
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVapor-phase hydrothermal construction of defective MoS2 for highly selective electrocatalytic hydrogenation of cinnamaldehyde†
Tianxing
Wu
 *a and
Miaomiao
Han
*b
*a and
Miaomiao
Han
*b
aNorthwest Institute for Non-ferrous Metal Research, Xi’an, 710016, P. R. China. E-mail: mybestteacher@163.com
bSchool of Science, Huzhou University, Huzhou, 313000, P. R. China. E-mail: mmhan@zjhu.edu.cn
First published on 31st August 2022
Abstract
Electrocatalytic selective hydrogenation of α,β-unsaturated aldehydes with water as the hydrogen donor is of great significance to produce fine chemicals. Here, we propose a facile vapor-phase hydrothermal (VPH) method to directly grow defective MoS2 nanoparticles on commercial carbon fiber cloth (MoS2/CFC) for electrocatalytic selective hydrogenation of cinnamaldehyde (CAL). The as-prepared MoS2/CFC exhibited the highest CAL conversion of 88.8% with a maximum TOF value of 12.8 mmol mmolMoS2−1 h−1 at −0.7 V vs. RHE. The high selectivity of CAL hydrogenation could be realized by adjusting the applied potentials, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O hydrogenation product cinnamyl alcohol (COL) was easier to form under lower applied potentials, whereas CC hydrogenation product hydrocinnamaldehyde (HCAL) was easier to achieve under higher applied potentials. Density functional theory (DFT) calculation findings demonstrated that the Mo vacancies in the defective MoS2/CFC facilitated the adsorption of hydrogen atoms, which benefited the generation of Hads and the proceeding of electrocatalytic hydrogenation. Additionally, the existence of Mo vacancies made the CO group more active than the CC group, and reduced the reaction energy of CAL hydrogenation toward COL. Combined with the experimental results and calculation data, we speculate that the real catalytic active center of the defective MoS2/CFC is the defective configuration of Mo vacancies. Furthermore, the high conversion and selectivity of furfural and benzaldehyde hydrogenation confirmed the universality of the defective MoS2/CFC catalyst in electrocatalytic hydrogenation reduction of unsaturated and saturated aldehydes.
O hydrogenation product cinnamyl alcohol (COL) was easier to form under lower applied potentials, whereas CC hydrogenation product hydrocinnamaldehyde (HCAL) was easier to achieve under higher applied potentials. Density functional theory (DFT) calculation findings demonstrated that the Mo vacancies in the defective MoS2/CFC facilitated the adsorption of hydrogen atoms, which benefited the generation of Hads and the proceeding of electrocatalytic hydrogenation. Additionally, the existence of Mo vacancies made the CO group more active than the CC group, and reduced the reaction energy of CAL hydrogenation toward COL. Combined with the experimental results and calculation data, we speculate that the real catalytic active center of the defective MoS2/CFC is the defective configuration of Mo vacancies. Furthermore, the high conversion and selectivity of furfural and benzaldehyde hydrogenation confirmed the universality of the defective MoS2/CFC catalyst in electrocatalytic hydrogenation reduction of unsaturated and saturated aldehydes.
1. Introduction
As an old and rich discipline, electrocatalysis has been shown to be a powerful method to perform organic synthesis reactions under green, mild and safe conditions.1 Among which, electrocatalytic hydrogenation (ECH) is an economical and environmentally friendly approach to realize the hydrogenation of organics containing unsaturated chemical bonds using water as the hydrogen donor.2–4 ECH utilizes chemisorbed hydrogen (Hads) via proton or water reduction,5,6 which avoids the difficult activation of low-soluble H2 in solvents, and thereby the elevated pressure and temperature that are usually desired in thermocatalysis.7,8The selective hydrogenation of α,β-unsaturated aldehydes into the corresponding unsaturated alcohols is significant for the production of fine chemicals.9 As a classical model of α,β-unsaturated aldehydes, the hydrogenation of cinnamaldehyde (CAL) has been widely investigated.10–14 As shown in Scheme 1, the CAL hydrogenation products include not only the desired half-hydrogenated cinnamyl alcohol (COL), but also half-hydrogenated hydrocinnamaldehyde (HCAL) and full-hydrogenated hydrocinnamyl alcohol (HCOL). However, the two adjacent unsaturated CC and CO bonds of CAL make the hydrogenation reaction complex and uncontrollable.15 From the perspective of thermodynamics, chemical reactions tend to occur on the CC bond because the CC bond energy (615 kJ mol−1) is lower than the CO bond energy (715 kJ mol−1), and the change of free energy enthalpy of CC bond hydrogenation is 35 kJ mol−1 smaller than that of CO bond hydrogenation. Therefore, achieving highly selective hydrogenation of the CO bond is extremely challenging.16,17 Recently, Zhang and co-workers developed a facile strategy to synthesize hollow CoS2 and CoS2−x nanocapsules for electrocatalytic selective transfer hydrogenation of CAL with water to synthesize HCAL and HCOL.9 Unfortunately, this report did not realize the high selectivity of CO hydrogenation. Wei and co-workers reported the highly selective electrochemical hydrogenation of CAL to COL on the RuO2–SnO2–TiO2/Ti electrode.18 However, a fly in the ointment is the introduction of the precious metal Ru, which limits the large-scale production and application of electrocatalysts.
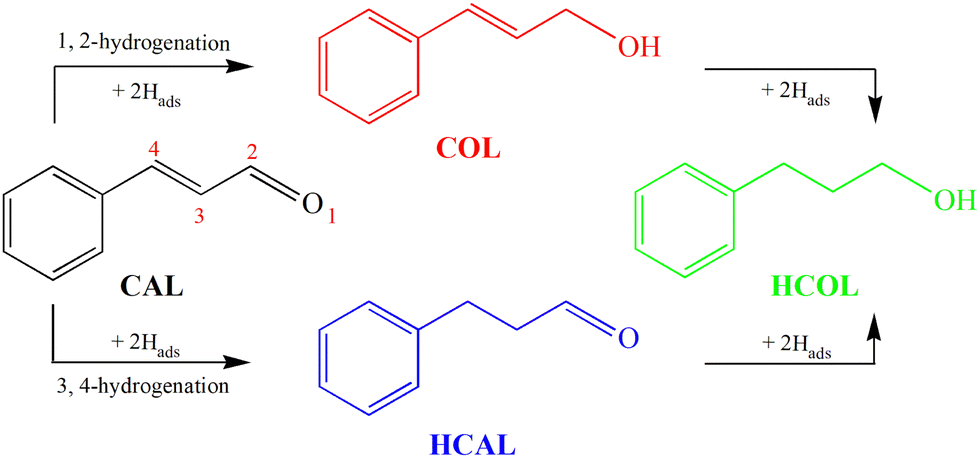 | ||
| Scheme 1 The hydrogenation pathways of cinnamaldehyde (CAL). | ||
ECH is challenging due to the existence of the competing reaction hydrogen evolution reaction (HER),2 which consumes Hads and reduces the faradaic efficiency of ECH.19 Hence, electrocatalysts that afford active and selective ECH but prohibit the HER are highly desirable.20,21 Enhancing the adsorption of carbonyl is a widely used strategy to improve CO hydrogenation.18 Precious metals (e.g., Ag, Pd, Pt, etc.) display the active ECH of carbonyl with a low onset potential;19,22 however, they suffer from poor faradaic efficiency, high cost, and low earth-abundance.23 Thus, the development of efficient, high-performance, and non-precious electrocatalysts is urgently required.
Molybdenum disulfide (MoS2), a conventional HER electrocatalyst,24–26 has strengthened surface chemisorption that is expected to stabilize Hads toward hydrogenation rather than self-recombination to H2. Furthermore, MoS2 can effectively activate unsaturated bonds (e.g., CO), enabling a rapid elementary step on the catalyst surface.23 For MoS2, the preferentially exposed basal plane is the thermodynamically stable (002) plane rather than the active edge planes.27,28 To circumvent this disadvantage, the engineering defect structure on the basal planes can be expected to increase the exposure of active edge sites by forming cracks on the surfaces,29 which may dramatically improve the electrocatalytic performance.
Herein, we report a vapor-phase hydrothermal (VPH) approach to in situ grow defective MoS2 nanoparticles using commercial carbon fiber cloth (CFC) with controllably adsorbed Mo6+ as the substrate and excess thiourea as the sulfur source at 180 °C for 24 h (MoS2/CFC). The as-prepared MoS2/CFC without further treatment was directly used as the electrode for ECH in 0.1 M phosphate buffer electrolyte (pH = 7.0), and exhibited highly selective hydrogenation of CAL to HCAL and COL. The high selectivity of CAL hydrogenation could be realized by adjusting the applied potentials, the hydrogenation of CAL to COL was easier to achieve under lower applied potential, whereas the hydrogenation of CAL to HCAL was easier to achieve under higher applied potential. DFT calculations demonstrated that the Mo vacancies in the defective MoS2 facilitated the adsorption of hydrogen atoms, which benefited the generation of Hads and the proceeding electrocatalytic hydrogenation. Besides, the Mo vacancies made the CO group more active than the CC group, and reduced the reaction energy of CAL hydrogenation toward COL. Furthermore, the high conversion and selectivity of other unsaturated aldehyde such as FAL and saturated aldehyde such as benzaldehyde hydrogenation confirmed the universality of the defective MoS2/CFC catalyst in the electrocatalytic hydrogenation reduction of aldehydes.
2. Experimental section
2.1 Materials
Sodium molybdate (Na2MoO4·2H2O), thiourea, sodium hydrogen phosphate (Na2HPO4·12H2O), sodium dihydrogen phosphate (NaH2PO4·2H2O), tetrahydrofuran and anhydrous ethanol were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Cinnamaldehyde (CAL), furfural (FAL) and benzaldehyde were purchased from Aladdin Ltd (Shanghai, China). All the chemicals were used as received without further purification. Commercial carbon fiber cloth (CFC) was purchased from Shanghai Hesen Electric Co., Ltd, which was washed with deionized water and anhydrous ethanol several times, respectively, then dried in air and cut into different sizes for further use.2.2 Vapor-phase hydrothermal construction of defective MoS2 on CFC
In a typical synthesis, 0.50 g Na2MoO4·2H2O powders were fully dissolved in 50 ml deionized water, and then a piece of cleaned CFC substrate (3.0 cm × 3.0 cm) was immersed in the above solution for 24 h to adequately adsorb Mo6+. Subsequently, the Mo6+-adsorbed CFC was dried at room temperature and then placed on a Teflon holder. The VPH reaction was performed in a Teflon-lined stainless steel autoclave (100 ml) with Mo6+-adsorbed CFC located above 20 ml thiourea solution (0.35 g thiourea/20 ml deionized water).30–32 The VPH reaction was performed at 180 °C for 24 h. After the autoclave was cooled down to room temperature, the product was collected and immersed into 0.1 M HCl for 2 h to remove surface adsorbed ammonia generated from the decomposition of thiourea, then rinsed with deionized water several times, and dried at 60 °C under vacuum. The obtained product was denoted as MoS2/CFC. The loading amount of MoS2 on the CFC substrate was determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES, 6300, Thermo Fisher Scientific, USA) after digestion by aqua regia through hydrothermal reaction under 180 °C for 48 h, and revealed a MoS2 loading of 0.56 mg cm−2.For comparison, bulk MoS2 was synthesized through a liquid-phase hydrothermal reaction. 0.50 g Na2MoO4·2H2O and 0.35 g thiourea were fully dissolved in 20 ml deionized water. The hydrothermal reaction was performed at 180 °C for 24 h.
2.3 Characterizations
The morphological properties of the samples were investigated using a field emission scanning electron microscope (FESEM, Hitachi SU8020) operated at an accelerating voltage of 3.0 kV. The microstructures of the samples were examined by a high-resolution transmission electron microscope (HRTEM, FEI Tecnai G2 F30 S-TWIN) with an acceleration voltage of 300 kV. X-Ray diffraction (XRD) patterns of the samples were recorded on a Philips X-Pert Pro X-ray diffractometer with Cu Kα radiation (λKα1 = 1.5418 Å). XPS analysis of the samples was performed on an ESCALAB 250 X-ray photoelectron spectrometer (Thermo, America) equipped with Al Kα1,2 monochromatized radiation at 1486.6 eV X-ray source. ICP measurements were performed by inductively coupled plasma atomic emission spectroscopy (ICP-AES, 6300, Thermo Fisher Scientific, USA).2.4 Electrochemical measurements
The electrochemical measurements were performed with a CHI 760E electrochemical workstation (CH Instruments, Inc., Shanghai, China), and a typical three-electrode divided H-type cell was used. The cathodic compartment and the anodic compartment were divided by a piece of Nafion 115 proton exchange membrane. Both the compartments contained 20 ml 0.1 M phosphate buffer electrolyte (pH = 7.0). If without extra statement, the electrolyte in the cathodic compartment maintained continuous stirring during all the electrochemical measurements. An Ag/AgCl electrode as the reference electrode was placed in the cathodic compartment, and a piece of pre-cleaned CFC (2.0 cm × 3.0 cm) as the counter electrode was placed in the anodic compartment. The as-prepared MoS2/CFC (1.0 cm × 2.0 cm) was directly used as the working electrode, and the effective geometric surface area of the working electrode was maintained at 1.0 cm × 1.0 cm. For bulk MoS2 powders, the working electrodes were prepared as follows: 6.0 mg as-prepared MoS2 powders were dispersed in 1000 μl of the mixed solution comprising 500 μl of deionized water, 490 μl of ethanol, and 10 μl of 5 wt% Nafion solution. The suspension was sonicated for 20 min to obtain a homogeneous ink. Then, 100 μl of MoS2 ink was dropped onto a cleaned CFC (1.0 × 2.0 cm, the MoS2 covering geometric area was 1.0 × 1.0 cm2) and then dried at room temperature. The areal loading mass of MoS2 on the CFC was about 0.60 mg.Electrochemical impedance spectroscopy (EIS) measurements were carried out in a frequency range of 100 kHz to 0.01 Hz at an open circuit voltage with an amplitude of 5 mV. Linear sweep voltammetry (LSV) curves were obtained at a scan rate of 5.0 mV s−1.
0.5 ml tetrahydrofuran containing 33 μl (0.25 mmol) cinnamaldehyde was added into the cathodic compartment and stirred for 10 min until the electrolyte was emulsified. LSV and EIS measurements followed. All the potentials were converted to the reversible hydrogen electrode (RHE) scale through calibration with the following Nernst equation: E (V vs. RHE) = E (V vs. Ag/AgCl) + 0.059 × pH + 0.197. The hydrogenation process was evaluated by chronoamperometry under different constant potentials for 5 h.
Another two aldehyde molecules, furfural (FAL) and benzaldehyde, were also investigated. For furfural, 21 μl (0.25 mmol) furfural was directly added into the cathodic compartment. For benzaldehyde, 0.5 ml tetrahydrofuran containing 26 μl (0.25 mmol) benzaldehyde was added into the cathodic compartment. Tetrahydrofuran was used because of its excellent solubility toward organic compounds and good stability during electrolysis.
2.5 Product analysis
After electrolysis reactions, 5.0 ml electrolyte solution was taken from the cathodic compartment and 3.0 ml ethyl acetate was added to extract the organics. The extracted organics were subsequently analyzed by gas chromatography-mass spectrometry (GC-MS, Agilent GC7890 + MS5977B). For cinnamaldehyde and benzaldehyde, the initial GC temperature was 100 °C for 2 min, and then increased at 10 °C min−1 to 300 °C where it was held for 2 min. For furfural, the initial GC temperature was 30 °C for 2 min, and then increased at 5 °C min−1 to 300 °C where it was held for 2 min.The conversion (%) of the reactant was calculated based on the following equation:
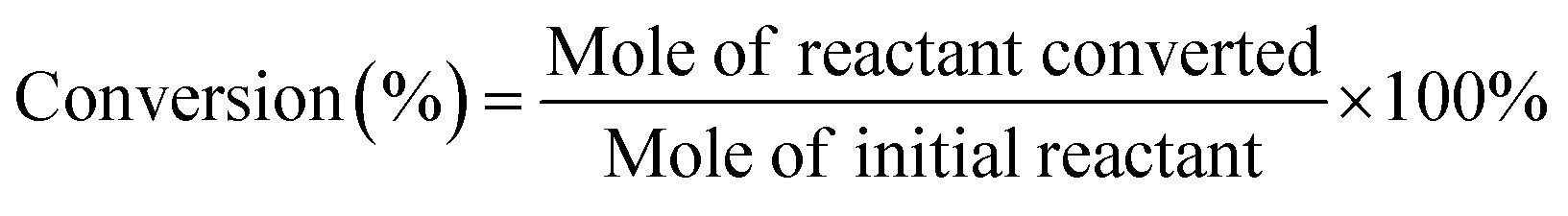 | (1) |
The product selectivity (%) was calculated by the following equation:
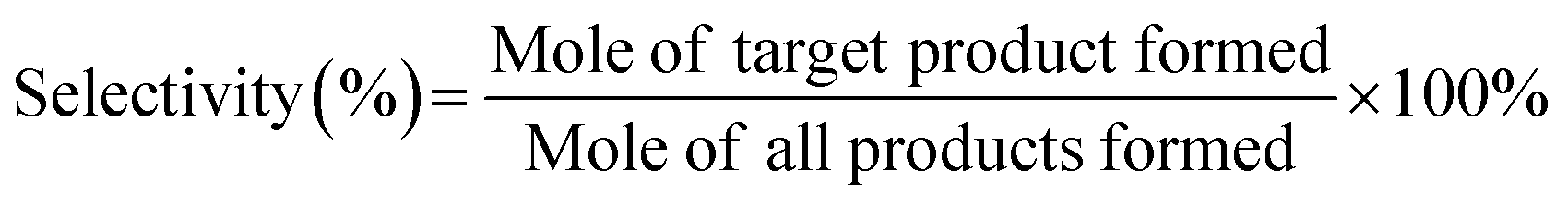 | (2) |
The faradaic efficiency (FE, %) of products formation was calculated using the following equation:
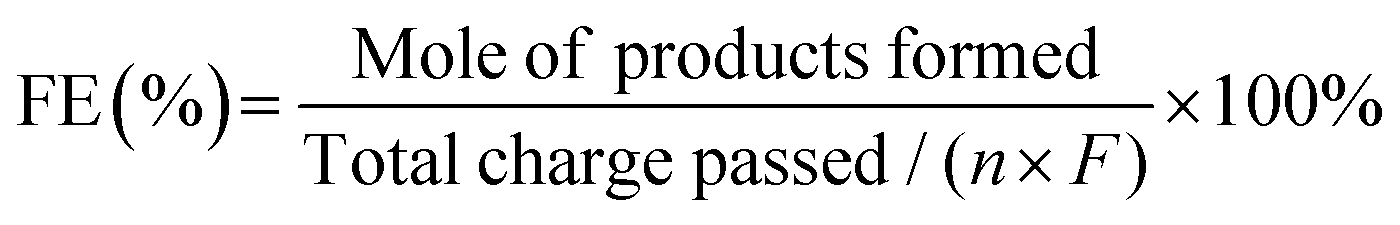 | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1).
485 C mol−1).
The turnover frequency (TOF) values of MoS2/CFC catalysts in electrocatalytic hydrogenation were calculated with the following equation while assuming all MoS2 components are available active sites and involved in the electrocatalytic hydrogenation process:
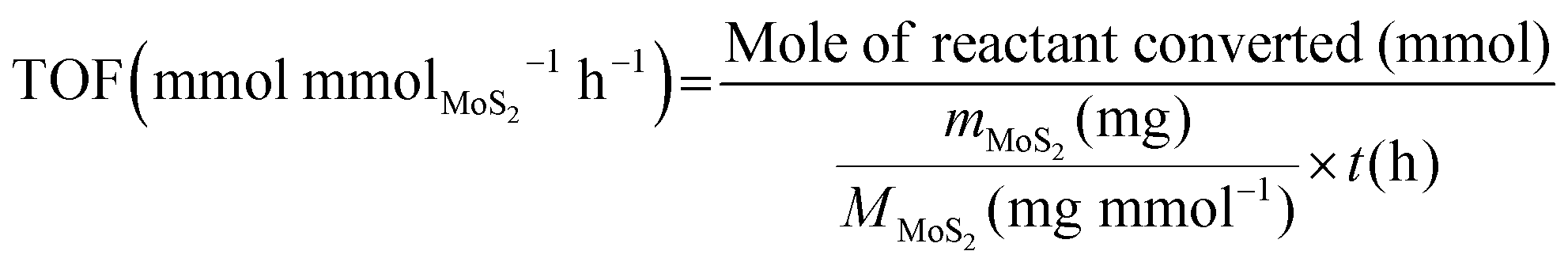 | (4) |
2.6 Theoretical calculations
All the first-principle calculations were performed within the framework of DFT as implemented in the Vienna Ab initio Simulation Package (VASP).33–35 The projector augmented wave (PAW) method has been used to describe the inert core electrons.36 A cut off energy of 400 eV was used for the expansion of the wave functions. The electronic exchange–correlation effects were described with Perdew–Burke–Ernzerhof generalized gradient approximation (PBE–GGA) functional.37 The basic carbon model was built using a periodic graphene structure containing 72 C atoms. A slab of MoS2(002) surface without defects, with Mo vacancies or Mo/S vacancies was built for the adsorption calculations, respectively. A vacuum of 20 Å in the z-direction is used. The gamma (Γ) cantered 2 × 2 × 1 Monkhorst-Pack k-point sampling was used throughout.38 The Fermi level was slightly broadened using a Fermi-Dirac smearing of 50 meV. All relaxations were carried out until the force of the system converges to 0.05 eV Å−1. The van der Waals interactions were considered using the DFT-D3 empirical correction.3. Results and discussion
In this work, commercial carbon fiber cloth (Fig. S1, ESI†) with controllably adsorbed Mo6+ was used as the substrate for VPH growth of MoS2 nanoparticles (MoS2/CFC). After the VPH reaction at 180 °C for 24 h (Fig. S2, ESI†), the obtained products still exhibited the configuration from the original CFC framework (Fig. S3, ESI†). Fig. 1a shows a single fiber in which the growth occurs on the surface, and the high-magnification image (Fig. 1b) confirms the presence of irregularly shaped Mo-based nanoparticles with an average diameter of less than 50 nm uniformly grown on the surface of the CFC. The microstructure of Mo-based species was further investigated by TEM characterization (Fig. 1c). The nanoparticle morphology is not clear in the TEM image since the Mo-based nanoparticles were ultrasonically dispersed in ethanol during the TEM sample preparation, which caused the morphological destruction. The high-resolution TEM (HRTEM) images shown in Fig. 1d and e indicate that the lattice fringe spacings are 2.7 Å and 6.3 Å, respectively. Interplanar spacing of 2.7 Å is consistent with the d spacing of (100) planes of hexagonal MoS2, while, the interlayer spacing of 6.3 Å observed from the curled edge is slightly larger than the layer-to-layer spacing of 6.15 Å in bulk MoS2. It is worth noting that many crystal fringes along the curled edge are discontinuous, which can be attributed to the existence of rich defects,25 as indexed in the white dotted circular area. The defect-rich structure resulted from the excess thiourea, which can be adsorbed on the surface of primary nanocrystallites, partially hindering the oriented crystal growth and leading to the formation of a defect-rich structure with quasiperiodic configuration.25 The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) analysis (Fig. 1f) and the corresponding elemental mapping images (Fig. 1g–i) show the presence and uniform distribution of Mo and S elements, corresponding to the obtained MoS2. Possibly owing to the low content and poor crystallinity of the formed MoS2 nanoparticles, only diffraction peaks at 24.7° and 43.5° of hexagonal graphite carbon (JCPDS No. 13-0148) in the XRD patterns (Fig. S4, ESI†) can be observed for the MoS2/CFC. | ||
| Fig. 1 MoS2/CFC: (a) low-magnification SEM image; (b) high-magnification SEM image; (c) TEM image; (d) HRTEM image; (e) high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and (f–i) corresponding elemental mapping images. | ||
In order to analyze the surface chemical and oxidation states of the Mo6+-adsorbed CFC and as-prepared defective MoS2/CFC, X-ray photoelectron spectroscopy (XPS) was carried out. The peaks related to Mo in the survey spectrum (Fig. S5a, ESI†) indicate the adsorption of Mo6+ ions on the CFC substrate. The high resolution Mo 3d spectrum (Fig. S5b, ESI†) shows two characteristic peaks at the binding energies of 231.9 and 235.1 eV, which can be attributed to the orbitals of MoVI 3d5/2 and MoVI 3d3/2, respectively. Fig. S5c (ESI†) shows the high resolution S 2p spectrum, confirming no S-related species in the Mo6+-adsorbed CFC. As shown in Fig. S5d (ESI†), the O 1s peak located at 530.0 eV corresponds to the binding energy of Mo–O bonds,39 which comes from the original reagent Na2MoO4. The peaks located at 532.3 eV and 534.6 eV can be attributed to C–O and H–O bonds. After the VPH reaction, the signal of S 2p was observed from the survey spectrum (Fig. 2a) of the obtained MoS2/CFC. Detailed compositional analysis (Table S1, ESI†) reveals that the atomic ratio of Mo:S is 1:2.94, giving the direct evidence that the existence of unsaturated sulfur atoms in the defective MoS2/CFC. As shown in Fig. 2b, two characteristic peaks arising from Mo 3d5/2 and Mo 3d3/2 orbitals are located at 229.0 and 232.3 eV, respectively, which corresponds to the +4 oxidation state, suggesting the dominance of MoIV in the product.40 A small S 2s peak is centered at 226.2 eV, whereas the S 2p region (Fig. 2c) exhibits primarily a single doublet with the 2p3/2 peak at 161.8 eV, which is consistent with the −2 oxidation state of sulfur.40 Two peaks of 163.5 and 164.4 eV represent the exposed sulfur atoms.41 The high resolution O 1s peak located at 530.0 eV disappeared in the obtained MoS2/CFC (Fig. 2d), which can be attributed to the breaking of Mo–O bonds. In conclusion, the XPS measurement results show that the unsaturated sulfur atoms in the defective MoS2/CFC produce a large number of Mo vacancies.
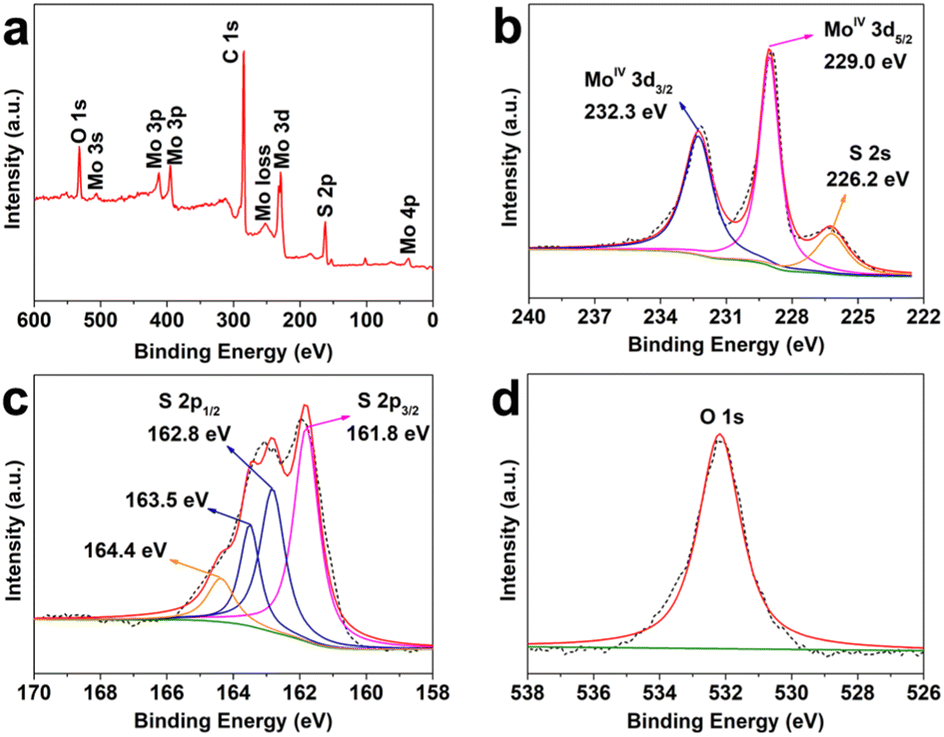 | ||
| Fig. 2 (a) Surface survey XPS spectrum of MoS2/CFC. High-resolution XPS spectra of (b) Mo 3d, (c) S 2p and (d) O 1s. | ||
CAL was first selected as a classical model to evaluate the selective hydrogenation performance of α,β-unsaturated aldehydes over the as-prepared defective MoS2/CFC. As shown in the LSV curves (Fig. 3a), the addition of CAL into the electrolyte slightly decreased the current density, which was because the electrocatalytic hydrogenation of CAL was more sluggish than hydrogen evolution by water decomposition, and the occupation of active sites by CAL molecules hindered hydrogen adsorption and evolution.18 In the EIS spectra (Fig. 3b), the high-frequency intercept on the real axis represents the Ohmic series resistance (Rs), and the semicircle in the high-frequency region corresponds to the charge–transfer resistance (Rct) of the electrolyte/electrode interface, which strongly influences the activity of an electrocatalyst.31 Clearly, the Rs and Rct values after the addition of the CAL precursor were smaller than that for pure water decomposition, indicating the higher electrocatalytic hydrogenation activity of the defective MoS2/CFC toward the CAL precursor.
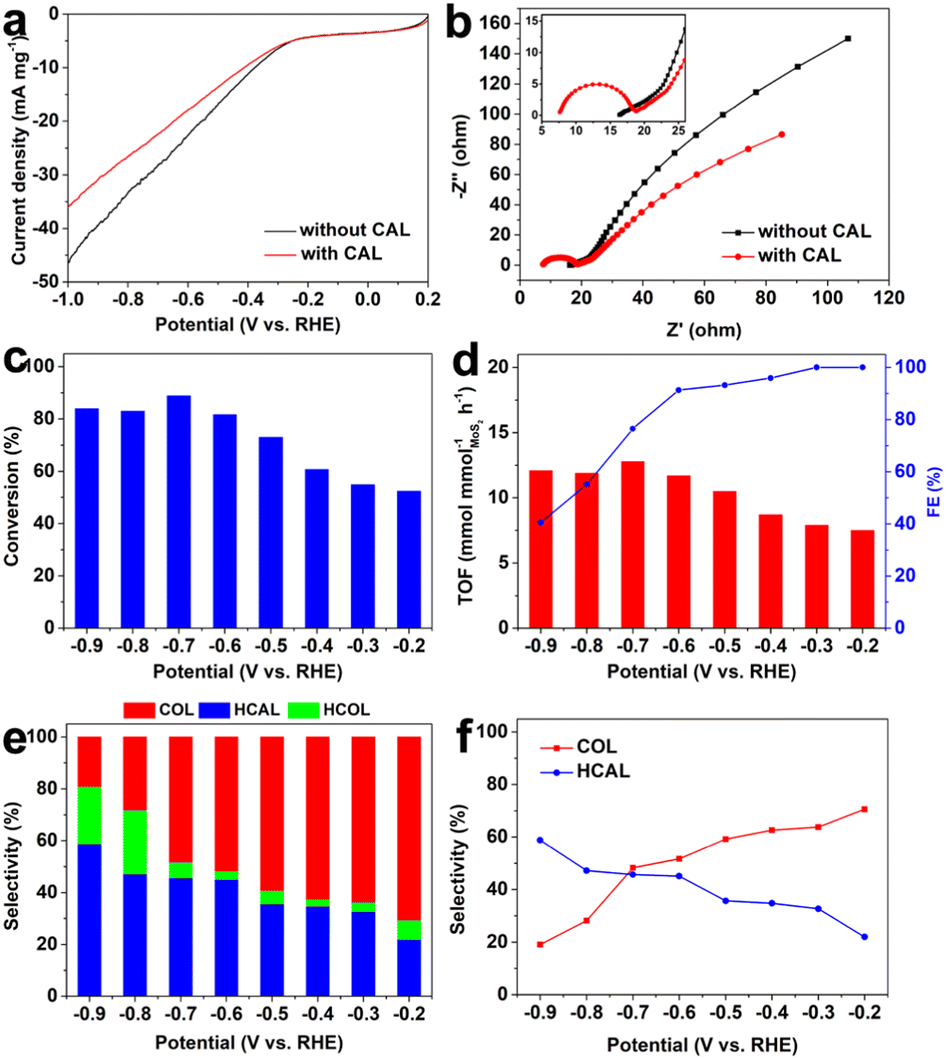 | ||
| Fig. 3 (a) LSV curves and (b) EIS spectra of MoS2/CFC in 0.1 M PBS electrolyte (pH = 7.0) with and without CAL; Potential-dependent performance of CAL hydrogenation over MoS2/CFC: (c) conversion, (d) TOF and FE, (e) and (f) selectivity. | ||
Subsequently, the potential-dependent results showed excellent CAL conversion yield, turnover frequency, faradaic efficiency and product selectivity over the defective MoS2/CFC (Fig. 3c–f, Fig. S6, S7 and Table S2, ESI†). As shown in Fig. 3c, the conversion of CAL increased with the increase of applied potentials, and reached a maximum value of 88.8% at −0.7 V vs. RHE. However, when the potential became more negative, the conversion decreased, mainly due to the competitive HER process. When considering the intrinsic activity of electrocatalysts, the most important figure of merit is the turnover frequency (TOF), which is the number of moles of products generated per unit time per active site.42 As shown in Fig. 3d, the variation tendency of TOF was consistent with the conversion in Fig. 3c, and reached a maximum value of 12.8 mmol mmolMoS2−1 h−1 at −0.7 V vs. RHE. The FE values of CAL hydrogenation decreased with the increase of applied potentials (Fig. 3d), attributing to the enhanced HER activity. The detailed FE values for COL, HCAL and HCOL are shown in Fig. S7 (ESI†). Fig. 3e and f are the histogram and the corresponding point plot of product selectivity with voltage change, respectively. From which it can be seen clearly that, with the increase of applied potentials from −0.2 V to −0.9 V vs. RHE, the selectivity of COL gradually decreased from 70.6% to 19.1%, whereas the selectivity of HCAL gradually increased from 22.0% to 58.8%. In other words, the hydrogenation of CAL to COL was easier to achieve under lower applied potentials, whereas the hydrogenation of CAL to HCAL was easier to achieve under higher applied potentials. Meanwhile, the selectivity of HCOL also increased with the increase of applied potential, which was because the HCAL and COL were more easily transformed into fully hydrogenated HCOL under higher applied potentials. As we know, obtaining a high COL yield under low voltage is challenging because the CO bond is thermodynamically and kinetical more difficult to be hydrogenated than the CC bond. In this work, the high selectivity of CAL hydrogenation can be achieved by adjusting the applied potentials. Compared with other representative reports based on electrocatalytic CAL selective hydrogenation, the defective MoS2/CFC showed an excellent performance (Table S3, ESI†).
To clarify the mechanism of the high selectivity of CAL hydrogenation over the defective MoS2/CFC, we explored the adsorption of reactants and products over defective MoS2 and graphite carbon substrate by means of DFT calculation, where bulk MoS2 without defects acted as a reference for a better understanding of defective MoS2. As mentioned above, excess thiourea was a prerequisite for the formation of the defect-rich structure. Therefore, we only considered the following two defective configurations: Mo vacancies and Mo/S vacancies with a stoichiometric atomic ratio of 1:2. The adsorption of hydrogen was investigated firstly. The adsorption configurations and energies of hydrogen atom over graphite carbon, MoS2(002) surface, and MoS2(002) surface with Mo vacancies or Mo/S vacancies were compared. As shown in Fig. S8 (ESI†), the corresponding adsorption energies of the hydrogen atom were calculated as 147.92, 174.98, −62.40 and −70.40 kJ mol−1, respectively. Clearly, the adsorption of hydrogen atom onto the (002) surface of MoS2 with Mo vacancies or Mo/S vacancies was easier than that of graphite carbon and bulk MoS2(002) surface without defects, which benefited the generation of Hads and the proceeding of electrocatalytic hydrogenation.
Subsequently, the adsorption of a CAL molecule and its hydrogenation products HCAL and COL over the Mo-vacancy MoS2(002) surface was explored. After optimizing the most stable adsorption configurations, a CAL molecule was adsorbed on the MoS2(002) surface through a parallel configuration with an adsorption energy of −90.43 kJ mol−1 (Fig. 4a). In detail, a CAL molecule was bound to the MoS2(002) surface through a phenyl group, CC bond, and CO bond. After the CC bond was fully hydrogenated with Hads, the resulting HCAL was interacted with the MoS2(002) surface through the phenyl group and the CO bond with an adsorption energy of −56.14 kJ mol−1 (Fig. 4b). On the other hand, when the CO bond was fully hydrogenated with Hads, the obtained COL was attracted onto the MoS2(002) surface via the phenyl group, CC bond and –OH group with an adsorption energy of −93.22 kJ mol−1 (Fig. 4c). If assuming the binding energy (Eb) of the same functional group remains constant in different compounds, Eb,CC and Eb,CO can be roughly estimated from the calculated adsorption energy (ΔEads) of CAL, HCAL, COL, and POL (n-propanol, adsorbed mainly through –OH group, Fig. 4d) by the following equations:18
| Eb,CC = ΔEads,CAL − ΔEads,HCAL | (5) |
| Eb,CO = ΔEads,CAL − ΔEads,COL + ΔEads,POL | (6) |
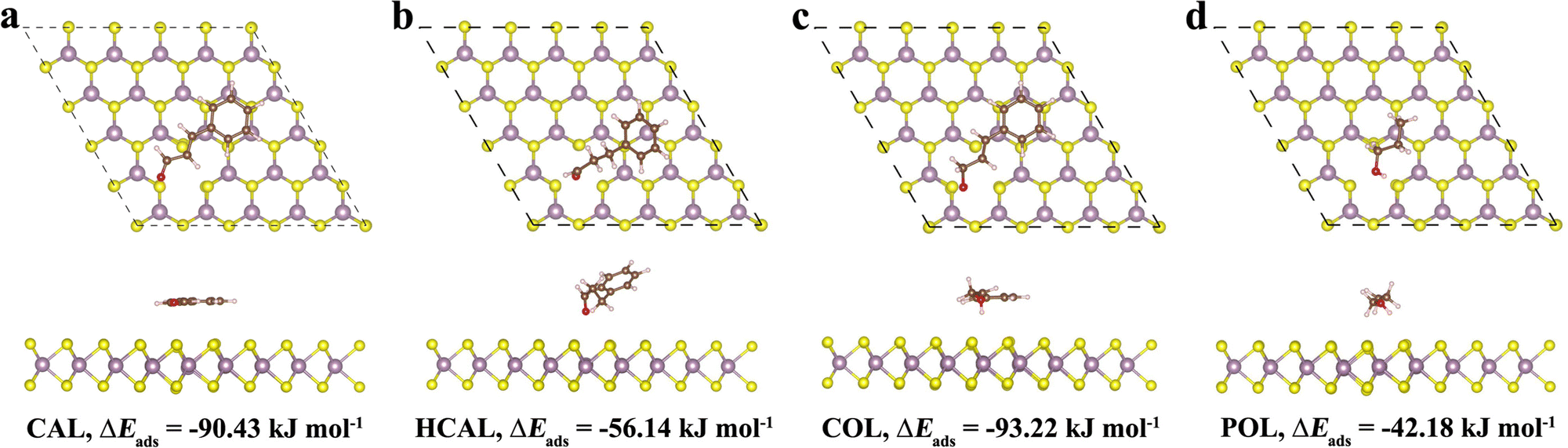 | ||
| Fig. 4 Adsorption configurations and energies of different reacting species on the Mo-vacancy MoS2(002) surface (brown sphere: C, white sphere: H, red sphere: O, yellow sphere: S, and purple sphere: Mo). | ||
As listed in Table 1, the calculated binding energy of the CO group of CAL (Eb,CO = −39.39 kJ mol−1) was more negative than that of the CC group (Eb,CC = −34.29 kJ mol−1), suggesting the CO group was more active than the CC group in CAL hydrogenation to generate COL over Mo-vacancy MoS2.
| Catalyst | ΔEads (kJ mol−1) | E b (kJ mol−1) | Reaction energy (kJ mol−1) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| H | CAL | HCAL | COL | POL | CC |
CO |
CAL → AHCAL | CAL → COL | |
| a Carbon: graphite carbon substrate CFC. b MoS2: bulk MoS2(002) surface without defects. c MoS2–MoVac: MoS2(002) surface with Mo vacancies. d MoS2–MoSVac: MoS2(002) surface with Mo/S vacancies. | |||||||||
| Carbona | 147.92 | −54.02 | −45.74 | −82.91 | −9.63 | −8.38 | 19.26 | −101.69 | −104.39 |
| MoS2b | 174.98 | −90.91 | −58.55 | −93.60 | −42.37 | −32.36 | −39.68 | −77.62 | −78.20 |
| MoS2–MoVacc | −62.40 | −90.43 | −56.14 | −93.22 | −42.18 | −34.29 | −39.39 | −75.79 | −83.88 |
| MoS2–MoSVacd | −70.40 | −85.32 | −64.23 | −89.94 | −47.09 | −21.09 | −42.47 | −88.88 | −80.12 |
Based on the most stable adsorption configurations of CAL, HCAL and COL on the Mo-vacancy MoS2(002) surface, we further calculated the reaction energies for the hydrogenation of CC (from CAL to HCAL) and CO (from CAL to COL). As shown in Fig. 5 and Table 1, the reaction energy change from CAL to COL (ΔG = −83.88 kJ mol−1) was more negative than that from CAL to HCAL (ΔG = −75.79 kJ mol−1), indicating the hydrogenation from CAL to COL was easier than that from CAL to HCAL. In other words, during the hydrogenation of CAL over Mo-vacancy MoS2, COL was easier to form under lower applied potentials, and HCAL was easier to form under higher applied potentials to overcome the reaction energy, according with our experimental results (Fig. 3e and f).
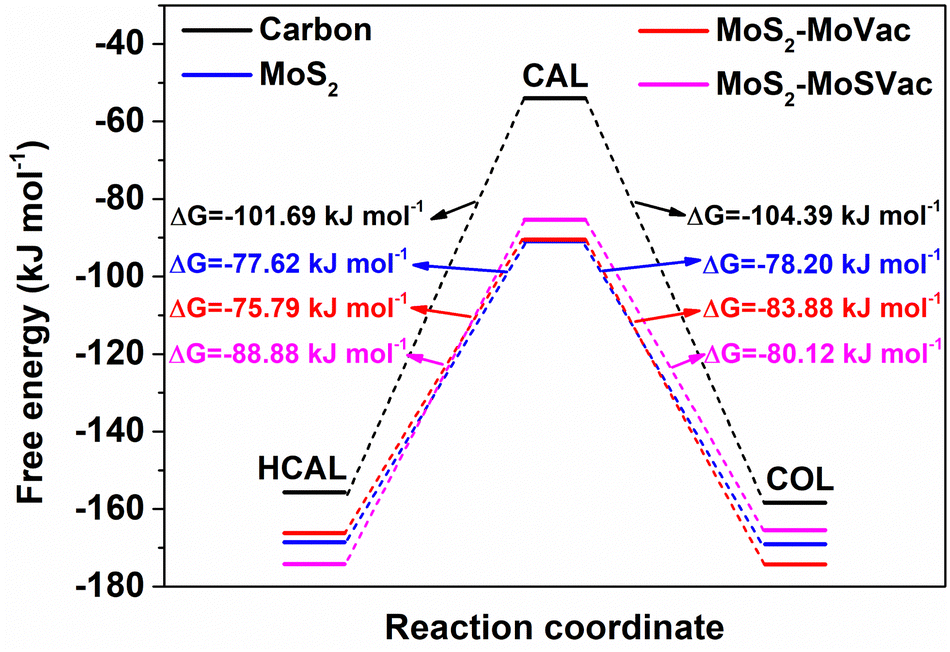 | ||
| Fig. 5 Reaction pathway and energy diagram of CAL hydrogenation that proceeded over graphite carbon (denoted as carbon), bulk MoS2(002) surface (denoted as MoS2), Mo-vacancy MoS2(002) surface (denoted as MoS2–MoVac), and Mo/S-vacancy MoS2(002) surface (denoted as MoS2–MoSVac). | ||
Besides, the adsorption energies of different reacting species, the binding energies of CC and CO, and the reaction energies for the hydrogenation of CC and CO over Mo/S vacancies MoS2(002) surface were calculated. As shown in Fig. S9 (ESI†), Fig. 5 and Table 1, although the calculated binding energy of the CO group of CAL (Eb,CO = −42.47 kJ mol−1) was much more negative than that of the CC group (Eb,CC = −21.09 kJ mol−1), the reaction energy change from CAL to HCAL (ΔG = −88.88 kJ mol−1) was more negative than that from CAL to COL (ΔG = −80.12 kJ mol−1), indicating the hydrogenation from CAL to HCAL was easier than that from CAL to COL. The calculation data was not according with our experimental results, suggesting that Mo/S vacancies were not the real catalytic active centers of the defective MoS2/CFC in this work.
Meanwhile, the adsorption energies of different reacting species, the binding energies of CC and CO, and the reaction energies for the hydrogenation of CC and CO over the bulk MoS2(002) surface without defects were calculated. As shown in Fig. S10 (ESI†), Fig. 5 and Table 1, although the calculated binding energy of the CO group of CAL (Eb,CO = −39.68 kJ mol−1) was more negative than that of CC group (Eb,CC = −32.36 kJ mol−1), the reaction energy changes from CAL to COL (ΔG = −78.20 kJ mol−1) and from CAL to HCAL (ΔG = −77.62 kJ mol−1) were almost identical, suggesting no significant difference in product selectivity of CAL hydrogenation. To confirm the calculation results, bulk MoS2 without defects was synthesized using stoichiometric Mo and S sources. As shown in Fig. S11a (ESI†), the obtained bulk MoS2 are nanosheets with a uniform lateral size of about 200 nm. Two peaks in the low-angle region (9.2° and 17.8°) in the XRD pattern (Fig. S11b, ESI†) correspond to the oxygen-incorporated MoS2, which could be further proved by the enlarged interlayer spacing of 9.5 Å and the uniform distribution of O element (Fig. S11c–h, ESI†).24 The XPS measurement results show that the atom ratio of Mo:S in the bulk MoS2 is 1:2.01 (Table S1 and Fig. S12, ESI†). The electrocatalytic hydrogenation performance of CAL over the bulk MoS2 was also investigated. As shown in Fig. S13 (ESI†), the conversion of CAL increased with the increase of applied potentials. However, the selectivity between COL and HCAL remained almost identical, which corresponded to the calculation results.
In order to explore the catalytic effect of CFC substrate in the defective MoS2/CFC in electrocatalytic CAL hydrogenation, contrast experiments were carried out. Bare CFC without MoS2 loading was used as an electrode directly to catalyze CAL hydrogenation under −0.7 V vs. RHE, exhibiting an 60.0% conversion of CAL and an 100.0% selectivity of HCAL. By contrast, the defective MoS2/CFC displayed an 88.8% conversion of CAL, an 45.7% selectivity of HCAL and an 48.3% selectivity of COL under the identical conditions, respectively. These results clearly indicated that the hydrogenation of CO was more active on the defective MoS2, whereas the hydrogenation of CC was more active on graphite carbon. The calculation results (Fig. S14, ESI,†Fig. 5 and Table 1) indicated that although the reaction energy change from CAL to COL (ΔG = −104.39 kJ mol−1) was more negative than that from CAL to HCAL (ΔG = −101.69 kJ mol−1) on graphite carbon, the calculated binding energy of the CC group of CAL (Eb,CC = −8.38 kJ mol−1) was much more negative than that of the CO group (Eb,CO = 19.26 kJ mol−1). Therefore, CC was more active in CAL hydrogenation to generate HCAL.
The changes in the structure and composition of the defective MoS2/CFC after ECH measurements were investigated to evaluate the stability of the catalyst. The XRD pattern (Fig. S15, ESI†) only displayed two main diffraction peaks of graphite carbon, attributed to the low content and poor crystallinity of the loaded MoS2 nanoparticles. The SEM images (Fig. S16a and b, ESI†) showed no obvious changes, which can be further confirmed by TEM characterization (Fig. S16c–i, ESI†). Fig. S17 (ESI†) showed the XPS spectra of MoS2/CFC after ECH. Compared to the initial MoS2/CFC (Fig. 2), both Mo 3d, S 2s and S 2p peaks shifted to higher binding energies, indicating that both Mo and S gave electrons to other components during the ECH process. Here, CAL molecules captured electrons to proceed the hydrogenation reduction reaction, suggesting that Mo and S served as “electron donors”, CAL served as an “electron acceptor”, respectively, and the electrons transferred from Mo and S to CAL during the ECH process. In addition, MoS2/CFC exhibited high stability for the electrocatalytic hydrogenation of CAL (Fig. S18, ESI†).
Furthermore, other typical unsaturated aldehydes such as furfural (FAL) were selected to evaluate the selective hydrogenation performance over the as-prepared defective MoS2/CFC. As shown in Fig. S19 (ESI†), Fig. 6 and Table S4 (ESI†), furfuryl alcohol (FOL) was the only hydrogenation product in a wide potential range from −0.2 to −0.9 V vs. RHE, indicating 100.0% hydrogenation selectivity. The conversion of FAL and TOF value increased with the increase of applied potentials, and reached a maximum value of 92.6% and 13.3 mmol mmolMoS2−1 h−1 at −0.8 V vs. RHE. The corresponding FE values of FAL hydrogenation generally decreased with the increase of applied potentials, attributing to the enhanced HER activity.
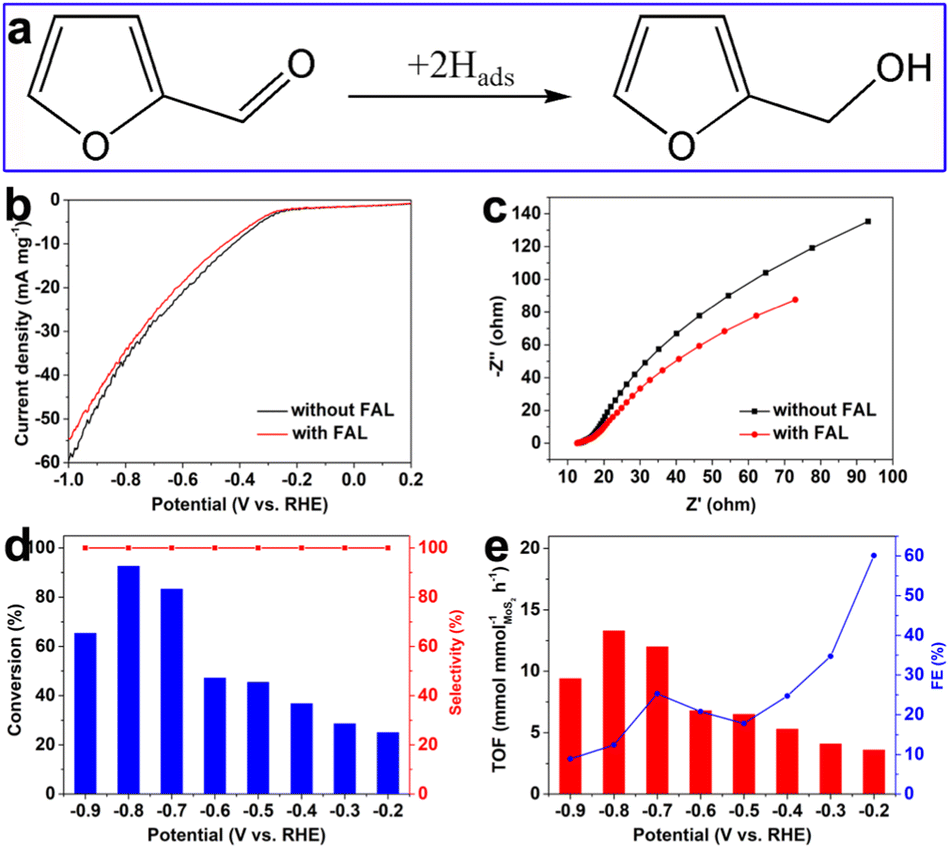 | ||
| Fig. 6 (a) The hydrogenation pathways of furfural (FAL), (b) LSV curves and (c) EIS spectra of MoS2/CFC in 0.1 M PBS electrolyte (pH = 7.0) with and without FAL; potential-dependent performance of FAL hydrogenation over MoS2/CFC: (d) conversion and selectivity, (e) TOF and FE. | ||
In addition, the hydrogenation performance of typical saturated aldehyde such as benzaldehyde over the as-prepared defective MoS2/CFC was also explored. As shown in Fig. S20 (ESI†), Fig. 7 and Table S5 (ESI†), benzyl alcohol was the only hydrogenation product in a wide potential range from −0.3 to −1.0 V vs. RHE with a 100.0% hydrogenation selectivity. The conversion and TOF values increased with the increase of applied potentials, and the corresponding FE values gradually decreased with the increase of applied potentials, which was due to the enhanced competitive HER activity. These results confirmed the universality of the defective MoS2/CFC catalyst in the electrocatalytic hydrogenation reduction of aldehydes.
 | ||
| Fig. 7 (a) The hydrogenation pathways of benzaldehyde, (b) LSV curves and (c) EIS spectra of MoS2/CFC in 0.1 M PBS electrolyte (pH = 7.0) with and without benzaldehyde; potential-dependent performance of benzaldehyde hydrogenation over MoS2/CFC: (d) conversion and selectivity, (e) TOF and FE. | ||
4. Conclusions
In summary, defective MoS2 nanoparticles were successfully grown on commercial carbon fiber cloth (MoS2/CFC) via a simple vapor-phase hydrothermal approach and directly used as the electrode electrocatalyst without further treatment for ECH in 0.1 M phosphate buffer electrolyte (pH = 7.0). The experimental results suggested that the as-prepared MoS2/CFC exhibited the highest CAL conversion of 88.8% with a maximum TOF value of 12.8 mmol mmolMoS2−1 h−1 at −0.7 V vs. RHE. With the increase of applied potentials from −0.2 V to −0.9 V vs. RHE, the selectivity of COL gradually decreased from 70.6% to 19.1%, whereas the selectivity of HCAL gradually increased from 22.0% to 58.8%, indicating that the hydrogenation of CAL to COL was easier to achieve under a lower applied potential, whereas the hydrogenation of CAL to HCAL was easier to achieve under a higher applied potential. DFT calculations proved that the real catalytic active center was the Mo vacancies in the defective MoS2/CFC, which facilitated the adsorption of hydrogen atoms, benefiting the generation of Hads and the proceeding electrocatalytic hydrogenation. Furthermore, the ECH performance of the defective MoS2/CFC for other unsaturated aldehyde such as FAL and saturated aldehyde such as benzaldehyde hydrogenation was also explored, exhibiting the universality in ECH of unsaturated and saturated aldehydes. This is inspirational for electrocatalyst design through defect engineering in electrocatalytic organic synthesis, especially in electrocatalytic hydrogenation of unsaturated or saturated aldehydes and ketones to produce the corresponding alcohols.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the Key Research and Development Projects of Shaanxi Province (Grant No. 2021GY-126), the Key Science and Technology Project of Northwest Institute for Non-ferrous Metal Research (Grant No. YK2020-6), and the National Natural Science Foundation of China (Grant No. 61804154 and 12147219). We thank the LvLiang Cloud Computing Centre of China, TianHe-2 and Hefei advanced computing center for theoretical calculations and the C3S2 computing center in Huzhou University for calculation support.Notes and references
- M. C. Leech and K. Lam, A practical guide to electrosynthesis, Nat. Rev. Chem., 2022, 6, 275–286 CrossRef.
- S. A. Akhade, N. Singh, O. Y. Gutierrez, J. Lopez-Ruiz, H. Wang, J. D. Holladay, Y. Liu, A. Karkamkar, R. S. Weber, A. B. Padmaperuma, M. S. Lee, G. A. Whyatt, M. Elliott, J. E. Holladay, J. L. Male, J. A. Lercher, R. Rousseau and V. A. Glezakou, Electrocatalytic hydrogenation of biomass-derived organics: a review, Chem. Rev., 2020, 120, 11370–11419 CrossRef CAS PubMed.
- J. Song, Z. F. Huang, L. Pan, K. Li, X. Zhang, L. Wang and J. J. Zou, Review on selective hydrogenation of nitroarene by catalytic, photocatalytic and electrocatalytic reactions, Appl. Catal., B, 2018, 227, 386–408 CrossRef CAS.
- W. Xu, C. Yu, J. Chen and Z. Liu, Electrochemical hydrogenation of biomass-based furfural in aqueous media by Cu catalyst supported on N-doped hierarchically porous carbon, Appl. Catal., B, 2022, 305, 121062 CrossRef CAS.
- P. Zhang and L. Sun, Electrocatalytic hydrogenation and oxidation in aqueous conditions, Chin. J. Chem., 2020, 38, 996–1004 CrossRef CAS.
- X. Chong, C. Liu, Y. Huang, C. Huang and B. Zhang, Potential-tuned selective electrosynthesis of azoxy-, azo- and amino-aromatics over a CoP nanosheet cathode, Natl. Sci. Rev., 2019, 7, 285–295 CrossRef PubMed.
- X. H. Chadderdon, D. J. Chadderdon, J. E. Matthiesen, Y. Qiu, J. M. Carraher, J. P. Tessonnier and W. Li, Mechanisms of furfural reduction on metal electrodes: distinguishing pathways for selective hydrogenation of bioderived oxygenates, J. Am. Chem. Soc., 2017, 139, 14120–14128 CrossRef CAS PubMed.
- K. Li and Y. Sun, Electrocatalytic upgrading of biomass-derived intermediate compounds to value-added products, Chem. – Eur. J., 2018, 24, 18258–18270 CrossRef CAS.
- S. Han, Y. Shi, C. Wang, C. Liu and B. Zhang, Hollow cobalt sulfide nanocapsules for electrocatalytic selective transfer hydrogenation of cinnamaldehyde with water, Cell Rep. Phys. Sci., 2021, 2, 100337 CrossRef CAS.
- M. Zhao, K. Yuan, Y. Wang, G. Li, J. Guo, L. Gu, W. Hu, H. Zhao and Z. Tang, Metal-organic frameworks as selectivity regulators for hydrogenation reactions, Nature, 2016, 539, 76–80 CrossRef CAS PubMed.
- X. Wang, X. Liang, P. Geng and Q. Li, Recent advances in selective hydrogenation of cinnamaldehyde over supported metal-based catalysts, ACS Catal., 2020, 10, 2395–2412 CrossRef CAS.
- S. Bai, L. Bu, Q. Shao, X. Zhu and X. Huang, Multicomponent Pt-based zigzag nanowires as selectivity controllers for selective hydrogenation reactions, J. Am. Chem. Soc., 2018, 140, 8384–8387 CrossRef CAS PubMed.
- M. J. Torres, P. Sánchez, A. de Lucas-Consuegra and A. R. de la Osa, Electrocatalytic hydrogenation of cinnamaldehyde in a PEM cell: the role of sodium hydroxide and platinum loading, Mol. Catal., 2020, 492, 110936 CrossRef CAS.
- T. Wu, H. Meng and R. Dang, Amorphous Ta2O5-supported Ru as an efficient electrocatalyst for selective hydrogenation of cinnamaldehyde with water as the hydrogen source, Inorg. Chem. Front., 2021, 8, 4712–4719 RSC.
- H. Wang, S. Bai, Y. Pi, Q. Shao, Y. Tan and X. Huang, A strongly coupled ultrasmall Pt3Co nanoparticle-ultrathin Co(OH)2 nanosheet architecture enhances selective hydrogenation of α,β-unsaturated aldehydes, ACS Catal., 2019, 9, 154–159 CrossRef CAS.
- X. Lan and T. Wang, Highly selective catalysts for the hydrogenation of unsaturated aldehydes: a review, ACS Catal., 2020, 10, 2764–2790 CrossRef CAS.
- X. Lan, K. Xue and T. Wang, Combined synergetic and steric effects for highly selective hydrogenation of unsaturated aldehyde, J. Catal., 2019, 372, 49–60 CrossRef CAS.
- X. Huang, L. Zhang, C. Li, L. Tan and Z. Wei, High selective electrochemical hydrogenation of cinnamaldehyde to cinnamyl alcohol on RuO2–SnO2–TiO2/Ti electrode, ACS Catal., 2019, 9, 11307–11316 CrossRef CAS.
- Y. Kwon, K. J. P. Schouten, J. C. van der Waal, E. de Jong and M. T. M. Koper, Electrocatalytic conversion of furanic compounds, ACS Catal., 2016, 6, 6704–6717 CrossRef CAS.
- C. J. Bondue, F. Calle-Vallejo, M. C. Figueiredo and M. T. M. Koper, Structural principles to steer the selectivity of the electrocatalytic reduction of aliphatic ketones on platinum, Nat. Catal., 2019, 2, 243–250 CrossRef CAS.
- C. J. Bondue and M. T. M. Koper, Electrochemical reduction of the carbonyl functional group: the importance of adsorption geometry, molecular structure, and electrode surface structure, J. Am. Chem. Soc., 2019, 141, 12071–12078 CrossRef CAS PubMed.
- Y. Kwon, Y. Y. Birdja, S. Raoufmoghaddam and M. T. M. Koper, Electrocatalytic hydrogenation of 5-hydroxymethylfurfural in acidic solution, ChemSusChem, 2015, 8, 1745–1751 CrossRef CAS PubMed.
- J. Tan, W. Zhang, Y. Shu, H. Lu, Y. Tang and Q. Gao, Interlayer engineering of molybdenum disulfide toward efficient electrocatalytic hydrogenation, Sci. Bull., 2021, 66, 1003–1012 CrossRef CAS.
- J. Xie, J. Zhang, S. Li, F. Grote, X. Zhang, H. Zhang, R. Wang, Y. Lei, B. Pan and Y. Xie, Controllable disorder engineering in oxygen-incorporated MoS2 ultrathin nanosheets for efficient hydrogen evolution, J. Am. Chem. Soc., 2013, 135, 17881–17888 CrossRef CAS.
- J. Xie, H. Zhang, S. Li, R. Wang, X. Sun, M. Zhou, J. Zhou, X. W. Lou and Y. Xie, Defect-rich MoS2 ultrathin nanosheets with additional active edge sites for enhanced electrocatalytic hydrogen evolution, Adv. Mater., 2013, 25, 5807–5813 CrossRef CAS PubMed.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, MoS2 nanoparticles grown on graphene: an advanced catalyst for the hydrogen evolution reaction, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed.
- J. V. Lauritsen, J. Kibsgaard, S. Helveg, H. Topsøe, B. S. Clausen, E. Lægsgaard and F. Besenbacher, Size-dependent structure of MoS2 nanocrystals, Nat. Nanotechnol., 2007, 2, 53–58 CrossRef CAS PubMed.
- A. Albu-Yaron, M. Levy, R. Tenne, R. Popovitz-Biro, M. Weidenbach, M. Bar-Sadan, L. Houben, A. N. Enyashin, G. Seifert, D. Feuermann, E. A. Katz and J. M. Gordon, MoS2 hybrid nanostructures: from octahedral to quasi-spherical shells within individual nanoparticles, Angew. Chem., Int. Ed., 2011, 50, 1810–1814 CrossRef CAS.
- C. B. Roxlo, H. W. Deckman, J. Gland, S. D. Cameron and R. R. Chianelli, Edge surfaces in lithographically textured molybdenum disulfide, Science, 1987, 235, 1629–1631 CrossRef CAS PubMed.
- T. Wu, G. Wang, X. Zhu, P. Liu, X. Zhang, H. Zhang, Y. Zhang and H. Zhao, Growth and in situ transformation of TiO2 and HTiOF3 crystals on chitosan-polyvinyl alcohol co-polymer substrates under vapor phase hydrothermal conditions, Nano Res., 2016, 9, 745–754 CrossRef CAS.
- T. Wu, X. Zhu, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Vapor-phase hydrothermal growth of single crystalline NiS2 nanostructure film on carbon fiber cloth for electrocatalytic oxidation of alcohols to ketones and simultaneous H2 evolution, Nano Res., 2018, 11, 1004–1017 CrossRef CAS.
- T. Wu, M. Han, X. Zhu, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Experimental and theoretical understanding on electrochemical activation and inactivation processes of Nb3O7(OH) for ambient electrosynthesis of NH3, J. Mater. Chem. A, 2019, 7, 16969–16978 RSC.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Hafner, Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- Y. Sun, X. Hu, W. Luo and Y. Huang, Self-assembled hierarchical MoO2/graphene nanoarchitectures and their application as a high-performance anode material for lithium-ion batteries, ACS Nano, 2011, 5, 7100–7107 CrossRef CAS PubMed.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Engineering the surface structure of MoS2 to preferentially expose active edge sites for electrocatalysis, Nat. Mater., 2012, 11, 963–969 CrossRef CAS PubMed.
- W. J. Xie, X. Li and F. J. Zhang, Mo-vacancy induced high performance for photocatalytic hydrogen production over MoS2 nanosheets cocatalyst, Chem. Phys. Lett., 2020, 746, 137276 CrossRef CAS.
- J. Masa, C. Andronescu and W. Schuhmann, Electrocatalysis as the nexus for sustainable renewable energy: the gordian knot of activity, stability, and selectivity, Angew. Chem., Int. Ed., 2020, 59, 15298–15312 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional SEM images and electrochemical. See DOI: https://doi.org/10.1039/d2ma00842d |
| This journal is © The Royal Society of Chemistry 2022 |