
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeptide linked perylene bisimide and diketopyrrolopyrrole: design, synthesis and investigation of mechanofluorochromism†
D.
Venkateswarlu
ab,
T.
Swetha
a,
Rajamouli
Boddula
 a and
Surya Prakash
Singh
*ab
a and
Surya Prakash
Singh
*ab
aPolymers and Functional Materials Division, CSIR-Indian Institute of Chemical Technology, Uppal Road, Tarnaka, Hyderabad-500007, India. E-mail: spsingh@iict.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India
First published on 15th September 2022
Abstract
Herein, for the first time, we report the synthesis of perylenediimide (PDI-PEP) and diketopyrrolopyrrole (DPP-PEP) derivatives which are substituted with a peptide linker in a core and wings manner (here DPP acts as the core moiety and the peptide linker works as the wings). Both molecules showed intramolecular charge transfer: aggregation caused quenching (ACQ) and multi-colour mechanofluorochromism by adding various dopants. Upon doping with various coloured dopants, the molecules’ PL emission spectra in the solid-state were found to be 634 nm (pinkish-red) and 720 nm (light brown) for PDI-PEP and DPP-PEP, respectively. Compared to emission in solution, the solid-state emission was shifted towards a higher wavelength by ∼94 nm and ∼150 nm for PDI-PEP and DPP-PEP, respectively. These results will provide insights into the multicolour mechanofluorochromism through molecular conformation and interactions.
1. Introduction
Nowadays, the development of multicolour mechanofluorochromic smart materials is of great interest in the field of materials science. Mechanofluorochromic (MFC) materials are different from mechanochromic materials, and these will show both sample colour and emission colour change by mechanical force.1–3 The mechano-stimuli response of the solid-state materials was reported for several years in the field of security systems, mechanical sensors, optical storage, diagnosis applications and optoelectronic devices.4–8 However, a solid-state stimuli-response with the fluorescence of organic molecules involved in different emissions is an interesting field of luminescence. Stimuli response includes various aspects such as mechanochromic, mechanofluorochromic, mechanoluminescence, vapochromic, acidochromic, solvatochromic and thermochromic. Nevertheless, suitable designs to get a specific response are still scarce.6–12 It can be found in several available literature analyses that among them, few are very negotiable, in which the chromophores are arranged densely in solid form with suppression of metastable states. To avoid the drawbacks of the mechano-stimuli study of the organic molecules, a few structures have been designed and established for mechanochromic studies.13–18 Until now, various metalloorganic and organic luminescent probes have been reported with the MFC property and most of them are characterized by a mechanobathochromic shift only;19,20 however multichannel and multicolour switching need to be addressed. Laskar's group reported a D–π–A-based conjugated AIE oligomer (oTPETP) and studied its aggregation induced emission enhancement (AIEE) and mechanoluminescence. It showed strong emission properties in the solid state compared to the solution due to restricted intramolecular rotation.21 Cheng's group developed D–π–A IDM-DHP (indene-1,3-dionemethylene-1,4-dihydropyridine) derivatives with various alkyl chain lengths and studied the effect of the N-alkyl chains on their fluorescence properties in the aggregated state. These derivatives showed good AIEE properties in a THF/water mixture due to their highly distorted conformations. The IDM-DHP solids have shown reversible mechanofluorochromic (MFC) properties, with larger MFC spectral redshifts when used with long length of the N-alkyl chain due to weaker CH–π hydrogen bonds in the molecules and as a result looser molecular stacking.22 Bryce et al. reported a multifunctional and switchable material, i.e., a neutral multifunctional dinuclear novel Ir(III) complex containing a Schiff base ligand. It showed combined piezochromic luminescence (PCL) vapochromism and AIE. This Ir-complex resulted in a reversible colour change, faint red and bright orange with high contrast intensity by mechanical grinding within 10 s or high polarity volatile organic compounds.23 Neelakandan's group developed a new tripodal Schiff base compound, which resembles a six-petal flower. This compound shows aggregation-induced emission and reversible mechanofluorochromism and it has a good detection capability of fluoride ions in solution and cells.24In this scenario, developing novel MFC smart materials with unique properties is an interesting topic for researchers. In the last few decades, the development of diketopyrrolopyrrole (DPP) and perylene (PDI)-based fluorescent materials has been attractive due to their intense color, and their alkylated forms lead to good solubility. They are the main building blocks for efficient field-effect transistors, fluorescent molecular probes, and photovoltaic applications.25 Nevertheless, the stimuli response to fluorescent materials is studied very limitedly. Similarly, not much attention has been paid towards perylene moieties for AIE and ACQ applications employing dipole–dipole interaction as well as π–π stacking.26–31 The main driving force for this design is that the peripheral groups of the main core will tune the intermolecular π–π* and aliphatic interactions to get variable molecular backbone conformations, which will help the design of various functionalized materials.32
In this context, we have designed mechanofluorochromic molecules based on the PDI and DPP-innovative materials named PDI-PEP (tetrabenzyl 2,2′-((2,2′-((4,4′-(1,3,8,10-tetraoxo-2,9-di (tricosan-12-yl)-1,2,3,8,9,10-hexahydroanthra[2,1,9-def:6,5,10-d′e′f′] diisoquinoline-5,12-diyl) bis(benzoyl)) bis(azanediyl)) bis(acetyl)) bis(azanediyl)) disuccinate) and DPP-PEP (tetrabenzyl 2,2′-((2,2′-(((2,5-bis(2-ethylhexyl)-3,6-dioxo-2,3,5,6-tetrahydropyrrolo[3,4-c]pyrrole-1,4-diyl)bis(thiophene-5,2-diyl)) bis(azanediyl)) bis(acetyl)) bis(azanediyl)) disuccinate) with the incorporation of a peptide linker. The peptide molecules are capable of being smart stimuli-responsive materials, with mechano-responsive luminescence and aggregation-induced emission (AIE),33–35 as well as being responsible for improving the thermal response of the PDI-PEP and DPP-PEP. No literature reports are available based on such molecular design (PDI-PEP and DPP-PEP), and there are few reports on simple diketopyrrolopyrrole and perylene cores that can be found. In this paper, we have developed two smart materials PDI-PEP and DPP-PEP, well-characterized using spectroscopic techniques, and studied the absorption emission, aggregation, stimuli-response, cyclic voltammetry and TCSPC. The chemical structures of PDI-PEP and DPP-PEP are shown in Fig. 1.
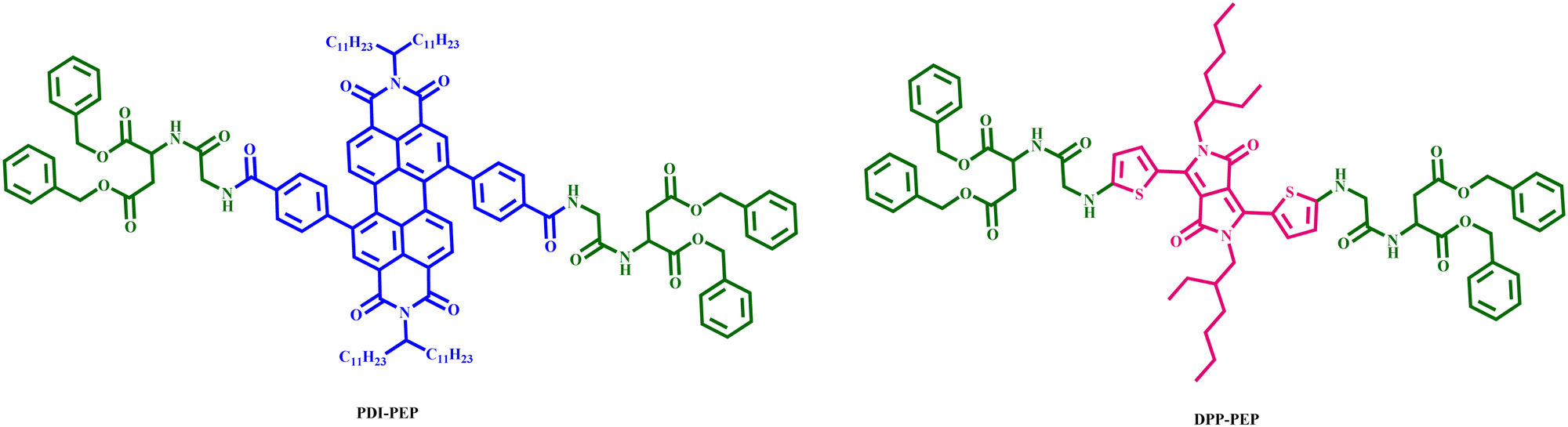 | ||
| Fig. 1 Chemical structure of (tetrabenzyl 2,2′-((2,2′-((4,4′-(1,3,8,10-tetraoxo-2,9-di(tricosan-12-yl)-1,2,3,8,9,10-hexahydroanthra[2,1,9-def:6,5,10-d′e′f′] diisoquinoline-5,12-diyl) bis(benzoyl)) bis(azanediyl)) bis(acetyl)) bis(azanediyl)) disuccinate) (PDI-PEP) and (tetrabenzyl 2,2′-((2,2′-(((2,5-bis(2-ethylhexyl)-3,6-dioxo-2,3,5,6-tetrahydropyrrolo[3,4-c]pyrrole-1,4-diyl)bis(thiophene-5,2-diyl)) bis(azanediyl)) bis(acetyl)) bis(azanediyl)) disuccinate) (DPP-PEP). | ||
2. Results and discussion
Synthesis of the molecules
The synthesis of the molecules started with the protection of a glycine followed by the condensation between the carboxylic group and amine to yield compound 3. TFA was used to deprotect compound 3 to get compound 4 (dibenzyl glycyl aspartate). The second reactant has been prepared using alkylated dibromo perylene/diketopyrrolopyrole, which undergoes Suzuki coupling with 4-bromobenzoic acid/(5-bromothiophen-2-yl) boronic acid to yield compounds 5 and 6. The coupled compounds 5 and 6 reacted with compound 4 in a toluene medium to afford the desired molecules (PDI-PEP and DPP-PEP) in good yields (Schemes 1 and 2).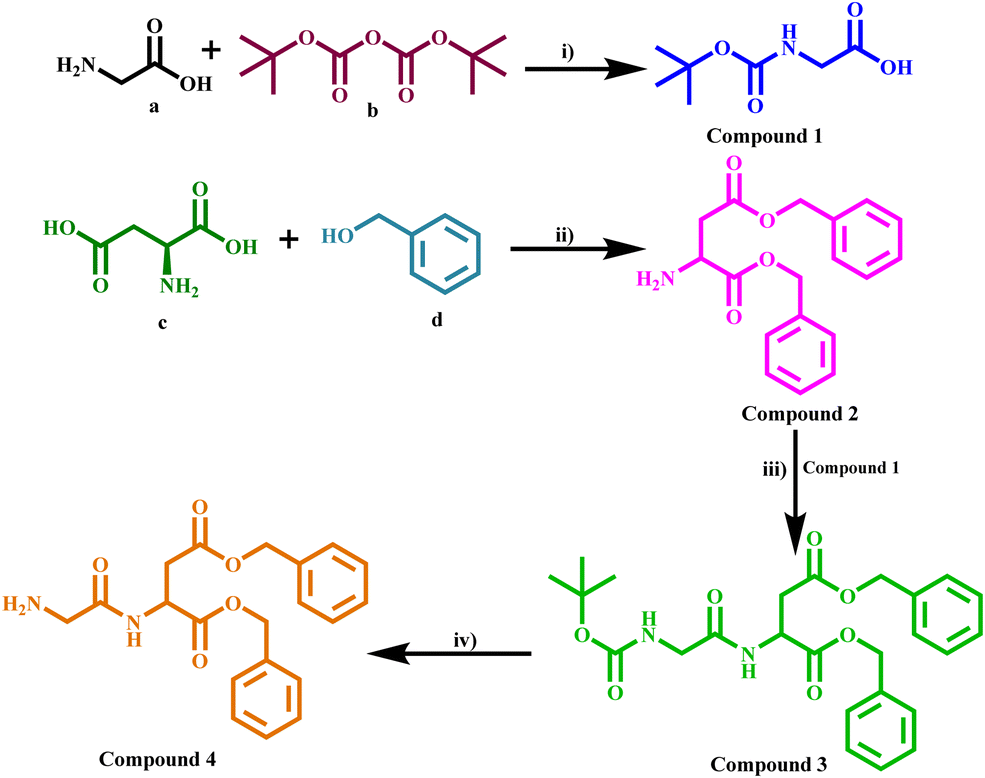 | ||
Scheme 1 Reagents and conditions: (i) 1,4 dioxane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (2:1), NaOH, RT, 2 h, yield: 80% (ii) PTSA, toluene, 8 h, yield: 90% (iii) DCC, hydroxy succinamide, N-methyl morpholine, RT, 14 h, yield: 70% (iv) TFA, DCM, 14 h. Yield: 50%. :water (2:1), NaOH, RT, 2 h, yield: 80% (ii) PTSA, toluene, 8 h, yield: 90% (iii) DCC, hydroxy succinamide, N-methyl morpholine, RT, 14 h, yield: 70% (iv) TFA, DCM, 14 h. Yield: 50%. | ||
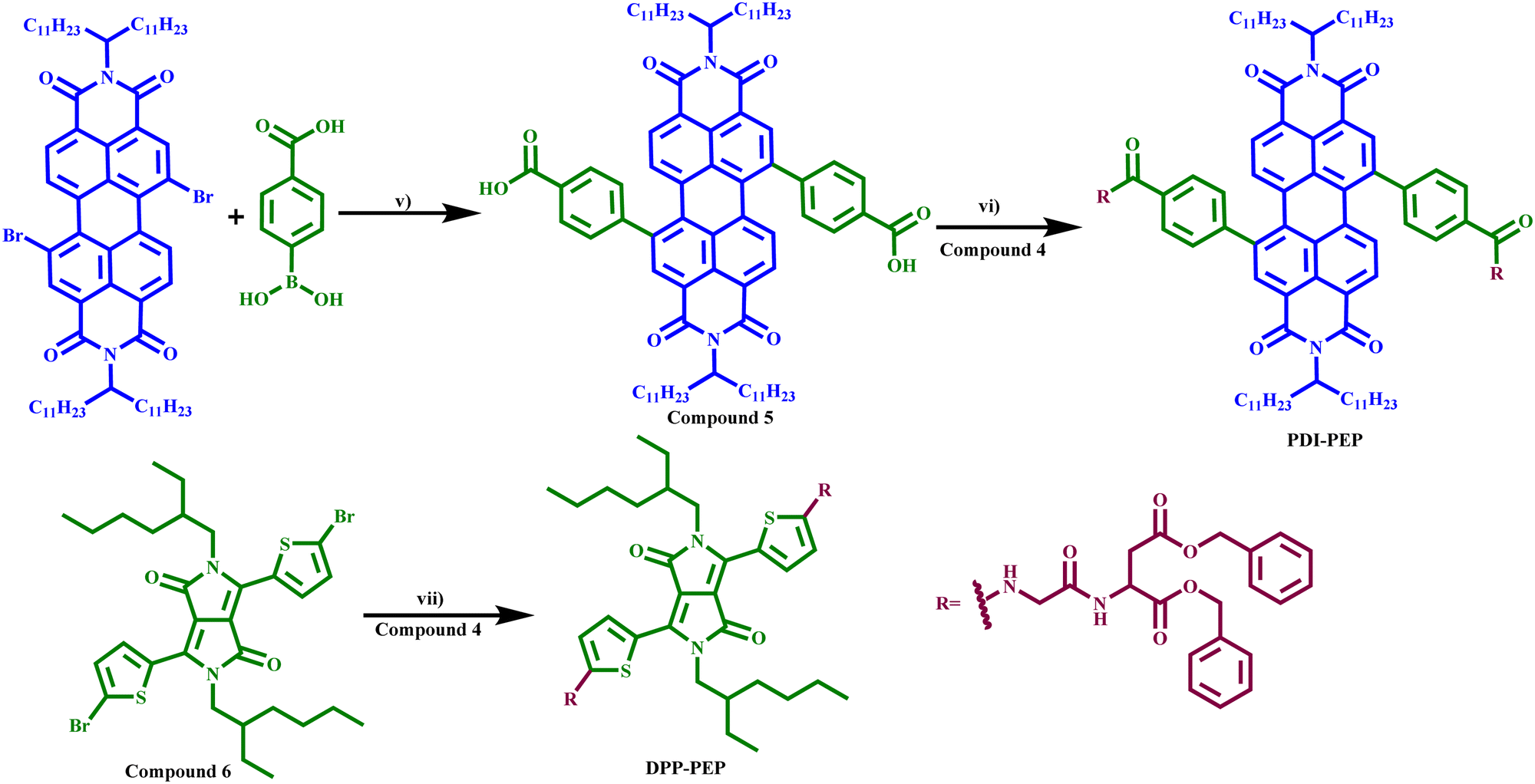 | ||
| Scheme 2 Reagents and conditions: (v) toluene, K2CO3, Pd(PPh3)2Cl2, 24 h, yield = 60%. (vi) DCC, hydroxy succinamide, N-methyl morpholine, RT, 24 h, yield: 80%. (vii) NaOtBu, toluene, triphenylphosphine, Pd2(dba)3, 24 h. Yield: 60%. | ||
Optical and electrochemical properties
A well-known factor is that the absorption and emission bands may vary using different solvents. To investigate this, we have carried out UV-visible and emission studies of PDI-PEP and DPP-PEP in various solvents (Fig. 2), and the corresponding data are tabulated in Table S1 (ESI†). As shown in Fig. 2a and b, a broad absorption band (λmax) was observed around 457 nm, 515 nm, and 518 nm, 560 nm, which originates from the intramolecular charge transfer transitions (ICT) of PDI-PEP and DPP-PEP, respectively. In addition, DPP-PEP exhibited an absorption band at around 350 nm, which is attributed to the localized π–π* transition. In the case of PDI-PEP, no such peak is observed due to the presence of high chromophore or p-cloud perylene group domination, which leads to strong and broadband coverage appearing in the range of 350 – 600 nm. In a comparison of the PDI-PEP and DPP-PEP, the DPP-PEP spectral ranges were shifted towards the bathochromic side, which is expected due to the presence of good donating capable nitrogen and oxygen groups. It also suggests that the possibility of intramolecular charge transfer in DPP-PEP is due to N, S, and keto being responsible for the charge transfer, which enhances the spectral shift. In the case of PDI-PEP, there is no such type of effective atom (donor–acceptor) present (Table S2, ESI†). To understand the absorption colour of the compound under normal light and 365 nm light, we have provided the images in Fig. S1 (ESI†).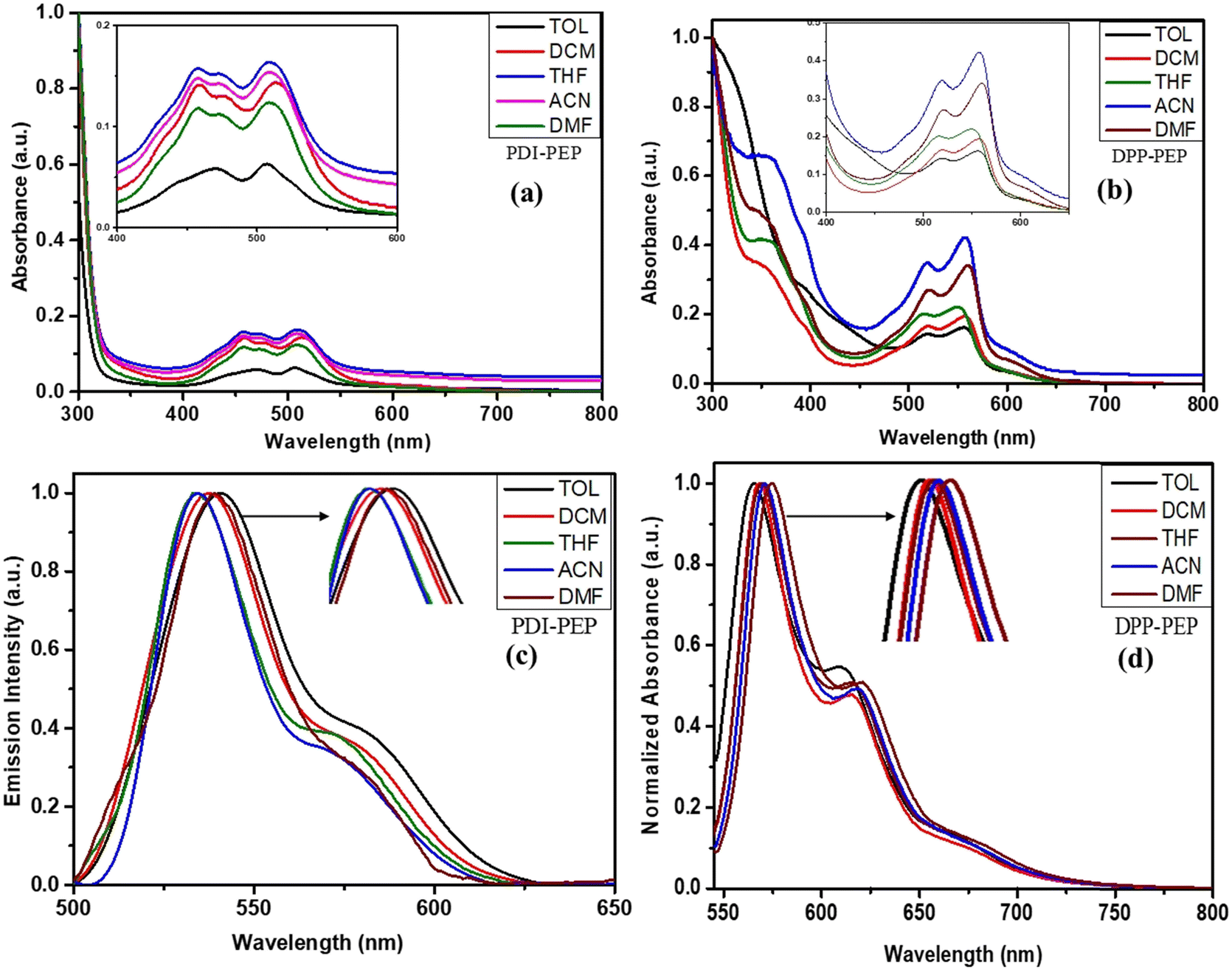 | ||
| Fig. 2 UV absorption of (a) PDI-PEP and (b) DPP-PEP, and emission spectra of PDI-PEP and DPP-PEP [(c) and (d)] in different solvents. | ||
To further understand the emission process of the molecules, we have measured the photoluminescence in different solvents. On increasing the polarity of the solvent, DPP-PEP shifted ∼10 nm more than PDI-PEP. DPP-PEP and PDI-PEP exhibited emissions in the range of 536–543 nm and 565–574 nm in different solvents, respectively (Fig. 2c and d). It was found that the average difference was ∼29 nm peak shift from PDI-PEP to DPP-PEP and this was expected due to the presence of the strain five-membered rings in the DPP-PEP and additionally embedded S and N atoms. These conclusions were made affecting the peak shifts towards higher wavelengths. The emission behavior of the molecules in the solid phase was measured, and detailed investigations are discussed in the mechanochromism portion.
To study the charge transfer process at a molecular level and estimate the photophysical properties and molecular orbitals, we have carried out TD-DFT (time-dependent density functional theory) studies. In the TD-DFT studies of DPP-PEP, the λmax is 520 nm with oscillation strength (f) = 1.116 originating from HOMO–LUMO (97%), and major absorption in PDI-PEP arises from HOMO to LUMO at 533 nm with oscillator strength (f) = 0.3174. The normalized simulated absorption spectra, experimental λmax, calculated λmax, (nm) and oscillator strengths (f) are shown in the ESI† (Fig. S2 and Tables S3 and S4).
Cyclic voltammetry (CV) studies reveal the energy level alignment i.e., highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). We have done CV measurements of PDI-PEP and DPP-PEP (Fig. S3a, ESI†) using the three-electrode conventional method. The oxidation potentials of PDI-PEP and DPP-PEP were observed to be −5.80 eV and −5.87 eV and the LUMOs were −3.63 eV and −3.73 eV, respectively. The schematic energy level diagram is depicted in Fig. S3b (ESI†). The corresponding data are provided in Table S1 (ESI†).
DFT (density functional theory) studies were carried out to determine the in-depth optical properties of PDI-PEP and DPP-PEP. The types of energy transformation in the electronic transitions, the isodensity surface plots of the frontier and important molecular orbitals are characterized, and the percentage contribution of groups to each molecular orbital is calculated at the B3LYP/6-311g(d,p) level in DCM solvent (Tables S5 and S6, ESI†). We have designated the fragments of the molecules as the diketopyrrolopyrrole moiety as DPP, thiophene as THIA, and the peptide linker part as LINKER in DPP-PEP. In the case of PDI-PEP, the perylene moiety is designated as PDI, the benzene moiety as BENZO, and the peptide linker part as LINKER for easy understanding. From the molecular orbitals’ geometries, it is clear that the HOMO, HOMO−1, and LUMO are contributing more to the PDI core (99%, 85%, and 96%), respectively, and HOMO−2 is delocalized almost equally on the PDI (40%) and BENZO (45%) part. HOMO−3 is majorly attributed to the BENZO core (97%). LUMO+1 and LUMO+3 are mostly spread on the linker (∼99%). LUMO+2 is almost equal on the PDI (34%), Benzo (48%), and LINKER (18%) in the PDI-PEP molecule. In the case of DPP-PEP, HOMO, HOMO−1 and HOMO−2 are located on the DPP (49%, 18%, 28%), THIA (39%, 52%, 28%) and LINKER (12%, 30%, 19%), respectively. HOMO−3 has been raised majorly from the DPP (99%) core. LUMO and LUMO+2 are similarly depicted on the DPP (56%, 50%), THIA (38%, 37%), and LINKER (6%, 14%), respectively. LUMO+1 is mostly on the LINKER part (93%) and minorly on the THIA (4%) and DPP (3%). The HOMO and LUMO molecular orbitals are shown in Fig. 3. The tuneable energy levels will be suitable for organic photovoltaics and light-emitting diodes.
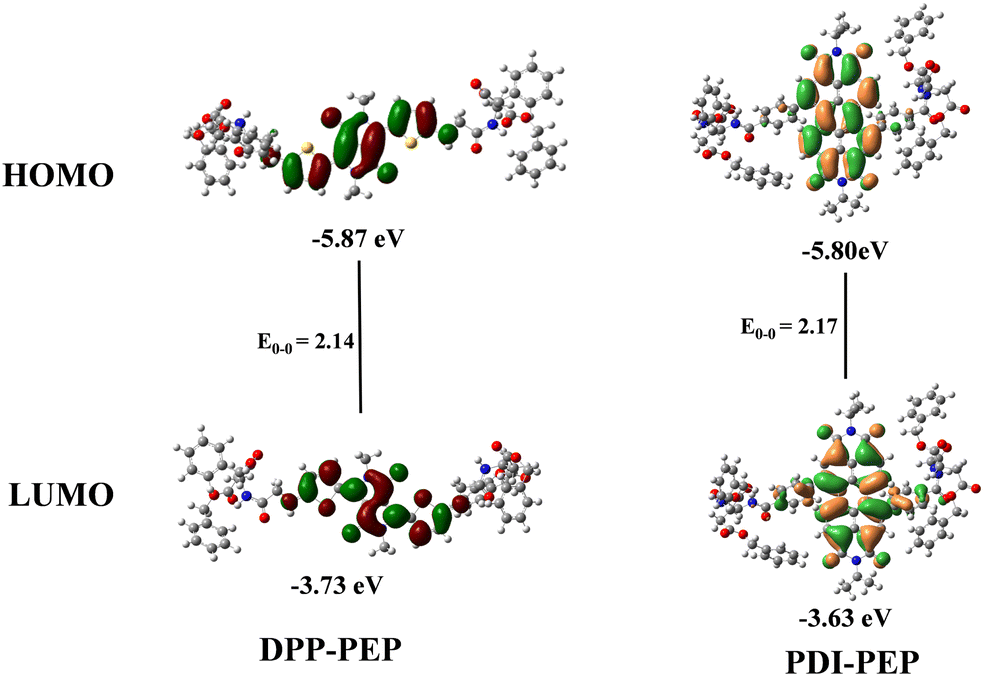 | ||
| Fig. 3 HOMO and LUMO molecular orbitals of the DPP-PEP and PDI-PEP. | ||
Aggregated emission process
Despite the solvent fluorescence emission, emission with the combination of THF in water at different percentage ratios from 10 to 90% was also measured. Usually, organic compounds show traditional aggregation-caused quenching (ACQ) fluorescence and have high luminous efficiency in the solution phase. However, their fluorescence intensities become low and this is expected due to strong π–π and hydrophobic interactions in the aggregation state. Tailoring the above, the currently synthesized PDI-PEP and DPP-PEP have shown aggregation-caused quenching processes (Fig. 4). A decrease in the emission intensity of PDI-PEP was noted from 10 to 40% (fw) gradually, and a sudden decrease at 50% was observed. Furthermore, 50–60% perceived a similar unexpected decrease in intensity and the rest of them are followed by regular decrement (Fig. 4a). A rapid decrease of the intensity is expected due to the generation of effective hydrophobic interactions with water at 50%. The decreases in the emission intensity or ACQ process led to a decrease in the quantum yield values. Similar observations are detected from the quantum yield analysis of PDI-PEP (Table S6, ESI†). To briefly understand this, the absorption of the molecules in THF in water (fw) is shown in Fig. S4(a) (ESI†) and in addition, shifting of the emission peaks can be seen in Fig. S4(b) (ESI†).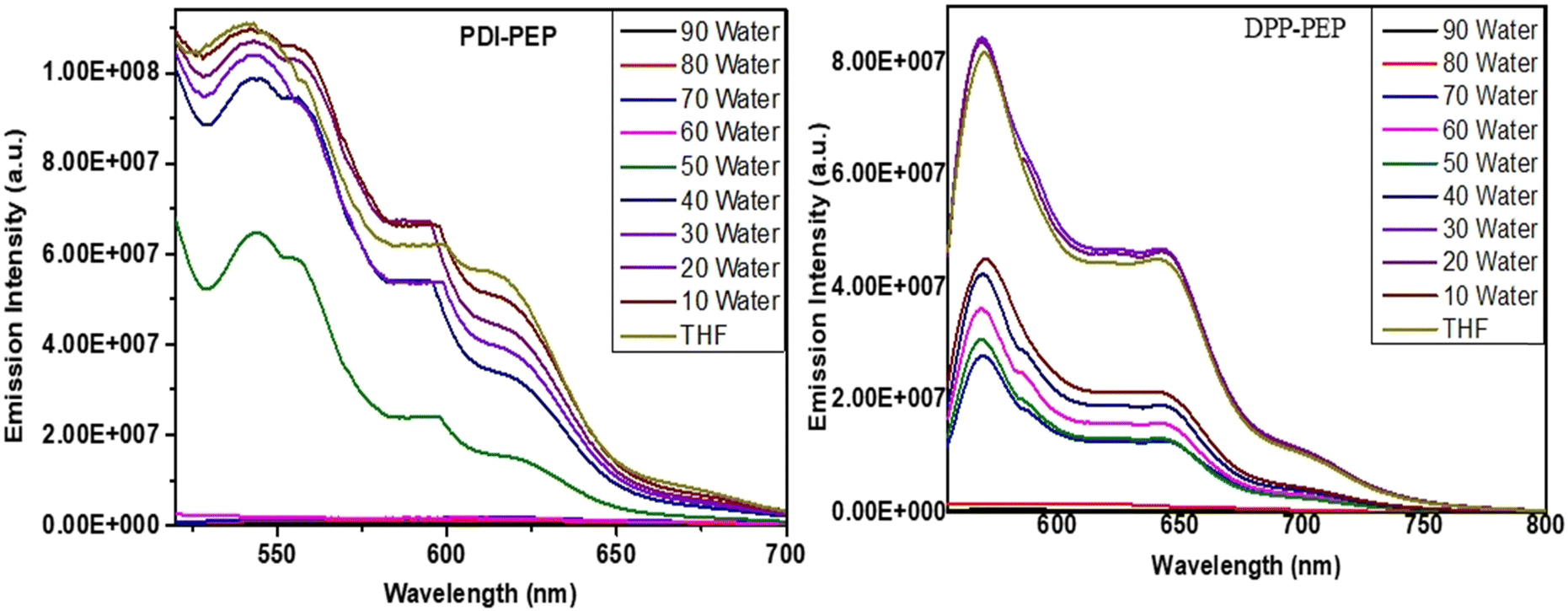 | ||
| Fig. 4 Solvent aggregation study in THF + water (fw %) of PDI-PEP and DPP-PEP. | ||
In the case of DPP-PEP (Fig. 4b), it expressed an increase of the intensity up to 30% due to aggregation. However, from 40 to 70%, a slight decrease in intensity was observed and there was a sharp decrease from 80 to 90%. Further quantum yield analyses expressed similar supportive results. The enhancement in DPP-PEP up to 30% (fw) indicates the limitation of intermolecular energy transfer (ICT) and enhancement of restriction of its intramolecular rotation in the molecules upon aggregation. Furthermore, there is no increment of intensity up to 90% (fw), revealing the absence of ICT. As compared to both the molecules, DPP-PEP showed aggregation even though it is less fraction of fw, but there are no such results found for PDI-PEP. Tailoring to the above discussion, it visibly indicates the presence of intramolecular charge transfer present in DPP-PEP (weak) more effectively than PDI-PEP. In addition, it can also be supported by the structural design of molecules where N, S, and keto are capable of intramolecular charge transfer in DPP-PEP whereas no/weak in PDI-PEP. As we discussed, primary information was observed from the UV absorption study to understand the intramolecular energy transfer effect in a molecule. Similarly, aggregation results supported the same. Quenching of the emission intensity can be attributed to π–π electronic coupling between perylene molecules while increasing the water fraction.36–38 The absorption and emission peaks of the molecules in THF in water (fw) can be seen in Fig. S4 and S5(a), (b) (ESI†).
Mechanofluorochromic analysis
Fluorescent molecules have significant luminous property changes while applying external stimuli. These properties can lead to applications in various fields where an external force is applied. The doping process will lead to good energy transfer via tunable energy levels of the organic materials.39 Similarly, external stimulus responsive molecules (PDI-PEP and DPP-PEP) are synthesized here and doped with yellow and orange dopants (Chemical structures are provided in Fig. S6, ESI†). Fig. 5 shows the images of the molecules before and after doping under normal and UV light. The PL emission of the molecules in the form of a solid was found to be 634 (pinkish-red) and 720 nm (light brown) for PDI-PEP and DPP-PEP, respectively. Compared to emission in solution, the solid state emission was shifted towards a higher wavelength of ∼94 nm and ∼150 nm for PDI-PEP and DPP-PEP, respectively. This suggests the aggregation of the molecules from solution to solid, which shows a large difference. This might be due to the presence of a large number of rotational groups in the molecule, which are highly restricted in a solid phase.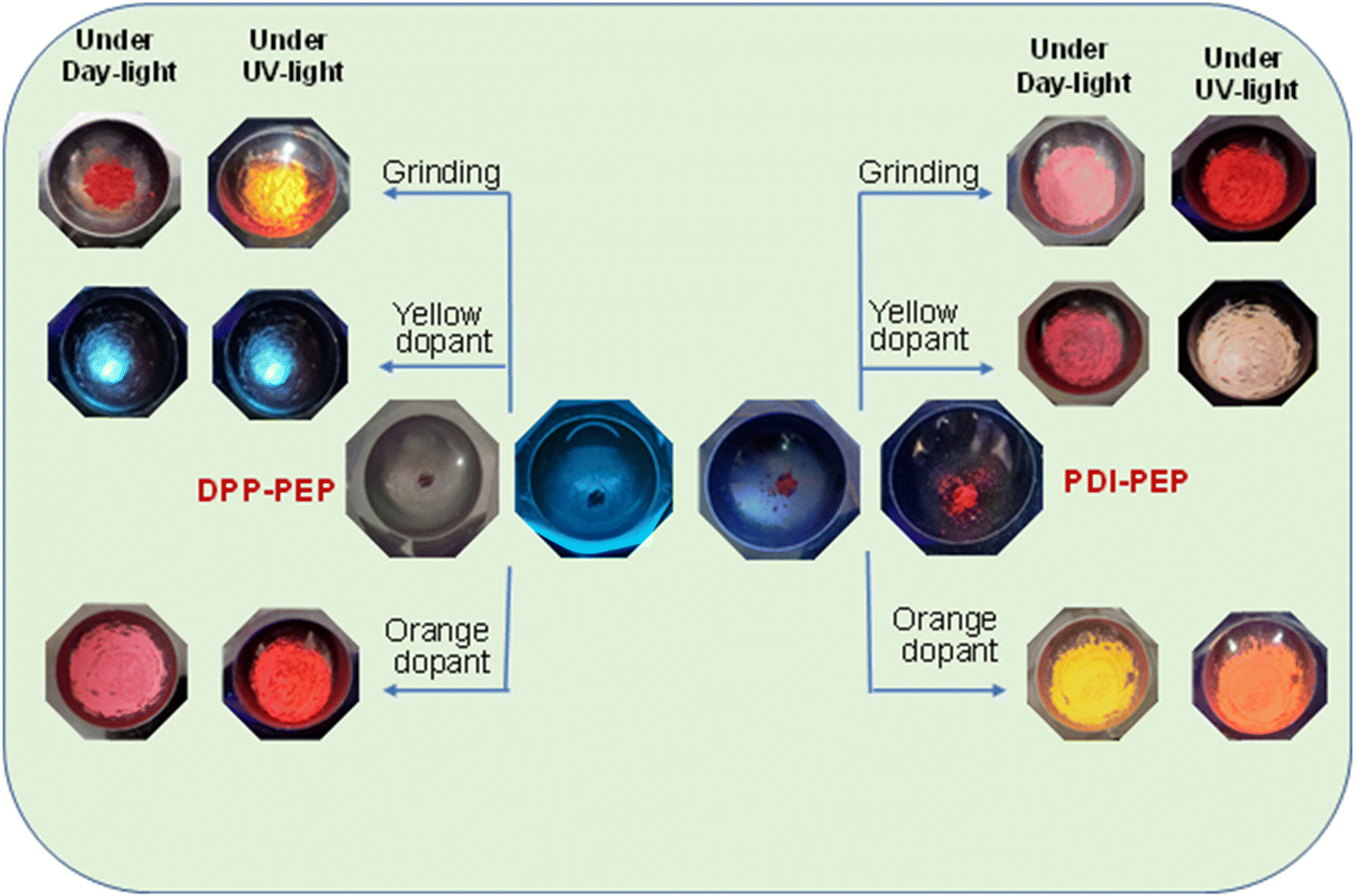 | ||
| Fig. 5 External stimuli response of the molecules under UV light and daylight of PDI-PEP and DPP-PEP with different dopants. | ||
The solid-phase of PDI-PEP was ground with the help of mortar and piston; the peak shift was observed as a hypsochromic shift from 634 to 629 nm (red). Further added yellow dopant (0.1%) leads to the change of the emission towards lower wavelength noticed from 634 to 598 nm (pale yellow), and the wavelength shift is 36 nm. Meanwhile, the orange dopant (0.1%) emission shifted towards a higher wavelength, noticed from 634 to 640 nm, changing the colour from pinkish-red to reddish-orange under UV light (Fig. 6). DPP-PEP showed yellowish colour under UV light while ground and showed emission at 718 nm. DPP-PEP doped with yellow dopant leads to bluish-green colour with emission at 616 nm, and the orange dopant results in 698 nm with pinkish-red colour. Comparing the DPP-PEP original solid and that after doping, the emission shifted to the blue region with orange and yellow dopants. However, PDI-PEP showed both blue and red shifts with doped yellow and orange dye, respectively. To understand the structural analysis of the molecules with doping, measured powder XRD analysis at 0–60° θ is shown in Fig. 7. The PDI-PEP and DPP-PEP compounds were both shown to be amorphous in nature even after doping of the dopant dye molecules; however, minor peak intensity changes were observed (Fig. S7, ESI†).
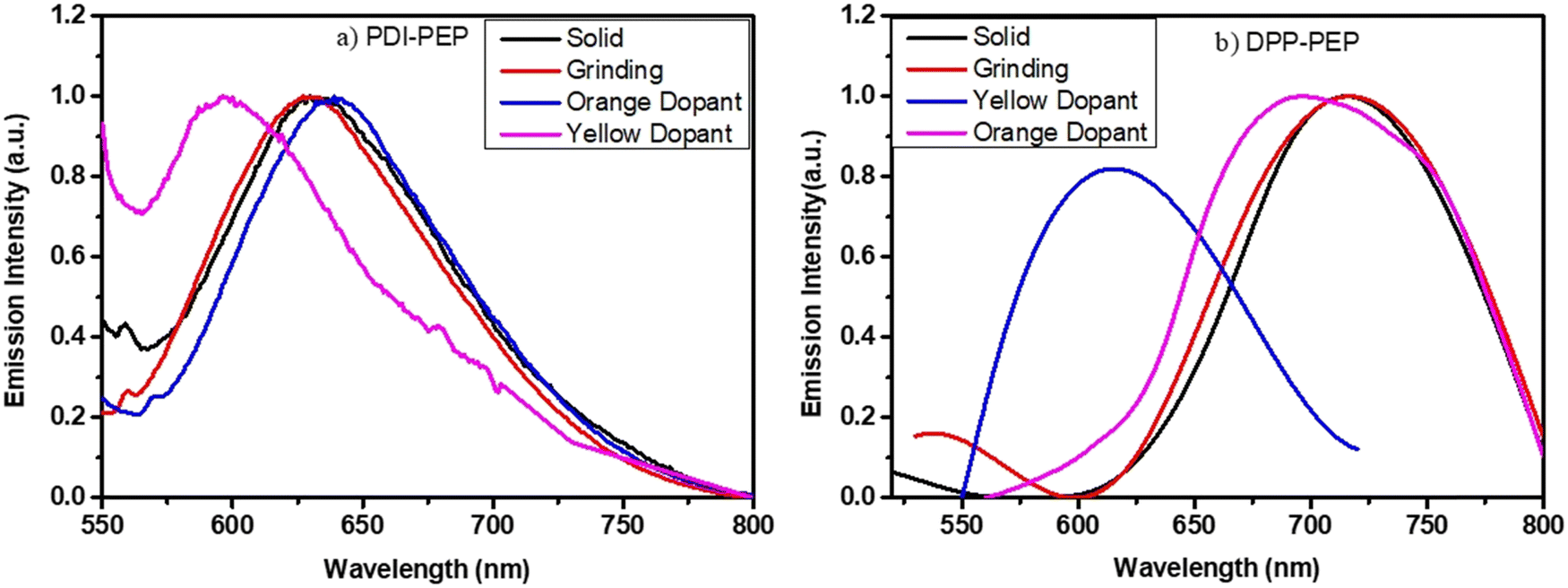 | ||
| Fig. 6 Mechanical stimuli response emission of the molecules with different dopants, (a) PDI-PEP and (b) DPP-PEP. | ||
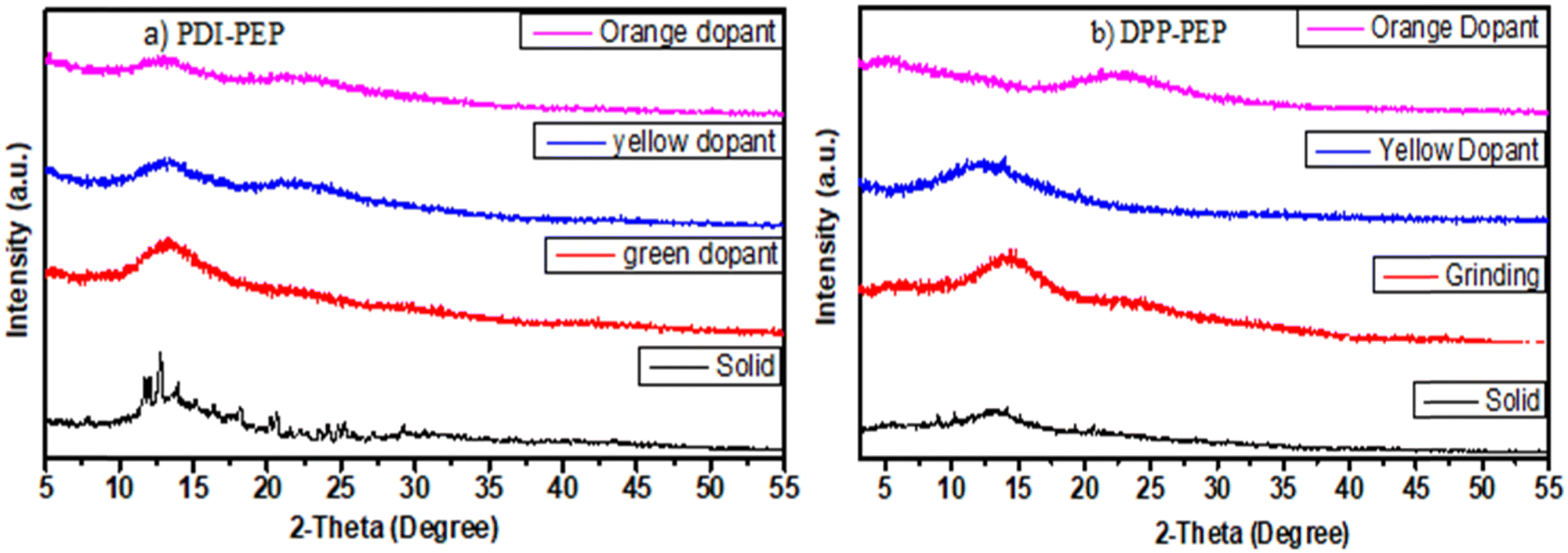 | ||
| Fig. 7 PXRD data of the molecules with different dopants, (a) PDI-PEP and (b) DPP-PEP. | ||
In addition to the doping of the molecules, here the halochromism effect was measured by using TFA. The halochromism effect was only observed in the case of DPP-PEP rather than PDI-PEP. TFA fuming was added to the ground sample, which showed greenish-yellow emission and after a few minutes, it changed to the original colour. Furthermore, no color change was found with ammonia. Fig. S8 (ESI†) indicates the interaction of the TFA with DPP-PEP in different time intervals and reaching reversible conversion. It exposes the presence of thiophene-S and its connected NH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, to form an intermolecular interaction with the TFA hydroxyl group. But the PDI-PEP (more flexible and non-hetero atom group present (benzene)) lacks similar interactions due to its structural arrangement variation to DPP-PEP. These results indicate that the present synthesized molecules are capable of mechanical stimuli response and can be utilized for further applications.
O, to form an intermolecular interaction with the TFA hydroxyl group. But the PDI-PEP (more flexible and non-hetero atom group present (benzene)) lacks similar interactions due to its structural arrangement variation to DPP-PEP. These results indicate that the present synthesized molecules are capable of mechanical stimuli response and can be utilized for further applications.
Time-correlated single-photon counting
To understand the energy transfer process of the molecules via excited states, in different structural arrangements and solvent ratios, time-correlated single-photon counting analyses were carried out. Fluorescence lifetime analysis in different solvents and THF + water (fw (vol%, 10–90%)) mixtures of both compounds was performed and shown in Fig. 8. The corresponding data are provided in the ESI† (Tables S8–S10). We have used the 484 nm laser pulse photo-excitation to calculate the DPP-PEP and PDI-PEP's lifetime decay and found bi-exponential decay kinetics at their corresponding emission maxima. The average lifetime (τavg) was calculated using the following equation.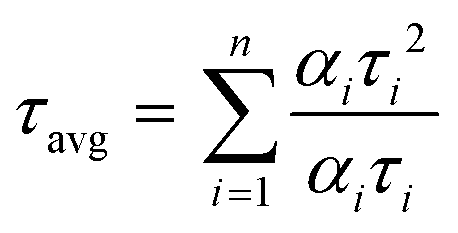 | (1) |
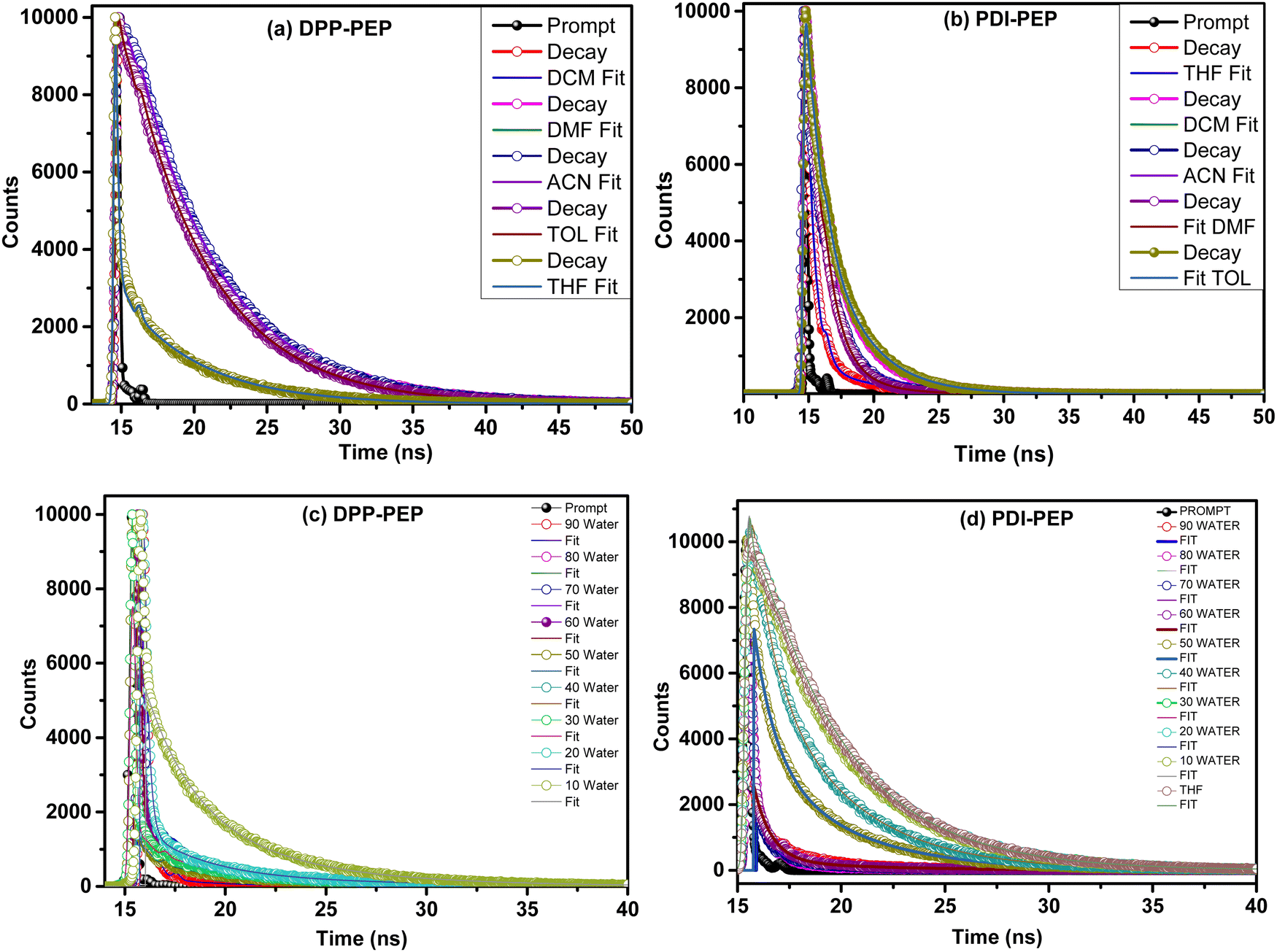 | ||
| Fig. 8 (a and b) Lifetime analysis of the DPP-PEP and PDI-PEP in different solvents; (c) DPP-PEP and (d) PDI-PEP in different THF + water (fw (vol%, 10–90%)) mixtures. | ||
The lifetime analysis of the DPP-PEP and PDI-PEP was carried out using various solvents and the life-time was found to be, for DPP-PEP, DCM (5.87 × 10−9 ns), THF (4.61 × 10−9 ns), TOL (5.63 × 10−9 ns), DMF (5.97 × 10−9 ns), ACN (5.84 × 10−9 ns), and for PDI-PEP, DCM (2.99 × 10−9 ns), THF (0.82 × 10−9 ns), TOL (3.13 × 10−9 ns), DMF (1.94 × 10−9 ns), ACN (3.81 × 10−9 ns). The PDI-PEP showed low lifetime values compared to the DPP-PEP, which might be due to the molecular conformation. The average fluorescence lifetime (τavg) is more for DPP-PEP at fw 50% and 40%, 6.07 × 10−9 ns and 5.93 × 10−9 ns, and in the case of PDI-DPP the τavg is high at fw 90% (5.46 × 10−9 ns) and 80% (6.72 × 10−9 ns), which might be due to intramolecular transfer decay of the excited state by non-radiative processes.
3. Experimental section
General information
Glycine, BOC-anhydride, aspartic acid, benzyl alcohol, PTSA, N-methyl morpholine, DCC, and HoSu were purchased from Merck. The solvents were purified by standard procedures and purged with nitrogen before use. Analytical-grade chemicals were used in this work without further purification. All reactions were performed under anhydrous conditions. 1H and 13C NMR spectra were measured with Avance ACP-300 or AMX2-500 spectrometers at 300 and 500 MHz, respectively, using TMS as the internal standard. UV-vis absorption spectra and fluorescence spectra were recorded using a quartz cell on a Shimadzu UV-vis spectrometer and a Fluorolog 3, J.Y. Horiba fluorescence spectrometer. Cyclic voltammetry studies were performed using a conventional three-electrode cell and a BAS100 electrochemical analyzer. The working electrode is a Pt rod, the counter electrode is a Pt wire, the reference electrode is silver/silver chloride, and the supporting electrolyte was 0.1 M TBAPF6.4. Conclusion
We have reported two novel optical materials for the first time by capping the PDI and DPP moieties with a peptide linker. The novel materials show aggregation caused quenching and multi-colour mechanofluorochromism via doping with different dopants. The two materials are highly fluorescent in solution as well as in the solid state. The emission spectra of the molecules in the solid-state were found to be 634 nm (pinkish-red) and 720 nm (light brown) for PDI-PEP and DPP-PEP, respectively. As compared to emission in solution, the solid was shifted towards a higher wavelength by ∼94 nm and ∼150 nm for PDI-PEP and DPP-PEP, respectively. Among them, DPP-PEP showed better mechanical stimuli responsive nature than the PDI-PEP, which may be due to the molecular conformation. They have demonstrated ACQ nature, which resulted in lower PLQY values. These results revealed that tailoring the molecular structure of the peptide-linked PDI and DPP derivatives is a good option for ML properties and they can tune the aggregation behavior of optoelectronic devices.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
D. V. and T. S. thank DST for providing fellowships. SPS acknowledges financial support from DST-SERB (EMR/2017/001506). CSIR-IICT Communication Number: IICT/Pubs./2022/051.References
- H. Sun, S. Liu, W. Lin, K. Y. Zhang, W. Lv, X. Huang, F. Huo, H. Yang, G. Jenkins, Q. Zhao and W. Huang, Nat. Commun., 2014, 5, 3601 CrossRef PubMed.
- T. Mutai, H. Satou and K. Araki, Nat. Mater., 2005, 4, 685 CrossRef CAS PubMed.
- J. Lee, O. Yarimaga, C. H. Lee, Y.-K. Choi and J.-M. Kim, Adv. Funct. Mater., 2011, 21, 1032 CrossRef CAS.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martinez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68 CrossRef CAS PubMed.
- A. L. Balch, Angew. Chem., Int. Ed., 2009, 48, 2641 CrossRef CAS PubMed.
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878 RSC.
- X. L. Luo, J. N. Li, C. H. Li, L. P. Heng, Y. Q. Dong, Z. P. Liu, Z. S. Bo and B. Z. Tang, Adv. Mater., 2011, 23, 3261 CrossRef CAS PubMed.
- L. Wang, K. Wang, B. Zou, K. Ye, H. Zhang and Y. Wang, Adv. Mater., 2015, 27, 2918 CrossRef CAS PubMed.
- Y. Gong, Y. Zhang, W. Z. Yuan, J. Z. Sun and Y. Zhang, J. Phys. Chem. C, 2014, 118, 10998 CrossRef CAS.
- T. Seki, K. Kobayashi, T. Mashimo and H. Ito, Chem. Commun., 2018, 54, 11136 RSC.
- D. Sun, S. Yuan, H. Wang, H.-F. Lu, S.-Y. Feng and D.-F. Sun, Chem. Commun., 2013, 49, 6152 RSC.
- H.-Y. Chen, C.-W. Lin, C.-T. Chen, C.-T. Chen, J. Golder, Y.-B. Lan and J.-K. Wang, Polym. Chem., 2017, 8, 3689 RSC.
- F. Ciardelli, G. Ruggeri and P. Andrea, Chem. Soc. Rev., 2013, 42, 857 RSC.
- S. Varghese and S. Das, J. Phys. Chem. Lett., 2011, 2, 863 CrossRef CAS PubMed.
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, J. Chem. Soc. Rev., 2012, 41, 3878 RSC.
- D.-H. Park, J. Hong, I. S. Park, C. W. Lee and J.-M. Kim, Adv. Funct. Mater., 2014, 24, 5186 CrossRef CAS.
- Z. Mao, Z. Y. Yang, Y. X. Mu, Y. Zhang, Y.-F. Wang, Z. G. Chi, C.-C. Lo, S. W. Liu, A. Lien and J. R. Xu, Angew. Chem., Int. Ed., 2015, 54, 6270 CrossRef CAS PubMed.
- K. Chung, M. S. Kwon, B. M. Leung, A. G. Wong-Foy, M. S. Kim, J. Kim, S. Takayama, J. Gierschner, A. J. Matzgerand and J. Kim, ACS Cent. Sci., 2015, 1, 94 CrossRef CAS PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC.
- S. Dineshkumar and I. R. Laskar, Polym. Chem., 2018, 9, 5123 RSC.
- Y. Liu, Y. Lei, M. Liu, F. Li, H. Xiao, J. Chen, X. Huang, W. Gao, H. Wu and Y. Cheng, J. Mater. Chem. C, 2016, 4, 5970 RSC.
- Y. Jiang, G. Li, D. Zhu, Z. Su and M. R. Bryce, J. Mater. Chem. C, 2017, 5, 12189 RSC.
- P. Marandi, N. Tyagi and P. P. Neelakandan, ChemPlusChem, 2022, 87, e202100555 CrossRef CAS PubMed.
- N. Mizoshita, T. Tani and S. Inagaki, Adv. Mater., 2012, 24, 3350 CrossRef CAS PubMed.
- Q. Zhao, S. Zhang, Y. Liu, J. Mei, S. Chen, P. Lu, A. Qin, Y. Ma, J. Zhi Sun and B. Z. Tang, J. Mater. Chem., 2012, 22, 7387 RSC.
- R. S. Szabadai, J. Roth-Barton, K. P. Ghiggino, J. M. White and D. J. D. Wilson, Aust. J. Chem., 2014, 67, 1330 CrossRef CAS.
- S. Yagai, S. Okamura, Y. Nakano, M. Yamauchi, K. Kishikawa, T. Karatsu, A. Kitamura, A. Ueno, D. Kuzuhara, H. Yamada, T. Seki and H. Ito, Nat. Commun., 2014, 5, 4013 CrossRef CAS PubMed.
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633 CrossRef CAS PubMed.
- T. Seki, Y. Takamatsu and H. Ito, J. Am. Chem. Soc., 2016, 138, 6252 CrossRef CAS PubMed.
- H. Naito, Y. Morisaki and Y. Chujo, Angew. Chem., Int. Ed., 2015, 54, 5084 CrossRef CAS PubMed.
- K. Zhang, Z. Liu, S. Ying, M. Chen, S. Xue, H. Zhang and W. Yang, RSC Adv., 2017, 7, 30610 RSC.
- C. L. Ho and W. Y. Wong, Coord. Chem. Rev., 2013, 257, 1614 CrossRef CAS.
- R. Boddula and S. P. Singh, J. Mater. Chem. C, 2021, 9, 12462 RSC.
- D. E. Clarke, E. T. Pashuck, S. Bertazzo, J. V. M. Weaver and M. M. Stevens, J. Am. Chem. Soc., 2017, 139(21), 7250 CrossRef CAS PubMed.
- S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339 CrossRef CAS PubMed.
- C. Huang, S. Barlow and S. R. Marder, J. Org. Chem., 2011, 76, 2386 CrossRef CAS PubMed.
- J. Feng, B. Liang, D. Wang, H. Wu, L. Xue and X. Li, Langmuir, 2008, 24, 11209 CrossRef CAS PubMed.
- W. Li, Q. Huang, Z. Mao, Q. Li, L. Jiang, Z. Xie, R. Xu, Z. Yang, J. Zhao, T. Yu, Y. Zhang, M. P. Aldred and Z. Chi, Angew. Chem., Int. Ed., 2018, 57, 12727 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, characterization techniques (NMR, mass spectra), AIE, ML, computational details, and lifetime data of PDI-PEP and DPP-PEP molecules. See DOI: https://doi.org/10.1039/d2ma00829g |
| This journal is © The Royal Society of Chemistry 2022 |