
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Robust CON-type zeolite nanocatalyst in methanol-to-olefins reaction: downsizing, recrystallisation and defect-healing treatments toward prolonged lifetime†
Masanori
Takemoto‡
 a,
Kenta
Iyoki‡
bc,
Yuki
Otsuka
b,
Hiroaki
Onozuka
d,
Anand
Chokkalingam
a,
Toshiyuki
Yokoi
e,
Susumu
Tsutsuminai
d,
Takahiko
Takewaki
d,
Toru
Wakahira
*ab and
Tatsuya
Okubo
*b
a,
Kenta
Iyoki‡
bc,
Yuki
Otsuka
b,
Hiroaki
Onozuka
d,
Anand
Chokkalingam
a,
Toshiyuki
Yokoi
e,
Susumu
Tsutsuminai
d,
Takahiko
Takewaki
d,
Toru
Wakahira
*ab and
Tatsuya
Okubo
*b
aInstitute of Engineering Innovation, School of Engineering, The University of Tokyo, 2-11-16 Yayoi, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: wakihara@chemsys.t.u-tokyo.ac.jp
bDepartment of Chemical System Engineering, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 13-8656, Japan. E-mail: okubo@chemsys.t.u-tokyo.ac.jp
cPrecursory Research for Embryonic Science and Technology (PRESTO) Japan Science and Technology Agency (JST), Kawaguchi, Saitama 332-0012, Japan
dInorganic Materials Laboratory, Science & Innovation Center Mitsubishi Chemical Corporation, 1000 Kamoshida-cho, Aoba-ku, Yokohama, 227-8502, Japan
eInstitute of Innovative Research, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama, 226-8503, Japan
First published on 17th May 2022
Abstract
CON-type zeolites have great potential as highly propene-selective catalysts in methanol-to-olefins reactions; however, their insufficient lifetime is a major limitation. This study demonstrates the remarkably prolonged lifetime of a CON-type zeolite in methanol-to-olefins reaction via a multistep strategy including post-milling recrystallisation and liquid-mediated healing of residual defects. Wet bead-milling for parent CON-type zeolite allows to yield nano-sized zeolite, partially composed of damaged parts. To remedy this, the milled zeolite undergoes a two-step hydrothermal treatment, where temperature and concentration of silicate species contained in alkaline solutions are carefully optimised. Furthermore, residual silanol defects in the recrystallised zeolite are healed via a liquid-mediated treatment using NH4F/TEAOH solution. The defect-healed zeolite nanoparticles showed a lifetime more than twice that of the parent zeolite owing to the increased external surface area and the reduced subnano-scale defects. This protocol could play a significant role in designing robust catalysts with controlled particle sizes and reduced subnano-scale defects.
1. Introduction
The methanol-to-olefins (MTO) reaction is a key technology for providing light olefins, important building blocks for various chemicals, from non-oil resources.1–3 The MTO catalysts with excellent stability and product selectivity are in high demand, and especially selective production of C3–C4 species among light olefins is of great interest.4–7 Zeolites and related materials are well-known catalysts that possess shape selectivity toward light olefins owing to their small pores in microporous frameworks.8,9 In MTO reactions over zeolites, methanol is catalytically converted into various chemicals, such as light olefins (C2–C4), bulky hydrocarbons (over-C5), and aromatics and/or paraffins.10,11 The product selectivities in an MTO reaction strongly depend on the shapes and sizes of the pores in zeolite frameworks. SAPO-34 (CHA-type in the three-letter code of the International Zeolite Association Structure Commission) and ZSM-5 (MFI-type) zeolites have been commercially used to yield propene in the MTO reaction.5 Recently, catalytic activities of RUB-13 (RTH-type)12 and MCM-22 (MWW-type)13 zeolites in the MTO reaction have been examined. However, such zeolites containing small- (8-membered rings, 8R) or medium- (10R) sized pores do not favour a high production selectivity toward C3–C4 species. Therefore, large-pore zeolites, such as zeolite Y (FAU-type), beta (*BEA-type), and MCM-68 (MSE-type),14 have been investigated as alternative catalysts in MTO reactions, with a high selectivity toward C3–C4 species.Recently, CON-type zeolites, comprising a three-dimensional channel with 12R, 10R, and 10R multipores, have been reported as promising acid catalysts in the MTO reaction to produce C3–C4 species with a high production selectivity.15 CIT-1, the first CON-type zeolite with pure polymorph B, was synthesised in a borosilicate framework16,17 and an aluminium-containing CON-type zeolite15 was also reported. Syntheses of CON-type zeolites with the enhanced catalytic performance have been demonstrated by means of heteroatom substitution,18 mesopore introduction19 and control of particle size and acid-site location.20 However, their catalytic performance is still insufficient, and the development of CON-type zeolite catalysts with high C3–C4 selectivities and long catalytic lifetimes remains a challenge. Coking, i.e. pore blockage by bulky hydrocarbon species evolved from methanol conversion, is a major and inevitable problem in MTO reactions over zeolite catalysts. Coking causes the deactivation of zeolite catalysts, necessitating frequent combustion cycles to recover the catalytic performance and leading to the degradation of zeolite structures. Therefore, the development of zeolite catalysts with long one-pass lifetimes is in high demand for practical applications.
Downsizing zeolite particles to increase their external surface area is a well-known strategy for improving their catalytic lifetime against coking.1,21 Top-down approaches via mechanical treatments, such as bead and planetary ball millings, are employed for the production of downsized zeolite nanoparticles.22 However, zeolite frameworks are generally degraded in such treatments, and milled zeolites suffer from pore blockage due to amorphisation. As a result, post-milling recrystallisation methods have been developed to recover the damaged parts of milled zeolites.23–25 It is still difficult to fully recover the damaged parts even after post-milling recrystallisation, and careful control of the reaction conditions is required.22,25,26 Therefore, the recrystallised zeolites possess increased defect sites in their frameworks. According to previous studies, the defect sites in the zeolite framework determine the product selectivity in the MTO reaction. Katia et al. reported that the internal defects in zeolites increase the amount of aromatics in the MTO reaction.27 Sazama et al. demonstrated that highly defective ZSM-5 shows a poor selectivity toward light olefins and a high selectivity toward C6+ hydrocarbons and butanes.28 Therefore, the synthesis of defect-free downsized zeolites to increase the catalytic lifetime and the selectivity of light olefins in an MTO reaction is of great interest.
Recently, we developed a novel liquid-mediated treatment using an aqueous solution of NH4F/TEAOH for healing the defect sites in zeolite frameworks via migration of the silicate species in the parent zeolites to produce extremely stable zeolites.29 Our motivation in the present work is to apply this method for developing nanosized CON-type zeolite, with a drastically reduced number of defect sites, which exhibit an excellent catalytic lifetime and a high selectivity toward C3–C4 olefin evolution. Fig. 1 shows a schematic of our strategy for synthesising fine defect-healed zeolite particles. An in-house-fabricated CON-type zeolite called the ‘parent zeolite’ was subjected to a wet bead-milling to yield fine particles, named as ‘milled zeolite’. The non-crystalline parts of the milled zeolite were recrystallised via a two-step hydrothermal treatment. Finally, residual defect sites were healed through a liquid-mediated treatment. The zeolites obtained through the two-step hydrothermal and NH4F/TEAOH treatments were named as ‘recrystallised zeolite’ and ‘defect-healed zeolite’, respectively. Catalytic tests were performed on the parent, the recrystallised, and the defect-healed zeolites in the MTO reaction.
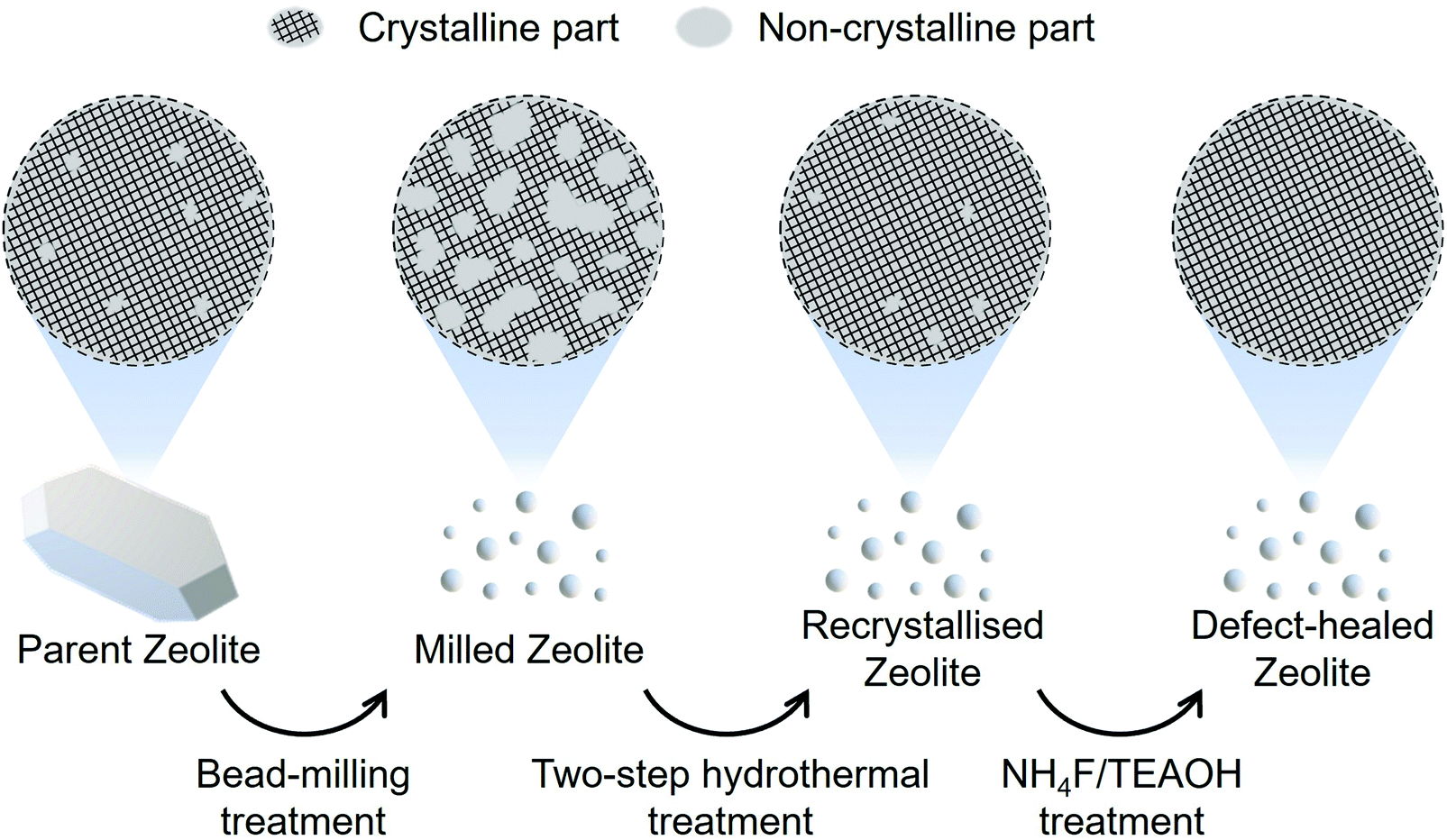 | ||
| Fig. 1 Schematic illustration of the synthesis of the defect-healed CON-type zeolite nanocatalyst using the presented protocol. | ||
2. Experimental section
2.1. Preparation of zeolites
The CON-type zeolite was synthesised following a previously reported method,30 using in-house-fabricated N,N,N-trimethyl-(−)-cis-myrtanylammonium hydroxide (TMMAOH) as an organic structure-directing agent (OSDA) and other raw materials, such as colloidal silica (Cataloid SI-30), aluminium sulfate, boric acid, and sodium hydroxide. The reactant composition was 2.0 SiO2:0.080 H3BO3:0.0025 Al2(SO4)3:0.20 NaOH: 0.40 TMMAOH: 42 H2O. Subsequently, 2 wt% of the fabricated silica-based CON-type zeolite was added as seed crystals to the mixture. After 96 h of hydrothermal treatment at 170 °C, the resultant mixture was separated into solid and liquid components by filtration, hereafter referred to as the parent zeolite and the supernatant, respectively. In the supernatant, silicate species were dissolved/dispersed as the main component (the mass ratio of Si was 1.23%). For the wet bead-milling, 20 g of the parent zeolite was mixed with 100 g of water and subjected to bead-milling using LMZ05 (Ashizawa Finetech). The rotor was spun at a speed of 9 m s−1 using 300 μm zirconia beads for 30 min. For the first recrystallisation, 0.2 g of the milled zeolite was dispersed into 2 g of the supernatant and sealed in an autoclave. The autoclave was placed in an oven set at 140–170 °C for 16–20 h and then cooled to room temperature. The resultant mixture was washed with water and dried overnight at 80 °C. To 0.2 g of the obtained sample, the procedure of the hydrothermal treatment using 2 g of the supernatant, washing and drying was applied as the second recrystallisation. For defect-healing, 2 g of the recrystallised zeolite was mixed with 4 g of an aqueous solution of NH4F and TEAOH, with a chemical composition of 0.1 TEAOH : 0.74 NH4F : 15.19 H2O. The mixture was transferred to an autoclave and heated at 170 °C for 24 h at a rotating speed of 22 rpm. The resultant mixture was washed with water and dried at 80 °C to yield a defect-healed zeolite. All zeolite samples were calcined at 600 °C for 6 h in air to eliminate OSDA cations.2.2. Characterisation
Powder X-ray diffraction (PXRD) patterns were obtained using a powder X-ray diffractometer (Ultima IV, Rigaku Corp.) with a Cu Kα radiation (λ = 0.15406 nm) at 40 kV and 40 mA over an angular range of 2θ = 3–50° with a scanning step and speed of 0.02° and 4° min−1, respectively. The relative crystallinities of the samples were calculated from the integrated peak intensities of the diffraction peaks corresponding to the 130, 510, 403, and 040 reflections. The parent zeolite was used as reference for 100% relative crystallinity. Nitrogen adsorption–desorption measurements were performed using an Autosorb-iQ2-MP (Anton Paar) device at the boiling point of liquid nitrogen (−195.8 °C). The samples were degassed at 350 °C for 6 h under vacuum. The chemical compositions of the samples were analysed using an inductively coupled plasma-atomic emission spectrometer (ICP-AES, Thermo Scientific iCAP-6300). Fourier transform infrared (FT-IR) spectroscopy was performed using an FT/IR-6600 spectrometer (JASCO) equipped with a mercury cadmium telluride detector. Prior to the measurement, the samples were preheated at 500 °C for 1 h under a N2 flow. After cooling for 30 min to 25 °C, the FT-IR spectra of the samples were recorded at a resolution of 4 cm−1 in the diffuse reflectance mode. Acidity of the samples was examined by temperature-programmed NH3 desorption (NH3-TPD). NH3-TPD profiles were recorded on a BELCAT II (MicrotracBEL Corp.) using a thermal conductivity detector (TCD). Solid-state magic-angle spinning nuclear magnetic resonance (MAS NMR) spectroscopy was performed using a JEOL ECA-600 spectrometer (14.1 T) equipped with an additional 1 kW power amplifier. The 27Al chemical shift was referenced to AlNH4(SO4)2·12H2O at −0.54 ppm, and the 29Si chemical shift was measured relative to polydimethylsiloxane (−34 ppm). The samples were spun at 15 kHz using a 4 mm ZrO2 rotor. The surfaces, morphologies, and sizes of the samples were observed using field-emission scanning electron (SEM, JSM-7000F or JSM-7400F, JEOL) and transmission electron microscopies (TEM, JEM-2000EX, JEOL).2.3. MTO reaction
MTO reactions were performed using a fixed-bed reactor equipped with an online gas chromatograph (GC-2014, Shimadzu). Catalytic reactions were conducted in a 6 mm quartz tubular flow microreactor loaded with a mixture of H-type zeolite (33 mg) and quartz sand (470 mg). At a temperature of 500 °C and at an absolute pressure of 0.1 MPa, the MTO reaction was started when methanol was introduced into the reactor using a microfeeder. 50% methanol diluted with N2 was used to achieve a weight hourly space velocity of 15 g-methanol g-catalyst−1 hr−1. The product stream was analysed using an automatic injection gas chromatograph connected directly to the reactor outlet using a heated transfer line.3. Results and discussion
3.1. Characterisation of the zeolites
Fig. 2(a) shows the scanning electron microscopy (SEM) images of the zeolite samples. The parent and the milled zeolite particles possessed prism-like and round morphologies, respectively. The recrystallised and the defect-healed zeolites possessed round morphologies similar to those of the milled zeolites. Fig. 2(b) shows a comparison of the size distributions of the zeolite samples. The parent zeolite had a broad size distribution in the range of 150–850 nm, and the size of the zeolite particles decreased to a range of 50–450 nm after the wet bead-milling. After the two-step hydrothermal and NH4F/TEAOH treatments, no significant change was observed in the zeolite particle size, and thus, the grain growth of fine particles was suppressed.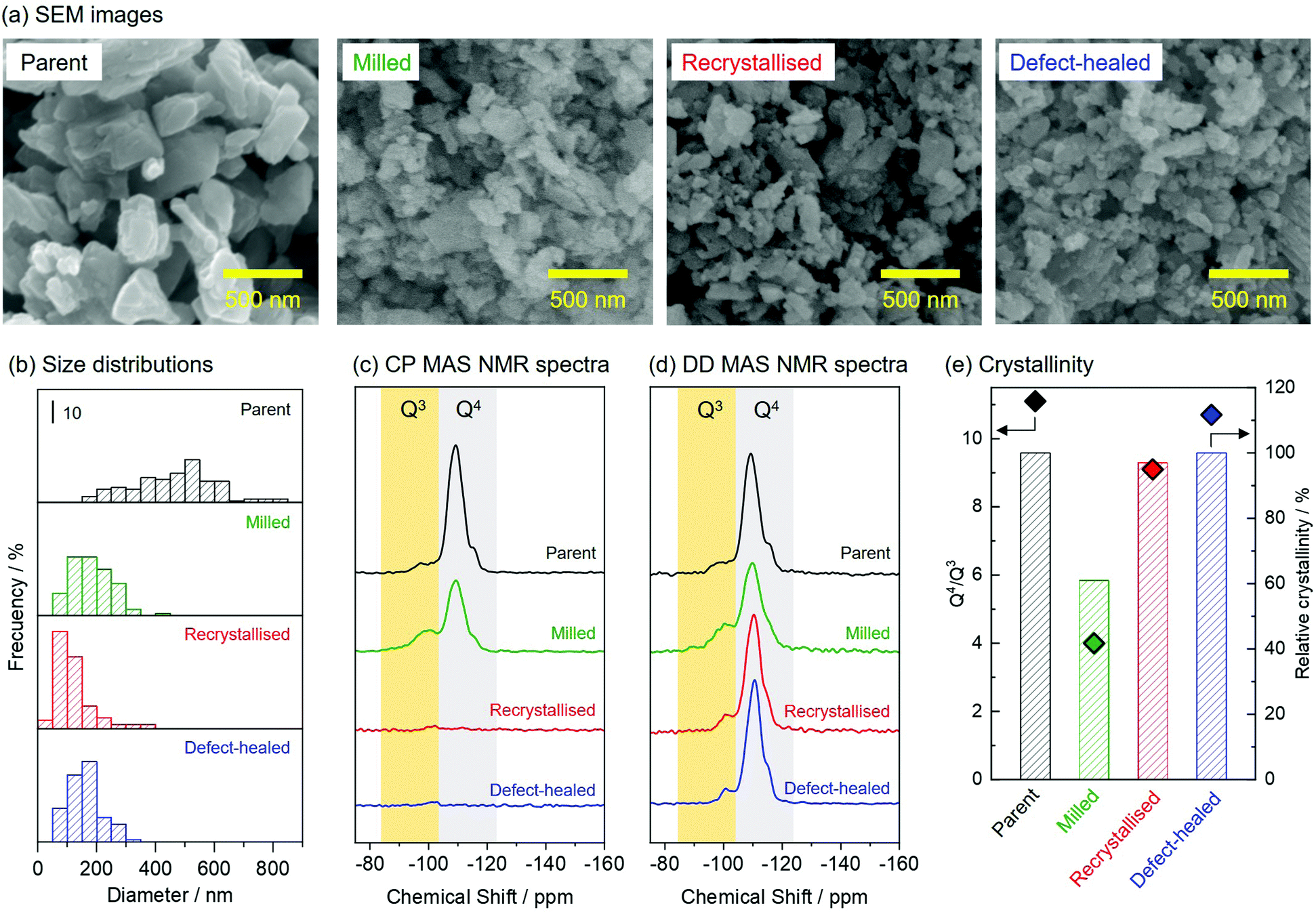 | ||
| Fig. 2 (a) SEM images, (b) particle size distributions, (c) and (d) 29Si CP and DD MAS NMR spectra, and (e) Q4/Q3 ratios and relative crystallinities of the zeolite samples. | ||
Fig. 2(c) and (d) shows the 29Si cross-polarisation magic-angle spinning (CP MAS) and dipolar decoupling (DD) nuclear magnetic resonance (MAS NMR) spectra of the zeolite samples. The CP MAS NMR spectrum of the parent zeolite contained two peaks at −100 and −110 ppm, which were attributed to Q3 and Q4 species, respectively.31 These peaks derived from Q3 and Q4 species could also be observed in the CP MAS NMR spectrum of the milled zeolite. Q3 and Q4 signals were barely observed in the CP MAS NMR spectra of the recrystallised and the defect-healed zeolites but were observed in the DD MAS NMR spectra. This confirmed that the parent and the milled zeolites had silanol groups as defect sites in their frameworks. The differences in the defective structures were quantitatively compared. Fig. 2(e) shows the Q4/Q3 ratios calculated from 29Si DD MAS NMR spectra (deconvoluted as shown in Fig. S1, ESI†) and the relative crystallinities of the zeolite samples estimated from their powder X-ray diffraction (PXRD) patterns (shown in Fig. S2, ESI†). After the wet bead-milling, both the Q4/Q3 ratio and the relative crystallinity decreased, indicating that the framework of the parent zeolite was degraded by the mechanical treatment. The transmission electron microscopy (TEM) image of the milled zeolite confirmed the formation of non-crystalline parts upon the wet bead-milling (Fig. S3, ESI†). It was found that two-step hydrothermal treatment on the milled zeolite under various conditions recovered the damaged parts without forming impurities (see Section 3.2.). Under optimised reaction conditions, the non-crystalline parts of the milled zeolite recrystallised, increasing the relative crystallinity from 61 to 97%. Finally, the relative crystallinity of the CON-zeolite increased to 100% after NH4F/TEAOH treatment, similar to that of the parent zeolite. The Q4/Q3 ratios increased during the two-step hydrothermal and NH4F/TEAOH treatments. Thus, this protocol allowed us to obtain fine particles of CON-zeolite with high crystallinity.
The physicochemical properties of the zeolite samples are listed in Table 1. The N2 adsorption–desorption isotherms for the zeolite samples were type I owing to the microporous features (Fig. S4, ESI†). All zeolites exhibited a high external surface area (>100 m2 g−1). The parent zeolite possessed a micropore volume of 0.22 cm3 g−1, which decreased after the wet bead-milling owing to the partial degradation of the zeolite framework. The micropore volume increased after the recrystallisation and NH4F/TEAOH treatments, indicating a successful recrystallisation of the damaged parts in the milled zeolite. ICP-AES measurement for the samples confirmed that four zeolite samples were obtained as aluminoborosilicate CON-type zeolites. In this study, a CON-type zeolite with a high silica content (Si/Al = 315) was used as the parent zeolite. Wet bead-milling promoted desilication, which decreased the Si/Al ratio of the milled zeolite down to 240. The recrystallised zeolite showed a further decrease to 201, suggesting a partial dissolution of the amorphous silica contained in the damaged parts of the milled zeolite. The NH4F/TEAOH treatment did not cause a significant change in the Si/Al ratio. It should be emphasised that this protocol yielded defect-healed CON-type zeolites with a Si/Al ratio as high as 202.
| S micro /m2 g−1 | S ext /m2 g−1 | V micro /cm3 g−1 | V total /cm3 g−1 | Si/Al | Si/B | |
|---|---|---|---|---|---|---|
| a Calculated by the t-plot method at P/P0 = 0.05–0.5. b Calculated as the molar volume of adsorbed nitrogen at P/P0 = 0.95. | ||||||
| Parent zeolite | 530 | 160 | 0.22 | 0.37 | 315 | 93.5 |
| Milled zeolite | 450 | 140 | 0.18 | 0.42 | 240 | 69.3 |
| Recrystallised zeolite | 390 | 260 | 0.16 | 0.52 | 201 | 367 |
| Defect-healed zeolite | 490 | 130 | 0.20 | 0.42 | 202 | 356 |
3.2. Optimising the hydrothermal conditions in the two-step recrystallisation process
In this study, the milled zeolite recrystallised in the alkaline solutions obtained in the synthesis of the parent zeolite30 (see Experimental Section). In post-milling recrystallisation, the reaction conditions should be carefully controlled to recover the microporous structures.23,25 We optimised the various reaction conditions for the two-step recrystallisation process, such as the reaction time, temperature, and concentration of supernatant, to yield nanosized CON-type zeolites with a high crystallinity and microporosity. Fig. 3(a) shows the PXRD patterns of the milled zeolite and three representative recrystallised zeolites, including one prepared under the optimised conditions. After the first recrystallisation treatment using the original supernatant at 200 °C for 20 h, the relative crystallinity of the CON-type zeolite increased from 61 to 84%, and no impurities were observed by PXRD; however, this was insufficient. To further improve the relative crystallinity, a second recrystallisation treatment was performed on the first recrystallisation sample; however, this promoted the formation of cristobalite impurities (the enlarged view of PXRD patterns was shown in Fig. S5, ESI†), resulting in the formation of a poorly recrystallised zeolite (RC = 65%). The SEM images of the recrystallised zeolites are shown in Fig. 3(b). The recrystallised zeolite obtained after the first recrystallisation retained the round morphology of the milled zeolite. The SEM images of the zeolite obtained after the second recrystallisation showed cristobalite crystals with rod-like shapes in addition to the round particles, suggesting that the decrease in crystallinity of the zeolite after the second recrystallisation was caused by the excessive dissolution of the zeolite. The reaction conditions in the two-step hydrothermal treatment were optimised for the recrystallisation of the milled zeolite. Table S1, ESI† summarises the products obtained after two-step hydrothermal treatments performed at various temperatures, relative crystallinities of the recrystallised zeolites, and solid yields. Among the tested conditions, recrystallised zeolites showed improved crystallinities (83 and 89%) compared to milled zeolites (61%) in two cases (first temp. = 170 °C and second temp.= 140 °C; first temp. = 200 °C and second temp.= 170 °C). The PXRD patterns of the other tested conditions indicated the formation of impurities, such as layered silicate and cristobalite (Fig. S6, ESI†). The inset in Fig. S6, ESI† shows clearer cristobalite diffraction peaks at relatively high temperatures (170 and 200 °C) of the second recrystallisation treatment, suggesting an excessive dissolution of the zeolite at such high temperatures.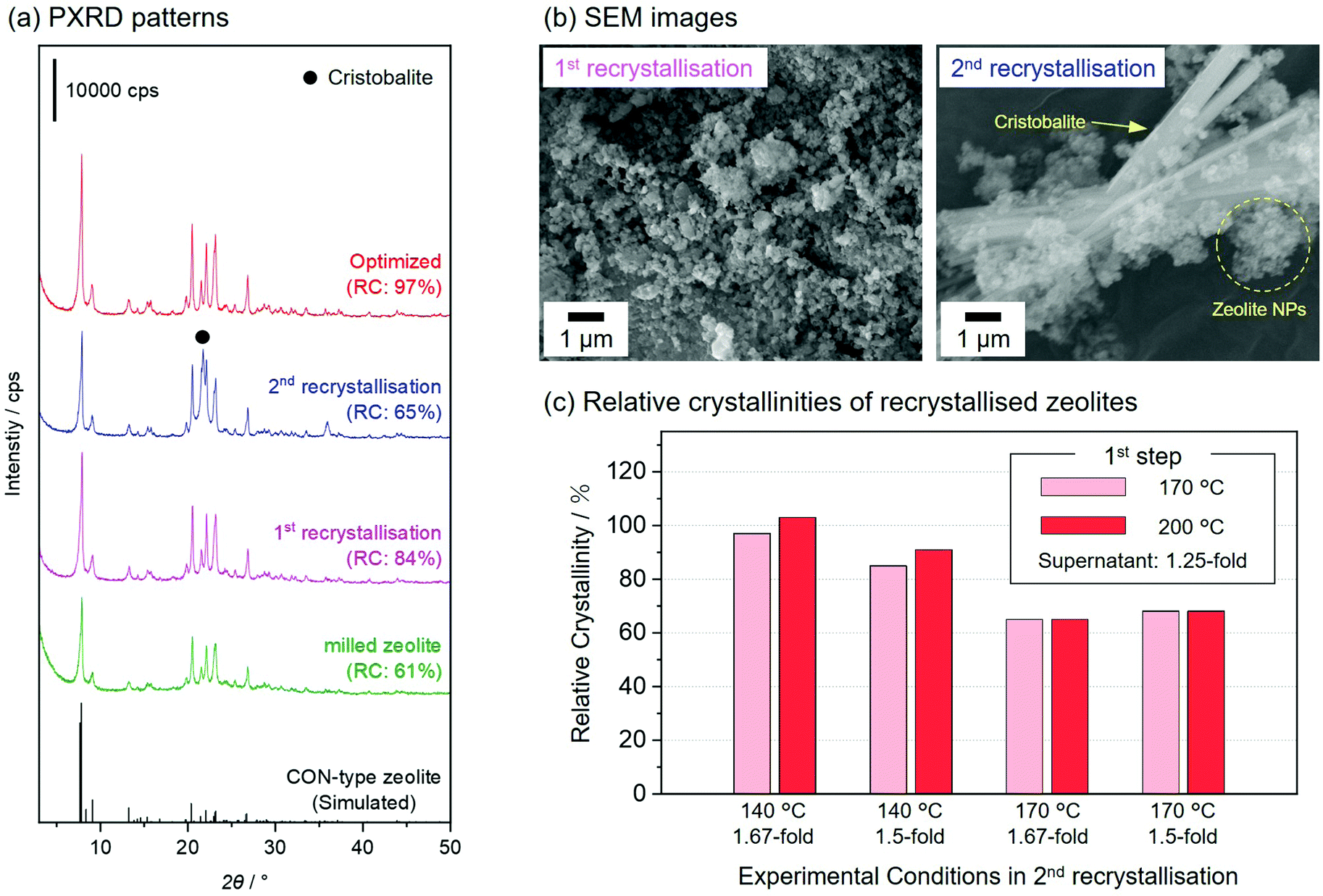 | ||
| Fig. 3 (a) PXRD patterns of milled zeolite (green) and representative recrystallised zeolites including one prepared under optimised conditions (red; optimised). Other samples were subjected to post-milling recrystallisation reactions using the original supernatant at 200 °C for 20 h one (first recrystallisation) or two (second recrystallisation) times, respectively. (b) SEM images of recrystallised zeolites after the first and second recrystallisation. (c) Relative crystallinity of zeolites recrystallised at various temperatures. Details of the reaction conditions are given in Table S2 (ESI).† | ||
We found that highly concentrated supernatants, prepared by the evaporation of water from the original supernatant obtained in the synthesis of the parent CON-type zeolite,30 further improved the crystallinity of the recrystallised zeolites in the two-step hydrothermal reaction. Fig. 3(c) shows the relative crystallinities of recrystallised zeolites via two-step recrystallisation treatments using concentrated supernatants (ranging from 1.25- to 1.67-fold concentrated). Zeolites recrystallised using a 1.67-fold concentrated supernatant at 140 °C in the second recrystallisation showed higher relative crystallinities (97 and 103%) than those recrystallised using a 1.5-fold concentrated supernatant (85 and 91%). Large amounts of silicate species were dissolved/dispersed into the reaction media using a highly concentrated supernatant, suppressing the dissolution of the milled zeolite throughout the post-milling recrystallisation. It is noteworthy that the relative crystallinities of the recrystallised zeolites did not improve when the reaction temperature was 170 °C in the second recrystallisation, despite the use of highly concentrated supernatants. This was because the high temperature excessively promoted the dissolution of zeolites and the formation of impurities (Fig. S7, ESI†). Optimal conditions for recrystallisation (first and second recrystallisations at 170 and 140 °C in 1.25- and 1.67-fold concentrated supernatants, respectively) resulted in pure and highly recrystallised (RC = 97%) CON-type zeolite nanoparticles with the highest solid yield (71%) among the tested conditions (Table S2, ESI†). In this study, a recrystallised zeolite with a relative crystallinity of 97% underwent a defect-healing treatment that increased the relative crystallinity to 100%, as shown in Fig. 2. In contrast, the defect-healed sample prepared without post-milling recrystallisation had a relative crystallinity of 72% (Fig. S8, ESI†). Thus, a carefully controlled two-step recrystallisation treatment was required for synthesising nanosized CON-type zeolites with high crystallinity.
3.3. MTO reaction over zeolites
Fig. 4 shows the results of the catalytic tests for the MTO reaction over the parent, the recrystallised, and the defect-healed zeolites. The conversion rates of methanol over the zeolite samples are compared in Fig. 4(a). The three zeolites exhibited conversion rates of 100% in the initial stage (<6 h). However, the parent zeolite was rapidly deactivated, and its conversion rate decreased from 100% after 7 h, owing to a coke formation in its microporous structure. The recrystallised zeolite showed an increased lifetime of 9 h owing to its increased external surface area. In contrast, the defect-healed zeolite exhibited a remarkably increased lifetime of 15 h. Compared to the previously reported results for the CON-type zeolite catalysts for an MTO reaction19 synthesised via hydrothermal and alkaline treatments, the defect-healed zeolite in this study exhibited a 100% conversion rate for a longer time, under similar reaction conditions. Fig. 4(b) and (c) shows the product selectivity for MTO reactions over the parent, recrystallised, and defect-healed zeolites. The three zeolite samples produced ethene (C2), propene (C3), butene (C4), bulky hydrocarbons (over-C5), aromatics, and paraffins under the present conditions. At the initial stage (<6 h), all three samples showed a high selectivity toward propene (C3) as the main product (higher than 40%). Furthermore, the product selectivities toward ethene (C2) of the three zeolites were as low as 5% during the catalytic tests. The three zeolites exhibited a high selectivity of more than 60% toward C3–C4 olefins, and the formation selectivities of the three zeolites toward light olefins (ethene, propene, and butene) were almost identical. Nevertheless, the selectivity toward over-C5 species increased after 7 h over the parent zeolite. An increase in the selectivity toward over-C5 species was also observed in the recrystallised and the defect-healed zeolites. It is well-known that a reduction in the acid density suppresses the coke formation.32 As shown in Table 1, the parent zeolite had a high silica content (Si/Al = 315) compared to those of the recrystallised and the defect-healed zeolites. The increased acid densities for the recrystallised and the defect-healed zeolites were also observed in NH3-TPD measurement (Fig. S9, ESI†). Although the acid densities of the parent zeolite increased after the two-step recrystallisation and defect-healing processes, the recrystallised and the defect-healed zeolites exhibited prolonged lifetimes. This could be explained by the effect of the reduced diffusion path length originating from the reduction of the particle sizes.21 Furthermore, it should be noted that the defect-healed zeolite exhibited a longer lifetime (15 h) than that of the recrystallised zeolite (9 h), even though there was no significant difference in their particle sizes and chemical compositions (Si/Al and Si/B ratios).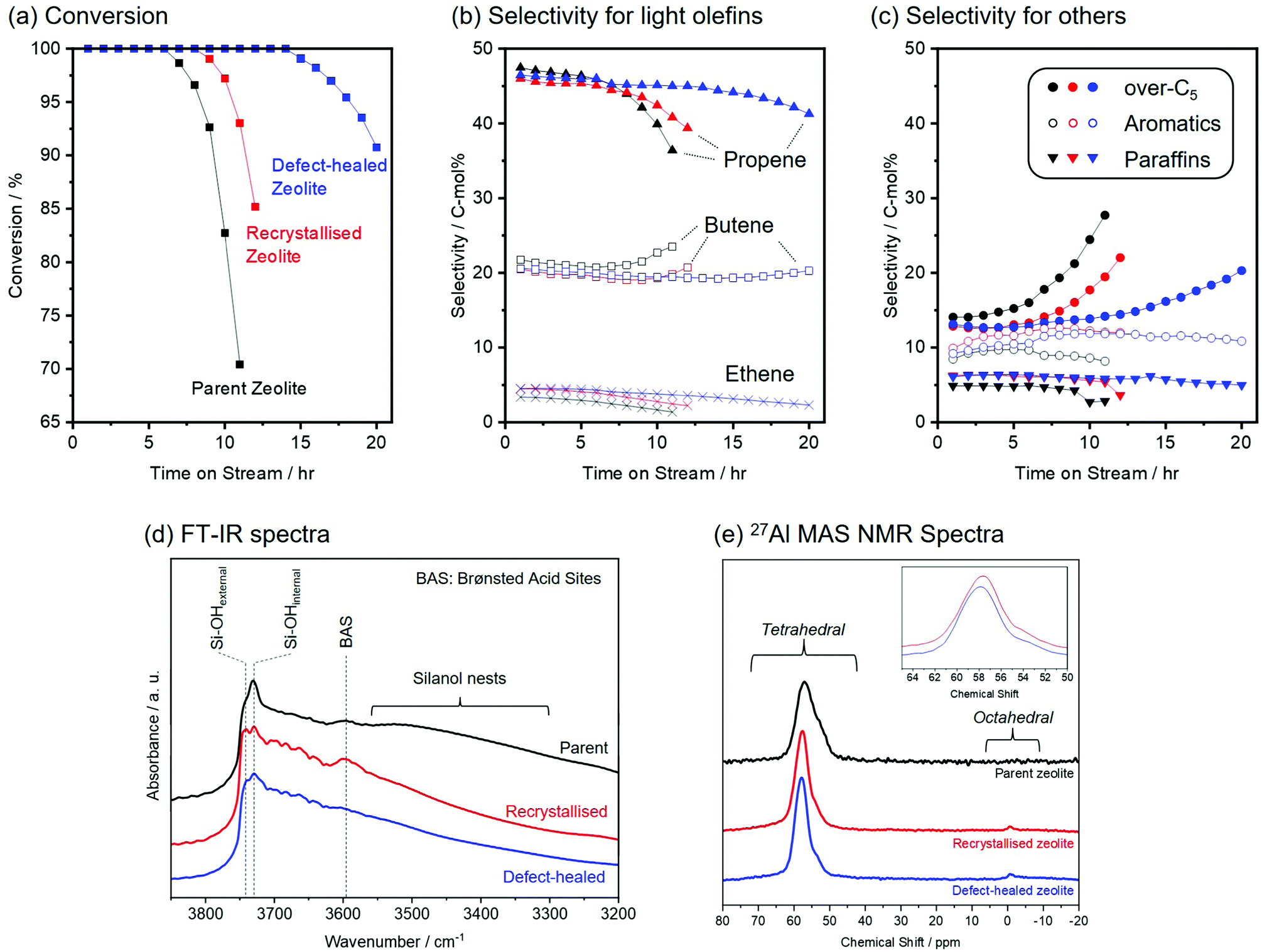 | ||
| Fig. 4 Results of the MTO reaction over the parent zeolite (black), recrystallised zeolite (red) and defect-healed zeolite (blue). (a) Methanol conversion rate and (b) and (c) selectivity toward the products. (d) FT-IR spectra and (e) 27Al MAS NMR spectra of the zeolite samples. | ||
Fig. 4(d) shows the Fourier-transform infrared (FT-IR) spectra of zeolite catalysts. In the spectrum of the parent zeolite, the FT-IR band and the shoulder peak at 3730 and 3741 cm−1 corresponded to silanol groups located on the internal and external surfaces, respectively.33 Furthermore, small and broad bands at 3600 and 3560–3260 cm−1 were observed, corresponding to the Brønsted acid sites formed by the OH group located between the Si and Al atoms and silanol nests, respectively.27 In addition to these bands, the recrystallised and the defect-healed zeolites showed FT-IR bands originating from Al–OH and B–OH in the region of 3710–3620 cm−1 (shown in Fig. S10, ESI†). In the FT-IR spectra of the recrystallised and the defect-healed zeolites, the relative peak intensities of external silanol groups with respect to those of the internal ones were higher than those of the parent zeolite. This corresponded to the downsizing of the CON-zeolite particles. The bands corresponding to silanol nests disappeared in the FT-IR spectra of the recrystallised and the defect-healed zeolites, further demonstrating the removal of defective silanol groups. It should be noted that the removal of silanol groups was also confirmed by 29Si MAS NMR and PXRD analyses (Fig. 2). In the system of large-pore zeolites, the dual-cycle concept proposed by Olsbye et al.8,34 has been widely accepted, wherein the MTO reaction occurs via olefin- and aromatic-based cycles. In the olefin-based cycle, higher alkenes are catalytically converted into olefins. Barbera et al. proposed that the silanol defects in a zeolite framework favour an aromatic-based cycle,27 which involves the formation of benzene, toluene, ethylbenzene, and xylene, which are well-known coke precursors.35 The longer lifetime of the defect-healed zeolite compared to that of the recrystallised zeolite can be attributed to the efficient removal of silanol groups, leading to the suppression of the aromatic-based cycle in this case. However, other factors and environments of Al species might also play a role in enhancing the lifetime of CON-type zeolites in this study. The changes in the Al environments were carefully investigated using spectroscopy.
Fig. 4(e) shows the 27Al MAS NMR spectra of the parent, the recrystallised, and the defect-healed zeolites. In the 27Al MAS NMR spectrum of the parent zeolite, a peak at ∼60 ppm, associated with tetrahedrally coordinated Al (Altet) in the zeolite framework, was observed. The 27Al MAS NMR spectra of the recrystallised and the defect-healed zeolites exhibited two peaks at approximately 60 and 0 ppm. The small peak at approximately 0 ppm originated from octahedrally coordinated Al (Aloct) in the external framework.36 The integrated ratios of Altet/Aloct in the recrystallised and the defect-healed zeolites were similar. Notably, the peak originating from Altet species in the 27Al MAS NMR spectrum of the defect-healed zeolite shifted to a lower field compared to that in the 27Al MAS NMR spectrum of the recrystallised zeolite (shown in Fig. 4(e) inset). This peak shift suggested that Altet was located in different environments in the recrystallised and the defect-healed zeolites. In the normalised FT-IR spectra of the recrystallised and the defect-healed zeolites (shown in Fig. S10, ESI†), these spectra do not entirely overlap, supporting the interpretation of 27Al MAS NMR spectroscopy. In the previous reports on CON-type zeolite catalysts in the MTO reaction, the peak shifts derived from the tetrahedral Al species in 27Al MAS NMR spectra also affected the catalytic performance.15,18,19 In the system of ZSM-5, it has been demonstrated that the reaction pathway for the MTO reaction is determined by aluminium siting in the zeolite framework.37 Although further investigation is required, the features derived from different environments suggest the possibility of controlling atomic-scale sites and/or distribution of Al species using the presented protocol.
4. Conclusions
This study demonstrated the synthesis of a robust CON-type aluminoborosilicate zeolite catalyst for the MTO reaction. Parent CON-type zeolite underwent wet bead-milling, resulting in the formation of nanosized particles (milled zeolite), partially composed of non-crystalline parts. Through careful optimisation of the treatment conditions in the post-milling two-step recrystallisation, the damaged (amorphous) parts of the milled zeolite were recrystallised without forming any impurities. The resultant defect sites in the recrystallised zeolite were healed via an NH4F/TEAOH treatment, as confirmed by PXRD and 29Si MAS NMR spectroscopy. The catalytic tests for the MTO reaction were performed in the parent, recrystallised, and defect-healed zeolites. The defect-healed zeolite exhibited a significantly longer lifetime than those of other zeolites, which was twice that of the parent zeolite. This was due to the dual effects of downsizing zeolite particles and the efficient removal of the defect sites in their frameworks. The presented protocol realised the development of excellent zeolite nanocatalysts by combining unique techniques to improve their catalytic performance and provided the possibility of controlling inorganic frameworks at the atomic scale.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the Japan Technological Research Association of Artificial Photo-synthetic Chemical Process (ARPChem), the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant-in-Aid for Transformative Research Areas (A) (grant number: JP20A206/20H05880), and New Energy and Industrial Technology Development Organization (NEDO) under the Moonshot Project. K. I. would like to thank JST PRESTO for financial support (Grant No. JPMJPR21N3).Notes and references
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922 CrossRef CAS.
- K. Hemelsoet, J. Van der Mynsbrugge, K. De Wispelaere, M. Waroquier and V. Van Speybroeck, ChemPhysChem, 2013, 14, 1526 CrossRef CAS PubMed.
- P. Dugkhuntod and C. Wattanakit, Catalysts, 2020, 10, 245 CrossRef CAS.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613 CrossRef CAS PubMed.
- M. A. Ali, S. Ahmed, N. Al-Baghli, Z. Malaibari, A. Abutaleb and A. Yousef, Catal. Lett., 2019, 149, 3395 CrossRef CAS.
- S. Standl and O. Hinrichsen, Catalysts, 2018, 8, 626 CrossRef.
- L. Lin, M. Fan, A. M. Sheveleva, X. Han, Z. Tang, J. H. Carter, I. da Silva, C. M. A. Parlett, F. Tuna, E. J. L. McInnes, G. Sastre, S. Rudić, H. Cavaye, S. F. Parker, Y. Cheng, L. L. Daemen, A. J. Ramirez-Cuesta, M. P. Attfield, Y. Liu, C. C. Tang, B. Han and S. Yang, Nat. Commun., 2021, 12, 822 CrossRef CAS PubMed.
- U. Olsbye, S. Svelle, M. Bjørgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810 CrossRef CAS PubMed.
- M. Yang, D. Fan, Y. Wei, P. Tian and Z. Liu, Adv. Mater., 2019, 31, 1902181 CrossRef CAS PubMed.
- C. D. Chang and A. J. Silvestri, J. Catal., 1977, 47, 249 CrossRef CAS.
- S. N. Khadzhiev, M. V. Magomedova and E. G. Peresypkina, Pet. Chem., 2014, 54, 245 CrossRef CAS.
- L. Zhang, S. Wang, D. Shi, Z. Qin, P. Wang, G. Wang, J. Li, M. Dong, W. Fan and J. Wang, Catal. Sci. Technol., 2020, 10, 1835 RSC.
- J. Chen, T. Liang, J. Li, S. Wang, Z. Qin, P. Wang, L. Huang, W. Fan and J. Wang, ACS Catal., 2016, 6, 2299 CrossRef CAS.
- D. L. Dorset, S. C. Weston and S. S. Dhingra, J. Phys. Chem. B, 2006, 110, 2045 CrossRef CAS PubMed.
- M. Yoshioka, T. Yokoi and T. Tatsumi, ACS Catal., 2015, 5, 4268 CrossRef CAS.
- R. F. Lobo and M. E. Davis, J. Am. Chem. Soc., 1995, 117, 3766 CrossRef CAS.
- R. F. Lobo, S. I. Zones and M. E. Davis, Stud. Surf. Sci. Catal., 1994, 84, 461 CrossRef CAS.
- S. Park, G. Sato, T. Nishitoba, J. N. Kondo and T. Yokoi, Catal. Today, 2020, 352, 175 CrossRef CAS.
- S. Park, G. Sato, H. Onozuka, S. Tsutsuminai, M. Koike, K. Kuroda, H. Gies, J. N. Kondo and T. Yokoi, Catal. Sci. Technol., 2020, 10, 4293 RSC.
- M. Sawada, T. Matsumoto, R. Osuga, S. Yasuda, S. Park, Y. Wang, J. N. Kondo, H. Onozuka, S. Tsutsuminai and T. Yokoi, Ind. Eng. Chem. Res., 2022, 61, 1733 CrossRef CAS.
- G. Yang, Y. Wei, S. Xu, J. Chen, J. Li, Z. Liu, J. Yu and R. Xu, J. Phys. Chem. C, 2013, 117, 8214 CrossRef CAS.
- T. Wakihara and J. Tatami, J. Jpn. Pet. Inst., 2013, 56, 206 CrossRef CAS.
- T. Wakihara, K. Sato, S. Inagaki, J. Tatami, K. Komeya, T. Meguro and Y. Kubota, ACS Appl. Mater. Interfaces, 2010, 2, 2715 CrossRef CAS.
- Z. Liu, N. Nomura, D. Nishioka, Y. Hotta, T. Matsuo, K. Oshima, Y. Yanaba, T. Yoshikawa, K. Ohara, S. Kohara, T. Takewaki, T. Okubo and T. Wakihara, Chem. Commun., 2015, 51, 12567 RSC.
- H. Yamada, T. Iida, Z. Liu, Y. Naraki, K. Ohara, S. Kohara, T. Okubo and T. Wakihara, Cryst. Growth Des., 2016, 16, 3389 CrossRef CAS.
- T. Wakihara, R. Ichikawa, J. Tatami, A. Endo, K. Yoshida, Y. Sasaki, K. Komeya and T. Meguro, Cryst. Growth Des., 2011, 11, 955 CrossRef CAS.
- K. Barbera, F. Bonino, S. Bordiga, T. V. W. Janssens and P. Beato, J. Catal., 2011, 280, 196 CrossRef CAS.
- P. Sazama, B. Wichterlova, J. Dedecek, Z. Tvaruzkova, Z. Musilova, L. Palumbo, S. Sklenak and O. Gonsiorova, Microporous Mesoporous Mater., 2011, 143, 87 CrossRef CAS.
- K. Iyoki, K. Kikumasa, T. Onishi, Y. Yonezawa, A. Chokkalingam, Y. Yanaba, T. Matsumoto, R. Osuga, S. P. Elangovan, J. N. Kondo, A. Endo, T. Okubo and T. Wakihara, J. Am. Chem. Soc., 2020, 142, 3931 CrossRef CAS PubMed.
- A. Chokkalingam, K. Iyoki, N. Hoshikawa, H. Onozuka, W. Chaikittisilp, S. Tsutsuminai, T. Takewaki, T. Wakihara and T. Okubo, React. Chem. Eng., 2020, 5, 2260 RSC.
- T. Ikeda, S. Inagaki, T. Hanaoka and Y. Kubota, J. Phys. Chem. C, 2010, 114, 19641 CrossRef CAS.
- W. Dai, G. Cao, L. Yang, G. Wu, M. Dyballa, M. Hunger, N. Guan and L. Li, Catal. Sci. Technol., 2017, 7, 607 RSC.
- A. Zecchina, S. Bordiga, G. Spoto, L. Marchese, G. Petrini, G. Leofanti and M. Padovan, J. Phys. Chem., 1992, 96, 4991 CrossRef CAS.
- M. Bjørgen, S. Svelle, F. Joensen, J. Nerlov, S. Kolboe, F. Bonino, L. Palumbo, S. Bordiga and U. Olsbye, J. Catal., 2007, 249, 195 CrossRef.
- F. L. Bleken, K. Barbera, F. Bonino, U. Olsbye, K. P. Lillerud, S. Bordiga, P. Beato, T. V. W. Janssens and S. Svelle, J. Catal., 2013, 307, 62 CrossRef CAS.
- C. A. Fyfe, G. C. Gobbi, J. Klinowski, J. M. Thomas and S. Ramdas, Nature, 1982, 296, 530 CrossRef CAS.
- T. Liang, J. Chen, Z. Qin, J. Li, P. Wang, S. Wang, G. Wang, M. Dong, W. Fan and J. Wang, ACS Catal., 2016, 6, 7311 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Analysis of zeolites prepared in the present protocol (29Si DD MAS NMR spectra, XRD, TEM, N2 adsorption–desorption isotherm, FT-IR spectra, NH3-TPD and solid yield). See DOI: https://doi.org/10.1039/d2ma00343k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |