
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIon transport in composite polymer electrolytes
Jialong
Fu
,
Zhuo
Li
,
Xiaoyan
Zhou
and
Xin
Guo
 *
*
State Key Laboratory of Material Processing and Die & Mould Technology, School of Materials Science and Engineering, Huazhong University of Science and Technology, Wuhan 430074, P. R. China. E-mail: xguo@hust.edu.cn
First published on 1st April 2022
Abstract
Owing to their high flexibility, remarkable processability, and favorable interfacial contacts with electrodes, polymer-based electrolytes are highly valued for solid-state batteries with high energy density and safety. Among all the polymer-based electrolytes, composite polymer electrolytes, consisting ofpolymer matrix and inorganic fillers, have shown great prospects, due to their high ionic conductivities and appropriate mechanical strength. In this review, we focus on the mechanisms of ion transport in composite polymer electrolytes. Three domains co-exist in a typical composite solid electrolyte, namely, the polymer matrix, the inorganic fillers and the interfacial regions between the polymer matrix and the inorganic fillers; the ion transport in the above three domains is elaborated on the basis of recent studies. Finally, challenges and perspectives are put forward for developing novel composite polymer electrolytes for high-performance lithium-ion batteries. This review aims to provide guidance for the development of composite solid electrolytes with high ionic conductivity.
 Jialong Fu | Jialong Fu, PhD student in the School of Material Science and Engineering, Huazhong University of Science and Technology (HUST), Wuhan, China. His research interests are solid electrolytes and solid-state batteries. |
 Zhuo Li | Zhuo Li, postdoctoral researcher in the School of Material Science and Engineering, HUST. He received his PhD degree in 2020 from HUST. His research interests are solid electrolytes and solid-state batteries. |
 Xiaoyan Zhou | Xiaoyan Zhou, PhD student in the School of Material Science and Engineering, HUST. Her research interests are solid electrolytes and solid-state batteries. |
 Xin Guo | Xin Guo, Distinguished professor in the School of Materials Science and Engineering, HUST. Before joining HUST, from 2002 to 2012, he was a senior scientist at the Research Center Juelich, Germany; from 1998 to 2002, he was working at the Max Planck Institute for Solid State Research, Stuttgart, Germany. In 2005, he received the Ross Coffin Purdy Award from the American Ceramic Society. His research focuses are: solid electrolytes and solid-state batteries; neuromorphic devices, systems and artificial intelligence; gas sensors and artificial olfaction; defects in complex oxides. |
1 Introduction
Environmental pollution and undesirable climate changes caused by the use and consumption of fossil fuels are becoming increasingly serious, causing severe problems to human life.1,2 It is necessary to develop devices for green and sustainable energy storage and conversion. Among them, lithium-ion batteries have received much attention because of their high energy density and long cycle life.3,4 Non-aqueous liquid electrolytes with high conductivity and excellent contacts with electrodes are widely used in commercial lithium-ion batteries. However, organic liquid solvents suffer from serious safety problems during cycling, due to their poor thermal stability, high effumability and flammability;5,6 these problems become more severe for the state-of-the-art lithium-ion batteries with high energy density, especially when high-voltage positive electrode materials are employed.7,8Replacing liquid electrolytes with solid electrolytes has been recognized as a promising approach to solving the above problems.9–12 Solid electrolytes can be roughly classified into two categories: inorganic electrolytes and polymer electrolytes. Inorganic electrolytes (Li7La3Zr2O12 (LLZO), Li1.5Al0.5Ge1.5(PO4)3 (LAGP), Li10GeP2S12 (LGPS), etc.) have some significant advantages, such as high ionic conductivity (10−4 to 10−2 S cm−1), high ionic transference number (∼1), wide electrochemical window (>6 V), excellent thermal stability and high mechanical strength.13–17 However, high impedance and polarization during cycling, originating from poor interfacial contacts between electrolytes and electrodes, seriously limit their applications.18,19 In contrast, solid polymer electrolytes have the advantages of high flexibility, remarkable processability, and favorable interfacial contacts with electrodes, therefore, are emerging as the most promising candidates for solid-state batteries.20–25 However, compared with liquid electrolytes and solid inorganic electrolytes, solid polymer electrolytes have lower ionic conductivities at room temperature, which counteracts their beneficial effects.26,27 Combining inorganic and polymer electrolytes to form composite polymer electrolytes is able to overcome their shortcomings, while making use of their advantages. Therefore, inorganic fillers are introduced into polymer matrices to improve the ionic conductivity of polymer electrolytes.28–32
Fast ionic conduction is most important for composite polymer electrolytes. However, the ion transport mechanisms in composite polymer electrolytes are complex and controversial, because varied states of electrolytes are associated with a number of factors, such as temperature, structures of polymer matrices, composite structures formed by polymer matrices and inorganic fillers.33,34 Hence, this review focuses on the ion transport mechanisms in composite polymer electrolytes; contributions of the polymer phase, inorganic phase, and the interfacial regions between the polymer matrix and inorganic fillers to the whole ionic conduction are elaborated. This review aims to make a comprehensive understanding of ion transport in composite polymer electrolytes, and challenges and prospects for the large-scale application of composite polymer electrolytes in solid-state lithium-ion batteries with high safety and excellent performances are also presented.
2 Ion transport pathways
As illustrated in Fig. 1, three transporting pathways for lithium ions are feasible in a typical composite polymer electrolyte: (i) polymer phase with low-crystallization, (ii) interfacial regions between inorganic fillers and the polymer matrix, and (iii) ionically conductive fillers. The mechanisms of ion transport are analyzed in detail in the following sections. | ||
| Fig. 1 Possible lithium–ion transport pathways in a composite polymer electrolyte. | ||
2.1 Ion transport in a polymer matrix
In order to be used as electrolytes, a polymer should meet some essential criteria, including cation solvation ability, high dielectric constant and excellent segment flexibility. Some linear polymers with sequential polar groups (e.g. –O–, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, –S–, –N–, –P–, –CO, –CN, etc.) were chosen as matrices for polymer-based electrolytes.37–40 Polar groups in the polymer matrix are beneficial for the dissolution of lithium salts and adsorption of anions; meanwhile, flexible liner polymer segments favor the ion transport. In general, lithium-ion transport in the polymer phase mainly occurs in amorphous regions above the glass transition temperature (Tg).41,42
O, –S–, –N–, –P–, –CO, –CN, etc.) were chosen as matrices for polymer-based electrolytes.37–40 Polar groups in the polymer matrix are beneficial for the dissolution of lithium salts and adsorption of anions; meanwhile, flexible liner polymer segments favor the ion transport. In general, lithium-ion transport in the polymer phase mainly occurs in amorphous regions above the glass transition temperature (Tg).41,42
The free-volume model and derived theories were widely accepted to reveal how lithium ions are transported in amorphous regions. For example, in PEO-based polymers, the transport of lithium ions is described as the motion of the lithium cations between complexation sites, assisted by the segmental motion of PEO. As shown in Fig. 2,43 the polar ether-oxygen (EO) groups have a high donor number for lithium ions, and the excellent flexibility of polymer chains significantly promotes lithium-ion transport. According to the free-volume model, polymer chains undergo local segmental motions in a quasi-liquid behavior as if there is some free volume around them.44,45 Lithium ions are coordinated by the oxygen atoms of EO units on the segmental PEO chains, similar to the complexation of lithium ions and organic carbonates in liquid electrolytes. With the breaking/forming of lithium–oxygen (Li–O) combinations, ion transport occurs by intra-chain or inter-chain hopping in PEO-based electrolytes under electric fields. As the ligands gradually replace the solvation of lithium ions, the successive segmental rearrangements lead to remote replacements of lithium ions.
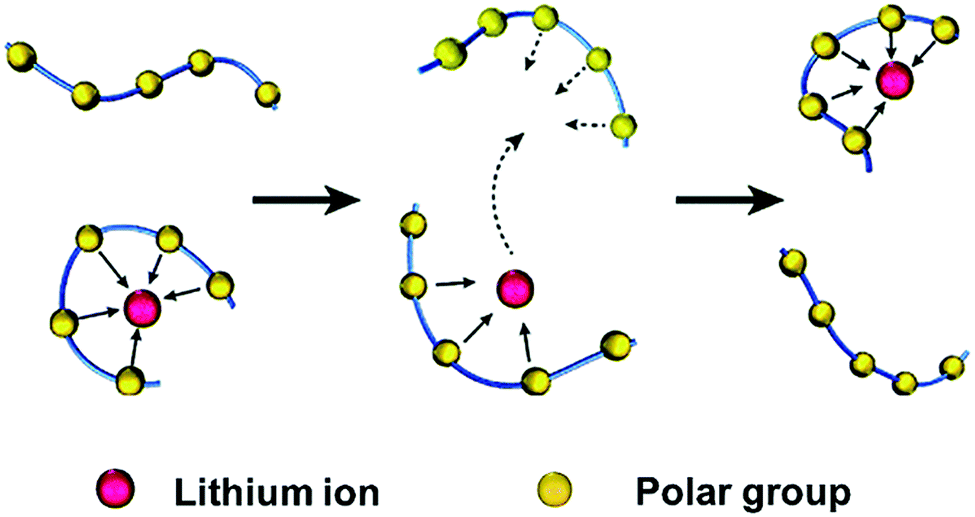 | ||
| Fig. 2 Schematic illustration of lithium-ion transport in amorphous regions of polymers. Reproduced with permission from ref. 43. Copyright 2016, the Royal Society of Chemistry. | ||
The ionic conductivity in amorphous regions obeys the Vogel–Tammann–Fulcher (VTF) model: σ ∼ exp[−B/(T − T0)], where B is the apparent activation energy (Ea/k), T0 is the equilibrium glass-transition temperature (Tg − 50 °C), and T is the given temperature.46 It is clear that the ionic conductivity of polymer electrolytes could be improved by lowering the Tg value. A similar mechanism of ion transport can be applied to other linear polymers with sequential polar groups (e.g. –CO in polycarbonate, –CN in polyacrylonitrile, –NR in polyamide, etc.).47
In addition to amorphous regions, there are a certain amount of crystallized regions in polymers, where the transport of lithium ions is less dependent on the segmental motion and could be mainly described by the Arrhenius equation. Gadjourova et al.48 observed the ion transport in crystallized regions, which are motionless, ordered environments composed of folded PEO chains. A couple of PEO chains fold to form cylindrical tunnels in the crystallized regions of PEO6:LiXF6 (X = P, As and Sb). Lithium ions are located at the coordination sites (–O– in PEO), while XF6 anions are uncoordinated and dispersed among the cylindrical tunnels. Under an electric field, lithium ions prefer to transport along the cylindrical tunnels via the neighboring coordination sites rather than migrate with the segmental motion of polymer chains.49 However, it is widely recognized that the ion transport kinetics in crystallized regions is much slower than that in amorphous regions, and therefore, the contribution of crystallized regions to lithium-ion transport in polymer matrices is negligible.
(1) Decreasing crystallinity and Tg. The conductivity and Tg have long been considered to be closely linked together in polymer electrolytes, which can be explained by the VTF model mentioned above. Solid polymer electrolytes barely conduct ions near or below Tg because of the dull segment motion. The addition of inorganic fillers disrupts the arrangement of conducting segments, decreases the crystallinity, and lowers the Tg values of polymers, thus boosting active segmental motion and thereby increasing the ionic conductivity of the composite polymer electrolytes. Croce et al.50,51 dispersed TiO2 and Al2O3 in PEO–LiClO4 (10 wt%), and the basic oxygen atoms in the inorganic fillers act as cross-linking centers for PEO segments, thereby reducing the reorganization tendency of the polymer chains and promoting preferred lithium-ion transport at the boundaries of the filler particles, which significantly changed the dynamics of polymer chains and thus promoted the segmental conductivity. Ao et al.52 applied CeO2 nanoparticles as fillers in the PEO-LiN(CF3SO2)2 (LiTFSI) system; the introduction of inorganic fillers reduced the crystalline regions of the polymer matrix and provided more amorphous regions for fast ion transport, as shown in Fig. 3a.
 | ||
| Fig. 3 (a) Schematic structures of different polymer-based electrolytes: PEO-LiTFSI with CeO2 nanoparticles and filler-free PEO-LiTFSI. Reproduced with permission from ref. 52. Copyright 2021, Elsevier. (b) Schematic of ion–ion and ion–polymer interactions in different polymer-based electrolytes: PEG-LiClO4 solid polymer electrolyte (i and ii) and PEG–α-Al2O3–LiClO4 composite polymer electrolyte (iii and iv). Reproduced with permission from ref. 55. Copyright 1998, American Chemical Society. (c) Schematic model of interactions between the PEO-LiSO3CF3 composite system and the surfaces of Al2O3 ceramic with different states: (i) acidic, (ii) neutral, and (iii) basic. Reproduced with permission from ref. 58. Copyright 2001, Elsevier. (d) Schematic structures of the C2H2F2LiClO4 cluster (i) and C2H2F2Li2LaTiClO7 cluster (ii); (e) experimental and theoretical IR spectra of the PVDF–LiClO4 complex (i) and the PVDF–LiClO4–LLTO complex (ii). Reproduced with permission from ref. 61. Copyright 2020, The Polymer Society of Korea and Springer. | ||
(2) Lewis acid–base effect. However, more and more researchers found that adding inorganic fillers into polymers did not obviously lead to a decreased Tg value, but the ionic conductivity of the composite polymer electrolyte still increased significantly. With the existence of inorganic fillers, the ion transport was not governed by the dynamics of polymer main chains, as suggested by Jayathilaka et al.,53 the increased ionic mobility was largely responsible for the conductivity enhancement. Dissanayake et al.54 found that the presence of filler particles enhanced the ionic conductivity substantially, and the degree of enhancement depended on the nature of the filler surface groups. The Lewis acid–base interactions between the filler surface groups and ionic species are capable of increasing the fraction of ‘free’ lithium ions, thus improving the ionic conductivity as well as the lithium ion transference number over a wide temperature range. Wieczorek et al.55 modified the poly(ethylene glycol) (PEG)/LiClO4 polymer with α-Al2O3 to verify the Lewis acid–base theory; FT-IR revealed decreased ion–polymer interactions and a low ionic aggregate fraction in the α-Al2O3/PEG/LiClO4 composite polymer electrolyte. Acidic centers on the fillers competed with Lewis acidic Li+ ions or ether oxygen atoms in PEG (Fig. 3b), leading to increased viscosity of the polymer and dissociations of LiClO4 salts, thereby improving the ionic conductivity. Mewill et al.56 found that the Lewis acid–base interactions between ceramic fillers and anions were responsible for the increased cation dissociation and consequently enhanced ion transport, even in a single-ion-conducting polymer electrolyte in which anions were covalently bound to the polymer matrix and could not move freely.
Furthermore, the ionic conductivity and lithium-ion transference number were found to be related to the acid surface groups of fillers.57 Croce et al.58 investigated the transport behaviors of PEO–LiSO3CF3 composite electrolytes with acidic, neutral and basic ceramic fillers (Fig. 3c); in the acidic case: (1) OH groups on the acidic ceramic surfaces favor both the filler-lithium salt and filler–polymer interactions via hydrogen bonding, increasing the salt dissociation and the local fraction of the PEO amorphous phase, thus enhancing the lithium ion transference number and ionic conductivity; (2) in the neutral case: there are fewer active hydrogens on the surface of the particles, resulting in weaker interactions between OH groups with lithium salt anions and PEO segments and leading to less enhancement of the lithium-ion transference number and the ionic conductivity; (3) in the basic case: there is no macroscopic change of the ionic transport properties in the composite electrolyte with respect to the ceramic-free polymer electrolyte, due to little filler–lithium salt and filler–polymer interactions. Therefore, ceramic fillers with Lewis acidic surface groups are more positive in improving the ionic conductivity and the lithium-ion transference number. Zhao et al.59 developed an anion-immobilized solid composite electrolyte consisting of PEO, LiTFSI and Al-doped Li6.75La3Zr1.75Ta0.25O12 (Al-LLZTO); in the electrolyte, Lewis basic anions (TFSI−) were immobilized by Lewis acidic groups on the surfaces of the Al-LLZTO particles, thus facilitating the dissociation of lithium salts and enhancing the ion transport.
Some theoretical simulations were also applied to understand the interactions between inorganic fillers and polymer–lithium salt complex. Kasemagi et al.60 studied the movement of lithium ions in the PEO–Al2O3–LiBF4 system by means of molecular dynamics (MD) simulations. According to their simulation, large amounts of lithium ions were released in the polymer matrix due to the Lewis acid–base effect, which increased the ionic conductivity. P. Sivaraj et al.61 studied the mechanism of lithium-ion transport in a LixLa1−xTiO3(LLTO)-PVDF-LiClO4 composite polymer electrolyte by DFT simulation and infrared spectroscopy analysis. To reveal the interactions between the LLTO nanoparticles and the PVDF polymer matrix, two models were constructed to represent the possible exiting forms of lithium salts in composite polymer electrolytes. As shown in Fig. 3d, the polymer matrix–lithium salt system (PVDF–LiClO4) was represented by the cluster of C2H2LiClO4, while the organic–inorganic composite system (PVDF–LiClO4–LLTO) was represented by the cluster of C2H2Li2LaTiClO7. As shown in Fig. 3e, the infrared spectroscopy analysis and calculated DFT results similarly indicated that the introduction of LLTO nanofillers into a lithium salt–polymer matrix system reduced the coordination bonding between Li+ and ClO4−. Therefore, the mobility of lithium ions was enhanced in the composite polymer electrolyte.
2.2 Ion transport through inorganic fillers
Although the content of inorganic fillers in most composite polymer electrolytes is low, the inorganic phase does exist in the whole complex system, and possibly provides extra pathways for ion transport. Inorganic fillers can be classified into two categories: passive (non-ionically conductive) fillers and active (ionically conductive) ones. Since passive fillers do not contain lithium ions, they are ionically insulating and lithium ions cannot transport through them. However, active fillers, which can be used independently as inorganic ceramic electrolytes, possess high ionic conductivity, possibly providing extra ion-transport pathways in composite polymer electrolytes.Garnet-structured, perovskite-structured, NASICON-structured, sulfide-type, halide-type solid electrolytes are widely employed as active fillers, and lithium-ion transport in these crystal structures occurs either via the vacancy mechanism or the interstitial mechanism.62–66 The vacancy mechanism normally relies on Schottky defects, the configuration of cation vacancies accompanied by anion vacancies. Due to this particular structure, a large number of vacancies are available for lithium ions to migrate through active fillers.67 As for the interstitial mechanism, it is mainly related to Frenkel defects, the configuration of anion vacancies accompanied by cation interstitials, which can be divided into two categories: direct interstitial diffusion and interstitial knock-off diffusion.68 In the former case, interstitial lithium ions move directly to an adjacent interstitial site. However, in the latter case, the migration of interstitial lithium ions is indirect; interstitial atoms occupy the sites of matrix atoms, and the removed matrix atoms migrate to other adjacent interstitial sites.
Using the advanced solid-state-NMR technique (ssNMR), Zheng et al.69 revealed that the cubic Li7La3Zr2O12 (LLZO) ceramic phase was the best choice for lithium-ion transport in the PEO–LiClO4–LLZO composite polymer electrolyte, as shown in Fig. 4a. The degree of replacement of lithium ions in LiClO4, interfaces between LLZO and the PEO matrix, and the dispersive LLZO after cycling were compared according to the quantitative analyses of the ssNMR spectra. The peak intensity of LLZO increased significantly, while there was a little increment in the peak intensities of the interfaces and the polymeric phase. These results showed that the lithium ions were mainly transported through the LLZO ceramic particles, and the contribution of the polymer phase to the ionic conduction was small. However, whether the lithium ions are transported preferentially through the ceramic phase depends on serval factors. Chen et al.70 prepared a series of composite electrolytes with different contents of Li6.4La3Zr1.4Ta0.6O12 (LLZTO), which was from “ceramic-in-polymer” (CIP) to “polymer-in-ceramic” (PIC). They found that when the LLZTO content was low, the ionic conduction mainly occurred in the PEO phase; while the ion transport occurred in the ceramic phase, when the LLZTO content reached a certain threshold. By means of 7Li magic angle spinning (MAS) nuclear magnetic resonance (NMR), Li et al.71 studied ion transport in PEO–LiTFSI–Li10GeP2S12 (LGPS) composite polymer electrolytes with a gradual increase in the content of inorganic fillers. As illustrated in Fig. 4c, lithium-ion transport in the composites was composition-dependent. The ion transport mainly occurred in the polymer phase when the content of LGPS was low (i.e., 10 wt%), while a large extent of lithium-ion transport in aggregated LGPS particles was detected when the content of LGPS was high (i.e., 50 wt% and 90 wt%). In addition, the interfacial barrier formed between active fillers and the polymer matrix affects the transport pathway of lithium ions as well. He et al.72 found that the increased current density caused a reduced lithium ion flux through the LLZO particles in the PEO–LiTFSI–LLZO (5 wt%) composite, because higher current densities enhanced the ion transport barrier between LLZO and the polymer matrix, and then confined the transfer of lithium ions from the polymer phase to LLZO.
 | ||
| Fig. 4 (a) Schematic illustration of the possible ion-transport pathways in the PEO–LLZO–LiClO4 composite electrolyte; (b) quantitative analysis of the 6Li amount in LiClO4, interfaces between LLZO and the PEO matrix, and dispersive LLZO before and after cycling. Reproduced with permission from ref. 69. Copyright 2016, Wiley-VCH GmbH. (c) The change of lithium-ion transport pathways in PEO–LiTFSI–LGPS composite electrolytes with an increase in the ceramic content. Reproduced with permission from ref. 71. Copyright 2021, American Chemical Society. (d) Schematic illustration of the lithium-ion transport pathways in PEO–LLZO–LiTFSI composite electrolytes with varying concentrations of inorganic fillers (5 to 20 wt%) and the corresponding 6Li NMR spectra. Reproduced with permission from ref. 73. Copyright 2018, American Chemical Society. | ||
As active fillers have higher ionic conductivity than polymer matrices, there should be a great improvement in the ionic conductivity with an increase in the filler content, and the ionic conductivity is expected to be even close to that of the active fillers, when the content of active fillers is high enough. However, in most composite polymer electrolyte systems, the optimal content of active fillers is kept at a low level, and the ionic conductivity shows an abnormal decrease with a further increase of active fillers. Some research studies were conducted to explain such a confusing phenomenon. Zheng et al.73 utilized the solid-state-NMR technique to reveal the ionic transport pathways in the PEO–LiTFSI–LLZO composite electrolyte, where the LLZO content ranged from 5 to 50 wt%. As illustrated in Fig. 4d, PEO–LiTFSI–LLZO (5 wt%) showed a reduction in ionic conductivity, because diffusely distributed LLZO particles blocked the lithium-ion transport through the polymer matrix. The enhancement of the ionic conductivity in PEO–LiTFSI–LLZO (20 wt%) was attributed to the increased lithium-ion concentration coming from decomposed LLZO particles. PEO–LiTFSI–LLZO (50 wt%) showed notably lower ionic conductivity, as compared with other composites, which could be explained by two factors: aggregated LLZO particles almost completely blocked the ionic transport pathways through the PEO matrix and the ion transport in the aggregated LLZO particles was slow, as ceramic particles were diluted by polymers. In other words, the ionic conduction in the polymer phase and the inorganic phase got in the way of each other when the content of active fillers was high.
2.3 Ion transport through interfaces between the polymer matrix and inorganic fillers
As inorganic fillers are generally small in size and are uniformly dispersed in the polymer matrix, there are plenty of non-negligible interfacial regions in the whole composite polymer electrolyte. What is more, due to the huge difference in the physical and chemical properties between the polymer matrix and inorganic ceramic fillers, the condition of the interfacial regions is complicated.74 Surfaces of inorganic fillers are defect-rich and highly reactive, easy to interact with other components, resulting in complex interfacial regions.75–77 For composite electrolytes consisting of active inorganic fillers and the polymer matrix, there exists an additional case. Compared with the complex of the polymer matrix and lithium salts, active fillers are lithium-rich, and the state of lithium ions in the interfacial regions is greatly different from those existing in the bulk of the polymer matrix and the active fillers.78 These differences obviously affect the conduction patterns of lithium ions, and some recent research studies provide conclusive evidence to confirm this.Using the tracer-exchange Li NMR technique, Zheng et al.79 revealed that lithium ions are transported mainly through the interfacial regions in the Li10GeP2S12 (LGPS)–LiTFSI–PEO composite electrolyte, as shown in Fig. 5a and b. Similarly, Yan et al.80 discovered the conduction of lithium ions along the organic–inorganic interfaces in the composite solid electrolyte (Fig. 5c) consisting of a three-dimensional porous Li1.3Al0.3Ti1.7(PO4)3 framework (p-LATP), in situ polymerized poly(ethylene glycol) methyl ether acrylate (P(PEGMEA)), and lithium difluoro(oxalato)borate (LiDFOB). The solid-state NMR and unidirectional galvanostatic polarization (Fig. 5d) were combined to reveal the migration pathways of lithium ions. The results of 6Li resonance showed that the ionic conduction in the p-LATP/P(PEGMEA) interfacial regions, as reflected by the 6Li content, had a huge improvement in the cycled electrolyte compared with that in the pristine state, even exceeding that in the P(PEGMEA) matrix. Similarly, with the synchrotron X-ray nanotomography, Zaman et al.81 reconstructed the accessible particle surface area in PEO–LiClO4–Li7.5La3Zr2Al0.25O12 (Al-LLZO) composite polymer electrolytes with different Al-LLZO contents. Images of the reconstructed accessible particle surface areas of the composite polymer electrolytes with 5 to 50 vol% Al-LLZO contents are shown in Fig. 5e, and the accessible surface area was reduced because of the agglomeration of fillers as the inorganic content increased. After normalizing the surface area with the volume of fillers in each electrolyte, they found that the maximal normalized surface area was realized at 15 vol%, which was correlated with the highest ionic conductivity, and the normalized surface area and the ionic conductivity decreased with the increase of the inorganic content, as shown in Fig. 5f. This relationship corroborated that the interfacial regions between the polymer matrix and the inorganic fillers seriously affected ion transport.
 | ||
| Fig. 5 6Li ssNMR spectra of the PEO-LGPS-LiTFSI composite electrolyte before (a) and after (b) 6Li → 7Li tracer-exchange. Reproduced with permission from ref. 79. Copyright 2019, the American Chemical Society. (c) Schematic illustration of lithium-ion-transport pathways in the P(PEGMEA)-p-LATP-LiDFOB complex; (d) 6Li ssNMR spectra of the pristine and cycled composite polymer electrolyte. Reproduced with permission from ref. 80. Copyright 2021, Wiley-VCH GmbH. (e) Reconstructed images of composite polymer electrolytes with different Al-LLZO contents; (f) relationship between theoretically calculated, experimentally measured normalized surface areas and experimentally measured ionic conductivity. Reproduced with permission from ref. 81. Copyright 2019, the Royal Society of Chemistry. (g) Schematic illustration of possible lithium-ion-transport pathways in the PAN–LLZO–LiClO4 composite electrolyte; (h) 6Li ssNMR spectra of the filler-free PAN–LiClO4, the LLZO fillers, and the PAN–LLZO–LiClO4 composite electrolyte; (i) comparison of 6Li ssNMR spectra between the pristine and cycled composite polymer electrolytes. Reproduced with permission from ref. 84. Copyright 2017, the American Chemical Society. (j) Evolution of the normalized lithium-ion concentration from 0 to 300 ns; (k) distribution of the normalized lithium-ion concentrations from the surface to the bulk center of a Ga-LLZO nanoparticle from 0 to 300 ns (the lithium-ion concentrations are normalized to their initial bulk values); (l) schematic illustration of the fast lithium-ion-transport pathway consisting of the interlinked space-charge regions. Reproduced with permission from ref. 88. Copyright 2019, American Chemical Society. | ||
The enhanced ionic conduction by adding inorganic ceramic fillers can be attributed to building ionic pathways at the interfaces between inorganic fillers and the polymer matrix.82,83 Yang et al.84 added 5 wt% of doped or undoped LLZO nanofillers (corresponding to 2 vol%) into the PAN–LiClO4 complex to fabricate composite polymer electrolytes. The electrolytes with Al- and Ta-doped active fillers were found to show ionic conductivities rather similar to that with undoped LLZO fillers, demonstrating that the ionic conductivity of the composite electrolytes was not dominated by the bulk conductivity of fillers. NMR measurements showed that the lithium-ion diffusion occurred preferentially in the modified regions at the LLZO/polymer interfaces, as shown in Fig. 5g–i. The authors speculated that the LLZO nanofillers could interact with the PAN matrix or lithium salts by acidic or basic groups on the LLZO surfaces by the Lewis acid–base interaction, to promote the dissociation of lithium salts and weaken the interactions between PAN and lithium ions, thus producing more free lithium ions on the LLZO surfaces and favoring lithium-ion transport through the interfacial regions. Yang et al.85 found that hydrogen-treated LLTO nanofillers intensified the deprotonation of CH2 groups and the dehydrofluorination of PVDF units in the PVDF-HFP matrix. These processes weakened the electron-drawing binding energy between fluorine and lithium ions, thus releasing more free lithium ions and favoring ion transport through the interfacial regions.
The space-charge effect is another reliable explanation for ion transport in the interfacial regions.86 Because of different lithium-ion concentrations and energy levels between active inorganic fillers and the polymer matrix, driven by the free energy in the interfacial regions, lithium-rich space-charge regions would spontaneously form and act as fast transport pathways for lithium ions.87 Li et al.88 developed a lithium-ion conduction model for the composite consisting of a PEO matrix and Li6.25Ga0.25La3Zr2O12 (Ga-LLZO) particles by combining the phase-field method, the random resistor model and the Monte Carlo simulation. Lithium salt was absent in the complex system, and thus the Lewis acid–base effect was avoided. According to the phase-field calculation, a space-charge region (thickness ∼3 nm) was established between a Ga-LLZO particle and the PEO matrix, as shown in Fig. 5j and k. The space-charge region provided a new kinetic pathway for ion transport. Moreover, when the content of the Ga-LLZO particles was increased beyond a percolation threshold, the space-charge regions were connected to each other to form continuous conductive pathways, or effective percolation conductive networks were constructed, as illustrated in Fig. 5l, and then the ionic conduction was greatly enhanced. However, when the Ga-LLZO content was further increased, the Ga-LLZO nanoparticles agglomerated; as a result, the continuous ionic conduction pathways were blocked, and the ionic conductivity decreased. The Monte Carlo simulation disclosed the ion transport in composites with different Ga-LLZO contents (Fig. 5i).
Since the interfacial regions between ceramic fillers and the polymer matrix have a significant contribution to the whole ionic conduction, the particle size, associated with the surface area of fillers, plays an important role in the enhancement of ion transport.89,90 Zhang et al.91 investigated the size effect of the LLZTO fillers on the ionic conduction in composite polymer electrolytes, and discovered that the percolation threshold decreased and the ionic conductivity increased with a decrease in the particle size. Therefore, smaller particles lead to an increase of the coherent interfacial conduction pathways and the consequent enhancement of the ionic conductivity in the case of the same amount of inorganic fillers. Utilizing these acquired interfaces, designing and constructing continuous ion-transport pathways can greatly improve the ionic conductivity of composite polymer electrolytes.
3 Conclusions and perspectives
In this review, the ion-transport mechanisms of the three co-existing domains in composite solid electrolytes, namely the polymer phase, the inorganic phase and the interfacial regions between the polymer matrix and the inorganic fillers, are discussed.(1) In the polymer phase, ion transport is associated with the polar segmental motion in amorphous regions. The incorporation of inorganic fillers into the polymer matrix influences ion transport in several aspects. First, the addition of inorganic fillers decreases the crystallinity and Tg of the polymer phase, which increases the conductive amorphous regions and speeds up the segmental motion. Second, lithium salts are easier to dissociate and release more free lithium ions into the polymer matrix due to the Lewis acid–base effect.
(2) In the inorganic phase, ion transport occurs via several mechanisms, including the vacancy mechanism, direct interstitial diffusion and interstitial knock-off diffusion, similar to the ion-transport mechanisms in ceramic electrolytes.
(3) Lithium-rich regions are spontaneously formed at the interfaces between the polymer matrix and the inorganic fillers, due to strong interactions between two vastly different phases. The lithium-rich interfacial regions are fast transport pathways for lithium ions, in which the space-charge effect and the Lewis acid–base effect are critical.
It is worth noting that the dominating ion transport pathway depends on the content of inorganic fillers. That is, when the filler content is low, ionic conduction mainly occurs in the polymer matrix; when the filler content increases to the percolation-onset threshold, effective percolation networks are constructed by highly conductive interfacial regions, and lithium ions are transported mainly through the interfaces; when the filler content increases to exceed the percolation–disruption threshold, a lot of filler particles agglomerate and continuous percolation networks are gradually blocked, and then lithium ions are transported mainly through the active fillers with high ionic conductivity.
The evolution of electrolytes from the liquid-state to the solid-state is inevitable, and composite polymer electrolytes consisting of the polymer matrix and inorganic fillers show better prospects for real applications, due to their excellent comprehensive properties among all electrolytes. However, the application of composite polymer electrolytes is still in its infancy, mostly because the properties of composite polymer electrolytes still need improvements. For this purpose, we propose several directions for future research studies as follows:
(1) Developing new materials or new structures. The ionic conductivities of composite polymer electrolytes at room temperature are generally lower than those of liquid electrolytes; new materials or structures with high ionic conductivities should be explored. The movement of lithium ions in polymers is mainly related to the creeping of polar chain segments above the Tg values; polymer structural engineering, including copolymerizing, cross-linking, grafting, etc., can suppress the crystallization of polymers and enhance the mobility of the chain segments to improve the ionic conductivity. In addition, novel polymers with continuous polar segments and good mobility of chain segments at room temperature contribute to high ionic conductivity as well.
(2) Optimizing organic and inorganic components in composite solid electrolytes. The properties, dimensions, and content of inorganic fillers can greatly affect the ionic transport behavior in composite polymer electrolytes. On one hand, inorganic fillers can improve the mobility of polymer segments, which is beneficial for the migration of lithium ions that are associated with the creeping of the polymer segments. On the other hand, the lithium-rich regions formed between the inorganic fillers and the polymer matrix are fast transport channels for lithium ions. By controlling factors such as the proportion, size, and dispersion of inorganic fillers, efficient interfacial conductive percolation networks can be constructed to greatly enhance ionic conduction. Since active inorganic fillers possess high ionic conductivity, three-dimensional structures of active inorganic fillers provide “highways” for ionic conduction and facilitate the formation of interfacial conductive percolation networks. All in all, by compounding two compounds to construct uniformly dispersed and continuous conductive networks, composite polymer electrolytes with excellent comprehensive performance can be developed.
(3) Developing and utilizing advanced characterization techniques and numerical simulations to disclose the mechanisms of lithium-ion transport. Some ionic conduction theories have been established based on imperfect computational methods and characterization techniques, which are difficult to adapt to current complex polymer electrolyte systems, especially for the complicated interfacial regions, namely, inorganic fillers/polymers, polymers/polymers, and inorganic fillers/inorganic fillers in various composite polymer electrolytes. Nowadays, computational techniques and in situ/operando characterization techniques have made great progress, which makes it possible to build large and elaborate theoretical models and observe in situ the behavior of components in composite polymer electrolytes. Clarifying and unifying the ion-transport mechanism in composite polymer electrolytes by combining and utilizing these techniques wisely are significant for the development of composite polymer electrolytes with excellent performances.
Author contributions
The manuscript was written through the contributions from all authors. All authors have given approval to the final version of the manuscript. Jialong Fu, Zhuo Li and Xiaoyan Zhou contributed equally to this paper.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors would like to express their appreciation to the National Natural Science Foundation of China and the Israeli Science Foundation for funding this research within the framework of the joint NSFC-ISF grant # 51961145302. This work is also supported by the China Postdoctoral Science Foundation funded project (Grant # 2020M682403).References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- Y. Zheng, J. Wang, B. Yu, W. Zhang, J. Chen, J. Qiao and J. Zhang, Chem. Soc. Rev., 2017, 46, 1427–1463 RSC.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- Y. Tian, G. Zeng, A. Rutt, T. Shi, H. Kim, J. Wang, J. Koettgen, Y. Sun, B. Ouyang, T. Chen, Z. Lun, Z. Rong, K. Persson and G. Ceder, Chem. Rev., 2021, 121, 1623–1669 CrossRef CAS PubMed.
- K. Liu, Y. Liu, D. Lin, A. Pei and Y. Cui, Sci. Adv., 2018, 4, eaas9820 CrossRef PubMed.
- J. Kalhoff, G. G. Eshetu, D. Bresser and S. Passerini, ChemSusChem, 2015, 8, 2154–2175 CrossRef CAS PubMed.
- W. Li, B. Song and A. Manthiram, Chem. Soc. Rev., 2017, 46, 3006–3059 RSC.
- W. He, W. Guo, H. Wu, L. Lin, Q. Liu, X. Han, Q. Xie, P. Liu, H. Zheng, L. Wang, X. Yu and D.-L. Peng, Adv. Mater., 2021, 33, 2005937 CrossRef CAS PubMed.
- Q. Zhao, X. Liu, S. Stalin, K. Khan and L. A. Archer, Nat. Energy, 2019, 4, 365–373 CrossRef CAS.
- Q. Wang, J.-F. Wu, Z. Lu, F. Ciucci, W. K. Pang and X. Guo, Adv. Funct. Mater., 2019, 29, 1904232 CrossRef.
- Z. Li, H.-X. Xie, X.-Y. Zhang and X. Guo, J. Mater. Chem. A, 2020, 8, 3892–3900 RSC.
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS.
- W. J. Kwon, H. Kim, K.-N. Jung, W. Cho, S. H. Kim, J.-W. Lee and M.-S. Park, J. Mater. Chem. A, 2017, 5, 6257–6262 RSC.
- K. Nie, S. Wu, J. Wang, X. Sun, Z. Yan, J. Qiu, Q. Yang, R. Xiao, X. Yu, H. Li, L. Chen and X. Huang, ACS Appl. Mater. Interfaces, 2021, 13, 38384–38393 CrossRef CAS PubMed.
- J.-F. Wu, B.-W. Pu, D. Wang, S.-Q. Shi, N. Zhao, X.-X. Guo and X. Guo, ACS Appl. Mater. Interfaces, 2019, 11, 898–905 CrossRef CAS.
- R. DeWees and H. Wang, ChemSusChem, 2019, 12, 3713–3725 CrossRef CAS PubMed.
- J. C. Bachman, S. Muy, A. Grimaud, H.-H. Chang, N. Pour, S. F. Lux, O. Paschos, F. Maglia, S. Lupart, P. Lamp, L. Giordano and Y. Shao-Horn, Chem. Rev., 2016, 116, 140–162 CrossRef CAS PubMed.
- K. Shi, Z. Wan, L. Yang, Y. Zhang, Y. Huang, S. Su, H. Xia, K. Jiang, L. Shen, Y. Hu, S. Zhang, J. Yu, F. Ren, Y.-B. He and F. Kang, Angew. Chem., Int. Ed., 2020, 59, 11784–11788 CrossRef CAS PubMed.
- J. van den Broek, S. Afyon and J. L. M. Rupp, Adv. Energy Mater., 2016, 6, 1600736 CrossRef.
- L. Yue, J. Ma, J. Zhang, J. Zhao, S. Dong, Z. Liu, G. Cui and L. Chen, Energy Storage Mater., 2016, 5, 139–164 CrossRef.
- X. Wang, R. Kerr, F. Chen, N. Goujon, J. M. Pringle, D. Mecerreyes, M. Forsyth and P. C. Howlett, Adv. Mater., 2020, 32, 1905219 CrossRef CAS PubMed.
- Y. Guo, X. Qu, Z. Hu, J. Zhu, W. Niu and X. Liu, J. Mater. Chem. A, 2021, 9, 13597–13607 RSC.
- C. Sun, Z. Wang, L. Yin, S. Xu, Z. A. Ghazi, Y. Shi, B. An, Z. Sun, H.-M. Cheng and F. Li, Nano Energy, 2020, 75, 104976 CrossRef CAS.
- J. Zhang, J. Zhao, L. Yue, Q. Wang, J. Chai, Z. Liu, X. Zhou, H. Li, Y. Guo, G. Cui and L. Chen, Adv. Energy Mater., 2015, 5, 1501082 CrossRef.
- T. Dong, J. Zhang, G. Xu, J. Chai, H. Du, L. Wang, H. Wen, X. Zang, A. Du, Q. Jia, X. Zhou and G. Cui, Energ. Environ. Sci., 2018, 11, 1197–1203 RSC.
- D. Zhou, D. Shanmukaraj, A. Tkacheva, M. Armand and G. Wang, Chem, 2019, 5, 2326–2352 CAS.
- L. Song, Y. Chen and J. W. Evans, J. Electrochem. Soc., 1997, 144, 3797–3800 CrossRef CAS.
- J. Zhang, X. Zang, H. Wen, T. Dong, J. Chai, Y. Li, B. Chen, J. Zhao, S. Dong, J. Ma, L. Yue, Z. Liu, X. Guo, G. Cui and L. Chen, J. Mater. Chem. A, 2017, 5, 4940–4948 RSC.
- Z. Lv, Q. Zhou, S. Zhang, S. Dong, Q. Wang, L. Huang, K. Chen and G. Cui, Energy Storage Mater., 2021, 37, 215–223 CrossRef.
- H. He, Y. Chai, X. Zhang, P. Shi, J. Fan, Q. Xu and Y. Min, J. Mater. Chem. A, 2021, 9, 9214–9227 RSC.
- R. Xu, Y. Xiao, R. Zhang, X.-B. Cheng, C.-Z. Zhao, X.-Q. Zhang, C. Yan, Q. Zhang and J.-Q. Huang, Adv. Mater., 2019, 31, 1808392 CrossRef PubMed.
- Z. Sun, Y. Li, S. Zhang, L. Shi, H. Wu, H. Bu and S. Ding, J. Mater. Chem. A, 2019, 7, 11069–11076 RSC.
- Z. Shen, Y. Cheng, S. Sun, X. Ke, L. Liu and Z. Shi, Carbon Energy, 2021, 3, 482–508 CrossRef CAS.
- F. Lv, Z. Wang, L. Shi, J. Zhu, K. Edström, J. Mindemark and S. Yuan, J. Power Sources, 2019, 441, 227175 CrossRef CAS.
- D. E. Fenton, J. M. Parker and P. V. Wright, Polymer, 1973, 14, 589 CrossRef CAS.
- C. Berthier, W. Gorecki, M. Minier, M. B. Armand, J. M. Chabagno and P. Rigaud, Solid State Ionics, 1983, 11, 91–95 CrossRef CAS.
- H.-S. Min, J.-M. Ko and D.-W. Kim, J. Power Sources, 2003, 119–121, 469–472 CrossRef CAS.
- J. Chai, Z. Liu, J. Ma, J. Wang, X. Liu, H. Liu, J. Zhang, G. Cui and L. Chen, Adv. Sci., 2017, 4, 1600377 CrossRef PubMed.
- Y. Wang, S. Chen, Z. Li, C. Peng, Y. Li and W. Feng, Energy Storage Mater., 2022, 45, 474–483 CrossRef.
- M. Jia, P. Wen, Z. Wang, Y. Zhao, Y. Liu, J. Lin, M. Chen and X. Lin, Adv. Funct. Mater., 2021, 31, 2101736 CrossRef CAS.
- Z. Xue, D. He and X. Xie, J. Mater. Chem. A, 2015, 3, 19218–19253 RSC.
- S. K. Fullerton-Shirey and J. K. Maranas, Macromolecules, 2009, 42, 2142–2156 CrossRef CAS.
- R. Chen, W. Qu, X. Guo, L. Li and F. Wu, Mater. Horiz., 2016, 3, 487–516 RSC.
- Y. Zheng, Y. Yao, J. Ou, M. Li, D. Luo, H. Dou, Z. Li, K. Amine, A. Yu and Z. Chen, Chem. Soc. Rev., 2020, 49, 8790–8839 RSC.
- D. Bresser, S. Lyonnard, C. Iojoiu, L. Picard and S. Passerini, Mol. Syst. Des. Eng., 2019, 4, 779–792 RSC.
- D. M. Pesko, Y. Jung, A. L. Hasan, M. A. Webb, G. W. Coates, T. F. Miller and N. P. Balsara, Solid State Ionics, 2016, 289, 118–124 CrossRef CAS.
- J. Mindemark, M. J. Lacey, T. Bowden and D. Brandell, Prog. Polym. Sci., 2018, 81, 114–143 CrossRef CAS.
- Z. Gadjourova, Y. G. Andreev, D. P. Tunstall and P. G. Bruce, Nature, 2001, 412, 520–523 CrossRef CAS PubMed.
- Z. Stoeva, I. Martin-Litas, E. Staunton, Y. G. Andreev and P. G. Bruce, J. Am. Chem. Soc., 2003, 125, 4619–4626 CrossRef CAS PubMed.
- F. Croce, R. Curini, A. Martinelli, L. Persi, F. Ronci, B. Scrosati and R. Caminiti, J. Phys. Chem. B, 1999, 103, 10632–10638 CrossRef CAS.
- F. Croce, G. B. Appetecchi, L. Persi and B. Scrosati, Nature, 1998, 394, 456–458 CrossRef CAS.
- X. Ao, X. Wang, J. Tan, S. Zhang, C. Su, L. Dong, M. Tang, Z. Wang, B. Tian and H. Wang, Nano Energy, 2021, 79, 105475 CrossRef CAS.
- P. A. R. D. Jayathilaka, M. A. K. L. Dissanayake, I. Albinsson and B. E. Mellander, Electrochimi. Acta, 2002, 47, 3257–3268 CrossRef CAS.
- M. A. K. Lakshman Dissanayake, Ionics, 2004, 10, 221–225 CrossRef.
- W. Wieczorek, P. Lipka, G. Żukowska and H. Wyciślik, J. Phys. Chem. B, 1998, 102, 6968–6974 CrossRef CAS.
- L. C. Merrill, X. C. Chen, Y. Zhang, H. O. Ford, K. Lou, Y. Zhang, G. Yang, Y. Wang, Y. Wang, J. L. Schaefer and N. J. Dudney, ACS Appl. Energy Mater., 2020, 3, 8871–8881 CrossRef CAS.
- M. Soweizy, M. Zahedifar and M. Karimi, J. Mater. Sci.: Mater. El., 2020, 31, 9614–9621 CAS.
- F. Croce, L. Persi, B. Scrosati, F. Serraino-Fiory, E. Plichta and M. A. Hendrickson, Electrochimi. Acta, 2001, 46, 2457–2461 CrossRef CAS.
- C.-Z. Zhao, X.-Q. Zhang, X.-B. Cheng, R. Zhang, R. Xu, P.-Y. Chen, H.-J. Peng, J.-Q. Huang and Q. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 11069–11074 CrossRef CAS PubMed.
- S. Mogurampelly and V. Ganesan, Macromolecules, 2015, 48, 2773–2786 CrossRef CAS.
- P. Sivaraj, K. P. Abhilash, B. Nalini, P. Perumal, K. Somasundaram and P. C. Selvin, Macromol. Res., 2020, 28, 739–750 CrossRef CAS.
- S. Qian, H. Chen, Z. Wu, D. Li, X. Liu, Y. Tang and S. Zhang, Batteries Supercaps, 2021, 4, 39–59 CrossRef CAS.
- M. Park, X. Zhang, M. Chung, G. B. Less and A. M. Sastry, J. Power Sources, 2010, 195, 7904–7929 CrossRef CAS.
- S. Ramakumar, C. Deviannapoorani, L. Dhivya, L. S. Shankar and R. Murugan, Prog. Mater. Sci., 2017, 88, 325–411 CrossRef CAS.
- Y. Liu, B. Xu, W. Zhang, L. Li, Y. Lin and C. Nan, Small, 2020, 16, 1902813 CrossRef CAS PubMed.
- T. Famprikis, P. Canepa, J. A. Dawson, M. S. Islam and C. Masquelier, Nat. Mater., 2019, 18, 1278–1291 CrossRef CAS PubMed.
- Z. Gao, H. Sun, L. Fu, F. Ye, Y. Zhang, W. Luo and Y. Huang, Adv. Mater., 2018, 30, 1705702 CrossRef.
- V. Thangadurai, S. Narayanan and D. Pinzaru, Chem. Soc. Rev., 2014, 43, 4714–4727 RSC.
- J. Zheng, M. Tang and Y.-Y. Hu, Angew. Chem., Int. Ed., 2016, 55, 12538–12542 CrossRef CAS PubMed.
- L. Chen, Y. Li, S.-P. Li, L.-Z. Fan, C.-W. Nan and J. B. Goodenough, Nano Energy, 2018, 46, 176–184 CrossRef CAS.
- M. Li, M. Kolek, J. E. Frerichs, W. Sun, X. Hou, M. R. Hansen, M. Winter and P. Bieker, ACS Sustainable Chem. Eng., 2021, 9, 11314–11322 CrossRef CAS.
- J. He, H. Chen, D. Wang, Q. Zhang, G. Zhong and Z. Peng, J. Phys. Chem. Lett., 2022, 13, 1500–1505 CrossRef CAS PubMed.
- J. Zheng and Y.-Y. Hu, ACS Appl. Mater. Interfaces, 2018, 10, 4113–4120 CrossRef CAS PubMed.
- R. Usiskin and J. Maier, Adv. Energy Mater., 2021, 11, 2001455 CrossRef CAS.
- Z. Zou, Y. Li, Z. Lu, D. Wang, Y. Cui, B. Guo, Y. Li, X. Liang, J. Feng, H. Li, C.-W. Nan, M. Armand, L. Chen, K. Xu and S. Shi, Chem. Rev., 2020, 120, 4169–4221 CrossRef CAS PubMed.
- Y. Tominaga, S. Igawa, S. Asai and M. Sumita, Electrochim. Acta, 2005, 50, 3949–3954 CrossRef CAS.
- P. Ma, Y. Fang, X. Zhou, Y. Shi, H. Y. Yang and Y. Lin, J. Phys. Chem. Lett., 2021, 12, 642–649 CrossRef CAS PubMed.
- Y. Huang, X. Mei and Y. Guo, J. Appl. Polym. Sci., 2022, 52143 CrossRef CAS.
- J. Zheng, P. Wang, H. Liu and Y.-Y. Hu, ACS Appl. Energy Mater., 2019, 2, 1452–1459 CrossRef CAS.
- Y. Yan, J. Ju, S. Dong, Y. Wang, L. Huang, L. Cui, F. Jiang, Q. Wang, Y. Zhang and G. Cui, Adv. Sci., 2021, 8, 2003887 CrossRef CAS.
- W. Zaman, N. Hortance, M. B. Dixit, V. De Andrade and K. B. Hatzell, J. Mater. Chem. A, 2019, 7, 23914–23921 RSC.
- N. Wu, P.-H. Chien, Y. Qian, Y. Li, H. Xu, N. S. Grundish, B. Xu, H. Jin, Y.-Y. Hu, G. Yu and J. B. Goodenough, Angew. Chem., Int. Ed., 2020, 59, 4131–4137 CrossRef CAS PubMed.
- N. Wu, P.-H. Chien, Y. Li, A. Dolocan, H. Xu, B. Xu, N. S. Grundish, H. Jin, Y.-Y. Hu and J. B. Goodenough, J. Am. Chem. Soc., 2020, 142, 2497–2505 CrossRef CAS PubMed.
- T. Yang, J. Zheng, Q. Cheng, Y.-Y. Hu and C. K. Chan, ACS Appl. Mater. Interfaces, 2017, 9, 21773–21780 CrossRef CAS PubMed.
- H. Yang, M. Abdullah, J. Bright, W. Hu, K. Kittilstved, Y. Xu, C. Wang, X. Zhang and N. Wu, J. Power Sources, 2021, 495, 229796 CrossRef CAS.
- J. Maier, Phys. Status Solidi B, 1984, 123, K89–K91 CrossRef CAS.
- D. Brogioli, F. Langer, R. Kun and F. La Mantia, ACS Appl. Mater. Interfaces, 2019, 11, 11999–12007 CrossRef CAS.
- Z. Li, H.-M. Huang, J.-K. Zhu, J.-F. Wu, H. Yang, L. Wei and X. Guo, ACS Appl. Mater. Interfaces, 2019, 11, 784–791 CrossRef CAS PubMed.
- F. Wang, P. Zhang, G. Wang, A. S. Nia, M. Yu and X. Feng, Small Sci., 2022, 2, 2100080 CrossRef.
- H. Gao, N. S. Grundish, Y. Zhao, A. Zhou and J. B. Goodenough, Energy Mater. Adv., 2021, 2021, 1932952 Search PubMed.
- J. Zhang, N. Zhao, M. Zhang, Y. Li, P. K. Chu, X. Guo, Z. Di, X. Wang and H. Li, Nano Energy, 2016, 28, 447–454 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |