
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluoride-philic reduced graphene oxide–fluorophore anion sensors†
Akhila A.
Kumaran
a,
Anjali
Chithrambattu
a,
Balaraman
Vedhanarayanan
 b,
Suresh Babu Adukamparai
Rajukrishnan
c,
Vakayil K.
Praveen
bd and
Renuka Neeroli
Kizhakayil
*a
b,
Suresh Babu Adukamparai
Rajukrishnan
c,
Vakayil K.
Praveen
bd and
Renuka Neeroli
Kizhakayil
*a
aAdvanced Materials Research Centre, Department of Chemistry, University of Calicut, Kerala 673 635, India. E-mail: renuka@uoc.ac.in; nkrenu@gmail.com
bPhotosciences and Photonics Section, Chemical Sciences and Technology Division, CSIR-National Institute for Interdisciplinary Science and Technology (CSIR-NIIST), Thiruvananthapuram, Kerala 695019, India
cDepartment of Chemistry, Anna University, Tamil Nadu 600025, India
dAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India
First published on 21st July 2022
Abstract
The detection and quantification of the fluoride ion, one of the most significant anions, have attracted much research interest because of its striking role in oral/bone health and clinical treatment of osteoporosis. A set of F− ion sensors have been reported here, which operate through the fluorescence turn-on response of reduced graphene oxide (rGO)–fluorophore noncovalent conjugate. The developed sensing systems perform well at neutral pH in aqueous solutions, and the response towards F− ions was initiated within seconds. The high specificity for these optical sensors towards F− ions, among a set of significant competing anions, was notable. For the three fluorescent organic dyes selected for study, tetraphenylporphyrin (DTPP), curcumin (DCURN), and coumarin (DCMN), the low-level detection (LOD) ability increased with an increase in π-interactions between rGO and the fluorophore. A fall in LOD to the attomolar level could be achieved for the rGO-DTPP system. The turn-on fluorescence strategy was extended further to develop solid-state sensor strips for F− ion detection at the attomolar level. The fluoride-philic nature of rGO–fluorophore systems was traced by systematic investigations using FT-IR, XRD, and XPS techniques, which revealed that the interaction between the most electronegative F− ion and the rGO in the sensor unit leads to the formation of the stable compound graphite fluoride, and this conversion in turn switches on the quenched fluorescence of the fluorophore.
Introduction
Graphene, the first successfully prepared 2D material, has attracted extensive research interest due to its excellent physico-chemical properties and widespread applications.1 The inherent fine features of this wonderful material can be further modulated by functionalisation with suitable molecular entities.2 Either covalent or noncovalent, these modifications augment the covetable features of graphene manyfold and widen the horizons of this valuable material at the application level. Among these, noncovalent functionalisation of graphene offers the possibility of attaching functional moieties to the latter without disturbing the π-conjugation network of graphene, unlike covalent functionalisation. Noncovalent functionalisation of graphenic systems is mainly based on van der Waals forces and π–π interactions using polymers and molecules with extended π-systems.2d,3 In partially reduced graphenic systems, ionic interactions and H-bonding are also often involved. Interactions involving π-systems are most relevant in the context of the design and fabrication of nanodevices based on graphene, as they can induce dramatic effects in the structure and properties of the system, including dispersibility, mechanical strength, stability, flexibility, electrocatalytic properties, and so on, ensuring extended applications.4–8A parameter that is affected drastically due to the interactions mentioned above is the photophysical properties of molecules bounded with graphene. Under photoexcitation, graphene, an established energy/electron acceptor, quenches the fluorescence of the organic molecule through photoinduced energy/electron transfer, paving the way to excellent chemosensors.8d The low interaction energy <50 kJ mol−1, involved in π–π or CH–π interactions, allows the noncovalent graphene–fluorophore system to be reversible or kinetically labile and enable facile replacement of the fluorophore from graphene, which is manifested as a change in the optical response of the pair. Oxygen-functionalised graphenic systems, viz., graphene oxide (GO) and reduced graphene oxide (rGO), are able to further modulate the above-mentioned interactions. Optical sensors based on noncovalently bound fluorophore–graphenic systems are an ever-expanding area that exploits the fluorescence quenching property of graphenic materials. Exchange of the fluorophore on the carbon framework with a competing analyte that releases the fluorophore realises a turn-on sensor. This strategy has given rise to highly selective and sensitive fluorimetric sensors.9,10 In the present work, the noncovalent assemblies formed between reduced graphene oxide (rGO) and fluorescent organic dyes, 5,10,15,20-tetraphenylporphyrin (DTPP), curcumin (DCURN), and coumarin (DCMN) (Fig. 1), are utilised to develop F− ion sensors manifested through the fluorescence turn-on response, with high selectivity among a group of common anions.
 | ||
| Fig. 1 The molecular structures of the fluorescent organic dyes selected for the development of fluoride ion sensors by noncovalently conjugating with rGO. | ||
Amongst the various biologically important anions studied, F− ions have attracted fundamental research interest owing to their discrete properties, like high charge density and hard Lewis basic nature. As the smallest anion, the F− ion plays a decisive role in environmental, biological, medical, and industrial fields. F− ions are easily absorbed and accumulated by the body. In daily life, F− ions are supplemented through drinking water and toothpaste due to their potential role in preventing dental caries and treating osteoporosis. However, high and excessive exposure to F− is hazardous to plants, marine life, animals, and humans. Acute, high-level exposure to fluoride leads to gastric and kidney problems, abdominal pain, seizures, and muscle spasms. Many diseases, including Down's syndrome, low intelligence, thyroid problems, cardiovascular diseases, neurological problems, immunotoxic effects, mitochondrial dysfunction, and even cancer, are also likely. The permissible dose of tolerance of F− ions in drinking water is 1.5 parts per million (ppm), ∼78.9 μM, as per WHO guidelines. Various methods, including optical,11 chemodosimetric,12 and electroanalytical,13 have been widely investigated.
The rGO–fluorophore units studied herein responded to the presence of F− ions at extremely low concentrations. The response was almost instantaneous, occurring in <5 s. The observed limit of detection (LOD) is 251.76 attomolar, 35.50 femtomolar, and 25.22 picomolar, for rGO-DTPP, rGO-DCURN, and rGO-DCMN, respectively. It is worth mentioning that an attomolar level of F− ion detection is unprecedented in the literature. The environmentally benign nature of the fluorophores selected is another added advantage. The pH of the solutions was between 6.5 and 7.0, which ensures ionisation of species like acetic acid, carbonic acid, etc. (precursors to competing anions). Besides, interference from hydroxides will be negligible at this pH. Carbonate and acetate ions of similar basicity to F− ions could not evoke an appreciable response, which is claimed as a merit of the sensor. The results obtained herein, along with our previous work,14,15 reveal the selective affinity (or universal selectivity) of such rGO–fluorophore turn-on sensors to F− ions. Systematic investigation shows that the strong interaction between the most electronegative F− ion and rGO that leads to the formation of a stable compound, graphite fluoride, serves as the driving force for the fluoride-philic nature of the aforementioned systems.
Experimental section
Materials
All the chemicals used in this study were purchased from commercial suppliers and used as received. The fluorophores 5,10,15,20-tetraphenylporphyrin, curcumin, and coumarin used in this study were sourced from Sigma-Aldrich. Fluorophore stock solutions were prepared by initially dissolving the required amount of dye in a DMF–ethanol mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) and subsequently diluting it with deionised water. Graphite powder was purchased from Acros Organics. Hydrazine hydrate and potassium permanganate were purchased from Merck. A modified Hummers' method was adopted to obtain rGO from graphite powder using a hydrazine hydrate mediated route. The details of the synthesis are given elsewhere.10a Stock solutions of the analyte anions (fluoride, chloride, bromide, iodide, nitrate, nitrite, bicarbonate, carbonate, acetate, and sulfate, procured from Merck as either sodium or potassium salts) were prepared by dissolving the required amount of their salts in deionized water. The salts were selected in such a manner that the counter cations of the anions are inert to the sensor.
:1 ratio) and subsequently diluting it with deionised water. Graphite powder was purchased from Acros Organics. Hydrazine hydrate and potassium permanganate were purchased from Merck. A modified Hummers' method was adopted to obtain rGO from graphite powder using a hydrazine hydrate mediated route. The details of the synthesis are given elsewhere.10a Stock solutions of the analyte anions (fluoride, chloride, bromide, iodide, nitrate, nitrite, bicarbonate, carbonate, acetate, and sulfate, procured from Merck as either sodium or potassium salts) were prepared by dissolving the required amount of their salts in deionized water. The salts were selected in such a manner that the counter cations of the anions are inert to the sensor.
Measurements
The electronic absorption spectra were recorded on a JASCO V-550 UV/vis spectrophotometer and the fluorescence spectra were recorded on a PerkinElmer LS-55 fluorescence spectrometer. Optical studies in solution state were carried out using a 1 cm quartz cuvette. The fluorescence spectra were recorded by exciting the samples at the respective excitation wavelengths of the fluorophores. Fluorescence lifetime measurements were carried out using a Horiba (model DeltaFlex) time-correlated single photon counting system. The lifetime values (τ), normalized pre-exponential value (α), and average lifetime (τav) were determined using EzTime decay analysis software. The quality of the fit was judged by fitting parameters, such as χ2 (<1.1), as well as visual inspection of the residuals. Infrared spectra were recorded on a JASCO FT/IR-4100 Fourier Transform Infrared Spectrometer using the KBr pellet method. X-ray photoelectron spectroscopy (XPS, PHI 5000 VersaProbe II, ULVAC-PHI Inc., USA) equipped with a micro-focused (200 μm, 15 kV) monochromatic Al-Kα X-ray source (1486.6 eV) was used for identifying the elements in the carbon material. Both survey spectra and narrow scans (high-resolution spectra) were recorded. Survey scans were recorded with an X-ray source with power of 50 W and pass energy of 187.85 eV. High-resolution spectra of the major elements were recorded at 46.95 eV pass energy. XPS data were processed using PHI's Multipak software.X-ray diffraction (XRD) studies were performed on a Rigaku MiniFlex 600 diffractometer with Cu Kα radiation (λ = 1.54 Å). Samples for XRD analysis were prepared by drop casting an aqueous solution rGO–F(fluorophore), followed by the complete removal of the solvents under a high vacuum. Scanning electron microscopy (SEM) images were obtained using a Zeiss EVO18cryo SEM with a secondary electron detector and an accelerating voltage of 15 kV after sputtering with gold–palladium alloy. Samples were prepared by dropcasting the aqueous solution of rGO-F(DTPP) over smooth aluminum foil and drying in air overnight.
Solution-state detection of fluoride ions using rGO–fluorophore
2.5 mL of 10−12 M fluorophore solution (initially dissolved in 1:1 DMF–ethanol mixture and subsequently diluted with water) was taken in a cuvette, and aliquots of rGO dispersion were added to the above solution so as to suppress the optical signal fluorescence of the dye. An rGO dispersion in deionised water of concentration 83 mg mL−1 was used throughout the experiment, which was subjected to ultrasonication for 15 min prior to the experiment. The interaction between the fluorophore and rGO was monitored using UV/vis absorption and fluorescence spectroscopy. The emission spectra were recorded after each addition of rGO dispersion, and a decrease in the emission intensity of the fluorophore was noted. The rGO–fluorophore noncovalent conjugate thus prepared constitutes the sensor unit here. The pH of the solutions of rGO-DTPP, rGO-DCURN, and rGO-DCMN sensors were determined as 6.59, 6.63, and 6.73, respectively. The sensor systems were stable for up to two weeks, after which a slight decrease in efficiency was noted due to factors like aggregation and oxidation of rGO. The selectivity of the sensors was checked by adding solutions of different anions prepared in deionised water to the rGO-fluorophore solution (2.5 mL fluorophore and 20–60 μL rGO dispersion). Quantitative determination of F− ions was done by measuring the emission intensity of the system after administering various concentrations of ions, prepared by dissolving requisite amounts of salt in deionized water.
rGO–fluorophore sensing strips for the detection of fluoride ions
A solid-phase filter paper-based sensor strip was developed using the most efficient sensor system, rGO-DTPP. The test strips were obtained by immersing fine-quality paper strips in rGO-DTPP solution for 1 h followed by drying at 60 °C for 1 h. The sensing ability of the sensing strips was tested under UV light illumination (365 nm) after adding the requisite amount of F− ion solution to the strip. The performance of the strip was found to be satisfactory for about two weeks.Results and discussion
rGO–fluorophore noncovalent conjugate for the selective detection of fluoride ions
Characterisation of the prepared rGO samples using XRD analysis, combined with FTIR spectral data and Raman spectral analysis, indicate a functionalised carbon core in the material (Fig. S1, ESI†). As is evident from the Raman spectral data, a significant amount of sp3 defects was noted, with an ID/IG ratio of 0.92. A C/O ratio of 2.7 is confirmed for the system from the XPS results, with 73 and 27% of carbon and oxygen, respectively (Fig. S2, ESI†). C 1s binding energies at 284.7, 286.9, and 287.77 eV identify C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–O, and CO functionalities indicating a graphenic system functionalised with carbonyl and hydroxyl groups. The detailed characterisation of rGO is described elsewhere.16
C, C–O, and CO functionalities indicating a graphenic system functionalised with carbonyl and hydroxyl groups. The detailed characterisation of rGO is described elsewhere.16
The sensing unit is constituted by combining the fluorophore and rGO. We first conducted a study on the detection of F− ions using an rGO-DTPP noncovalent conjugate. For this purpose, the interaction between rGO and DTPP in an aqueous solution was studied using UV/vis absorption spectroscopy, a tool recognized for exploring the interactions in such systems (Fig. 2a). The Soret band of DTPP, arising due to the a1u(π) to eg*(π) transition, appears at 414 nm, and the four less intense peaks corresponding to the a2u(π) to eg*(π) transition (Q bands) are seen at 517, 555, 592, and 650 nm, which are characteristic of the free-base monomer. rGO had a remarkable influence on the absorbance of DTPP, as the absorbance intensity decreases with the addition of rGO (with rGO dispersion as the reference). However, with an increasing concentration of rGO, the absorption peak of DTPP undergoes a marginal change in absorbance wavelength, an observation justifying the noncovalent linkage of porphyrin with the rGO in the ground state17 where the major contribution arises from π–π interactions of the two moieties.18Fig. 2b shows the fluorescence spectrum of DTPP that exhibits emission peaks at 655 and 717 nm when excited at 410 nm in an aqueous solution. As is evident from the figure, noticeable emission quenching is observed upon adding rGO to the fluorophore solution. These results agree with studies based on other fluorophore–graphenic systems reported previously.19–24 The formation of a non-emissive complex between DTPP and rGO in the ground state is further evidenced by time-resolved fluorescence lifetime studies. The fluorescence decay profiles subjected to bi-exponential fitting provided average lifetime values of 10.29 and 10.20 ns for DTPP and rGO-DTPP, respectively, indicating static quenching (Fig. S3, ESI†). The constant lifetime values of DTPP in the absence and presence of rGO indicate that static quenching operates in the process, as rGO and DTPP form a non-emissive complex in the ground state.17 If dynamic quenching operates, the excited state kinetics of DTPP will be altered, leading to a change in fluorescence lifetime, which is not observed here.
 | ||
| Fig. 2 (a) UV/vis absorption and (b) fluorescence spectra of DTPP (10−12 M) in the absence and presence of rGO (40 μL), λex = 410 nm. The inset of (b) shows a photograph of aqueous solutions of DTPP (10−12 M) and DTPP (10−12 M) + rGO (40 μL) under the illumination of 365 nm UV light. (c) The fluorescence response of rGO-DTPP towards various anions. The concentration of F− ions is 10 μM (100 μL) and that of other ions is 50 μM (100 μL). | ||
We then tested the response of the rGO-DTPP noncovalent conjugate towards various anions. It was observed that among the different anions (Cl−, Br−, I−, F−, SO42−, CH3COO−, NO2−, NO3−, CO32−, and HCO3−) studied, only F− responds to the system, by recovering the rGO-mediated quenched fluorescence of DTPP. The change in fluorescence was noticeable in <5 s (Fig. S4, ESI†). Fig. 2c displays the selectivity of the sensor towards F− ions, illustrating the ability of the rGO-DTPP system to identify F− ions selectively through a fluorimetric response. Interference-free detection of F− in the presence of other competing anions is also evident from the figure, as a mixture of anions devoid of F− ions produces a marginal enhancement in the fluorescence of rGO-DTPP.
It was noted that the fluorescence of the sensor probe, rGO-DTPP, was restored with successive introduction of increasing amounts of F− ions (Fig. 3). The concentration range that evoked such response was significantly wide, from micromolar to commendably low concentrations like the attomolar, ensuring the utility of the system as an F− ion sensor in a wide range of concentrations. The results on picomolar, femtomolar, and attomolar concentrations of F− ions are provided in Fig. 3. The fluorescence variation of the system (λem@655 nm) with F− ion concentration was found to be perfectly linear in all the concentration regions studied, which allow quantitative determination of the F− ion (Fig. 3b, d and f). To the best of our knowledge, rGO-DTPP, with a limit of detection (LOD) of 251.76 attomolar, is superior in sensitivity (in terms of its LOD) to other F− ion sensors reported previously.11a,25–34 Another attractive feature of the present system is the wide linear range of fluorescence variation with the concentration of F− ions, which enabled the highly selective quantification of F− ions over a wide range of concentrations. This is a noteworthy feature, as most sensors lack a wide range of sensing applications. The lowest concentration of F− ions that showed a noticeable enhancement in the fluorescence of the sensor (rGO-DTPP) experimentally was 38.85 attomolar (LOD = 38.85 × 10−18 mol L−1).
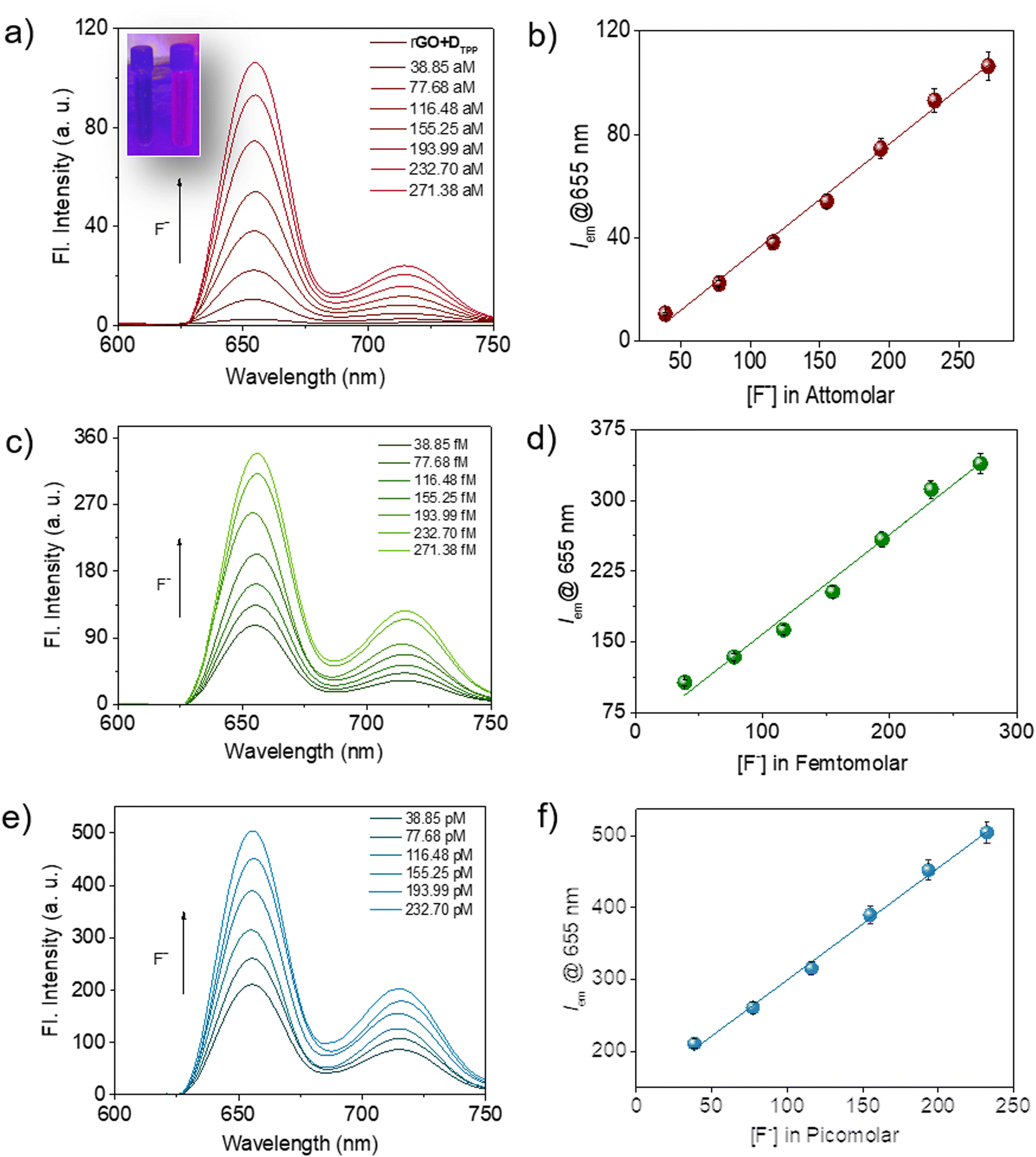 | ||
| Fig. 3 Restoration of quenched fluorescence of rGO-DTPP on adding F− ions in (a) attomolar, (c) femtomolar, and (e) picomolar concentration levels (λex = 410 nm). (b, d and f) Plots showing the variation in fluorescence response (λem@655 nm) with the concentration of F− ions. Inset of (a) shows a photograph of rGO-DTPP (DTPP: 10−12 M, rGO: 40 μL) aqueous solution in the absence and presence of F− ions (10−18 M) under illumination with 365 nm UV light. | ||
Solid-state fluoride ion sensor
To simplify the detection strategy, a solid-state paper sensor strip was developed using the most efficient (in terms of LOD) solution-state F− ion sensor system, rGO-DTPP. The test strips were obtained after immersing the paper strips in rGO-DTPP aqueous solution for 1 h, then drying at 60 °C. The performance of the solid-state sensor for detecting F− ions is shown in Fig. 4. The sensor strip was monitored under 365 UV light illumination after adding an attomolar concentration of F− ions. The appearance of light pink luminescence of DTPP is noted, which indicates the potential of the strip to respond to F− ions at this low concentration. The results demonstrate the potential of rGO-DTPP-based paper strips with simple operation and rapid response for onsite applications for monitoring F− ions at low concentration levels. | ||
| Fig. 4 Image of rGO-DTPP-coated paper strip sensor (left) and strip treated with F− ions (right) under 365 nm UV light illumination. | ||
rGO–curcumin and rGO–coumarin systems as fluoride ion sensors
The noncovalent interactions of rGO with the other two fluorophores, DCURN and DCMN, also resulted in similar fluorescence quenching properties. As shown in Fig. S5a (ESI†), the fluorescence of DCURN observed at 545 nm (λex = 400 nm) showed quenching in the presence of rGO. Similar behaviour is also displayed by DCMN, which showed quenching of emission at 396 nm (λex = 265 nm) in the presence of rGO (Fig. S5b, ESI†). These two systems yielded an interesting observation when evaluated for their anion sensing efficiency. Both these systems respond to the same anion, F−, while all other anions were inert in affecting the optical response in the sensors, analogous to the rGO-DTPP system. Fig. 5 summarises the response of the three systems towards various anions, thus showing the specificity of the systems for F− ions. rGO-DCURN could sense F− ions at concentrations down to the femtomolar range, while rGO-DCMN could detect F− ions at a picomolar level (Fig. S6 and S7, ESI†). Quantitative determination of F− ions was also enabled by these two systems. The LOD values obtained were 35.50 femtomolar and 25.22 picomolar for rGO-DCURN and rGO-DCMN, respectively. The sensitivities achieved were significant compared to several other reported F− ion sensors. Among rGO-DTPP, rGO-DCURN, and rGO-DCMN, rGO-DTPP was most sensitive, with an LOD of 251.76 attomolar, a value unprecedented in the literature.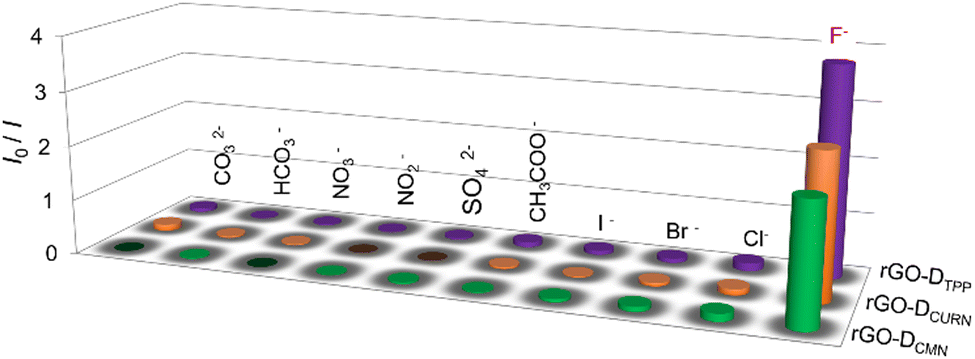 | ||
| Fig. 5 Plot showing selective response of rGO–fluorophore systems towards F− ions. | ||
Fluoride-philic nature of rGO–fluorophore systems
All the rGO–fluorophore fluorimetric sensors studied are selective towards F− ions among the selected anions (Table 1). This result should be read along with our previously reported work on rGO–dye hybrids, which have also shown selectivity towards F− ions (Table 1).14,15 This observation prompted us to gain insight into the factors determining the selectivity of these turn-on rGO–fluorophore anion sensors. To this end, a detailed investigation was conducted using the material obtained after the complete restoration of the fluorescence of the rGO–fluorophore upon adding a sufficient amount of F− ions. This experiment was conducted under the conditions for fluoride detection in the micromolar range. Typically, 80 μL (1 μM) of KF solution was added to a mixture of 2.5 mL (1 pM) fluorophore solution and 60 μL of rGO dispersion. After equilibration, the solid was collected via centrifugation and washed for several cycles with methanol, followed by a final rinse with water. The remaining material was expected to be a combination of F− ions and rGO, devoid of the fluorophores used. The systems thus obtained are labelled as rGO–F(fluorophore), in which the name of the corresponding fluorophore is given in parentheses to identify the respective samples.Fig. 6a presents the SEM image of a representative system, rGO-F(DTPP), revealing piled carbon layers after fluoride ion incorporation. Fig. 6b displays the FT-IR spectra of the various rGO–F(fluorophore) materials. The figure also shows the FT-IR spectrum of graphite fluoride for a meaningful comparison. Comparing the obtained FT-IR spectra with that of rGO (Fig. S1b, ESI†) indicates that the rGO–F(fluorophore) materials are no longer pure rGO. An additional band is observed in the rGO-F(fluorophore) systems at ∼1220 cm−1 corresponding to C–F vibration, as in the case of graphite fluoride,35 and indicates the formation of C–F covalent bond upon the interaction of rGO with F− ions.36,37
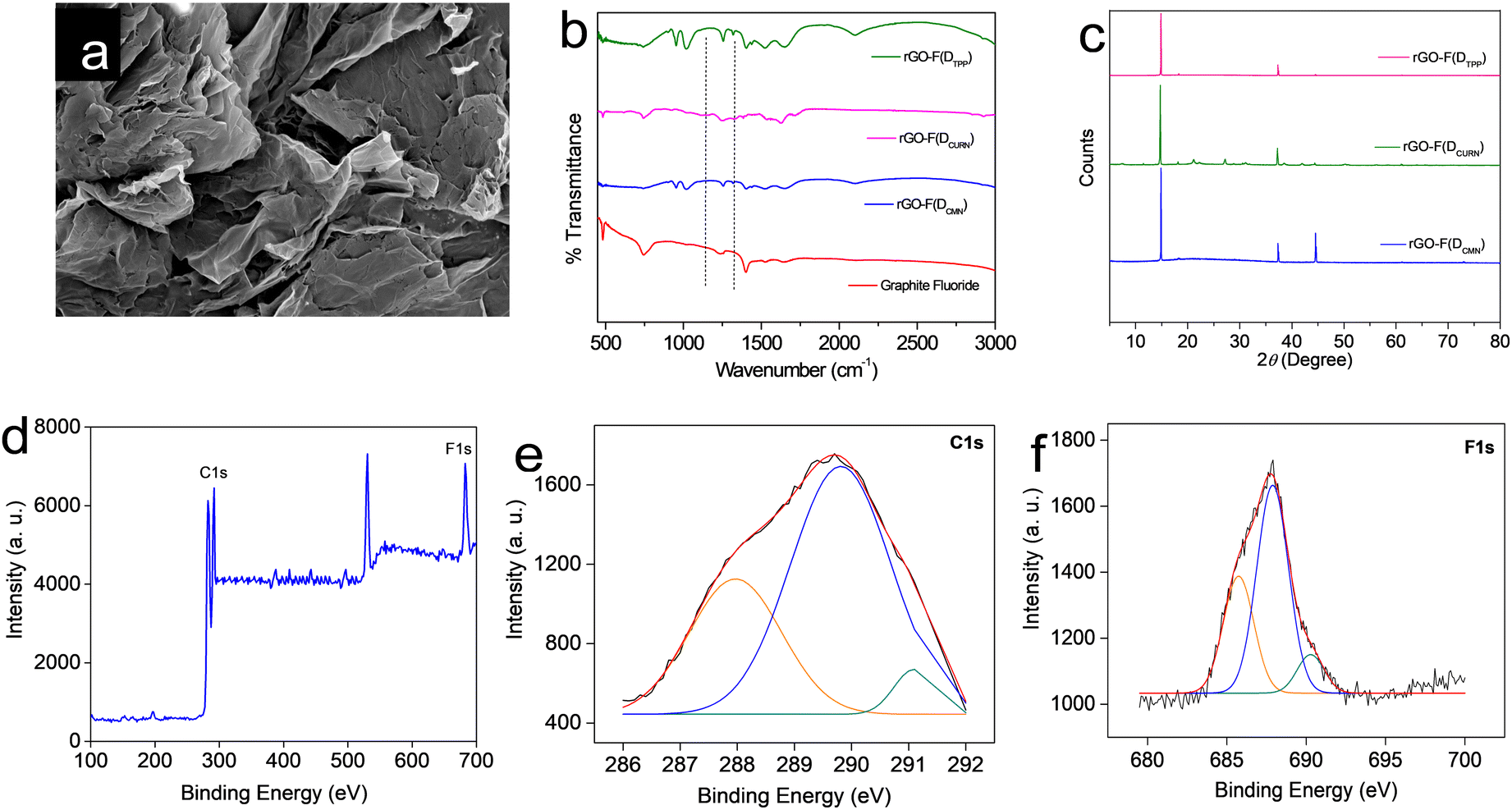 | ||
| Fig. 6 (a) SEM image of rGO-F(DTPP). (b) A comparison of FT-IR spectra of rGO–F(fluorophore) systems with that of graphite fluoride. (c) XRD patterns of rGO–F(fluorophores) systems. (d) Survey XPS spectrum of rGO-F(DTPP). XPS (e) C1s and (f) F1s spectra of rGO-F(DTPP). | ||
Further confirmation of the formation of the C–F bond and conversion of rGO into a stable compound is obtained from the powder XRD patterns of the rGO–F(fluorophore) systems (Fig. 6c). The characteristic peak of rGO observed at ∼25° (Fig. S1a, ESI†) is not observed in these systems. Loss of graphene character and change to a crystalline graphitic nature are evident from the diffraction patterns. The maximum intense XRD peak observed at 2θ = 14–16° is indexed to the (001) reflection of the (CFx)n stacked structure in graphite fluoride.38 The position of the aforementioned peak depends on the degree of fluorination,38,39 the increase in the intensity of which suggests an increasing F/C ratio.38,40 A theoretical study by Rimsza and co-workers indicates that the position of the said peak at 2θ = 14.8° corresponding to a d-spacing of 5.98 Å, as observed herein, signifies a hexagonal crystal structure in graphite fluoride, in which the direction of the C–F bond alternates from one side of the fluorinated graphite layer to the other side between each adjacent C atom, giving rise to a distinct chair conformation for each ring.38 The peak noted at 2θ = 44°, which is more prominent in the rGO-F(DTPP) sample, accounts for the diffraction from the (101) plane in hexagonal graphitic carbon.41
Hence, it is inferred that the high affinity of F− ions towards rGO–fluorophore systems leads to the formation of a stable compound, which is the driving force behind the fluoride-philic nature of rGO–fluorophore sensor systems. This inference is further substantiated by the XPS spectrum of rGO-F(DTPP), chosen as a representative case. The survey XPS spectrum of the system is provided in Fig. 6d. Atomic percentages of the elements present (C – 65.2%, F – 17.3%, and O – 17.5%) assign the CF0.26O0.27 formula to rGO-F(DTPP). Two distinct peaks are observed for C 1s electrons. The peak at ∼284 eV explains carbons bound to other carbons and oxygen species, while the region at ∼290 eV accounts for carbons bound to fluorine atoms. Peaks at 287.9, 289.2, and 291.2 eV represent binding energy values of C1s spectra of semi-ionic C–F, C–F covalent, and CF2 bands, respectively (Fig. 6e).37 A comparison of relative areas in the XPS spectra shows that the C–F state dominates in the system. Peaks in the spectrum with binding energies in the range 684–689 eV correspond to the binding energy values of fluorine in fluorinated graphite.35 The fluorine 1s spectra of semi-ionic C–F bond, C–F, and C–F2 correspond to energy values 686.8, 688.1, and 689.0 eV (Fig. 6f).37 Fluorination of carbon layers is supposed to be at the expense of oxygen bound to carbon. A decrease in oxygen content in graphite fluoride samples compared to the starting graphenic materials upon fluorine incorporation is attributed to the substitution of oxygen functional groups like hydroxyl by the fluoride ions.42 Atomic percentages of oxygen derived from XPS analysis, viz., rGO (27%) (Fig. S2, ESI†) and rGO-F(DTPP) (17.5%) (Fig. 6d), validate this suggestion. The F− ion, being the smallest and most electronegative entity with a high charge density, can exert strong interaction between F− and rGO paving the way to the fluoride-philic nature of noncovalently bound rGO–fluorophore sensor systems.
Thus, the restoration of fluorescence or the release of fluorophores from the rGO–fluorophore complex by F− ions that displace hydroxyl groups with high specificity is attributed to the strong interaction of F− ions with rGO that results in the formation of graphite fluoride, which eventually releases the noncovalently bounded fluorophore, leading to fluorescence enhancement. The detection strategy involved in the process is presented in Scheme 1, taking DTPP as a representative fluorophore.
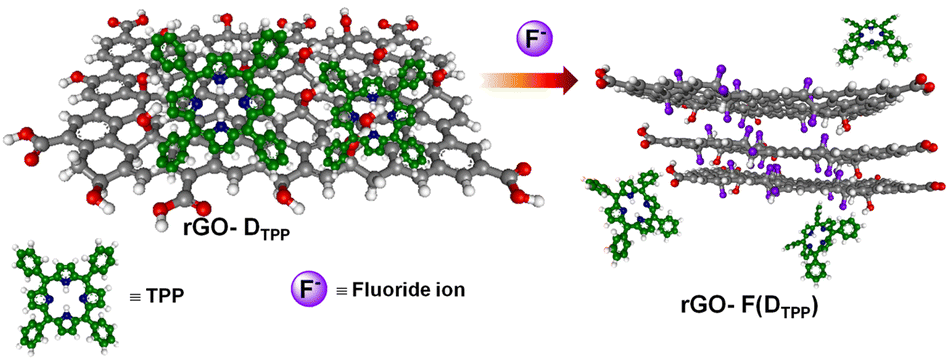 | ||
| Scheme 1 Schematic of F− ion sensing strategy of rGO–fluorophore system. | ||
It can also be seen that, despite the presence of an extended π network, the two fluorophores, Rhodamine 6G and fluorescein, do not exhibit the expected high sensitivity (low LODs) in F− ion detection in the presence of rGO compared to other molecules studied in this work. It is important to note that a combined computational and experimental study by Yu and co-workers proved the strong interactions between the Rhodamine 6G molecule and graphenic systems that involve oxygen-containing functional groups on graphene.43 Similar types of interactions could operate in the case of an rGO–fluorescein system.9a,44 These strong interactions obviously hinder the facile displacement of the fluorophores from rGO in the presence of F− ions, as it reduces the density of the functional groups that serve as the displacement sites for fluorine attachment. This explains the lower sensitivity of rGO–Rhodamine 6G and rGO–fluorescein systems in F− ion detection, despite their extended π-conjugation. This aspect also confirms the involvement of oxygen functionalities in the formation of graphite fluoride reported in the present study.
Conclusions
In summary, rGO–fluorophore noncovalent conjugates, specific for the fluorimetric detection of F− ions in aqueous solutions at lower concentrations, are reported. A turn-on fluorescence sensing modality is realised for these sensors in the presence of the analyte of concern. For the rGO–fluorophore systems studied, tetraphenyl porphyrin (rGO-DTPP), curcumin (rGO-DCURN), and coumarin (rGO-DCMN), the sensitivity order in terms of LOD values of the sensors was rGO-DTPP > rGO-DCURN > rGO-DCMN, giving the impression that the sensitivity increased with the degree of conjugation of the aromatic framework of the fluorophore selected. The LOD values achieved were 38.85 aM, 35.50 fM, and 25.22 pM for rGO-DTPP > rGO-DCURN > rGO-DCMN, respectively. To simplify the detection strategy, a solid-state paper sensor strip was developed using the rGO–fluorophore system immobilised on a paper strip, which showed sensitivity towards F− ions at attomolar concentration. Characterisation of the material obtained after the complete restoration of the fluorescence of the rGO–fluorophore system upon adding a sufficient amount of F− ions using FT-IR, XRD, and XPS revealed that the interaction between the most electronegative F− ion and rGO, resulting in the formation of stable graphite fluoride, justifies the fluoride-philic nature of the aforementioned systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. A. K. thanks for a research fellowship from the Government of Kerala and University of Calicut, Kerala for providing research facilities. A. C. acknowledges the Government of Kerala for the financial assistance through E Grant. V. K. P. is grateful to the Council of Scientific and Industrial Research (CSIR) and Department of Science and Technology (DST), Government of India, Nano Mission (DST/NM/TUE/EE-02/2019) for the financial support. R. N. K. gratefully acknowledges DST, Government of India, for the research facilities provided under FIST.Notes and references
- (a) C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS PubMed; (b) A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed; (c) K. S. Novoselov, V. I. Falko, L. Colombo, P. R. Gellert, M. G. Schwa and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed; (d) W. Ren and H. M. Cheng, Nat. Nanotech., 2014, 9, 726–730 CrossRef CAS PubMed; (e) X. J. Lee, B. Y. Z. Hiew, K. C. Lai, L. Y. Lee, S. Gan, T.-G. Suchithra and S. Rigby, J. Taiwan Inst. Chem. Eng., 2019, 98, 163–180 CrossRef CAS; (f) B. Nassef, G. Nassef and M. A. E. Daha, Int. J. Mater. Eng., 2020, 10, 1–12 CrossRef; (g) Wei Zheng, Xin Zhao and W. Fu, ACS Appl. Mater. Interfaces, 2021, 13, 9561–9579 CrossRef CAS PubMed.
- (a) T. Kuila, S. Bose, A. K. Mishra, P. Khanra, N. H. Kim and J. H. Lee, Prog. Mater. Sci., 2012, 57, 1061–1105 CrossRef CAS; (b) A. Criado, M. Melchionna, S. Marchesan and M. Prato, Angew. Chem., Int. Ed., 2015, 54, 10734–10750 CrossRef CAS PubMed; (c) G. Bottari, M. A. Herranz, L. Wibmer, M. Volland, L. Rodrıguez-Perez, D. M. Guldi, A. Hirsch, N. Martın, F. D’Souza and T. Torres, Chem. Soc. Rev., 2017, 46, 4464–4500 RSC; (d) B. Vedhanarayanan, V. K. Praveen, G. Das and A. Ajayaghosh, NPG Asia Mater., 2018, 10, 107–126 CrossRef CAS.
- V. Georgakilas, J. N. Tiwari, K. C. Kemp, J. A. Perman, A. B. Bourlinos, K. S. Kim and R. Zboril, Chem. Rev., 2016, 116, 5464–5519 CrossRef CAS PubMed.
- E.-Y. Choi, T. H. Han, J. Hong, J. E. Kim, S. H. Lee, H. W. Kim and S. O. Kim, J. Mater. Chem., 2010, 20, 1907–1912 RSC.
- T. Kim, H. Lee, J. Kim and K. S. Suh, ACS Nano, 2010, 4, 1612–1618 CrossRef CAS PubMed.
- V. Georgakilas, A. Demeslis, E. Ntararas, A. Kouloumpis, K. Dimos, D. Gournis, M. Kocman, M. Otyepka and R. Zbořil, Adv. Funct. Mater., 2015, 25, 1481–1487 CrossRef CAS.
- (a) E. Bi, Y. Su, H. Chen, X. Yang, M. Yin, F. Ye, Z. Li and L. Han, RSC Adv., 2015, 5, 9075–9078 RSC; (b) R. Mo, Z. Lei, K. Sun and D. Rooney, Adv. Mater., 2014, 26, 2084–2088 CrossRef CAS PubMed; (c) X. Zeng, W. Tu, J. Li, J. Bao and Z. Dai, ACS Appl. Mater. Interfaces, 2014, 6, 16197–16203 CrossRef CAS PubMed; (d) H. Xia, C. Hong, B. Li, B. Zhao, Z. Lin, M. Zheng, S. V. Savilov and S. M. Aldoshin, Adv. Funct. Mater., 2015, 25, 627–635 CrossRef CAS; (e) S. Shah, P. T. Yin, T. M. Uehara, S. T. D. Chueng, L. Yang and K. B. Lee, Adv. Mater., 2014, 26, 3673–3680 CrossRef CAS PubMed.
- (a) T.-H. Kim, S. Shah, L. Yang, P. T. Yin, M. K. Hossain, B. Conley, J.-W. Choi and K.-B. Lee, ACS Nano, 2015, 9, 3780–3790 CrossRef CAS PubMed; (b) J. Park, B. Kim, J. Han, J. Oh, S. Park, S. Ryu, S. Jung, J.-Y. Shin, B. S. Lee and B. H. Hong, ACS Nano, 2015, 9, 4987–4999 CrossRef CAS PubMed; (c) J. N. Tiwari, V. Vij, K. C. Kemp and K. S. Kim, ACS Nano, 2016, 10, 46–80 CrossRef CAS PubMed; (d) S. Suguna, C. I. David, J. Prabhu and R. Nandhakumar, Mater. Adv., 2021, 2, 6197–6212 RSC.
- (a) S. T. Huang, Y. Shi, N. B. Li and H. Q. Luo, Analyst, 2012, 137, 2593–2599 RSC; (b) S. Basiruddin and S. K. Swain, Mater. Sci. Eng., C, 2016, 58, 103–109 CrossRef CAS PubMed; (c) R. Mitra and A. Saha, ACS Sustainable Chem. Eng., 2017, 5, 604–615 CrossRef CAS; (d) D. J. Dmonte, A. Pandiyarajan, N. Bhuvanesh, S. Suresh and R. Nandhakumar, Mater. Lett., 2018, 227, 154–157 CrossRef CAS.
- (a) A. K. Akhila and N. K. Renuka, New J. Chem., 2019, 43, 1001–1008 RSC; (b) X. Chen, X. Wang, Z. Lu, H. Luo, L. Dong, Z. Ji, F. Xu, D. Huo and C. Hou, Sen. Actuators, B, 2020, 311, 127898 CrossRef CAS; (c) M. Anju and N. K. Renuka, Nano-Struct. Nano-Objects, 2019, 17, 194–217 CrossRef.
- (a) B. Sui, B. Kim, Y. Zhang, A. Frazer and K. D. Belfield, ACS Appl. Mater. Interfaces, 2013, 5, 2920–2923 CrossRef CAS PubMed; (b) Y. Xiong, J. Wu, Q. Wang, J. Xu, S. Fang, J. Chen and M. Duan, Talanta, 2017, 174, 372–379 CrossRef CAS PubMed; (c) P. Singh, A. A. Prabhune, C. S. P. Tripathi and D. Guin, ACS Sustainable Chem. Eng., 2017, 5, 982–987 CrossRef CAS; (d) S. Juanjuan, W. Linlin and H. Yangfeng, Anal. Methods, 2019, 11, 2585–2590 RSC; (e) A. Chatterjee, N. Pan, T. K. Maji, S. S. Pasha, S. Singh, S. A. Ahmed, J. T. Al-Thakafy and S. K. Pal, ACS Sustainable Chem. Eng., 2021, 9, 7160–7170 CrossRef CAS.
- (a) P. Chen, W. Bai and Y. Bao, J. Mater. Chem. C, 2019, 7, 11731–11746 RSC; (b) Y. Yang, Q. Zhao, W. Feng and F. Li, Chem. Rev., 2013, 113, 192–270 CrossRef CAS PubMed; (c) Y. Bao, Q. Li, B. Liu, F. Du, J. Tian, H. Wang, Y. Wang and R. Bai, Chem. Commun., 2012, 48, 118–120 RSC; (d) Y. Li, Y. Duan, J. Zheng, J. Li, W. Zhao, S. Yang and R. Yang, Anal. Chem., 2013, 85, 11456–11463 CrossRef CAS PubMed; (e) T. Wang, N. Zhang, R. Bai and Y. Bao, J. Mater. Chem. C, 2018, 6, 266–270 RSC; (f) Y. Shen, X. Zhang, Y. Zhang, H. Li and Y. Chen, Sen. Actuators, B, 2018, 258, 544–549 CrossRef CAS; (g) A. Feng, Y. Jia, L. Huang, L. Wang, G. Zhou, S. Wang and P. Liu, Spectrochim. Acta, Part A, 2019, 220, 117108 CrossRef CAS PubMed; (h) G. Jiang, X. Liu, Y. Wu, J. Wang, X. Dong, G. Zhang, Y. Li and X. Fan, RSC Adv., 2016, 6, 59400–59404 RSC.
- (a) R. Appiah-Ntiamoah, B. T. Gadisa and H. Kim, New J. Chem., 2018, 42, 11341–11350 RSC; (b) N. Spano, V. Guccini, M. Ciulu, I. Floris, V. M. Nurchi, A. Panzanelli, M. I. Pilo and G. Sanna, Arab. J. Chem., 2018, 11, 492–500 CrossRef CAS; (c) M. N. Biyareh, A. R. Rezvani, K. Dashtian, M. Montazerozohori, M. Ghaedi, A. M. Asl and J. White, IEEE Sens. J., 2018, 19, 413–425 Search PubMed.
- M. Anju and N. K. Renuka, Mater. Res. Bull., 2019, 110, 50–56 CrossRef.
- M. Anju, A. K. Akhila and N. K. Renuka, Nano-Struct. Nano-Objects, 2020, 24, 100606 CrossRef CAS.
- A. K. Akhila, A. R. Suresh Babu, A. A. Anappara and N. K. Renuka, Spectrochim. Acta, Part A, 2022, 266, 120408 CrossRef PubMed.
- Y. Xu, L. Zhao, H. Bai, W. Hong, C. Li and G. Shi, J. Am. Chem. Soc., 2009, 131, 13490–13497 CrossRef CAS PubMed.
- M. Wang, Q. Wang, W. Zhu, Y. Yang, H. Zhou, F. Zhang, L. Zhou, J. M. Razal, G. G. Wallace and J. Chen, Green Energy Environ., 2017, 2, 285–293 CrossRef.
- J. Geng and H.-T. Jung, J. Phys. Chem. C, 2010, 114, 8227–8234 CrossRef CAS.
- Z. Zhang, H. Huang, X. Yang and L. Zang, J. Phys. Chem. Lett., 2011, 2, 2897–2905 CrossRef CAS.
- J. Tang, L. Niu, J. Liu, Y. Wang, Z. Huang, S. Xie, L. Huang, Q. Xu, Y. Wang and L. A. Belfiore, Mater. Sci. Eng., C, 2014, 34, 186–192 CrossRef CAS PubMed.
- G. Xi, X. Wang and T. Chen, Int. J. Nanomed., 2016, 11, 1537–1547 CrossRef CAS PubMed.
- S. Srivastava, T. D. Senguttuvan and B. K. Gupta, J. Vac. Sci. Technol. B, 2018, 36, 04G104 CrossRef.
- X. Wu, Y. Xing, K. Zeng, K. Huber and J. X. Zhao, Langmuir, 2018, 34, 603–611 CrossRef CAS PubMed.
- V. V. Kumar, D. Ramadevi, V. M. Ankathi, T. K. Pradhan and K. Basavaiah, Microchem. J., 2020, 157, 105028 CrossRef CAS.
- Y. Kang, J. W. Kampf and M. E. Meyerhoff, Anal. Chim. Acta, 2007, 598, 295–303 CrossRef CAS PubMed.
- Y. Liu, Q. Ouyang, H. Li, Z. Zhang and Q. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 18314–18321 CrossRef CAS PubMed.
- S. Chakraborty and A. P. D. Ilagan, Philipp. J. Sci., 2020, 149, 27–33 Search PubMed.
- K. Chansaenpak, A. Kamkaew, O. Weeranantanapan, K. Suttisintong and G. Tumcharern, Sensors, 2018, 18, 2042 CrossRef PubMed.
- L. Ma, T. Leng, K. Wang, C. Wang, Y. Shen and W. Zhu, Tetrahedron, 2017, 73, 1306–1310 CrossRef CAS.
- X. Cao, W. Lin, Q. Yu and J. Wang, Org. Lett., 2011, 13, 6098–6101 CrossRef CAS PubMed.
- Y. Zhou, J. F. Zhang and J. Yoon, Chem. Rev., 2014, 114, 5511–5571 CrossRef CAS PubMed.
- A. Bamesberger, C. Schwartz, Q. Song, W. Han, Z. Wang and H. Cao, New J. Chem., 2014, 38, 884–888 RSC.
- P. G. Sutariya, N. R. Modi, A. Pandya, B. K. Joshi, K. V. Joshi and S. K. Menon, Analyst, 2012, 137, 5491–5494 RSC.
- W. Feng, P. Long, Y. Feng and Y. Li, Adv. Sci., 2016, 3, 1500413 CrossRef PubMed.
- M. Jahanshahi, E. Kowsari, V. Haddadi-Asl, M. Khoobi, B. Bazri, M. Aryafard, J. H. Lee, F. B. Kadumudi, S. Talebian and N. Kamaly, Appl. Surf. Sci., 2020, 515, 146071 CrossRef CAS.
- W. Kang and S. Li, RSC Adv., 2018, 8, 23459–23467 RSC.
- J. M. Rimsza, B. J. Walder and T. M. Alam, J. Phys. Chem. C, 2021, 125, 2699–2712 CrossRef CAS.
- M. A. Rodriguez, M. R. Keenan and G. Nagasubramanian, J. Appl. Crystallogr., 2007, 40, 1097–1104 CrossRef CAS.
- R. Hagiwara and Y. Sato, Structures of Highly Fluorinated Compounds of Layered Carbon, in New Fluorinated Carbons: Fundamentals and Applications, ed. A. Tressaud, Elsevier, 2017, pp. 283–303 Search PubMed.
- A. Ramya, B. Manoj and A. N. Mohan, Asian J. Chem., 2016, 28, 1031 CrossRef CAS.
- V. Mazánek, O. Jankovský, J. Luxa, D. Sedmidubský, Z. Janoušek, F. Šembera, M. Mikulics and Z. Sofer, Nanoscale, 2015, 7, 13646–13655 RSC.
- K. Zhang, S. Yu, B. Jv and W. Zheng, Phys. Chem. Chem. Phys., 2016, 18, 28418–28427 RSC.
- M. D. Sharma, S. S. Rayalu, S. D. Kolev and R. J. Krupadam, Sci. Rep., 2021, 11, 17321 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures of rGO and rGO–Ffuorophore systems. See DOI: https://doi.org/10.1039/d2ma00393g |
| This journal is © The Royal Society of Chemistry 2022 |