
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSea-urchin-like iron-cobalt phosphide as an advanced anode material for lithium ion batteries†
Prakash Kumar
Pathak‡
,
Ved Prakash
Joshi‡
,
Nitish
Kumar
 and
Rahul R.
Salunkhe
*
and
Rahul R.
Salunkhe
*
Materials Research Laboratory, Department of Physics, Indian Institute of Technology Jammu, Jammu and Kashmir 181121, India. E-mail: rahul.salunkhe@iitjammu.ac.in
First published on 28th July 2022
Abstract
Lithium-ion batteries (LIBs) are the most advanced and well-developed renewable energy storage solutions over the last three decades. Resourceful and cost-effective transition metal phosphides (TMPs) have gained immense attention for various energy storage devices due to their excellent electrochemical properties. Here, we report a bimetallic phosphide (iron-cobalt phosphide, FeCoP) synthesized by a simple hydrothermal method followed by a low-temperature phosphorization. We have used this material as the anode for LIBs with lithium foil as the counter and reference and 1 M lithium hexafluorophosphate (LiPF6) in a mixture of EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMC (1:1) as the electrolyte. The electrochemical tests reveal that the device exhibited an excellent discharge capacity of 1653.4 mA h g−1 at a current density of 100 mA g−1. The device retained about 80% of its performance even after 1000 cycles at a current density of 5 A g−1. The performance of the synthesized material shows that TMPs are of great use for the application of LIBs.
:DMC (1:1) as the electrolyte. The electrochemical tests reveal that the device exhibited an excellent discharge capacity of 1653.4 mA h g−1 at a current density of 100 mA g−1. The device retained about 80% of its performance even after 1000 cycles at a current density of 5 A g−1. The performance of the synthesized material shows that TMPs are of great use for the application of LIBs.
Introduction
The rising demand for energy storage devices has derived immense research interest in developing reliable energy storage materials with high capacity and cyclability. Lithium-ion batteries (LIBs) are a well-established rechargeable devices used in most of our electronic gadgets for the last two decades.1–3 The conventional LIBs consist of a transition metal oxide cathode, an organic electrolyte with lithium salt as a source of Li+ ions, and graphite as the anode material.4–6 The commercially utilized graphite anodes have a limited capacity of 372 mA h g−1, which must be improved to fulfill various electronic and electric vehicle demands. Although tremendous research has made LIB technology more advanced, the development of anode materials still lags behind since most of the works are focused on cathode and electrolyte developments.Recently, transition metal phosphides (TMPs) have earned enormous attention in energy storage applications due to their excellent electrocatalytic activity.7–9 TMPs are favorable for battery materials due to their metalloid characteristic and superior chemical properties. They possess high electronic conductivity, making them kinetically favorable for fast electron/ion transfer.10–12 This is because the phosphorus (P) atom has high electronegativity, which significantly restricts the electron delocalization in the metal and thus increases the electronic conductivity of the material.13 Also, all the TMPs exhibit very high gravimetric capacities compared to graphite anodes.14 Most TMPs in their original form suffer from poor cyclability and faded capacity due to the extreme volume changes during lithium-ion coupling charge transfer reactions. However, nanostructuring and composite formation have been reported as possible solutions to reduce the volume expansion during cycling.14,15 An early report by Nazar et al. has shown that CoP3 is efficient in the uptake and extraction of lithium with a reversible capacity of 400 mA h g−1 at an average potential of 0.9 V vs. Li/Li+.16 Another interesting study by Li et al. reported phosphorous-rich CuP2 nanowires as an anode material for LIBs, which exhibited a discharge capacity of 945 mA h g−1 even after 100 cycles, displaying 88% capacity retention. Here, the nanostructuring of the material helped in improved performance.17 Moreover, compositing with carbon materials further improves capacity retention, as reported by Zhu et al. in their seminal report.18 The ultrafine cobalt phosphide (CoP) nanoparticles embedded in a nitrogen (N2)-doped carbon matrix helped shorten the diffusion path for Li-ions and reduce the volume expansions during cycling. The composite of CoP nanoparticles and a carbon matrix as an anode for LIBs exhibited a discharge capacity of 522.6 mA h g−1 after 750 cycles. Indeed, CoP have significant potential if tuned efficiently to reveal high capacities without compromising cycling stability. Further doping and composite formation strategies must be examined to develop better anode materials for LIBs.
The reaction mechanism between the metal phosphide MPx (M = Mn, V, Sn, Ni, Cu, Fe, Co, Zn, Ge, Se, Mo, Ga, In; x = 1, 2, 3, 4) and lithium occurs via a conversion reaction, which involves a redox reaction of metallic nanoparticles and matrix formation of Li3P, as shown in eqn (1).17,19
| MPx + 3xLi → M0 + xLi3P | (1) |
The reaction mechanism involves one P atom from metal phosphide that can react with three lithium ions to form a Li3P compound that shows an excellent theoretical specific capacity of 2596 mA h g−1, much superior to graphite, indicating that the phosphorus-rich materials have good potential for LIBs.20,21 However, single metal phosphides generally suffer from high volumetric expansion and alloying during electrochemical reactions, leading to capacity fading and pulverization.22–26 Such a scenario can be buffered by incorporating the metal phosphides/chalcogenides with a carbon matrix or introducing other transition metals to form bimetallic TMPs. The synergistic effect of precursor transition metals boost the reaction kinetics and charge transfer owing to improved interfacial interactions and better structural stability during the volume change. Li et al.27 reported the substantial synergistic effect of iron-doped nickel phosphate, which enhances the electrocatalytic activity for water oxidation. A similar doping strategy was adopted by Song et al.28 for the application of OER/ORR and zinc-air batteries (ZABs). The cobalt-doped iron phosphate exhibits high electrocatalytic activity in this work and delivers an increased power density ZAB. Thus, the strategic doping of a transition metal into a single metal compound can be of great use for energy storage applications. Although researchers have used various single and bimetallic phosphides for battery applications, reports discussing Fe doping/compositing CoP as an anode material for LIBs are still scarce.
Herein we report the hydrothermal synthesis of iron-cobalt phosphide (FeCoP) for the anode material of LIBs. We have used the traditional hydrothermal method followed by low-temperature phosphorization. This material has grown in a sea urchin-like morphology and exhibits a good surface area of 29.3 m2 g−1. The half cell measurements using lithium foil have demonstrated remarkable discharge capacity and cycling stability due to Fe incorporation in the primary material.
Experimental
Chemicals
Cobalt(II) chloride (CoCl2, purity 97%), urea and sodium hypophosphite monohydrate (NaH2PO2), ethylene carbonate (EC), dimethyl carbonate (DMC), N-methyl-2-pyrrolidone (NMP) and lithium hexafluorophosphate (LiPF6) were purchased from Sigma-Aldrich. Iron(III) chloride (FeCl3) and deionized water (DI) were purchased from S D fine-chem ltd., and methanol (AR) was obtained from TCI Co. Ltd. Polyvinylidene fluoride (PVDF) filter membranes (0.45 μm) were purchased from Merck Millipore.Material synthesis
The FeCoP was synthesized by a simple hydrothermal method followed by phosphorization. Firstly, 1 mM CoCl2, 1 mM FeCl3, and 5 mM urea were mixed in 75 mL DI water and stirred for 30 minutes. The solution was transferred into a 100 mL Teflon-lined hydrothermal autoclave and kept at 120 °C for 12 h. After cooling down, the solid product was obtained through filtration. This powder was further washed several times with DI water and methanol. Finally, the washed product was kept for drying at 80 °C overnight. The obtained powders were set for phosphorization in a horizontal split hinge tube furnace with temperature and gas flow controllers. The obtained product and NaH2PO2·H2O were placed in two separate boats such that NaH2PO2·H2O is upstream of the N2 flow. The furnace was heated to 300 °C for 3 h at the heating rate of 5 °C min−1 under continuous N2 flow. The brief synthesis process is shown in Fig. 1.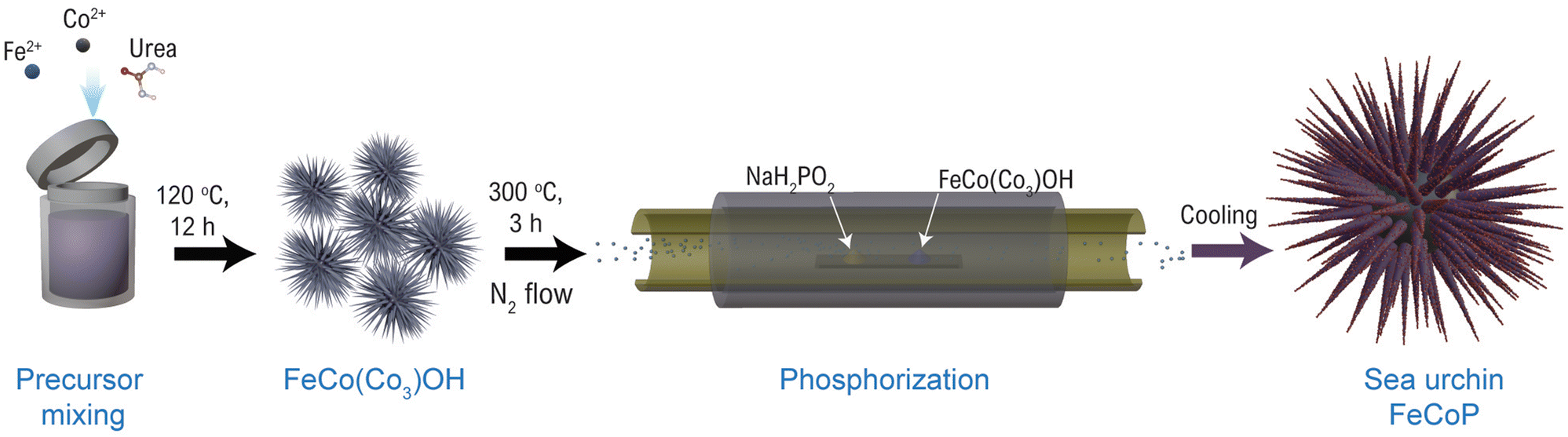 | ||
| Fig. 1 Schematic illustration for various steps for the synthesis of FeCoP sea urchins. | ||
Material characterizations
The morphology of the samples was studied using field emission scanning electron microscopy (FESEM, JEOL, JSM 7900F). The energy dispersive spectroscopy (EDS) mapping was analyzed using an EDS detector (Oxford instrumentation) equipped with FESEM. The material's crystal structure was determined by powder X-ray diffraction (PXRD; Malvern Panalytical Empyrean) with Cu Kα (λ = 1.5406 Å). The high-resolution transmission electron microscopy (HRTEM) studies were done using a JEOL JEM-ARM200F NEOARM at 200 keV. The surface area of the samples was studied by a surface area and pore size analyzer (Quanta Chrome Autosorb-iQ) using N2 at 77 K. The materials' surface chemistry and bonding information were analyzed using an X-ray photoelectron spectroscopy (XPS) instrument (Model Nexa from Thermofisher Scientific).Electrochemical measurements
The electrodes were prepared as per our earlier report.29 The active material, PVDF, and carbon black were mixed in a ratio of 8:1:1 in NMP to form a homogeneous slurry. The slurry was coated on the current collector using the doctor blade technique and left to dry under a vacuum at 110 °C for 12 h. For the uniform thickness of the material, the coated current collector was passed through a hot rolling press and then punched out in a disc of 16 mm with the help of a disc cutter and assembled in a CR2032 coin cell with a 19 mm polypyrene (PP) separator and metallic lithium disc of 16 mm as the counter and reference electrode. The coin cell was assembled in an N2-filled glove box. The electrolyte was prepared by mixing 1 M LiPF6 in a mixture of EC:DMC (1:1 by volume). The cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) studies were performed on a CHI 1150C electrochemical workstation. The charge-discharge studies were conducted on a potentiostat Neware battery testing machine.
Results and discussion
The material was studied extensively before and after phosphorization by FESEM images, as shown in Fig. S1a–f (ESI†). The material morphology remained unaffected by the annealing at 300 °C. The FeCoP shows sea urchin-like particles grown as displayed in low and high-resolution FESEM images (Fig. 2a and b). Furthermore, the crystallinity and structural information were examined by PXRD measurements. Fig. 2c shows the XRD pattern of FeCoP powder, and the diffraction peaks at 31.7°, 35.5°, 36.5°, 46.3°, 48.3°, 52.3°, 56.2°, 56.9°, and 77.1° can be indexed to the (011), (200), (111), (112), (211), (103), (020), (013) and (222) planes of orthorhombic CoP (COD Ref. No. 96-900-8929), respectively.30,31 The peak positions are slightly shifted (± 0.1–0.3°) from the indexed position, possibly due to the incorporation of Fe in the CoP lattice. Also, the XRD pattern shows low crystallinity due to the semiconductive nature of phosphorous. Furthermore, the crystal structure was verified using an HRTEM image, as displayed in Fig. 2d. The marked planes (211), (111), and (011) match with the XRD spectra planes. Additionally, the elemental mapping of the prepared material was studied by EDS, as shown in Fig. 2e and f. The overlapped FESEM image used for data acquisition of EDS is shown in Fig. 2e. Moreover, the uniform distributions of the elements cobalt (Co), iron (Fe), phosphorous (P), and oxygen (O) are shown in Fig. 2f.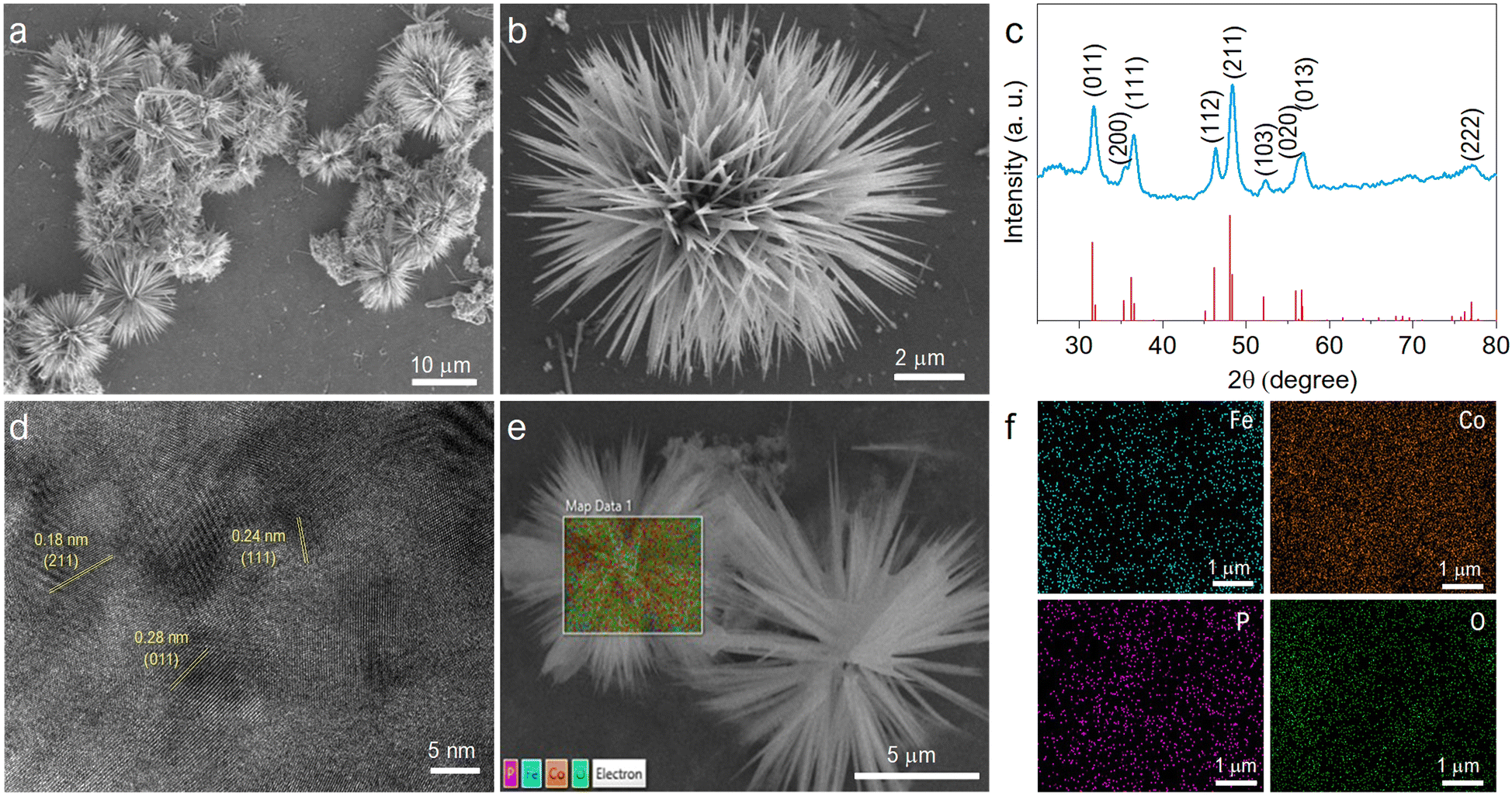 | ||
| Fig. 2 Structural and morphological studies. FESEM images of sea urchin-like morphology at (a) low and (b) high magnification. (c) PXRD spectra confirming that the material crystallizes in the CoP structure. (d) HRTEM image showing the orientation of various planes matching with planes in the XRD spectra. (e) EDS overlapped image and (f) mapping for different elements including Fe, Co, P and O. | ||
The EDS mappings before and after phosphorization confirm the presence of P after the treatment, indicating successful completion of the process (Fig. S2a and b, ESI†). The chemical composition of the material was characterized by XPS survey and narrow region scans. Fig. 3a shows the survey spectrum of FeCoP, indicating the signals from Co, Fe, P, and Oxygen (O). The high-resolution spectra for Fe 2p in Fig. 3b show two typical peaks at 724.8 eV and 711.0 eV assigned to Fe 2p1/2 and Fe 2p3/2, with spin-energy separation of 13.8 eV, while the peaks at 729.68 eV and 714.28 eV correspond to the satellite peaks. The narrow region spectra for Co are shown in Fig. 3c. The peaks at 781.58 eV and 797.78 eV are attributed to the Co 2p3/2 and Co 2p1/2 doublet, whereas the peaks at 786.78 eV and 803.28 eV are assigned to satellite peaks, respectively.32 A small peak at 778.78 eV indicates the Co–P bond.33 The spectrum shown in Fig. 3d shows the signals from element P. The peak at 133.38 eV corresponds to the P–O/P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds from oxidized P species, while the other peak at 129.18 eV is indexed to P 2p1/2. Compared to metallic Fe, Co, and elemental P species, these elements' binding energies (BE) appear to be shifted a little, which can be explained as the partial charge transfer between the metal and P atoms.
O bonds from oxidized P species, while the other peak at 129.18 eV is indexed to P 2p1/2. Compared to metallic Fe, Co, and elemental P species, these elements' binding energies (BE) appear to be shifted a little, which can be explained as the partial charge transfer between the metal and P atoms.
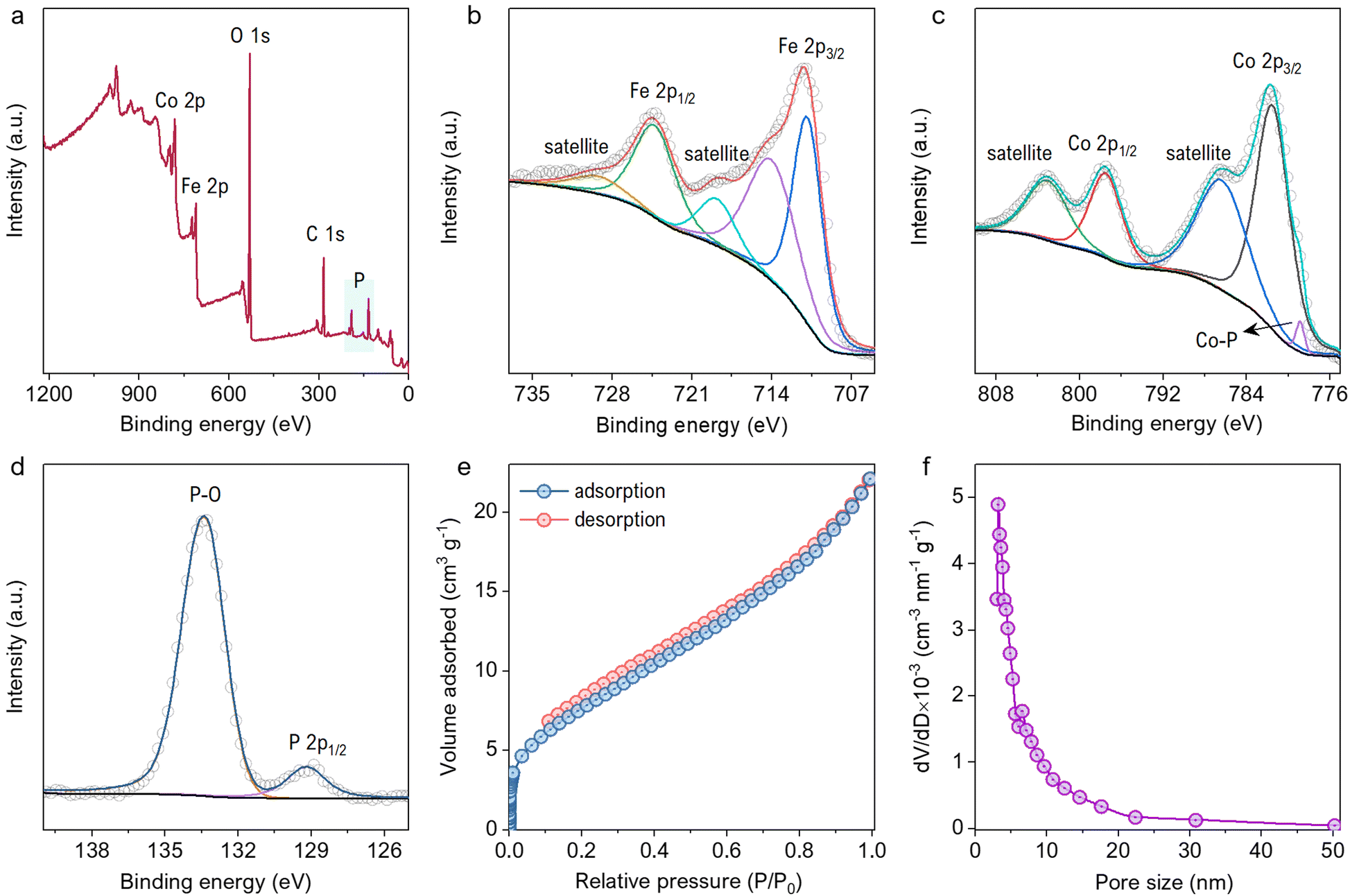 | ||
| Fig. 3 (a) XPS survey spectra showing the presence of P, C, O, Fe, and Co. (b) Narrow region high-resolution spectra for Fe 2p, (c) Co 2p, (d) and P 2p. (e) Adsorption–desorption isotherms for the FeCoP sample at 77 K, and (f) BJH model pore size distribution of the same sample. | ||
Moreover, the positive BE shift of the Co and Fe 2p orbitals tends to increase electron transfer capacity, while the negative change of the P 2p orbitals indicates increased electron-donation potency.33 The surface area and porosity studies were carried out by investigating N2 adsorption–desorption isotherms and BJH (Barrett–Joyner–Halenda) model pore size distributions, as displayed in Fig. 3e and f. The BET surface area was calculated to be 29.3 m2 g−1 for FeCoP sea urchins. The BJH pore size distributions predict mesopores varying from 3 to 50 nm that contribute to the good surface area of FeCoP.
The electrochemical performance of the as-synthesized material was studied in a half cell packed using a CR2032 type coin cell with a mass loading of about 1.1 mg cm−2. The active material was used as a working electrode, while lithium foil acts as the counter and the reference electrode. The electrochemical tests were performed in 1 M LiPF6 electrolyte. Fig. 4a shows the CV curve of the cell in the potential range of 0–3 V at a scan rate of 1 mV s−1. The peaks in the CV curves show the lithiation process of the metal phosphide as per the literature reported for various TMPs.18,34,35 The peak observed at 0.58 V corresponds to the reduction of metal phosphide. The area enclosed by the CV curve is reduced for the second cycle compared to the first due to forming an irreversible solid electrolyte interface (SEI) layer and the electrolyte decomposition.36 The subsequent CV cycles show an almost overlapping curve indicating high reversibility of the material.
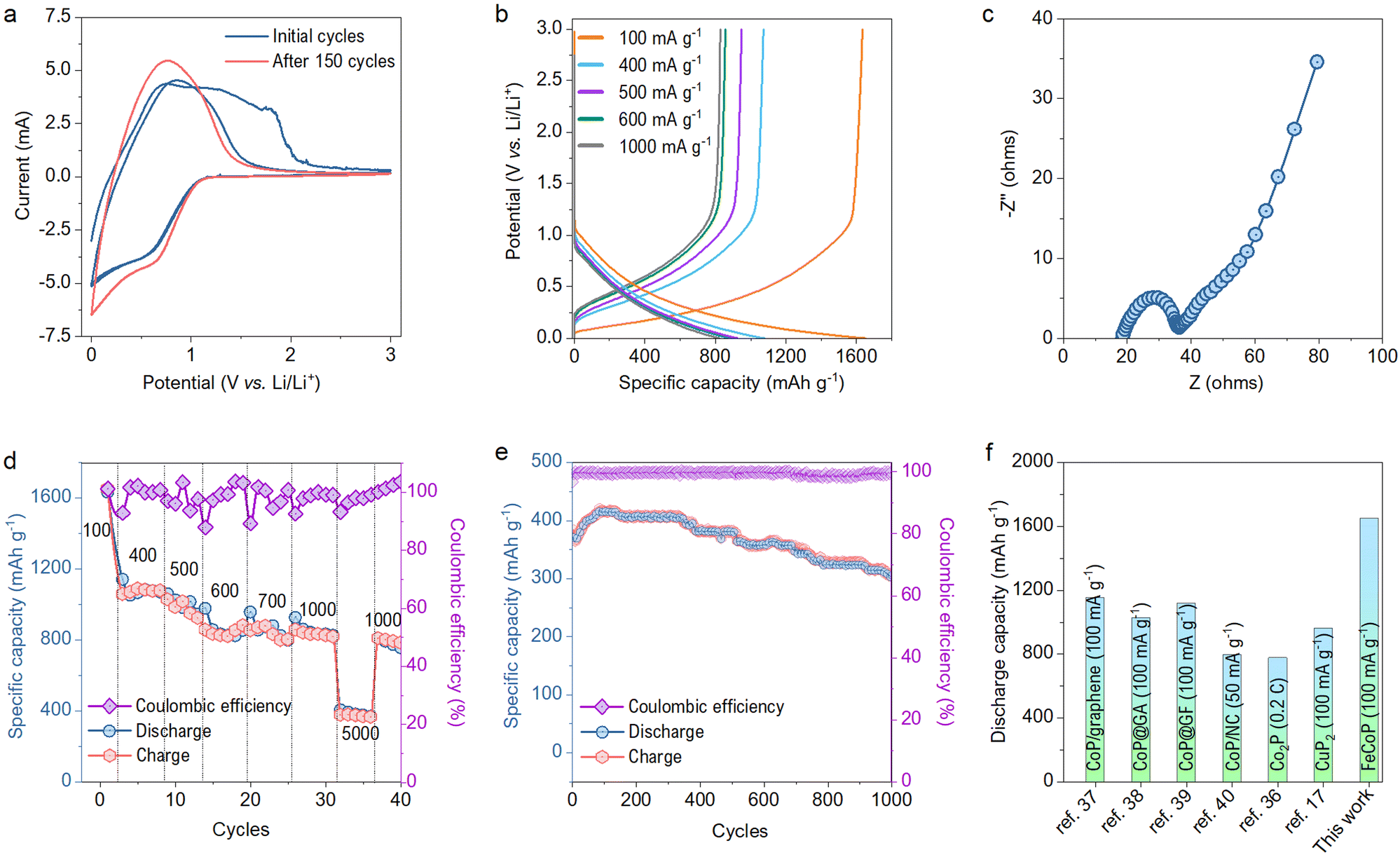 | ||
| Fig. 4 Battery test results. (a) CV profiles for the half-cell at a scan rate of 1 mV s−1. (b) Galvanostatic charge–discharge profiles at varying current densities. (c) Nyquist plot showing real vs imaginary impedance variation with input frequencies. (d) Rate capability measurements and coulombic efficiency at different current densities ranging from 100 mA g−1 to 5000 mA g−1. (e) Cyclic life testing and corresponding coulombic efficiency at the current density of 5000 mA g−1. (f) Comparison of this work with previously reported TMPs and TMPs/composites. | ||
The charge–discharge profiles at different current densities are plotted in Fig. 4b. The sharp changes in the charge–discharge curves follow intense current changes in the CV profiles. The packed half cell shows a charge capacity of 1634.5 mA h g−1 and discharge capacity of 1653.4 mA h g−1 at the current density of 100 mA g−1. The capacity obtained by FeCoP is higher than that of the previously reported metal phosphides compared in Table S1 (ESI†). The reaction kinetics at the electrode/electrolyte interfaces was studied extensively using EIS measurement in the frequency range of 1 mHz to 1 MHz before and after the cyclability test of 150 cycles. The Nyquist plots before the cyclability test are shown in Fig. 4c, obtained with semicircles in the high-frequency region. The device offers the electrode resistance of 18.36 Ω, and the semicircle region indicates the charge transfer resistance (Rct) of 18.13 Ω at the electrode-electrolyte interface.
In addition, the assembled device's rate capability was studied at various current densities. As shown in Fig. 4d, the device exhibits a specific discharge capacity of 1091.2, 1030.45 884.25, 881, 820.85 and 380.1 mA h g−1 at the current densities of 400, 500, 600, 700, 1000 and 5000 mA g−1 respectively. Furthermore, a cycling stability test was performed for 1000 cycles at a current density of 5 A g−1, as displayed in Fig. 4e. The device exhibited an excellent coulombic efficiency of nearly 99%. Also, the specific discharge capacity after the 1000th cycle is 306 mA h g−1 (80% of the first cycle), showing a minimal capacity fading rate. The EIS measurement was again performed after the 150th cycle, and Rct has decreased to 13.32 Ω, due to the formation of diffusive channels after a long cycling test as shown in Fig. S3 (ESI†). Interestingly, even after 150 cycles, an increment of 28 mA h g−1 to the initial capacity has been observed. This increment can be explained based on the reduced Rct, which indicates the easy ion intercalation due to the already developed SEI layer.
Moreover, we have compared our specific discharge capacity with the various TMPs and their composites with carbon-based materials, as shown in Fig. 4f. Our material has exhibited much better performance than previous literature reports.17,36–40 Thus, incorporating Fe in the CoP network significantly boosted the capacity and provided robust structural stability to the parent CoP.
Conclusions
We have synthesized a FeCoP via a simple hydrothermal method followed by phosphorization under a N2 atmosphere. The synthesized bimetallic TMP has exhibited an excellent electrochemical behavior and outperformed previously reported metal phosphides. The material manifests a high specific discharge capacity of 1653.4 mA h g−1 at a current density of 100 mA g−1. Also, the device shows minimal capacity fading even after 1000 cycles and delivers an excellent coulombic efficiency near 99%. Through this work, FeCoP has demonstrated a high potential of being an anode substitute for conventional LIBs. We hope that this work will provide a new approach toward developing bimetallic and trimetallic phosphides for improved metal ion battery anodes.Author contributions
PKP synthesized and characterized the materials and their electrochemical analysis. NK and VPJ have analyzed material characterization and electrochemical analysis. RRS conceived the idea, and designed, and supervised the project. All the authors contributed to writing this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
RRS is grateful for the SERB funding support from the grants SB/S2/RJN-023/2017 and CRG/2020/003199.References
- J. B. Goodenough and K. S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- J. B. Goodenough, Acc. Chem. Res., 2013, 46, 1053–1061 CrossRef CAS PubMed.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- B. N. Loeffler, D. Bresser, S. Passerini and M. Copley, Johnson Matthey Technol. Rev., 2015, 59, 34–44 CrossRef.
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X. Q. Yang and J. G. Zhang, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- M. Weiss, R. Ruess, J. Kasnatscheew, Y. Levartovsky, N. R. Levy, P. Minnmann, L. Stolz, T. Waldmann, M. W. Mehrens, D. Aurbach, M. Winter, Y. Ein-Eli and J. Janek, Adv. Energy Mater., 2021, 11, 2101126 CrossRef CAS.
- E. J. Popczun, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Angew. Chem., Int. Ed., 2014, 53, 5427–5430 CrossRef CAS PubMed.
- J. Wang, Z. Liu, Y. Zheng, L. Cui, W. Yang and J. Liu, J. Mater. Chem. A, 2017, 5, 22913–22932 RSC.
- W. Xu, T. Wang, H. Wang, S. Zhu, Y. Liang, Z. Cui, X. Yang and A. Inoue, Energy Storage Mater., 2019, 17, 300–308 CrossRef.
- X. Wang, H. M. Kim, Y. Xiao and Y. K. Sun, J. Mater. Chem. A, 2016, 4, 14915–14931 RSC.
- H. Liang, C. Xia, Q. Jiang, A. N. Gandi, U. Schwingenschlögl and H. N. Alshareef, Nano Energy, 2017, 35, 331–340 CrossRef CAS.
- Z. Wang, X. Xu, Z. Liu, D. Zhang, J. Yuan and J. Liu, Chem. – Eur. J., 2021, 27, 13494–13512 CrossRef CAS PubMed.
- M. Du, B. Qiu, Q. Zhu, M. Xing and J. Zhang, Res. Chem. Intermed., 2018, 44, 7847–7859 CrossRef CAS.
- J. W. Hall, N. Membreno, J. Wu, H. Celio, R. A. Jones and K. J. Stevenson, J. Am. Chem. Soc., 2012, 134, 5532–5535 CrossRef CAS PubMed.
- X. Wang, H.-M. Kim, Y. Xiao and Y. K. Sun, J. Mater. Chem. A, 2016, 4, 14915–14931 RSC.
- V. Pralong, D. C. S. Souza, K. T. Leung and L. F. Nazar, Electrochem. Commun., 2002, 4, 516–520 CrossRef CAS.
- G. A. Li, C. Y. Wang, W. C. Chang and H. Y. Tuan, ACS Nano, 2016, 10, 8632–8644 CrossRef CAS PubMed.
- K. Zhu, J. Liu, S. Li, L. Liu, L. Yang, S. Liu, H. Wang and T. Xie, Adv. Mater. Interfaces, 2017, 4, 1700377 CrossRef.
- W. Liu, H. Zhi and X. Yu, Energy Storage Mater., 2019, 16, 290–322 CrossRef.
- L. Wang, X. He, J. Li, W. Sun, J. Gao, J. Guo and C. Jiang, Angew. Chem., Int. Ed., 2012, 51, 9034–9037 CrossRef CAS PubMed.
- J. Sun, G. Zheng, H. W. Lee, N. Liu, H. Wang, H. Yao, W. Yang and Y. Cui, Nano Lett., 2014, 14, 4573–4580 CrossRef CAS PubMed.
- K. Wang, Y. Wang, Y. Zhang, F. Liu, J. Shi, S. Liu, X. Xie, G. Cao and A. Pan, Nanoscale, 2020, 12, 12623–12631 RSC.
- J. Yu, H. Zhang, C. Yang, Y. Xie, J. Shen, Y. Lin, C. Zheng and H. Huang, Chem. Commun., 2022, 58, 2307–2310 RSC.
- L. Cheng, S. Chen, Q. Zhang, Y. Li, J. Chen and Y. Lou, J. Energy Storage, 2020, 31, 101663 CrossRef.
- Y. Chen, W. Zhang, D. Zhou, H. Tian, D. Su, C. Wang, D. Stockdale, F. Kang, B. Li and G. Wang, ACS Nano, 2019, 13, 4731–4741 CrossRef CAS PubMed.
- R. Dai, W. Sun, L. P. Lv, M. Wu, H. Liu, G. Wang and Y. Wang, Small, 2017, 13, 1700521 CrossRef PubMed.
- Y. Li and C. Zhao, Chem. Mater., 2016, 28, 5659–5666 CrossRef CAS.
- L. Song, T. Zheng, L. Zheng, B. Lu, H. Chen, Q. He, W. Zheng, Y. Hou, J. Lian, Y. Wu, J. Chen, Z. Ye and J. Lu, Appl. Catal., B, 2022, 300, 120712 CrossRef CAS.
- N. Kumar, T. A. Wani, P. K. Pathak, A. Bera and R. R. Salunkhe, Sustainable Energy Fuels, 2022, 6, 1762–1769 RSC.
- Y. Li, F. Li, Y. Zhao, S. N. Li, J. H. Zeng, H. C. Yao and Y. Chen, J. Mater. Chem. A, 2019, 7, 20658–20666 RSC.
- C. Lin, P. Wang, H. Jin, J. Zhao, D. Chen, S. Liu, C. Zhang and S. Mu, Dalton Trans., 2019, 48, 16555–16561 RSC.
- M. Zhuang, X. Ou, Y. Dou, L. Zhang, Q. Zhang, R. Wu, Y. Ding, M. Shao and Z. Luo, Nano Lett., 2016, 16, 4691–4698 CrossRef CAS PubMed.
- Y. Lian, H. Sun, X. Wang, P. Qi, Q. Mu, Y. Chen, J. Ye, X. Zhao, Z. Deng and Y. Peng, Chem. Sci., 2019, 10, 464–474 RSC.
- N. Wang, Z. Bai, Z. Fang, X. Zhang, X. Xu, Y. Du, L. Liu, S. Dou and G. Yu, ACS Mater. Lett., 2019, 1, 265–271 CrossRef CAS.
- H. Gao, F. Yang, Y. Zheng, Q. Zhang, J. Hao, S. Zhang, H. Zheng, J. Chen, H. Liu and Z. Guo, ACS Appl. Mater. Interfaces, 2019, 11, 5373–5379 CrossRef CAS PubMed.
- D. Yang, J. Zhu, X. Rui, H. Tan, R. Cai, H. E. Hoster, D. Y. W. Yu, H. H. Hng and Q. Yan, ACS Appl. Mater. Interfaces, 2013, 5, 1093–1099 CrossRef CAS PubMed.
- Y. Yang, Y. Jiang, W. Fu, X. Z. Liao, Y. S. He, W. Tang, F. M. Alamgir and Z. F. Ma, Dalton Trans., 2019, 48, 7778–7785 RSC.
- H. Gao, F. Yang, Y. Zheng, Q. Zhang, J. Hao, S. Zhang, H. Zheng, J. Chen, H. Liu and Z. Guo, ACS Appl. Mater. Interfaces, 2019, 11, 5373–5379 CrossRef CAS PubMed.
- J. Wang, G. Zhu, Z. Zhang, Y. Wang, H. Wang, J. Bai and G. Wang, Chem. – Eur. J., 2021, 27, 1713–1723 CrossRef CAS PubMed.
- K. Zhu, J. Liu, S. Li, L. Liu, L. Yang, S. Liu, H. Wang and T. Xie, Adv. Mater. Interfaces, 2017, 4, 1700377 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: FESEM and EDS data before phosphorization, Nyquist plot after 150 cycles and table of performance comparison. See DOI: https://doi.org/10.1039/d2ma00610c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |