
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLocally-ordered A-site vacancy assisted photoluminescence enhancement in simply rare-earth doped perovskite oxide†
Lei
Xia
 a,
Jiyang
Xie
*ab and
Wanbiao
Hu
*ab
a,
Jiyang
Xie
*ab and
Wanbiao
Hu
*ab
aKey Laboratory of LCR Materials and Devices of Yunnan Province, National Center for International Research on Photoelectric and Energy Materials, School of Materials and Energy, Yunnan University, Kunming 650091, P. R. China. E-mail: xiejiyang@ynu.edu.cn; huwanbiao@ynu.edu.cn
bElectron Microscopy Center, Yunnan University, Kunming 650091, P. R. China
First published on 4th October 2022
Abstract
Perovskite oxide based phosphors are widely used in luminous applications. Enhanced photoluminescence (PL) performances in terms of, for example, combining multiple activators, introducing absorption channels, and constructing defect states have been pursued over the long term. Herein, we reveal a new route to improving PL excitation and emission via forming locally-ordered A-site vacancies in simple rare-earth-doped perovskite oxide. An A-site Sm3+ activator and deliberate A-site vacancy (VA) are chosen to synthesize orthorhombically structured Ca1–1.5xSmx□0.5xTiO3 (VA-CST-x) perovskite phosphors, which demonstrate a 2.1-fold larger PL intensity than CaTiO3:Sm3+ phosphors without A-site vacancies (CST-x). VA-CST-x brings about weak superlattice satellites with the reflection vector 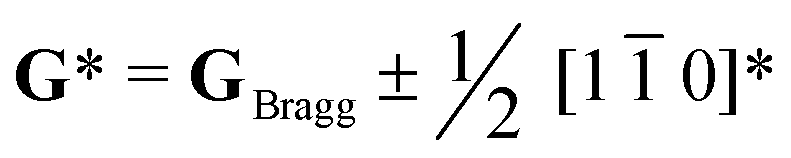 and apparent diffuse streaking along the crystallographic a and b directions, which combines with integrated differential phase contrast (iDPC)-STEM to suggest the formation of local A-site vacancy ordering along the 〈1
and apparent diffuse streaking along the crystallographic a and b directions, which combines with integrated differential phase contrast (iDPC)-STEM to suggest the formation of local A-site vacancy ordering along the 〈1 ![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0〉* direction. As a result of the Sm3+ dopants and VA, PL excitation (408 nm) and emissions (565, 600, 647, 710 nm) are greatly upgraded in terms of enabling charge exchanges between [Ca/Sm/VAOn] (n < 12) and [CaO12] complex clusters. In conclusion, this work confirms the formation of locally-ordered A-site vacancies in rare-earth-doped perovskite, which could be a new route for enhancing the photoluminescence property.
0〉* direction. As a result of the Sm3+ dopants and VA, PL excitation (408 nm) and emissions (565, 600, 647, 710 nm) are greatly upgraded in terms of enabling charge exchanges between [Ca/Sm/VAOn] (n < 12) and [CaO12] complex clusters. In conclusion, this work confirms the formation of locally-ordered A-site vacancies in rare-earth-doped perovskite, which could be a new route for enhancing the photoluminescence property.
1. Introduction
Phosphor-converting light-emitting diodes (pc-LEDs) are emerging as an essential solid-state light source for next-generation lighting and display systems because of their significant merits, e.g. high efficiency, low energy consumption, long lifetime and environmental compatibility.1,2 As the core of pc-LEDs, phosphors have been extensively explored for gaining desirable photoluminescence (PL) performances.3–7 Rare-earth (Ln3+) doped phosphors are extremely intriguing in this regard with large Stokes/anti-Stokes shifts, sharp band emissions, superior photostability and upconversion capability etc.8–10 A general pathway to achieve excellent phosphors involves compositional and structural modulations of the Ln3+ dopant ions and the matrix materials simultaneously.11 It is well known that all their luminescent properties are subject to intraconfigurational f–f transitions of Ln3+, which, however, are Laporte forbidden.12 Fortunately, when a certain number of uneven components of the crystal field and local structural perturbations are incorporated, partial intraconfigurational transitions could become allowed.13Perovskite-structured oxides, due to the coupled multiple energy landscapes in, for example, electric, strain, polarization and A/B-site crystal fields for modulating crystal symmetry, have been considered a crucial sort of PL matrix in this respect.14–17 Moreover, rather high tunability on structures, compositions, and properties is offered, even if only for simple ABO3-type perovskites.18,19 Great efforts have been made to improve the PL properties of Ln3+-doped ABO3-perovskite phosphors, such as excitation/emission intensity, luminescent lifetime, and thermal stability. Different routes, e.g. co-doping, size control, annealing, surface modification and core–shell structures have been explored.19–21 Simply doping Ln3+ into perovskites as well as other structures has been the most extensive action, which indeed brings about optimization of the PL property and also many new properties. Beyond single doping with Ln3+, co-doping Pr3+ and Bi3+ in CaTiO3 could greatly enhance UV excitation in the spectral range of 370–390 nm for PL enhancement.22 Some non-Ln3+ ions, enabling multiple-site occupations in matrix crystals (e.g. Cr3+)23 or giving rise to intense new absorption channels (e.g. Mn2+, Bi3+),24 could prominently upgrade PL emissions and efficiencies. With regard to responsible principles that are generally accepted, these doping effects, without doubt, could suppress non-radiative recombinations while radiative recombination rates are increased in terms of localizing charge carriers.25
When significantly increased densities of photonic states via heteroionic doping that directly correlates with PL improvements are emphasized and thoroughly attributed, the underlying variations in the local structures e.g. lattice distortion, defects, ordering–disordering etc. are rarely addressed. As a matter of the fact, whatever the most extensive Ln3+ or other ionic substitutions, e.g. Na+, Ag+ in A-sites or Al3+, Mg2+, Zn2+ for B-site substitutions,22 defect states are inevitably encountered but their roles are seemingly neglected. Unlike A3+B3+O3-type perovskite, LaInO3, for example, exhibits a broadband self-trapped exciton emission, where the doped Ln3+ ions do not require charge compensation, and can be excited through host sensitization.26 The induced complex structures and charges mentioned in the more general perovskite A2+B4+O3 usually result in abundant trap-assisted non-radiative recombinations, and their rates can be reduced through local recrystallization and/or chemical passivation of the defects under certain circumstances.27 Apart from ABO3-structured perovskite materials, ABO4 structures e.g. CaMoO4 of scheelite type are also promising phosphors. Much effort has been put into the PL of CaMoO4 doped with different Ln3+ ions, e.g. Eu3+-doped CaMoO4 in particular, has been investigated extensively as a red-emitting phosphor.28–30 Despite the improved red emissions (mainly in systems involving Al3+, Eu3+ or Mn2+/4+) or afterglows that are attributed to the formation of trapping centers generally being passable,31–34 heteroionic doping-induced structural variations have not been inferred. As a more extensive question, how the local structures, e.g. lattice distortion, defects, and ordering–disordering, contribute to the PL phenomenon as well as the associated PL mechanism has not yet been established, but is highly necessary and timely.
Here in this work, we reveal a new route to improving PL excitation and emission by forming locally-ordered A-site vacancies in a simple Ln3+-doped perovskite oxide. Orthorhombically structured Ca1–1.5xSmx□0.5xTiO3 (VA-CST-x) perovskite phosphors are synthesized by choosing an A-site Sm3+ activator and deliberately introducing an A-site vacancy (VA). Structural and spectroscopic characterizations are indicative of the weak superlattice structure along the 〈1 0〉* direction with the formation of local VA ordering. This enables charge exchanges between [Ca/Sm/VAOn] (n < 12) and [CaO12] complex clusters for enhanced PL properties. The relation between the formation of local VA ordering and PL enhancement is comprehensively studied.
2. Experimental procedure
2.1 Sample preparation
A-Site Sm3+-doped CaTiO3 phosphors, e.g. both Ca1–1.5xSmx□0.5xTiO3 (VA-CST-x) and Ca1−xSmxTiO3 (CST-x), were prepared by a solid-state reaction method. The raw materials used for the experiments were Sm2O3 (Sinoreagent, 99.99%), TiO2 (Sinoreagent, 99.9%), and CaCO3 (Aladdin, 99.99%) powders that had been stored long-term at 150 °C to avoid water absorption. The raw materials were weighted stoichiometrically and mixed thoroughly for 1 h in an agate mortar. The milled mixtures were dried and transferred into corundum crucibles and pre-calcined at 1250 °C for 4 h. Afterwards, the pre-calcined mixtures were ground into powders. The powders were sintered at 1450 °C for 4 h in air. Finally, the naturally cooled samples were ground thoroughly to obtain the phosphor samples.2.2 Structure and property characterizations
Phase purity and structure were examined by powder X-ray diffraction (XRD) (Rigaku SmartLab SE, Cu-Ka radiation) in a 2θ range from 10 to 90°. In order to detect the low-level impurity phase, an integral linear detector was employed with a very slow scanning step of 0.01 during the XRD measurements. The atomic-structure information was observed by a high-resolution scanning transmission electron microscope (STEM) equipped with a double-aberration corrector in the objective and condenser lens system (Thermofisher FEI Spectra 300). The integrated differential phase contrast (iDPC) STEM technique was performed to identify the atomic and defect arrangements. The optical absorption spectra were measured on a UV-vis spectrometer (PerkinElmer Lambda 650S). The photoluminescence properties e.g. excitation/emission spectra and lifetimes, were recorded on a fluorescence spectrometer (Edinburgh Instruments FS5) equipped with a xenon lamp. Color coordinates were calculated with CIE1931. Additional emission spectra in the temperature range from 20 °C to 300 °C were recorded using an FL970 fluorescence spectrometer (Shanghai Techcomp Instrument Ltd).3. Results
3.1 Enhanced photoluminescent by A-site vacancy design
It has long been recognized that photoluminescence (PL) emission could be influenced and modulated by the cation vacancies in a solid phosphor, but apparent experimental confirmations have not yet been established. To demonstrate clear evidence in this respect, a series of Sm3+-doped CaTiO3 perovskite based phosphors with different Sm3+ doping levels (x = 0.01, 0.02, 0.05 and 0.1) with the creation of A-site cation vacancies, i.e. Ca1–1.5xSmx□0.5xTiO3 (VA-CST-x), and without any vacancies, i.e. Ca1−xSmxTiO3 (CST-x), were therefore designed and a direct comparison of the PL emission was explored, as shown in Fig. 1. The photoluminescence excitation (PLE) spectra (Fig. 1a and c) were monitored at λem = 647 nm, which mainly show seven excitation peaks that are related to the 4f–4f transition of Sm3+ ions: 6H5/2 → 4D3/2 (365 nm), 6H5/2 → 4D1/2 (379 nm), strongest 6H5/2 → 4F7/2 (408 nm), 6H5/2 → (6P, 4P)5/2 (421 nm), 6H5/2 → 4G9/2 (441 nm), 6H5/2 → 4I13/2 (464 nm) and 6H5/2 → 4I11/2 (481 nm).35 The emission spectra of VA-CST-x (Fig. 1b) and CST-x (Fig. 1d) under a maximum excitation of λex = 408 nm consist of four bands with different intensities ranging from 550 to 750 nm, all corresponding to the characteristic emission of Sm3+ ions: 565 nm for 4G5/2 → 6H5/2, 600 nm for 4G5/2 → 6H7/2, 647 nm for the strongest 4G5/2 → 6H9/2, and 710 nm for 4G5/2 → 6H11/2.36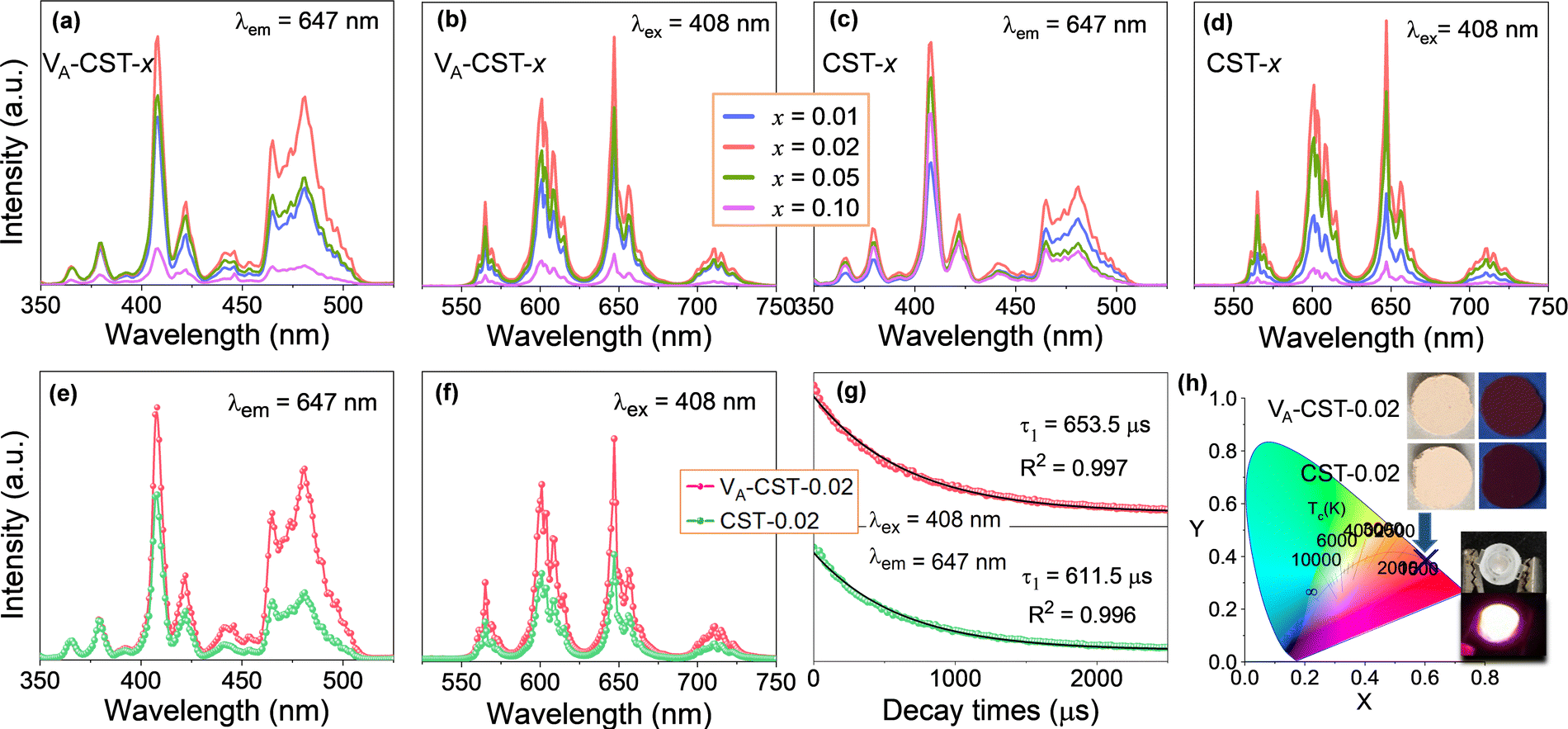 | ||
| Fig. 1 Photoluminescent properties of A-site Sm3+-doped CaTiO3 phosphors with A-site vacancy (VA-CST-x) and without vacancy (CST-x) (x = 0.01, 0.02, 0.05 and 0.1). (a) Excitation spectra of VA-CST-x; (b) emission spectra of VA-CST-x; (c) excitation spectra of CST-x; (d) emission spectra of CST-x; (e) excitation spectra of VA-CST-0.02 and CST-0.02; (f) emission spectra of VA-CST-0.02 and CST-0.02; (g) PL decay curves of VA-CST-0.02 and CST-0.02; (h) CIE chromaticity coordinates of VA-CST-0.02 and CST-0.02 excited at 408 nm. | ||
It is quite clear that these excitations and emissions are all associated with the luminescence energy level characteristics of rare-earth Sm3+ dopants. Nevertheless, the emission and excitation intensities for both VA-CST-x and CST-x phosphors are strongly dependent on the Sm3+-doping levels (x), i.e. they first increase and subsequently decrease with increasing x, reaching a maximum at x = 0.02. Furthermore, when taking the strongest excitation and emission of both phosphors (x = 0.02) into consideration, it is interesting that VA-CST-0.02 displays much higher intensities in both PL excitation (Fig. 1e) and emission (Fig. 1f) than those of CST-0.02. In particular, much stronger excitations for VA-CST-x in the range of 460–520 nm are presented (Fig. 1a and e). To conclude, both phosphors should be subject to a similar luminescence generation and quenching process, which, however, is significantly governed by the deliberately introduced A-site cation vacancies.
As a key feature of fluorescent materials, the luminescence lifetimes in terms of the room-temperature decay curves for both VA-CST-0.02 and CST-0.02 phosphors were further investigated. The curves were recorded with excitation at 408 nm and monitoring at 647 nm, as shown in Fig. 1g. It was found that the luminescence attenuation curves can be well fitted by the following first-order exponential function:37
It = A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(t/τ) exp(t/τ) | (1) |
The orange-red emission of CaTiO3:Sm3+ phosphors from the PL emission spectra (Fig. 1b, d and f) suggests a potential application in LEDs. As greatly needed, the main and most important features, i.e. the chromaticity coordinates and color purity (CP), were investigated. Fig. 1h depicts the Commission International de l’Eclairage (CIE) 1931 chromaticity coordinates (x, y) of VA-CST-0.02 and CST-0.02 phosphors excited by 408 nm, which are calculated to be (0.6164, 0.3829) and (0.6109, 0.3878), respectively. This means that the emission colors are located in the reddish-orange region of the standard CIE diagram, consistent with the results from the PL emission spectra. Along with the practical lighting effect, the luminescence features for both phosphors under sunlight and 365 nm UV light (top inset of Fig. 1h), as well as lighting a real LED device (bottom inset of Fig. 1h) using the VA-CST-0.02 phosphor excited by a 390 nm UV chip are demonstrated, revealing quite promising luminescence performance.
To evaluate the color purity (CP) of the phosphors, the following equation is adopted:38
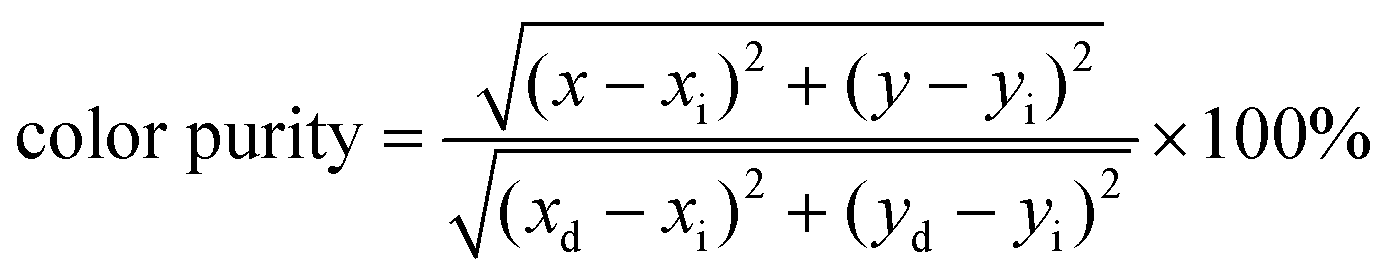 | (2) |
| Sample | Chromaticity coordinates | Color purity (CP) (%) | |
|---|---|---|---|
| x | y | ||
| VA-CST-0.02 | 0.6164 | 0.3829 | 78.23 |
| CST-0.02 | 0.6109 | 0.3878 | 77.00 |
Photoluminescence thermal stability, another technological parameter that is extremely important for phosphors,39 was further explored. To this end, the VA-CST-0.02 phosphor showing the best PL property, and also its two strongest transitions, i.e.4G5/2 → 6H7/2 (600 nm) and 4G5/2 → 6H9/2 (647 nm), were mainly used for the study (Fig. 2). It is generally documented that the PL emission intensity for a phosphor would experience a reduction all the way with rising temperatures due to the substantial increase in the non-radiative relaxation rate.40 However, with increasing measurement temperatures, the PL intensity of the two transitions (600 and 647 nm) for VA-CST-0.02 shows a similar trend, i.e. it first increases and then decreases, giving rise to a maximum at 100 °C. Such an anti-thermal-quenching (anti-TQ) property, therefore, cannot simply be attributed to non-radiative relaxation; instead, A-site vacancy assisted excitation that may compensate for non-radiative hopping at high temperatures could be responsible. In other words, such vacancies may form defect energy levels or new luminescence centers, and they will act as energy traps to improve the luminescence thermal stability of phosphors.41,42 The existence of intermediate energy states inside the band gap was confirmed by characterization with UV-vis absorption spectra (which will be addressed later). As a consequence, the VA-CST-0.02 phosphor could maintain a rather high PL emission intensity under high-temperature photoexcitation, i.e. maximum intensities of 130% (100 °C), and 100% (140–145 °C) as indicated in the insets to Fig. 2, compared to that at room temperature, indicative of great superiority in thermal stability for high-temperature phosphors.
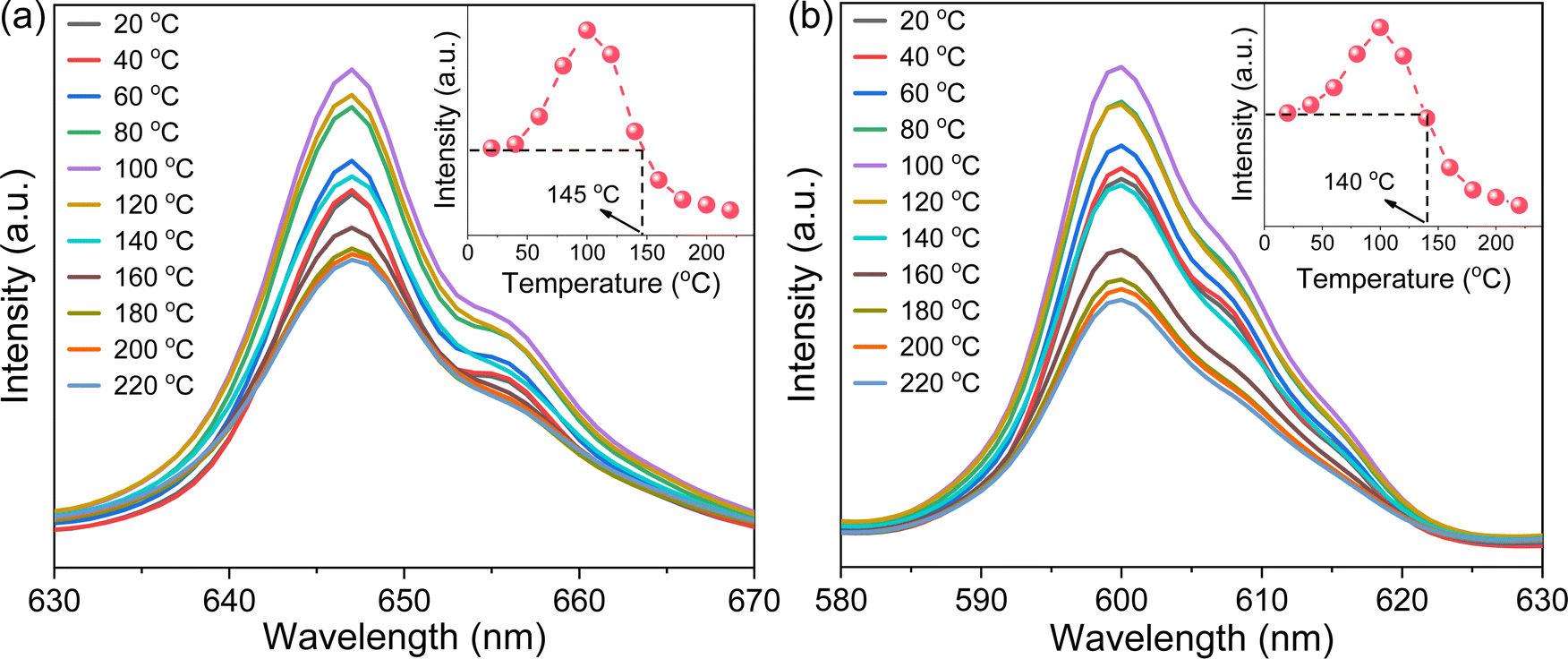 | ||
| Fig. 2 Photoluminescence thermal stability of the VA-CST-0.02 phosphor characterized by the PL emission intensity as a function of temperature. (a) The strongest PL emission spectra (4G5/2 → 6H9/2 centered at 647 nm); (b) the second strongest PL emission spectra (4G5/2 → 6H7/2 centered at 600 nm). The insets to a and b summarize the intensity of the emission centers as a function of temperature. Note that the highest intensities for both series of spectra are normalized to clearly demonstrate the variation in intensity with temperature. | ||
3.2 Phase structure and A-site vacancy ordering
As A-site cation vacancies were deliberately designed and their significant impacts on the photoluminescence property were steadily revealed, the structural features of vacancies were further explored. From the XRD patterns in the form of logarithmic intensity (Fig. 3a), both CST-0.02 and VA-CST-0.02 form in a single phase without any impurities, and crystallize in an orthorhombic structure with a space group of Pbnm. Moreover, the chemical composition determinations at a relatively high doping level i.e. VA-CST-0.1 through careful SEM-EDS (Fig. S2, ESI†) suggest that all the cations, especially the dopants Sm3+, are uniformly distributed, again confirming their successful incorporation. XRD structural Rietveld refinement (Fig. 3b) indicates that Sm3+ doping leads to lattice changes, as clearly evidenced by the refined lattice parameters (Fig. 3c). The existence of an A-site vacancy apparently affects the lattice constants a, b, c as well as their variations being obviously different to the normal Sm3+ doping (CST-0.02). The A-site vacancies in VA-CST-0.02, on the one hand, result in smaller lattice constants a, b, c than those of CST-0.02, and on the other hand, cause anisotropic elongation (for b) or shortening (for a and c), which would bring about serious lattice distortion and further influence the PL property.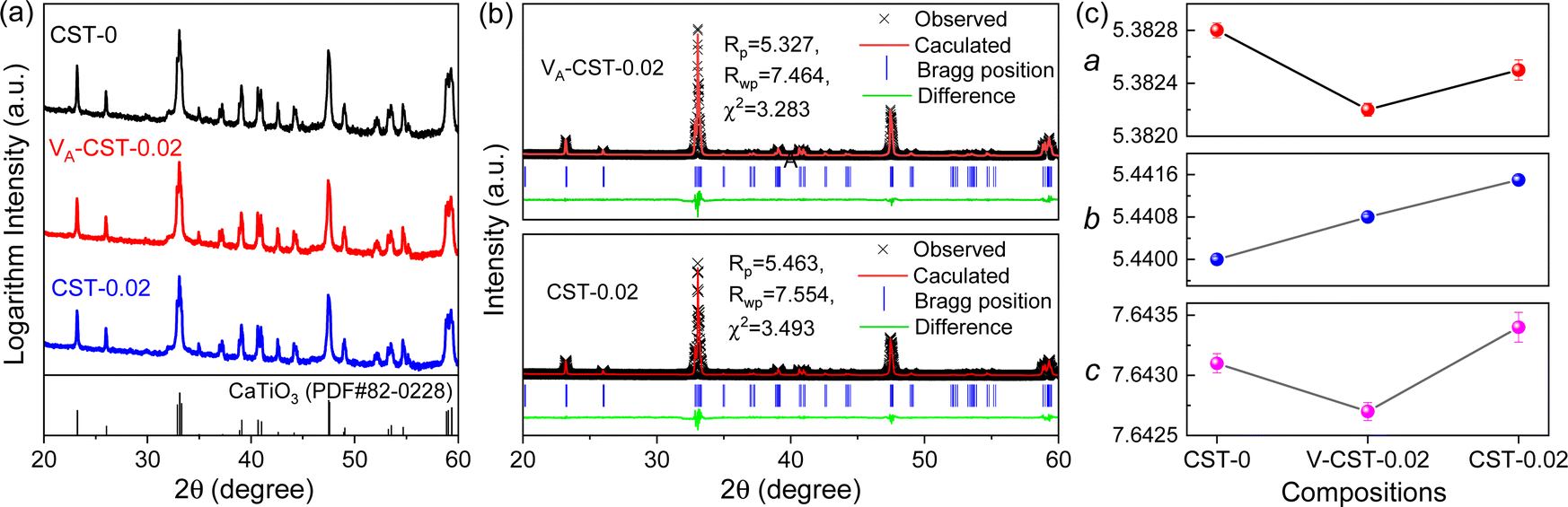 | ||
| Fig. 3 Phase and structural characterizations of the undoped and A-site Sm3+-doped CaTiO3. (a) Measured XRD patterns, which are presented in the form of logarithmic intensity to clearly demonstrate the tiny diffraction lines; (b) Rietveld-refined XRD patterns; (c) unit cell parameters (a, b, c) as a function of composition. | ||
To further clarify the status and structural features of A-site vacancies at the atomic level, scanning transmission electron microscopy coupled high-angle annular dark field (STEM-HAADF) was performed on VA-CST-0.02. Fig. 4a shows the STEM-HADDF atomic distributions imaged along the [001] orientation, which clearly present rather regular atomic arrangements for both main cations Ca and Ti (Fig. 4b). A further Fourier transform (FFT) pattern (Fig. 4c) indicates, in addition to the normal Bragg diffractions of orthorhombic CaTiO3 (S.G.: Pbnm), the presence of apparent diffuse streaking along the crystallographic a and b directions (highlighted by the dashed arrows). What is most striking is the appearance of extra diffraction spots, e.g. (3/2 1/2 0), (1/2 −1/2 0) and (−1/2 −3/2 0), which can be ascribed to weak superlattice satellites with the reflection vector 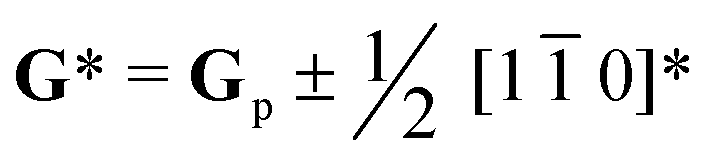 , where Gp represents the Bragg reflections of the CaTiO3 average structure. As a consequence, with regard to the underlying local ordering, these superlattice reflections along the 〈1 0〉* direction of reciprocal space must be associated with the local A-site ordering, i.e. cations and/or vacancies. The integrated differential phase contrast (iDPC)-STEM technique was further explored for confirmation. As shown in Fig. 4d, there are indeed clear discrepancies in different columns of the atoms, which correspond to local ordering structures, i.e. higher VCa (Ca vacancies) and lower VCa as well as isolated Ti atoms (diagnosed in Fig. 4e). It is clear that A-site (Ca2+) vacancies tend to form a superlattice-like structure, i.e. …lower VA–Ti – higher VA–Ti – lower VA–Ti – higher VA…. The distribution of higher VA, lower VA and Ti isolated atoms can be seen clearly from the iDPC image and intensity profiles (Fig. S3, ESI†). This is very consistent with the ordered arrangements of superlattice-like structure where extra diffraction spots appear in the FFT image. (1/2 −1/2 0) corresponds to the regular arrangement of Ti – higher VA–Ti – lower VA–Ti. (3/2 1/2 0) and (−1/2 −3/2 0) should correspond to lattice planes of higher VA and lower VA, respectively. To clearly trace the structural fluctuations, Fig. 3f shows the structures around the dopants Sm3+ and VA in CaTiO3:Sm3+. The introduction of SmCa and VA will cause at least VA-neighbored (or connected) sixteen CaO12 polyhedrons, eight TiO6 octahedra, and two 1SmCa-in-8TiO6 clusters to move toward the VA. When the A-site vacancies are introduced, orthorhombic anisotropy may drive the Sm3+ to occupy the A sites in 〈1 1 0〉 directions, and nearby, the vacancies tend to situate along 〈1 0〉 directions to compensate for the charges and local distortions, which therefore expands the distance between Sm3+ ions. The production of A-site vacancies naturally leads to overall lattice contraction i.e. smaller lattice constants a, b, c than those without A-site vacancies (CST-x), but anisotropic occupancies for Sm3+ dopants and A-site vacancies would result in discrepant changes for a and b, rightly analogous to the structural refinement (Fig. 3c).
, where Gp represents the Bragg reflections of the CaTiO3 average structure. As a consequence, with regard to the underlying local ordering, these superlattice reflections along the 〈1 0〉* direction of reciprocal space must be associated with the local A-site ordering, i.e. cations and/or vacancies. The integrated differential phase contrast (iDPC)-STEM technique was further explored for confirmation. As shown in Fig. 4d, there are indeed clear discrepancies in different columns of the atoms, which correspond to local ordering structures, i.e. higher VCa (Ca vacancies) and lower VCa as well as isolated Ti atoms (diagnosed in Fig. 4e). It is clear that A-site (Ca2+) vacancies tend to form a superlattice-like structure, i.e. …lower VA–Ti – higher VA–Ti – lower VA–Ti – higher VA…. The distribution of higher VA, lower VA and Ti isolated atoms can be seen clearly from the iDPC image and intensity profiles (Fig. S3, ESI†). This is very consistent with the ordered arrangements of superlattice-like structure where extra diffraction spots appear in the FFT image. (1/2 −1/2 0) corresponds to the regular arrangement of Ti – higher VA–Ti – lower VA–Ti. (3/2 1/2 0) and (−1/2 −3/2 0) should correspond to lattice planes of higher VA and lower VA, respectively. To clearly trace the structural fluctuations, Fig. 3f shows the structures around the dopants Sm3+ and VA in CaTiO3:Sm3+. The introduction of SmCa and VA will cause at least VA-neighbored (or connected) sixteen CaO12 polyhedrons, eight TiO6 octahedra, and two 1SmCa-in-8TiO6 clusters to move toward the VA. When the A-site vacancies are introduced, orthorhombic anisotropy may drive the Sm3+ to occupy the A sites in 〈1 1 0〉 directions, and nearby, the vacancies tend to situate along 〈1 0〉 directions to compensate for the charges and local distortions, which therefore expands the distance between Sm3+ ions. The production of A-site vacancies naturally leads to overall lattice contraction i.e. smaller lattice constants a, b, c than those without A-site vacancies (CST-x), but anisotropic occupancies for Sm3+ dopants and A-site vacancies would result in discrepant changes for a and b, rightly analogous to the structural refinement (Fig. 3c).
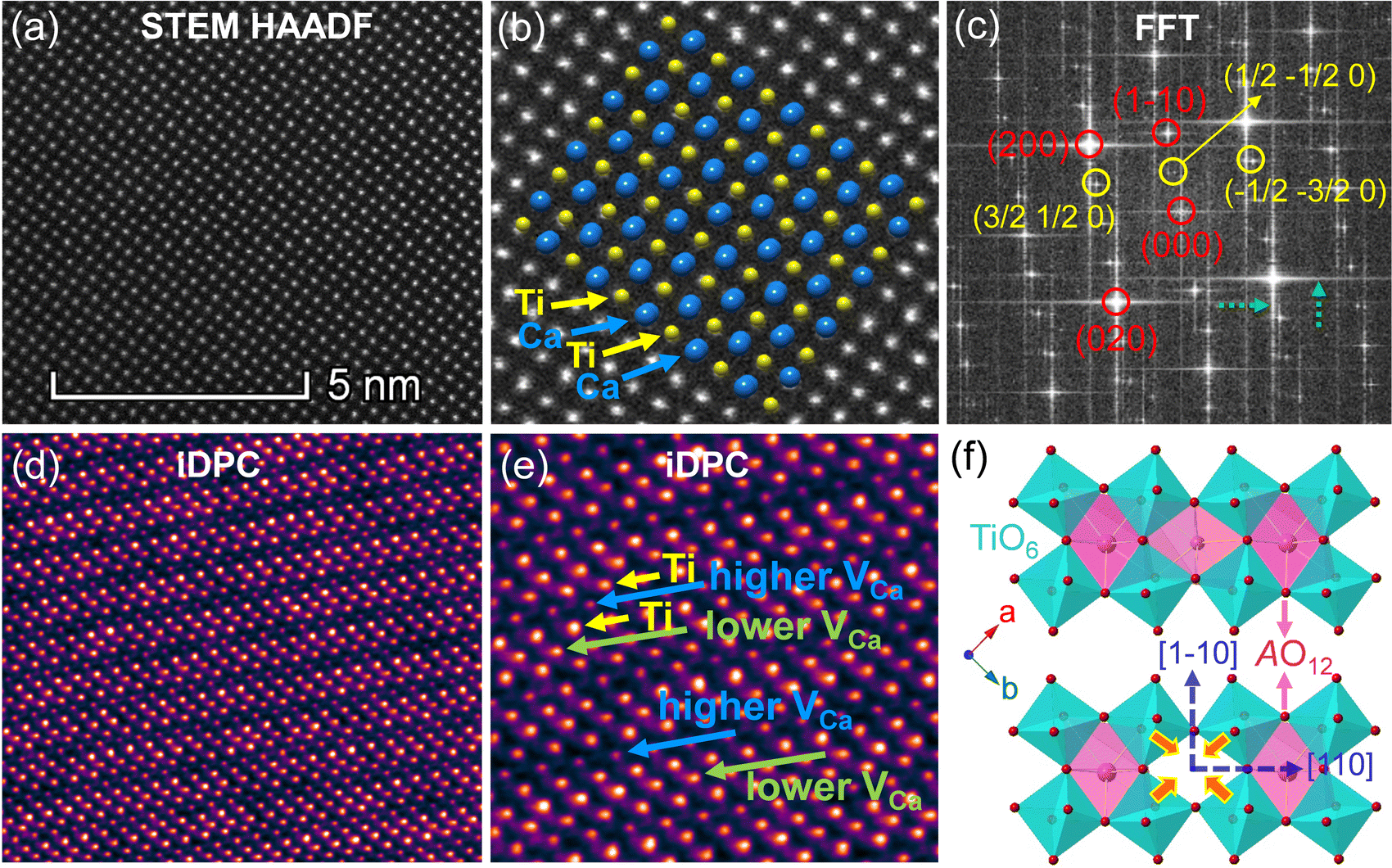 | ||
| Fig. 4 Atomic-scale structural characterizations of VA-CST-0.02. (a) STEM-HADDF image in the [001] zone axis; (b) STEM-HADDF image with atomic arrangements; (c) FFT pattern of STEM-HADDF image; (d) iDPC-STEM image in the [001] zone; (e) partial enlargement of (d); (f) the structures around dopants Sm3+ and VA in CST-x and VA-CST-x. | ||
When the A-site Sm3+ and vacancies are introduced, an apparent local ordering–disordering could occur in the A site. As crucial evidence for this, UV-Vis absorption spectra that confirm the existence of complex clusters in terms of the different electronic levels were collected (Fig. 5). According to the Wood–Tauc method with a direct band transition of (αhν)2 − (hν) curves for Pbnm-structured CaTiO3, where α is the absorbance and h is the Planck constant, the optical band gaps are obtained as 3.52 ± 0.02 eV. Introducing A-site Sm3+ only slightly red-shifts the absorption edge, but obviously upgrades the visible-region absorption tails, which are further enhanced simultaneously with A-site Sm3+ and vacancies, i.e. in VA-CST-0.02. This suggests the existence of intermediary states inside the band gap, consistent with the interpretation that the exponential optical absorption edge is controlled by the degree of structural ordering–disordering. The local disordering is related to a change from the regular 12-fold coordination [CaO12] to the lower-fold coordination [Ca/Sm/VAOn] (n < 12), which has been proven by experimental XANES for the Ca K-edge in CaTiO3 with [TiO5·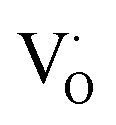 ] and/or [CaO11·
] and/or [CaO11· ] complex clusters.43 It is documented that the modified lattice in the A-site can strongly affect the intensity of PL emission due to charge exchanges between [CaOn] (n < 12) and [CaO12] complex clusters. The different extents of structural disordering in VA-CST-0.02 and CST-0.02 would bring about distinct PL phenomena. The presence of different A-site vacancy orderings (lower VA and higher VA) causes structural disordering and atomic displacement, yielding gradients in local charges among the lattice clusters. As a result of enhancing the intermediate levels in the band gap that is generally essential for PL emission, VA-CST-0.02 could present an enhanced PL intensity.
] complex clusters.43 It is documented that the modified lattice in the A-site can strongly affect the intensity of PL emission due to charge exchanges between [CaOn] (n < 12) and [CaO12] complex clusters. The different extents of structural disordering in VA-CST-0.02 and CST-0.02 would bring about distinct PL phenomena. The presence of different A-site vacancy orderings (lower VA and higher VA) causes structural disordering and atomic displacement, yielding gradients in local charges among the lattice clusters. As a result of enhancing the intermediate levels in the band gap that is generally essential for PL emission, VA-CST-0.02 could present an enhanced PL intensity.
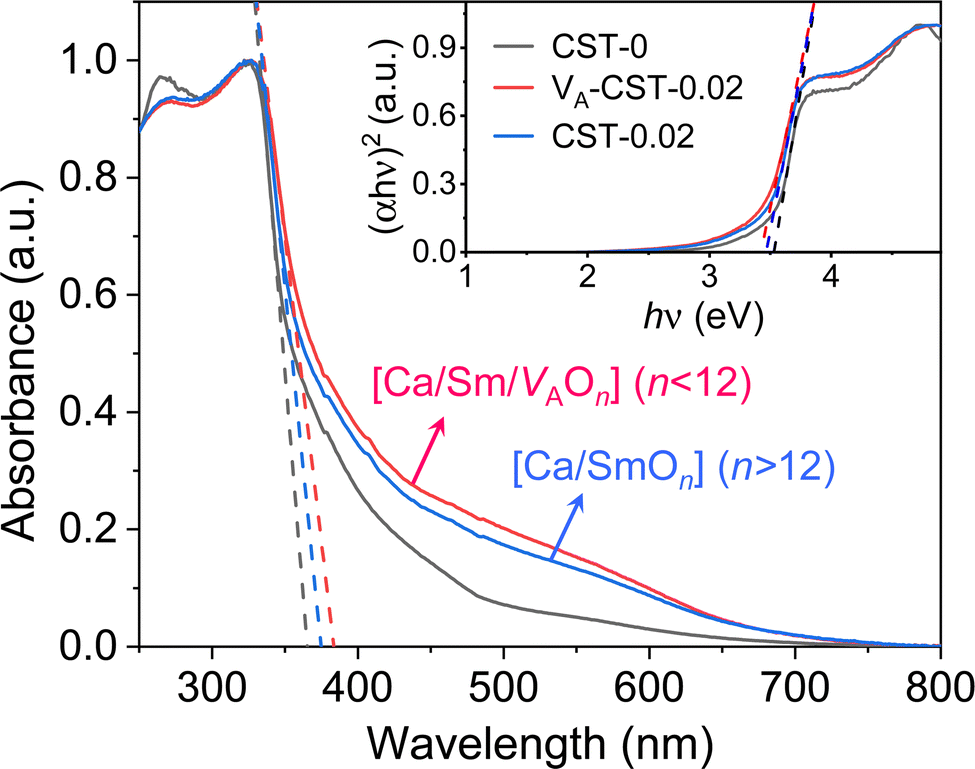 | ||
| Fig. 5 UV-Vis absorption spectra of CaTiO3:Sm3+. Inset shows plots of (αhν)2 − (hν) based on the direct optical band gap. | ||
4. Discussion
Luminescence quenching occurs in both VA-CST-x and CST-x phosphors with an Sm3+ doping level greater than x = 0.02 (Fig. 1). Upon experiencing the local structural changes that cause the differently enhanced PL emission for VA-CST-x, the energy transfer between rare earth ions in the transmission of non-radiative energy that is usually responsible for PL quenching should be taken into account, which is directly determined by the distance between active dopants, i.e. Sm3+ ions. The critical distance (Rc) associated with PL quenching can be estimated with the following equation suggested by Blasse et al.:44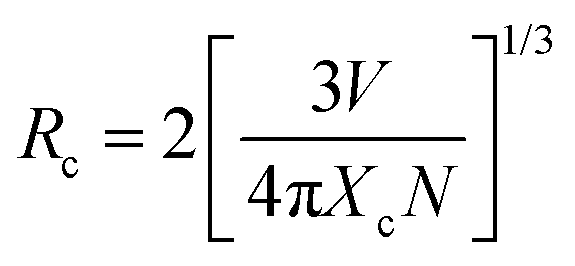 | (3) |
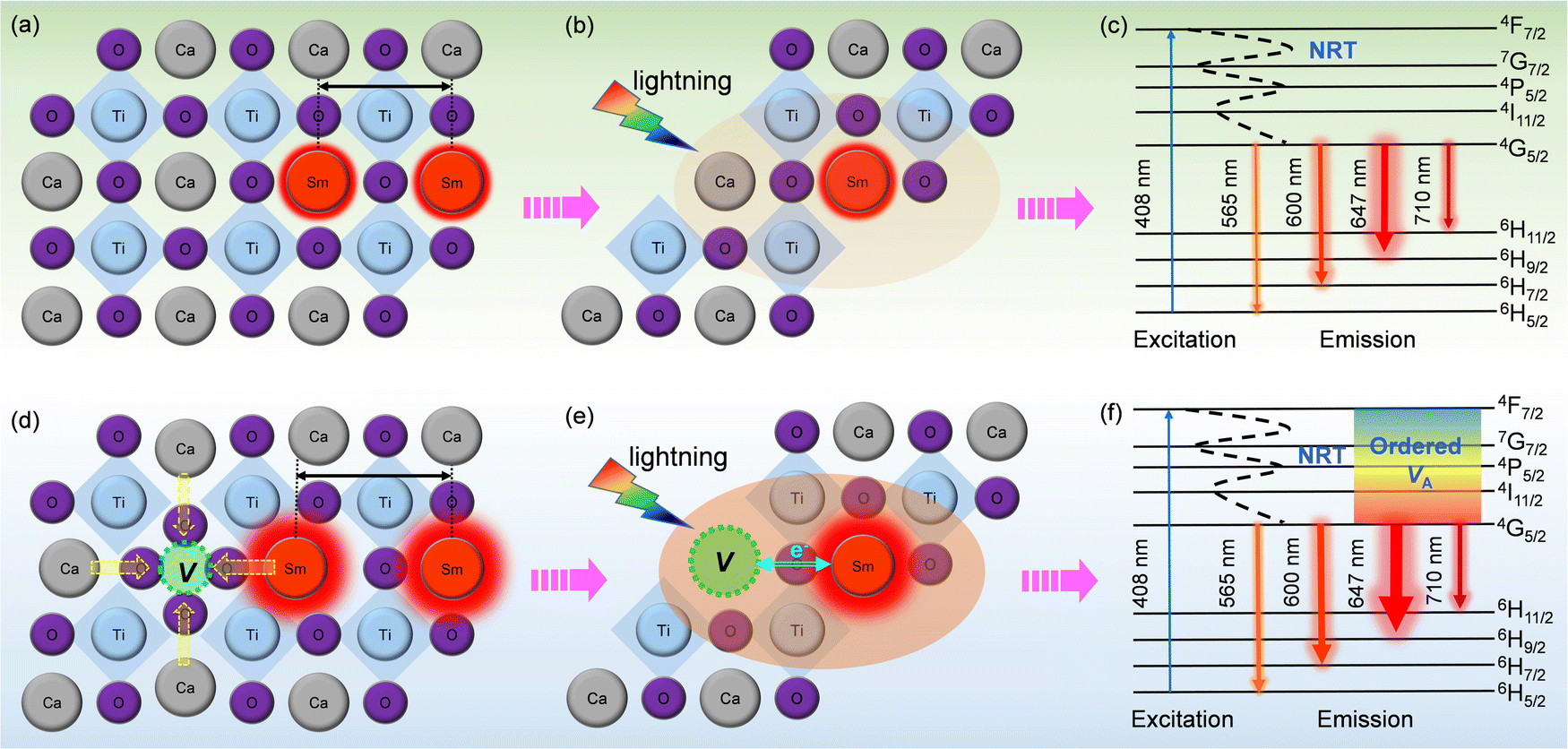 | ||
| Fig. 6 Schematic diagram of PL emission mechanisms. (a–c) CST-0.02; (d–f) VA-CST-0.02. “NRT” in c and f denotes “non-radiative transition”. | ||
To clarify the relationship between structure and photoluminescence as well as the photoluminescence mechanism involved, a simplified energy level diagram describing the dopant Sm3+ ions and the attraction of the A-site vacancy is given. As Sm3+ is the sole PL-active ion and the distance between them is so large, non-radiative transition (NRT) is responsible for PL emission for both CST-0.02 and VA-CST-0.02. Specifically, as shown in Fig. 6c and f, under light excitation (408 nm), the electrons of Sm3+ doped in the CaTiO3 host can be excited from the ground level to an excitation state, e.g. the 4F7/2 level. These electrons subsequently return into the lower stable energy level 4G5/2 through non-radiative transition, and finally return to the lower energy states 6H(5,7,9,11)/2 through radiative transition in terms of continuously transferring energy and orange-red light emission (Fig. 6c and f). Both CST-0.02 and VA-CST-0.02 should experience a similar NRT where the PL emissions have been enhanced by structural distortion or overall disordering, as indicated by the UV-Vis spectra (Fig. 5), but what is quite different is that for VA-CST-0.02, the locally ordered A-site vacancies, i.e. lower VA and higher VA revealed in Fig. 4, can further upgrade PL emission due to exchanges of charge between [Ca/Sm/VAOn] (n < 12) and [CaO12] complex clusters (Fig. 6e and f), where the ions around the vacancies will contract toward the vacancy direction, and the distance between rare-earth Sm3+ ions will be enlarged, weakening the interactions between the active luminescence ions (Fig. 6d).
5. Conclusion and outlook
To conclude, the enhancement in PL properties in rare-earth-doped perovskite was realized by the formation of locally-ordered A-site vacancies (VA). We have designed and successfully synthesized orthorhombically structured Ca1–1.5xSmx□0.5xTiO3 (VA-CST-x) perovskite phosphors with an A-site Sm3+ activator and deliberate VA, which demonstrate a 2.1-fold larger PL intensity than CaTiO3:Sm3+ phosphors without A-site vacancies (CST-x). Structural and spectral analysis results indicate that weak superlattice satellites with the reflection vector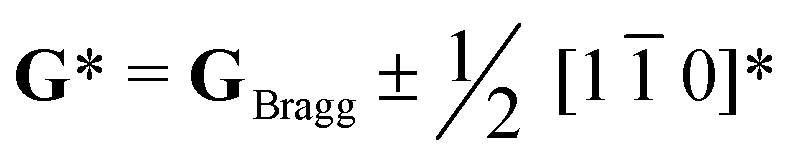 and apparent diffuse streaking along crystallographic a and b directions form in VA-CST-x. Further combination with iDPC-STEM analysis reveals local A-site vacancy ordering develops along the 〈1 0〉* direction. The PL intensities for both excitation and emission spectra, except for the contributions from normal non-radiative transitions, are additionally upgraded significantly in terms of the charge exchanges enabled between [Ca/Sm/VAOn] (n < 12) and [CaO12] complex clusters. The findings herein confirm the complicated local structures in rare-earth-doped perovskite oxide which could have a great influence on PL performance and, more importantly, they reveal a new route for forming locally-ordered A-site vacancies for improved PL property.
and apparent diffuse streaking along crystallographic a and b directions form in VA-CST-x. Further combination with iDPC-STEM analysis reveals local A-site vacancy ordering develops along the 〈1 0〉* direction. The PL intensities for both excitation and emission spectra, except for the contributions from normal non-radiative transitions, are additionally upgraded significantly in terms of the charge exchanges enabled between [Ca/Sm/VAOn] (n < 12) and [CaO12] complex clusters. The findings herein confirm the complicated local structures in rare-earth-doped perovskite oxide which could have a great influence on PL performance and, more importantly, they reveal a new route for forming locally-ordered A-site vacancies for improved PL property.
Data availability
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.Author contributions
Lei Xia conducted the experiments, analyzed the data and writing. Jiyang Xie carried out the XRD Rietveld refinement and TEM structure characterizations. Wanbiao Hu provided frequent input in the interpretation of all results, and revised the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the Natural Science Foundation of China (Grant No. 22175150 and U2002217) and the Key R&D program of Yunnan Province (Grant No. 2018BA068).References
- C. Feldmann, T. Justel, C. R. Ronda and P. J. Schmidt, Adv. Funct. Mater., 2003, 13, 511–516 CrossRef CAS.
- Y. Zhao, D. Peng, G. Bai, Y. Huang, S. Xu and J. Hao, Adv. Funct. Mater., 2021, 31, 2010265 CrossRef CAS.
- C. Cheng, L. Ning, X. Ke, M. S. Molokeev, Z. Wang, G. Zhou, Y.-C. Chuang and Z. Xia, Adv. Opt. Mater., 2019, 8, 1901608 CrossRef.
- J. Jin, Y. Peng, Y. Xu, K. Han, A. Zhang, X.-B. Yang and Z. Xia, Chem. Mater., 2022, 34, 5717–5725 CrossRef CAS.
- X. Zhou, J. Qiao and Z. Xia, Chem. Mater., 2021, 33, 1083–1098 CrossRef CAS.
- Q. Zhang, J. Li, W. Jiang, L. Lin, J. Ding, M. G. Brik, M. S. Molokeev, H. Y. Ni and M. Wu, J. Mater. Chem. C, 2021, 9, 11292–11298 RSC.
- W. U. Khan, L. Zhou, X. Li, W. Zhou, D. Khan, S.-I. Niaz and M. Wu, Chem. Eng. J., 2021, 410, 128455 CrossRef.
- Z. Xia and L. Quanlin, Prog. Mater. Sci., 2016, 84, 59–117 CrossRef CAS.
- W. Bin, Im, N. George, J. Kurzman, S. Brinkley, A. Mikhailovsky, J. Hu, B. F. Chmelka, S. P. DenBaars and R. Seshadri, Adv. Mater., 2011, 23, 2300–2305 CrossRef PubMed.
- J. Wang, R. Deng, M. A. MacDonald, B. Chen, J. Yuan, F. Wang, D. Chi, T. S. A. Hor, P. Zhang, G. Liu, Y. Han and X. Liu, Nat. Mater., 2014, 13, 157–162 CrossRef CAS PubMed.
- Z. Wang, J. Ha, Y. H. Kim, W. Bin Im, J. McKittrick and S. P. Ong, Joule, 2018, 2, 914–926 CrossRef CAS.
- F. Wang and X. Liu, Chem. Soc. Rev., 2009, 38, 976–989 RSC.
- Z. Dong, H. Chen, M. Qi, J. Shen, W. Liu, E.-J. Guo, D. Li, Y. Zhang and Z. Wu, Laser Photonics Rev., 2022, 16, 2100454 CrossRef CAS.
- J. Zhang, J. Wang, D. Gao, H. Liu, J. Xie and H. Wanbiao, J. Eur. Ceram. Soc., 2021, 41, 352–359 CrossRef CAS.
- L. Xia, T. Hu, H. Liu, J. Xie, S. U. Asif, F. Xiong and W. Hu, J. Alloys Compd., 2020, 845, 15613 CrossRef.
- D. Gao, J. Xie, J. Wang and W. Hu, J. Mater. Chem. C, 2022, 10, 665–671 RSC.
- K. Lyu, E. Song and Z. Xia, J. Mater. Chem. C, 2022, 10, 9636–9643 RSC.
- M. Chen, Q. Xiong, Z. Liu, K. Qiu and X. Xiao, Ceram. Int., 2020, 46, 12111–12119 CrossRef CAS.
- G. K. Ribeiro, F. S. Vicente, M. I. B. Bernardi and A. Mesquita, J. Alloys Compd., 2016, 688, 497–503 CrossRef CAS.
- X. Zhang, J. Zhang, X. Zhang, M. Wang, H. Zhao, S. Lu and X.-J. Wang, J. Phys. Chem. C, 2007, 111, 18044–18048 CrossRef CAS.
- R. F. Goncalves, A. R. F. Lima, M. J. Godinho, A. P. Moura, J. Espinosa, E. Longo and A. P. A. Marques, Ceram. Int., 2015, 41, 12841–12848 CrossRef CAS.
- J. Kaur, V. Dubey, Y. Parganiha, D. Singh and N. S. Suryanarayana, Res. Chem. Intermed., 2015, 41, 3597–3621 CrossRef CAS.
- D. Liu, G. Li, P. Dang, Q. Zhang, Y. Wei, H. Lian, M. Shang, C. C. Lin and J. Lin, Angew. Chem., Int. Ed., 2021, 60, 14644–14649 CrossRef CAS PubMed.
- H. Arfin, J. Kaur, T. Sheikh, S. Chakraborty and A. Nag, Angew. Chem., Int. Ed., 2020, 59, 11307–11311 CrossRef CAS PubMed.
- H. Dong, C. Zhang, W. Nie, S. Duan, C. N. Saggau, M. Tang, M. Zhu, Y. Zhao, L. Ma and O. G. Schmidt, Angew. Chem., Int. Ed., 2022, 61, e202115875 Search PubMed.
- T. Hu, L. Xia, W. Liu, J. Xie, Z. Jiang, F. Xiong and W. Hu, J. Mater. Chem. C, 2021, 9, 13410–13419 RSC.
- Y. Yamada and Y. Kanemitsu, J. Lumin., 2013, 133, 30–34 CrossRef CAS.
- S. K. Gupta, M. Sahu, P. S. Ghosh, D. Tyagi, M. K. Saxena and R. M. Kadam, Dalton Trans., 2015, 44, 18957–18969 RSC.
- B. P. Singh, A. K. Parchur, R. S. Ningthoujam, A. A. Ansari, P. Singh and S. B. Rai, Dalton Trans., 2014, 43, 4779–4789 RSC.
- Q. Zhang and Z. Xia, RSC Adv., 2014, 4, 53237–53244 RSC.
- Z. Wang, P. Cheng, Y. Liu, Y. Zhou, Q. Zhou and J. Guo, Opt. Mater., 2014, 37, 277–280 CrossRef CAS.
- T. Li, M. Shen, L. Fang, F. Zheng and X. Wu, J. Alloys Compd., 2009, 474, 330–333 CrossRef CAS.
- Z. Wang, Z. Yang, Z. Yang, Q. Wei, Q. Zhou, L. Ma and X. Wang, Inorg. Chem., 2019, 58, 456–461 CrossRef CAS PubMed.
- Z. Wang, T. Li, J. Li, Y. Ye, Q. Zhou, L. Jiang and H. Tang, Chem. Commun., 2022, 58, 4596–4598 RSC.
- X. Xu, J. Zhao, H. Feng, Z. Gao, L. Guan, Y. Yang, F. Wang, D. Wang and X. Li, J. Lumin., 2020, 226, 117356 CrossRef CAS.
- Y. V. Baklanova, L. G. Maksimova, O. A. Lipina, A. P. Tyutyunnik and V. G. Zubkov, J. Lumin., 2020, 224, 117315 CrossRef CAS.
- D. L. Dexter and J. H. Schulman, J. Chem. Phys., 1954, 22, 1063–1070 CrossRef CAS.
- G. Zhu, Z. Li, F. Zhou, M. Gao, C. Wang and S. Xin, J. Lumin., 2019, 211, 76–81 CrossRef CAS.
- G. Zheng, W. Xiao, J. Wu, X. Liu, H. Masai and J. Qiu, Adv. Sci., 2022, 9, 2105713 CrossRef CAS PubMed.
- H. Tang, X. Zhang, L. Cheng, L. Jiang, X. Mi, Q. Liu, J. Xie and Y. Wang, J. Lumin., 2019, 214, 116532 CrossRef CAS.
- M. Zhao, Z. Yang, L. Ning and Z. Xia, Adv. Mater., 2021, 33, 2101428 CrossRef CAS PubMed.
- T. Hu, L. Ning, Y. Gao, J. Qiao, E. Song, Z. Chen, Y. Zhou, J. Wang, M. S. Molokeev, X. Ke, Z. Xia and Q. Zhang, Light: Sci. Appl., 2021, 10, 56 CrossRef CAS PubMed.
- J. Milanez, A. T. de Figueiredo, S. de Lazaro, V. M. Longo, R. Erlo, V. R. Mastelaro, R. W. A. Franco, E. Longo and J. A. Varela, J. Appl. Phys., 2009, 106, 043526 CrossRef.
- G. Blasse, Phys. Lett., 1968, 28A, 444–445 CrossRef.
- B. Yu, Y. Li, Y. Wang and L. Geng, J. Lumin., 2020, 220, 116978 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00843b |
| This journal is © The Royal Society of Chemistry 2022 |