DOI:
10.1039/D2MA00585A
(Paper)
Mater. Adv., 2022,
3, 7072-7079
Urea-assisted synthesis of carbon-doped BiNbO4 with oxygen vacancies and visible light photocatalytic applications†
Received
25th May 2022
, Accepted 14th July 2022
First published on 20th July 2022
Abstract
Although carbon doping in simple metal oxides like TiO2 and ZnO has been proven to increase photocatalytic dye degradation in visible light, it's unclear if carbon doping in complex BiNbO4 would have the same effect. For the first time, carbon doped BiNbO4 (U-BiNbO4) has been synthesized with urea as the carbon source utilizing a simple sol–gel process. The creation of oxygen vacancies and their energy levels in BiNbO4's electronic band structure are controlled by the decomposition of urea. X-Ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), and carrier scavenging tests verified the presence of surface oxygen vacancies and carbon doping. Both pure BiNbO4 and U-BiNbO4 are multi-crystalline structures with orthorhombic and triclinic phases. When compared to pure BiNbO4, U-BiNbO4 degraded cationic methylene blue (MB, 75.7 ± 3.5%) and brilliant green dyes (BG, 86.3 3.5%) more efficiently. The creation of carbon modulated oxygen vacancies and the lowering of the bandgap (from 3.19 eV to 2.95 eV for BiNbO4 and U-BiNbO4) were also postulated as dye degradation mechanisms. By employing a synthesis method of alternate low- and high-oxygen partial pressures, this novel process might be exploited to construct high-performance Bi-based photocatalysts.
Introduction
Photocatalysis as a technology has attracted attention for the degradation of organic and inorganic pollutants, air purification and applications in self-cleaning surfaces. Since the pioneering discovery in 1972, titanium dioxide (TiO2) has been one of the most promising materials because of its low cost, chemical stability, non-toxicity and robust performance. However, the main drawback is associated with its wide band gap (3.2 eV) and its ability to absorb only UV light.1 In order to overcome this issue, new promising materials have been developed to satisfy the demand in efficient visible light utilization. Among others, BiNbO4 has been recognized and fabricated in a number of studies.2–7 The experimental band gap of BiNbO4 is 3.0 eV, which is smaller than that for TiO2, but still wide for efficient visible light absorption. In order to reduce the band gap non-metal doping has been proposed and effectively utilized to prepare visible-light-sensitive catalysts. For instance, a carbon (C) doping agent was successfully introduced in a crystal lattice in a number of works where urea, oleylamine, sucrose, etc. were used as a source of C.8–14 Dyes have been efficiently used as prototyping pollutants. There are very few manuscripts published on the dye degradation of BiNbO4. A comparison of the dye degradation performances by BiNbO4 are summarized in Table 1. It can be concluded that doped BiNbO4 shows a higher degradation rate compared to pristine BiNbO4 under the same experimental conditions. Although not exhaustive, it can be seen that only one paper was released on methylene blue (MB) degradation (240 min) and no papers were published on brilliant green (BG) degradation.
Table 1 The literature overview of the doped-BiNbO4 degradation performance towards cationic and anionic dyes
| Material |
Dopant |
Light intensity |
Catalyst amount |
Dye type |
Dye mass |
Degradation time |
Total degradation percentage (%) |
| BiNbO4, Ag3PO4/BiNbO424 |
Ag3PO4 |
350 W/xenon lamp |
15 mg |
RhB |
20 mg L−1 |
60 min |
14.9, 98.8 |
| BiNbO4, Ag/Ag2O/BiNbO425 |
Ag/Ag2O |
48 W/100 mW cm−2 |
15 mg |
MB, RhB, AR1 |
1 g L−1 |
MB: 240 min, RhB: 240 min, AR1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 min :25 min |
36.5, 84.2, 5.9, 55.6, 0, 88.3 |
| BiNbO4, C3N4/BiNbO426 |
C3N4 |
300 W/xenon lamp |
15 mg |
RhB |
10 mg L−1 |
40 min |
25.1, 100.0 |
Previously, by DFT calculations15 and by hands-on experiments to prepare C-doped TiO2,16 C-doped SnO217 and C-doped ZnO,18 it was shown that under oxygen-poor conditions oxygen vacancies are stabilized. However, under oxygen-rich conditions, the site occupancy of carbon is more complicated which depends on the redox potentials of the cations in the semiconductor. For example, for C-doped TiO2 carbon goes to the metal site (interstitial and substitutional).15 Conversely, for p-type C-doped ZnO the most stable structure is a C–O cluster.19 It was also shown that carbon in C-doped mono-clinic BiVO4 does not lead to oxygen substitution (by carbon).20 Urea is a low-cost and abundant material widely available in industry. It is an active molecular precursor produced by thermal treatment which can be utilized to form graphitic carbon nitride (g-C3N4). As a result of thermal decomposition, urea is converted to by-products such as cyanuric acid and ammonia gas. Therefore, urea may be a potential precursor for the solid state synthesis of C-doped BiNbO4.21–23 By simultaneously applying oxygen-rich and oxygen-deficient conditions, which originate from the thermal decomposition of urea, we fabricated carbon-doped BiNbO4 (referred to throughout the text as U-BiNbO4). The enhanced presence of C and oxygen vacancies was confirmed by X-ray photo-electron spectroscopy (XPS). The photocatalytic properties of pristine BiNbO4 and U-BiNbO4 were investigated toward the visible-light degradation of cationic MB and BG dyes. Electron, hole, and ˙OH scavenging experiments were performed and a mechanism of dye degradation was proposed based on (i) reduced band gap and (ii) C and oxygen vacancies.
Experimental
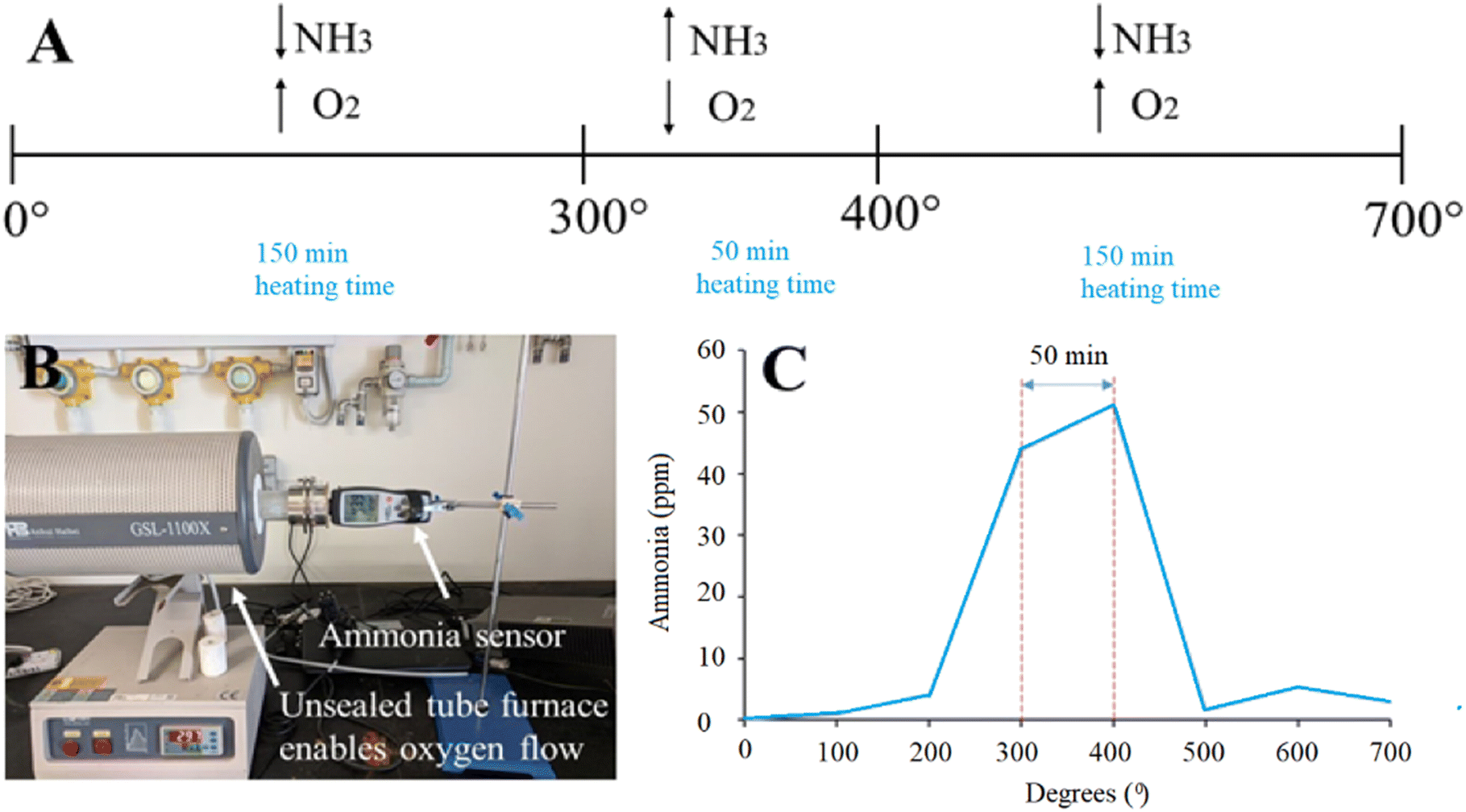
Schematics of the annealing process with a combination of high- and low-oxygen partial pressure is shown in Fig. 1 (panel A). The unique synthesis method included a tube furnace unsealed from one side and connected to an ammonia sensor on the other side to monitor the concentration of gaseous ammonia (NH3), which is shown in Fig. 1 (panel B). The material was annealed at 700 °C with a heating rate of 2 degrees per min, which allowed us to measure the amount of produced ammonia. The particles were calcined at 700 °C with a heating rate of 2 degrees per min. The largest presence of ammonia was recorded at 300–400 °C (40–50 ppm), corresponding to oxygen deficient conditions. For other experiments, the amount of produced ammonia was low (below 5 ppm), which corresponds to oxygen-rich conditions (Fig. 1, panel C). As a result of the unique synthesis process, oxygen vacancies are formed, which are controlled by an oxygen-poor atmosphere.
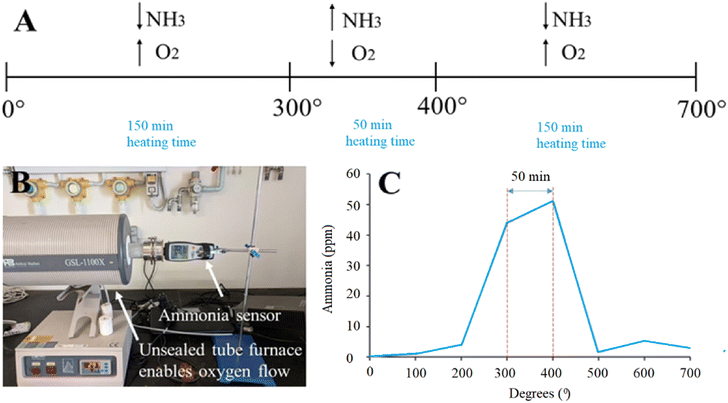 |
| | Fig. 1 (A) Schematic of the annealing process of U-BiNbO4 in the tube furnace. Oxygen-poor conditions were applied between 300–400 °C, and oxygen-rich conditions were maintained below 300 °C and above 400 °C, respectively. (B) Set up with a tube furnace and ammonia sensor. The tube furnace is unsealed from one side to allow free oxygen flow. (C) A graph showing the recorded ammonia concentration (ppm) vs. annealing temperature (°C). | |
Preparation
Reagents
Bismuth nitrate pentahydrate (Bi(NO3)3·5H2O, 98%, Sigma-Aldrich) and niobium pentachloride (NbCl5, 99%, Sigma-Aldrich) were employed as precursors to provide bismuth and niobium sources, respectively. Nitric acid (HNO3, 70%, Sigma-Aldrich) and ethanol (C2H5OH, 96%, Sigma-Aldrich) were purchased and used as solvents. Urea (CH4N2O, 98%, Mw = 60.06) was purchased from Sigma-Aldrich. Ethylenediaminetetraacetic acid (C10H16N2O8, 98%, Sigma-Aldrich), isopropanol (C3H8O, 99.5%, Sigma-Aldrich) and p-benzoquinone (C6H4O2, 98%, Sigma-Aldrich) were purchased as scavengers for holes, ˙OH radicals and electrons, respectively. Methylene Blue (C16H18ClN3S, dye content 82%) and Brilliant Green (C27H33N2HO4S, dye content 95%) were also purchased from Sigma-Aldrich and used as prototyping pollutants. Deionized water (DI, 50 KΩ) was used throughout all experiments.
BiNbO4 nanoparticles (NPs)
BiNbO4 particles were synthesized by using a sol–gel method. First, 1.14 g of bismuth nitrate pentahydrate and 0.64 g of niobium chloride were separately mixed with 10 mL of ethanol each under strong magnetic stirring (solutions A and B, respectively). This was followed by the heating of solution A at 100 °C for 60 min and solution B at 70 °C for 30 min, respectively. Second, both solutions were mixed together and 0.5 mL of nitric acid was added to the mixture dropwise to make the solution precipitate (solution C). Third, the as-prepared solution C was continuously stirred for 30 min and kept in an oven at 90 °C for 24 hours until it dried. Finally, the dried material was placed in an alumina crucible and annealed in a tube furnace. Then, it was heated to 700 °C at a rate of 2 °C min−1 and annealed for 60 min at 700 °C.
U-BiNbO4 catalyst
The U-BiNbO4 composite was prepared by using the same pathway as BiNbO4 with the addition of the following steps. Urea (2 g) was dissolved separately in 10 mL of ethanol (20 °C) as a source of carbon (urea has a solubility in ethanol of 100 g L−1) and added into solution C. After 60 min of stirring, the solution was stored in an oven at 90 °C for 24 hours to dry. At the final step it was annealed in a tube furnace at 700 °C for 60 min at a heating rate of 2 °C min−1.
Instrumentation and characterization
A Prizmatix Ultra High Power LED lamp and Techno Digm UVR 400/600 lamp were used for photo-degradation tests as sources of visible and UV light, respectively. X-Ray diffraction (XRD) tests were performed in a Bruker D8 Advance X-ray diffractometer with Cu Kα radiation of 0.154 nm wavelength to evaluate the powder's composition and phase. The scanning angle was set from 20° to 35°. UV-Vis absorbance measurements were carried out and collected by using a PG Instruments TIIO + UV-Vis spectrophotometer. Particle size and the corresponding morphology were evaluated by scanning electron microscope (SEM) (JEOL JSM-7600F) coupled with an energy dispersive X-ray spectrum (EDS). X-Ray photoelectron spectroscopy (XPS) tests were performed using a Thermo Fischer Scientific Theta Probe system. Using a micro-Raman device (Renishaw Raman spectrometer) and a He–Ne laser, RL633 (633 nm, 1.5 m diameter spot size), micro-Raman scattering tests were carried out in the spectral region of 50 to 1000 cm−1. The annealing of the powder was performed in a tube furnace (Anhui Haibei GSL-1100X) and the light intensity was recorded using an Optical Power Meter PM100D from Thorlabs. The amount of gaseous ammonia was measured using an XIN Air Quality Monitor Meter.
Photocatalytic performance test
The photocatalytic performance of pristine BiNbO4 and U-BiNbO4 oxides was investigated by photocatalytic degradation of MB and BG dyes under visible irradiation. The Prizmatix Ultra High Power LED lamp was used as a visible light source. The light intensity was set at 100 mW cm−2. The experimental conditions were as follows: 15 mg of the photocatalyst was dispersed in 15 mL of a dye aqueous solution to reach a concentration of 1 g L−1 and the suspension was sonicated for 60 min. Before exposure to visible light the suspension was stirred for 30 min under dark conditions to reach adsorption–desorption equilibrium. Subsequently, the solution was transferred into 1.5 mL tubes and a photocatalytic reaction was initiated. The visible light exposure was set to 240 min. Every 60 min the suspension was collected and transferred into a UV-Vis cuvette. Finally, the UV-Vis absorption response was recorded.
Results and discussion
Characterization of the photocatalyst
X-Ray diffraction (XRD) patterns of pristine and U-BiNbO4 indicate the presence of a multi-crystalline structure with orthorhombic and triclinic phases as shown in Fig. 2. The BiNbO4 catalyst lattice parameters are a = 7.59 Å, b = 5.48 Å, and c = 7.95 Å. The U-BiNbO4 lattice parameters are a = 7.52 Å, b = 5.39 Å, and c = 7.91 Å. The corresponding peaks are denoted with O for orthorhombic and T for triclinic, respectively (the space group is P1). Notably, the (121) and (040) triclinic peaks exhibit a shift to a higher angle from 28.01° and 30.36° for pristine BiNbO4 to 28.20° and 30.49° for U-BiNbO4, respectively. The decrease in lattice parameters can be ascribed to the carbon influence (from a urea source) leading to oxygen vacancies.15,18 Since the ionic radius of O2− is larger than the ionic radius of C4+ (126 pm and 30 pm, respectively),27,28 the lattice shrinks due to the formation of oxygen vacancies (even after considering the volume expansion resulting from the interstitial carbon dopant). A larger peak shift was observed for the (121) versus (040) facet (0.188° versus 0.127°, respectively). This is evidence of the larger number of oxygen vacancies presented along (121) than the (040) facet, which can potentially contribute to a more pronounced photocatalytic activity of U-BiNbO4. Compared to the pristine BiNbO4, the U-BiNbO4 does not exhibit the (031) triclinic peak and small peak ascribed to the Bi2O3 phase. In summary, the XRD results suggest that carbon doping occurs along the (121) and (040) planes.
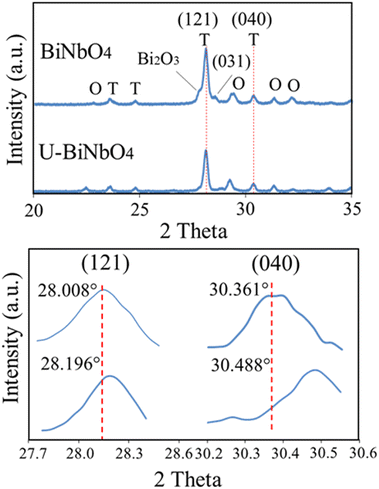 |
| | Fig. 2 XRD profiles for pristine BiNbO4 and U-BiNbO4, respectively. Bottom panel displays (121) and (040) triclinic peaks shift to a higher angle when urea is introduced. | |
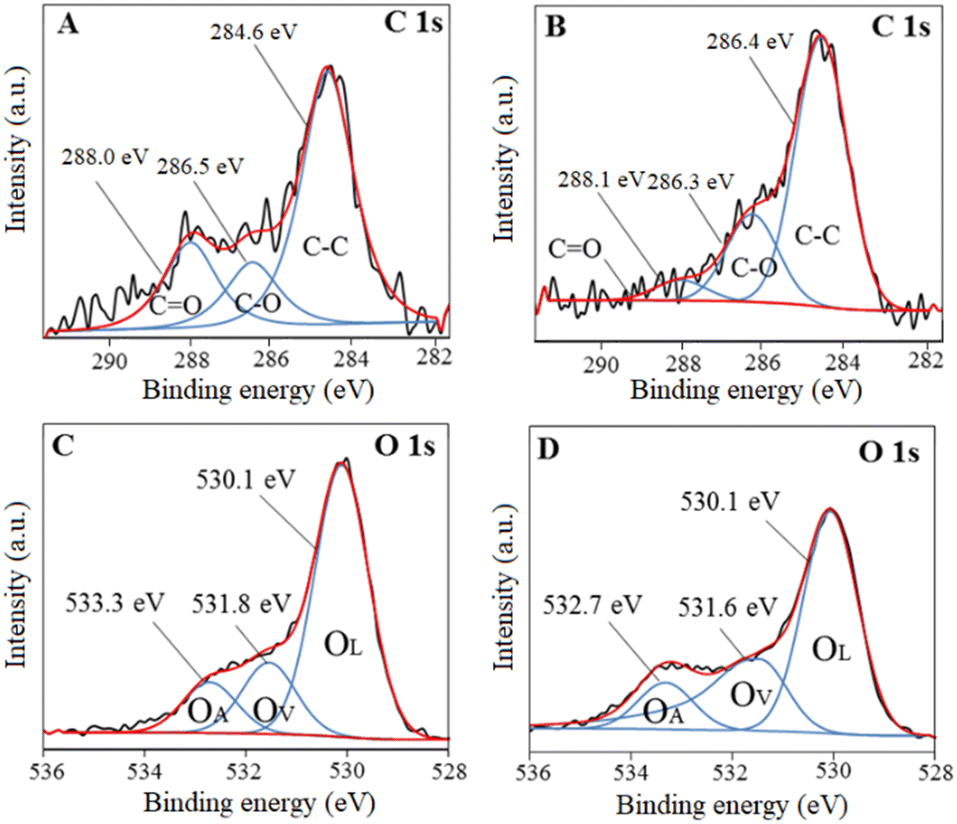
X-Ray photoelectron spectroscopy (XPS) analysis was carried out to determine the chemical states existing in BiNbO4 and U-BiNbO4. Fig. 3 displays C1s and O1s chemical states for pristine BiNbO4 and U-BiNbO4, respectively. The curve fitting analysis of the C1s spectrum affords three major peaks at 284.6, 286.5 eV and 288.0 eV for pristine BiNbO4, and 284.6 eV, 286.3 eV and 288.1 eV for U-BiNbO4, respectively. The peak at 284.6 eV originates from adventitious carbon, while the peaks at 286.3 eV and 288.1 eV are attributed to the C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, respectively.16 Compared to pristine BiNbO4, the C1s peaks of U-BiNbO4 are shifted, indicating a different chemical environment, which is due to C doping. The presence of surface carbon for BiNbO4 and U-BiNbO4 is 25.25 atomic% and 31.12 atomic%, respectively. The O1s peak was fitted into three components at 530.1 eV, 531.8 eV and 533.3 eV for pristine BiNbO4 and 530.1 eV, 531.6 eV and 532.7 eV for U-BiNbO4, respectively. These peaks were ascribed to lattice oxygen (OL), the region of surface oxygen vacancy (OV) and chemisorbed oxygen species (OA), respectively,29,30 while the signal at 531.6 eV is also attributed to surface hydroxyls. The ratios of surface oxygen vacancies to the total oxygen are 8.60 atomic% and 8.84 atomic% for pristine BiNbO4 and U-BiNbO4, respectively. Oxygen vacancies were probed further using Raman spectroscopy (Fig. S1 in the ESI†). When compared to pristine BiNbO4, the Raman active modes of U-BiNbO4 shifted and broadened somewhat to a higher wavenumber, which is compatible with the reported observations of oxygen vacancies.31 The formation of oxygen vacancies was further confirmed by using Raman spectroscopy. Therefore, it further confirmed the role of C doping in BiNbO4.
O bonds, respectively.16 Compared to pristine BiNbO4, the C1s peaks of U-BiNbO4 are shifted, indicating a different chemical environment, which is due to C doping. The presence of surface carbon for BiNbO4 and U-BiNbO4 is 25.25 atomic% and 31.12 atomic%, respectively. The O1s peak was fitted into three components at 530.1 eV, 531.8 eV and 533.3 eV for pristine BiNbO4 and 530.1 eV, 531.6 eV and 532.7 eV for U-BiNbO4, respectively. These peaks were ascribed to lattice oxygen (OL), the region of surface oxygen vacancy (OV) and chemisorbed oxygen species (OA), respectively,29,30 while the signal at 531.6 eV is also attributed to surface hydroxyls. The ratios of surface oxygen vacancies to the total oxygen are 8.60 atomic% and 8.84 atomic% for pristine BiNbO4 and U-BiNbO4, respectively. Oxygen vacancies were probed further using Raman spectroscopy (Fig. S1 in the ESI†). When compared to pristine BiNbO4, the Raman active modes of U-BiNbO4 shifted and broadened somewhat to a higher wavenumber, which is compatible with the reported observations of oxygen vacancies.31 The formation of oxygen vacancies was further confirmed by using Raman spectroscopy. Therefore, it further confirmed the role of C doping in BiNbO4.
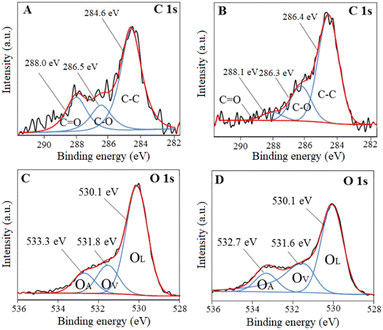 |
| | Fig. 3 XPS spectra of C1s and O1s for pristine BiNbO4 (panels A and C) and U-BiNbO4 (panels B and D), respectively. | |
The XPS spectrum of Bi4f and Nb3d for pristine BiNbO4 and U-BiNbO4 catalysts are shown in Fig. S2 in the ESI.† In summary, the elemental composition of BiNbO4 is 25.25% of C, 44.02% of O, 14.59% of Bi and 14.05% of Nb. The elemental composition of U-BiNbO4 is 31.12% of C, 48.56% of O, 8.04% of Bi and 17.7% of Nb. Enhancement of surface C and O atomic percentages were further validated by EDS elemental mapping as shown in Fig. S3 (in the ESI†) in which samples were deposited on top of a piece of Cu foil for the characterization to avoid C conductive adhesive. The presence of surface carbon for BiNbO4 and U-BiNbO4 is 25.40 atomic% and 38.30 atomic% and oxygen for BiNbO4 and U-BiNbO4 is 45.40 atomic% and 36.5 atomic%, respectively. As a result, it was established that urea doping increases carbon concentration, which was further demonstrated by an increase of C% in CHNS analysis, which can be seen from Table S1 (in the ESI†), in the XPS survey spectra (Fig. S4, ESI†) and has been explained thoroughly by Chaudhari et al.32 This proves that urea reaction significantly contributed for C doping and contribution for N doping is negligible.
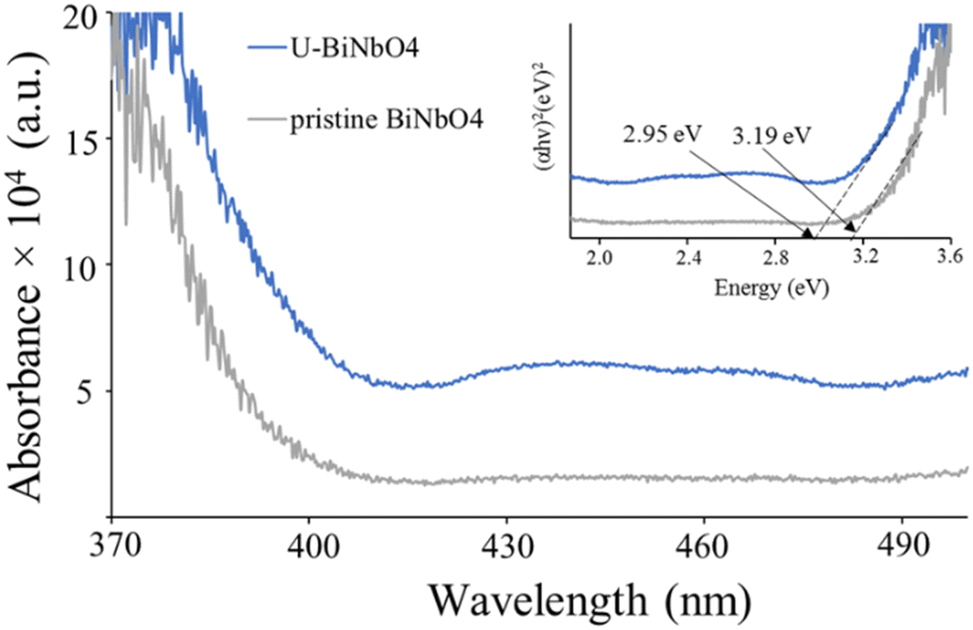
Fig. 4 shows the absorbance spectra and the corresponding Kubelka–Munk plot (inset) of U-BiNbO4 and pristine BiNbO4. The band gap was found by extrapolating the linear portion of the absorption edge to the energy (eV) axis. The values were determined to be 2.95 eV and 3.19 eV for U-BiNbO4 and pristine BiNbO4, respectively. The reduced bandgap of U-BiNbO4 is attributed to the presence of oxygen vacancies as it makes it easier to transport electrons from the valence band to the conduction band. The morphology and size of pristine and U-BiNbO4 catalysts were examined using a Scanning Electron Microscope (SEM) and are shown in Fig. 5. Noticeably, the morphology and particle size do not differ much for doped and pristine samples with a sheet-like form of 200–400 nm in size. Additionally, slight agglomeration was observed leading to a larger structure of a few micrometers.
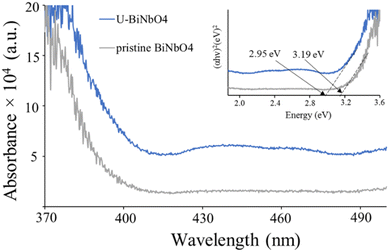 |
| | Fig. 4 UV-Vis absorbance spectroscopy of pristine BiNbO4 and U-BiNbO4. The inset of the plot shows band gap values from the corresponding Kubelka–Munk plot of pristine BiNbO4 and U-BiNbO4. | |
 |
| | Fig. 5 SEM representative images of pristine BiNbO4 (panels A and B) and U-BiNbO4 (panels C and D). | |
Photocatalytic degradation of methylene blue (MB) and brilliant green (BG) under visible light
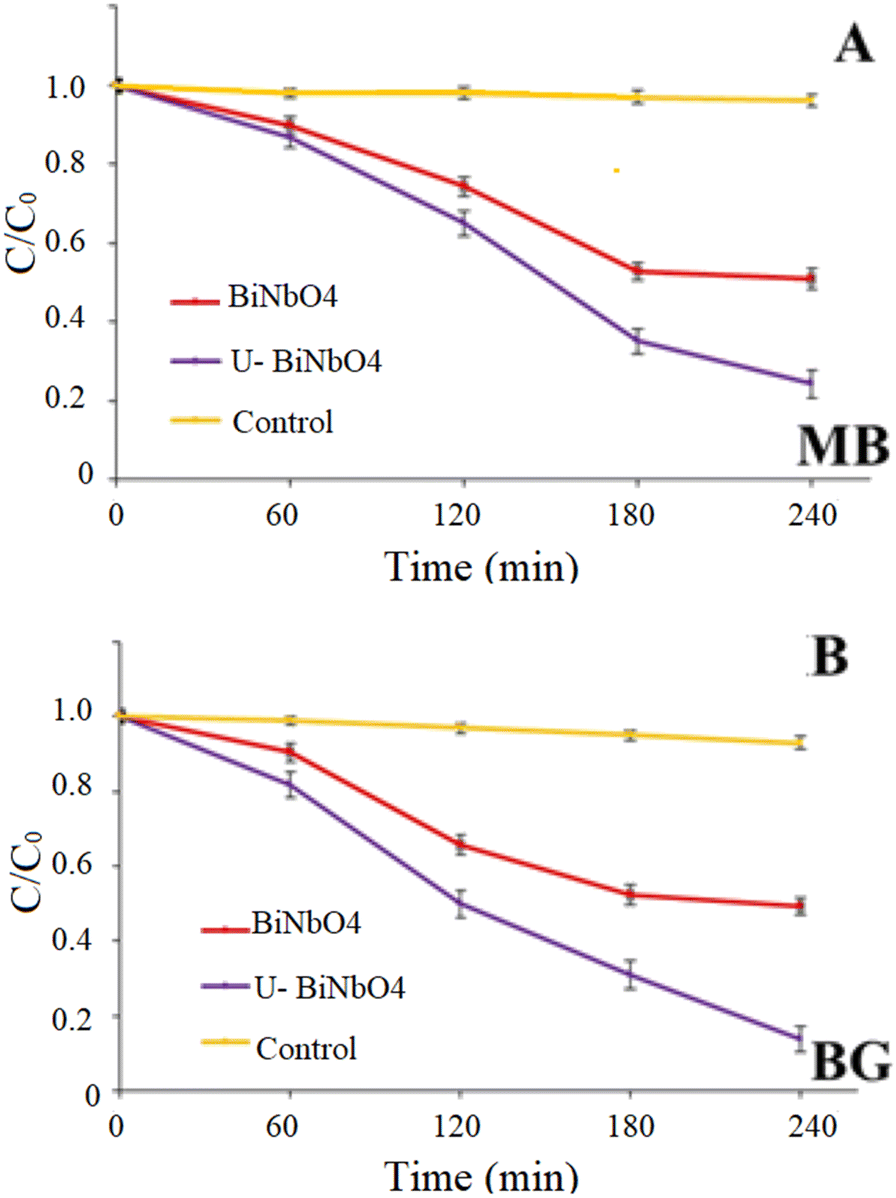
Fig. 6 shows photocatalytic degradation of MB and BG dyes in the presence of pristine BiNbO4 and U-BiNbO4 under visible light irradiation. The solution pH at which the degradation reaction was carried out was pH = 11.5 in MB and pH = 11.3 in BG. The absorbance decay as a function of wavelength for MB and BG in the presence of U-BiNbO4 is shown in Fig. S5 (in the ESI†). The light intensity was kept at 1 sun (100 mW cm−2) and the kinetics was monitored using a UV-Vis technique. The total degradation time was set for 240 min with every 60 min period when the absorption properties of the suspension were evaluated. See Table 1 for a comparison to earlier efforts. The average and standard deviation values were based on n = 3 trials.
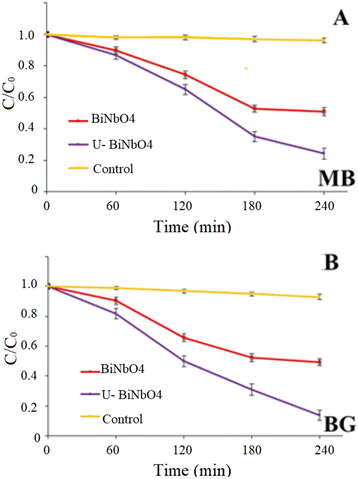 |
| | Fig. 6 Photocatalytic degradation and first-order kinetic plots of MB and BG dyes by pristine BiNbO4 and U-BiNbO4 under visible light. | |
The best degradation rate of MB was ascribed to U-BiNbO4 (75.7 ± 3.5%), followed by pristine BiNbO4 (49.0 ± 2.6%). In aqueous solution MB shows a maximum absorption peak at 664 nm and a shoulder peak at 615 nm, which correspond to monomers and dimers, respectively.33 Throughout the degradation both peaks exhibited a slight shift toward the blue region which is evidence of the N-demethylation reaction.33,34 The best BG removal rate was also attributed to U-BiNbO4 (86.3 ± 3.5%), followed by pristine BiNbO4 (51.0 ± 2.3%). The main peak is located at 619 nm and showed a gradual decrease within 240 min. The decay was accompanied by a noticeable hypsochromic shift toward the blue region (from 619 nm to 603 nm) proving the N-demethylation degradation pathway, which is shown in Fig. S5 in the ESI.†
Fig. S6 (in the ESI†) represents the degradation rates of MB and BG against different amounts of U-BiNbO4 catalyst. The amount was varied from 10 to 45 mg, and the irradiation time was set for 240 min. It can be seen that for MB the best decomposition rate was ascribed to 10 and 15 mg of the catalyst (49.6 ± 2.6% and 75.7 ± 3.5%, respectively). With higher catalyst loading the degradation rate drastically drops to 28.8 ± 2.0% and 15.1 ± 1.7% for 30 and 45 mg, respectively. BG degrades rapidly in the presence of 15 mg of catalyst (86.3 ± 2.9%), followed by 30 and 45 mg (67.2 ± 2.8% and 63.7 ± 2.5%, respectively). Higher loading and the accompanying drop in degradation efficiency can be explained by particle–particle interactions. When the number of particles in the system increases, it causes the formation of a higher number of free radicals participating in the dye degradation process. At the same time, beyond a certain limit of catalyst amount, the turbidity of the solution prevents visible light from penetrating and decreases the overall efficiency in the catalytic system.35
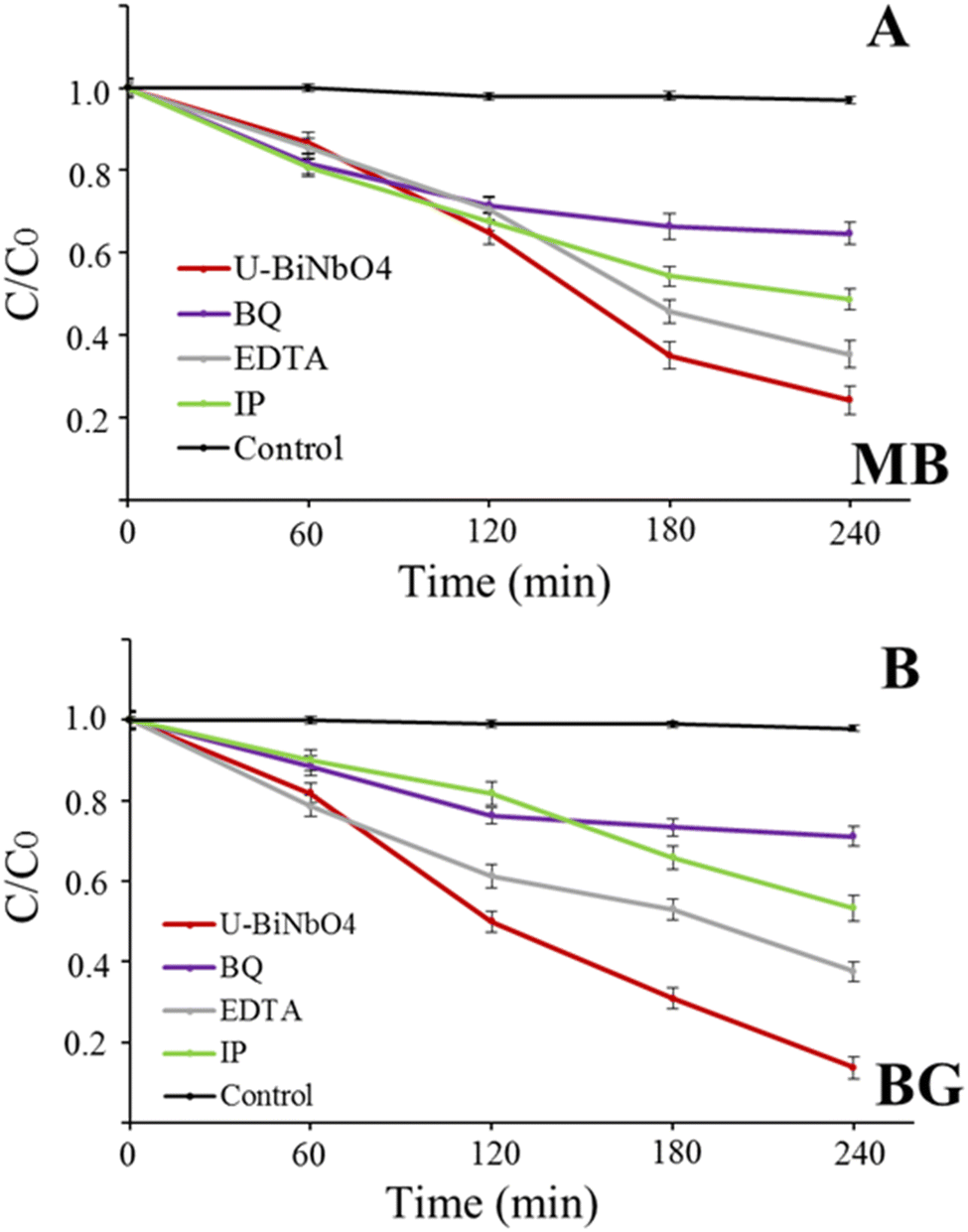
In order to identify the degradation mechanism of MB and BG dyes in the presence of U-BiNbO4, hydroxyl radicals (˙OH), electron and hole scavenging experiments were executed and the results are shown in Fig. 7. EDTA, p-benzoquinone (BQ), and isopropanol (IP) were used to scavenge holes, electrons and ˙OH radicals, respectively. For both dyes the reaction kinetics significantly decreased in the presence of BQ, highlighting the dominant role of electrons in the degradation process (35.3 ± 2.6% for MB and 28.8 ± 2.4% for BG, respectively). The hydroxyl radical is another important specie that affects the degradation rate (51.3 ± 2.6% for MB and 46.6 ± 3.2% for BG, respectively), while holes participate the least in the degradation process.
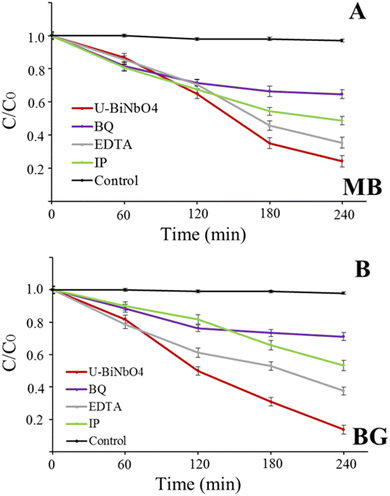 |
| | Fig. 7 Photocatalytic degradation with the U-BiNbO4 catalyst in the presence of carrier scavengers (BQ, EDTA, IP) for (A) MB and (B) BG. | |
Proposed mechanism of photocatalytic activity
The values of the valence band (VB) and the conduction band (CB) can be found from the measured band gap of U-BiNbO4 (Eg = 2.95 eV), the electronegativity of the catalyst (χ = 6.26 eV)34 and the parameter relating the reference of the electrode's redox level to the absolute vacuum scale (E0 = 4.5 eV). The following empirical eqn (1) and (2) can be used:
Based on the above parameters, the values of VB and CB vs. NHE are 3.235 eV and 0.285 eV, respectively. The schematic photocatalytic mechanism of MB and BG degradation is shown in Fig. 8. Based on the scavenging experiments from Fig. 7, photoelectrons are the primary reactive species affecting the degradation process, followed by ˙OH radicals and photo-holes. The proposed sequence of chemical reactions is the following:13,16,25,36
| | | U-BiNbO4 + hν → U-BiNbO4 + e− + h+ | (3) |
| | | e− + H2O2 + H+ → H2O + ˙OH | (4) |
| | | ˙OH + H2O + h+ → H2O2 + H+ | (5) |
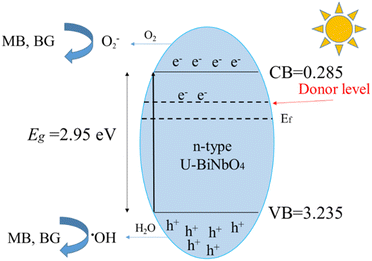 |
| | Fig. 8 Schematic photocatalytic mechanism of MB and BG degradation. | |
The enhanced photocatalytic properties of U-BiNbO4 can be ascribed to the cumulative effect of carbon doping and oxygen vacancies.37 As the band gap of U-BiNbO4 (compared to pristine BiNbO4) reduced, the electrical resistivity drops. Additionally, the increased amount of oxygen vacancies makes U-BiNbO4 a more efficient n-type semiconductor, acting as an electron donor, and helping to inhibit electron–hole recombination.9,16 Secondly, doping with carbon changes the flat potential of U-BiNbO4, leading to the formation of a potential barrier.16 The potential barrier yields transfer of photoelectrons to carbon species, promoting electron–hole separation. At the next stage, hydrogen at the surface of the catalyst reacts with photoelectrons to produce hydroxyl radicals (˙OH, from reaction (4). Photo-holes react with water and ˙OH, yielding a large amount of H2O2 (from reaction (5)). Thus, degradation of MB and BG dyes is the combination of photocatalytic (by photoelectrons and photo-holes) and photo-oxidative (by ˙OH and H2O2) pathways.
However, the effects of semiconductor band gaps, band edge positions (as reported by R. Yang et al. for carbon-doped BiVO437), and photogenerated electron–hole pair recombination rates on the generation rates of the aforementioned active radicals have not yet been thoroughly investigated. In order to forecast charge transport in photoelectrochemical systems that incorporate electrode entropy, electrolyte chemistry and electronic structures, our team is now extending its existing charge transport prediction models for general electrochemical cells to photo-electrochemical systems.38
Conclusions
In this work we employed urea-assisted synthesis to prepare a carbon-doped BiNbO4 catalyst via a facile sol–gel method. This approach has been previously shown only for TiO2 and ZnO.15,16 Gaseous ammonia, which originated from the thermal decomposition of urea, allowed us to apply a combination of oxygen-poor (300–400 °C) and oxygen-rich atmospheres (below 300 °C and above 400 °C). The unique synthesis conditions lead to (1) enhanced presence of carbon (from 25.25 atomic% for pristine BiNbO4 to 31.12 atomic% for U-BiNbO4) and (2) enhanced presence of oxygen vacancies (from 8.60 atomic% for pristine BiNbO4 to 8.84 atomic% for U-BiNbO4). Tuned properties of the U-BiNbO4 catalyst resulted in superior properties toward the degradation of cationic MB and BG pollutants under visible light (75.7 ± 3.5% and 86.3 ± 3.5% after 4 hours, respectively). The excellent photocatalytic performance was ascribed to the role of carbon and the resultant oxygen vacancies by promoting electron–hole separation. Based on carrier scavenger experiments, photoelectrons are the major species affecting degradation kinetics, followed by ˙OH radicals and photo-holes. The combination of oxygen-rich and oxygen-poor atmospheres can be an effective tool to enhance the photocatalytic properties of Bi-based catalysts.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The authors would like to acknowledge the financial assistance from the Agency for Science, Technology and Research (A*STAR) (Singapore), under the AME Individual Research Grant (Award No. A20E7c0108) and the Ministry of Education (Singapore), under Tier 2 program (Award No. MOE2018-T2-1-163).
References
- S. Akel, R. Dillert, N. O. Balayeva, R. Boughaled, J. Koch, M. El Azzouzi and D. W. Bahnemann, Catalysts, 2018, 8, 647 CrossRef.
- M. Bakiro, S. H. Ahmed and A. Alzamly, J. Environ. Chem. Eng., 2020, 8, 104461 CrossRef CAS.
- S. S. Dunkle and K. S. Suslick, J. Phys. Chem. C, 2009, 113, 10341–10345 CrossRef CAS.
- S. Xiong, Y. Liu, T. Li, F. Li and W. Cao, Ceram. Int., 2020, 46, 21790–21793 CrossRef CAS.
- H.-F. Zhai, A.-D. Li, J.-Z. Kong, X.-F. Li, J. Zhao, B.-L. Guo, J. Yin, Z.-S. Li and D. Wu, J. Solid State Chem., 2013, 202, 6–14 CrossRef CAS.
- J. Zhao, Q. He, X. Zhang, J. Yang, B. Yao, Q. Zhang and X. Yu, Integr. Ferroelectr., 2016, 176, 37–53 CrossRef CAS.
- N. Zhuk, S. Shugurov, M. Krzhizhanovskaya, V. Belyy, N. Sekushin, B. Makeev, S. Nekipelov, D. Beznosikov and Y. A. Busargina, J. Alloys Compd., 2020, 822, 153619 CrossRef CAS.
- Q.-C. Cao, X.-B. Ding, F. Li, Y.-H. Qin and C. Wang, J. Colloid Interface Sci., 2020, 576, 139–146 CrossRef CAS PubMed.
- S. Cho, J.-W. Jang, J. S. Lee and K.-H. Lee, CrystEngComm, 2010, 12, 3929–3935 RSC.
- V. M. Naik, D. B. Gunjal, A. H. Gore, S. P. Pawar, S. T. Mahanwar, P. V. Anbhule and G. B. Kolekar, Diamond Relat. Mater., 2018, 88, 262–268 CrossRef CAS.
- Y. Shi, G. Liu, L. Wang and H. Zhang, J. Colloid Interface Sci., 2019, 557, 336–348 CrossRef CAS PubMed.
- G. Sun, L. Ma, J. Ran, B. Li, X. Shen and H. Tong, Electrochim. Acta, 2016, 194, 168–178 CrossRef CAS.
- X. Wu, S. Yin, Q. Dong, C. Guo, H. Li, T. Kimura and T. Sato, Appl. Catal., B, 2013, 142, 450–457 CrossRef.
- Y. Yang, D. Ni, Y. Yao, Y. Zhong, Y. Ma and J. Yao, RSC Adv., 2015, 5, 93635–93643 RSC.
- C. Di Valentin, G. Pacchioni and A. Selloni, Chem. Mater., 2005, 17, 6656–6665 CrossRef CAS.
- J. Lu, Y. Wang, J. Huang, J. Fei, L. Cao and C. Li, Dyes Pigm., 2017, 144, 203–211 CrossRef CAS.
- K. Mallikarjuna, G. A. R. Bari, S. P. Vattikuti and H. Kim, Int. J. Hydrogen Energy, 2020, 45, 32789–32796 CrossRef CAS.
- X. Zhang, J. Qin, R. Hao, L. Wang, X. Shen, R. Yu, S. Limpanart, M. Ma and R. Liu, J. Phys. Chem. C, 2015, 119, 20544–20554 CrossRef CAS.
- S. T. Tan, X. Sun, Z. Yu, P. Wu, G. Q. Lo and D. L. Kwong, Appl. Phys. Lett., 2007, 91, 072101 CrossRef.
- W.-J. Yin, S.-H. Wei, M. M. Al-Jassim, J. Turner and Y. Yan, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 155102 CrossRef.
- J. Liu, T. Zhang, Z. Wang, G. Dawson and W. Chen, J. Mater. Chem., 2011, 21, 14398–14401 RSC.
- F. Dong, L. Wu, Y. Sun, M. Fu, Z. Wu and S. Lee, J. Mater. Chem., 2011, 21, 15171–15174 RSC.
- Y. Zhang, J. Liu, G. Wu and W. Chen, Nanoscale, 2012, 4, 5300–5303 RSC.
- N. Li, S. Miao, X. Zheng, J. Lai, S. Lv, X. Gu, M. Zhang, J. Yang and S. Cui, Ceram. Int., 2019, 45, 24260–24268 CrossRef CAS.
- A. Lebedev, F. Anariba, X. Li, D. Seng Hwee Leng and P. Wu, Sol. Energy, 2019, 178, 257–267 CrossRef CAS.
- J. Hao, Q. Wang and Z. Zhao, J. Photochem. Photobiol., A, 2017, 335, 94–101 CrossRef CAS.
- E. Keve and A. Skapski, J. Solid State Chem., 1973, 8, 159–165 CrossRef CAS.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- D. Li, W. Wang, D. Jiang, Y. Zheng and X. Li, RSC Adv., 2015, 5, 14374–14381 RSC.
- J.-M. Wu, Y. Chen, L. Pan, P. Wang, Y. Cui, D. Kong, L. Wang, X. Zhang and J.-J. Zou, Appl. Catal., B, 2018, 221, 187–195 CrossRef CAS.
- H. Mai, T. Lu, Q. Sun, J. Langley, N. Cox, F. Kremer, K. Catchpole, H. Chen, Z. Yi and T. J. Frankcombe, J. Mater. Chem. A, 2021, 9, 13182–13191 RSC.
- N. K. Chaudhari, M. Y. Song and J.-S. Yu, Sci. Rep., 2014, 4, 1–10 Search PubMed.
- R. Zuo, G. Du, W. Zhang, L. Liu, Y. Liu, L. Mei and Z. Li, Adv. Mater. Sci. Eng., 2014, 2014, 7 Search PubMed.
- F. Wang, S. Min, Y. Han and L. Feng, Superlattices Microstruct., 2010, 48, 170–180 CrossRef CAS.
-
A. Kumar, A Review on the Factors Affecting the Photocatalytic Degradation of Hazardous Materials, 2017 Search PubMed.
- A. Lebedev, G. H.-H. Ma, F. Anariba, X. Li, T. S. K. Lim, F. C. Ng and P. Wu, Sol. Energy, 2018, 160, 298–302 CrossRef CAS.
- R. Yang, R. Zhu, Y. Fan, L. Hu and Q. Chen, RSC Adv., 2019, 9, 14004–14010 RSC.
- P. Wu and H. L. Senevirathna, Electrochim. Acta, 2022, 410, 140051 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a