
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultifaceted ligand design facilitates chemical- or peptide-mediated linking of hollow gold nanoshells with tuned interparticle distance, interference and cytotoxicities†
Yu-Chen
Wang
a,
Gabriel
Théberge-Julien
 b,
Jean-Claude
Tardif
bc,
Éric
Rhéaume
*bc,
Frédéric
Lesage
*d and
Ashok
Kakkar
*a
b,
Jean-Claude
Tardif
bc,
Éric
Rhéaume
*bc,
Frédéric
Lesage
*d and
Ashok
Kakkar
*a
aDepartment of Chemistry, McGill University, 801 Sherbrooke St. West, Montreal, QC H3A 0B8, Canada. E-mail: ashok.kakkar@mcgill.ca
bResearch Center, Montreal Heart Institute, 5000 Belanger Street, Montreal, QC H1T 1C8, Canada. E-mail: eric.rheaume@icm-mhi.org
cDepartment of Medicine, Université de Montréal, Montreal, Quebec H3T 1J4, Canada
dDepartment of Electrical Engineering, Ecole Polytechnique de Montreal, C.P. 6079 succ. Centre-ville, Montreal, QC H3C 3A7, Canada. E-mail: frederic.lesage@polymtl.ca
First published on 1st August 2022
Abstract
We report a versatile methodology to covalently link hollow gold nanoshells (HAuNS) through modular design of multi-tasking ligands and bio-adaptable chemistry. The biocompatible ligand composition includes strategically placed two polyethylene glycol (PEG) chains, protected thiol-terminated tetraethylene glycol, and a reactive functional group, on a core. HAuNS are functionalized through an in situ one-pot deprotection/thiol-Au binding. The ligand-functionalized HAuNS with surface exposed COOH or OH entities are employed in constructing linked-HAuNS conjugated through a short chemical- or a longer bio-spacer (GPLGVRG peptide), in which (i) the length of the PEG chains plays an important role in minimizing oligomerization during covalent linking of HAuNS; (ii) inter-particle distance and interference of HAuNS surface plasmon resonance are regulated through chemical/peptide junctions, with UV-Vis-NIR absorption maxima red-shifted in chem-linked HAuNS; and (iii) chem-linked HAuNS-to-monomer conversion leads to amplification of the photoacoustic signal. Ligand stabilized monomeric and linked-HAuNS are less cytotoxic than citrate protected HAuNS. The synthetic tools and facile chemistry described here provide opportunities in designing linked metal nanoparticles for broad applications in biology.
Introduction
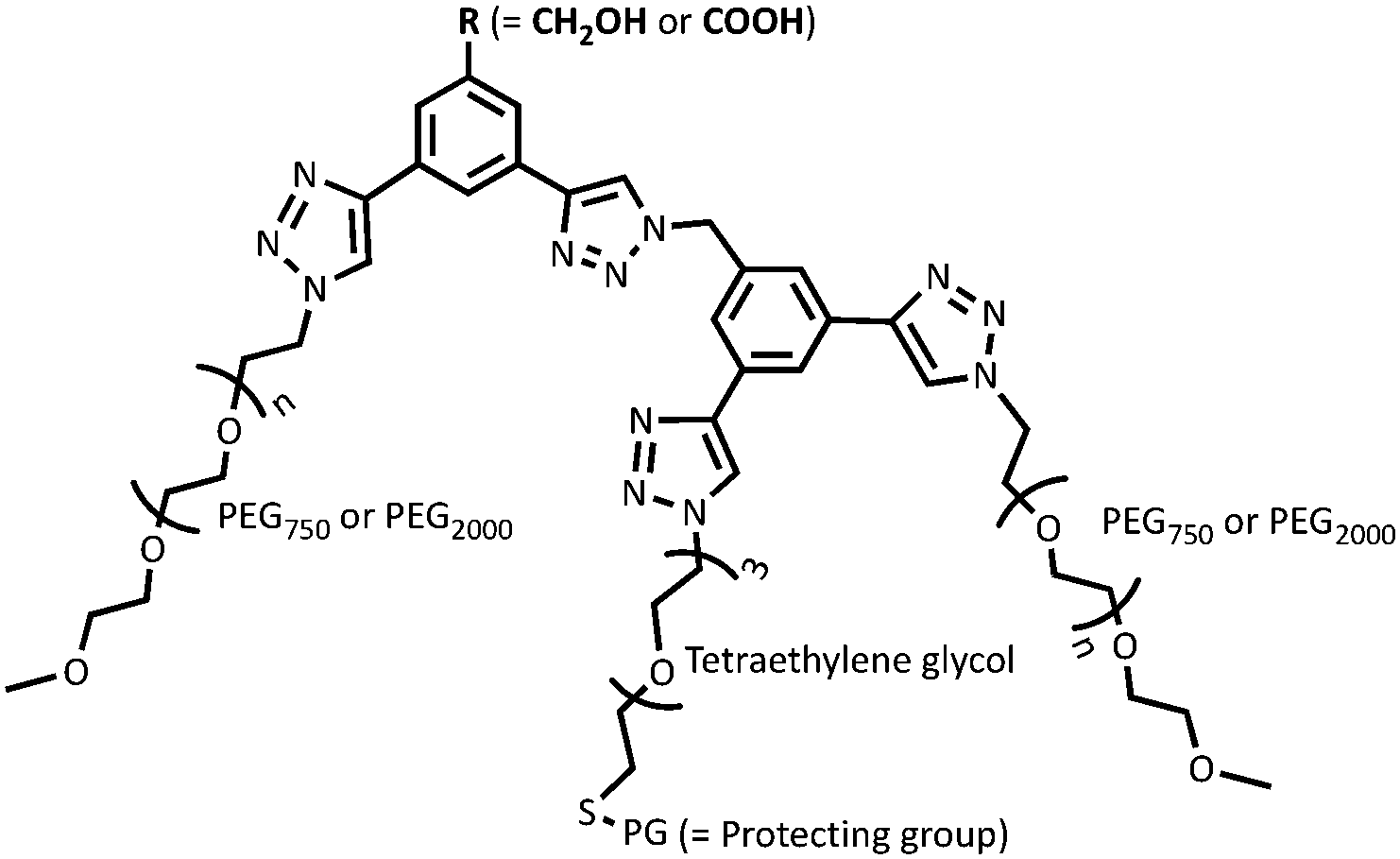
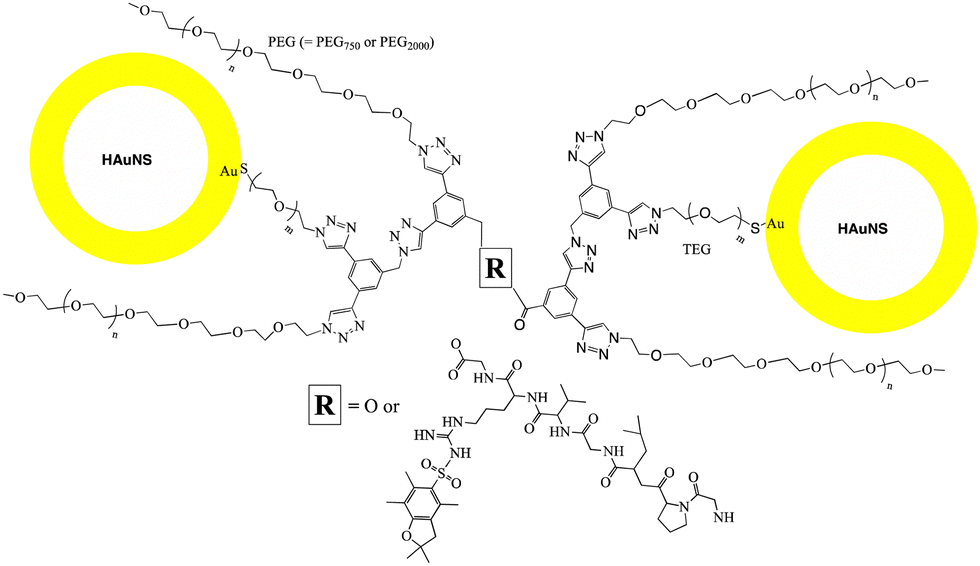
Hollow gold nanoshells (HAuNS) that offer a unique combination of properties including localized surface plasmon resonance, high optical absorption and scattering cross-section etc.,1–3 are ideally suited for a variety of applications such as photothermal therapy, surface-enhanced Raman scattering (SERS) and photoacoustic imaging.4–7 HAuNS’ surface can also be customized to offer an advantageous platform in designing a diverse range of nanomaterials.8,9 The colloidal stability of HAuNS is strongly influenced by the capping agents, which can vary from simple citrate ions to polymeric structures (poly(vinylpyrrolidone) for example).2,3,10,11 Surface modification of HAuNS is not only essential for their stabilization, but also provides easy access to introduce additional functions. HAuNS stabilized with a layer of citrate ions can be substituted with desired molecules through ligand displacement chemistry.12–17 Tailoring the surface of HAuNS can help expand their scope to the needs of a wide array of applications. For example, in nanomedicine, surface modification with poly(ethylene glycol) (PEG) is commonly used to enhance water solubility and biocompatibility of HAuNS.18,19 Functionalization with PEG prevents HAuNS from being cleared by the reticuloendothelial system, increasing their circulation.20 Other surface moieties that have been used to decorate HAuNS include antibodies,21 aptamers, peptide fragments, dextran,22 hyaluronic acid,23 as well as large macromolecules such as dendrimers.24In this study, we designed and synthetically articulated multi-tasking ligands, which were tailored to include two PEG arms, a tetraethylene glycol (TEG) unit with a protected-thiol end group in the middle of these two PEG arms, and terminal functional groups (COOH or OH) on the core (Fig. 1). Upon conjugation of these ligands to HAuNS, the long-chain PEG arms can help enhance biocompatibility, water solubility, as well as provide stability to HAuNS through steric shielding. To probe this stabilizing effect, two PEG lengths (PEG750 and PEG2000) were investigated. The COOH and OH functionalities could be used to couple HAuNS into extended metal nanoparticles using biologically relevant esterification and amidation reactions. Free thiol groups are known to be water and oxygen sensitive,25,26 and this can interfere in the surface functionalization process through S–S bond formation in solution. To address this issue, we utilized an in situ one-pot surface functionalization methodology that employs trityl-protected thiols.27 Such functionalized HAuNS can be isolated, dried and re-suspended in varied solvents.
 | ||
| Fig. 1 Ligand design for linking HAuNS. | ||
Surface plasmon resonance, a collective oscillation of conduction electrons with incident light in noble metals such as gold, has intrigued the scientific community for years due to its applications in a variety of fields.28–36 There has been tremendous effort devoted to understanding its coupling, for example (i) in nanomatryoshkas,37–39 where it is hybridized between nanoparticle inner core and the outer thin shell;40 and (ii) when metal nanoparticles come in close proximity to form an extended metal nanoparticle (EMN), leading to constructive or destructive interference.41–44 Nanomatryoshkas and EMNs have offered opportunities in designing nanomaterials for applications in photo-thermal therapy, bio- and chemical-sensing, and advanced optics and electronics.40,45,46 Hollow gold nanoshells are ideal for such applications,2–4,47–49 and theoretical understanding of the surface plasmon resonance behavior of metal nanoparticles including gold nanoshells has been ascertained using plasmon hybridization method.50 Coupling of discrete surface plasmon resonances, as in linked-HAuNS, has significant advantages in tuning nanoparticle behavior by tailoring inter-particle distance.51,52 It opens up new possibilities for exploring the design of molecular probes for applications in areas including theranostics.53
Due to the immense potential of linked metal nanoparticles in designing novel functional materials, a tremendous effort has been devoted to developing methodologies for their construction. Attempts that have mainly focused on metal nanoparticle dimer synthesis in general, have included using deoxyribose nucleic acid (DNA) base pairing, electrostatic interactions, hydrogen bonding, coordination chemistry, and through covalent bonds.53–56 Despite this elegant work, significant challenges still remain in developing chemical routes that could streamline linking metal nanoparticles57 and hollow gold nanoshells in particular, and provide a platform to evaluate their properties.
Using our synthetically articulated ligands, we have developed a versatile methodology to design covalently linked HAuNS through (i) a chemical spacer; and (ii) a small peptide, GPLGVRG (biological spacer), a targeting moiety which can be cleaved by matrix metalloproteinase (MMP-2) enzyme, generally over-expressed, for example, at atherosclerotic disease plaques.58 Through an evaluation of these linked HAuNS, we demonstrate that (i) the composition of the ligand plays a significant role in limiting oligomer formation; (ii) the length of the spacer strongly influences the interaction between linked-HAuNS; and (iii) upon reversal to monomers in chemically linked HAuNS, an amplification of the photoacoustic signal is observed. Finally, we show that the ligand-functionalized hollow gold nanoshell monomers and their linked analogs have lower cytotoxicities than the corresponding citrate-capped nanoshells.
Results and discussion
To explore the role of interparticle distance on the properties of metal nanoparticles, we investigated two different strategies for linking HAuNS: (i) through optimized separation of the monomers by conjugation through the designed ligands bound to the surface of HAuNS (chemical-spacer); and (ii) introducing a peptide linker with sequence GPLGVRG between the functionalized HAuNS monomers (bio-spacer), which will increase inter-HAuNS distance. The ligands were designed to provide steric protection to individual HAuNS, which could lead to controlled chemical reactions between monomers, and prevent oligomerization. Increase in the bulk of the multi-tasking ligand reduces the number of such molecules that can be attached to HAuNS. It leads to a decrease in the number of terminal chemical entities on the functionalized HAuNS surface monomers which will undergo a reaction for covalent linking.We envisioned that in managing and enhancing the efficacy in linking HAuNS, tailoring the ligands with fine-tuned size and composition will be a crucial parameter. In addition to providing steric protection and stability to each gold nanoshell, the ligand should allow flexibility through its exposed complementary terminal moieties for linking via chemical or biological spacers. Important features of the two complementary ligands employed in this study are shown in Fig. 1. Each has two long-chain poly(ethylene glycol)methyl ether arms with average molecular weight either 750 (PEG750) or 2000 (PEG2000), and located at ∼120° from each other. This spatial arrangement and steric hindrance of the two PEG arms will maximize HAuNS stability through firm anchoring of the ligands onto the nanoparticle surface. For this purpose, we designed a third arm between the two PEGs, based on tetraethylene glycol (TEG) with a protected SH group at the end. The latter upon deprotection would covalently link the ligands through Au–S bond, as well as provide additional flexibility through TEG.
Four different ligands in which average PEG molecular weight was 750 or 2000, were designed for this study: OH-CH2-Core-(PEG750)2-(TEGSR) (Ligand I), OH-CH2-Core-(PEG2000)2-(TEGSR) (Ligand II); COOH-Core-(PEG750)2-(TEGSR) (Ligand III) and COOH-Core-(PEG2000)2-(TEGSR) (Ligand IV). Two different lengths of PEG help examine the degree of steric protection around HAuNS, which is needed in inhibiting the formation of metal nanoparticle oligomers. To synthesize linked metal structures, we chose terminal hydroxyl and carboxylic acid groups, which can be bonded together through ester bond formation, as well as provide chemical pathways to link a bifunctional peptide via ester/amide bonds. The reasons for the selection of these functional groups also include versatility of esterification and amidation reactions that are frequently used in biology.
Multi-tasking ligands
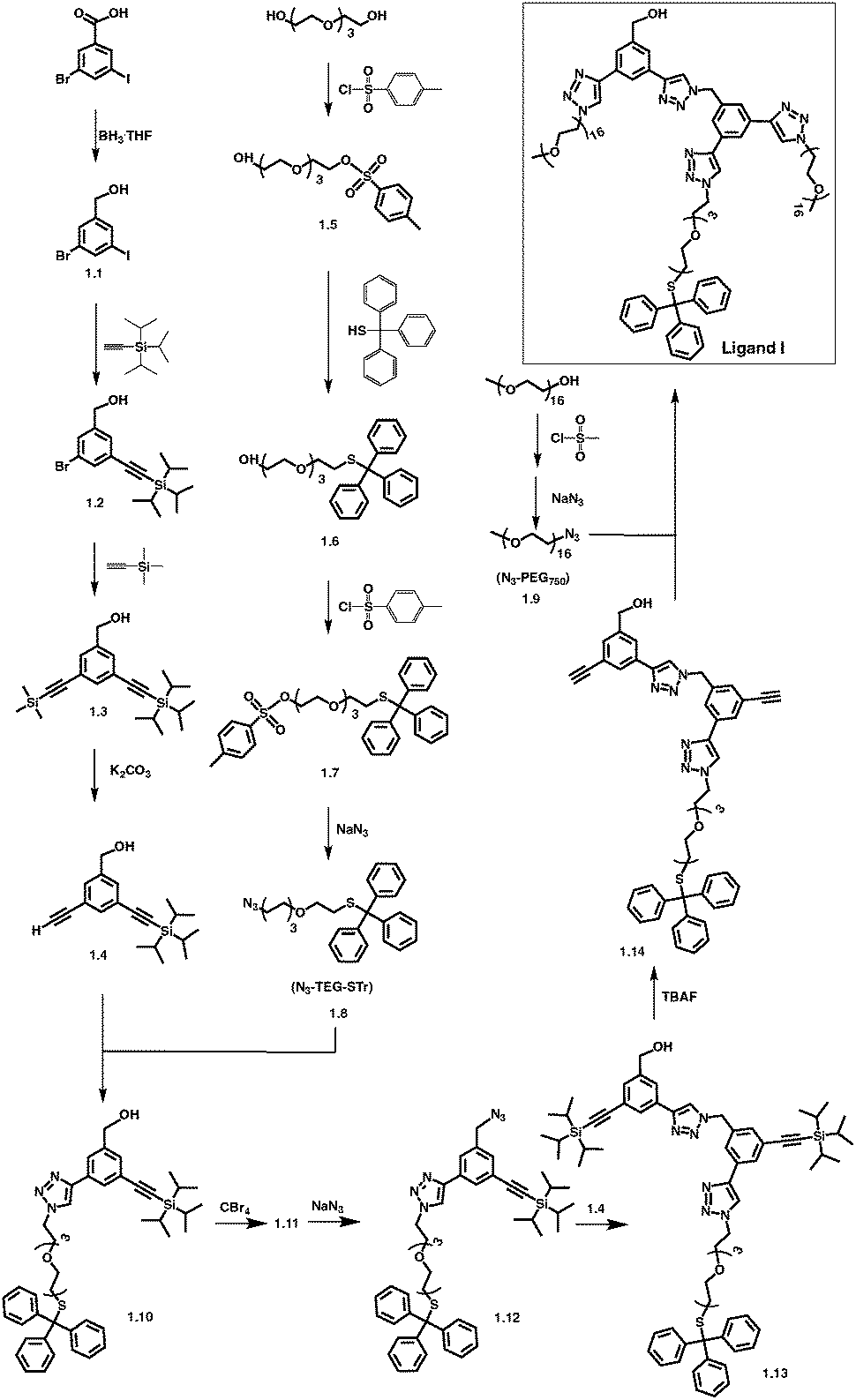
The design of overarching tri-armed ligands (Fig. 1) was achieved through a judicious choice of the core, and build-up of the structure on it through efficient coupling reactions.59,60 For example, the synthesis of OH-terminated branched ligand, OH-CH2-Core-(PEG750)2-(TEGSR) (Ligand I, Scheme 1) began with the reduction of the commercially available 3-bromo-5-iodo-benzoic acid to give compound 1.1. Its chemical structure was confirmed by the appearance of a singlet at 4.64 ppm, corresponding to the benzylic protons (OH–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2–Bz, s, 2H) in the 1H NMR spectrum. Sonogashira couplings on 1.1 first with (i) triisopropylsilyl(TiPS)-acetylene (singlet at 1.21 ppm for the TiPS protons); and then (ii) trimethylsilyl(TMS)-acetylene (additional singlet at 0.24 ppm for the TMS protons), gave compounds 1.2 and 1.3. The deprotection of the trimethylsilyl group yielded free ethynyl group (1.4), which was accompanied by the appearance of a singlet at 3.07 ppm.
2–Bz, s, 2H) in the 1H NMR spectrum. Sonogashira couplings on 1.1 first with (i) triisopropylsilyl(TiPS)-acetylene (singlet at 1.21 ppm for the TiPS protons); and then (ii) trimethylsilyl(TMS)-acetylene (additional singlet at 0.24 ppm for the TMS protons), gave compounds 1.2 and 1.3. The deprotection of the trimethylsilyl group yielded free ethynyl group (1.4), which was accompanied by the appearance of a singlet at 3.07 ppm.
 | ||
| Scheme 1 Synthesis of OH-CH2-Core-(PEG750)2-(TEGSR) (Ligand I). For complete synthetic details see ESI.† | ||
Monotosylation of tetraethylene glycol (TEG) (1.5),61 was followed by replacement with trityl group to yield the protected thiol-terminated TEG (1.6). The singlet for the methyl protons from the tosyl group in 1.5 at 2.45 ppm, and two doublets at 7.78 for the four protons of tosyl in its 1H NMR spectrum, disappeared. Instead, a multiplet for the 15 protons of the trityl group appeared between 7.24–7.48 ppm, suggesting the replacement of tosyl with trityl group. Additionally, we observed an upfield shift of the triplet resonance, corresponding to C2–O–tosyl, from 4.16 ppm to 2.43 ppm, caused by the influence of less electron-withdrawing effect of S-trityl compared to O-tosyl group. 13C NMR was consistent with the 1H NMR results, in which the appearance of ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) -Ph3 at 31.6 ppm and the disappearance of Bz–H3 at 21.4 ppm, also indicated the replacement of tosyl by trityl group in 1.6.
-Ph3 at 31.6 ppm and the disappearance of Bz–H3 at 21.4 ppm, also indicated the replacement of tosyl by trityl group in 1.6.
The tosylation at the other end of 1.6 led to the appearance of a doublet at 7.78 ppm for the two aromatic protons on the tosyl group in its 1H NMR spectrum, while the peaks for the other two aromatic protons overlapped with that for the similar protons on trityl at 7.20–7.44 ppm, with an increase in proton integration from 15 to 17. In addition, a triplet at 4.14 ppm indicated the presence of methylene protons next to O-tosyl, and the integration for the protons at 2.40–2.44 ppm increased from 3 to 5. It implied that the protons for SO2–Bz–C3 and C2–S–CPh3, were overlapping in 1.7. In its 13C NMR spectrum, the peaks for tosyl and trityl groups (126.6 to 144.8 ppm), and a peak at 21.6 ppm for the terminal methyl of the tosyl group were observed.
Azidation reaction on 1.7 led to the formation of 1.8 and followed the loss of peaks in the aromatic region, as well as a singlet at 2.43 ppm (3H, C3–Bz–SO2) which overlapped with the methylene protons next to S-trityl on the tosyl group. In addition, the peak for the ethylene protons shifted upfield from 4.14 ppm (t, 2H, –CH2–C2-tosyl) to 3.31–3.37 ppm (m, 2H, –CH2–C2–N3), which is due to less electron-withdrawing effect of the azide. Azidation of PEG750 was similarly carried out by first reacting it with mesyl chloride, followed by the reaction with sodium azide to give 1.9.62
The CuI-catalyzed alkyne-azide click reaction between 1.4 and 1.8 gave compound 1.10, the 1H NMR of which showed that the singlet at 3.07 ppm, corresponding to ethynyl proton had disappeared, and a singlet for the proton on the triazole ring appeared at 8.02 ppm. The aromatic protons on the adjacent carbon of the more electron-withdrawing triazole ring were relocated to 7.78 and 7.87 ppm from 7.46 and 7.52 ppm. The multiplet at 3.34–3.36 ppm, corresponding to the ethylene protons next to an azide, shifted lower field to 4.49 ppm; and the ethylene protons at β carbon (m, 3.34–3.68 ppm), moved lower field (3.84 ppm). In the 13C NMR, the peaks at 77.9 and 82.7 ppm, corresponding to the ethynyl carbons, disappeared.
The bromination of 1.10 led to the formation of 1.11, in which benzylic protons showed a small downfield shift from 4.67 to 4.70 ppm. In the 13C NMR spectrum of 1.11 the peak for the benzylic carbon shifted from 63.2 to 32.5 ppm. Its azidation with NaN3 yielded 1–12, in which the resonance for the benzylic protons moved upfield from 4.69 to 4.55 ppm in its 1H NMR, and the benzylic carbon moved from 32.5 to 53.6 ppm in the 13C NMR spectrum.
Subsequently, a click reaction between 1.4 and 1.12 led to the formation of 1.13 and was accompanied by the disappearance of the peak at 3.07 ppm and appearance of a singlet at 8.52 ppm. The singlet for the benzylic protons moved downfield from 4.55 to 5.75 ppm, suggesting that the azide, has been replaced with a triazole ring. In the 13C NMR spectrum, the carbon peaks for the ethynyl group at 91.7 and 105.7 ppm disappeared and were replaced with the peaks corresponding to the triazole carbons at 143.8 and 146.6 ppm.
The triisopropylsilyl protective group on acetylene in 1.13 was removed with TBAF and was confirmed by the disappearance of the singlet at 1.17 ppm corresponding to TiPs protons. Instead, two singlets at 3.66 and 3.74 ppm corresponding to the ethynyl protons were observed. The 13C NMR spectrum of 1.14 was consistent with the results from its 1H NMR and showed that peaks for the triisoproprylsilyl carbons at 11.1 and 18.2 ppm had disappeared; and the peaks for the acetylene carbons shifted from 106.6 and 107.3, to 78.2 and 79.2 ppm.
Finally, N3-PEG750 (1.9) was reacted with 1.14 in a CuAAC reaction, leading to the formation of Ligand I (OH-CH2-Core-(PEG750)2-(TEGSR), Scheme 1). As expected, peaks for the ethynyl protons in 1.14 at 3.66 and 3.74 ppm disappeared, and two singlets for the protons on the triazole rings appeared at 8.31 and 8.45 ppm in Ligand I. The thiol protecting group was left intact during the whole synthetic multi-step procedure.
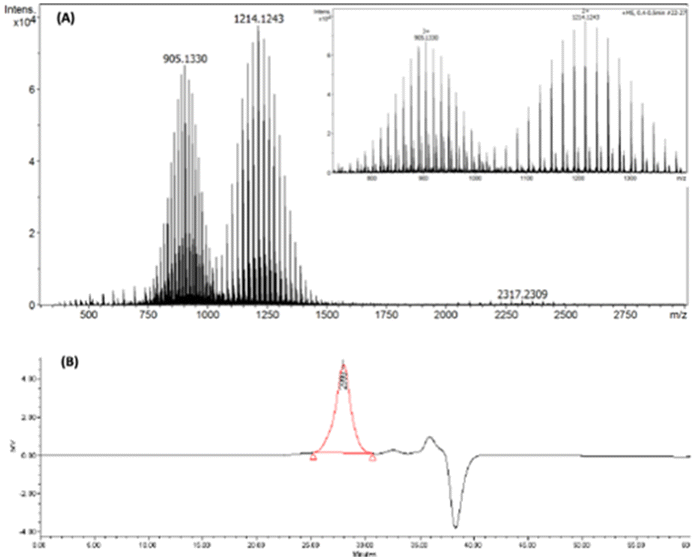
Additional confirmation for the synthesis of Ligand I was obtained from high resolution mass spectroscopy (HR-MS) and gel permeation chromatography (GPC). The HR-MS spectrum of Ligand I (Fig. 2(A)) contained two ion sets carrying three and two positive charges, and both showed the unique molecular weight distribution of PEG. GPC chromatogram (Fig. 2(B)) showed a single distribution. Monomethylated PEG750, PEG2000, PEG3400 and PEG5000 were used for calibration, and an average molecular weight (Mn) equal to 2910 with a polydispersity index (PDI) of 1.13 (close to HR-MS results) was obtained for Ligand I.
 | ||
| Fig. 2 Ligand I, (OH-CH2-Core-(PEG750)2-(TEGSR)): (A) high-resolution mass spectrum; and (B) GPC chromatogram. | ||
Using the same method (Scheme 1), the size of PEG moieties was easily modified in the synthesis of OH-CH2-Core-(PEG2000)2-(TEGSR) (Ligand II). The only change in the procedure was substituting N3-PEG750 (1.9) with N3-PEG2000. The successful synthesis was confirmed by 1H, 13C NMR, MALDI-TOF and GPC. For comparison, the 1H NMR integration on the –O–C2– region at 3.49–3.74 ppm (m) gave 144 protons for OH-CH2-Core-(PEG750)2-(TEGSR), and 312 protons for OH-CH2-Core-(PEG2000)2-(TEGSR). This increase in the number of protons is consistent with the increase in average number of repeat units of ethylene glycol from 16 in N3-PEG750 to 46 in N3-PEG2000. Using GPC, an Mn of 6935 with PDI of 1.09 was obtained.
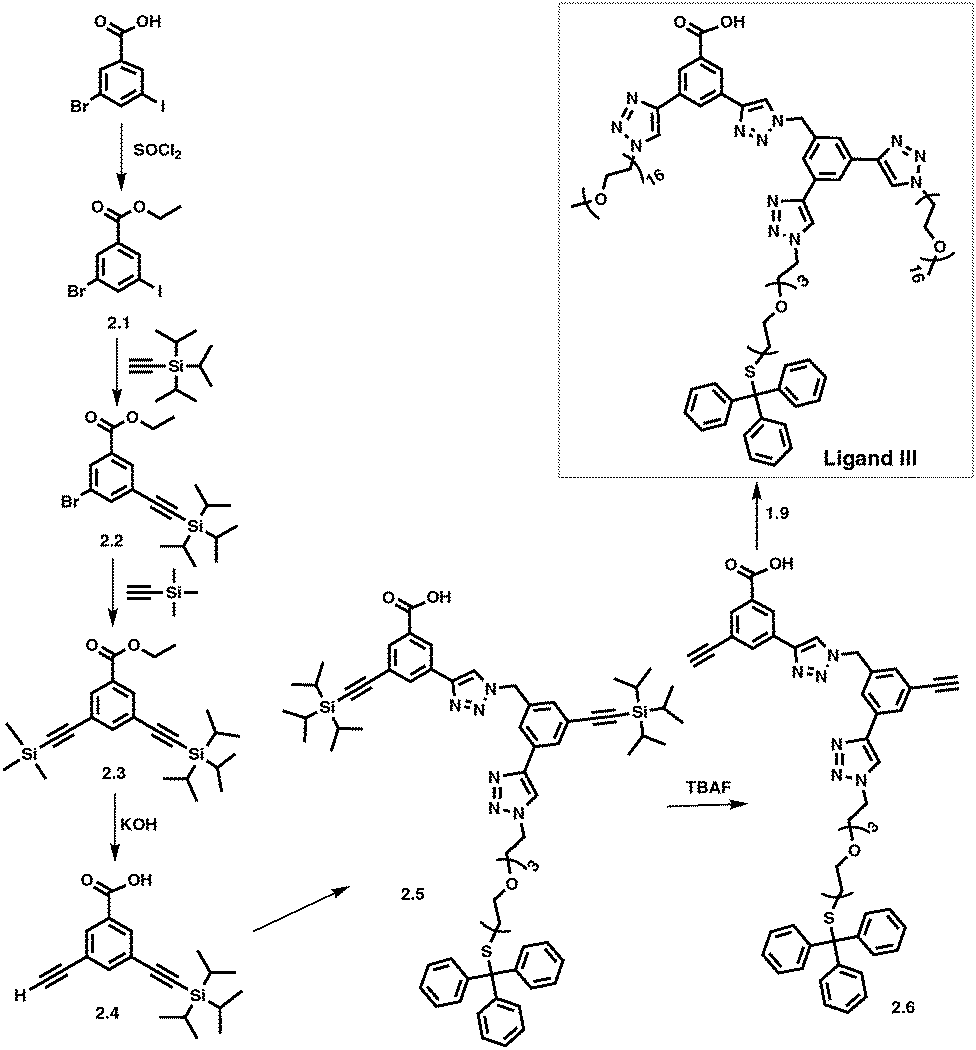
By adapting a similar methodology as described above for Ligands I and II (Scheme 1), COOH terminated ligands with two different PEG lengths, COOH-Core-(PEG750)2-(TEGSR) (Scheme 2, Ligand III) and COOH-Core-(PEG2000)2-(TEGSR) (Ligand IV) were prepared. Syntheses of compounds 2.1 to 2.4 were carried out by adaptation and modification of the literature procedures.63 Protection of the starting material, 3-bromo-5-iodobenzoic acid with an ethyl group (2.1) was confirmed with the appearance of a doublet at 1.30 and a quartet at 4.26 ppm in its 1H NMR spectrum. Sonogashira coupling on its (i) iodo end with TiPS-acetylene led to the appearance of a singlet for the TiPS protons at 1.15 ppm (2.2), and then (ii) TMS-acetylene (2.3), another singlet at 0.28 ppm. Deprotection of TMS and ethyl groups with KOH led to the synthesis of 2.4 in one pot. It was confirmed with the appearance of ethynyl proton at 3.86 ppm, and the disappearance of the peaks corresponding to ethyl and TMS protons.
 | ||
| Scheme 2 Synthesis of COOH-Core-(PEG750)2-(TEGSR) (Ligand III). For complete synthetic details see ESI.† | ||
CuI catalysed alkyne-azide coupling reaction (CuAAC) between compounds 2.4 and 1.8 led to the synthesis of 2.5 (Scheme 2), which was accompanied by the appearance of two singlets at 8.56 and 8.67 ppm corresponding to triazole ring protons, as well as a downfield shift in the singlet for the benzylic protons from 4.56 to 5.78 ppm. The successful TiPS deprotection on 2.5 was confirmed by the absence of peaks corresponding to TiPS protons, and the appearance of two singlets at 3.74 and 3.81 ppm for the ethynyl protons (2.6). Similarly, in the 13C NMR spectrum, peaks for TiPS carbons disappeared and those for the ethynyl carbons shifted upfield from 90.9, 91.4, 105.9 and 106.6 ppm to 79.2, 79.6, 82.1 and 82.6 ppm. The synthesis of COOH-Core-(PEG750)2-(TEGSR) (Scheme 2, Ligand III) was completed by the CuAAC reaction between 2.6 and N3-PEG750. In its 1H NMR spectrum, peaks corresponding to ethynyl protons in compound 2.6 disappeared, followed by the appearance of two singlets at 8.60 and 8.67 ppm, corresponding to triazole ring protons. In its 13C NMR, disappearance of the resonances corresponding to ethynyl carbons at 79.2, 79.6, 82.1 and 82.6 ppm were observed. Additional analyses through HR-MS showed unique distribution of PEG with two and three positive charges sets. GPC gave Mn equal to 2830 with PDI of 1.13. The synthesis of COOH-Core-(PEG2000)2-(TEGSR) (Ligand IV) was carried out using a similar procedure by substitution with N3-PEG2000, and characterized using a combination of techniques as described above.
One-pot in situ detritylation and ligand exchange on HAuNS
As mentioned earlier, free thiol chemical entities are reactive, and it is essential to minimize their dimerization during surface functionalization process. It can be achieved through protective groups such as trityl, which can be subsequently removed using a variety of methods.64,65 We carried out the ligand detritylation reaction using trifluoroacetic acid (TFA) and triethylsilane (TES), in which TES plays the role of a carbocation scavenger.66 Due to ligand bulkiness, the detritylation reaction was performed overnight for efficient deprotection, followed by in situ ligand to gold nanoshell binding. The reaction was carried out in a biphasic environment of water and DCM, and a change in solubility of the gold nanoshells was observed upon ligand attachment. The solubility of functionalized HAuNS in DCM increased, and this could be observed visually. The citrate-capped hollow gold nanoshells prefer the upper water layer (Fig. 3(A)), while the ligand-stabilized gold nanoshells (Fig. 3(B)) prefer to be in DCM. It showed that upon deprotection of the thiol and covalent anchoring of the resulting ligands in situ, the ligand functionalized gold nanoshells moved to the DCM layer, leaving the water fraction clear and colorless. | ||
| Fig. 3 Solubility of HAuNS: citrate-capped HAuNS before (A), after (B) ligand attachment (OH-CH2-Core-(PEG750)2-TEGS-HAuNS). | ||
Functionalized HAuNS were characterized using a combination of techniques including thermogravimetric analysis (TGA), UV-Vis-NIR spectroscopy, dynamic light scattering (DLS), and transmission electron microscopy (TEM). Thermogravimetric analyses of the functionalized HAuNS indicated that in situ detritylation reaction was efficient in covalently linking the desired ligands. The number of ligands per shell attached to HAuNS were found to be dependent on the length of polyethylene glycol chains (Table S1, ESI†). Ligand surface coverage, as expected, decreased from 58 and 63% for the smaller length PEG ligands (OH-CH2-Core-(PEG750)2-TEGS-HAuNS and COOH-Core-(PEG750)2-TEGS-HAuNS) to 35 and 49% for OH-CH2-Core-(PEG2000)2-TEGS-HAuNS and COOH-Core-(PEG2000)2-TEGS-HAuNS respectively. It should be noted that in our calculations it is assumed that the weight loss during the heating process was fully caused by ligand combustion for all the functionalized HAuNS.
UV-Vis-NIR spectroscopy can provide information related to the size, shape, concentration, and the agglomeration state of the functionalized HAuNS. Fig. 4 below shows the UV-Vis-NIR spectra of gold nanoshells decorated with the four ligands: OH-CH2-Core-(PEG750)2-(TEGSR) (Ligand I), OH-CH2-Core-(PEG2000)2-(TEGSR) (Ligand II); COOH-Core-(PEG750)2-(TEGSR) (Ligand III) and COOH-Core-(PEG2000)2-(TEGSR) (Ligand IV). The absorption of HAuNS before and after ligand attachment indicated no significant changes, suggesting that the multifunctional ligands are able to stabilize HAuNS, and the length of PEG does not seem to significantly affect the absorption characteristics of HAuNS.
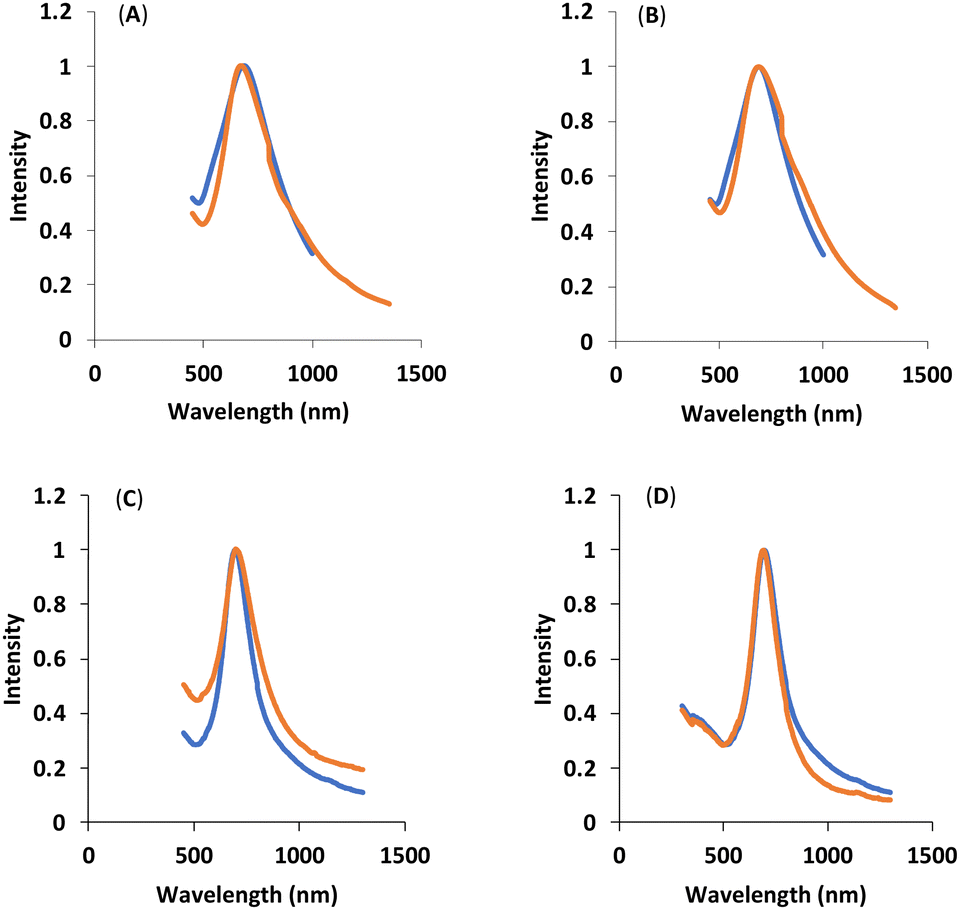 | ||
| Fig. 4 UV-Vis-NIR spectra: blue (citrate ions-capped HAuNS), orange (Ligand functionalized HAuNS): (A) OH-CH2-Core-(PEG750)2-TEGS-HAuNS, Ligand I-HAuNS; (B) COOH-Core-(PEG750)2-TEGS-HAuNS, Ligand II-HAuNS; (C) OH-CH2-Core-(PEG2000)2-TEGS-HAuNS, Ligand III-HAuNS; and (D) COOH-Core-(PEG2000)2-TEGS-HAuNS, Ligand IV-HAuNS. | ||
Dynamic light scattering (DLS) measurements were conducted to understand the hydrodynamic size changes caused by functionalization of HAuNS with ligands reported here, on two different batches of citrate stabilized HAuNS (Tables 1 and 2). In addition, zeta potentials can provide information about the surface charge and nanoparticle stability. In general, upon citrate replacement with Ligands I–IV, an increase in hydrodynamic diameter was observed in each case (Tables 1 and 2), which is related to the larger size of the ligands compared to small citrate ions.
| HAuNS | Hydrodynamic diameter (nm) | Zeta potential (mV) |
|---|---|---|
| Citrate stabilized HAuNS | 41.59 ± 0.99 | |
| OH-CH2-Core-(PEG750)2-TEGS-HAuNS | 49.25 ± 1.08 | −25.83 |
| COOH-Core-(PEG750)2-TEGS-HAuNS | 54.87 ± 2.06 | −36.78 |
| HAuNS | Hydrodynamic diameter (nm) | Zeta potential (mV) |
|---|---|---|
| Citrate stabilized HAuNS | 22.32 ± 2.02 | |
| OH-CH2-Core-(PEG2000)2-TEGS-HAuNS | 26.85 ± 2.58 | −6.36 |
| COOH-Core-(PEG2000)2-TEGS-HAuNS | 35.93 ± 1.81 | −14.6 |
In evaluating the effect of the ligands with the same PEG length but different terminal groups (carboxylic acid vs. hydroxyl), the hydrodynamic diameter difference between citrate terminated HAuNS and COOH-Core-(PEG)2-TEGS-HAuNS (Ligands II and IV-based HAuNS, Table 2) was found to be larger than with OH-CH2-Core-(PEG)2-TEGS-HAuNS (Ligands I and III-based HAuNS, Table 1). This may likely be due to dissociation of the carboxylic acid terminal group compared to hydroxyl (pKa of benzoic acid = 4.2; pKa of benzyl alcohol = 15.2) in these ligands.67,68 Carboxylic acid terminal group would also have a stronger affinity for water and counter ions, compared to hydroxyl. In other words, COOH-Core-(PEG)2-TEGS-HAuNS (Ligands II and IV-based HAuNS) may not be as ‘mobile’ as OH-CH2-Core-(PEG)2-TEGS-HAuNS (Ligands I and III-based HAuNS).
For the same PEG length, COOH-Core-(PEG)2-TEGS-HAuNS (Ligand II- and Ligand IV-based HAuNS) had higher surface charge than OH-CH2-Core-(PEG)2-TEGS-HAuNS (Ligand I- and Ligand III-based HAuNS, Tables 1 and 2), again suggesting dissociation of the carboxylic acid terminal group. For the same terminal groups on the ligand (COOH-core-(PEG750![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) or2000)2-TEGS-HAuNS; OH-CH2-Core-(PEG750or2000)2-TEGS-HAuNS), surface charge decreased when PEG chain length increased (Tables 1 and 2). This is consistent with earlier reports, in which it has been suggested that the longer PEG chains offer steric shield and decrease surface charge on gold nanoparticle surface.69 Based on both DLS and zeta potential results, we concluded that longer PEG chains better engulf nanoparticle surface, as well as limit the ability of ligand terminal groups to attract water and other ions.
or2000)2-TEGS-HAuNS; OH-CH2-Core-(PEG750or2000)2-TEGS-HAuNS), surface charge decreased when PEG chain length increased (Tables 1 and 2). This is consistent with earlier reports, in which it has been suggested that the longer PEG chains offer steric shield and decrease surface charge on gold nanoparticle surface.69 Based on both DLS and zeta potential results, we concluded that longer PEG chains better engulf nanoparticle surface, as well as limit the ability of ligand terminal groups to attract water and other ions.
HAuNS were subsequently analyzed using TEM, and the micrographs for citrate-capped HAuNS and typical ligand functionalized L-HAuNS (OH-CH2-Core-(PEG2000)2-TEGSHAuNS) and (COOH-Core-(PEG2000)2-TEGHAuNS) are shown in Fig. 5. The images depict that the HAuNS are intact and are coated with a layer of multifunctional ligands (Fig. 5(B) and (C)). The distribution of gold nanoshells can be affected by the interaction between HAuNS surface and carbon grid. We noticed that the ligand-functionalized L-HAuNS in general were distinctly isolated from each other as compared to the citrate-capped HAuNS (Fig. 5(A)). Such representations of functionalized gold nanoparticles in TEM have also been reported earlier.70–73 It suggested that ligand functionalized HAuNS were well stabilized through steric repulsions imposed by these ligands compared to citrate ions.
 | ||
| Fig. 5 TEM images of the citrate capped HAuNS (A); (OH-CH2-Core-(PEG2000)2-TEGS-HAuNS) (B); and COOH-Core-(PEG2000)2-TEG-HAuNS (C). | ||
Linked-HAuNS
As mentioned earlier, we explored two approaches to covalently bind HAuNS, (i) ChemLinked: in which a set of HAuNS were individually functionalized with complementary ligands (OH-CH2-Core-(PEG750or2000)2-TEGS-HAuNS) or (COOH-Core-(PEG750or2000)2-TEGS-HAuNS), followed by covalent bond formation via esterification; and (ii) BioLinked, in which functionalized HAuNS were bound through a bifunctional linker (a peptide, with sequence GPLGVRG). We used 1,4-dimethylpyridin-1-ium,4-methylbenzenesulfonate (DPTS) and N,N′-diisopropylcarbodiimide (DIC) as catalysts for Steglich esterification. DPTS allows high yield esterification under mild reaction conditions, and it is also tolerant of a small amount of water needed in these reactions.74,75
We chose esterification to chemically link surface functionalized nanoshells (OH-CH2-Core-(PEG750or2000)2-TEGS-HAuNS) and (COOH-Core-(PEG750or2000)2-TEGS-HAuNS) (Fig. 6), which required the individual ligand functionalized HAuNS to be dried before the reaction. Though gold nanoparticles can be placed under reduced pressure for an extended period of time during the drying process, HAuNS could be left for no more than 5 minutes. The esterification reaction was carried out in anhydrous DCM, as ligand-functionalized-HAuNS suspend well in this solvent.
 | ||
| Fig. 6 Cartoon representation of ChemLinked- and BioLinked-HAuNS. | ||
After stirring the reaction at room temperature under N2 overnight, a precipitate settled at the bottom of the reaction flask. After washing it with DCM multiple times, it still showed the tendency to gradually settle at the bottom of the flask in DCM. This suggested to us that the precipitate was from the desired linked-HAuNS, since monomeric HAuNS that would have precipitated due to excess charge on the surface brought on by reagents such as DPTS, would have resuspended after washings. DCM was subsequently used as a solvent of choice to remove any unreacted monomeric HAuNS using centrifugation. ChemLinked-HAuNS could be suspended in DMSO and DMF and were stable for a tested period of over a month, while they were stable in water and methanol only for a few days.
A peptide with sequence GPLGVRG was chosen for bio-linking HAuNS. To synthesize the desired BioLinked-HAuNS, (Fig. 6) amine group on the peptide was first protected with a fluorenylmethyloxycarbonyl (Fmoc) group. This was to prevent side reactions, including the peptide reacting with its own carboxylic acid end groups. To optimize conditions and enhance the efficacy of bio-linking functionalized HAuNS, a model reaction was first conducted. Benzyl alcohol and benzoic acid, due to their similarities to the core of the ligands, were chosen for the reaction with Fmoc protected peptide (Fmoc-P). Formation of the Fmoc-P-ester with benzyl alcohol was monitored using 1H NMR and HR-MS.
The 1H NMR spectrum was not very clear in the region below 6.5 ppm, possibly due to the strong affinity of Fmoc-P-ester chains to stick to each other. We therefore employed diffusion ordered spectroscopy (DOSY), to determine if there were multiple Fmoc-P-ester products or a mixture of Fmoc-P-ester and reactants. Compared to conventional 1H NMR spectroscopy, a magnetic field gradient is applied to the sample, as an extra spatial parameter in 2D DOSY NMR.76,77 Besides, electrophoretic mobilities are correlated with friction factors and effective charges. As a result, different structures have varied diffusion coefficients and are at different positions in an NMR tube. It lends DOSY the ability to determine structures of mixtures, reaction intermediates or even molecules with varied molecular weights.78,79 A comparison of the routine 1H and 2D DOSY NMR spectra (Fig. S19, ESI†) showed that the aromatic region was a good indicator for the completion of the reaction in which the presence of the peak for the aromatic proton from benzyl alcohol overlaps with that from Fmoc at 7.27–7.33 ppm (m, 7H).
Fmoc deprotection was carried out with piperidine, and reaction completion was confirmed using HR-MS and 1H NMR (Fig. S10, ESI†) in which the disappearance of Fmoc at 7.34 (t, 2H), 7.43 (t, 2H), 7.63 (s, 2H), 7.80 (s, H) and 7.82 ppm (s, H) indicated the removal of Fmoc. Amide-P-Ester was synthesized via amidation of the resulting P-Ester with benzoic acid, and the reaction completion was similarly confirmed using HR-MS and 1H NMR spectroscopies (Fig. S11, ESI†).
Synthesis of the BioLinked-HAuNS (Fig. 6) was subsequently carried out using conditions set by the above model reaction. Fmoc-P-O-CH2-Core-(PEG)2-TEGSR was synthesized via esterification between protected peptide, Fmoc-P, and OH-CH2-Core-(PEG)2-TEGSR. We found that the solubility difference between Fmoc-P-O-CH2-Core-(PEG)2-TEGSR and Fmoc-P was the best method to afford the pure product. HAuNS were subsequently functionalized using an in situ detritylation reaction for the removal of the protecting group on the thiol (SR), which then reacted with HAuNS, yielding ligand-anchored Fmoc-P-O-CH2-Core-(PEG)2-TEG-HAuNS. Peptide end was subsequently deprotected using piperidine in DMF to afford H2N-P-O-CH2-Core-(PEG)2-TEG-HAuNS. Amidation reaction was then carried out between H2N-P-O-CH2-Core-(PEG)2-TEG-HAuNS and COOH-Core-(PEG)2-TEGS-HAuNS using DPTS and DIC in dicholoromethane.80 Excess reagent was removed by washing with DCM, centrifugation and removal of supernatant.
Linked-HAuNS were visualized using transmission electron microscopy (TEM, Fig. 7). These micrographs together with those of monomers indicated that upon functionalization with sterically imposing ligands (I to IV), and upon covalently linking, HAuNS were far apart and few in number in each image, in contrast to citrate stabilized-HAuNS (Fig. 5 and 7). When the PEG chain length was shorter (PEG750), we observed the formation of dimers, as well as some trimers, for both ChemLinked-PEG750 and BioLinked-PEG750. When a longer chain PEG2000 was employed, only dimers for both ChemLinked-PEG2000 and BioLinked-PEG2000 were observed. The magnifications in Fig. 7 (see insets) showed that, in each, there is a layer of ligand surrounding HAuNS. For the ChemLinked, it seems that HAuNS were “fused”, while for BioLinked, we measured a space of ∼1 nm between the two HAuNS.
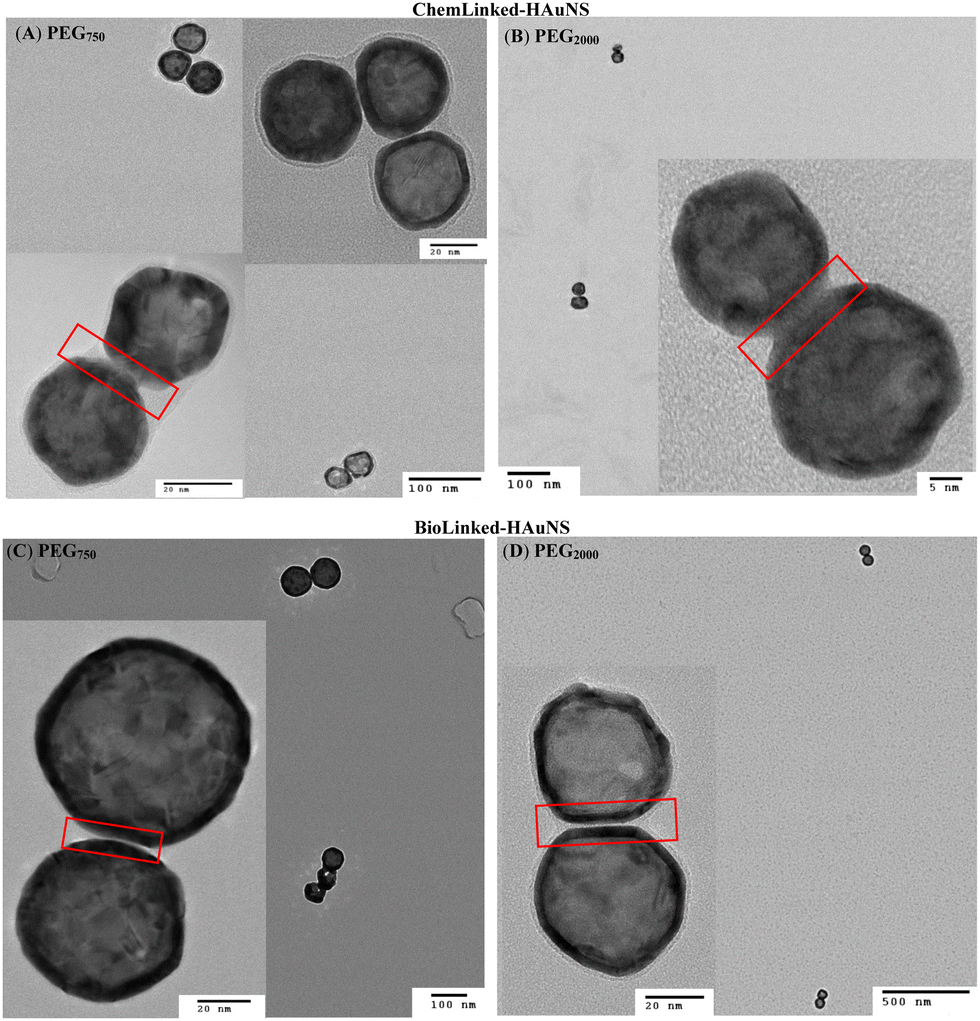 | ||
| Fig. 7 TEM images of ChemLinked- (A: PEG750, B: PEG2000) and BioLinked- (C: PEG750, D: PEG2000) HAuNS. | ||
The interparticle distance in BioLinked-HAuNS may be affected by TEM sample preparation which leads to drying and is a stochastic process. The measured values via TEM may not truly represent the BioLinked-HAuNS interparticle distance in solution, which may be shorter. To investigate this possibility, we carried out DLS measurements to determine their hydrodynamic sizes, and the data are presented in Table 3. In DLS analysis it is assumed that all particles in solution are spheres. However, the shape of the linked-HAuNS is expected to be that of dumbbell, which means that DLS measurements will represent size combinations of long and short sides of the HAuNS. In addition, as noted through TEM, ChemLinked-HAuNS-PEG750 and BioLinked-HAuNS-PEG750 contained both dimers and trimers, which must also be taken into consideration for data interpretation. The hydrodynamic size of trimer may be smaller than the dimer, and the presence of trimer may thus decrease the overall size.
| HAuNS | Hydrodynamic size (nm) |
|---|---|
| ChemLinked-HAuNS-PEG750 | 75.88 |
| ChemLinked-HAuNS-PEG2000 | 84.21 |
| BioLinked-HAuNS-PEG750 | 85.59 |
| BioLinked-HAuNS-PEG2000 | 133.30 |
As shown in Table 3, for the same linkages and different PEG lengths (Bio or ChemLinked-HAuNS-PEG750 and Bio or ChemLinked-HAuNS-PEG2000), the hydrodynamic size is higher by about 10 nm for the longer PEG for the ChemLinked, and ∼30 nm for the BioLinked HAuNS. If we take into account the population of trimers, and the contributions made by these, the size of ChemLinked-HAuNS-PEG750 < BioLinked-HAuNS-PEG750 ≈ ChemLinked-HAuNS-PEG2000 < BioLinked-HAuNS-PEG2000. The smaller size of ChemLinked-HAuNS-PEG750 may be expected due to the presence of trimers in solution. BioLinked-HAuNS are longer in comparison to ChemLinked-HAuNS. In general, the difference between the hydrodynamic sizes of the ChemLinked- and BioLinked-HAuNS was larger than expected. The length of peptide is estimated to be ∼5.89 Å via the model built by Harpaz et al.81 However, the size difference between ChemLinked- and BioLinked-HAuNS is larger (DLS measurements). We hypothesize that the increased size of the BioLinked-HAuNS may be due to the affinity of the peptide linker to solvent and counter ions.
UV-Vis-NIR study
We were subsequently interested in understanding the changes in photophysical properties of linked-HAuNS, especially compared to their monomeric analogs. UV-Vis-NIR spectra were obtained in DMSO for the ligand-functionalized HAuNS and the linked-HAuNS, and are shown in Fig. 8. The maxima for the UV-Vis-NIR absorptions for the linked-HAuNS are red shifted from those of the monomers. However, the shift is smaller for the BioLinked-HAuNS, as compared to ChemLinked-HAuNS of both PEG lengths. This could potentially be due to lower longitudinal mode coupling as particles get further apart from each other, as in the case of BioLinked-HAuNS. It is also possible that the PEG length on the surface of nanoparticles plays a role in separation distance between linked-HAuNS, which could explain the spectral shifts observed in the UV-Vis spectra in Fig. 8.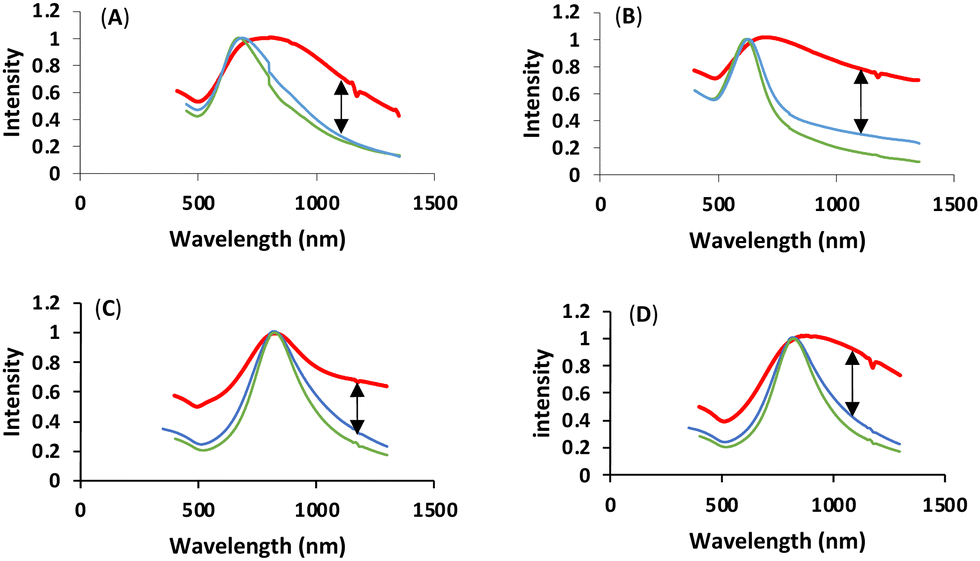 | ||
| Fig. 8 Normalized UV-Vis-NIR spectra, red (Linked-HAuNS), blue (HO-CH2-Core-(PEG)2-TEG-HAuNS) monomer, green (COOH-Core-(PEG)2-TEG-HAuNS): (A) ChemLinked-PEG750; (B) ChemLinked-HAuNS-PEG2000; (C) BioLinked-HAuNS-PEG750; (D) BioLinked-HAuNS-PEG2000. | ||
In addition, changes in the spectral profiles were also observed for all the linked-HAuNS and are shown by an arrow in Fig. 8. The UV-Vis-NIR absorption curves are strongly affected by the properties of HAuNS, including the aspect ratio of shell thickness to diameter, shape, as well as composition. We predicted that the linked-HAuNS would possess similar unique UV-Vis-NIR absorption to gold nanorods (AuNR), which have two surface plasmon resonances modes, longitudinal and transverse surface plasmon resonance.82 Longitudinal surface plasmon resonance occurs when the collective oscillation of polarized electron is caused by light along the length axis. As for the transverse mode, the collective oscillation is caused by light along with transverse direction.42 The profile changes in the UV-Vis-NIR absorption in linked-HAuNS, therefore, are likely due to both longitudinal and transverse surface plasmon resonance modes. The additional presence of trimers in the ChemLinked-PEG750 and BioLinked-PEG750 may also contribute to the observed UV-Vis-NIR profile changes.
Linked-HAuNS to monomer conversion
Since the surface plasmon resonances for the HAuNS monomers were different from the ChemLinked-HAuNS, as shown by the maxima shift in the UV-Vis-NIR spectra, we subsequently carried out a preliminary investigation of the reverse process, conversion to monomer. We were intrigued to see if this could help better understand the influence of interparticle distance, not only on surface plasmon resonance but also photoacoustic signal of the linked-HAuNS. After breaking the covalent bond, the HAuNS monomers should diffuse away from each other to minimize their free energy. We hypothesized that we could use laser and photoacoustic imaging studies to monitor the photoacoustic signal changes during this diffusion process, which will accompany interparticle distance increase. Model reactions were first conducted to choose the appropriate reagent for cleavage, before monitoring linked-HAuNS to monomer conversion with photoacoustic tomography. This initial evaluation is important because we needed to optimize the conversion at a rate in which we could monitor the photoacoustic signal change with time.Optimizing ChemLinked-HAuNS cleavage reaction
To determine the ideal reaction conditions for ester linkage cleavage (between the linked HAuNS), we chose benzyl benzoate as the model compound, and reagents including tetra-n-butylammonium fluoride (TBAF), KOH, and trifluoroacetic acid (TFA). The reactions were monitored via TLC for a period up to 48 hours. Workup and purification procedures subsequently led to the isolation of benzoic acid. The reaction mixture could be easily purified using the pKa differences between the products: benzoic acid (4.2) and benzyl alcohol (15.4). Purification process involved washing the organic phase, which contained the reactants, cleaving reagent and the product, with alkaline water. The water layer was then acidified with 0.1 N HCl(aq) to precipitate benzoic acid. We found that the yield after a reaction time of 48 hours was 28, 18 and 90% for TFA, TBAF and KOH, respectively. As a result, KOH was chosen as the cleaving reagent, and a kinetic study was subsequently conducted, and we found that 80% yield could be obtained after 4 hours, and a maximum of 90% after 24 hours.ChemLinked-HAuNS to monomer
We applied the above-mentioned optimized reaction conditions for the ester bond cleavage in linked-HAuNS, and the surface plasmon resonance changes during this process were monitored by photoacoustic imaging studies. The changes in photoacoustic signal were followed for 4 h, assuming that 80% of the dimer should have been cleaved. The interparticle distance between HAuNS should increase while they diffuse away from each other.For photoacoustic studies, we chose wavelengths: (i) 690 nm, since it is close to the monomer UV-Vis-NIR absorption maxima wavelength; and (ii) 720 nm, which is closer to that for the linked-HAuNS. The results of this study are shown in Fig. 9, in which images (a) and (b) were taken at t = 0 h, and (c) and (d) at 4 h after initiating the HAuNS dimer to monomer cleavage reaction. Brightness of the signal at t = 4 h was found to be much stronger than at 0 h, and it was further quantified using ImageJ software (Fig. 10). Our preliminary results suggest that upon cleaving the ester bond in linked-HAuNS, as expected, there is constructive interference of their photoacoustic signals as they diffuse away from each other. It is clear that further studies are warranted to better understand as to how the interparticle distance affects the photoacoustic signal through changes in absorption cross-section and light-to-heat conversion. However, our results do indicate that the photoacoustic signal is amplified during HAuNS dimer-to-monomer conversion. Autogenous cleavage process of HAuNS dimers by esterases could thus offer a platform to develop photoacoustic imaging contrast agents.
 | ||
| Fig. 9 Photoacoustic tomography for ChemLinked-HAuNS-PEG750 during conversion to monomers: t = 0 h, (a) wavelength = 690 nm and (b) 720 nm; t = 4 h, (c) wavelength = 690 nm and (d) 720 nm. | ||
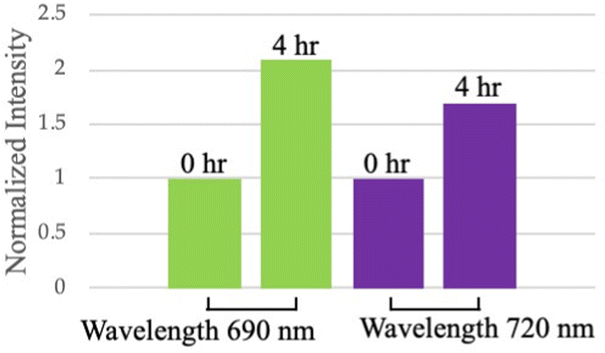 | ||
| Fig. 10 Quantification of photoacoustic tomography for ChemLinked-HAuNS-PEG750 at 0 and 4 h time intervals during dimer-to-monomer conversion. | ||
Cytotoxicity evaluation of Linked-HAuNS
Functionalized HAuNS reported in this study contain versatile biocompatible ligands with terminal groups (OH and COOH), which can be used to develop a variety of nanomaterials with broad biological applications. Since, to the best of our knowledge, cytotoxicity of linked-HAuNS has not been evaluated, we carried out such an analysis of the monomers and their linked-HAuNS analogs by exposing the 2H11 murine endothelial cells to varied concentrations. The results of these studies are shown in Fig. 11.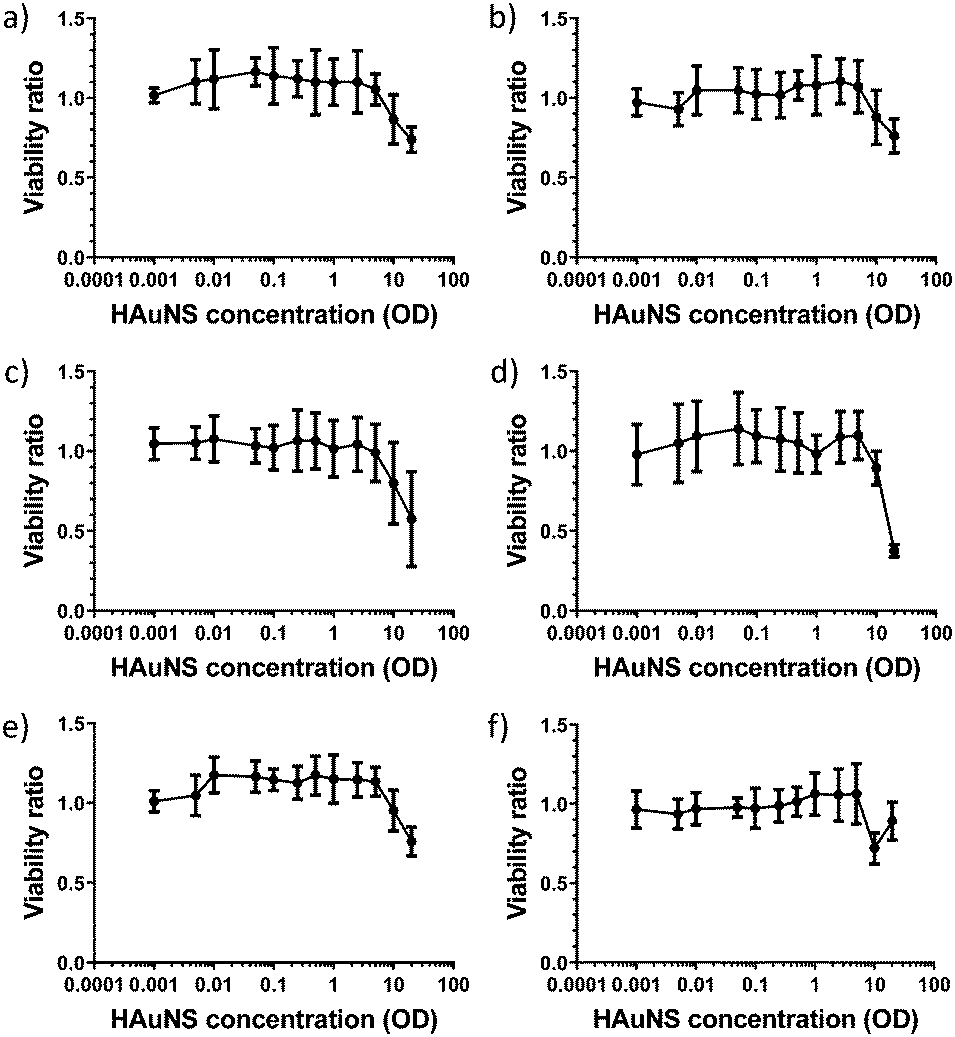 | ||
| Fig. 11 Cell viability evaluation of (a) ChemLinked-HAuNS-PEG750; (b) ChemLinked-HAuNS-PEG2000; (c) monomer, HO-CH2-Core-(PEG750)2-TEGS-HAuNS; (d) HOOC-Core-(PEG750)2-TEGS-HAuNS; (e) HO-CH2-Core-(PEG2000)2-TEGS-HAuNS; (f) HOOC-Core-(PEG2000)2-TEGS-HAuNS. | ||
The cell viabilities for the monomers, HO-CH2-Core-(PEG2000)2-TEGS-HAuNS and HOOC-Core-(PEG2000)2-TEGS-HAuNS, as well as those for the linked-HAuNS, remained unchanged from OD = 0.01 to 5; decreased slightly at concentration of OD = 10; and reached 75% at OD = 20. At OD = 20, the monomers decorated with shorter PEG chains (HO-CH2-Core-(PEG750)2-TEGS-HAuNS and HOOC-Core-(PEG750)2-TEGS-HAuNS; Fig. 11 panels c and d, respectively) showed slightly higher toxicities compared to the corresponding HAuNS decorated with PEG2000 (HO-CH2-Core-(PEG2000)2-TEGS-HAuNS and HOOC-Core-(PEG2000)2-TEGS-HAuNS; Fig. 11 panels e and f, respectively). The zeta potentials of HO-CH2-Core-(PEG750)2-TEGS-HAuNS and HOOC-Core-(PEG750)2-TEGS-HAuNS (Table 1) show increased negative charges compared to HO-CH2-Core-(PEG2000)2-TEGS-HAuNS and HOOC-Core-(PEG2000)2-TEGS-HAuNS (Table 2). There is contradictory literature regarding the uptake of nanoparticles as a function of their zeta potential which may result from the use of different cell models and/or conditions.83–88 It should also be noted that the ChemLinked-HAuNS and the corresponding monomers had lower cytotoxicities than citrate stabilized HAuNS, whose cell viability was lower than 75% at a concentration as low as OD = 0.5, as reported previously by us using the same 2H11 murine endothelial cell line.89 The added surface negative charge may indeed cause higher uptake by this cell line under our conditions of evaluation.
Conclusions
Hollow gold nanoshells have offered tremendous potential for a wide range of biomedical applications. We have developed a strategy to expand their scope through surface modification with ligands that enhance their stability and provide pathways to covalently link them with tuned chemical or biological junctions. Ligand composition, a crucial parameter for the purpose, is controlled through a modular synthetic methodology that facilitates such a rational design: (i) on a core with orthogonal moieties and its subsequent coupling with desired components through high-yield chemistry; and (ii) protection of thiol with a group that can survive varied reaction conditions/purification and can be easily removed for binding of the resulting thiol to gold nanoshells, in one pot reaction sequence. Our studies show that variations in (i) molecular weight of the biocompatible PEG arms, placed at pre-determined locations to enhance aqueous dispersion, stealth and steric protection to HAuNS, can be used to limit and prevent the formation of oligomeric species; and (ii) inter-particle distance can be controlled through chemical and biological spacers, which plays a key role in optical interference of linked HAuNS. The latter can be reversed by breaking the covalent link in HAuNS and is accompanied by an amplification of the photoacoustic signal. Surface functionalization of HAuNS (monomers and linked-HAuNS) with these multifarious ligands, lowers their cytotoxicities compared to those stabilized with citrate. The ease of linked-HAuNS fabrication, afforded by versatile synthetic tools reported here, offers a venue which could be further exploited for broad biological applications, as well as in assembling varied metallic nanostructures.Experimental
Syntheses of ligands shown in Schemes 1 and 2 are described in detail in the ESI.† Citrate capped hollow gold nanoshells (HAuNS) were prepared using an adaptation of the procedure reported earlier.12,19,49,89Materials
The following compounds were purchased and used as received: 3-bromo-5-iodobenzoic acid from AK Scientific (USA); tetraethylene glycol, poly(ethylene glycol) methyl ether MW = 750, and MW = 2000, p-toluene sulfonyl chloride (tosyl chloride), methanesulfonyl chloride (mesyl chloride), sodium hydroxide, triethylamine, diethyl amine, sodium azide, sodium ascorbate (Na-Ascorbate), copper(II)sulfate pentahydrate (CuSO4·5H2O), borane tetrahydrofuran, potassium carbonate, potassium thioacetate, thionyl chloride, tetrabromomethane (CBr4), triphenylphosphine (TPP), silver fluoride, triphenylsilane, bis(triphenylphosphine)palladium(II) dichloride and tetrabutyl ammonium fluoride (TBAF) from Sigma Aldrich (USA and Canada); magnesium sulfate form ACP chemicals; (triisopropylsilyl) acetylene; sodium sulfate from Thermo Fisher and (trimethylsilyl) acetylene from Oakwoods Chemicals (USA). The solvents methanol, ethanol and triethyl amine, diethyl amine, tetrahydrofuran (THF), acetone, ethyl acetate, acetonitrile (ACN), chloroform, dichloromethane (DCM), benzene, and toluene were purchased from Fisher scientific and ACP Chemicals and used as received. Dry solvents were obtained from drying columns and purged under nitrogen prior to use. Milli-Q Ultrapure water was doubly distilled by reverse osmosis though a Millipore RiOS8, followed by filtration through a Milli-Q Academic A10 filtration unit prior to use.Instrumentation
NMR spectral acquisitions were carried out on 300 and 500 MHz Mercury (Varian) instruments and operated using VNMRJ 2.2D (Chempack 5) and VNMRJ 2.3A (Chempack 5) software, as well as on an AV 500 MHz (Bruker) using a 5 mm Smart Probe. The chemical shifts are in ppm and reported relative to tetramethylsilane (TMS) as an internal standard for 1H, and 13C{1H}. Mass spectra analyses (MS-HR, ESI and APCI) were performed and analysed on an Exactive Plus Orbitrap-API (Thermo Scientific) high resolution mass spectrometer. TGA spectra were acquired on a Q500 thermogravimetric analyzer (TGA) from TA Instruments, and data were analyzed using TA Instrument Data Analysis software. The functionalized gold nanoshells were used as solution in dichloromethane (DCM). The samples were placed on platinum and heated to 30 °C with 20 °C min−1 rate and kept at 30 °C for 30 minutes to make sure DCM was completely removed before taking a measurement. Samples were then heated from 30 °C to 900 °C with 20 °C min−1 rate. For UV-Vis-NIR, all the measurements of monomers and dimers were carried out in DMSO. The samples were centrifuged for two hours at 4600 rpm. The supernatant was removed after centrifugation. Another two cycles of adding DMSO, and centrifuging were carried out to ensure that all the samples were in pure DMSO. UV-Vis-NIR spectra were acquired using Cary 5000 spectrometer (Agilent) and data were processed via Excel. Dynamic light scattering (DLS) was performed using Zetasizer Nano S90 (Malvern Panalytical, UK) with He–Ne laser set at a 90° incidence using Standard Operating Procedures (SOP) software. All the measurements were carried out at 25 °C by diluting L-HAuNS to concentration at 0.0375 mg mL−1 adjusted for optimal scattering detection condition. The aqueous solutions were sonicated for 2 minutes prior to being passed through PVDF membrane filters of 0.45 μm pore size and added to polystyrene cuvettes carefully in order to avoid air bubble formation. DLS acquisition lasted 2.5 minutes and averaged over 10 measurements and repeated the same measurement for three times for each sample. Zeta potential measurements were similarly averaged over 10 measurements. Transmission Electron Microscope (TEM) studies were performed in the Electron Microscopy Research (FEMR) facility using the FEI Tecnai 12 BioTwin at 120 kV with AMT XR80C CCD camera, and Philip CM200 at 200 kV with AMT XR40B CCD camera. Samples were first sonicated for 2 minutes before placing 10 μL sample solutions on a CF400-Cu carbon film of a 400 square-mesh copper grid and left for 30 seconds before washing which was performed by swirling the carbon side on a drop of water three times. Excess water was then removed with filter paper. TEM images were processed via ImageJ software.Cytotoxicity
Cytotoxicity of ligand functionalized gold nanoshells was examined using 2H11 murine cell line (CRL-2163, ATCC). 2H11 cells were detached and resuspended at a concentration of 104 cells per mL in Dulbecco's modified Eagle's medium (DMEM) 10% phosphate buffer saline (PBS), and 100 μL per well, were plated in 96 wells plates. After an overnight adhesion, the media in the well plates was aspirated and replaced with 90 μL of fresh media. Nanoparticles were diluted to various concentrations in PBS 10% DMSO and 10 μL of the dilutions was added to the cells. After an incubation for 24 hours at 37 °C, and 5% CO2, the media was again aspirated, cells were washed twice with 37 °C warmed PBS and the CellTiter 96® AQueous assay (Promega) was performed. 100 μL of a solution of phenol red-free DMEM, 10% FBS and 10% AQueous One Solution Reagent were added to each well. After one hour, the wells were monitored using a BioTek Synergy H1 (Agilent) plate-reader and confirmed the absorbance of 490 nm. The viability ratio was defined as the absorbance ratio between wells with the nanoparticles and control untreated wells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Natural Sciences and Engineering Research Council of Canada (NSERC), Canadian Institutes for Health Research (CIHR), Fonds de Recherche du Québec Nature et technologies (FRQNT, Quebec, Canada), and the Quebec Centre for Advanced Materials/Centre Québécois sur les Matériaux Fonctionnels (QCAM/CQMF) for financial support.Notes and references
- P. K. Jain, X. H. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578–1586 CrossRef CAS PubMed.
- V. W. K. Ng, R. Berti, F. Lesage and A. Kakkar, J. Mater. Chem. B, 2013, 1, 9–25 RSC.
- Y.-C. Wang, É. Rhéaume, F. Lesage and A. Kakkar, Molecules, 2018, 23(11), 2851 CrossRef PubMed.
- H. J. Lee, Y. Liu, J. Zhao, M. Zhou, R. R. Bouchard, T. Mitcham, M. Wallace, R. J. Stafford, C. Li, S. Gupta and M. P. Melancon, J. Controlled Release, 2013, 172, 152–158 CrossRef CAS PubMed.
- I. Grabowska-Jadach, D. Kalinowska, M. Drazd and M. Pietrzak, Biomed. Pharmacother., 2019, 111, 1147–1155 CrossRef CAS PubMed.
- Z. Huang, A. Zhang, Q. Zhang and D. Cui, J. Mater. Chem. B, 2019, 7, 3755–3774 RSC.
- K. Cai, W. Zhang, M. F. Foda, X. Li, J. Zhang, Y. Zhang, H. Liang, H. Li, H. Han and T. Zhai, Small, 2020, 16(37), 2002748 CrossRef CAS PubMed.
- S. A. Lindley and J. Z. Zhang, ACS Appl. Nano Mater., 2019, 2, 1072–1081 CrossRef CAS.
- Y.-H. Chiu, S. A. Lindley, C.-W. Tsao, M.-Y. Kup, J. K. Cooper, Y.-J. Hsu and J. Z. Zhang, J. Phys. Chem. C, 2020, 124, 11333–11339 CrossRef CAS.
- P. C. Ohara, D. V. Leff, J. R. Heath and W. M. Gelbart, Phys. Rev. Lett., 1995, 75, 3466–3469 CrossRef CAS PubMed.
- B. L. V. Prasad, C. M. Sorensen and K. J. Klabunde, Chem. Soc. Rev., 2008, 37, 1871–1883 RSC.
- A. M. Schwartzberg, T. Y. Olson, C. E. Talley and J. Z. Zhang, J. Phys. Chem. B, 2006, 110, 19935–19944 CrossRef CAS PubMed.
- R. Bardhan, S. Lal, A. Joshi and N. J. Halas, Acc. Chem. Res., 2011, 44, 936–946 CrossRef CAS PubMed.
- E. C. Dreaden, M. A. Mackey, X. Huang, B. Kang and M. A. El-Sayed, Chem. Soc. Rev., 2011, 40, 3391–3404 RSC.
- J. Nam, N. Won, H. Jin, H. Chung and S. Kim, J. Am. Chem. Soc., 2009, 131(38), 13639–13645 CrossRef CAS PubMed.
- A. M. Alkilany, S. R. Abulateefeh, K. K. Mills, A. I. Bani Yaseen, M. A. Hamaly, H. S. Alkhatib, K. M. Aiedeh and J. W. Stone, Langmuir, 2014, 30, 13799–13808 CrossRef CAS PubMed.
- S. E. Crawford, M. J. Hartman and J. E. Millstone, Acc. Chem. Res., 2019, 52(3), 695–703 CrossRef CAS PubMed.
- I. Fratoddi, I. Venditti, C. Cametti and M. V. Russo, J. Mater. Chem. B, 2014, 2, 4204–4220 RSC.
- V. W. K. Ng, P. K. Avti, M. Bedard, T. Lam, L. Rouleau, J.-C. Tardif, É. Rhéaume, F. Lesage and A. Kakkar, J. Mater. Chem. B, 2014, 2, 6334–6344 RSC.
- G. Prencipe, S. M. Tabakman, K. Welsher, Z. Liu, A. P. Goodwin, L. Zhang, J. Henry and H. Dai, J. Am. Chem. Soc., 2009, 131(13), 4783–4787 CrossRef CAS PubMed.
- M. P. Melancon, W. Lu, Z. Yang, R. Zhang, Z. Cheng, A. M. Elliot, J. Stafford, T. Olson, J. Z. Zhang and C. Li, Mol. Cancer Ther., 2008, 7, 1730–1739 CrossRef CAS PubMed.
- A. Topete, M. Alatorre-Meda, P. Iglesias, E. M. Villar-Alvarez, S. Barbosa, J. A. Costoya, P. Taboada and V. Mosquera, ACS Nano, 2014, 8, 2725–2738 CrossRef CAS PubMed.
- X. Liu, C. Gao, J. Gu, Y. Jiang, X. Yang, S. Li, W. Gao, T. An, H. Duan, J. Fu, Y. Wang and X. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 27622–27631 CrossRef CAS PubMed.
- J. P. Hermes, F. Sander, U. Fluch, T. Peterle, D. Thompson, R. Urbani, T. Pfohl and M. Mayor, J. Am. Chem. Soc., 2012, 134(36), 14674–14677 CrossRef CAS PubMed.
- H. M. Zakaria, A. Shah, M. Konieczny, J. A. Hoffmann, A. J. Nijdam and M. E. Reeves, Langmuir, 2013, 29, 7661–7673 CrossRef CAS PubMed.
- V. Chegel, O. Rachkov and A. Lopatynskyi, J. Phys. Chem. C, 2012, 116, 2683–2690 CrossRef CAS.
- Y.-C. Wang, PhD Thesis, Functional hollow gold nanoshells through ligand design: developing a synthetic protocol to linked gold nanostructures, McGill University, 2020 Search PubMed.
- V. Guerrero-Florez, S. C. Mendez-Sanchez, O. A. Patrón-Soberano, V. Rodríguez-González, D. Blach and F. Martínez O, J. Mater. Chem. B, 2020, 8, 2862–2875 RSC.
- X. Zhang, L. An, Q. Tian, J. Lin and S. Yang, J. Mater. Chem. B, 2020, 8, 4738–4747 RSC.
- R. B. M. Schasfoort, Introduction to surface plasmon resonance, Handbook of Surface Plasmon Resonance (2), RSC, London, 2017, ch. 1, p. 1 Search PubMed.
- M. P. Melancon, W. Lu, M. Zhong, M. Zhou, G. Liang, A. M. Elliott, J. D. Hazle, J. N. Myers, C. Li and R. Jason Stafford, Biomaterials, 2011, 32, 7600–7608 CrossRef CAS PubMed.
- P. Sharma, S. C. Brown, N. Bengtsson, Q. Zhang, G. A. Walter, S. R. Grobmyer, S. Santra, H. Jiang, E. W. Scott and B. M. Moudgil, Chem. Mater., 2008, 20, 6087–6094 CrossRef CAS PubMed.
- X. Li, Z. Xiaong, X. Xu, Y. Luo, C. Peng, M. Shen and X. Shi, ACS Appl. Mater. Interfaces, 2016, 8, 19883–19891 CrossRef CAS PubMed.
- X. Li, L. Xing, K. Zheng, P. Wei, L. Du, M. Shen and X. Shi, ACS Appl. Mater. Interfaces, 2017, 9, 5817–5827 CrossRef CAS PubMed.
- X. Li, L. Kong, W. Hu, C. Zhang, A. Pich, X. Shi, X. Wang and L. Xing, J. Adv. Res., 2022, 37, 255–266 CrossRef CAS PubMed.
- C. Li, L. Zhao, L. Jia, Z. Ouyang, Y. Gao, R. Guo, S. Song, X. Shi and X. Cao, J. Mater. Chem. B, 2022, 10, 3648–3656 RSC.
- B. De Angelis, N. Depalo, F. Petronella, C. Quintarelli, M. L. Curri, R. Pani, A. Calogero, F. Locatelli and L. De Sio, J. Mater. Chem. B, 2020, 8, 1823–1840 RSC.
- C. K. K. Choi, X. Zhou, Y. T. E. Chiu, H. Yang, J. Wang and C. H. J. Choi, Nanoscale, 2017, 9, 16968–16980 RSC.
- J. Zhou, Q. Xiong, J. Ma, J. Ren, P. B. Messersmith, P. Chen and H. Duan, ACS Nano, 2016, 10, 11066–11075 CrossRef CAS PubMed.
- C. Ayala-Orozco, C. Urban, S. Bishnoi, A. Urban, H. Charron, T. Mitchell, M. Shea, S. Nanda, R. Schiff, N. Halas and A. Joshi, J. Controlled Release, 2014, 191, 90–97 CrossRef CAS PubMed.
- H.-H. Jeong, E. Choi, E. Ellis and T.-C. Lee, J. Mater. Chem. B, 2019, 7, 3480–3496 RSC.
- E. Y. Kim, D. Kumar, G. Khang and D.-K. Lim, J. Mater. Chem. B, 2015, 3, 8433–8444 RSC.
- W. Rechberger, A. Hohenau, A. Leitner, J. R. Krenn, B. Lamprecht and F. R. Aussenegg, Opt. Commun., 2003, 220, 137–141 CrossRef CAS.
- K. Kokubo, M. K. E. Cabello, N. Sato and H. Sakurai, ChemNanoMat, 2020, 6(4), 524–528 CrossRef CAS.
- Y. Fang, X. Tian and Y. Huang, Chem. Phys. Lett., 2015, 619, 139–143 CrossRef CAS.
- W. P. McConnell, J. P. Novak, L. C. Brousseau, R. R. Fuierer, R. Tenent and D. L. Feldheim, J. Phys. Chem. B, 2000, 104, 8925–8930 CrossRef CAS.
- J. K. Kim and D.-J. Jang, New J. Chem., 2019, 43, 9732–9739 RSC.
- J. Wang and N. Li, J. Mater. Chem. B, 2017, 5, 8430–8445 RSC.
- M. Bedard, P. K. Avti, T. Lam, L. Rouleau, J.-C. Tardif, E. Rheaume, F. Lesage and A. Kakkar, J. Mater. Chem. B, 2015, 3, 1788–1800 RSC.
- P. Nordlander, C. Oubre, E. Prodan, K. Li and M. I. Stockman, Nano Lett., 2004, 4, 899–903 CrossRef CAS.
- Z. Zhang, P. Yang, H. Xu and H. Zheng, J. Appl. Phys., 2013, 113, 033102 CrossRef.
- J. M. Wessels, H.-G. Nothofer, W. E. Ford, F. von Wrochem, F. Scholz, T. Vossmeyer, A. Schroedter, H. Weller and A. Yasuda, J. Am. Chem. Soc., 2004, 126(10), 3349–3356 CrossRef CAS PubMed.
- R. Sardar, T. B. Heap and J. S. Shumaker-Parry, J. Am. Chem. Soc., 2007, 129(17), 5356–5357 CrossRef CAS PubMed.
- A. J. Mastroianni, S. A. Claridge and A. P. Alivisatos, J. Am. Chem. Soc., 2009, 131(24), 8455–8459 CrossRef CAS PubMed.
- H. Yao, H. Kojima, S. Sato and K. Kimura, Langmuir, 2004, 20, 10317–10323 CrossRef CAS PubMed.
- T. H. Galow, A. K. Boal and V. M. A. Rotello, Adv. Mater., 2000, 12, 576–579 CrossRef CAS.
- M. Zhang, Y. Xu, X. Peng, H. Chen and H. Wang, Chem. Commun., 2022, 58, 7932–7935 RSC.
- Z. Li, L. Li, H. R. Zielke, L. Cheng, R. Xiao, M. T. Crow, W. G. Stetler-Stevenson, J. Froehlich and E. G. Lakatta, Am. J. Pathol., 1996, 148, 121–128 CAS.
- U. Lehmann and A. D. Schlüter, Eur. J. Org. Chem., 2000, 3483–3487 CrossRef CAS.
- R. Hourani, M. Jain, D. Maysinger and A. Kakkar, Chem. – Eur. J., 2010, 16(21), 6164–6168 CrossRef CAS PubMed.
- O. Mikio, I. Yoshihisa, L. Yu, N. Satoshi, N. Satoko, W. Kazuhito and H. Tadao, Bull. Chem. Soc. Jpn., 1990, 63(4), 1260–1262 CrossRef.
- H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129(20), 6633–6639 CrossRef CAS PubMed.
- F. Ishiwari, K.-i Fukasawa, T. Sato, K. Nakazono, Y. Koyama and T. Takata, Chem. – Eur. J., 2011, 17(43), 12067–12075 CrossRef CAS PubMed.
- H. Kunz and H. Waldmann in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, p. 631 Search PubMed.
- C. E. Inman, S. M. Reed and J. E. Hutchison, Langmuir, 2004, 20, 9144–9150 CrossRef CAS PubMed.
- A. Mehta, R. Jaouhari, T. J. Benson and K. T. Douglas, Tetrahedron Lett., 1992, 33(37), 5441–5444 CrossRef CAS.
- C. A. Hollingsworth, P. G. Seybold and C. M. Hadad, Int. J. Quantum Chem., 2002, 90(4-5), 1396–1403 CrossRef CAS.
- R. J. Ouellette and J. D. Rawn, in Organic Chemistry, ed. R. J. Ouellette, and J. D. Rawn, Academic Press, 2nd edn, 2018, pp. 625–663 Search PubMed.
- J. Manson, D. Kumar, B. J. Meenan and D. Dixon, Gold Bull., 2011, 44(2), 99–105 CrossRef CAS.
- S. Maji, B. Cesur, Z. Zhang, B. G. De Geest and R. Hoogenboom, Polym. Chem., 2016, 7, 1705–1710 RSC.
- H.-J. Kim, K. Y. Chao, S. S. Hwang, D. H. Choi, M. J. Ko and K.-Y. Baek, RSC Adv., 2016, 6, 49206–49213 RSC.
- M. Fruhnert, F. Kretschmer, R. Geiss, I. Perevyazko, D. Cialla-May, M. Steinert, N. Janunts, D. Sivun, S. Hoeppener, M. D. Hager, T. Peretsch, U. S. Schubert and C. Rockstuhul, J. Phys. Chem. C, 2015, 119(31), 17809–17817 CrossRef CAS.
- C. Kruger, S. Agarwal and A. Greiner, J. Am. Chem. Soc., 2008, 130(9), 2710–2711 CrossRef PubMed.
- E. R. Zubarev, J. Xu, A. Sayyad and J. D. Gibson, J. Am. Chem. Soc., 2006, 128(15), 4958–4959 CrossRef CAS PubMed.
- B. Vanhaecht, M. N. Teerenstra, D. R. Suwier and C. E. Koning, J. Macromol. Sci., Part A: Pure Appl. Chem., 2000, 37, 633–648 CrossRef.
- A. Ambrus and D. Yang, Anal. Biochem., 2007, 367, 56–67 CrossRef CAS PubMed.
- C. S. Johnson, Prog. Nucl. Magn. Reson. Spectrosc., 1999, 34, 203–256 CrossRef CAS.
- R. Neufeld and D. Stalke, Chem. Sci., 2015, 6, 3354–3364 RSC.
- L. Wang, W. Amelung and S. Willbold, Environ. Sci. Technol., 2017, 51, 13256–13264 CrossRef CAS PubMed.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- Y. Harpaz, M. Gerstein and C. Chothia, Structure, 1994, 2, 641–649 CrossRef CAS PubMed.
- H. Chen, L. Shao, Q. Li and J. Wang, Chem. Soc. Rev., 2013, 42, 2679–2724 RSC.
- E. Fröhlich, Int. J. Nanomed., 2012, 7, 5577–5591 CrossRef PubMed.
- S. Patil, A. Sandberg, E. Heckert, W. Self and S. Seal, Biomaterials, 2007, 28(31), 4600–4607 CrossRef CAS PubMed.
- S. Borse, S. Joshi and A. Khan, RSC Adv., 2015, 5, 13402–13410 RSC.
- Y. Y. Khine, M. Callari, H. Lu and M. H. Stenzel, Macromol. Chem. Phys., 2016, 217, 2302–2309 CrossRef CAS.
- S. Jeon, J. Clavadetscher, D.-K. Lee, S. V. Chankeshwara, M. Bradley and W.-S. Cho, Nanomaterials, 2018, 8(12), 1028 CrossRef PubMed.
- X. Li, Z. Ouyang, H. Li, C. Hu, P. Saha, L. Xing, X. Shi and A. Pich, Bioact. Mater., 2021, 6(10), 3244–3253 CrossRef CAS PubMed.
- L. Rouleau, R. Berti, V. W. K. Ng, C. Matteau-Pelletier, T. Lam, P. Saboural, A. Kakkar, F. Lesage, E. Rhéaume and J.-C. Tardif, Contrast Media Mol. Imaging, 2013, 8(1), 27–39 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details and additional characterization data. See DOI: https://doi.org/10.1039/d2ma00624c |
| This journal is © The Royal Society of Chemistry 2022 |