
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface-enhanced luminescence of Cr3+-doped ZnAl2O4 and MgAl2O4 using Ag@SiO2 and Au@SiO2 core–shell nanoparticles†
Rodrigo A.
Valenzuela-Fernández
 *a,
Julien
Cardin
*b,
Xavier
Portier
b,
Christophe
Labbé
b,
Camilo
Segura
c,
Víctor
Vargas
a,
Antonio
Galdámez
a and
Igor O.
Osorio-Román
d
*a,
Julien
Cardin
*b,
Xavier
Portier
b,
Christophe
Labbé
b,
Camilo
Segura
c,
Víctor
Vargas
a,
Antonio
Galdámez
a and
Igor O.
Osorio-Román
d
aDepartamento de Química, Facultad de Ciencias, Universidad de Chile, P.O. Box 653, Chile. E-mail: rvalenzuelafer@uchile.cl
bCIMAP, CEA, CNRS, UMR6252, Normandie Université, ENSICAEN UNICAEN, 14050, Caen Cedex 4, France. E-mail: julien.cardin@ensicaen.fr
cCenter for Soft Matter Research, SMAT-C, Universidad de Santiago de Chile, Avenida Bernardo O'Higgins 3363, Santiago, Chile
dInstituto de Ciencias Químicas, Facultad de Ciencias, Universidad Austral de Chile, Isla Teja, P.O. Box 567, Valdivia, Chile
First published on 23rd May 2022
Abstract
In this study, we investigate chromium-doped spinels (CDSs) and their characteristic red photoluminescence (PL) due to the strong crystal field and octahedral coordination geometries of the Cr3+ ions. An increase in the luminescence efficiency of the CDSs caused by the interaction with plasmon excitation is studied to achieve more efficient luminescence. We successfully synthesise CDSs using the citrate sol–gel method and metallic nanoparticles (MNPs) using the classical citrate reduction method. In addition, we cover the MNPs with silica shells, which are used to modify the surface. The surface-enhanced effect exerted by the surfaces modified with Ag@SiO2 and Au@SiO2 core–shell nanoparticles on the PL of the two CDSs is studied. The silica shell of the MNPs is used as a separator between the MNPs@SiO2-modified surface and the CDS. An enhancement of the PL is found for the CDS coupled to MNPs compared with the uncoupled CDSs. The PL enhancement factors and lifetimes are also investigated. The interaction between the CDSs and MNPs is investigated, and several factors are found to influence the PL of the CDSs. The main mechanism explaining the PL enhancement is proposed. A better understanding of the surface-enhanced luminescence of the CDSs may lead to further improvements in these systems.
1. Introduction
Spinels such as ZnAl2O4 (ZAO) and MgAl2O4 (MAO) have advantageous electrical, mechanical, magnetic, optical, and catalytic properties.1–3 These oxides, doped with small amounts of Cr3+, can be found in nature as red cubic crystals4 and used as paint additives to lower the temperature of objects exposed to sunlight.5 Under controlled growth conditions, changing the synthesis method and parameters, such as temperature or dopant concentration, can lead to different sizes, morphologies, and luminescent properties of inorganic phosphors.6–8 Inorganic phosphors, such as spinels, consist of a crystalline host material doped with a small amount of rare earth elements or transition metal ions, such as Eu3+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 or Cr3+.3,6,8 Cr3+ is widely used as a dopant for the development of tunable lasers, displays, and bioimaging.10 Cr3+ has a 3d3 electronic configuration and exhibits photoluminescence (PL) emission from either the 2E or the 4T2 excited state. The emission depends on the strength of the crystal field environment surrounding the Cr3+ ion,11 which can be divided into two categories: the low-field host matrix resulting in a spin-allowed transition of 4T2 → 4A2 (green emission) or the high-field host matrix resulting in the R-lines spin-forbidden transition of 2E → 4A2 (red/pink emission).
9 or Cr3+.3,6,8 Cr3+ is widely used as a dopant for the development of tunable lasers, displays, and bioimaging.10 Cr3+ has a 3d3 electronic configuration and exhibits photoluminescence (PL) emission from either the 2E or the 4T2 excited state. The emission depends on the strength of the crystal field environment surrounding the Cr3+ ion,11 which can be divided into two categories: the low-field host matrix resulting in a spin-allowed transition of 4T2 → 4A2 (green emission) or the high-field host matrix resulting in the R-lines spin-forbidden transition of 2E → 4A2 (red/pink emission).
Many fundamental and applied studies have been conducted on the interaction of plasmon resonances generated in metallic nanostructures with luminescent centers12–16 or optical interactions. The latter can be studied by using techniques, such as surface-enhanced Raman scattering (SERS), surface-enhanced fluorescence (SEF), and metal-enhanced phosphorescence (MEP).17–22 SEF has been widely studied for organic molecules and some inorganic luminophores, such as quantum dots,23–25 whereas MEP has been studied using organic dyes19,20 and organometallic complexes at room temperature.21,22
In the previously mentioned studies luminescent compounds with excited-state lifetimes were used within the nanosecond time range. However, compounds with longer excited-state lifetimes exist, such as inorganic phosphors with variable lifetime ranges depending on their atomic composition.26 For instance, the excited-state lifetimes in some Cr3+-doped inorganic phosphors have been reported to be in the millisecond time range.27–29 To the best of our knowledge, surface-enhanced luminescence (SEL) of inorganic phosphors has not been studied. In this work, we report on the SEL of two chromium-doped spinels (CDSs) with lifetimes in the millisecond range at room temperature. We present the synthesis of two CDSs: ZnAl2O4:Cr3+ (ZAOC) and MgAl2O4:Cr3+ (MAOC), deposited on the surface of two types of core–shell metallic nanoparticles (MNPs), namely, Ag@SiO2 and Au@SiO2. Furthermore, the SEL of rare-earth-doped NPs coupled with MNPs and an inverse-opal-structured substrate show promising upconversion results.30,31 Finally, we study the effect of the core–shell MNP surfaces on the spectroscopic properties of absorption and PL by comparing with the corresponding properties of a similar surface without the core–shell MNPs.
2. Experimental
2.1. Materials
A Coors® alumina crucible (50 mL), Corning® glass 2947, Zn(NO3)2·6H2O (≥99.5% pure), Cr(NO3)3·9H2O (99%), anhydrous citric acid (≥99.5%), tetraethylorthosilicate (TEOS, 98%), KAuCl4 (98%), (3-aminopropyl)trimethoxysilane (APTMS, 97%), sodium silicate solution, poly(2-(dimethylamino)ethyl methacrylate) solution 20 wt% (PDMAEMA) and ammonium hydroxide solution (28.0–30.0 wt%) were purchased from Sigma-Aldrich (USA). Al(NO3)3·9H2O (98.5%), Mg(NO3)2·6H2O (>99%), AgNO3 (99.8%), absolute ethanol, 2-propanol, NaOH, HCl (37 wt%) and HNO3 (65 wt%) were acquired at Merck (Germany).2.2. Spinel synthesis
Spinels were synthesised by using the citrate sol–gel method described by Abdukayum et al.32 The molar ratio of citric acid to total metal ions was 1.5:1. ZAO and MAO, corresponded to spinels of zinc (Zn) and magnesium (Mg), respectively, with aluminum (Al) and a molar ratio of A:Al (A = Zn or Mg) of 1:2. ZAOC and MAOC corresponded Zn:Al:Cr and Mg:Al:Cr spinels, respectively, and both spinels had a molar ratio of 2:4:1.
Scheme 1 shows the synthetic steps for all the materials. The synthetic process for obtaining the spinels involved mixing citric acid and the metal ions in Milli-Q water at pH 5, and the pH was adjusted by adding an ammonia hydroxide solution. The mixture was then stirred vigorously for 2 h at room temperature. The mixture was heated at 70 °C to form a gel. Once the gel was formed, the product was dried at 130 °C for 3 h and heated again at 200 °C for 7 h. After this process, a powder was obtained, milled in an Agata mortar, and annealed in air in an alumina crucible at 1000 °C for 3 h. The final material was a white powder for both ZAO and MAO and a slightly pink powder for both ZAOC and MAOC.
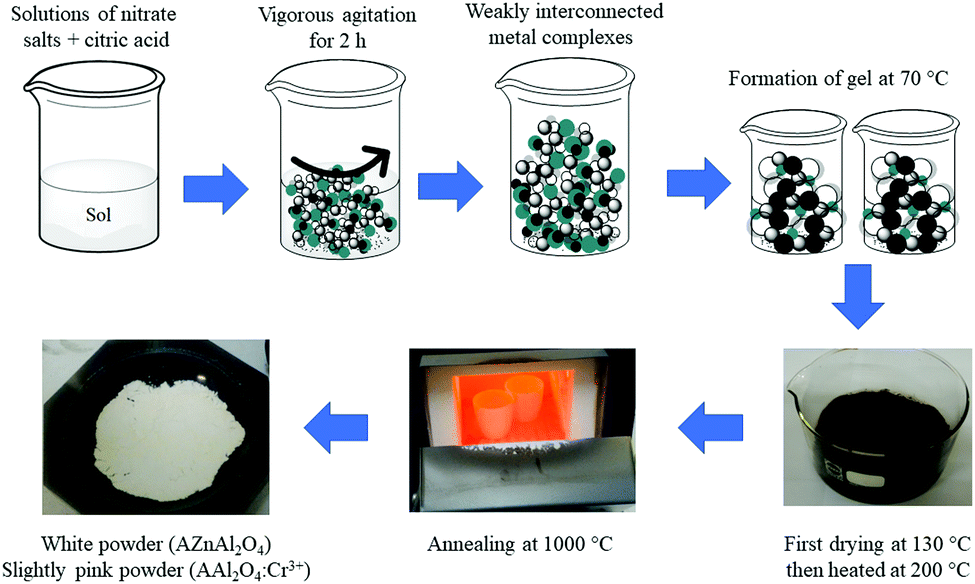 | ||
| Scheme 1 Representation of the citrate sol-gel synthesis. (A = Zn or Mg). | ||
2.3. Synthesis of Ag@SiO2 and Au@SiO2 core–shell NPs
Ag@SiO2 and Au@SiO2 core–shell MNPs were synthesized using the method described by Segura et al.33 with a few modifications. The synthesis of AgNPs (silver NPs) was based on the protocols described by Lee–Meisel.34 AgNO3 (18 mg) was dissolved in 100 mL of Milli-Q water, the solution was boiled; and 2 mL of trisodium citrate (38.8 mM) was added. This solution was boiled for 1 h until the colour of the solution changed from colourless to silver-grey, and then, it was cooled to room temperature. The final product was a colloidal suspension of AgNPs, which were centrifuged for 10 min at 1000 rpm. The supernatant was then collected for further analysis.For the AgNP coating, 45 mL of the colloidal supernatant was mixed with 200 mL of ethanol in an amber bottle. Then, 1.5 mL of 30% (w/v) ammonia and 300 μL of TEOS (40 mM) were added. The solution was agitated for 5 min and then allowed to settle for 60 h. After 60 h, the solution was centrifuged for 45 min at 5000 rpm, and the precipitated material (Ag@SiO2) was collected and dispersed in Milli-Q water.
AuNPs (gold NPs) were obtained by dissolving 20 mg of KAuCl4 in 100 mL of Milli-Q water. The solution was brought to a boil; and 5 mL of trisodium citrate (38.8 mM) was added. This solution was boiled for 1 h until the colour of the solution changed from colourless to wine red, and then, it was cooled to room temperature. The final product was a colloidal suspension of AuNPs, which were centrifuged for 10 min at 1000 rpm. The supernatant was then collected for further analysis.
For the AuNP coating, 50 mL of the colloidal solution of AuNPs was transferred to a 250 mL Erlenmeyer flask and 4 mL of APTMS (1 mM) was added. The solution was stirred for 30 min at room temperature. Then, the temperature was raised to 90 °C, and 6 mL of sodium silicate 0.54% (w/v) was added under constant stirring and left for 4 h. The colloidal solution was then cooled down to room temperature and centrifuged for 45 min at 5000 rpm. The precipitate material (Au@SiO2) was collected and dispersed in Milli-Q water.
2.4. Surface modifications
Ag@SiO2 and Au@SiO2 were deposited on a Corning® Glass 2947. First, the glass slides were cleaned using aqua regia for 3 h, rinsed with deionised water, and submerged in a 1 M NaOH solution for 30 min. Then, the glass slides (Scheme 2(A)) were rinsed with deionised water and dried. The dry glass was submerged in a 0.5 vol%. Solution of PDMAEMA for 15 min and then rinsed with deionised water. The active glass slides were dried and stored at room temperature. This process activates the glass surface with positive charges, which allows deposition of the Ag@SiO2 NPs that are negatively charged by electrostatic attraction on the glass slides. The glass slides with PDMAEMA were submerged in an Ag–colloidal solution for 12 h. The glass slides were washed, dried, and stored in a closed container in a moisture-free atmosphere. Scheme 2(B) shows a glass slide of an Ag@SiO2 NPs core–shell modified surface (AgMS).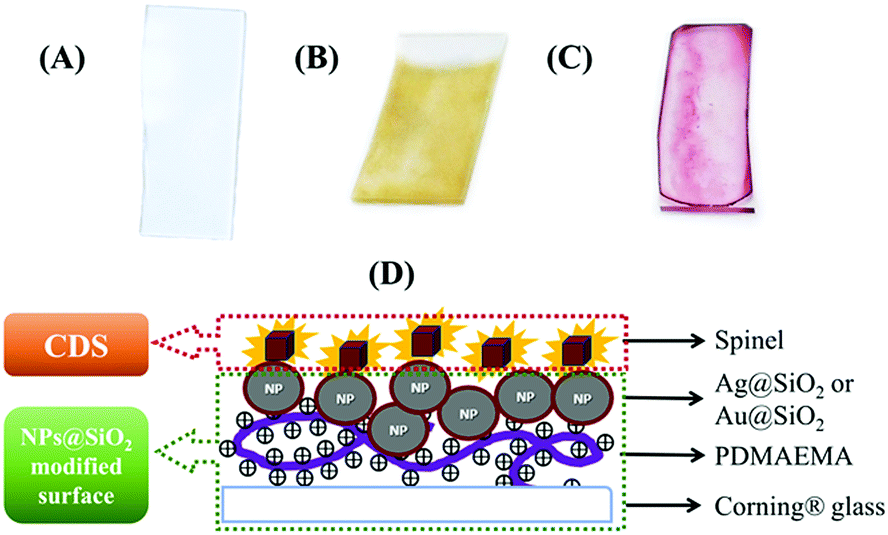 | ||
| Scheme 2 Surface modification schemes. (A) Cleaned Corning® glass; (B) Ag@SiO2; (C) Au@SiO2; (D) Scheme describing the ensemble formed with deposited spinels and MNPs-modified surfaces. | ||
For the Au@SiO2 deposition, we used dried PDMAEMA-functionalised glass slides and dropped 30 μL of the Au@SiO2 colloidal solution. The surfaces were then dried in a closed container at room temperature and stored for later use. Scheme 2(C) shows a glass slide of an Au@SiO2 NPs core–shell modified surface (AuMS).
2.5. Samples preparation for photoluminescence studies
For the samples used in the PL experiments, and the photoluminescence excitation (PLE) and time-resolved PL (TRPL) measurements, we prepared a 0.2% (w/v) dispersion of the spinels in 2-propanol. These suspensions were sonicated for 30 min and then allowed to rest for 10 min to allow the larger particles to settle. For the samples devoted to the PL and PLE measurements, we took an aliquot of the supernatant with a Pasteur pipette and dropped it onto the PDMAEMA-functionalised glass slides. For lifetime measurements, a 30 μL aliquot of the supernatant was added to the PDMAEMA-functionalised and MNP-functionalised glass slides by spin coating them at 1000 rpm for 30 s. Scheme 2(D) shows a schematic representation of the spinels deposited on the MNP-MS; henceforth, we refer to them as ensembles.2.6. Characterisation
Scanning electron microscopy-energy-dispersive X-ray spectroscopy (SEM-EDX) was performed using a Bruker Tescan Vega 3 scanning electron microscope equipped with a Quantax 400 EDS spectrometer. Cylindrical pellets were prepared by uniaxially pressing polycrystalline powder samples at approximately 5 × 108 Pa. They were coated with an Au–Pd sputtering system. The samples were mounted on double-sided carbon tape and adhered to an Al specimen holder for analysis.Powder X-ray diffraction (PXRD) patterns were collected at room temperature in the 2θ range of 5°–80° using a Bruker D8 ADVANCE powder diffractometer (Bruker, Billerica, MA, USA) with CuKα radiation (λ = 1.54178 Å). The PXRD patterns were indexed using the CHEKCELL computer program.
Conventional transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) were performed on the spinels using a double-corrected cold FEG ARM 200F microscope operated at 200 kV. TEM analyses of the Ag@SiO2 and Au@SiO2 core–shell MNPs were performed using a Hitachi HT7700 system operating at 120 kV (Hitachi, Tokyo, Japan). In both cases, the powder was mixed with n-butanol, and a drop of the resulting suspension was deposited on a copper grid (300 mesh) with a carbon film containing holes, and the grid was dried before loading it on the TEM specimen holder. The digitalised images were processed using DigitalMicrograph® software (GATAN).
Atomic force microscopy (AFM) was performed using a WiTec Alpha 3000 with a 75 kHz tip in tapping mode. AFM micrographs were taken, discarding edges and less homogeneous areas.
Continuous-wave (CW) PL measurements were performed at room temperature by using an Innova 90C argon laser (excitation wavelength 514 nm) at 100 mW, an incident angle of 45°, and a beam spot size of approximately 1 mm2 and chopped at 3 Hz. The emitted photons were collected into a light cone of 28° through a set of lenses and dispersed using a Horiba Jobin–Yvon Triax 180 monochromator. The detection was ensured by an R5108 Hamamatsu photomultiplier tube connected to an SR830 lock-in amplifier referenced at the excitation light-chopper frequency. SEL measurements were performed at room temperature with an ISS-PC1 photon counting spectrofluorimeter using an optical arrangement in which the emission was observed in the same plane as the excitation (front face). The excitation source was a violet laser diode emitting at 405 nm with an output power of 20 mW. A 570 nm longpass filter was used.
For the extinction spectra of the colloidal solutions, core–shell solutions, and core–shell MNPs deposited on glass, ultraviolet-visible studies were carried out using a double-beam Shimadzu UV-1800 spectrophotometer in specular transmission mode.
Lifetime measurements were performed at room temperature using a Chronos FD with an emission detector Model R928 PMT by Hamamatsu. The excitation light source was a 405 nm LED with a 530 nm long-pass filter. The emission detector wavelength ranged from 240 to 900 nm.
3. Results and discussion
3.1. SEM-EDS, X-ray diffraction and HRTEM analyses
SEM was used to analyse the powder samples. The SEM and backscattered electron (BSE) images are shown in the ESI,† (Fig. S1). Fig. S1 (ESI†) shows a homogeneous growth from a morphological point of view in the compounds and shows no apparent difference between one grain and another. Fig. 1 shows a typical SEM-EDX image of the ZAOC sample and reveals that the samples are homogeneous throughout the scanned area (217 × 212 μm). In addition, the elemental mapping images reveal that all the constituent atoms are uniformly distributed throughout the material.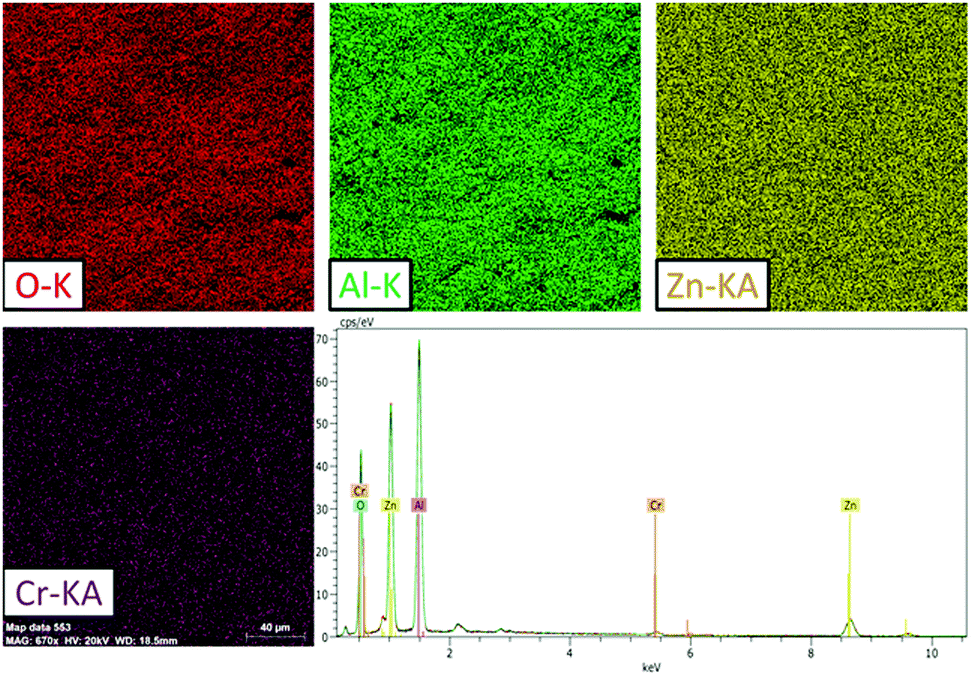 | ||
| Fig. 1 ZAOC SEM results: O, Al, Zn, and Cr EDS chemical mapping (20 kV) and typical EDS spectrum of the area shown by the chemical maps. | ||
The PXRD patterns are presented in Fig. 2. The PXRD patterns are fully consistent with those of the Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group, and the cell parameters are summarised in Table 1. All compounds crystallised in the well-known spinel-type structure according to the standard Joint Committee on Powder Diffraction (JCPDS) cards No. 01-070-8182 (ZAO and ZAOC) and No. 04-008-3488 (MAO and MAOC). Fig. 2 shows the reaction products were of the nominal composition of AAl2O4:Cr3+ (A = Zn, Mg). Additionally, there were only single phases within the detection limits, and secondary phases or impurity peaks were not detected. A comparison of the Cr-doped and undoped samples confirms that Cr3+ doping does not change the crystalline structure and that there is no apparent distortion caused by Cr3+.
m space group, and the cell parameters are summarised in Table 1. All compounds crystallised in the well-known spinel-type structure according to the standard Joint Committee on Powder Diffraction (JCPDS) cards No. 01-070-8182 (ZAO and ZAOC) and No. 04-008-3488 (MAO and MAOC). Fig. 2 shows the reaction products were of the nominal composition of AAl2O4:Cr3+ (A = Zn, Mg). Additionally, there were only single phases within the detection limits, and secondary phases or impurity peaks were not detected. A comparison of the Cr-doped and undoped samples confirms that Cr3+ doping does not change the crystalline structure and that there is no apparent distortion caused by Cr3+.
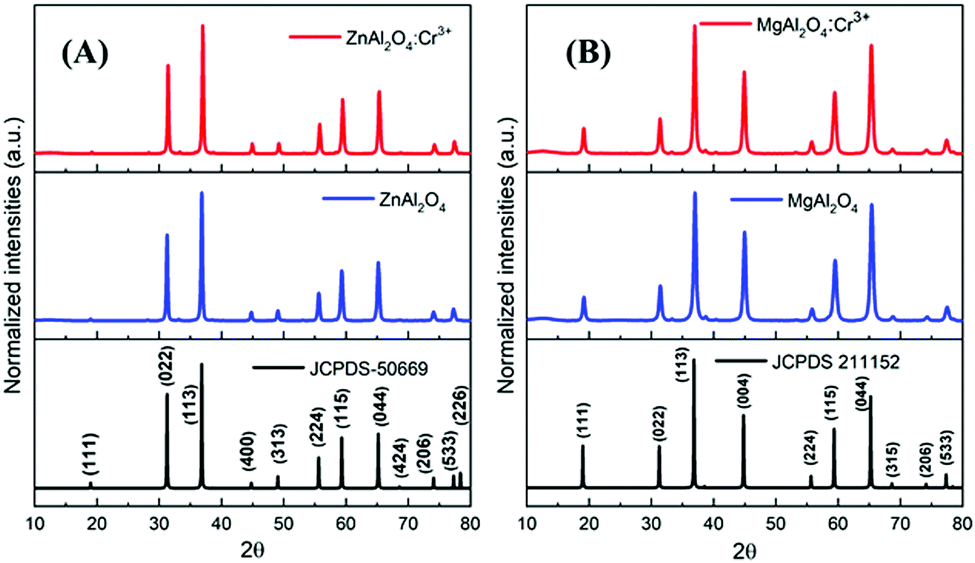 | ||
| Fig. 2 PXRD patterns of Cr-doped and undoped ZnAl2O4 (A) and MgAl2O4 (B). The bottom diagrams are the reference XRD data of the corresponding spinels. | ||
The crystallite sizes (t) were calculated using Scherrer's equation35
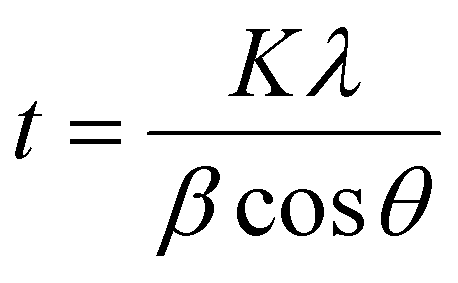 | (1) |
TEM and HRTEM images and the corresponding fast Fourier transform (FFT) patterns are displayed in Fig. 3 for all the samples. We observed that the ZAO and ZAOC spinels have larger grains (average 64.5 nm and 52.5 nm, respectively) in comparison to the MAO and MAOC spinels (average 17.2 nm and 16.0 nm, respectively). The ZAO and ZAOC crystallite sizes found from the TEM images differed from those estimated using the Scherrer equation (Table 1). The difference may stem from PXRD data that characterise a large volume of material being used for the Scherrer equation, and the TEM technique only considers the volume of a few grains (the grain size values were obtained by averaging 40 grains). A more probable explanation for the difference is the presence of stacking faults in the ZAO and ZAOC grains, leading to smaller sizes from PXRD than those observed using TEM. The grain sizes for MAO and MAOC (17.2 nm and 16.0 nm, respectively) obtained by using TEM were consistent with the crystallite sizes determined by using the Scherrer equation. The single-grain micrographs (Fig. 3(ii) and (iii)) show a crystalline ordering symmetry that is consistent with the spinel crystal structure. All the FFTs of the single-grain micrographs are in agreement with cubic symmetry (fcc to be more precise).36Fig. 4 shows the TEM images of single-crystal MNPs and core–shell MNP colloidal solutions. The AgNPs (Fig. 4(A)) had diameters of approximately 50 nm and the Ag@SiO2 (Fig. 4(B)) had diameters of about 100 nm, indicating that the silica shell thickness was approximately 25 nm. Alternatively, the diameters of the AuNPs (Fig. 4(C)) were less than 20 nm and the Au@SiO2 (Fig. 4(D)) had diameters of approximately 40 nm, indicating that the silica shell thickness was approximately 10 nm.
 | ||
Fig. 3 TEM and HRTEM images and the corresponding FFT for (A) ZAO; (B) ZAOC; (C) MAO; (D) MAOC. In each figure, (i) shows crystals; (ii) image of a crystal; (iii) shows a magnification of (ii), and (iv) is a FFT of (iii) (showing the following crystallographic projections [0![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1], [1 1], [1![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ], [110], [110], for ZAO, ZAOC, MAO and MAOC, respectively). ], [110], [110], for ZAO, ZAOC, MAO and MAOC, respectively). | ||
 | ||
| Fig. 4 Bright field TEM images of MNPs. (A) AgNPs; (B) Ag@SiO2; (C) AuNPs; (D) Au@SiO2. | ||
3.2. Photoluminescence
Fig. 5 shows the PL and PLE measurements of the CDSs ZAOC and MAOC. The PL and PLE spectra were also fit using the Lorentzian function (Fig. 5, red-dashed lines), and the data describing the measured bands are reported in Table S1 (ESI†). The ZAOC PL spectrum (Fig. 5(A), left-hand side) contains several narrow bands that are characteristic of emission from a Cr3+ with octahedral coordination geometry in a strong crystal field.37–39 Although the band with the highest intensity appears at 14124.3 cm−1 (708 nm), the most important band appears at 14534.8 cm−1 (688 nm) and corresponds to the R-line, called the zero-phonon line (ZPL). The ZPL is marked with a green arrow in Fig. 5, and is related to the 2E → 4A2 transition.6,37–50 The lower energy band next to the ZPL, at 14287.1 cm−1 (700 nm) corresponds to the N2-line.38,39,47–50 In addition, several studies37–39,48,49 indicated that the bands appearing at 13915.6 and 14124.3 cm−1 (719 and 708 nm) correspond to Stokes phonon sidebands (PSB), whereas others studies40,42,46,47,51 reported them as different N-lines. The bands appearing at 14757.3, 14990.9 and 15157.0 cm−1 (678, 667, and 660 nm, respectively) are anti-Stokes PSB.37–39,48,49 Both Stokes and anti-Stokes PSB are related to vibrionic transitions.37–39,48,49 The bands at lower energies of 13113.4, 13369.2, 13506.9, and 13651.8 cm−1 (763, 748, 740, and 733 nm, respectively) were also reported as N-lines,51 but there is evidence38,42,48 that these bands correspond to the 4T2 → 4A2 transition. Finally, the ZAOC PL spectrum also depicts small bands located at 13812.1, 14388.5, and 14684.3 cm−1 (724, 695, and 681 nm, respectively), marked with pink arrows. These bands correspond to N-lines as well and appear when the Cr3+ concentration is ∼0.2 mol%.4,41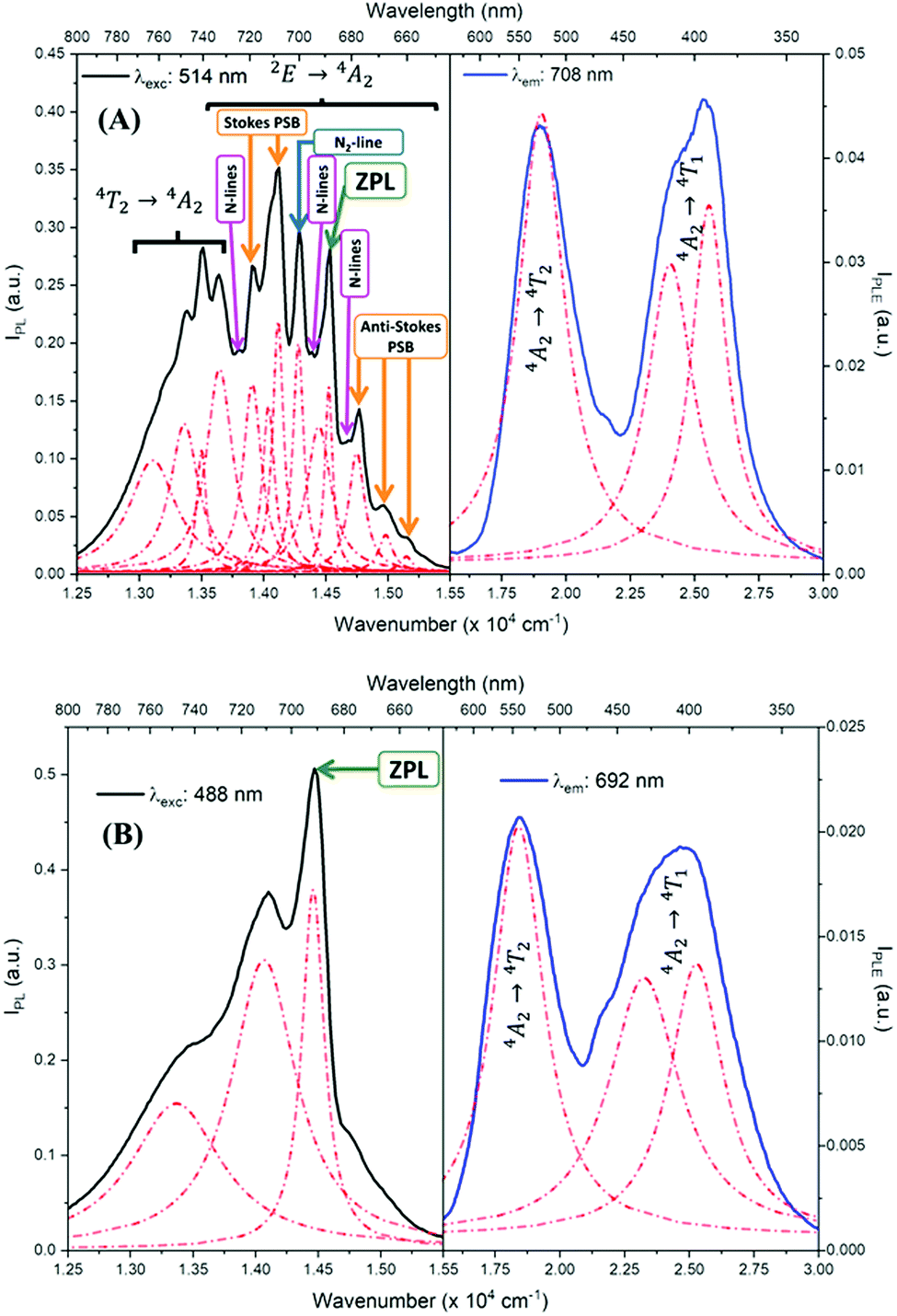 | ||
| Fig. 5 PL (black) and PLE (blue) spectra of (A) ZAOC and (B) MAOC. The red-dashed lines represent Lorentzian fits. The green arrow indicates the zero-phonon line, the blue arrow indicates the N2-line, and the orange arrows indicate Stokes and anti-Stokes phonon sidebands (PSB). The pink arrows indicate different N-lines that appear when the concentration of Cr3+ is ∼0.2 mol%. | ||
R- and N-lines originate at the atomic scale in the crystal of the compounds. In normal-type spinels, depending on the oxidation state, the cations can adopt octahedral or tetrahedral coordination geometry.52 In the ideal case, the Al atoms adopt octahedral geometries (16d positions), whereas the Zn or Mg atoms adopt tetrahedral geometries (8a positions). Because Cr atoms have a +3 oxidation state, they are more likely to adopt octahedral geometries.46,47,52 Nevertheless, occasional defects may occur in the crystal lattice, and the atoms conforming to normal-type spinels can exchange positions to adopt an inverse spinel, where some Al atoms occupy 8a positions and some Zn atoms occupy 16d positions.46,47 The R-lines observed in the ZAOC emission spectrum are associated with the 16d positions of the normal spinel, whereas the N-lines are associated with Cr3+ ions that are in inversion sites between Zn2+ and Al3+ within the first two coordination spheres.46,47 This explanation also applies in the 4T2 → 4A2 transition appearing at lower energies because the Cr3+ ions located in the inversion sites lie in weakly, tetrahedrally coordinated crystal field sites.38
In contrast, the intensities of R- and N-lines depend on the Cr3+ concentration in spinels.4 According to Loan et al.,6 when the Cr3+ concentration is approximately 0.1 mol%, the R-line (14534.8 cm−1) should be the most intense in the PL spectrum. However, when the Cr3+ concentration is approximately 8 mol%, the band with the highest intensity appears at 14124.3 cm−1, which is similar to the band found in the ZAOC PL spectrum. This result supports that the Cr3+ concentration in the CDS is 5 mol%. Furthermore, when the concentration exceeds 16 mol%, the lattice parameters increase slightly (not observed here), which implies a decrease in the crystal field environment around the octahedrally-coordinated Cr3+ ion, and the R- and N-lines sidebands disappear.
In MAOC PL spectrum (Fig. 5(B), left-hand side), the most intense band at 14471.8 cm−1 (691 nm) can be attributed to ZPL and is due to the 2E → 4A2 transition. The bands at 13495.3 and 14104.4 cm−1 (741 and 709 nm) can be ascribed to an R-line side band.53,54 The R-line is due to a Cr3+ transition when the cation has an octahedral coordination geometry in a strong crystal field, whereas the N-line is due to the perturbation of Cr3+ owing to the inversion of Mg2+ and Al3+ in the first two coordination spheres.4,10,53–55
The PLE spectra (Fig. 5, right-hand side) reveal two broad bands between 15500.0 and 30000.0 cm−1. For both spinels, the bands at approximately 18500.0 cm−1 are related to the 4A2 → 4T2 transition, and the bands at approximately 25000.0 cm−1 correspond to the 4A2 → 4T1 transition.10,42,45,46,53,55,56 At higher annealing temperatures, the 4A2 → 4T1 transition band appears with a higher intensity than that of the 4A2 → 4T2 transition band.55
Table S1 (ESI†) lists data from the fits. The ZAOC PL spectrum (Fig. 5(A), left-hand side) presents two doublets for the two bands that have shoulders. One doublet is at 14046.7 and 14124.2 cm−1, which is related to the R-side band as mentioned earlier, and the other one is the R-line, located at 14451.9 and 14532.1 cm−1. The doublet found in the R-line is well described in the literature as R1 and R2 bands, which are usually well resolved at low temperatures.4,37,41,43,45 The bands observed in the MAOC PL spectrum (Fig. 5(B), left-hand side) fit well three bands. In addition, in both PLE spectra (Fig. 5, right-hand side), the fit for the 4A2 → 4T1 transition band contained two bands around 23232.7 and 25355.1 cm−1, which is due to the 4T1 state splitting into the 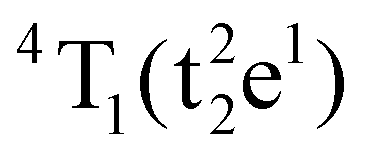 and
and 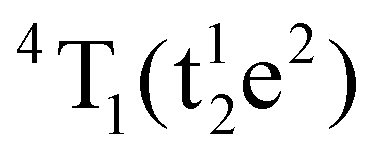 states. This phenomenon manifests as shoulders peaks in the PLE spectra.48,50,55,57
states. This phenomenon manifests as shoulders peaks in the PLE spectra.48,50,55,57
Using the observed excitation bands, we evaluated the crystal field splitting parameter, Dq, and Racah parameters B and C, according with the following equations:53,56
| 10Dq = ν1 | (2) |
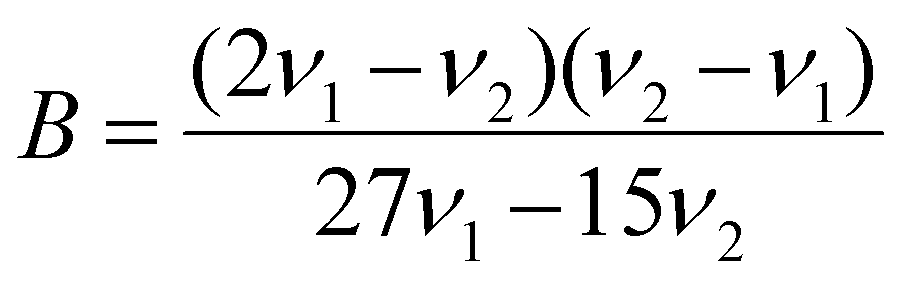 | (3) |
| 9B + 3C = ν3 | (4) |
The ν1, ν2, and ν3 frequencies represent 4A2 → 4T2, 4A2 → 4T1 and 2E → 4A22E → 4A2 transitions, respectively, in cm−1. For both doped spinels, the value used for ν3 was the energy of the zero-phonon line. The values obtained are listed in Table 2 and compared with values previously reported in the literature.46,53 The Dq/B values for both CDSs are similar and greater than 3, indicating that the Cr3+ ions are in strong crystal field sites because Dq/B < 2.3 for weak field sites and Dq/B > 2.3 for strong field sites. Additionally, the Dq/B values are in the expected range for Cr3+ in octahedral coordination geometry.46,53
3.3. Luminescence enhancement
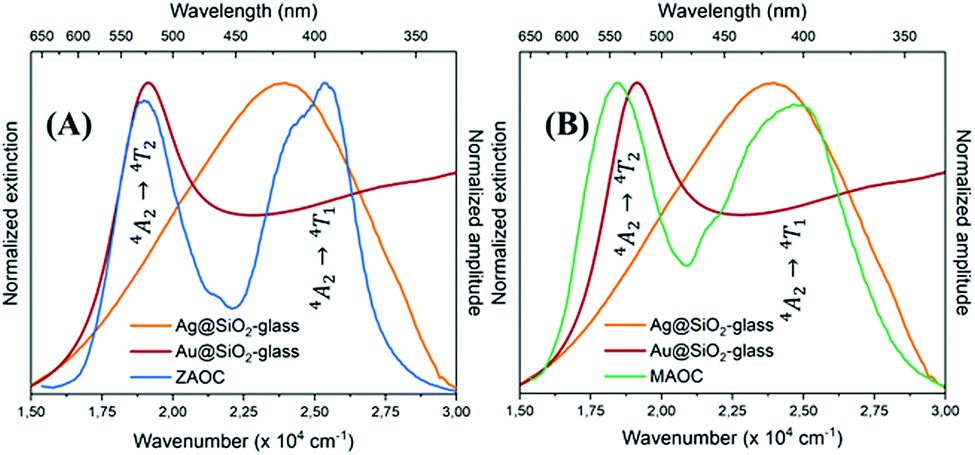 | ||
| Fig. 6 Normalised extinction spectra of AgMS (orange) and AuMS (red) with normalised PLE spectra of (A) ZAOC (blue) and (B) MAOC (green). | ||
Fig. 7 shows typical AFM images of MNPs@SiO2 MSs. The AgMS are more separated from each other (Fig. 7(A)) and are more homogeneously distributed compared to the AuMS (Fig. 7(B)), which are aggregated. This is a consequence of the preparation methods. The glass was dipped into the Ag@SiO2 colloidal solution, whereas the colloidal solution of Au@SiO2 was dropped onto the glass instead. Additionally, the height profiles of the AFM images are shown in Fig. 7. The AgMS has a height profile similar in size to the observed AgNP TEM image. In contrast, the AuMS has a higher height profile than the observed size of the AuNP by TEM image (Fig. 4(D)) because the Au@SiO2 particles aggregate on this surface.
 | ||
| Fig. 7 AFM images and height profiles of the silica coated MNPs on glass for (A) AgMS; (B) AuMS. For both MS, we can see the 2D AFM images (left); 3D AFM images (center); and height profiles (right). The lines on the 2D AFM images are related to the respective height profiles. | ||
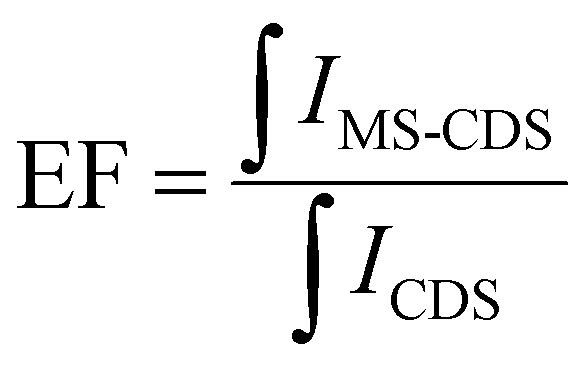 | (5) |
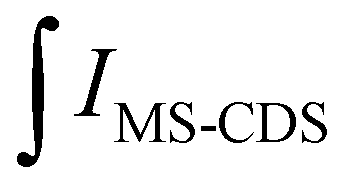 is the integral of the emission intensity for the ensemble formed by silica-coated MNP-MS and CDS and
is the integral of the emission intensity for the ensemble formed by silica-coated MNP-MS and CDS and 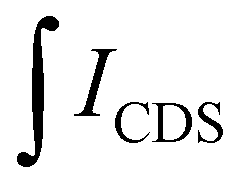 is the integral of the emission intensity of CDS. Fig. 8 shows SEL spectra of the CDSs at room temperature. This figure displays the ZPL previously identified in Fig. 5. Additionally, MNP-MSs do not produce a blue- or redshift in the PL spectra. For the ZAOC PL spectrum, there are two transitions; therefore, we calculated the EF for each transition. Moreover, the EF values found were approximately 2.4, which indicated that the MNP-MSs can enhance the luminescence and PL efficiency of the CDSs at room temperature and may enhance another luminescent inorganic phosphor.
is the integral of the emission intensity of CDS. Fig. 8 shows SEL spectra of the CDSs at room temperature. This figure displays the ZPL previously identified in Fig. 5. Additionally, MNP-MSs do not produce a blue- or redshift in the PL spectra. For the ZAOC PL spectrum, there are two transitions; therefore, we calculated the EF for each transition. Moreover, the EF values found were approximately 2.4, which indicated that the MNP-MSs can enhance the luminescence and PL efficiency of the CDSs at room temperature and may enhance another luminescent inorganic phosphor.
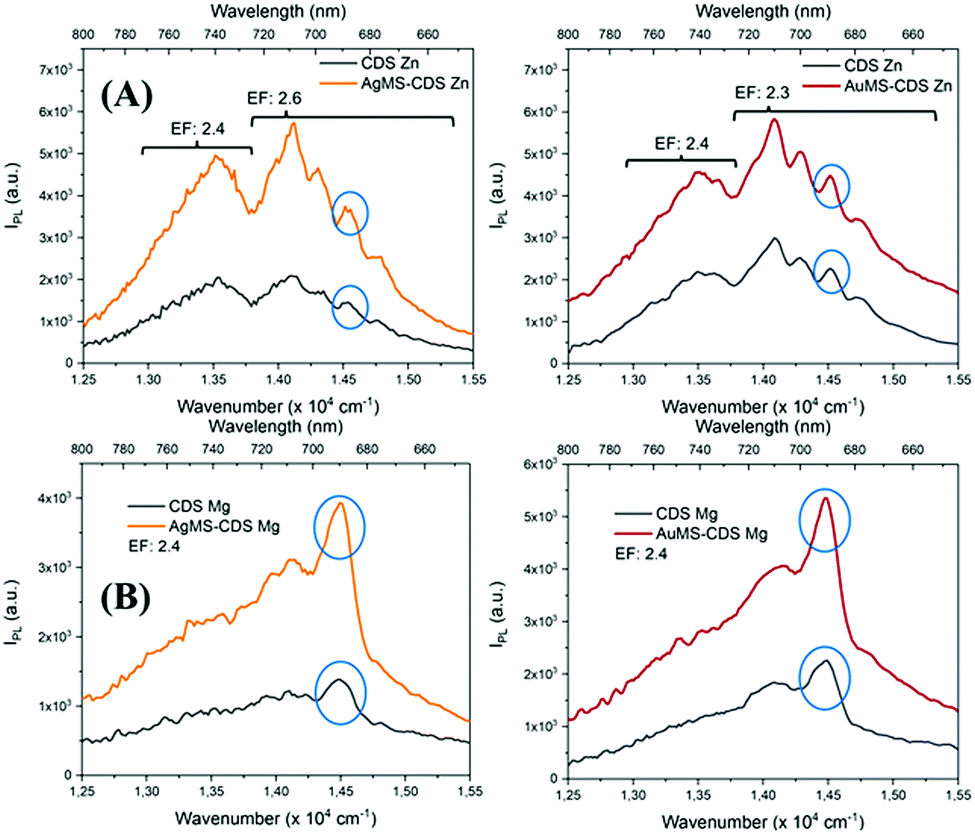 | ||
| Fig. 8 SEL spectra of (A) ZAOC and (B) MAOC on Ag@SiO2 (orange) and Au@SiO2 (red) MSs and their counterparts without MSs (black). The blue circle indicates the ZPL. | ||
3.4. Lifetimes
The frequency-domain (FD) model was used to measure the lifetimes, as previously described.70–72 For this technique, the excitation light intensity is modulated with varying angular frequency ω, and both excitation and modulated emission are recorded. The phase shift P and the relative modulation M of the emission are calculated from these data, and the lifetime is determined using models of time decay as single, multi, or average exponential. The resulting measured phase and the modulation values were analysed by using the multi-exponential time-dependent model given by eqn (6)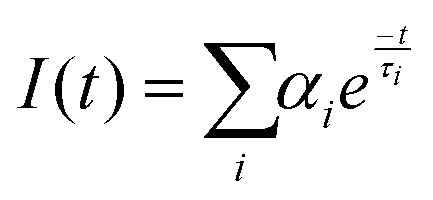 | (6) |
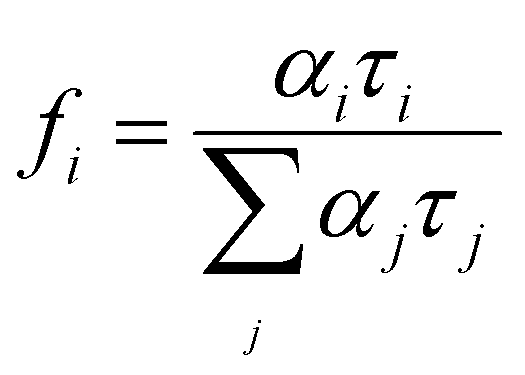 | (7) |
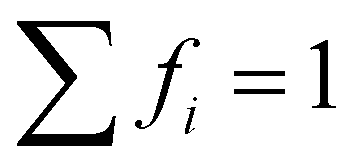 .73 The average lifetime is given by eqn (8)
.73 The average lifetime is given by eqn (8)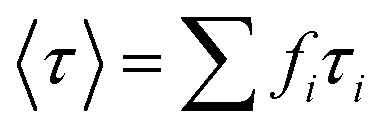 | (8) |
The FD measurement fits are shown in the ESI† (Fig. S3 and S4). The best fits were obtained with two-exponential decay models, leading to the smallest values of χ2 for ZAOC and MAOC with both MNPs@SiO2 ensembles at room temperature. Table 3 reports the lifetime results. For all the ensembles, the average lifetimes were in the millisecond range, with the shortest lifetimes for the MAOC ensembles. For the ZAOC sets, we compared the different lifetime values for the CDS with Ag@SiO2 or Au@SiO2 core–shell MNPs and without MNPs. We observed comparable lifetime values, with a higher value for the Ag@SiO2 ensemble than that of the Au@SiO2 ensemble. For the MAOC samples, we observed an increase in the lifetimes for both MNPs@SiO2 ensembles. In addition, either in the absence or presence of MNPs@SiO2, τ1 is larger than τ2 for both CDSs. The same trend was repeated for the fractional contributions, where f1 was larger than f2, except for the ZAOC-Au@SiO2 ensemble. Previous literature48,50,74,75 has shown that the emission of Cr3+ ions for different spinels is in the millisecond range, and the lifetime values obtained for these compounds were directly proportional to the annealing duration and temperature.38,50,75
| CDS | τ 1 [ms] | τ 2 [ms] | τ [ms] | f 1 | f 2 | α 1 | α 2 | χ 2 |
|---|---|---|---|---|---|---|---|---|
| ZAOC | ||||||||
| NP-free | 1.29(2) | 0.0201(5) | 1.09 | 0.840(2) | 0.160 | 65 | 792 | 2.14 |
| AgMS | 2.31(2) | 0.0186(10) | 1.55 | 0.670(1) | 0.330 | 29 | 1778 | 2.52 |
| AuMS | 1.38(2) | 0.0126(8) | 0.532 | 0.380(1) | 0.620 | 27.5 | 4937 | 1.18 |
| MAOC | ||||||||
| NP-free | 0.90(1) | 0.0182(10) | 0.725 | 0.802(1) | 0.198 | 89.2 | 1089 | 1.86 |
| AgMS | 0.910(6) | 0.0510(20) | 0.852 | 0.933(1) | 0.067 | 103 | 130 | 1.69 |
| AuMS | 1.33(2) | 0.00828(4) | 0.816 | 0.611(1) | 0.389 | 45.9 | 4693 | 1.97 |
3.5. Interaction between the CDSs and MNPs
When the MNPs are near luminescent emitters, the PL intensity is modified in two ways. First, the incident light excites the surface plasmon resonance (SPR) of the MNPs. Emitters localised in regions of near-field enhancement are subject to enhanced excitation light and manifest an increased excitation rate. Moreover, the radiative decay rate of the emitter is influenced by the modification of the localised density of states (LDOS) introduced by the MNPs through the well-known Purcell effect.76–78 Second, the emitters in an excited state can recombine through a nonradiative decay channel, emitting photons directly to the far-field (PL), or relax rapidly by exciting the localised plasmon resonance of the MNPs via the energy transfer channel (Förster energy transfer). The relaxation via the plasmon resonance increases with the shortening of the MNP-emitter distance and leads to radiative phenomena or nonradiative recombination. The radiative rate occurring with SPR is associated with the far-field radiation emitted (scattered) by the accelerating and decelerating charged particles. The ratio of this radiative rate to the total recombination rate usually increases with the NP size.79,80 The nonradiative rate (absorption) has been previously studied,81–83 where several mechanisms were shown to contribute to this rate: electron–electron (e–e) scattering, electron–phonon (e–p) scattering, electron-defects (e–d), and damping due to surface effect (e–s).For the ensembles, the CDS emitters were deposited on the surface of the MNPs@SiO2 core–shell which were located on a glass slide surface with SiO2 acting as a spacer between the MNPs and CDSs. This ensemble can be considered an emitter–MNP hybrid system, and the excitation and emission processes can be described as a donor (emitter)–acceptor (MNP) system. Scheme 3 shows this CDS-MNP system in two configurations with a larger shell thickness (Scheme 3(A)) of a few tens of nanometers and a smaller shell thickness (Scheme 3(B)) of a few nm.
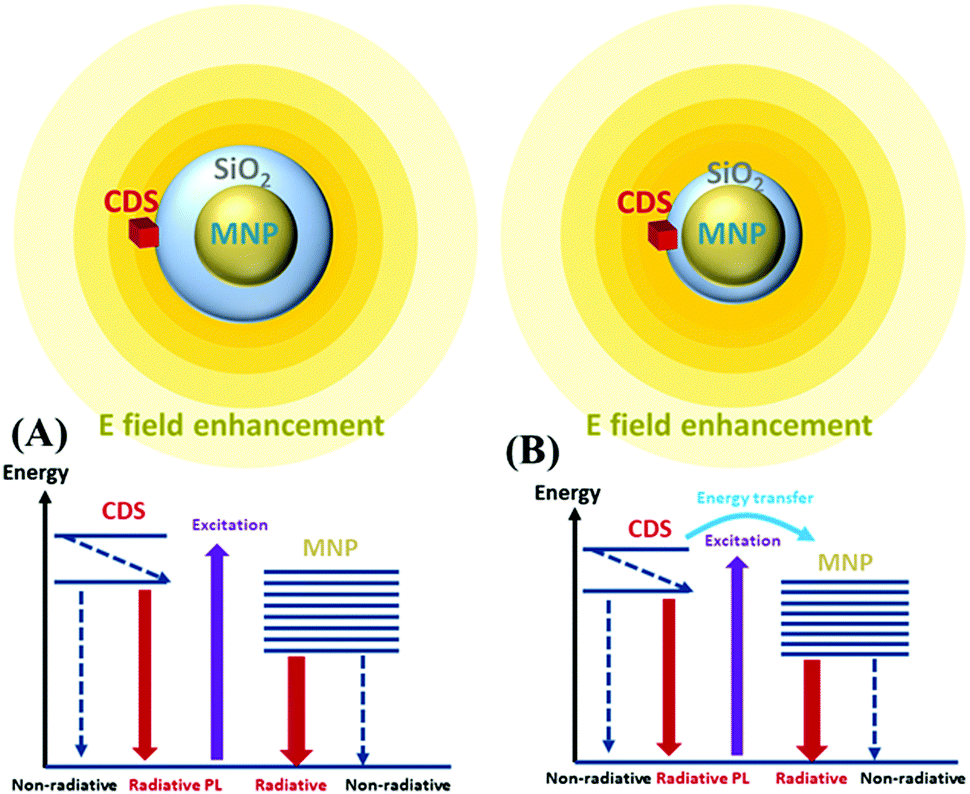 | ||
| Scheme 3 Representation of the donor-acceptor system with the excitation and emission processes of the CDS-MNP surface hybrid system. (A) a large shell thickness and (B) a short shell thickness are shown. | ||
We observed an enhancement in the luminescence of the CDSs in the presence of MNPs with an EF of ∼2.4. Moreover, we observed time decays with one slow component in the millisecond range and one fast component in the tens of nanosecond range, which are comparable for CDSs in the absence of MNPs and the ensembles. We examined the luminescence enhancement using the theoretical arguments developed for this subject. First, in the low photon flux approximation, the emission intensity, mainly due to the 2E → 4A2 transition, is proportional to the excitation rate Γexc (in s−1) and to the quantum yield (QY) according to eqn (9)
| IPL ∝ ΓexcQY | (9) |
An increase in PL emission in the presence of MNPs is then either related to an increase in Γexc, QY, or both. The excitation rate can be expressed as the product of the emitter emission (2E → 4A2 transition) cross-section σemi (in cm2) and photon flux ϕhν (in cm−2 s−1). The emission cross-section can be considered an intrinsic property that remains unchanged. On the other hand, in a medium with MNPs, the photon flux can locally fluctuate because of the localized electromagnetic field enhancement, leading to a locally enhanced photon flux, 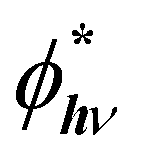 . In the case of the Ag@SiO2 and Au@SiO2 core–shell MNPs, it was reported84 that, depending on their surroundings, the refractive index, and shell thickness up to 40 nm, the electromagnetic field enhancement extends up to the external interface of the shell and beyond. In the ensemble, the QY can be strongly modified by the CDS–MNP distance, which is a key parameter because it influences the CDS–MNP interaction.
. In the case of the Ag@SiO2 and Au@SiO2 core–shell MNPs, it was reported84 that, depending on their surroundings, the refractive index, and shell thickness up to 40 nm, the electromagnetic field enhancement extends up to the external interface of the shell and beyond. In the ensemble, the QY can be strongly modified by the CDS–MNP distance, which is a key parameter because it influences the CDS–MNP interaction.
Two distinct situations are usually described according to the distance between emitters (CDSs) and MNPs, as shown in Scheme 3. The first situation occurs when CDSs are located far from the MNPs or when the MNPs are not on MSs. The intrinsic and nonperturbed quantum yield of CDSs (QY0) can be given by eqn (10)
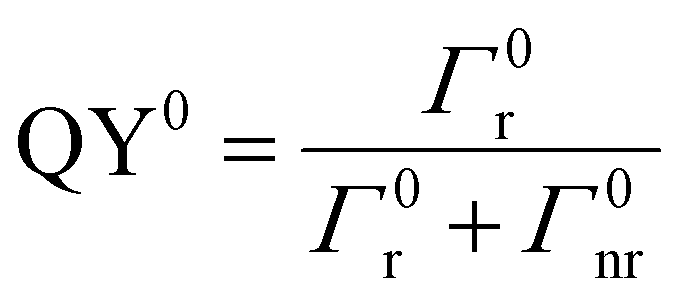 | (10) |
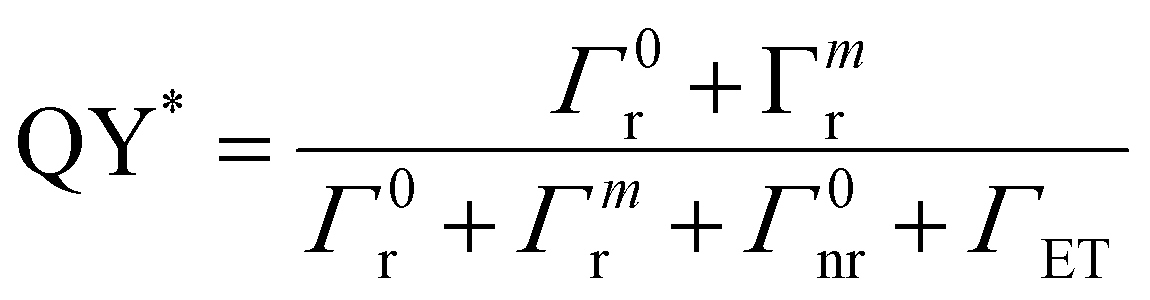 | (11) |
The CDS lifetimes (τ) in an MS without MNPs are described only as a function of Γ0r and Γ0nr given by eqn (12)
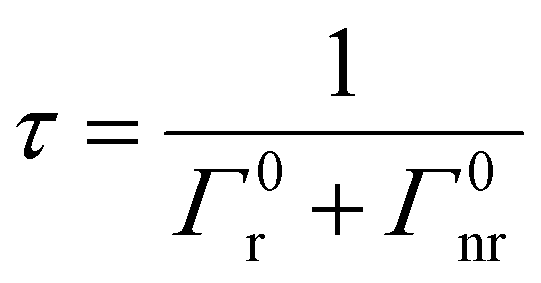 | (12) |
When the CDSs form an ensemble, eqn (12) can be rewritten as eqn (13)
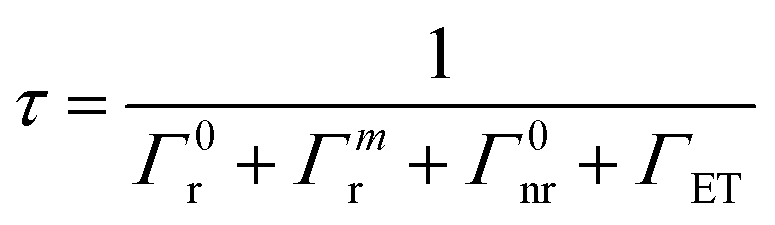 | (13) |
As mentioned before, assuming a low photon flux approximation, the emission intensity (PL) is proportional to Γexc times QY (eqn (9)). The increase in PL emission in the presence of MNPs can be attributed to either an increase in Γexc, an increase in QY, or both. However, the time decays found here has two components for both CDSs without MNPs and in an ensemble with an MS, while a modification of QY should result in different time constants, according to eqn (10) and (13). For this reason, based on our experimental results, we can assume that the increase in the PL is mainly due to an increased excitation rate, with no obvious change in quantum efficiency. Thus, the CDSs had unmodified quantum efficiencies and decay times but benefitted from the increase in the excitation rate induced by the presence of metal particles. In the CDS–MNP systems, the minimum distance between the CDSs and MNPs was obtained owing to the outer shell of SiO2 being 25 nm for Ag@SiO2 and 10 nm for Au@SiO2. This distance is approximately ten times larger than the optimal fluorophore MNP distance reported for SEL.85 For these reasons, we did not observe QY modification that would occur at short distances (∼1–2 nm), but rather an increased excitation rate that might be due to electromagnetic field enhancement, which takes place at a distance equivalent to the SiO2 shell thickness.
4. Conclusions
In this work, we investigated the SEL when CDS interacts with MNPs at room temperature. The citrate sol–gel synthesis yielded homogeneous and crystalline inorganic materials or spinels. The Cr3+ dopant in spinel matrices did not significantly change the crystal lattice parameters. The backscattered images and EDS elemental analyses showed a uniform distribution of Zn, Mg, Al, and Cr (concentration ∼5% mol) atoms in the samples.The Lorentzian fitting of the ZAOC PL spectrum showed a characteristic doublet of the R-line, whereas the Lorentzian fitting of the PLE spectra showed splitting of the 4A2 → 4T1 transition band. The most intense band found in the ZAOC PL spectrum was an R-line sideband at 14124.3 cm−1, instead of the R-line (zero phonon line) band at 14534.8 cm−1. Finally, the evaluation of the crystal field parameters indicated that the Cr3+ ions in the CDSs were in a strong crystal field environment.
We obtained PLE spectra for the CDSs deposited on a metallic silica-coated NP surface, at room temperature. We monitored the bands with the highest intensities and found that the enhancement factor values were approximately 2.4 for both MAOC and ZAOC spinels. However, the average lifetime values 〈τ〉 were not significantly altered when the CDSs were near Ag@SiO2 and Au@SiO2. Thus, the results showed that obtaining enhanced luminescence for compounds with lifetimes higher than the nanosecond range at room temperature is possible. We believe that this enhancement is governed by an enhancement of the local electromagnetic field in the vicinity of the core–shell MNPs rather than an energy transfer induced by QY modification. This study paves the way for new studies on PL enhancement based on the SEL effect using rare-earth-doped phosphor.
Author contributions
The manuscript was prepared and written with the contributions of all authors. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Rodrigo thanks ANID (ex-CONICYT) National PhD Scholarship 21150816. Camilo thanks ANID (ex-CONICYT) National PhD Scholarship 21150397. Antonio thanks to ANID-FONDECYT 1190856. Igor thanks to ANID-FONDECYT 1190246. The authors thank the Chilean-French International Associated Laboratory for ‘Multifunctional Molecules and Materials’ (LIAM3-CNRS N°1027).Notes and references
- F. Zerarga, A. Bouhemadou, R. Khenata and S. Bin-Omran, Solid State Sci., 2011, 13, 1638–1648 CrossRef CAS.
- G. Fan, J. Wang and F. Li, Catal. Commun., 2011, 15, 113–117 CrossRef CAS.
- C. Wang, A. H. Shen and Y. Liu, J. Lumin., 2020, 227, 117552 CrossRef CAS.
- D. L. Wood, G. F. Imbusch, R. M. Macfarlane, P. Kisliuk and D. M. Larkin, J. Chem. Phys., 1968, 48, 5255–5263 CrossRef CAS.
- P. Berdahl, S. S. Chen, H. Destaillats, T. W. Kirchstetter, R. M. Levinson and M. A. Zalich, Sol. Energy Mater. Sol. Cells, 2016, 157, 312–317 CrossRef CAS.
- T. T. Loan and N. N. Long, VNU J. Sci. Math. - Phys., 2018, 34, 1–7 Search PubMed.
- M. M. Golsheikh, A. M. Arabi and M. S. Afarani, Mater. Res. Express, 2019, 6, 125052 CrossRef CAS.
- D. Zhang, J. Chen, C. Du, B. Zhu, Q. Wang, Q. Shi, S. Cui and W. Wang, Front. Mater. Sci., 2020, 14, 73–80 CrossRef.
- Y.-C. Lin, M. Karlsson and M. Bettinelli, Top. Curr. Chem., 2016, 374, 21 CrossRef PubMed.
- C. Pratapkumar, S. C. Prashantha, H. Nagabhushana and D. M. Jnaneshwara, J. Sci. Adv. Mater. Devices, 2018, 3, 464–470 CrossRef.
- M. G. Brik, J. Papan, D. J. Jovanović and M. D. Dramićanin, J. Lumin., 2016, 177, 145–151 CrossRef CAS.
- K. Sokolov, G. Chumanov and T. M. Cotton, Anal. Chem., 1998, 70, 3898–3905 CrossRef CAS PubMed.
- J. Gersten and A. Nitzan, J. Chem. Phys., 1981, 75, 1139–1152 CrossRef CAS.
- T. Ming, L. Zhao, Z. Yang, H. Chen, L. Sun, J. Wang and C. Yan, Nano Lett., 2009, 9, 3896–3903 CrossRef CAS PubMed.
- S. A. Camacho, P. H.-B. Aoki, P. Albella, O. N. Oliveira, C. J.-L. Constantino and R. F. Aroca, J. Phys. Chem. C, 2016, 120, 20530–20535 CrossRef CAS.
- F. Tam, G. P. Goodrich, B. R. Johnson and N. J. Halas, Nano Lett., 2007, 7, 496–501 CrossRef CAS PubMed.
- C. D. Geddes and J. R. Lakowicz, J. Fluoresc., 2002, 12, 121–129 CrossRef.
- R. Aroca, G. J. Kovacs, C. A. Jennings, R. O. Loutfy and P. S. Vincett, Langmuir, 1988, 4, 518–521 CrossRef CAS.
- Y. Zhang, K. Aslan, S. N. Malyn and C. D. Geddes, Chem. Phys. Lett., 2006, 427, 432–437 CrossRef CAS.
- Y. Zhang, K. Aslan, M. J.-R. Previte, S. N. Malyn and C. D. Geddes, J. Phys. Chem. B, 2006, 110, 25108–25114 CrossRef CAS PubMed.
- M. J.-R. Previte, K. Aslan, Y. Zhang and C. D. Geddes, J. Phys. Chem. C, 2007, 111, 6051–6059 CrossRef CAS.
- M. Meng, F.-L. Zhang, J. Yi, L.-H. Lin, C.-L. Zhang, N. Bodappa, C.-Y. Li, S.-J. Zhang, R. F. Aroca, Z.-Q. Tian and J.-F. Li, Anal. Chem., 2018, 90, 10837–10842 CrossRef CAS PubMed.
- M. Ramírez-Maureira, V. Vargas, A. Riveros, P. J.-G. Goulet and I. O. Osorio-Román, Mater. Chem. Phys., 2015, 151, 351–356 CrossRef.
- E. Konstantinova, A. Zyubin, V. Slezhkin, V. Bryukhanov, K. Matveeva, E. Moiseeva and I. Samusev, Biophotonics—Riga 2017, 2017, 24 Search PubMed.
- K. Jia, L. Yuan, X. Zhou, L. Pan, P. Wang, W. Chen and X. Liu, RSC Adv., 2015, 5, 58163–58170 RSC.
- P. Vitta, P. Pobedinskas and A. Zukauskas, IEEE Photonics Technol. Lett., 2007, 19, 399–401 CAS.
- D. Chen, X. Chen, X. Li, H. Guo, S. Liu and X. Li, Opt. Lett., 2017, 42, 4950 CrossRef CAS PubMed.
- K. Tsuchiya, K. Sako, N. Ishiwada and T. Yokomori, Meas. Sci. Technol., 2020, 31, 065005 CrossRef CAS.
- A. Wadhwa, C. Wang, C. Wang, R. Ma, X. Qiao, X. Fan and G. Qian, J. Am. Ceram. Soc., 2018, 102, jace.16098 CrossRef.
- B. Shao, Z. Yang, Y. Wang, J. Li, J. Yang, J. Qiu and Z. Song, ACS Appl. Mater. Interfaces, 2015, 7, 25211–25218 CrossRef CAS PubMed.
- Y. Wang, Z. Yang, Y. Ma, Z. Chai, J. Qiu and Z. Song, J. Mater. Chem. C, 2017, 5, 8535–8544 RSC.
- A. Abdukayum, J.-T. Chen, Q. Zhao and X.-P. Yan, J. Am. Chem. Soc., 2013, 135, 14125–14133 CrossRef CAS PubMed.
- C. Segura, V. Vargas, R. A. Valenzuela-Fernández, C. S. Danna and I. O. Osorio-Román, ACS Appl. Energy Mater., 2020, 3, 7680–7688 CrossRef CAS.
- P. C. Lee and D. Meisel, J. Phys. Chem., 1982, 86, 3391–3395 CrossRef CAS.
- B. D. Cullity and S. R. Stock, Elements of X-Ray Diffraction, Pearson Education Limited, 3rd edn, 2014 Search PubMed.
- D. B. Williams and C. B. Carter, Transmission Electron Microscopy, Springer US, Boston, MA, 2009 Search PubMed.
- A. Bessière, S. Jacquart, K. Priolkar, A. Lecointre, B. Viana and D. Gourier, Opt. Express, 2011, 19, 10131–10137 CrossRef PubMed.
- D. N. Hebbar, S. G. Menon, K. S. Choudhari, S. A. Shivashankar, C. Santhosh and S. D. Kulkarni, J. Am. Ceram. Soc., 2018, 101, 800–811 CrossRef CAS.
- D. Zhang, Q. Guo, Y. Ren, C. Wang, Q. Shi, Q. Wang, X. Xiao, W. Wang and Q. Fan, J. Sol-Gel Sci. Technol., 2018, 85, 121–131 CrossRef CAS.
- K. Tanaka, K. Hirao, T. Ishihara and N. Soga, J. Ceram. Soc. Jpn., 1993, 101, 102–104 CrossRef CAS.
- H. M. Kahan and R. M. Macfarlane, J. Chem. Phys., 1971, 54, 5197–5205 CrossRef CAS.
- G. Rani, Powder Technol., 2017, 312, 354–359 CrossRef CAS.
- W. Mikenda and A. Preisinger, J. Lumin., 1981, 26, 67–83 CrossRef CAS.
- H. H. Luc, T. K. Nguyen, V. M. Nguyen, A. Suchocki, A. Kamińska, V. K. Le, V. H. Nguyen and T. T. Luong, Acta Phys. Pol., A, 2003, 104, 581–587 CrossRef CAS.
- W. Nie, F. M. Michel-Calendini, C. Linarès, G. Boulon and C. Daul, J. Lumin., 1990, 46, 177–190 CrossRef CAS.
- V. Singh, R. P.-S. Chakradhar, J. L. Rao and H.-Y. Kwak, J. Mater. Sci., 2011, 46, 2331–2337 CrossRef CAS.
- D. Zhang, Y. H. Qiu, Y. R. Xie, X. C. Zhou, Q. R. Wang, Q. Shi, S. H. Li and W. J. Wang, Mater. Des., 2017, 115, 37–45 CrossRef CAS.
- H. G. S. Karthik, S. G. Menon, D. Hebbar, K. S. Choudhari, C. Santhosh and S. D. Kulkarni, Mater. Res. Bull., 2019, 111, 294–300 CrossRef.
- S. G. Menon, D. N. Hebbar, S. D. Kulkarni, K. S. Choudhari and C. Santhosh, Mater. Res. Bull., 2017, 86, 63–71 CrossRef CAS.
- S. G. Menon, K. S. Choudhari, S. A. Shivashankar, C. Santhosh and S. D. Kulkarni, J. Alloys Compd., 2017, 728, 484–489 CrossRef CAS.
- T. T. Loan, L. H. Ha and N. N. Long, VNU J. Sci. Math. - Phys., 2010, 26, 37–42 Search PubMed.
- A. R. West, Solid State Chemistry and its Applications, Students Edition, John Wiley & Sons, Ltd, Chichester, West Sussex, United Kingdom, 2nd edn, 2014 Search PubMed.
- Y. Hao, S. Wang and K. Zhang, Mater. Chem. Phys., 2020, 253, 123323 CrossRef CAS.
- T. L. Phan, S. C. Yu, M. H. Phan and T. P.-J. Han, J. Korean Phys. Soc., 2004, 45, 63–66 CAS.
- N. T.-K. Chi, N. V. Quang, N. T. Tuan, N. D.-T. Kien, D. Q. Trung, P. T. Huy, P. D. Tam and D. H. Nguyen, J. Electron. Mater., 2019, 48, 5891–5899 CrossRef CAS.
- A. R. Molla, C. R. Kesavulu, R. P.-S. Chakradhar, A. Tarafder, S. K. Mohanty, J. L. Rao, B. Karmakar and S. K. Biswas, J. Alloys Compd., 2014, 583, 498–509 CrossRef CAS.
- S. Karthik, S. G. Menon, D. N. Hebbar, K. S. Choudhari, C. Santhosh and S. D. Kulkarni, Mater. Res. Bull., 2017, 94, 513–519 CrossRef.
- K.-C. Lee, S.-J. Lin, C.-H. Lin, C.-S. Tsai and Y.-J. Lu, Surf. Coat. Technol., 2008, 202, 5339–5342 CrossRef CAS.
- H. Baida, P. Billaud, S. Marhaba, D. Christofilos, E. Cottancin, A. Crut, J. Lermé, P. Maioli, M. Pellarin, M. Broyer, N. Del Fatti, F. Vallée, A. Sánchez-Iglesias, I. Pastoriza-Santos and L. M. Liz-Marzán, Nano Lett., 2009, 9, 3463–3469 CrossRef CAS PubMed.
- R. K. Chava, Noble Metal-Metal Oxide Hybrid Nanoparticles, Elsevier, 2019, pp. 499–516 Search PubMed.
- R. F. Aroca, Phys. Chem. Chem. Phys., 2013, 15, 5355 RSC.
- M. J.-R. Previte, K. Aslan, Y. Zhang and C. D. Geddes, Chem. Phys. Lett., 2006, 432, 610–615 CrossRef CAS PubMed.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou, D. Y. Wu, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464, 392–395 CrossRef CAS PubMed.
- O. Kulakovich, N. Strekal, A. Yaroshevich, S. Maskevich, S. Gaponenko, I. Nabiev, U. Woggon and M. Artemyev, Nano Lett., 2002, 2, 1449–1452 CrossRef CAS.
- V. Zucolotto, K. M. Gattás-Asfura, T. Tumolo, A. C. Perinotto, P. A. Antunes, C. J.-L. Constantino, M. S. Baptista, R. M. Leblanc and O. N. Oliveira, Appl. Surf. Sci., 2005, 246, 397–402 CrossRef CAS.
- K. Ray, R. Badugu and J. R. Lakowicz, Chem. Mater., 2007, 19, 5902–5909 CrossRef CAS PubMed.
- D. S. dos Santos, Jr. and R. F. Aroca, Analyst, 2007, 132, 450 RSC.
- R. F. Aroca, G. Y. Teo, H. Mohan, A. R. Guerrero, P. Albella and F. Moreno, J. Phys. Chem. C, 2011, 115, 20419–20424 CrossRef CAS.
- A. R. Guerrero and R. F. Aroca, Angew. Chem., Int. Ed., 2011, 50, 665–668 CrossRef CAS PubMed.
- J. R. Lakowicz, G. Laczko, H. Cherek, E. Gratton and M. Limkeman, Biophys. J., 1984, 46, 463–477 CrossRef CAS.
- J. R. Lakowicz, Y. Shen, S. D’Auria, J. Malicka, J. Fang, Z. Gryczynski and I. Gryczynski, Anal. Biochem., 2002, 301, 261–277 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, SpringerUS, Boston, MA, 3rd edn, 2006 Search PubMed.
- V. Vargas, J. Phys. Chem. A, 2004, 108, 281–288 CrossRef CAS.
- H. Aizawa, N. Ohishi, S. Ogawa, E. Watanabe, T. Katsumata, S. Komuro, T. Morikawa and E. Toba, Rev. Sci. Instrum., 2002, 73, 3089–3092 CrossRef CAS.
- P. Głuchowski, R. Pązik, D. Hreniak and W. Stręk, Chem. Phys., 2009, 358, 52–56 CrossRef.
- D. Kleppner, Phys. Rev. Lett., 1981, 47, 233–236 CrossRef CAS.
- E. Yablonovitch, Phys. Rev. Lett., 1987, 58, 2059–2062 CrossRef CAS PubMed.
- S. Haroche and D. Kleppner, Phys. Today, 1989, 42, 24–30 CrossRef CAS.
- C. Sönnichsen, T. Franzl, T. Wilk, G. von Plessen, J. Feldmann, O. Wilson and P. Mulvaney, Phys. Rev. Lett., 2002, 88, 774021 CrossRef PubMed.
- A. Alabastri, S. Tuccio, A. Giugni, A. Toma, C. Liberale, G. Das, F. De Angelis, E. Di Fabrizio and R. P. Zaccaria, Materials, 2013, 6, 4879–4910 CrossRef PubMed.
- S. Link and M. A. El-Sayed, Annu. Rev. Phys. Chem., 2003, 54, 331–366 CrossRef CAS PubMed.
- C. Voisin, N. Del Fatti, D. Christofilos and F. Vallee, J. Phys. Chem. B, 2001, 105, 2264–2280 CrossRef CAS.
- K. O. Aruda, M. Tagliazucchi, C. M. Sweeney, D. C. Hannah, G. C. Schatz and E. A. Weiss, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4212–4217 CrossRef CAS PubMed.
- J. L. Montaño-Priede, O. Peña-Rodríguez and U. Pal, J. Phys. Chem. C, 2017, 121, 23062–23071 CrossRef.
- R. Gill and E. C. Le Ru, Phys. Chem. Chem. Phys., 2011, 13, 16366 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: SEM and BSE images, extinction spectra of colloidal solutions, frequency domain intensities decays and data fits of emission and excitation spectra. See DOI: https://doi.org/10.1039/d2ma00217e |
| This journal is © The Royal Society of Chemistry 2022 |