Attia, N. Anton, J. Wallyn, Z. Omran and T. F. Vandamme, An overview of active and passive targeting strategies to improve the nanocarriers efficiency to tumour Sites, J. Pharm. Pharmacol., 2019, 71(8), 1185–1198, DOI:10.1111/jphp.13098.
- T. D. Clemons, R. Singh, A. Sorolla, N. Chaudhari, A. Hubbard and K. S. Iyer, Distinction between active and passive targeting of nanoparticles dictate their overall therapeutic efficacy, Langmuir, 2018, 34(50), 15343–15349, DOI:10.1021/acs.langmuir.8b02946.
- C. T. N. Pham, Nanotherapeutic approaches for the treatment of rheumatoid arthritis, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2011, 3(6), 607–619, DOI:10.1002/wnan.157.
- R. Mohammadinejad, M. Ashrafizadeh, A. Pardakhty, I. Uzieliene, J. Denkovskij, E. Bernotiene, L. Janssen, G. S. Lorite, S. Saarakkala and A. Mobasheri, Nanotechnological strategies for osteoarthritis diagnosis, monitoring, clinical management, and regenerative medicine: Recent advances and future opportunities, Curr. Rheumatol. Rep., 2020, 22(4), 1–17, DOI:10.1007/s11926-020-0884-z.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. D. P. Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H.-S. Shin, Nano based drug delivery systems: recent developments and future prospects, J. Nanobiotechnol., 2018, 16(1), 71, DOI:10.1186/s12951-018-0392-8.
- N. Hoshyar, S. Gray, H. Han and G. Bao, The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction, Nanomedicine, 2016, 11(6), 673–692, DOI:10.2217/nnm.16.5.
- G. Chen, B. Hao, D. Ju, M. Liu, H. Zhao, Z. Du and J. Xia, Pharmacokinetic and pharmacodynamic study of triptolide-loaded liposome hydrogel patch under microneedles on rats with collagen-induced arthritis, Acta Pharm. Sin. B, 2015, 5(6), 569–576, DOI:10.1016/j.apsb.2015.09.006.
- M. Vomero, C. Barbati, T. Colasanti, C. Perricone, L. Novelli, F. Ceccarelli, F. R. Spinelli, M. Di Franco, F. Conti, G. Valesini and C. Alessandri, Autophagy and rheumatoid arthritis: Current knowledges and future perspectives, Front. Immunol., 2018, 9, 1577, DOI:10.3389/fimmu.2018.01577.
- H. Cheng, A. Chawla, Y. Yang, Y. Li, J. Zhang, H. L. Jang and A. Khademhosseini, Development of nanomaterials for bone-targeted drug delivery, Drug Discovery Today, 2017, 22(9), 1336–1350, DOI:10.1016/j.drudis.2017.04.021.
- N. Y. Lin, C. Beyer, A. Gießl, T. Kireva, C. Scholtysek, S. Uderhardt, L. Enrique Munoz, C. Dees, A. Distler, S. Wirtz, G. Krönke, B. Spencer, O. Distler, G. Schett and J. H. W. Distler, Autophagy regulates TNFα-mediated joint destruction in experimental arthritis, Ann. Rheum. Dis., 2013, 72(5), 761–768, DOI:10.1136/annrheumdis-2012-201671.
- N. Y. Lin, A. Stefanica and J. H. W. Distler, Autophagy: A key pathway of TNF-induced inflammatory bone loss, Autophagy, 2013, 9(8), 1253–1255, DOI:10.4161/auto.25467.
- S. Onuora, Bone research: Autophagy is central to joint destruction in arthritis, Nat. Rev. Rheumatol., 2012, 8(11), 633, DOI:10.1038/nrrheum.2012.171.
- G. Li, J. W. Boyle, C. N. Ko, W. Zeng, V. K. W. Wong, J. B. Wan, P. W. H. Chan, D. L. Ma and C. H. Leung, Aurone derivatives as Vps34 inhibitors that modulate autophagy, Acta Pharm. Sin. B, 2019, 9(3), 537–544, DOI:10.1016/j.apsb.2019.01.016.
- Y. Ohsumi, Historical landmarks of autophagy research, Cell Res., 2014, 24(1), 9–23, DOI:10.1038/cr.2013.169.
- N. T. Cheng, H. Meng, L. F. Ma, L. Zhang, H. M. Yu, Z. Z. Wang and A. Guo, Role of autophagy in the progression of osteoarthritis: The autophagy inhibitor, 3-methyladenine, aggravates the severity of experimental osteoarthritis, Int. J. Mol. Med., 2017, 39(5), 1224–1232, DOI:10.3892/ijmm.2017.2934.
- X. Yu, R. Zhang, L. Lei, Q. Song and X. Li, High drug payload nanoparticles formed from dexamethasone-peptide conjugates for the treatment of endotoxin-induced uveitis in rabbit, Int. J. Nanomed., 2019, 14, 591–603, DOI:10.2147/IJN.S179118.
- L. Zhu, H. Wang, Y. Wu, Z. He, Y. Qin and Q. Shen, The autophagy level is increased in the synovial tissues of patients with active rheumatoid arthritis and is correlated with disease severity, Mediators Inflamm., 2017, 2017, 7623145, DOI:10.1155/2017/7623145.
- S. Tavakol, M. Ashrafizadeh, S. Deng, M. Azarian, A. Abdoli, M. Motavaf, D. Poormoghadam, H. Khanbabaei, E. Ghasemipour Afshar, A. Mandegary, A. Pardakhty, C. T. Yap, R. Mohammadinejad and A. Prem Kumar, Autophagy Modulators: Mechanistic Aspects and Drug Delivery Systems, Biomolecules, 2019, 9(10), 1–41, DOI:10.3390/biom9100530.
- S. Li, J. W. Chen, X. Xie, J. Tian, C. Deng, J. Wang, H. N. Gan and F. Li, Autophagy inhibitor regulates apoptosis and proliferation of synovial fibroblasts through the inhibition of PI3K/AKT pathway in collagen-induced arthritis rat model, Am. J. Transl. Res., 2017, 9(5), 2065–2076, DOI:10.1136/annrheumdis-2017-eular.2044.
- S. W. Ha, M. Neale Weitzmann and G. R. Beck, Bioactive silica nanoparticles promote osteoblast differentiation through stimulation of autophagy and direct association with LC3 and P62, ACS Nano, 2014, 8(6), 5898–5910, DOI:10.1021/nn5009879.
- L. Jia, S. L. Hao and W. X. Yang, Nanoparticles induce autophagy via MTOR pathway inhibition and reactive oxygen species generation, Nanomedicine, 2020, 15(14), 1419–1435, DOI:10.2217/nnm-2019-0387.
- Q. Wang, X. Qin, J. Fang and X. Sun, Nanomedicines for the treatment of rheumatoid arthritis: State of art and potential therapeutic strategies, Acta Pharm. Sin. B, 2021, 11(5), 1158–1174, DOI:10.1016/j.apsb.2021.03.013.
- J. Hwang, K. Rodgers, J. C. Oliver and T. Schluep, α-Methylprednisolone conjugated cyclodextrin polymer-based nanoparticles for rheumatoid arthritis therapy, Int. J. Nanomed., 2008, 3(3), 359–371 CAS.
- J. C. Fernandes, H. Wang, C. Jreyssaty, M. Benderdour, P. Lavigne, X. Qiu, F. M. Winnik, X. Zhang, K. Dai and Q. Shi, Bone-protective effects of nonviral gene therapy with folate-chitosan DNA nanoparticle containing interleukin-1 receptor antagonist gene in rats with adjuvant-induced arthritis, Mol. Ther., 2008, 16(7), 1243–1251, DOI:10.1038/mt.2008.99.
- C. M. Chan, C. Y. Hsiao, H. J. Li, J. Y. Fang, D. C. Chang and C. F. Hung, The inhibitory effects of gold nanoparticles on VEGF-A-induced cell migration in choroid-retina endothelial cells, Int. J. Mol. Sci., 2020, 21(1), 1–11, DOI:10.3390/ijms21010109.
- N. C. Honakeri, S. J. Malode, R. M. Kulkarni and N. P. Shetti, Electrochemical behavior of diclofenac sodium at coreshell nanostructure modified electrode and its analysis in human urine and pharmaceutical samples, Sens. Int., 2020, 1(February), 100002, DOI:10.1016/j.sintl.2020.100002.
- L. Patras, B. Sylvester, L. Luput, A. Sesarman, E. Licarete, A. Porfire, D. Muntean, D. M. Drotar, A. D. Rusu, A. L. Nagy, C. Catoi, I. Tomuta, L. Vlase, M. Banciu and M. Achim, Liposomal prednisolone phosphate potentiates the antitumor activity of liposomal 5-fluorouracil in C26 murine colon carcinoma in vivo, Cancer Biol. Ther., 2017, 18(8), 616–626, DOI:10.1080/15384047.2017.1345392.
- S. Dolati, S. Sadreddini, D. Rostamzadeh, M. Ahmadi, F. Jadidi-Niaragh and M. Yousefi, Utilization of nanoparticle technology in rheumatoid arthritis treatment, Biomed. Pharmacother., 2016, 80, 30–41, DOI:10.1016/j.biopha.2016.03.004.
- Y. Cheng, Z. Xu, M. Ma and T. Xu, Dendrimers as drug carriers: Applications in different routes of drug administration, J. Pharm. Sci., 2008, 97(1), 123–143, DOI:10.1002/jps.21079.
- K. A. Howard, S. R. Paludan, M. A. Behlke, F. Besenbacher, B. Deleuran and J. Kjems, Chitosan/SiRNA nanoparticle-mediated TNF-α knockdown in peritoneal macrophages for anti-inflammatory treatment in a murine arthritis model, Mol. Ther., 2009, 17(1), 162–168, DOI:10.1038/mt.2008.220.
- G. Cappellano, C. Comi, A. Chiocchetti and U. Dianzani, Exploiting PLGA-based biocompatible nanoparticles for next-generation tolerogenic vaccines against autoimmune disease, Int. J. Mol. Sci., 2019, 20(1), 1–16, DOI:10.3390/ijms20010204.
- N. Butoescu, C. A. Seemayer, M. Foti, O. Jordan and E. Doelker, Dexamethasone-containing PLGA superparamagnetic microparticles as carriers for the local treatment of arthritis, Biomaterials, 2009, 30(9), 1772–1780, DOI:10.1016/j.biomaterials.2008.12.017.
- R. I. Scheinman, R. Trivedi, S. Vermillion and U. B. Kompella, Functionalized STAT1 SiRNA nanoparticles regress rheumatoid arthritis in a mouse model, Nanomedicine, 2011, 6(10), 1669–1682, DOI:10.2217/nnm.11.90.
- Y. Chen and D. J. Klionsky, The regulation of autophagy – Unanswered questions, J. Cell Sci., 2011, 124(2), 161–170, DOI:10.1242/jcs.064576.
- A. L. Alvers, M. S. Wood, D. Hu, A. C. Kaywell, W. A. Dunn and J. P. Aris, Autophagy is required for extension of yeast chronological life span by rapamycin, Autophagy, 2009, 5(6), 847–849, DOI:10.4161/auto.8824.
- H. Tang, S. Sebti, R. Titone, Y. Zhou, C. Isidoro, T. S. Ross, H. Hibshoosh, G. Xiao, M. Packer, Y. Xie and B. Levine, Decreased BECN1 MRNA expression in human breast cancer is associated with estrogen receptor-negative subtypes and poor prognosis, EBioMedicine, 2015, 2(3), 255–263, DOI:10.1016/j.ebiom.2015.01.008.
- S. Metzger, C. Walter, O. Riess, R. A. C. Roos, J. E. Nielsen, D. Craufurd, R. I. of the E. H. D. Network and H. P. Nguyen, The V471A polymorphism in autophagy-related gene ATG7 modifies age at onset specifically in Italian Huntington disease patients, PLoS One, 2013, 8(7), e68951 CrossRef CAS PubMed.
- C. Ruckenstuhl, C. Netzberger, I. Entfellner, D. Carmona-Gutierrez, T. Kickenweiz, S. Stekovic, C. Gleixner, C. Schmid, L. Klug, A. G. Sorgo, T. Eisenberg, S. Büttner, G. Mariño, R. Koziel, P. Jansen-Dürr, K.-U. Fröhlich, G. Kroemer and F. Madeo, Lifespan extension by methionine restriction requires autophagy-dependent vacuolar acidification, PLoS Genet., 2014, 10(5), e1004347, DOI:10.1371/journal.pgen.1004347.
- L. R. Lapierre, C. D. De Magalhaes Filho, P. R. McQuary, C. C. Chu, O. Visvikis, J. T. Chang, S. Gelino, B. Ong, A. E. Davis, J. E. Irazoqui, A. Dillin and M. Hansen, The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans, Nat. Commun., 2013, 4(1), 1–8, DOI:10.1038/ncomms3267.
- Y. Mei, M. D. Thompson, R. A. Cohen and X. Y. Tong, Autophagy and oxidative stress in cardiovascular diseases, Biochim. Biophys. Acta, Mol. Basis Dis., 2015, 1852(2), 243–251, DOI:10.1016/j.bbadis.2014.05.005.
- S. Periyasamy-Thandavan, M. Jiang, P. Schoenlein and Z. Dong, Autophagy: Molecular machinery, regulation, and implications for renal pathophysiology, Am. J. Physiol. Renal Physiol., 2009, 297(2), F244–F256, DOI:10.1152/ajprenal.00033.2009.
- A. Krishnamurthy, A. J. Ytterberg, M. Sun, K. Sakuraba, J. Steen, V. Joshua, N. K. Tarasova, V. Malmström, H. Wähämaa, B. Réthi and A. I. Catrina, Citrullination controls dendritic cell transdifferentiation into osteoclasts, J. Immunol., 2019, 202(11), 3143–3150, DOI:10.4049/jimmunol.1800534.
- J. M. Ireland and E. R. Unanue, Processing of proteins in autophagy vesicles of antigen-presenting cells generates citrullinated peptides recognized by the immune system, Autophagy, 2012, 8(3), 429–430, DOI:10.4161/auto.19261.
- W. Cheng, D. Wu, Q. Zuo, Z. Wang and W. Fan, Ginsenoside Rb1 prevents interleukin-1 beta induced inflammation and apoptosis in human articular chondrocytes, Int. Orthop., 2013, 37(10), 2065–2070, DOI:10.1007/s00264-013-1990-6.
- J. S. Rockel and M. Kapoor, Autophagy: Controlling cell fate in rheumatic diseases, Nat. Rev. Rheumatol., 2016, 12(9), 517–531, DOI:10.1038/nrrheum.2016.92.
- J. F. Xue, Z. M. Shi, J. Zou and X. L. Li, Inhibition of PI3K/AKT/MTOR signaling pathway promotes autophagy of articular chondrocytes and attenuates inflammatory response in rats with osteoarthritis, Biomed. Pharmacother., 2017, 89, 1252–1261, DOI:10.1016/j.biopha.2017.01.130.
- I. Tanida, T. Ueno and E. Kominami, LC3 and autophagy, Methods Mol. Biol., 2008, 445(2), 77–88, DOI:10.1007/978-1-59745-157-4_4.
- K. J. Petherick, O. J. L. Conway, C. Mpamhanga, S. A. Osborne, A. Kamal, B. Saxty and I. G. Ganley, Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (MTOR)-dependent autophagy, J. Biol. Chem., 2015, 290(18), 11376–11383, DOI:10.1074/jbc.C114.627778.
- H. Yan, Suppression of experimental arthritis through AMP-activated protein kinase activation and autophagy modulation, J. Rheum. Dis. Treat., 2015, 1(1), 1–18, DOI:10.23937/2469-5726/1510005.
- J. H. Cha, W. H. Yang, W. Xia, Y. Wei, L. C. Chan, S. O. Lim, C. W. Li, T. Kim, S. S. Chang, H. H. Lee, J. L. Hsu, H. L. Wang, C. W. Kuo, W. C. Chang, S. Hadad, C. A. Purdie, A. M. McCoy, S. Cai, Y. Tu, J. K. Litton, E. A. Mittendorf, S. L. Moulder, W. F. Symmans, A. M. Thompson, H. Piwnica-Worms, C. H. Chen, K. H. Khoo and M. C. Hung, Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1, Mol. Cell, 2018, 71(4), 606–620, DOI:10.1016/j.molcel.2018.07.030.
- K. I. Paraskevas, Statin treatment for rheumatoid arthritis: A promising novel indication, Clin. Rheumatol., 2008, 27(3), 281–287, DOI:10.1007/s10067-007-0806-8.
- Y. Kabeya, N. Mizushima, T. Ueno, A. Yamamoto, T. Kirisako, T. Noda, E. Kominami, Y. Ohsumi and T. Yoshimori, Erratum: LC3, a Mammalian homolog of yeast Apg8p, is localized in autophagosome membranes after processing (EMBO Journal (2000) 19 (5720–5728)), EMBO J., 2003, 22(17), 4577, DOI:10.1093/emboj/cdg454.
- N. Mizushima, T. Yoshimori and B. Levine, Methods in Mammalian autophagy research, Cell, 2010, 140(3), 313–326, DOI:10.1016/j.cell.2010.01.028.
- M. B. Goldring and M. Otero, Inflammation in osteoarthritis, Curr. Opin. Rheumatol., 2011, 23(5), 471–478, DOI:10.1097/BOR.0b013e328349c2b1.
- S. A. Jones, K. H. G. Mills and J. Harris, Autophagy and inflammatory diseases, Immunol. Cell Biol., 2013, 91(3), 250–258, DOI:10.1038/icb.2012.82.
- D. L. Lacey, E. Timms, H. L. Tan, M. J. Kelley, C. R. Dunstan, T. Burgess, R. Elliott, A. Colombero, G. Elliott, S. Scully, H. Hsu, J. Sullivan, N. Hawkins, E. Davy, C. Capparelli, A. Eli, Y. X. Qian, S. Kaufman, I. Sarosi, V. Shalhoub, G. Senaldi, J. Guo, J. Delaney and W. J. Boyle, Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation, Cell, 1998, 93(2), 165–176, DOI:10.1016/S0092-8674(00)81569-X.
- J. Keffer, L. Probert, H. Cazlaris, S. Georgopoulos, E. Kaslaris, D. Kioussis and G. Kollias, Transgenic mice expressing human tumour necrosis factor: A predictive genetic model of arthritis, EMBO J., 1991, 10(13), 4025–4031, DOI:10.1002/j.1460-2075.1991.tb04978.x.
- R. Felix, M. G. Cecchini, W. Hofstetter, P. R. Elford, A. Stutzer and H. Fleisch, Rapid publication: Impairment of macrophage colony-stimulating factor production and lack of resident bone marrow macrophages in the osteopetrotic Op/Op mouse, J. Bone Miner. Res., 1990, 5(7), 781–789, DOI:10.1002/jbmr.5650050716.
- Y. Ge, M. Huang and Y. M. Yao, Autophagy and proinflammatory cytokines: Interactions and clinical implications, Cytokine Growth Factor Rev., 2018, 43(July), 38–46, DOI:10.1016/j.cytogfr.2018.07.001.
- L. Morviducci, F. Rota, L. Rizza, P. Di Giacinto, S. Ramponi, M. R. Nardone, C. Tubili, A. Lenzi, P. Zuppi and R. Baldelli, Everolimus is a new anti-cancer molecule: Metabolic side effects as lipid disorders and hyperglycemia, Diabetes Res. Clin. Pract., 2018, 143, 428–431, DOI:10.1016/j.diabres.2018.04.001.
| This journal is © The Royal Society of Chemistry 2022 |